 Nicholas J. Priebe
Nicholas J. Priebe Aaron W. McGee
Aaron W. McGee- 1Section of Neurobiology, School of Biological Sciences, University of Texas at Austin, Austin, TX, USA
- 2Developmental Neuroscience Program, Saban Research Institute, Children’s Hospital of Los Angeles, Department of Pediatrics, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
Genetic programs controlling ontogeny drive many of the essential connectivity patterns within the brain. Yet it is activity, derived from the experience of interacting with the world, that sculpts the precise circuitry of the central nervous system. Such experience-dependent plasticity has been observed throughout the brain but has been most extensively studied in the neocortex. A prime example of this refinement of neural circuitry is found in primary visual cortex (V1), where functional connectivity changes have been observed both during development and in adulthood. The mouse visual system has become a predominant model for investigating the principles that underlie experience-dependent plasticity, given the general conservation of visual neural circuitry across mammals as well as the powerful tools and techniques recently developed for use in rodent. The genetic tractability of mice has permitted the identification of signaling pathways that translate experience-driven activity patterns into changes in circuitry. Further, the accessibility of visual cortex has allowed neural activity to be manipulated with optogenetics and observed with genetically-encoded calcium sensors. Consequently, mouse visual cortex has become one of the dominant platforms to study experience-dependent plasticity.
The establishment of normal primary visual cortex (V1) binocularity and depth perception (stereopsis) in humans depends critically on visual experience, particularly during development (McKee et al., 2003). Disrupting concordant vision between both eyes early in life generates amblyopia, a visual deficiency that cannot be explained by alterations in retinal function of the affected eye (Hubel and Wiesel, 1965; Lepard, 1975; Kiorpes et al., 1998). Amblyopia can arise due to either a difference in depth of focus between the two eyes (anisometropia) or from the eyes not properly moving in parallel (strabismus), and it is thought to occur in 1–5% of the human population (Webber and Wood, 2005). Amblyopia results in a number of deficits in spatial vision, including lower visual acuity and depth perception (Levi et al., 1979; Harwerth and Levi, 1983; McKee et al., 2003). While patching the non-affected eye is current standard of care for improving function of the affected eye, this approach is less effective after adolescence, a time in life characterized by the close of what is termed the critical period for brain circuit plasticity. For each sensory system there exists a developmental period in which experience has a remarkable role in shaping cortical connectivity and beyond which this influence is mostly lost (Simons and Land, 1987; Lendvai et al., 2000; Zhang et al., 2002; de Villers-Sidani et al., 2007; Poo and Isaacson, 2007). Understanding the mechanisms that both govern and drive experience-dependent plasticity during the critical period, as well as those that control the timing of the critical period, could provide therapeutic interventions to improve recovery from amblyopia and other neurodevelopmental disorders.
Conservation of Neural Circuitry for Vision
The functional convergence of right and left eye information occurs in V1; binocular integration within V1 has become the primary platform for studying experience-dependent plasticity. Normally, the information from the two eyes is combined in V1 to generate a three-dimensional representation of the visual world: because the two eyes are horizontally offset they signal distinct perspectives on the visual scene, and those distinct signals are used to compute the distances of objects in the world. The monocular signals from the two retinae leave the eye via the optic tract and cross at the optic chiasm. In mammals, the axons of neurons located in the nasal portion of the retina cross the midline in the optic chiasm and project to subcortical targets on the contralateral side via the optic tract. Neurons from the temporal portion of the retina, in contrast, project to ipsilateral subcortical targets. This specific crossing pattern ensures that animals with frontally-positioned eyes (e.g., cat, ferret, primate) will have signals from both eyes for corresponding regions of the retinae. Importantly, the projections from the contralateral and ipsilateral eyes innervate separate sections of their subcortical targets. For example, retinal ganglion cell axons that innervate the visual thalamus lateral geniculate nucleus (LGN), provide inputs to separate portions of the LGN, and thus the LGN relay cells that project to V1 are monocular. The binocularity observed in V1 is therefore primarily due to a mixing of monocular inputs from the LGN relay cells.
Because experience drives similar changes in both the functional response properties of cortical neurons and the anatomical projections to visual cortex from the thalamus across mammals (Antonini and Stryker, 1993; Antonini et al., 1999), the ease of accessibility and genetics, as well as the compendium of available tools, techniques, and resources for mouse has led to it becoming a standard system to investigate both the governing principles and mechanisms necessary for activity-dependent plasticity. That said, while the mouse has a number of advantages as a model system, it is important to note that in addition to similarities, there also exist large differences between rodents and other mammals that have been studied previously. One of the primary differences is the positioning of the two eyes (Figure 1). In the rodent the eyes are positioned laterally, in contrast to the frontal location of human eyes. This hemi-panoramic vision has consequences for studying cortical binocularity, as the visual world seen by both eyes in front of the mouse is small, covering only the central 50° (Drager, 1978), compared to 135° in man. Therefore, much of the mouse visual system is devoted to monocular—rather than binocular—vision. This difference in eye placement is evident at the optic chiasm: in man approximately 45% of retinal ganglion cell axons project to the ipsilateral LGN, whereas in the mouse only 4% of retinal ganglion cell axons project to the ipsilateral LGN (Dräger, 1974; Godement et al., 1984). Additionally, the anatomical organization of the LGN is distinct in the human and mouse (Figure 1). The human LGN contains multiple segregated eye-specific laminae, whereas the mouse LGN is not laminar but dominated by the contralateral eye with only a small ipsilateral patch (Dräger, 1974). Finally, the functional and anatomical organization of eye-specific signals in primary visual cortex (V1) differs between primates and mice. In primates, V1 is well characterized by a regular columnar organization for ocular dominance (OD) (Hubel and Wiesel, 1977; Adams et al., 2007). V1 neurons across cortical layers share preference for one eye over the other eye, and this ocular preference changes gradually at regular intervals across the surface of cortex. Because of this organization, primate V1 neurons near one another share functional selectivity. In mice, however, no such columnar organization has been observed, and V1 neurons near one another have little functional relationship to each other (Gordon et al., 1996; Antonini et al., 1999).
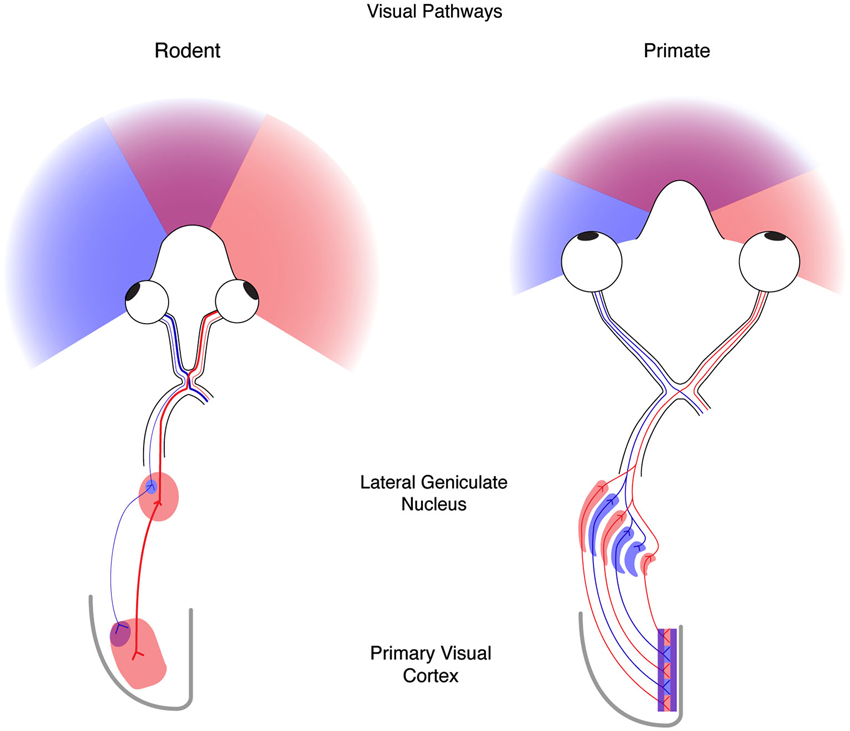
Figure 1. The mouse and human visual systems share basic similarities but differ in complexity. Right, a schematic of the rodent visual system. The eyes in the rodent are positioned laterally resulting in hemi-panoramic vision that includes a narrow central binocular zone (purple) flanked by regions of monocular vison (blue and red). Retinal ganglion cells (RGCs) with receptive fields in the binocular zone from the ipsilateral eye (blue) send a minor projection to a discrete patch in the lateral geniculate nucleus (LGN), whereas the contralateral eye (red) provides the predominant innervation to the LGN. Thalamocortical projections from these two regions converge on the binocular zone (purple) in primary visual cortex (V1). Left, a schematic of the human visual system. Forward facing eyes provide for a more expansive zone of binocular vision. Retinal ganglion cells from the two eyes send similar projections to the LGN that are distributed to eye-specific laminae. Thalamocortical projections similarly converge on V1 (purple) but also maintain evident regions of enrichment termed ocular dominance columns.
Despite these differences in functional architecture across mammals, visual experience sculpts the selectivity of neurons in all mammals examined to date. The effects of activity on neural circuitry are particularly pronounced within the developmental critical period. During the critical period, the functional response properties of neurons, particularly OD, may be manipulated by perturbing the incoming signals from the periphery. Changes resulting from such manipulation are durable, generally persisting through adulthood. Hubel and Wiesel demonstrated OD plasticity in cat by occluding one eye (monocular deprivation, MD) (Wiesel and Hubel, 1963) or disrupting the alignment of the two eyes (strabismus) (Hubel and Wiesel, 1965) during the developmental critical period. After MD, V1 neurons responded strongly to the open eye and weakly to the closed eye; after strabismus, V1 neurons were far less binocular than in normal animals. This decrease in binocularity arises in part from the disruption of normal synaptic integration of binocular inputs by simple cells in visual cortex (Scholl et al., 2013b). In concert with these functional changes, anatomical correlates of experience-dependent plasticity have also been observed. The LGN relay cells that provide inputs to V1 neurons undergo a period of refinement during development in the cat and the primate (Rakic, 1976; Hubel et al., 1977; LeVay et al., 1978; Löwel, 1994). Initially, the right and left eye thalamocortical projections intermix in layer IV, but over the course of development these projections become increasingly patchy and periodic.
These same patterns of activity-dependent changes during the critical period have not only been observed in mouse V1, but in all mammals in which they have been tested (e.g., rabbit: (Van Sluyters and Stewart, 1974), rat: (Maffei et al., 1992) cat: (Wiesel and Hubel, 1963) sheep: (Martin et al., 1979) hamster: (Emerson et al., 1982) macaque: (Hubel et al., 1977) marmoset: (DeBruyn and Casagrande, 1981)). Both MD and strabismus generate changes in the functional response properties of V1 neurons, causing V1 neurons to be more sensitive to the open eye in MD (Wiesel and Hubel, 1963), and less binocular following disruptions of simultaneous patterned activity from the two eyes (Hubel and Wiesel, 1965; Gordon and Stryker, 1996). Monocular deprivation also causes anatomical shifts in the thalamocortical projection, enhancing the growth of the thalamocortical axonal arbors associated with the open eye (Antonini et al., 1999).
Genetic Dissection of OD Plasticity
Many specific genes have been identified as necessary for OD plasticity in mice (Figure 2). The products of such genes are known to operate at different locations within the neuron, from components of the postsynaptic density (Taha and Stryker, 2002; Taha et al., 2002; Sawtell et al., 2003; Ranson et al., 2013) to transcription factors in the nucleus (Pham et al., 1999; Mower et al., 2002) and proteins redistributed to the dendritic compartment that regulate protein stability and turnover (Tagawa et al., 2005; McCurry et al., 2010; Shepherd and Bear, 2011). These genes can be broadly categorized into two groups: (1) necessary pieces of the neural machinery to drive changes in the strength of synaptic connections; and (2) controllers of when and how much plasticity is induced.
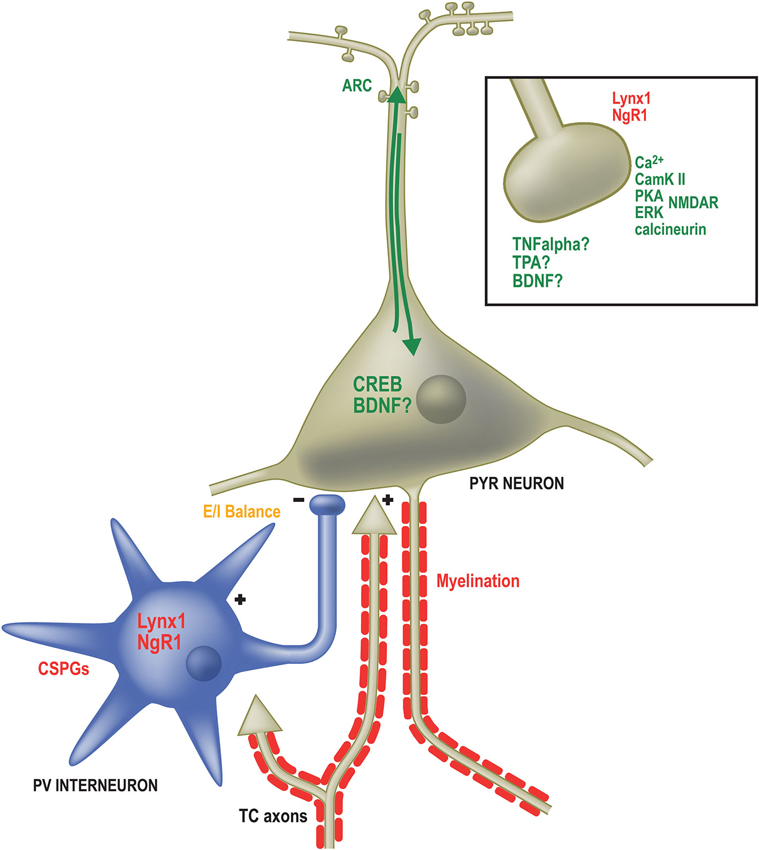
Figure 2. Factors governing the expression and duration of OD plasticity operate in numerous subcellular locations. Genes required for OD plasticity (green text) are present both at sites of synaptic contact as well as the somatodendritic compartment. Calcium signaling (Ca2+) through the NMDA receptor (NMDAR) in excitatory pyramidal (PYR) neurons activates several proteins required for OD plasticity including Calmodulin-dependent protein Kinase 2a (CamKII), Protein Kinase A (PKA), Extracellular signal-Regulated Kinase (ERK) and the phosphatase calcineurin. Likewise, Tumor Necrosis Factor alpha (TNFα), Tissue plasminogen activator (TPA) and Brain-Derived Neurotrophic Factor (BDNF) are all required for OD plasticity and may function at excitatory synapses, such as those on dendritic spines (boxed inset). Proteins restricting OD plasticity (red text) to the critical period may also function at synapses, including Nogo Receptor 1 and Lynx1. Calcium-dependent signaling proteins result in the activation of the activity-dependent transcription factor calcium/cyclic AMP binding element (CREB) as well as the immediate early gene activity-regulated cytoskeletal associated protein (ARC). Several extracellular factors are required to close the critical period and inhibit further OD plasticity. These include Chondroitin Sulfate Proteoglycans (CSPGs) that surround Parvalbumin-positive inhibitory neurons and inhibitors associated with myelin membranes. The proper balance of excitatory and inhibitory neurotransmission (E/I balance) is essential both for opening and potentially closing the critical period (orange text). Multiple approaches (not shown) that affect E/I balance affect OD plasticity.
The genes required for OD plasticity overlap with those that have been implicated in other forms of plasticity, particularly long-term potentiation (LTP) and long-term depression (LTD) (but see Rao et al., 2004). A critical synaptic factor that appears to be the first step in OD plasticity is activity at the N-methyl-D-aspartate (NMDA) receptor, which is required for synaptic plasticity and OD plasticity (Bear et al., 1990; Sawtell et al., 2003). Because the NMDA receptor (NMDAR) is voltage-gated, opening only when the neuron is already depolarized, it signals the coincident activation of incoming synaptic inputs and the activation of the neuron itself. N-methyl-D-aspartate channels are permeable to sodium, potassium and, importantly, allow the influx of calcium. It is this calcium influx that initiates the signaling cascade that eventually leads to changes in synaptic weight and that is required for normal OD plasticity.
Indeed, the calcium influx triggers a number of molecular pathways required for OD plasticity. It has been demonstrated previously that disrupting the interaction between incoming calcium and CaMKII (Taha et al., 2002), cAMP (Beaver et al., 2001; Fischer et al., 2004) or calcineurin (Yang et al., 2005) interferes with OD plasticity during the critical period. These initial calcium-driven signals lead directly or indirectly, through additional kinases such as Extracellular signal-regulated kinase (ERK; Di Cristo et al., 2001), to the activation of activity-dependent regulators of gene expression, including the calcium/cyclic AMP binding element (CREB; Pham et al., 1999, 2001). Thus, perturbing the calcium signaling pathway by weakening or eliminating a step in the cascade diminishes both synaptic plasticity and OD plasticity, providing strong evidence that synaptic modifications are a central and necessary component for the functional changes in selectivity of neurons in V1 during the critical period (Silva, 2003; Taha and Stryker, 2005).
Genetic and Circuit Regulation of the Critical Period
In parallel with the molecular signals necessary to drive plasticity, an additional set of genes governs the timing of the critical period. While the ecological benefit of constraining plasticity to a narrow time window (P20–P32 in mice) is unclear, the conditions required for plasticity are now being uncovered. Opening the critical period requires a discrete maturation of inhibitory cortical circuitry (Levelt and Hübener, 2012). The differentiation of inhibitory neurons expressing the calcium binding protein parvalbumin (PV) precedes the onset of the critical period (Huang et al., 1999), and it has been demonstrated that OD plasticity may be induced earlier in mouse V1 by artificially increasing inhibition (Fagiolini and Hensch, 2000; Iwai et al., 2003). Indeed, increasing levels of Brain-Derived Neurotrophic Factor (BDNF), which may accelerate the maturation of inhibitory circuitry, drive a precious critical period for OD plasticity in mouse V1 (Hanover et al., 1999; Huang et al., 1999). Reducing the amount of GABAA mediated inhibition in cortex, either by deleting GAD 65 (glutamic acid decarboxylase 65 kD), an enzyme required for synthesis of the inhibitory neurotransmitter GABA, or deleting the gene NARP, a pentraxin molecule required for normal excitatory drive onto inhibitory neurons during development, prevents opening of the critical period (Fagiolini and Hensch, 2000; Gu et al., 2013). Another method to delay inhibitory neuron development, and thus the critical period, is to dark-rear animals (Huang et al., 1999). Only once those animals are moved into normal lighting conditions does the critical period open. Thus, the amount of cortical inhibition, particularly inhibition mediated by PV interneurons, appears to be an essential factor in controlling the opening of the critical period for OD plasticity.
Extracellular signals play a critical role at the closure of the critical period. For example, the distribution of perineuronal nets (PNNs), which contain chondroitin sulfate proteoglycans (CSPGs) that are components of the extracellular matrix that inhibit axonal growth, plateaus at the end of the critical period (Pizzorusso et al., 2002). The distribution of myelination in visual cortex also plateaus as the critical period closes (McGee et al., 2005) and intracortical synaptogenesis begins to decline (Morales et al., 2002). Two genes related to these alterations to the extracellular environment of visual cortex are required to close the critical period. Nogo receptor 1 (NgR1) is a neuronal receptor both for CSPGs as well as several inhibitors of neurite outgrowth associated with myelin membranes (McGee and Strittmatter, 2003; Dickendesher et al., 2012). Mice that lack NgR1 continue to display OD critical period plasticity into adulthood (McGee et al., 2005). The cartilage link protein (Crtl1) also plays an essential role in closing the critical period for OD plasticity. CRT1 is a neuronal product that triggers the formation of the PNNs (Carulli et al., 2010). Normally CRT1 is upregulated in V1 as the critical period closes; mice lacking Crtl1 retain OD plasticity into adulthood like the NgR1 mutant mice (Carulli et al., 2010). In addition to these two proteins that interact with the extracellular matrix, a third gene, Lynx1, an important regulator of cholinergic tone that increases at the end of the critical period. Mice lacking Lynx1 continue to display OD plasticity into adulthood, indicating that cholinergic signaling also plays a role in closing the critical period (Morishita et al., 2010).
Upon closure of the critical period, OD plasticity is attenuated but not absent in V1. Partial shifts in OD can still be detected by single-unit recordings, though these require longer periods of MD (e.g., 6+ days in adults vs. 4 days during the critical period) (Hofer et al., 2006). During the critical period, OD plasticity appears to proceed in two stages that overlap considerably: a weakening of responses to the deprived eye followed by a homeostatic strengthening of the non-deprived eye (Frenkel and Bear, 2004; Hofer et al., 2006). This latter homeostatic component of OD plasticity requires Tumor Necrosis Factor alpha (TNFα; Kaneko et al., 2008). Intriguingly, adult plasticity is primarily confined to a slow strengthening of the non-deprived eye by a distinct mechanism that is largely independent of TNFα but requires CaMKII (Ranson et al., 2012).
Reactivating Visual Plasticity in the Adult
One major focus of research into OD plasticity has been to understand how, and whether, plasticity may be enhanced in adults to improve recovery from neurological disorders. The first approach demonstrating that the critical period for visual plasticity could be reopened involved injecting immature astrocytes into adult cat visual cortex (Muller and Best, 1989). Several pharmacologic and environmental manipulations subsequently have been reported to restore developmental OD plasticity to the adult visual system of rats and mice. One approach has been to disrupt the extracellular signals that prevent synaptogenesis and neurite outgrowth. Injection of chondroitinase ABC degrades the CSPGs present in PNNs surrounding PV interneurons. This treatments yields modest OD plasticity (Pizzorusso et al., 2002). How loss of these PNNs affects the function of PV interneurons or impacts cortical circuitry is not yet clear. An alternative approach has been to alter the activity of inhibitory interneurons, and thus the balance between excitation and inhibition in V1. Several strategies have been employed to do this, including direct injection of immature inhibitory neurons (Southwell et al., 2010), dark exposure (He et al., 2006), administration of fluoxetine (Maya Vetencourt et al., 2008), and environmental enrichment (Sale et al., 2007). Direct reduction of overall cortical inhibition by infusing GABAa antagonists also partially restores OD plasticity (Harauzov et al., 2010). The degree to which these approaches may affect excitatory to inhibitory balance is not yet known (Morishita and Hensch, 2008).
Classic genetics, pharmacology, and environmental manipulations have revealed important aspects of both the regulation and mechanisms of OD plasticity in mouse. The combination of sophisticated tools for manipulating and measuring neuronal function in mice is now permitting the dissection of the progression of experience-dependent plasticity through the cortical circuit with greater cell-type specificity and temporal precision. For example, a recent study revealed that OD plasticity requires a decrease in inhibitory drive from a specific inhibitory cell type (Kuhlman et al., 2013). In this study, Kuhlman et al. discover with cell-attached recordings in vivo that an early event following MD during the critical period is a paradoxical increase in neuronal responsiveness of pyramidal (PYR) neurons in layer (L) 2/3 to visual stimulation of either eye. This disinhibition results from a decrease in excitatory drive onto L2/3 PV neurons from L4 and is only observed with MD during the critical period. Interestingly, decreasing the activity specifically of PV neurons with designer receptors exclusively activated by designer drugs (DREADDs) (Armbruster et al., 2007; Ferguson et al., 2010) in concert with MD in adult mice results in visual plasticity indistinguishable from what is observed during the critical period. These experiments are a compelling demonstration of the utility of emerging techniques available for mouse to investigate how plasticity may originate and propagate through cortical circuitry. These available genetic and molecular tools will permit experiments in the mouse that are very difficult, at a minimum, to undertake in other animal model systems.
OD Plasticity and Acuity
Short periods of MD (2–4 days) during the critical period in both mouse and cat shift OD, whereas longer MD (long-term MD, LTMD, 10 or more days) results in poor acuity in the deprived eye (Giffin and Mitchell, 1978; Prusky and Douglas, 2003). LTMD throughout the critical period has been employed as a model of amblyopia in cats and rodents for decades. The effects of LTMD on acuity may stem from a combination of changes in the periphery as well as in cortical circuitry. Lid closure can cause changes in the shape of the eye (Wallman et al., 1978), potentially disrupting optics, thus creating either myopia or hyperopia in one eye (Kiorpes and Wallman, 1995). Unequal refractive error in the eyes can then lead to changes in the cortical circuitry (e.g., Kiorpes et al., 1998). One model is that loss of cortical responsiveness to the deprived eye reduces visual acuity and the subsequent close of the critical period consolidates this visual impairment. Approaches that reactivate developmental visual plasticity, particularly when any anisometropia is corrected, may therefore be expected to improve recovery from LTMD.
Several manipulations in rodents that enhance OD plasticity also improve visual acuity following LTMD (Morishita and Hensch, 2008). Treatment with chondroitinase ABC to block extracellular signals, and environmental enrichment in combination with briefly closing the previously non-deprived eye (reverse suture), restores visual acuity in the deprived eye to normal (Pizzorusso et al., 2006; Sale et al., 2007), as does dark exposure, administration of fluoxetine, and deletion of either the Lynx1 or NgR1 gene (He et al., 2006; Morishita and Hensch, 2008; Morishita et al., 2010; Stephany et al., 2014). This string of correlation has led to the model that OD plasticity and the recovery of acuity in rodents following LTMD are linked. However, genetic dissection of the requirement for NgR1 to close the critical period reveals these facets of visual plasticity are dissociable. While completely abolishing expression of NgR1 permits both OD plasticity and recovery of acuity after LTMD, restricting deletion of NgR1 to PV maintains developmental OD plasticity in the adult but is not sufficient to improve acuity after LTMD (Stephany et al., 2014). The ability to make such specific, targeted changes in protein expression illustrates the power that the mouse model can provide to our understanding of cortical neural circuitry.
Autism and OD Plasticity
It is the hope that understanding the conditions that support critical period plasticity will eventually yield therapeutic approaches for acutely reactivating developmental plasticity, aiding in the correction of amblyopia as well as the spectrum of neurologic disorders, including autism (LeBlanc and Fagiolini, 2011), brain injury (Maurer and Hensch, 2012), and perhaps even prevention of neurodegeneration. In this regard, the sensitivity of the mouse cortex to visual disruption is particularly useful for exploring how genes implicated in syndromic forms of neurodevelopmental disorders may alter the relationship between experience and neural circuit refinement in the developing brain.
For example, OD plasticity has been examined in mouse models of Fragile X syndrome (FXS; Dölen et al., 2007) and Angelman’s syndrome (Yashiro et al., 2009; Sato and Stryker, 2010). Fragile X syndrome is a leading cause of developmental mental impairment and although symptoms vary in severity and expression, characteristic deficits include reduced intellectual abilities, hyperactivity, increased seizure susceptibility, and impaired visuo-spatial processing (Pfeiffer and Huber, 2009). Mice lacking a functional gene for fragile X mental retardation 1 (FMR1) phenocopy some aspects of FXS and have deficits in OD plasticity. Whereas MD during the developmental critical period decreases deprived eye responses in normal (wildtype) mice, FMR1 mutants exhibit a potentiation of open eye responses similar to the visual plasticity resident in the adult visual system (Frenkel and Bear, 2004; Dölen et al., 2007). Whether FMR1 mutant mice are responsive to LTMD is as yet unknown. Interestingly, FMR1 mutant mice also display an imbalance of neocortical excitation and inhibition (Gibson et al., 2008).
Angelman’s syndrome is caused by mutations that disrupt expression of ubiquitin E3 ligase (UBE3A), a gene sensitive to genomic imprinting (Kishino et al., 1997; Matsuura et al., 1997). Symptoms of Angelman’s syndrome include mental impairment, seizures and behavioral abnormalities (Clayton-Smith and Laan, 2003). Ubiquitin E3 ligase mutant mice do not exhibit OD plasticity with short (3-day) MD during the critical period as measured by either visually-evoked potentials or optical imaging of intrinsic signals (Yashiro et al., 2009; Sato and Stryker, 2010), but instead display limited OD plasticity with LTMD both during the critical period and as adults. Ubiquitin E3 ligase mutant mice also possess a deficit in the balance of excitatory and inhibitory cortical neurotransmission (Wallace et al., 2012). This phenotype is reminiscent of the mice mutant for GAD65 (above) in which the maturation of inhibitory cortical circuitry is impaired (Fagiolini and Hensch, 2000). Whether enhancing inhibition rescues visual plasticity in the UBE3A mice, akin to the effects of diazepam on GAD65 mutants, not been reported.
As both FMR1 and UBE3A mutant mice display aberrant E/I balance, these associated deficits in experience-dependent visual plasticity may share a common circuit-level dysfunction. OD plasticity was evaluated in the both FMR1 and UBE3A mutants with visually-evoked potentials (Dölen et al., 2007; Yashiro et al., 2009) and optical imaging of intrinsic signals (Sato and Stryker, 2010), techniques with less temporal and spatial specificity than either single-unit recordings or emerging approaches to study OD plasticity such as cell-attached recordings in vivo and calcium imaging (Kuhlman et al., 2013). As recent studies have begun to dissect with greater precision the interaction between components of the cortical circuitry that drive OD plasticity, this model may continue to improve as a useful framework for understanding if mutations in other genes also linked to syndromic forms of autism spectrum disorders, including neuroligin 3 (NLGN3), Src Homology-3 domain and multiple ankyrin repeat domains protein 3 (SHANK3), and Methyl CpG binding protein 2 (MECP2), interfere with experience-dependent plasticity conserved within neocortex.
Directions of Future Vision Research in Mouse
A compendium of tools are now available for selectively expressing or deleting genes with various drivers of Cre recombinase (CRE), manipulating the activity specific neuronal populations with optogenetics, and measuring the activity of populations of neurons with genetically-encoded calcium indicators. These techniques are essential tools to dissect how experiences shape cortical circuitry. For example, by combining specific CRE drivers (Madisen et al., 2012) with CRE-dependent genetically-encoded calcium indicators, it may be possible to monitor plasticity during MD in specific cortical layers or subsets of interneurons with chronic calcium imaging in vivo. Similar experiments could then be performed on various mutant mice that lack OD plasticity in order to determine how and where plasticity is disrupted by these mutations, as well as within which neuronal populations these genes operate.
Importantly, the utility of the mouse is not restricted to OD plasticity. The mouse may serve as a model system for examining several outstanding questions in vision research. Several characteristics of visual circuitry are conserved between mouse and carnivores, including linear vs. nonlinear spatial summation, contrast-invariant tuning, and selectivity for stimulus parameters such as orientation and spatial frequency (Niell and Stryker, 2008). Thus, although mouse V1 lacks OD columns and possesses relatively poor spatial vision, it may nonetheless serve as a beneficial model system for investigating these properties of visual circuitry and potentially others, such as including disparity tuning (Scholl et al., 2013a) and/or simple and conserved relationships and connectivity between V1 and higher visual areas (Marshel et al., 2011). Overall, despite its small size and relatively simple architecture, the mouse visual system will continue to offer unique advantages for studying how experience shapes neural circuitry, allowing the field to ask—and answer—key questions with far-reaching relevance.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, D. L., Sincich, L. C., and Horton, J. C. (2007). Complete pattern of ocular dominance columns in human primary visual cortex. J. Neurosci. 27, 10391–10403. doi: 10.1523/jneurosci.2923-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Antonini, A., Fagiolini, M., and Stryker, M. P. (1999). Anatomical correlates of functional plasticity in mouse visual cortex. J. Neurosci. 19, 4388–4406.
Antonini, A., and Stryker, M. P. (1993). Development of individual geniculocortical arbors in cat striate cortex and effects of binocular impulse blockade. J. Neurosci. 13, 3549–3573.
Armbruster, B. N., Li, X., Pausch, M. H., Herlitze, S., and Roth, B. L. (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U S A 104, 5163–5168. doi: 10.1073/pnas.0700293104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bear, M. F., Kleinschmidt, A., Gu, Q. A., and Singer, W. (1990). Disruption of experience-dependent synaptic modifications in striate cortex by infusion of an NMDA receptor antagonist. J. Neurosci. 10, 909–925.
Beaver, C. J., Ji, Q., Fischer, Q. S., and Daw, N. W. (2001). Cyclic AMP-dependent protein kinase mediates ocular dominance shifts in cat visual cortex. Nat. Neurosci. 4, 159–163. doi: 10.1038/83985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carulli, D., Pizzorusso, T., Kwok, J. C., Putignano, E., Poli, A., Forostyak, S., et al. (2010). Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain 133, 2331–2347. doi: 10.1093/brain/awq145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clayton-Smith, J., and Laan, L. (2003). Angelman syndrome: a review of the clinical and genetic aspects. J. Med. Genet. 40, 87–95. doi: 10.1136/jmg.40.2.87
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeBruyn, E. J., and Casagrande, V. A. (1981). Demonstration of ocular dominance columns in a New World primate by means of monocular deprivation. Brain Res. 207, 453–458. doi: 10.1016/0006-8993(81)90378-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Villers-Sidani, E., Chang, E. F., Bao, S., and Merzenich, M. M. (2007). Critical period window for spectral tuning defined in the primary auditory cortex (A1) in the rat. J. Neurosci. 27, 180–189. doi: 10.1523/jneurosci.3227-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dickendesher, T. L., Baldwin, K. T., Mironova, Y. A., Koriyama, Y., Raiker, S. J., Askew, K. L., et al. (2012). NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat. Neurosci. 15, 703–712. doi: 10.1038/nn.3070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Cristo, G., Berardi, N., Cancedda, L., Pizzorusso, T., Putignano, E., Ratto, G. M., et al. (2001). Requirement of ERK activation for visual cortical plasticity. Science 292, 2337–2340. doi: 10.1126/science.1059075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dölen, G., Osterweil, E., Rao, B. S., Smith, G. B., Auerbach, B. D., Chattarji, S., et al. (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. doi: 10.1016/j.neuron.2007.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dräger, U. C. (1974). Autoradiography of tritiated proline and fucose transported transneuronally from the eye to the visual cortex in pigmented and albino mice. Brain Res. 82, 284–292. doi: 10.1016/0006-8993(74)90607-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Emerson, V. F., Chalupa, L. M., Thompson, I. D., and Talbot, R. J. (1982). Behavioural, physiological and anatomical consequences of monocular deprivation in the golden hamster (Mesocricetus auratus). Exp. Brain Res. 45, 168–178. doi: 10.1007/bf00235776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fagiolini, M., and Hensch, T. K. (2000). Inhibitory threshold for critical-period activation in primary visual cortex. Nature 404, 183–186. doi: 10.1038/35004582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferguson, S. M., Eskenazi, D., Ishikawa, M., Wanat, M. J., Phillips, P. E., Dong, Y., et al. (2010). Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat. Neurosci. 14, 22–24. doi: 10.1038/nn.2703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischer, Q. S., Beaver, C. J., Yang, Y., Rao, Y., Jakobsdottir, K. B., Storm, D. R., et al. (2004). Requirement for the RIIbeta isoform of PKA, but not calcium-stimulated adenylyl cyclase, in visual cortical plasticity. J. Neurosci. 24, 9049–9058. doi: 10.1523/jneurosci.2409-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frenkel, M. Y., and Bear, M. F. (2004). How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron 44, 917–923. doi: 10.1016/j.neuron.2004.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626. doi: 10.1152/jn.90752.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Giffin, F., and Mitchell, D. E. (1978). The rate of recovery of vision after early monocular deprivation in kittens. J. Physiol. 274, 511–537.
Godement, P., Salaün, J., and Imbert, M. (1984). Prenatal and postnatal development of retinogeniculate and retinocollicular projections in the mouse. J. Comp. Neurol. 230, 552–575. doi: 10.1002/cne.902300406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gordon, J. A., Cioffi, D., Silva, A. J., and Stryker, M. P. (1996). Deficient plasticity in the primary visual cortex of alpha-calcium/calmodulin-dependent protein kinase II mutant mice. Neuron 17, 491–499. doi: 10.1016/s0896-6273(00)80181-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gordon, J. A., and Stryker, M. P. (1996). Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J. Neurosci. 16, 3274–3286.
Gu, Y., Huang, S., Chang, M. C., Worley, P., Kirkwood, A., and Quinlan, E. M. (2013). Obligatory role for the immediate early gene NARP in critical period plasticity. Neuron 79, 335–346. doi: 10.1016/j.neuron.2013.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanover, J. L., Huang, Z. J., Tonegawa, S., and Stryker, M. P. (1999). Brain-derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J. Neurosci. 19, RC40.
Harauzov, A., Spolidoro, M., Di Cristo, G., De Pasquale, R., Cancedda, L., Pizzorusso, T., et al. (2010). Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J. Neurosci. 30, 361–371. doi: 10.1523/JNEUROSCI.2233-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harwerth, R. S., and Levi, D. M. (1983). Psychophysical studies on the binocular processes of amblyopes. Am. J. Optom. Physiol. Opt. 60, 454–463. doi: 10.1097/00006324-198306000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, H. Y., Hodos, W., and Quinlan, E. M. (2006). Visual deprivation reactivates rapid ocular dominance plasticity in adult visual cortex. J. Neurosci. 26, 2951–2955. doi: 10.1523/jneurosci.5554-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hofer, S. B., Mrsic-Flogel, T. D., Bonhoeffer, T., and Hübener, M. (2006). Prior experience enhances plasticity in adult visual cortex. Nat. Neurosci. 9, 127–132. doi: 10.1038/nn1610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., et al. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755. doi: 10.1016/s0092-8674(00)81509-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hubel, D. H., and Wiesel, T. N. (1965). Binocular interactions in striate cortex of kittens reared with artificial squint. J. Neurophysiol. 28, 1041–1059.
Hubel, D. H., and Wiesel, T. N. (1977). Ferrier lecture: functional architecture of macaque visual cortex. Proc. R. Soc. Lond. B Biol. Sci. 198, 1–59. doi: 10.1098/rspb.1977.0085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hubel, D. H., Wiesel, T. N., and LeVay, S. (1977). Plasticity of ocular dominance columns in monkey striate cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 278, 377–409. doi: 10.1098/rstb.1977.0050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iwai, Y., Fagiolini, M., Obata, K., and Hensch, T. K. (2003). Rapid critical period induction by tonic inhibition in visual cortex. J. Neurosci. 23, 6695–6702.
Kaneko, M., Stellwagen, D., Malenka, R. C., and Stryker, M. P. (2008). Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 58, 673–680. doi: 10.1016/j.neuron.2008.04.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kiorpes, L., Kiper, D. C., O’Keefe, L. P., Cavanaugh, J. R., and Movshon, J. A. (1998). Neuronal correlates of amblyopia in the visual cortex of macaque monkeys with experimental strabismus and anisometropia. J. Neurosci. 18, 6411–6424.
Kiorpes, L., and Wallman, J. (1995). Does experimentally-induced amblyopia cause hyperopia in monkeys? Vision Res. 35, 1289–1297. doi: 10.1016/0042-6989(94)00239-i
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73. doi: 10.1038/ng0197-70
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuhlman, S. J., Olivas, N. D., Tring, E., Ikrar, T., Xu, X., and Trachtenberg, J. T. (2013). A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature 501, 543–546. doi: 10.1038/nature12485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
LeBlanc, J. J., and Fagiolini, M. (2011). Autism: a “critical period” disorder? Neural Plast. 2011:921680. doi: 10.1155/2011/921680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lendvai, B., Stern, E. A., Chen, B., and Svoboda, K. (2000). Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404, 876–881. doi: 10.1038/35009107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lepard, C. W. (1975). Comparative changes in the error of refraction between fixing and amblyopic eyes during growth and development. Am. J. Ophthalmol. 80, 485–490.
LeVay, S., Stryker, M. P., and Shatz, C. J. (1978). Ocular dominance columns and their development in layer IV of the cat’s visual cortex: a quantitative study. J. Comp. Neurol. 179, 223–244. doi: 10.1002/cne.901790113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levelt, C. N., and Hübener, M. (2012). Critical-period plasticity in the visual cortex. Annu. Rev. Neurosci. 35, 309–330. doi: 10.1146/annurev-neuro-061010-113813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levi, D. M., Harwerth, R. S., and Manny, R. E. (1979). Suprathreshold spatial frequency detection and binocular interaction in strabismic and anisometropic amblyopia. Invest. Ophthalmol. Vis. Sci. 18, 714–725.
Löwel, S. (1994). Ocular dominance column development: strabismus changes the spacing of adjacent columns in cat visual cortex. J. Neurosci. 14, 7451–7468.
Madisen, L., Mao, T., Koch, H., Zhuo, J. M., Berenyi, A., Fujisawa, S., et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 15, 793–802. doi: 10.1038/nn.3078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maffei, L., Berardi, N., Domenici, L., Parisi, V., and Pizzorusso, T. (1992). Nerve growth factor (NGF) prevents the shift in ocular dominance distribution of visual cortical neurons in monocularly deprived rats. J. Neurosci. 12, 4651–4662.
Marshel, J. H., Garrett, M. E., Nauhaus, I., and Callaway, E. M. (2011). Functional specialization of seven mouse visual cortical areas. Neuron 72, 1040–1054. doi: 10.1016/j.neuron.2011.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin, K. A., Ramachandran, V. S., Rao, V. M., and Whitteridge, D. (1979). Changes in ocular dominance induced in monocularly deprived lambs by stimulation with rotating gratings. Nature 277, 391–393. doi: 10.1038/277391a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J., Jiang, Y. H., Benton, C. S., et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15, 74–77. doi: 10.1038/ng0197-74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maurer, D., and Hensch, T. K. (2012). Amblyopia: background to the special issue on stroke recovery. Dev. Psychobiol. 54, 224–238. doi: 10.1002/dev.21022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maya Vetencourt, J. F., Sale, A., Viegi, A., Baroncelli, L., De Pasquale, R., O’Leary, O. F., et al. (2008). The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 320, 385–388. doi: 10.1126/science.1150516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCurry, C. L., Shepherd, J. D., Tropea, D., Wang, K. H., Bear, M. F., and Sur, M. (2010). Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat. Neurosci. 13, 450–457. doi: 10.1038/nn.2508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McGee, A. W., and Strittmatter, S. M. (2003). The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 26, 193–198. doi: 10.1016/s0166-2236(03)00062-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McGee, A. W., Yang, Y., Fischer, Q. S., Daw, N. W., and Strittmatter, S. M. (2005). Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science 309, 2222–2226. doi: 10.1126/science.1114362
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McKee, S. P., Levi, D. M., and Movshon, J. A. (2003). The pattern of visual deficits in amblyopia. J. Vis. 3, 380–405. doi: 10.1167/3.5.5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morales, B., Choi, S. Y., and Kirkwood, A. (2002). Dark rearing alters the development of GABAergic transmission in visual cortex. J. Neurosci. 22, 8084–8090.
Morishita, H., and Hensch, T. K. (2008). Critical period revisited: impact on vision. Curr. Opin. Neurobiol. 18, 101–107. doi: 10.1016/j.conb.2008.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morishita, H., Miwa, J. M., Heintz, N., and Hensch, T. K. (2010). Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science 330, 1238–1240. doi: 10.1126/science.1195320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mower, A. F., Liao, D. S., Nestler, E. J., Neve, R. L., and Ramoa, A. S. (2002). cAMP/Ca2+ response element-binding protein function is essential for ocular dominance plasticity. J. Neurosci. 22, 2237–2245.
Muller, C. M., and Best, J. (1989). Ocular dominance plasticity in adult cat visual cortex after transplantation of cultured astrocytes. Nature 342, 427–430. doi: 10.1038/342427a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niell, C. M., and Stryker, M. P. (2008). Highly selective receptive fields in mouse visual cortex. J. Neurosci. 28, 7520–7536. doi: 10.1523/JNEUROSCI.0623-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pfeiffer, B. E., and Huber, K. M. (2009). The state of synapses in fragile X syndrome. Neuroscientist 15, 549–567. doi: 10.1177/1073858409333075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pham, T. A., Impey, S., Storm, D. R., and Stryker, M. P. (1999). CRE-mediated gene transcription in neocortical neuronal plasticity during the developmental critical period. Neuron 22, 63–72. doi: 10.1016/s0896-6273(00)80679-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pham, T. A., Rubenstein, J. L., Silva, A. J., Storm, D. R., and Stryker, M. P. (2001). The CRE/CREB pathway is transiently expressed in thalamic circuit development and contributes to refinement of retinogeniculate axons. Neuron 31, 409–420. doi: 10.1016/s0896-6273(01)00381-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J. W., and Maffei, L. (2002). Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298, 1248–1251. doi: 10.1126/science.1072699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pizzorusso, T., Medini, P., Landi, S., Baldini, S., Berardi, N., and Maffei, L. (2006). Structural and functional recovery from early monocular deprivation in adult rats. Proc. Natl. Acad. Sci. U S A 103, 8517–8522. doi: 10.1073/pnas.0602657103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poo, C., and Isaacson, J. S. (2007). An early critical period for long-term plasticity and structural modification of sensory synapses in olfactory cortex. J. Neurosci. 27, 7553–7558. doi: 10.1523/jneurosci.1786-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prusky, G. T., and Douglas, R. M. (2003). Developmental plasticity of mouse visual acuity. Eur. J. Neurosci. 17, 167–173. doi: 10.1046/j.1460-9568.2003.02420.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rakic, P. (1976). Prenatal genesis of connections subserving ocular dominance in the rhesus monkey. Nature 261, 467–471. doi: 10.1038/261467a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ranson, A., Cheetham, C. E., Fox, K., and Sengpiel, F. (2012). Homeostatic plasticity mechanisms are required for juvenile, but not adult, ocular dominance plasticity. Proc. Natl. Acad. Sci. U S A 109, 1311–1316. doi: 10.1073/pnas.1112204109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ranson, A., Sengpiel, F., and Fox, K. (2013). The role of GluA1 in ocular dominance plasticity in the mouse visual cortex. J. Neurosci. 33, 15220–15225. doi: 10.1523/JNEUROSCI.2078-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rao, Y., Fischer, Q. S., Yang, Y., McKnight, G. S., LaRue, A., and Daw, N. W. (2004). Reduced ocular dominance plasticity and long-term potentiation in the developing visual cortex of protein kinase A RII alpha mutant mice. Eur. J. Neurosci. 20, 837–842. doi: 10.1111/j.1460-9568.2004.03499.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sale, A., Maya Vetencourt, J. F., Medini, P., Cenni, M. C., Baroncelli, L., De Pasquale, R., et al. (2007). Environmental enrichment in adulthood promotes amblyopia recovery through a reduction of intracortical inhibition. Nat. Neurosci. 10, 679–681. doi: 10.1038/nn1899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sato, M., and Stryker, M. P. (2010). Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. U S A 107, 5611–5616. doi: 10.1073/pnas.1001281107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sawtell, N. B., Frenkel, M. Y., Philpot, B. D., Nakazawa, K., Tonegawa, S., and Bear, M. F. (2003). NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron 38, 977–985. doi: 10.1016/s0896-6273(03)00323-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scholl, B., Burge, J., and Priebe, N. J. (2013a). Binocular integration and disparity selectivity in mouse primary visual cortex. J. Neurophysiol. 109, 3013–3024. doi: 10.1152/jn.01021.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scholl, B., Tan, A. Y., and Priebe, N. J. (2013b). Strabismus disrupts binocular synaptic integration in primary visual cortex. J. Neurosci. 33, 17108–17122. doi: 10.1523/JNEUROSCI.1831-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shepherd, J. D., and Bear, M. F. (2011). New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 14, 279–284. doi: 10.1038/nn.2708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silva, A. J. (2003). Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. J. Neurobiol. 54, 224–237. doi: 10.1002/neu.10169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simons, D. J., and Land, P. W. (1987). Early experience of tactile stimulation influences organization of somatic sensory cortex. Nature 326, 694–697. doi: 10.1038/326694a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Southwell, D. G., Froemke, R. C., Alvarez-Buylla, A., Stryker, M. P., and Gandhi, S. P. (2010). Cortical plasticity induced by inhibitory neuron transplantation. Science 327, 1145–1148. doi: 10.1126/science.1183962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stephany, C.-E., Chan, L. L. H., Parivash, S. N., Dorton, H. M., Piechowica, M., Qiu, S., et al. (2014). Plasticity of binocularity and visual acuity are differentially limited by nogo receptor. J. Neurosci. 34, 11631–11640. doi: 10.1523/JNEUROSCI.0545-14.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tagawa, Y., Kanold, P. O., Majdan, M., and Shatz, C. J. (2005). Multiple periods of functional ocular dominance plasticity in mouse visual cortex. Nat. Neurosci. 8, 380–388. doi: 10.1038/nn1410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taha, S., Hanover, J. L., Silva, A. J., and Stryker, M. P. (2002). Autophosphorylation of alphaCaMKII is required for ocular dominance plasticity. Neuron 36, 483–491. doi: 10.1016/s0896-6273(02)00966-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taha, S., and Stryker, M. P. (2002). Rapid ocular dominance plasticity requires cortical but not geniculate protein synthesis. Neuron 34, 425–436. doi: 10.1016/s0896-6273(02)00673-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taha, S. A., and Stryker, M. P. (2005). Molecular substrates of plasticity in the developing visual cortex. Prog. Brain Res. 147, 103–114. doi: 10.1016/S0079-6123(04)47008-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Sluyters, R. C., and Stewart, D. L. (1974). Binocular neurons of the rabbit’s visual cortex: effects of monocular sensory deprivation. Exp. Brain Res. 19, 196–204. doi: 10.1007/bf00238534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wallace, M. L., Burette, A. C., Weinberg, R. J., and Philpot, B. D. (2012). Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74, 793–800. doi: 10.1016/j.neuron.2012.03.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wallman, J., Turkel, J., and Trachtman, J. (1978). Extreme myopia produced by modest change in early visual experience. Science 201, 1249–1251. doi: 10.1126/science.694514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Webber, A. L., and Wood, J. (2005). Amblyopia: prevalence, natural history, functional effects and treatment. Clin. Exp. Optom. 88, 365–375. doi: 10.1111/j.1444-0938.2005.tb05102.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wiesel, T. N., and Hubel, D. H. (1963). Single cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017.
Yang, Y., Fischer, Q. S., Zhang, Y., Baumgärtel, K., Mansuy, I. M., and Daw, N. W. (2005). Reversible blockade of experience-dependent plasticity by calcineurin in mouse visual cortex. Nat. Neurosci. 8, 791–796. doi: 10.1038/nn1464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yashiro, K., Riday, T. T., Condon, K. H., Roberts, A. C., Bernardo, D. R., Prakash, R., et al. (2009). Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783. doi: 10.1038/nn.2327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, L. I., Bao, S., and Merzenich, M. M. (2002). Disruption of primary auditory cortex by synchronous auditory inputs during a critical period. Proc. Natl. Acad. Sci. U S A 99, 2309–2314. doi: 10.1073/pnas.261707398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ocular dominance plasticity, visual cortex, binocularity, inhibition, development
Citation: Priebe NJ and McGee AW (2014) Mouse vision as a gateway for understanding how experience shapes neural circuits. Front. Neural Circuits 8:123. doi: 10.3389/fncir.2014.00123
Received: 31 July 2014; Accepted: 18 September 2014;
Published online: 02 October 2014.
Edited by:
Andrea Benucci, RIKEN Brain Science Institute, JapanReviewed by:
Tim Murphy, The University of British Columbia, CanadaDennis Eckmeier, Cold Spring Harbor Laboratory, USA
Prakash Kara, Medical University of South Carolina, USA
Copyright © 2014 Priebe and McGee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron W. McGee, Developmental Neuroscience Program, Saban Research Institute, Children’s Hospital of Los Angeles, Department of Pediatrics, Keck School of Medicine, University of Southern California, 4650 W Sunset Blvd, Los Angeles, CA 90027, USA e-mail:YW1jZ2VlQHVzYy5lZHU=