 Marta S. Vismer
Marta S. Vismer Patrick A. Forcelli
Patrick A. Forcelli Mark D. Skopin
Mark D. Skopin Karen Gale
Karen Gale Mohamad Z. Koubeissi
Mohamad Z. Koubeissi- 1Department of Neurology, The George Washington University, Washington, DC, USA
- 2Department of Pharmacology, Georgetown University, Washington, DC, USA
Understanding neural network behavior is essential to shed light on epileptogenesis and seizure propagation. The interconnectivity and plasticity of mammalian limbic and neocortical brain regions provide the substrate for the hypersynchrony and hyperexcitability associated with seizure activity. Recurrent unprovoked seizures are the hallmark of epilepsy, and limbic epilepsy is the most common type of medically-intractable focal epilepsy in adolescents and adults that necessitates surgical evaluation. In this review, we describe the role and relationships among the piriform (PIRC), perirhinal (PRC), and entorhinal cortex (ERC) in seizure-generation and epilepsy. The inherent function, anatomy, and histological composition of these cortical regions are discussed. In addition, the neurotransmitters, intrinsic and extrinsic connections, and the interaction of these regions are described. Furthermore, we provide evidence based on clinical research and animal models that suggest that these cortical regions may act as key seizure-trigger zones and, even, epileptogenesis.
Introduction
The rhinal and piriform cortices are highly epileptogenic regions that are broadly interconnected with other limbic and cortical areas. The functional architecture of these regions involves oscillatory network activity commonly associated with memory processing. As a result, the unique anatomical organization and connections of these regions can also lead to pathological hypersynchronization resulting in seizures. For example, the piriform cortex (PIRC) is part of the primary olfactory cortex, and odor can be a powerful trigger for memory (Neville and Haberly, 2003). The long-range projections of piriform cortex, including interconnections with amygdala, hippocampus, and rhinal cortex place this structure as a crucial hub in the ability of an odor to “trigger a feeling,” stir an autobiographical memory, or trigger seizure activity throughout the limbic system. The rhinal cortex is divided into the entorhinal (ERC) and perirhinal cortices (PRC), which serve as the major cortical communication relays for the hippocampus. Information flow from the rhinal cortex, through the hippocampus, and back to the rhinal cortex is a perfect example of a reverberatory, Hebbian cell assembly. Under normal conditions, this architecture plays a crucial role in the processing of declarative memory. Under pathological conditions, the rhinal-hippocampal loop tends toward excessive propagation and loop-gain amplification, which is a hallmark of temporal lobe epilepsy (Cataldi et al., 2013). Herein, we review: (1) the normal anatomy and functional connectivity of these structures with an emphasis on the features that make them particularly prone to seizure initiation, (2) evidence for epilepsy-related pathology in these sites, and (3) evidence supporting the notion that these features make these sites critical “trigger zones” for seizure initiation in the temporal lobe.
Piriform Cortex (PIRC)
PIRC Functions
The PIRC is a part of olfactory cortex and its typical activities are olfactory processing and memory coding (Neville and Haberly, 2003). The posterior PIRC is activated in tasks that require categorization of odors (Howard et al., 2009). Closely associated with the PIRC is the endopiriform nucleus (EPN), a large mass of multipolar cells located within the PIRC and has been implicated in seizure generation and propagation (Piredda and Gale, 1985, 1986; Demir et al., 1998, 1999, 2001; Behan and Haberly, 1999; Mraovitch, 2003; Laufs et al., 2011; Vaughan and Jackson, 2014). Although its functions remain largely unknown, the EPN may have a role in integrating gustatory and olfactory sensory information (Sugai et al., 2012). Furthermore, PIRC and EPN may also produce olfactory and gustatory semiology when affected by ictal discharges.
Pirc Anatomy and Histology
The PIRC is a large, cytoarchitectonically homogeneous area that extends over the ventrolateral surface of the forebrain, and constitutes a major component of primary olfactory cortex (Neville and Haberly, 2003). The PIRC consists of three layers: superficial plexiform layer (layer I), cell body layer (layer II), and layer III containing soma and fibers (Sperber et al., 1998). Adjacent to layer III is the well-defined EPN comprised of multipolar cells (Tseng and Haberly, 1989). The EPN is also referred as layer IV of the PIRC since it spans the entire rostro-caudal dimension of the PIRC. The dorsal region of this nucleus consists of densely packed cells while the ventral region has more diffusely arranged cells (Krettek and Price, 1977). The ventro-rostral aspect of the PIRC encompasses a functionally defined and histologically distinct chemoconvulsant trigger zone, located deep within the anterior PIRC, named area tempestas (AT) (Piredda and Gale, 1985, 1986; Ekstrand et al., 2001). Compared to the rest of the PIRC, studies have demonstrated that AT possesses a decreased number of GAT-1 immunoreactivity with decreased density of GABAergic inputs at the axonal initial segments along with decreased concentration of cholecystokinin positive interneurons (Ekstrand et al., 2001). EPN shares a number of properties with the PIRC such as excitatory discharges, hyperexcitability, and high speed of signal propagation. Together these features lead to a high potential for epileptogenecity (Hoffman and Haberly, 1991, 1996; Demir et al., 1998, 2001). Moreover, both the PIRC and EPN are exceedingly vulnerable to excitotoxic injury, and represent one of the earliest sites of neuronal loss after status epileptics (Scholl et al., 2013). As discussed below, the extensive interconnections of the PIRC/EPN with other highly excitable brain regions may allow for the generation and spread of epileptiform activity within PIRC/EPN and throughout the brain (Löscher and Ebert, 1996).
PIRC Connections
The integrative connections between PIRC and several limbic nuclei likely facilitate propagation of ictal activity between these structures (Figure 1). Afferents from the ERC constitute the most dense connections of PIRC (Burwell and Amaral, 1998). In turn, the PIRC and EPN send efferent projections to the ERC (Luskin and Price, 1983a; Agster and Burwell, 2009). There is also a direct connection from the PIRC to the amygdala (Veening, 1978; Wakefield, 1980) and a diffuse polysynaptic connection from the amygdala to the PIRC via the EPN (Krettek and Price, 1977), which may have significant implications for temporal lobe epilepsy (TLE). There are also pathways from the PIRC to the subiculum (Krettek and Price, 1977; Luskin and Price, 1983a) and to areas 35 and 36 of PRC (Agster and Burwell, 2009).
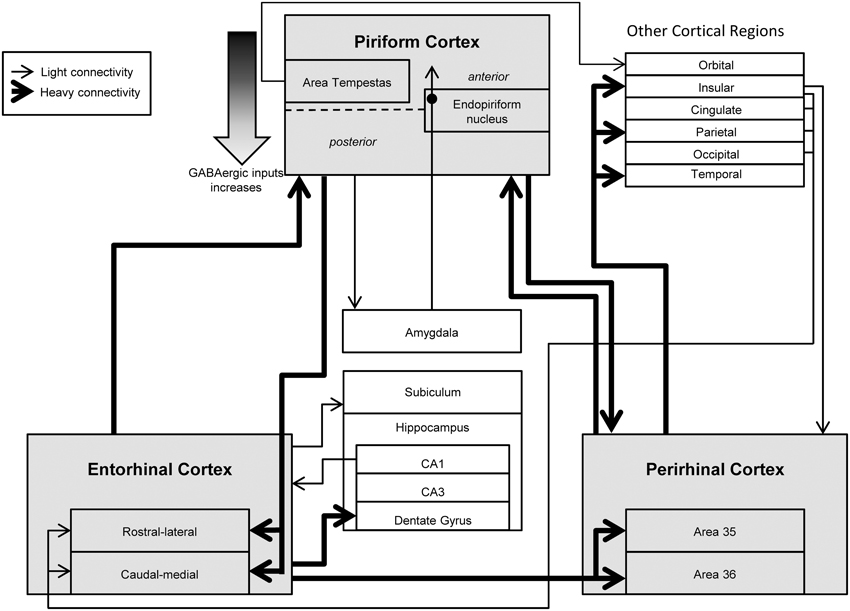
Figure 1. Overview of the functional connectivity of piriform, perirhinal, and entorhinal cortices. Many studies have investigated the connectivity of the piriform (PIRC), perirhinal (PRC), and entorhinal cortices (ERC) along with their relationship with other cortical and limbic structures. These regions are highly interconnected with each other and mediate bidirectional information from higher processing regions. Abnormal activity from these regions is hypothesized to cause hyperexcitability and excessive propagation associated with temporal lobe epilepsy along with perceptual disturbances such as hallucinations and auras.
There are significant differences in the connectivity of the anterior and posterior subregions of the PIRC (Haberly, 2001; Schwabe et al., 2004), suggesting functional specialization within the PIRC (Illig, 2005), which may be of particular importance to the functional topography related to seizure initiation. For example, local circuit GABAergic transmission appears to be stronger in the posterior sub-region of the PIRC and become increasingly less prominent more anteriorly (Löscher and Ebert, 1996; Haberly, 2001), Interestingly, AT, crucial to seizure generation (Piredda and Gale, 1985), is located in the anterior aspect of the PIRC, and has low density of GABAergic inputs to the axon initial segments (Ekstrand et al., 2001). Unlike the rest of the PIRC that receives olfactory input from mitral cells only, the AT also receives direct projections from tufted cells in the olfactory bulb (Ekstrand et al., 2001) and provides most input from the PIRC to ventrolateral orbital cortex. These connections are both directly and via di- and trisynaptic connections incorporating pre-endopiriform nucleus and submedial and mediodorsal thalamus (Ekstrand et al., 2001; Illig, 2005). This interconnectivity may be important for seizure propagation and generalization.
Projections from the PIRC and EPN are very diffuse. Single neurons within posterior PIRC send axonal arbors to the insula, PRC, ERC, amygdala, and olfactory tubercule, with a tendency to have no overlap with the arborization patches created by neighboring neurons (Johnson et al., 2000). Projections of the EPN parallel those of the PIRC, targeting ERC, PRC, insular, orbital, and other neocortical structures (Behan and Haberly, 1999). The EPN is the major route for information transfer along the rostrocaudal axis of PIRC/EPN and may facilitate seizure propagation longitudinally within this structure (Behan and Haberly, 1999). Moreover, EPN may play an especially important role in the spread of activity from EPN and PIRC to other cortical regions, because the EPN has a more diffuse topographic and laminar organization of axon terminals than does PIRC (Luskin and Price, 1983a,b; Behan and Haberly, 1999). The extensive interconnectivity of EPN and PIRC with other epileptogenic sites within the temporal lobe (e.g., ERC, PRC) may provide a loop for positive feedback of activity and subsequent generalization of epileptic discharges.
Perirhinal Cortex (PRC)
PRC Functions
The PRC is involved in complex memory functions (Rosen et al., 1992; Zola-Morgan et al., 1994; Wiig et al., 1996), such as object recognition, sensory representation, and spatial orientation (Murray and Richmond, 2001). Damage to PRC results in deficits in complex visual discrimination (Ryan et al., 2012), object recognition memory (Meunier et al., 1993; Zola-Morgan et al., 1994), and attention to visual stimuli (Bucci and Burwell, 2004). The PRC represents a component of the ventral visual stream, with interconnections to secondary visual regions being more robust than those with primary visual cortex (Agster and Burwell, 2009). This may support its involvement in higher visual information processing and memory (Barbeau et al., 2005) and may account for complex visual hallucinations in some temporal lobe seizures. Induction of theta activity in the PRC network results in the recall of vivid memories (Barbeau et al., 2005) and therefore, can represent a substrate for visual auras that accompany some TLE. PRC damage may also account for the feeling of déjà vu reported by a subset of TLE patients during the aura of their seizures (Martin et al., 2012). Extensive damage to PRC in epilepsy may explain the cognitive decline, including primarily memory deficits (Shatskikh et al., 2006) that occur in some patients with chronic TLE.
PRC Anatomy and Histology
The human PRC is located along the collateral sulcus in the ventromedial portion of the temporal lobe. Laterally, it borders the ERC with which it comprises the parahippocampal gyrus (Insausti et al., 1987). On the basis of cytoarchitectural, immunohistochemical, and physiological criteria, the PRC is further subdivided into two parallel cortical bands, areas 35 medially, and 36 laterally (Insausti et al., 1987; De Curtis and Paré, 2004; Saleem et al., 2007).
PRC Connections
Afferents
Both Areas 35 and 36 of PRC receive substantial input from ERC (Figure 1). In addition to this input, PRC receives projections from ventral temporal association areas, PIRC, and other limbic-cortical areas (e.g., insula). The relative abundance and distribution of these inputs varies as a function of subregions of PRC (i.e., Area 35 vs. 36) (Burwell and Amaral, 1998).
Efferents
The PRC projects back to many of the same regions it receives input (Luskin and Price, 1983a,b; Burwell and Amaral, 1998; Agster and Burwell, 2009). For example, the PIRC, insula, and subcortical limbic structures all have reciprocal connections with the PRC. Just as the pattern of afferents differs between Areas 35 and 36, the patterns of efferents also differ (Agster and Burwell, 2009). The PRC may represent a route by which seizure activity can propagate to distal cortex due to the heavy connections with the insula and sensory domains within frontal, temporal and parietal cortices. These projections may also help explain sensory symptoms associated with TLE including gustatory or autonomic symptoms related to insular cortex activation. There is a high degree of specialization within various subregions of the PRC (e.g., rostral PRC projects heavily to somatosensory cortex, while caudal PRC projects heavily to visual cortex) (Agster and Burwell, 2009). A relatively underexplored question is the degree to which topographic activation of the PRC may be associated with different patterns of propagation and/or symptoms associated with TLE.
The PRC is a “gate-keeping” area that mediates bidirectional information transfer between temporal neocortex and ERC (Pelletier et al., 2004). It is worth noting that while most communication takes place through a slow, stepwise progression through cortical areas, a minor portion of axons bypass the PRC (De Curtis and Paré, 2004) directly activating the lateral ERC. Moreover, some ERC axons extend to area 35 and the temporal neocortex. These projections may allow for spread of pathological excitatory activity by circumventing the PRC gate. In conditions such as chronic epilepsy, the reciprocal interconnectivity of PRC with other hyperexcitable structures positions the PRC as a relay region within the limbic network for the generation and propagation of epileptiform activity.
Entorhinal Cortex (ERC)
ERC Functions
The ERC, located in the anterior parahippocampal gyrus, serves as the major interface between the hippocampus and sensory cortices (Insausti et al., 1987). Its close connection with cortical regions involved in the processing of visual and spatial information suggests that the ERC is an important substrate for visuo-spatial functioning (Burwell and Amaral, 1998; Killian et al., 2012). Moreover, hippocampal memory function depends on an intact ERC (Lu et al., 2013) and appears to be well-conserved across species. For example, human neuroimaging studies have found correlations between parahippocampal activity and spatial working memory (Courtney et al., 1996), and with learning and recall of topographic information (Aguirre et al., 1996). In monkeys, single neurons within the ERC fire in response to visual input from specific locations in the environment, forming memories of spatial environments (Rolls and O'Mara, 1995). Moreover, “grid neurons” of the ERC have been well-described in rodents and primates (Sargolini et al., 2006; Killian et al., 2012; Navratilova et al., 2012; Lu et al., 2013). As with PIRC and PRC, the extensive connections between ERC and other limbic brain regions suggest it is a major relay for seizure activity.
ERC Anatomy and Histology
In humans, the ERC, consisting of Brodmann areas 28 and 34, is situated bilaterally between the sulcus semiannularis and sulcus rhinalis in the rostral parahippocampal gyrus (Leichnetz and Astruc, 1976; Amaral et al., 1987). It is an allocortical structure, consisting of six layers, with a striking paucity of cells in layer IV (lamina dessicans), which separates the outer cell-rich layers (V and VI) from the inner cell-rich layers (II, and III) (Amaral et al., 1987; Insausti et al., 1995). Layer III may be particularly important for seizure propagation as it consists of pyramidal projection neurons that target the hippocampus and other regions. For a detailed review of ERC anatomy and cytoarchitectonics, see Insausti (1993).
Like the PRC, the ERC is divided in two major sub-fields, the rostral-lateral (LERC) and the caudal-medial ERC (MERC) (Dolorfo and Amaral, 1998; Uva et al., 2004). These two divisions differ in their histology, morphology and physiology. The LERC has a number of morphological inhomogeneities and the neurons are organized in patches surrounded by thick radially-oriented bundles of fibers. Conversely, the MERC is more reminiscent of the neocortex, with a distinctly laminar appearance and columnar organization of neurons and fibers (Amaral et al., 1987). As will be discussed in the later sections, this laminar structure may have a special role in signal amplification and epileptogenesis.
ERC Connections
Afferents
A major input to both LERC and MERC originates within PIRC and accounts for about one-third of the input to these regions (Figure 1). Other inputs originates in temporal and frontal cortices. Less abundant sources of input include insular, cingulate, parietal, and occipital cortex, with the relative abundance of these inputs varying between the MERC and LERC.
Efferents
In addition to its rostro-caudal organization, the ERC is also organized into bands that project to different septo-temporal levels of the dentate gyrus (Dolorfo and Amaral, 1998; Agster and Burwell, 2009). As other structures discussed in this review, the distribution of efferent projections differs between subregions of ERC. PIRC is the major recipient of input from both subdivisions of ERC of projections to inferotemporal, retrosplenial, posterior parietal, frontal, and insular cortices originating in medial and lateral domains. Similar to PIRC and PRC, many of the connections from ERC are reciprocated (Biella et al., 2002a,b). This ability for bidirectional information transfer, while presumably important for normal information processing, is yet another example of a cortico-cortical loop that can amplify and propagate seizure discharges.
The hippocampus is a key area to seizure propagation and has a series of parallel ERC-hippocampus-ERC loops that underlie memory functions (Andersen et al., 1971; Acsády and Káli, 2007; Kibler and Durand, 2011). In the TLE, one of the most common types of medically intractable epilepsy (Engel et al., 2003, 2012), ictal synchronization within hippocampus is a pre-requisite to spread of activity to other cortical areas (Bartolomei et al., 2001; Pallud et al., 2008; Umeoka et al., 2012) and the hippcampal-ERC communication may serve as a critical substrate for propagation of epileptic activity of ERC origin to hippocampus.
Intrinsic Circuitry
Despite histological data that strongly suggest the presence of substantial fiber tracts between the ERC and PRC, the functional strength of connections in the healthy brain is debatable (Biella et al., 2002a). Previously, the PRC was considered to be a relay nucleus for the bidirectional communication between the neocortex and the ERC. However, more recent data suggests the PRC is an important center for feed-forward inhibition of the ERC (Finch et al., 1988; Biella et al., 2002a; De Curtis and Paré, 2004; Pelletier et al., 2004). Extracellular recordings and optical imaging studies demonstrated that PRC–ERC and ERC–PRC interactions produce only limited neuronal activation with a relatively low probability for the transfer of neocortical inputs to ERC. This highly restricted and controlled information transfer is essential in a recurrent circuit to prevent loop-gain amplification. Therefore, in the healthy brain, the excitatory activity of the ERC is controlled by inhibitory interactions in the network that are activated by extrinsic projections. The PRC may act as a “gate” for spread of excitatory activity between the ERC and neocortex. While conditions of normal hyperexcitability can facilitate the propagation of olfactory-driven activity from the ERC to the PRC (Federico and MacVicar, 1996; Kelly and McIntyre, 1996), malfunction of intrinsic control mechanisms can result in hyperexcitability of the ERC and, consequently, epileptic seizures.
Summary of Functional Anatomy
Together, PIRC/EPN, PRC, and ERC form a unique and widely connected network that is ideally organized to engage the hippocampus (and the rest of the limbic system) in rhythmic activity. While this rhythmic activity is a part of normal brain function, malfunctions in the circuitry can easily lead to pathological synchronization and seizure spread. Seizures do not propagate randomly through the brain, but rather utilize circuits that normally support highly controlled recurrent activity. Through this network, activity can spread to distal cortical and subcortical regions along pathways that are also used in normal motor system (Haberly and Bower, 1989; Wilson and Stevenson, 2003; McIntyre and Gilby, 2006; Howard et al., 2009).
In the sections above, we have described the anatomical features that position PIRC/EPN, PRC and ERC as sites through which seizure discharges can amplify and propagate. While these regions are capable of autonomous generation of ictal discharges, the importance of their interconnections is underscored by data demonstrating a significant reduction in ictal activity when the connections between these regions are disrupted (De Guzman et al., 2004). These observations imply that the communication between the ictogenic structures is a key component for amplification and synchronization of ictal bursts and may have particular relevance to understanding the effects of temporal lobe resection for the treatment of epilepsy. Even “selective” amygdalohippocampectomy (aside from radiofrequency lesions) involve resection of the anterior aspects of the middle and inferior temporal gyrus, disrupting many connections intrinsic to the temporal lobe in addition to removing temporal cortical tissue, amygdala, and hippocampus.
Below we discuss conditions that may lead to failures in the intrinsic gating properties of these structures. These pathologies and subsequent aberrant gating are hypothesized to underlie the emergence of spontaneous seizures in chronic epilepsy.
Morphological and Histological Changes due to Epilepsy
The transition from normal network activity to hypersynchronous activity characteristic of a seizure may indicate a subtle change in the interconnections or functions within PIRC, PRC, ERC, or the broader limbic network. A common histopathological finding in TLE is neuronal loss and inflammation. It is possible to speculate that these changes may result in the network perturbations necessary for epilepsy or the comorbid memory and cognitive deficits. However, the degree to which these histopathological findings are a cause or an effect of recurrent seizures remains a matter of debate. In the section below, we will discuss evidence for neuropathology in PIRC, PRC, and ERC in the context of epilepsy and epileptogenic insults.
Neuroimaging
In patients with TLE, volumetric analyses reveal volume loss of the anterior temporal lobe on the epileptic side (Lencz et al., 1992; Jutila et al., 2001; Bonilha et al., 2003; Martin et al., 2012), bilateral reduction in temporal neocortical gray matter, and reduction in temporal white matter (Lee et al., 1998). Changes in volume of temporal cortex appear to be specific to TLE, and are not seen in extratemporal epilepsy (Jutila et al., 2001; Bonilha et al., 2003). Analysis of substructures within the TLE has also reveal volumetric changes, including in the hippocampus, PRC, and ERC (Bonilha et al., 2003). In one study, 52% of patients with medically intractable TLE have reduction in ipsilateral ERC volume (Jutila et al., 2001). Moreover, the extent of atrophy in ERC correlates with ipsilateral hippocampal volume and the duration of epilepsy (Jutila et al., 2001).
While it remains controversial if damage to piriform and rhinal structures leads to epilepsy, or rather if the recurrent seizures characteristic of epilepsy lead to damage, some patients have atrophic tissue in the temporal lobe at what appears to be the site of seizure origin. For example, in some patients with TLE, ERC appears to display epileptiform activity before the hippocampus, but only in patients with atrophy of ERC (Bartolomei et al., 2005). Interestingly, the degree of atrophy in ERC correlates with hypersynchronization between ERC and hippocampus. These data suggest that the atrophic ERC may be the epicenter of epileptogenic activity with dissemination to other limbic structures via an elaborate network of neuronal connections of the ERC.
Histopathology
Morphological changes, such as neuronal loss and reactive gliosis, are prominent both in hippocampus and extrahippocampal areas (e.g., ERC, PRC) in patients with TLE (Du et al., 1993; Jung et al., 2009; Scholl et al., 2013). Animal models parallel these findings with ERC, PIRC, and PRC showing some of the most severe damage as soon as 72 h after status epilepticus (Scholl et al., 2013). Even sublayers of these structures show differential vulnerability. For example, there is a profound and relatively selective loss of neurons within layer III of the ERC in patients with TLE (Du et al., 1993). In animal models, loss of neurons within layer III of ERC can occur as soon as 24 h after injury, and progresses with time (Drexel et al., 2012). Morphological changes in the CA1, CA3, and the hilus of the dentate gyrus showed a continuous degeneration of neurons up to 7 days following experimental status epilepticus (Langer et al., 2011). Synaptic reorganization and atrophy of layer III of the ERC produces a robust change in evoked and spontaneous activity in parts of the ERC and hippocampus that result in increased excitability and susceptibility to reentrant activation of the hippocampal–ERC loop via disinhibition of local networks in CA1 (Empson and Heinemann, 1995).
Interestingly, neuronal loss in layer III shows a marked sparing of parvalbumin-expressing interneurons within the medial ERC (Du et al., 1995) with predominate loss of glutamatergic neurons (Drexel et al., 2012) suggesting that a simple depletion of inhibition is insufficient to account for the hyperexcitable phenotype. However, others have described depletions of GABAergic neurons and a reduction in the number of GABAergic synapses after status epilepticus (Kumar and Buckmaster, 2006) suggesting that loss of inhibitory tone may contribute to a hyperexcitable state permissive to seizures. In addition, low-threshold-spiking (LTS) and fast-spiking (FS) interneurons within the medial entorhinal cortex creates a recurrent inhibitory network which is central to stability in grid formation (Couey et al., 2013).
In addition to the cell loss associated with excitotoxic seizure injury (i.e., status epilepticus), the activation and proliferation of microglia may contribute to secondary injury. For example, within the rodent hippocampus, prolonged seizure activity can trigger the proliferation and differentiation of neural progenitor cells (Parent et al., 1997, 2002), resulting in the aberrant and ectopic neurogenesis in the hilus of the hippocampus. However, in extrahippocampal sites (i.e., PIRC, ERC) cellular proliferation and differentiation manifests as increased numbers of astrocytes and oligodendrocytes (Jung et al., 2009). The production of chemokines by prolonged seizures may be of particular importance for triggering aberrant neurogenesis in the hippocampus, reactive gliosis, and astrocytosis in the temporal cortex (Jung et al., 2009).
As soon as 5 h after generalized seizures, a marked elevation in the expression of IL-1β can be detected in the endothelial cells of the blood vessels in the PIRC (Mraovitch, 2003). IL-1β is a modulator of inflammatory response and apoptosis (Darlington et al., 1986; Hartung et al., 1989). The effect of IL-1β and TNF-α on hyperexcitability and seizure development have been shown in several studies (Vezzani et al., 2011). However, some evidence suggest that TNF-α may decrease glutamate-mediated excitotoxicity via NF-kappaB-dependent up regulation of K2.2 channels (Dolga et al., 2008). In the PRC, astrocytes and microglia also express IL-1β and display vascular endothelial expression of TNF-α, another vital component of inflammatory response (Darlington et al., 1986). Within 6 weeks after generalized seizures, histopathological changes such as reactive astrocytosis and irregular microvascular proliferation appear in the allocortex including the PIRC, PRC, and ERC. Perivascular infiltration of C3 positive leukocytes has also been observed (Jamali et al., 2006). These changes are suggestive of inflammation and remodeling and provide a link between the epilepsy-induced inflammatory vascular processes, gliosis, and subsequent allocortical atrophy (Mraovitch, 2003).
Reactive astrocytes and microglia are other markers of inflammatory process and appear in the areas of neuronal deterioration as early as 24 h after the induction of kainate-induced animal model of status epilepticus. Inflammatory cells continue to rise, becoming prominent after 8 days with their number peaking at 30 days and subsequently declining almost to the baseline by 3 month after status; the initial rise in these cells mirrors the time course of spontaneous seizures in this model (Drexel et al., 2012).
Markers of neuronal remodeling and synaptic plasticity, such as polysialylated neural cell adhesion molecule (PSA-NCAM), are increased in ERC and hippocampus from human patients with TLE (Mikkonen et al., 1998) and in rodent models of TLE (Pekcec et al., 2008; Rossi et al., 2013). Changes in neuromodulatory systems and co-transmitters also occur in TLE with striking increases in NPY and somatostatin immunoreactivity and dysregulation of NPY receptor expression in PIRC and ERC (Kato et al., 1983; Vezzani et al., 1996; Kang et al., 2000; Jamali et al., 2006). In TLE and its models, it is difficult to determine if changes in plasticity represents a compensatory or decompensatory response to initial injury. For a more detailed review of the role of inflammation in TLE, see Vezzani et al. (2011).
In summary, epileptic seizures are associated with significant alterations in the histology of the PRC, PIRC, and ERC. Cell loss, inflammation, and altered gene expression may shift the balance between inhibition and excitation to create a pro-epileptic environment. Because these structures gate information transfer within the temporal lobe and extratemporal targets, atrophy in these regions may facilitate seizure propagation throughout the forebrain.
Epileptogenecity
Piriform Cortex—Area Tempestas
The deep anterior piriform cortex contains a highly circumscribed and functionally-defined chemoconvulsant trigger zone called area tempestas (AT). From this site, unilateral microinjection of picomole amounts of GABA receptor antagonists or glutamate receptor agonists can trigger limbic motor seizures in rats and non-human primates (Piredda and Gale, 1985, 1986). More recently, neuroimaging approaches have found focal fMRI activation and reduced GABAA receptor binding in the anterior piriform cortex of human patients with focal epilepsy (Laufs et al., 2011). Not only is activation of AT sufficient to trigger complex partial seizures in animals, it also appears to be necessary for the response to systemically applied chemoconvulsants (i.e., bicuculline) (Piredda and Gale, 1985, 1986) and nerve agents (i.e., soman) (Myhrer et al., 2006, 2008). Enhancement of GABA-mediated inhibition within this structure markedly attenuates seizure activity evoked by systemic chemoconvulsant administration (Piredda and Gale, 1985, 1986). In adult animals, AT is generally accepted as a zone of high epileptogenicity, while in immature rats, there was no difference between the rate of kindling from AT and PIRC (Sperber et al., 1998). These findings suggest that developmental changes in excitability within these subregions may modulate their epileptogenicity.
Some studies suggest a crucial role for the deep layers of the PIRC and/or the EPN in the induction of epileptiform bursting in slice preparations (Hoffman and Haberly, 1991, 1993; Doherty et al., 2000). While in vitro studies have demonstrated that PIRC/EPN slices are highly excitable across the entire anteroposterior axis, in vivo studies demonstrate that the anterior PIRC/EPN has the highest susceptibility to seizure initiation (Piredda and Gale, 1985). The differences between these findings may be explained by the nature of the slice model utilized in which the entire slice was disinhibited by reduction in extracellular chloride (Demir et al., 1999).
Seizure propagation from AT requires both the posterior PIRC and PRC; pharmacological inhibition of these structures significantly attenuates the severity of seizures evoked from AT (Halonen et al., 1994). Likewise, blockade of excitatory neurotransmission within PRC suppressed amygdala kindled seizures (Holmes et al., 1992). Together, convergent evidence from experiments in acute slices, pharmacological manipulation of the rat brain in vivo, and human neuroimaging provide evidence that PIRC, and in particular, AT, is a crucial site for seizure initiation, and PRC is a key relay for seizure propagation within the limbic network. In the sections below we will outline additional evidence from pharmacological and kindling studies supporting these conclusions.
Kindling
Animal epilepsy model studies using kindling suggest that PIRC and PRC have particularly low thresholds for epileptogenesis; kindling from these structures occurs at a faster rate (i.e., with fewer stimulations necessary to trigger Stage 5 seizures) as compared to other forebrain structures (McIntyre and Plant, 1993; McIntyre et al., 1999). Interestingly, the faster rate of kindling from PIRC/PRC was seen in both kindling-prone and kindling-resistant rat strains (McIntyre et al., 1999). Within PIRC, cells in Layer III have the lowest afterdischarge threshold, but electrode placement within the EPN results in the fastest rate of kindling (Löscher and Ebert, 1996). PIRC lesions have only mild effects on kindling: Lesions to the anterior PIRC have no effects, lesions to the posterior PIRC increase afterdischarge thresholds but do not alter kindling rate, and lesions to central PIRC increase the number of Stage 3 seizures before animals fully kindled (Wahnschaffe et al., 1993; Schwabe et al., 2000). Low frequency stimulation of the central PIRC has a similar effect of hindering generalization of focal seizures (Yang et al., 2006) and decreasing the duration and stage of seizures in fully kindled animals. Thus, the PIRC is readily capable of kindling and plays an important role in development and generalization of seizures of different origins, which may have tremendous therapeutic potential. The PIRC may be the site of interictal discharge origination within the limbic circuitry of kindled animals—regardless of the site of kindling (Kairiss et al., 1984). Within PRC, kindling not only occurs with the fewest stimulations of any site within the forebrain, but the latency from stimulation to behavioral seizures is very brief. This brief latency, along with extensive projections from PRC to frontal cortex led McIntyre and Kelly to propose that PRC is a crucial relay that clonic motor convulsions are triggered after limbic seizures (Kelly and McIntyre, 1996). Supporting this hypothesis, damage to perirhinal cortex prevents motor seizures evoked by hippocampal kindling (Kelly and McIntyre, 1996). While both the PIRC and PRC are highly susceptible to generating epileptiform discharges, the PRC is generally involved in generating seizures in naïve animals, though the role of epilepsy trigger zone shifts to PIRC as a result of the process of epileptogenesis (McIntyre and Plant, 1993).
Epileptogenesis relies heavily on synaptic plasticity. For example, tetanic stimulation of the white matter of the PRC and Schaffer collaterals of the hippocampus suggesting reciprocal long-term potentiation between the hyperexcitable structures pertinent in epilepsy (Supcun et al., 2012). Short and long-term plasticity also implicate changes at the receptor level and refer to the alteration in both number and composition of certain receptor groups leading to decreased inhibition of some brain structures and increasing their propensity to produce excitatory bursts. An increase in the density of α-1 adrenergic receptors (Gundlach et al., 1995), modifications in the composition of NMDA and AMPA glutamate receptors (Supcun et al., 2012), and changes in morphology and number of gluzinergic (zinc-containing glutaminergic) terminals in PRC and PIRC (Galvis-Alonso et al., 2004) occur as part of the changes of synaptic plasticity elicited by induction of long term potentiation. These changes are believed to play a role in epilepsy by facilitation of further seizures. Garcia-Cairasco and colleagues used acoustic kindling to observe long-term morphological changes (Garcia-Cairasco et al., 1996). In the groups that underwent long-term kindling and 60 days of recovery, staining revealed mossy fiber sprouting in the ventral hippocampus and a significant change in the optical density of PRC in susceptible animals as compared to resistant ones suggesting that permanent structural changes are present soon after the initial seizure. Changes on the synaptic and cellular level collectively create an imbalance between excitation and inhibition in PRC and PIRC which are predisposed to excitation and have the infrastructure for synchronization of neuronal activity that amplifies and propagates excitatory signals throughout the neocortex resulting in TLE with its other comorbidities.
Hippocampal kindling also induces long-term changes in the dynamics within the amygdala-piriform-perirhinal slice. After hippocampal kindling, spontaneous discharges in the slice originated within PIRC, whereas in slices from control animals, discharges originated within PRC. Unlike amygdala kindling, which produced strictly unilateral changes in excitability, kindling of dorsal hippocampus generated bilateral alterations. Even partial hippocampal kindling was linked to presence of spontaneous spikes in the PRC under hyperexcitable conditions (McIntyre et al., 2000). These findings show a change in the region of seizure onset are consistent with in vivo observations that early in epilepsy seizures began with focal onset, while later seizures had more diffuse onset (including hippocampus, amygdala, piriform and entorhinal cortices) (Bertram, 1997). This provides evidence for the hypothesis that epileptogenesis is an active process, initiated in hyperexcitable trigger zones that progressively involves more structures with wider network distribution over time and emphasis should be placed on early therapeutic intervention. As discussed in the section on histopathology, this process is accompanied by structural and functional changes in the seizure network, including loss of functional complexity in the interneurons of epilepsy trigger zones (Gavrilovici et al., 2012).
PIRC and PRC Pharmacological Studies
NMDA Receptor
The role of N-methyl-D-aspartate (NMDA) receptors in the generation and propagation of seizures differs between anterior PIRC (i.e., AT), posterior PIRC, and the PRC. Within the AT, blockade of NMDA receptors prevents bicuculline-evoked seizures (Halonen et al., 1994; Fornai et al., 2005) and attenuates amygdala-kindled seizures (Holmes et al., 1992). Similarly, within the PRC, NMDA receptor activation is necessary for seizure propagation. In contrast, blockade of NMDA receptors in posterior PIRC was without effect on AT-evoked seizure propagation (Piredda and Gale, 1986; Halonen et al., 1994; Tortorella et al., 1997). The pattern differs slightly in the non-human primate: blockade of NMDA receptors within AT using 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo (f)-quinoxaline (NBQX) or 3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid (CPP) does not significantly protect against AT-evoked complex partial seizures (Malkova et al., 2015). Seizures evoked by soman, a nerve agent inhibitor of acetylcholinesterase, show some resemblance with AT-evoked seizures in terms of the profile of NMDA receptor involvement across regions. For example, NMDA receptor blockade in the posterior PIRC offered no protection against soman-induced seizures, while NMDA receptor blockade in the PRC more than doubled the latency to seizure onset after soman exposure (Myhrer et al., 2010). This pattern differs with respect to NMDA receptor antagonism in AT, which is without effect against soman administration (Myhrer et al., 2008). In the ex vivo guinea pig brain, seizure activity evoked with the potassium channel blocker 4-aminopyridine (4-AP) is actually worsened by blockade of NMDA receptors. Finally, in slice models, blockade of NMDA-receptor mediated neurotransmission attenuates ictal and interictal-like activity evoked by 4-AP (Salah and Perkins, 2011), by high extracellular potassium concentrations (Traynelis and Dingledine, 1988), and spontaneous and evoked discharges from pilocarpine (Benini et al., 2011). Thus, NMDA mediated neurotransmission plays a pivotal role in the propagation of seizure activity within the PIRC/PRC network.
AMPA
Alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor mediated neurotransmission is critical for seizure initiation and propagation within anterior and posterior PIRC along with PRC. For example, systemic administration of the AMPA receptor antagonist, NBQX, produced a dose-dependent decrease in the severity of seizures evoked from AT (Tortorella et al., 1997). Moreover, microinjection of NBQX into either AT, posterior PIRC or PRC antagonized AT-evoked seizures with doses as low as 500 pmol. Unlike the role of NMDA receptors, the role of AMPA receptors in this circuit is conserved between rats and monkeys: AMPA receptor blockade within AT suppressed AT-evoked seizures in the monkey (Malkova et al., 2015). Blockade of AMPA receptors within PRC is also effective against soman-evoked seizures (Myhrer et al., 2010). In the guinea pig model, blockade of AMPA receptors significantly attenuated seizures triggered by 4-AP within piriform cortex, entorhinal cortex, and hippocampus (Carriero et al., 2010).
AMPA receptor desensitization within AT appears to play a pivotal role in controlling the duration of seizure activity. For example, co-application of cyclothiazide (which blocks AMPA receptor desensitization) and bicuculline triggers a self-sustaining status epilepticus (Fornai et al., 2005). Moreover, overexpression of slow desensitizing (GluR1 flip), but not fast desensitizing (GluR1 flop) AMPA receptors within AT significantly increased the severity of bicuculline evoked seizures. In all cases, AT-evoked status epilepticus was insensitive to NMDA receptor blockade but was prevented by AMPA blockade (Fornai et al., 2005). Within the monkey, given the relative propensity of AT-evoked seizures to result in status epilepticus, and the insensitivity of AT-evoked seizures to NMDA receptor activation, it could be speculated that AMPA receptors within PIRC may be slow to desensitize.
At the level of synaptic physiology, AMPA antagonism with DNQX can eliminate epileptiform events in the slice (Hoffman and Haberly, 1996). This effect is associated with decreased buildup in multiunit activity preceding epileptiform discharges. Interestingly, this effect occurs at a concentration below that required to completely block monosynaptic EPSPs suggesting that the disruption in epileptiform activity by AMPA blockade results from disruption of a multisynaptic process. EPSPs mediated by AMPA receptors may be the basis for the epileptiform events in the PIRC. In contrast to NMDA receptors, which contribute but are not indispensable for seizure generation, AMPA receptors appear to be a key component to triggering seizures.
Together, the experimental data discussed above indicate that AMPA-mediated (as compared to NMDA-mediated) neurotransmission plays a relatively larger role in seizure generation within the PIRC/PRC network.
GABA
GABA-mediated (in particular GABAA) neurotransmission plays a critical role in controlling hyperexcitability of neuronal circuits. Disinhibition of hyperexcitable regions by loss of GABAergic inhibition has been long proposed as a mechanism of seizure propagation (Prince and Wilder, 1967). As previously discussed, epileptogenic insults can change the balance between excitation and inhibition within PIRC/PRC in several ways, including by loss of inhibitory neurons. Multiple studies have demonstrated the pivotal role for GABAA-mediated neurotransmission in PIRC and PRC for seizure control. For example, picomole amounts of GABA antagonist administered within AT or in posterior PIRC can trigger limbic motor seizure in rat (Piredda and Gale, 1985). In the ex vivo guinea pig brain, microinjection of bicuculline in the anterior PIRC results in interictal spiking and spontaneous epileptiform potentials (De Curtis et al., 1994). Interestingly, in a PIRC slice model of epileptiform activity evoked by 4-AP blockade of GABAA mediated transmission abolishes ictal-like discharges, but leaves periodic interictal activity intact (Panuccio et al., 2012).
Activation of GABAA receptors by pretreatment of PIRC or PRC with a muscimol protected animals from development of AT-evoked seizures (Halonen et al., 1994). The effect of muscimol was unique to PIRC and PRC with no seizure prevention observed when it was placed into areas such as amygdala or hippocampus (Halonen et al., 1994). This suggests that proper functioning of GABAA receptors is critical for prevention of ictal activity in PIRC and PRC, two areas that are hyperexcitable under normal conditions and serving as epilepsy trigger zones (Giacchino et al., 1984). These data demonstrate that ERC is capable of producing focal epileptiform discharges, but also has the necessary functional connections to facilitate propagation of these discharges to other brain areas. Febrile seizures upregulate the expression of GABAA receptors in neonatal granule cells. The increase in GABAAreceptors causes a reversal in the direction of granule cell migration. Furthermore, preventing GABA-mediated action by using bumetanide, which inhibits that Na,+-K+-2-Cl− co-transporter, susceptibility to limbic seizures is reduced (Fujiwara-Tsukamoto et al., 2003; Koyama et al., 2012).
ERC function is significantly altered by epileptogenic insults. For example, unilateral damage to ERC caused by amino-oxyacetic acid (AOAA) produced long-lasting (1 month–1.5 year) changes in extracellular responses to white matter stimulation in the superficial layers of the medial ERC. Extracellular responses in slices from AOAA treated animals were prolonged and repetitive and intracellular recordings from residual principal cells revealed prolonged, repetitive excitatory postsynaptic potentials (EPSPs). This pattern differed significantly from the brief EPSPs and single discharges seen in slices from controls. In neurons located in the deep layers of ERC, the initial synaptic response to white matter stimulation did not differ between AOAA and control treated animals. However, in a subset of deep neurons in AOAA treated animals, repetitive action potentials were seen after a brief delay (Scharfman et al., 1998). All together, these changes are consistent with those needed to facilitate the generation of sustained synchronous activity.
ERC Pharmacological Studies
As in PIRC/PRC, glutamatergic and GABAergic neurotransmission within ERC are essential for seizure propagation. However, relatively fewer studies have directly manipulated ERC neurotransmitter signaling in vivo. In the sections below, we will outline the in vivo and in vitro pharmacological studies that have established ERC as a key site for seizure origin and propagation.
Glutamatergic Transmission
The role of glutamatergic neurotransmission within ERC is very similar to its role in the PIRC. Not only glutamate antagonists have a differential effect on epileptiform activity depending on the type of receptor blocked, but the effect of the antagonists also differ based on the type of discharges. Glutamatergic transmission has a function in both seizure generation in the deep layers and seizure propagation in the superficial layers of the ERC. Moreover, the process of kindling induces long-lasting changes in glutamatergic neurotransmission within ERC (Cincotta et al., 1991; Lee et al., 1994; Prince et al., 1995; Behr et al., 2001), along with other limbic regions.
AMPA
In the slice, the role of AMPA-receptor mediated neurotransmission is consistent across ages and methods of inducing epileptiform activity. AMPA antagonists suppress seizure-like events caused by increases in extracellular potassium, bath application of 4-AP, decrease in extracellular magnesium concentration (Borck and Jefferys, 1999; Luhmann et al., 2000) and pilocarpine application (Nagao et al., 1996). Moreover, AMPA antagonists also attenuate a subset of interictal discharges (Avoli et al., 2013). Ex vivo, in the isolated guinea pig brain, AMPA receptor blockade completely suppresses seizure activity evoked either electrically from lateral ERC (Federico and MacVicar, 1996) or by 4-AP perfusion (Carriero et al., 2010).
While the effects of in vivo manipulation of glutamatergic neurotransmission within ERC on seizure propagation remain understudied, the compelling slice and ex vivo data above suggest an instrumental role for AMPA-receptor mediated glutamatergic signaling in seizure generation within ERC.
NMDA
In contrast to AMPA-receptor mediated transmission, NMDA receptor mediated signaling does not appear to play as consistently vital a role in seizure propagation within ERC. NMDA receptor antagonists may have no effect in slice models (Lücke et al., 1995; Avoli et al., 1996; Nagao et al., 1996; Luhmann et al., 2000), and in the ex vivo guinea pig model (Federico and MacVicar, 1996). However, other authors have found that NMDA antagonists decrease the probability and speed of propagation, but do not abolish the propagation completely (Avoli et al., 1994).
Some studies have suggested that NMDA-receptor-mediated transmission plays a particularly important role in the genesis of late epileptiform events, as compared to early events (Zhang et al., 1994). In slices taken from rats that had received status epilepticus, blockade of NMDA receptors partially normalized neuronal activity (Bear et al., 1996; Fountain et al., 1998). As with AMPA-mediated transmission, there is a dearth of data regarding effects of focal entorhinal pharmacological manipulations on seizure propagation. In conclusion, glutamatergic transmission in particular mediated by AMPA receptors is vital for seizure genesis within ERC and propagation both within ERC and to other structures.
GABA
A failure of the inhibitory control mechanisms that usually dominate interactions between PRC and ERC may underlie seizure initiation and propagation. In the ex vivo guinea pig brain, disinhibition by application of bicuculline with concentrations as low as 20 μM resulted in spontaneous seizures that spread to the amygdala more rapidly than electrically evoked seizures (Federico and MacVicar, 1996). Moreover, when compared to electrically-evoked seizures, the distribution area of bicuculline-induced seizures was greater, involving the posterior PIRC and larger areas over the medial amygdala. Both the speed of propagation and the extent of seizures were proportional to the concentration of bicuculline. In this preparation, interictal activity is first detected in ERC, before propagating to other regions and progressing to fast ictal discharges (Uva et al., 2004). Interestingly, during the onset of seizure activity in this model, principal (excitatory) neurons are silent—ictal activity is generated by hypersynchronous and sustained firing of interneurons (Gnatkovsky et al., 2008).
In addition to effects on generation of ictal discharges, blockade of GABA-mediated transmission in the ERC stimulates generation of interictal-like discharges, which consisted of an initial paroxysmal depolarizing shift followed by afterdischarges. The initial component of the interictal discharges, namely, the paroxysmal depolarizing shifts can be blocked in a progressive fashion, while the second component, the afterdischarges, are abolished strictly in all-or-none manner (Jones and Lambert, 1990). Interictal-like discharges represent an intrinsic electrophysiological property of the ERC and support the hypothesis that this region is highly epileptogenic. Similarly, spontaneous sharp waves in the temporal cortex were abolished by administration of GABA antagonists bicuculline (Köhling et al., 1998) or μ-receptor agonists (Louvel et al., 2001), while removal of antagonists restored paroxysmal depolarizing activity (Köhling et al., 1998), effectively suggesting that the synchronization currents were mediated solely by GABA transmission.
The epileptogenecity is associated with changes on the molecular, cellular, and system levels that facilitate generation and propagation of seizures. The implication of these changes can be measured in part as a change in the latency to seizure generation under stimulating conditions. The latency to occurrence of the first seizure-like events after brain slices from SE-subjected animals were placed in Mg2+-free artificial cerebrospinal fluid was significantly reduced at 4 and 8 weeks. This provides an evidence for escalation of brain excitability and neuronal hypersynchrony due to adaptations in brain plasticity, predisposing to recurrent epileptic seizures (Holtkamp et al., 2011). Exposure to a convulsant such as 4-AP produces lasting changes in the ERC suggesting plasticity that renders the area epileptogenic. Moreover, different network interactions within the hippocampus-ERC loop characterize control and epileptic slices in the pilocarpine model of epilepsy. For example, ERC-driven ictal discharges in pilocarpine-treated slices occur throughout the experiment and spread to the CA1/subicular area via the temporoammonic path (D'Antuono et al., 2002). These functional changes caused by seizure-induced cell damage lead to the development of epileptic seizures. The process is facilitated by a disinhibition of the ERC due to loss of feed-forward inhibition from the PRC and hippocampal outputs and possibly sustained by the reverberant activity between ERC and CA1 networks that are excited via the temporoammonic path (D'Antuono et al., 2002).
In conclusion, pharmacological means have demonstrated that ERC is an area that is highly susceptible to ictal discharges when exposed to low doses of excitatory compounds.
Conclusion
In this review, we have described the functional neuroanatomy, pharmacology, and physiology of three limbic-cortical areas: ERC, PRC, and PIRC. These regions are highly interconnected with each other and with other limbic structures, and have features that make them particularly prone to the genesis and propagation of ictal discharges. While a great deal of attention has been paid to subcortical limbic structures in epilepsy (e.g., amygdala, hippocampus), the data reviewed herein clearly underscore the importance of these cortical regions for epileptogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was made possible by the startup funds from the George Washington University and pilot grant from Clinical and Translational Science Institute at Children's National in Washington, DC. Both sources of funding are for MZK.
Dedication
We dedicate this article in memory of Dr. Karen Gale, Ph.D. who passed away on August 21, 2014. In addition to her extraordinary contributions to neuroscience, she was most notably the first to discover the region of the brain that triggers epileptic seizures called the Area Tempestas. A remarkable teacher, mentor, and advocate, Dr. Gale helped develop the Interdisciplinary Program in Neuroscience and Georgetown Women in Medicine, both at Georgetown University. Her passion and commitment to the scientific community at all levels will be remembered.
Abbreviations
4-AP, 4-aminopyridine; AOAA, amino-oxyacetic acid; AMPA, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; AT, area tempestas; CPP, 3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid; EPN, endopiriform nucleus; EPSP, excitatory postsynaptic potentials; ERC, entorhinal cortex; GABA, gamma aminobutyric acid; NBQX, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo (f)-quinoxaline; NMDA, N-methyl-D-aspartate; PIRC, piriform cortex; PRC, perirhinal cortex; PSA-NCAM, polysialylated neural cell adhesion molecule; TLE, temporal lobe epilepsy.
References
Acsády, L., and Káli, S. (2007). Models, structure, function: the transformation of cortical signals in the dentate gyrus. Prog. Brain Res. 163, 577–599. doi: 10.1016/S0079-6123(07)63031-3
Agster, K. L., and Burwell, R. D. (2009). Cortical efferents of the perirhinal, postrhinal, and entorhinal cortices of the rat. Hippocampus 19, 1159–1186. doi: 10.1002/hipo.20578
Aguirre, G. K., Detre, J. A., Alsop, D. C., and D'Esposito, M. (1996). The parahippocampus subserves topographical learning in man. Cereb. Cortex N.Y. 6, 823–829.
Amaral, D. G., Insausti, R., and Cowan, W. M. (1987). The entorhinal cortex of the monkey: I. Cytoarchitectonic organization. J. Comp. Neurol. 264, 326–355. doi: 10.1002/cne.902640305
Andersen, P., Bliss, T. V., and Skrede, K. K. (1971). Lamellar organization of hippocampal pathways. Exp. Brain Res. 13, 222–238.
Avoli, M., Barbarosie, M., Lücke, A., Nagao, T., Lopantsev, V., and Köhling, R. (1996). Synchronous GABA-mediated potentials and epileptiform discharges in the rat limbic system in vitro. J. Neurosci. Off. J. Soc. Neurosci. 16, 3912–3924.
Avoli, M., Mattia, D., Siniscalchi, A., Perreault, P., and Tomaiuolo, F. (1994). Pharmacology and electrophysiology of a synchronous GABA-mediated potential in the human neocortex. Neuroscience 62, 655–666.
Avoli, M., Panuccio, G., Herrington, R., D'Antuono, M., de Guzman, P., and Lévesque, M. (2013). Two different interictal spike patterns anticipate ictal activity in vitro. Neurobiol. Dis. 52, 168–176. doi: 10.1016/j.nbd.2012.12.004
Barbeau, E., Wendling, F., Régis, J., Duncan, R., Poncet, M., Chauvel, P., et al. (2005). Recollection of vivid memories after perirhinal region stimulations: synchronization in the theta range of spatially distributed brain areas. Neuropsychologia 43, 1329–1337. doi: 10.1016/j.neuropsychologia.2004.11.025
Bartolomei, F., Khalil, M., Wendling, F., Sontheimer, A., Régis, J., Ranjeva, J.-P., et al. (2005). Entorhinal cortex involvement in human mesial temporal lobe epilepsy: an electrophysiologic and volumetric study. Epilepsia 46, 677–687. doi: 10.1111/j.1528-1167.2005.43804.x
Bartolomei, F., Wendling, F., Bellanger, J. J., Régis, J., and Chauvel, P. (2001). Neural networks involving the medial temporal structures in temporal lobe epilepsy. Clin. Neurophysiol.Off. J. Int. Fed. Clin. Neurophysiol. 112, 1746–1760. doi: 10.1016/S1388-2457(01)00591-0
Bear, J., Fountain, N. B., and Lothman, E. W. (1996). Responses of the superficial entorhinal cortex in vitro in slices from naive and chronically epileptic rats. J. Neurophysiol. 76, 2928–2940.
Behan, M., and Haberly, L. B. (1999). Intrinsic and efferent connections of the endopiriform nucleus in rat. J. Comp. Neurol. 408, 532–548.
Behr, J., Heinemann, U., and Mody, I. (2001). Kindling induces transient NMDA receptor-mediated facilitation of high-frequency input in the rat dentate gyrus. J. Neurophysiol. 85, 2195–2202.
Benini, R., Longo, D., Biagini, G., and Avoli, M. (2011). Perirhinal cortex hyperexcitability in pilocarpine-treated epileptic rats. Hippocampus 21, 702–713. doi: 10.1002/hipo.20785
Bertram, E. H. (1997). Functional anatomy of spontaneous seizures in a rat model of limbic epilepsy. Epilepsia 38, 95–105.
Biella, G., Uva, L., and de Curtis, M. (2002a). Propagation of neuronal activity along the neocortical-perirhinal-entorhinal pathway in the guinea pig. J. Neurosci. Off. J. Soc. Neurosci. 22, 9972–9979.
Biella, G., Uva, L., Hofmann, U. G., and de Curtis, M. (2002b). Associative interactions within the superficial layers of the entorhinal cortex of the guinea pig. J. Neurophysiol. 88, 1159–1165. doi: 10.1152/jn.00022.2002
Bonilha, L., Kobayashi, E., Rorden, C., Cendes, F., and Li, L. M. (2003). Medial temporal lobe atrophy in patients with refractory temporal lobe epilepsy. J. Neurol. Neurosurg. Psychiatr. 74, 1627–1630. doi: 10.1136/jnnp.74.12.1627
Borck, C., and Jefferys, J. G. (1999). Seizure-like events in disinhibited ventral slices of adult rat hippocampus. J. Neurophysiol. 82, 2130–2142.
Bucci, D. J., and Burwell, R. D. (2004). Deficits in attentional orienting following damage to the perirhinal or postrhinal cortices. Behav. Neurosci. 118, 1117–1122. doi: 10.1037/0735-7044.118.5.1117
Burwell, R. D., and Amaral, D. G. (1998). Cortical afferents of the perirhinal, postrhinal, and entorhinal cortices of the rat. J. Comp. Neurol. 398, 179–205.
Carriero, G., Uva, L., Gnatkovsky, V., Avoli, M., and de Curtis, M. (2010). Independent epileptiform discharge patterns in the olfactory and limbic areas of the in vitro isolated Guinea pig brain during 4-aminopyridine treatment. J. Neurophysiol. 103, 2728–2736. doi: 10.1152/jn.00862.2009
Cataldi, M., Avoli, M., and de Villers-Sidani, E. (2013). Resting state networks in temporal lobe epilepsy. Epilepsia 54, 2048–2059. doi: 10.1111/epi.12400
Cincotta, M., Young, N. A., and Beart, P. M. (1991). Unilateral up-regulation of glutamate receptors in limbic regions of amygdaloid-kindled rats. Exp. Brain Res. 85, 650–658.
Couey, J. J., Witoelar, A., Zhang, S.-J., Zheng, K., Ye, J., Dunn, B., et al. (2013). Recurrent inhibitory circuitry as a mechanism for grid formation. Nat. Neurosci. 16, 318–324. doi: 10.1038/nn.3310
Courtney, S. M., Ungerleider, L. G., Keil, K., and Haxby, J. V. (1996). Object and spatial visual working memory activate separate neural systems in human cortex. Cereb Cortex N.Y. 6, 39–49.
D'Antuono, M., Benini, R., Biagini, G., D'Arcangelo, G., Barbarosie, M., Tancredi, V., et al. (2002). Limbic network interactions leading to hyperexcitability in a model of temporal lobe epilepsy. J. Neurophysiol. 87, 634–639. doi: 10.1152/jn.00351.2001
Darlington, G. J., Wilson, D. R., and Lachman, L. B. (1986). Monocyte-conditioned medium, interleukin-1, and tumor necrosis factor stimulate the acute phase response in human hepatoma cells in vitro. J. Cell Biol. 103, 787–793.
De Curtis, M., Biella, G., Forti, M., and Panzica, F. (1994). Multifocal spontaneous epileptic activity induced by restricted bicuculline ejection in the piriform cortex of the isolated guinea pig brain. J. Neurophysiol. 71, 2463–2476.
De Curtis, M., and Paré, D. (2004). The rhinal cortices: a wall of inhibition between the neocortex and the hippocampus. Prog Neurobiol. 74, 101–110. doi: 10.1016/j.pneurobio.2004.08.005
De Guzman, P., D'Antuono, M., and Avoli, M. (2004). Initiation of electrographic seizures by neuronal networks in entorhinal and perirhinal cortices in vitro. Neuroscience 123, 875–886. doi: 10.1016/j.neuroscience.2003.11.013
Demir, R., Haberly, L. B., and Jackson, M. B. (1998). Voltage imaging of epileptiform activity in slices from rat piriform cortex: onset and propagation. J. Neurophysiol. 80, 2727–2742.
Demir, R., Haberly, L. B., and Jackson, M. B. (1999). Sustained and accelerating activity at two discrete sites generate epileptiform discharges in slices of piriform cortex. J. Neurosci. Off. J. Soc. Neurosci. 19, 1294–1306.
Demir, R., Haberly, L. B., and Jackson, M. B. (2001). Epileptiform discharges with in-vivo-like features in slices of rat piriform cortex with longitudinal association fibers. J. Neurophysiol. 86, 2445–2460.
Doherty, J., Gale, K., and Eagles, D. A. (2000). Evoked epileptiform discharges in the rat anterior piriform cortex: generation and local propagation. Brain Res. 861, 77–87. doi: 10.1016/S0006-8993(00)02000-X
Dolga, A. M., Granic, I., Blank, T., Knaus, H.-G., Spiess, J., Luiten, P. G. M., et al. (2008). TNF-alpha-mediates neuroprotection against glutamate-induced excitotoxicity via NF-kappaB-dependent up-regulation of K2.2 channels. J. Neurochem. 107, 1158–1167. doi: 10.1111/j.1471-4159.2008.05701.x
Dolorfo, C. L., and Amaral, D. G. (1998). Entorhinal cortex of the rat: organization of intrinsic connections. J. Comp. Neurol. 398, 49–82.
Drexel, M., Preidt, A. P., and Sperk, G. (2012). Sequel of spontaneous seizures after kainic acid-induced status epilepticus and associated neuropathological changes in the subiculum and entorhinal cortex. Neuropharmacology 63, 806–817. doi: 10.1016/j.neuropharm.2012.06.009
Du, F., Eid, T., Lothman, E. W., Köhler, C., and Schwarcz, R. (1995). Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 15, 6301–6313.
Du, F., Whetsell, W. O. Jr. Abou-Khalil, B., Blumenkopf, B., Lothman, E. W., and Schwarcz, R. (1993). Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 16, 223–233.
Ekstrand, J. J., Domroese, M. E., Johnson, D. M., Feig, S. L., Knodel, S. M., Behan, M., et al. (2001). A new subdivision of anterior piriform cortex and associated deep nucleus with novel features of interest for olfaction and epilepsy. J. Comp. Neurol. 434, 289–307. doi: 10.1002/cne.1178
Empson, R. M., and Heinemann, U. (1995). The perforant path projection to hippocampal area CA1 in the rat hippocampal-entorhinal cortex combined slice. J. Physiol. 484, 707–720.
Engel, J. Jr. McDermott, M. P., Wiebe, S., Langfitt, J. T., Stern, J. M., Dewar, S., et al. (2012). Early surgical therapy for drug-resistant temporal lobe epilepsy: a randomized trial. J. Am. Med. Assoc. 307, 922–930. doi: 10.1001/jama.2012.220
Engel, J. Jr. Wiebe, S., French, J., Sperling, M., Williamson, P., Spencer, D., et al. (2003). Practice parameter: temporal lobe and localized neocortical resections for epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology, in association with the American Epilepsy Society and the American Association of Neurological Surgeons. Neurology 60, 538–547. doi: 10.1212/01.WNL.0000055086.35806.2D
Federico, P., and MacVicar, B. A. (1996). Imaging the induction and spread of seizure activity in the isolated brain of the guinea pig: the roles of GABA and glutamate receptors. J. Neurophysiol. 76, 3471–3492.
Finch, D. M., Tan, A. M., and Isokawa-Akesson, M. (1988). Feedforward inhibition of the rat entorhinal cortex and subicular complex. J. Neurosci. Off. J. Soc. Neurosci. 8, 2213–2226.
Fornai, F., Busceti, C. L., Kondratyev, A., and Gale, K. (2005). AMPA receptor desensitization as a determinant of vulnerability to focally evoked status epilepticus. Eur. J. Neurosci. 21, 455–463. doi: 10.1111/j.1460-9568.2005.03873.x
Fountain, N. B., Bear, J., Bertram, E. H. III., and Lothman, E. W. (1998). Responses of deep entorhinal cortex are epileptiform in an electrogenic rat model of chronic temporal lobe epilepsy. J. Neurophysiol. 80, 230–240.
Fujiwara-Tsukamoto, Y., Isomura, Y., Nambu, A., and Takada, M. (2003). Excitatory GABA input directly drives seizure-like rhythmic synchronization in mature hippocampal CA1 pyramidal cells. Neuroscience 119, 265–275. doi: 10.1016/S0306-4522(03)00102-7
Galvis-Alonso, O. Y., Cortes De Oliveira, J. A., and Garcia-Cairasco, N. (2004). Limbic epileptogenicity, cell loss and axonal reorganization induced by audiogenic and amygdala kindling in wistar audiogenic rats (WAR strain). Neuroscience 125, 787–802. doi: 10.1016/j.neuroscience.2004.01.042
Garcia-Cairasco, N., Wakamatsu, H., Oliveira, J. A., Gomes, E. L., Del Bel, E. A., and Mello, L. E. (1996). Neuroethological and morphological (Neo-Timm staining) correlates of limbic recruitment during the development of audiogenic kindling in seizure susceptible Wistar rats. Epilepsy Res. 26, 177–192.
Gavrilovici, C., Pollock, E., Everest, M., and Poulter, M. O. (2012). The loss of interneuron functional diversity in the piriform cortex after induction of experimental epilepsy. Neurobiol. Dis. 48, 317–328. doi: 10.1016/j.nbd.2012.07.002
Giacchino, J. L., Somjen, G. G., Frush, D. P., and McNamara, J. O. (1984). Lateral entorhinal cortical kindling can be established without potentiation of the entorhinal-granule cell synapse. Exp. Neurol. 86, 483–492.
Gnatkovsky, V., Librizzi, L., Trombin, F., and de Curtis, M. (2008). Fast activity at seizure onset is mediated by inhibitory circuits in the entorhinal cortex in vitro. Ann. Neurol. 64, 674–686. doi: 10.1002/ana.21519
Gundlach, A. L., Burazin, T. C., Jenkins, T. A., and Berkovic, S. F. (1995). Spatiotemporal alterations of central alpha 1-adrenergic receptor binding sites following amygdaloid kindling seizures in the rat: autoradiographic studies using [3H]prazosin. Brain Res. 672, 214–227.
Haberly, L. B. (2001). Parallel-distributed processing in olfactory cortex: new insights from morphological and physiological analysis of neuronal circuitry. Chem. Senses. 26, 551–576. doi: 10.1093/chemse/26.5.551
Haberly, L. B., and Bower, J. M. (1989). Olfactory cortex: model circuit for study of associative memory? Trends Neurosci. 12, 258–264.
Halonen, T., Tortorella, A., Zrebeet, H., and Gale, K. (1994). Posterior piriform and perirhinal cortex relay seizures evoked from the area tempestas: role of excitatory and inhibitory amino acid receptors. Brain Res. 652, 145–148.
Hartung, H. P., Schäfer, B., Heininger, K., and Toyka, K. V. (1989). Recombinant interleukin-1 beta stimulates eicosanoid production in rat primary culture astrocytes. Brain Res. 489, 113–119.
Hoffman, W. H., and Haberly, L. B. (1991). Bursting-induced epileptiform EPSPs in slices of piriform cortex are generated by deep cells. J. Neurosci. Off. J. Soc. Neurosci. 11, 2021–2031.
Hoffman, W. H., and Haberly, L. B. (1993). Role of synaptic excitation in the generation of bursting-induced epileptiform potentials in the endopiriform nucleus and piriform cortex. J. Neurophysiol. 70, 2550–2561.
Hoffman, W. H., and Haberly, L. B. (1996). Kindling-induced epileptiform potentials in piriform cortex slices originate in the underlying endopiriform nucleus. J. Neurophysiol. 76, 1430–1438.
Holmes, K. H., Bilkey, D. K., and Laverty, R. (1992). The infusion of an NMDA antagonist into perirhinal cortex suppresses amygdala-kindled seizures. Brain Res. 587, 285–290.
Holtkamp, M., Buchheim, K., Elsner, M., Matzen, J., Weissinger, F., and Meierkord, H. (2011). Status epilepticus induces increasing neuronal excitability and hypersynchrony as revealed by optical imaging. Neurobiol. Dis. 43, 220–227. doi: 10.1016/j.nbd.2011.03.014
Howard, J. D., Plailly, J., Grueschow, M., Haynes, J.-D., and Gottfried, J. A. (2009). Odor quality coding and categorization in human posterior piriform cortex. Nat. Neurosci. 12, 932–938. doi: 10.1038/nn.2324
Illig, K. R. (2005). Projections from orbitofrontal cortex to anterior piriform cortex in the rat suggest a role in olfactory information processing. J. Comp. Neurol. 488, 224–231. doi: 10.1002/cne.20595
Insausti, R. (1993). Comparative anatomy of the entorhinal cortex and hippocampus in mammals. Hippocampus 3, 19–26.
Insausti, R., Amaral, D. G., and Cowan, W. M. (1987). The entorhinal cortex of the monkey: II. Cortical afferents. J. Comp. Neurol. 264, 356–395. doi: 10.1002/cne.902640306
Insausti, R., Tuñón, T., Sobreviela, T., Insausti, A. M., and Gonzalo, L. M. (1995). The human entorhinal cortex: a cytoarchitectonic analysis. J. Comp. Neurol. 355, 171–198. doi: 10.1002/cne.903550203
Jamali, S., Bartolomei, F., Robaglia-Schlupp, A., Massacrier, A., Peragut, J.-C., Régis, J., et al. (2006). Large-scale expression study of human mesial temporal lobe epilepsy: evidence for dysregulation of the neurotransmission and complement systems in the entorhinal cortex. Brain J. Neurol. 129, 625–641. doi: 10.1093/brain/awl001
Johnson, D. M., Illig, K. R., Behan, M., and Haberly, L. B. (2000). New features of connectivity in piriform cortex visualized by intracellular injection of pyramidal cells suggest that “primary” olfactory cortex functions like “association” cortex in other sensory systems. J. Neurosci. Off. J. Soc. Neurosci. 20, 6974–6982.
Jones, R. S., and Lambert, J. D. (1990). Synchronous discharges in the rat entorhinal cortex in vitro: site of initiation and the role of excitatory amino acid receptors. Neuroscience 34, 657–670.
Jung, K.-H., Chu, K., Lee, S.-T., Kim, J.-H., Kang, K.-M., Song, E.-C., et al. (2009). Region-specific plasticity in the epileptic rat brain: a hippocampal and extrahippocampal analysis. Epilepsia 50, 537–549. doi: 10.1111/j.1528-1167.2008.01718.x
Jutila, L., Ylinen, A., Partanen, K., Alafuzoff, I., Mervaala, E., Partanen, J., et al. (2001). MR volumetry of the entorhinal, perirhinal, and temporopolar cortices in drug-refractory temporal lobe epilepsy. AJNR Am. J. Neuroradiol. 22, 1490–1501.
Kairiss, E. W., Racine, R. J., and Smith, G. K. (1984). The development of the interictal spike during kindling in the rat. Brain Res. 322, 101–110.
Kang, T. C., Park, S. K., Do, S. G., Suh, J. G., Jo, S. M., Oh, Y. S., et al. (2000). The over-expression of somatostatin in the gerbil entorhinal cortex induced by seizure. Brain Res. 882, 55–61. doi: 10.1016/S0006-8993(00)02824-9
Kato, N., Higuchi, T., Friesen, H. G., and Wada, J. A. (1983). Changes of immunoreactive somatostatin and beta-endorphin content in rat brain after amygdaloid kindling. Life Sci. 32, 2415–2422.
Kelly, M. E., and McIntyre, D. C. (1996). Perirhinal cortex involvement in limbic kindled seizures. Epilepsy Res. 26, 233–243.
Kibler, A. B., and Durand, D. M. (2011). Orthogonal wave propagation of epileptiform activity in the planar mouse hippocampus in vitro. Epilepsia 52, 1590–1600. doi: 10.1111/j.1528-1167.2011.03125.x
Killian, N. J., Jutras, M. J., and Buffalo, E. A. (2012). A map of visual space in the primate entorhinal cortex. Nature 491, 761–764. doi: 10.1038/nature11587
Köhling, R., Lücke, A., Straub, H., Speckmann, E. J., Tuxhorn, I., Wolf, P., et al. (1998). Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain J. Neurol. 121(Pt 6), 1073–1087.
Koyama, R., Tao, K., Sasaki, T., Ichikawa, J., Miyamoto, D., Muramatsu, R., et al. (2012). GABAergic excitation after febrile seizures induces ectopic granule cells and adult epilepsy. Nat. Med. 18, 1271–1278. doi: 10.1038/nm.2850
Krettek, J. E., and Price, J. L. (1977). Projections from the amygdaloid complex and adjacent olfactory structures to the entorhinal cortex and to the subiculum in the rat and cat. J. Comp. Neurol. 172, 723–752. doi: 10.1002/cne.901720409
Kumar, S. S., and Buckmaster, P. S. (2006). Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 26, 4613–4623. doi: 10.1523/JNEUROSCI.0064-06.2006
Langer, M., Brandt, C., Zellinger, C., and Löscher, W. (2011). Therapeutic window of opportunity for the neuroprotective effect of valproate versus the competitive AMPA receptor antagonist NS following status epilepticus in rats. Neuropharmacology 61, 1033–1047. doi: 10.1016/j.neuropharm.2011.06.015
Laufs, H., Richardson, M. P., Salek-Haddadi, A., Vollmar, C., Duncan, J. S., Gale, K., et al. (2011). Converging, P. E. T., and fMRI evidence for a common area involved in human focal epilepsies. Neurology 77, 904–910. doi: 10.1212/WNL.0b013e31822c90f2
Lee, J. W., Andermann, F., Dubeau, F., Bernasconi, A., MacDonald, D., Evans, A., et al. (1998). Morphometric analysis of the temporal lobe in temporal lobe epilepsy. Epilepsia 39, 727–736.
Lee, S., Miskovsky, J., Williamson, J., Howells, R., Devinsky, O., Lothman, E., et al. (1994). Changes in glutamate receptor and proenkephalin gene expression after kindled seizures. Brain Res. Mol. Brain Res. 24, 34–42.
Leichnetz, G. R., and Astruc, J. (1976). The squirrel monkey entorhinal cortex: architecture and medial frontal afferents. Brain Res. Bull. 1, 351–358.
Lencz, T., McCarthy, G., Bronen, R. A., Scott, T. M., Inserni, J. A., Sass, K. J., et al. (1992). Quantitative magnetic resonance imaging in temporal lobe epilepsy: relationship to neuropathology and neuropsychological function. Ann. Neurol. 31, 629–637. doi: 10.1002/ana.410310610
Löscher, W., and Ebert, U. (1996). The role of the piriform cortex in kindling. Prog. Neurobiol. 50, 427–481.
Louvel, J., Papatheodoropoulos, C., Siniscalchi, A., Kurcewicz, I., Pumain, R., Devaux, B., et al. (2001). GABA-mediated synchronization in the human neocortex: elevations in extracellular potassium and presynaptic mechanisms. Neuroscience 105, 803–813. doi: 10.1016/S0306-4522(01)00247-0
Lücke, A., Nagao, T., Köhling, R., and Avoli, M. (1995). Synchronous potentials and elevations in [K+]o in the adult rat entorhinal cortex maintained in vitro. Neurosci. Lett. 185, 155–158.
Lu, L., Leutgeb, J. K., Tsao, A., Henriksen, E. J., Leutgeb, S., Barnes, C. A., et al. (2013). Impaired hippocampal rate coding after lesions of the lateral entorhinal cortex. Nat. Neurosci. 16, 1085–1093. doi: 10.1038/nn.3462
Luhmann, H. J., Dzhala, V. I., and Ben-Ari, Y. (2000). Generation and propagation of 4-AP-induced epileptiform activity in neonatal intact limbic structures in vitro. Eur. J. Neurosci. 12, 2757–2768. doi: 10.1046/j.1460-9568.2000.00156.x
Luskin, M. B., and Price, J. L. (1983a). The laminar distribution of intracortical fibers originating in the olfactory cortex of the rat. J. Comp. Neurol. 216, 292–302. doi: 10.1002/cne.902160306
Luskin, M. B., and Price, J. L. (1983b). The topographic organization of associational fibers of the olfactory system in the rat, including centrifugal fibers to the olfactory bulb. J. Comp. Neurol. 216, 264–291. doi: 10.1002/cne.902160305
Malkova, L., Forcelli, P. A., Wellman, L. L., Dybdal, D., Dubach, M. F., and Gale, K. (2015). Blockade of glutamatergic transmission in perirhinal cortex impairs object recognition memory in macaques. J. Neurosci. Off. J. Soc. Neurosci. 35, 5043–5050. doi: 10.1523/JNEUROSCI.4307-14.2015
Martin, C. B., Mirsattari, S. M., Pruessner, J. C., Pietrantonio, S., Burneo, J. G., Hayman-Abello, B., et al. (2012). Déjà vu in unilateral temporal-lobe epilepsy is associated with selective familiarity impairments on experimental tasks of recognition memory. Neuropsychologia 50, 2981–2991. doi: 10.1016/j.neuropsychologia.2012.07.030
McIntyre, D. C., and Gilby, K. L. (2006). Parahippocampal networks, intractability, and the chronic epilepsy of kindling. Adv. Neurol. 97, 77–83.
McIntyre, D. C., Kelly, M. E., and Dufresne, C. (1999). FAST and SLOW amygdala kindling rat strains: comparison of amygdala, hippocampal, piriform and perirhinal cortex kindling. Epilepsy Res. 35, 197–209.
McIntyre, D. C., and Plant, J. R. (1993). Long-lasting changes in the origin of spontaneous discharges from amygdala-kindled rats: piriform vs. perirhinal cortex in vitro. Brain Res. 624, 268–276.
McIntyre, D. C., Plant, J. R., and Kelly, M. E. (2000). Dorsal hippocampal kindling produces long-lasting changes in the origin of spontaneous discharges in the piriform versus perirhinal cortex in vitro. Epilepsy Res. 39, 191–200. doi: 10.1016/S0920-1211(99)00120-5
Meunier, M., Bachevalier, J., Mishkin, M., and Murray, E. A. (1993). Effects on visual recognition of combined and separate ablations of the entorhinal and perirhinal cortex in rhesus monkeys. J. Neurosci. Off. J. Soc. Neurosci. 13, 5418–5432.
Mikkonen, M., Soininen, H., Kälviänen, R., Tapiola, T., Ylinen, A., Vapalahti, M., et al. (1998). Remodeling of neuronal circuitries in human temporal lobe epilepsy: increased expression of highly polysialylated neural cell adhesion molecule in the hippocampus and the entorhinal cortex. Ann. Neurol. 44, 923–934. doi: 10.1002/ana.410440611
Mraovitch, S. (2003). Isocortical hyperemia and allocortical inflammation and atrophy following generalized convulsive seizures of thalamic origin in the rat. Cell. Mol. Neurobiol. 23, 773–791. doi: 10.1023/A:1025057004447
Murray, E. A., and Richmond, B. J. (2001). Role of perirhinal cortex in object perception, memory, and associations. Curr. Opin. Neurobiol. 11, 188–193. doi: 10.1016/S0959-4388(00)00195-1
Myhrer, T., Enger, S., and Aas, P. (2008). Anticonvulsant efficacy of drugs with cholinergic and/or glutamatergic antagonism microinfused into area tempestas of rats exposed to soman. Neurochem. Res. 33, 348–354. doi: 10.1007/s11064-007-9429-3
Myhrer, T., Enger, S., and Aas, P. (2010). Roles of perirhinal and posterior piriform cortices in control and generation of seizures: a microinfusion study in rats exposed to soman. Neurotoxicology 31, 147–153. doi: 10.1016/j.neuro.2009.10.002
Myhrer, T., Nguyen, N. H. T., Enger, S., and Aas, P. (2006). Anticonvulsant effects of GABA(A) modulators microinfused into area tempestas or substantia nigra in rats exposed to soman. Arch. Toxicol. 80, 502–507. doi: 10.1007/s00204-006-0070-8
Nagao, T., Alonso, A., and Avoli, M. (1996). Epileptiform activity induced by pilocarpine in the rat hippocampal-entorhinal slice preparation. Neuroscience 72, 399–408.
Navratilova, Z., Giocomo, L. M., Fellous, J.-M., Hasselmo, M. E., and McNaughton, B. L. (2012). Phase precession and variable spatial scaling in a periodic attractor map model of medial entorhinal grid cells with realistic after-spike dynamics. Hippocampus 22, 772–789. doi: 10.1002/hipo.20939
Neville, K. R., and Haberly, L. B. (2003). Beta and gamma oscillations in the olfactory system of the urethane-anesthetized rat. J. Neurophysiol. 90, 3921–3930. doi: 10.1152/jn.00475.2003
Pallud, J., Devaux, B., and Depaulis, A. (2008). Changes in spontaneous epileptic activity after selective intrahippocampal transection in a model of chronic mesial temporal lobe epilepsy]. Neurochirurgie 54, 135–140. doi: 10.1016/j.neuchi.2008.02.002
Panuccio, G., Sanchez, G., Lévesque, M., Salami, P., de Curtis, M., and Avoli, M. (2012). On the ictogenic properties of the piriform cortex in vitro. Epilepsia 53, 459–468. doi: 10.1111/j.1528-1167.2012.03408.x
Parent, J. M., Valentin, V. V., and Lowenstein, D. H. (2002). Prolonged seizures increase proliferating neuroblasts in the adult rat subventricular zone-olfactory bulb pathway. J. Neurosci. Off. J. Soc. Neurosci. 22, 3174–3188.
Parent, J. M., Yu, T. W., Leibowitz, R. T., Geschwind, D. H., Sloviter, R. S., and Lowenstein, D. H. (1997). Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 17, 3727–3738.
Pekcec, A., Fuest, C., Mühlenhoff, M., Gerardy-Schahn, R., and Potschka, H. (2008). Targeting epileptogenesis-associated induction of neurogenesis by enzymatic depolysialylation of NCAM counteracts spatial learning dysfunction but fails to impact epilepsy development. J. Neurochem. 105, 389–400. doi: 10.1111/j.1471-4159.2007.05172.x
Pelletier, J. G., Apergis, J., and Paré, D. (2004). Low-probability transmission of neocortical and entorhinal impulses through the perirhinal cortex. J. Neurophysiol. 91, 2079–2089. doi: 10.1152/jn.01197.2003
Piredda, S., and Gale, K. (1985). A crucial epileptogenic site in the deep prepiriform cortex. Nature 317, 623–625.
Piredda, S., and Gale, K. (1986). Role of excitatory amino acid transmission in the genesis of seizures elicited from the deep prepiriform cortex. Brain Res. 377, 205–210.
Prince, D. A., and Wilder, B. J. (1967). Control mechanisms in cortical epileptogenic foci. “Surround” inhibition. Arch. Neurol. 16, 194–202.
Prince, H. K., Conn, P. J., Blackstone, C. D., Huganir, R. L., and Levey, A. I. (1995). Down-regulation of AMPA receptor subunit GluR2 in amygdaloid kindling. J. Neurochem. 64, 462–465.
Rolls, E. T., and O'Mara, S. M. (1995). View-responsive neurons in the primate hippocampal complex. Hippocampus 5, 409–424. doi: 10.1002/hipo.450050504
Rosen, J. B., Hitchcock, J. M., Miserendino, M. J., Falls, W. A., Campeau, S., and Davis, M. (1992). Lesions of the perirhinal cortex but not of the frontal, medial prefrontal, visual, or insular cortex block fear-potentiated startle using a visual conditioned stimulus. J. Neurosci. Off. J. Soc. Neurosci. 12, 4624–4633.
Rossi, A. R., Angelo, M. F., Villarreal, A., Lukin, J., and Ramos, A. J. (2013). Gabapentin administration reduces reactive gliosis and neurodegeneration after pilocarpine-induced status epilepticus. PLoS ONE 8:e78516. doi: 10.1371/journal.pone.0078516
Ryan, L., Cardoza, J. A., Barense, M. D., Kawa, K. H., Wallentin-Flores, J., Arnold, W. T., et al. (2012). Age-related impairment in a complex object discrimination task that engages perirhinal cortex. Hippocampus 22, 1978–1989. doi: 10.1002/hipo.22069
Salah, A., and Perkins, K. L. (2011). Persistent ictal-like activity in rat entorhinal/perirhinal cortex following washout of 4-aminopyridine. Epilepsy Res. 94, 163–176. doi: 10.1016/j.eplepsyres.2011.01.017
Saleem, K. S., Price, J. L., and Hashikawa, T. (2007). Cytoarchitectonic and chemoarchitectonic subdivisions of the perirhinal and parahippocampal cortices in macaque monkeys. J. Comp. Neurol. 500, 973–1006. doi: 10.1002/cne.21141
Sargolini, F., Fyhn, M., Hafting, T., McNaughton, B. L., Witter, M. P., Moser, M.-B., et al. (2006). Conjunctive representation of position, direction, and velocity in entorhinal cortex. Science 312, 758–762. doi: 10.1126/science.1125572
Scharfman, H. E., Goodman, J. H., Du, F., and Schwarcz, R. (1998). Chronic changes in synaptic responses of entorhinal and hippocampal neurons after amino-oxyacetic acid (AOAA)-induced entorhinal cortical neuron loss. J. Neurophysiol. 80, 3031–3046.
Scholl, E. A., Dudek, F. E., and Ekstrand, J. J. (2013). Neuronal degeneration is observed in multiple regions outside the hippocampus after lithium pilocarpine-induced status epilepticus in the immature rat. Neuroscience 252, 45–59. doi: 10.1016/j.neuroscience.2013.07.045
Schwabe, K., Ebert, U., and Löscher, W. (2000). Bilateral lesions of the central but not anterior or posterior parts of the piriform cortex retard amygdala kindling in rats. Neuroscience 101, 513–521. doi: 10.1016/S0306-4522(00)00407-3
Schwabe, K., Ebert, U., and Löscher, W. (2004). The central piriform cortex: anatomical connections and anticonvulsant effect of GABA elevation in the kindling model. Neuroscience 126, 727–741. doi: 10.1016/j.neuroscience.2004.04.022
Shatskikh, T. N., Raghavendra, M., Zhao, Q., Cui, Z., and Holmes, G. L. (2006). Electrical induction of spikes in the hippocampus impairs recognition capacity and spatial memory in rats. Epilepsy Behav. 9, 549–556. doi: 10.1016/j.yebeh.2006.08.014
Sperber, E. F., Velísková, J., Benenati, B., and Moshé, S. L. (1998). Enhanced epileptogenicity of area tempestas in the immature rat. Dev. Neurosci. 20, 540–545.
Sugai, T., Yamamoto, R., Yoshimura, H., and Kato, N. (2012). Multimodal cross-talk of olfactory and gustatory information in the endopiriform nucleus in rats. Chem. Senses. 37, 681–688. doi: 10.1093/chemse/bjs046
Supcun, B., Ghadiri, M. K., Zeraati, M., Stummer, W., Speckmann, E.-J., and Gorji, A. (2012). The effects of tetanic stimulation on plasticity of remote synapses in the hippocampus-perirhinal cortex-amygdala network. Synap. N.Y. 66, 965–974. doi: 10.1002/syn.21591
Tortorella, A., Halonen, T., Sahibzada, N., and Gale, K. (1997). A crucial role of the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype of glutamate receptors in piriform and perirhinal cortex for the initiation and propagation of limbic motor seizures. J. Pharmacol. Exp. Ther. 280, 1401–1405.
Traynelis, S. F., and Dingledine, R. (1988). Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J. Neurophysiol. 59, 259–276.
Tseng, G. F., and Haberly, L. B. (1989). Deep neurons in piriform cortex. I. Morphology and synaptically evoked responses including a unique high-amplitude paired shock facilitation. J. Neurophysiol. 62, 369–385.
Umeoka, S. C., Lüders, H. O., Turnbull, J. P., Koubeissi, M. Z., and Maciunas, R. J. (2012). Requirement of longitudinal synchrony of epileptiform discharges in the hippocampus for seizure generation: a pilot study. J. Neurosurg. 116, 513–524. doi: 10.3171/2011.10.JNS11261
Uva, L., Grüschke, S., Biella, G., De Curtis, M., and Witter, M. P. (2004). Cytoarchitectonic characterization of the parahippocampal region of the guinea pig. J. Comp. Neurol. 474, 289–303. doi: 10.1002/cne.20121
Vaughan, D. N., and Jackson, G. D. (2014). The piriform cortex and human focal epilepsy. Front. Neurol. 5:259. doi: 10.3389/fneur.2014.00259
Veening, J. G. (1978). Cortical afferents of the amygdaloid complex in the rat: an HRP study. Neurosci. Lett. 8, 191–195.
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Vezzani, A., Monhemius, R., Tutka, P., Milani, R., and Samanin, R. (1996). Functional activation of somatostatin- and neuropeptide Y-containing neurons in the entorhinal cortex of chronically epileptic rats. Neuroscience 75, 551–557.
Wahnschaffe, U., Ebert, U., and Löscher, W. (1993). The effects of lesions of the posterior piriform cortex on amygdala kindling in the rat. Brain Res. 615, 295–303.
Wakefield, C. (1980). The topographical organization and laminar origin of some cortico-amygdaloid connections. Neurosci. Lett. 20, 21–24.
Wiig, K. A., Cooper, L. N., and Bear, M. F. (1996). Temporally graded retrograde amnesia following separate and combined lesions of the perirhinal cortex and fornix in the rat. Learn Mem Cold Spring Harb. 3, 313–325.
Wilson, D. A., and Stevenson, R. J. (2003). Olfactory perceptual learning: the critical role of memory in odor discrimination. Neurosci. Biobehav. Rev. 27, 307–328. doi: 10.1016/S0149-7634(03)00050-2
Yang, L.-X., Jin, C.-L., Zhu-Ge, Z.-B., Wang, S., Wei, E.-Q., Bruce, I. C., et al. (2006). Unilateral low-frequency stimulation of central piriform cortex delays seizure development induced by amygdaloid kindling in rats. Neuroscience 138, 1089–1096. doi: 10.1016/j.neuroscience.2005.12.006
Zhang, C. L., Gloveli, T., and Heinemann, U. (1994). Effects of NMDA- and AMPA-receptor antagonists on different forms of epileptiform activity in rat temporal cortex slices. Epilepsia 35(Suppl. 5), S68–S73.
Keywords: piriform cortex, area tempestas, perirhinal cortex, entorhinal cortex, epileptogenesis, temporal lobe epilepsy
Citation: Vismer MS, Forcelli PA, Skopin MD, Gale K and Koubeissi MZ (2015) The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circuits 9:27. doi: 10.3389/fncir.2015.00027
Received: 03 November 2014; Accepted: 15 May 2015;
Published: 29 May 2015.
Edited by:
Takao K. Hensch, Harvard University, USAReviewed by:
Igor Timofeev, Laval University, CanadaFrances Jensen, University of Pennsylvania, USA
Copyright © 2015 Vismer, Forcelli, Skopin, Gale and Koubeissi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohamad Z. Koubeissi, Department of Neurology, Medical Faculty Associates, Epilepsy Center, The George Washington University, 2150 Pennsylvania Ave., NW; #9-405, Washington, DC 20037, USA,bWtvdWJlaXNzaUBtZmEuZ3d1LmVkdQ==