 Qiang Wang
Qiang Wang Wangming Zhang
Wangming Zhang- The National Key Clinic Specialty, Guangdong Provincial Key Laboratory on Brain Function Repair and Regeneration, The Engineering Technology Research Center of Education Ministry of China, Department of Neurosurgery, Zhujiang Hospital, Southern Medical University, Guangzhou, China
The emergence of L-DOPA-induced dyskinesia (LID) in patients with Parkinson disease (PD) could be due to maladaptive plasticity of corticostriatal synapses in response to L-DOPA treatment. A series of recent studies has revealed that LID is associated with marked morphological plasticity of striatal dendritic spines, particularly cell type-specific structural plasticity of medium spiny neurons (MSNs) in the striatum. In addition, evidence demonstrating the occurrence of plastic adaptations, including aberrant morphological and functional features, in multiple components of cortico-basal ganglionic circuitry, such as primary motor cortex (M1) and basal ganglia (BG) output nuclei. These adaptations have been implicated in the pathophysiology of LID. Here, we briefly review recent studies that have addressed maladaptive plastic changes within the cortico-BG loop in dyskinetic animal models of PD and patients with PD.
Introduction
Parkinson’s disease (PD) is characterized by severe, progressive degeneration of nigrostriatal dopamine (DA) neurons, which results in motor deficits, including akinesia, rigidity, tremor and postural dysfunction. These clinical manifestations can be ameliorated by pharmacological stimulation of DA biosynthesis with exogenous supplementation of L-DOPA (i.e., levodopa), the metabolic precursor to DA. Unfortunately, however, most patients who take L-DOPA also experience adverse secondary effects, including L-DOPA-induced dyskinesia (LID; Voon et al., 2009).
The striatum is the principal recipient of cortical efferents within the basal ganglia (BG). Hence, it serves as a main entryway of information from the neocortex to the BG in the BG-thalamo-cortical network. Furthermore, the striatum, which is composed of functionally and anatomically distinct dorsal and ventral divisions, is also the primary target of DA neurons of the substantia nigra pars compacta. In recent decades, impaired striatal function due to maladaptive synaptic plasticity has been implicated mechanistically in several movement disorders, including LID (Picconi et al., 2003; Paillé et al., 2010).
Recent methodological breakthroughs, such as bacterial artificial chromosome (BAC) and optogenetic techniques, have enabled researchers to not only mimic features of motor disorders in a controlled manner in relation to the extent of nigrostriatal degeneration, but also to further delineate and examine the functions of the direct- and indirect-pathway medium spiny neurons (MSNs; also known as spiny projection neurons) in the striatum (Cui et al., 2013; Fieblinger et al., 2014).
Apart from affecting corticostriatal synaptic plasticity, L-DOPA induced adaptions elsewhere in cortico-basal ganglionic circuitry have come to light (Cenci and Lundblad, 2006). Indeed, several studies have produced evidence suggesting that maladaptive synaptic plasticity processes throughout the cortico-basal ganglionic circuitry may be of critical importance to the pathophysiology of LID (Prescott et al., 2014; Ueno et al., 2014). The latest seminal studies addressing these plastic changes in dyskinetic animal models and patients are discussed below.
DA-Dependent Synaptic Plasticity in Corticostriatal Synapses
The neuronal population in the striatum consists in large majority (~95%) of MSNs, which are projection cells characterized by the spiny cytoarchitecture of their dendritic trees, where synaptic plasticity occurs (Cenci et al., 2011). Striatal MSNs can be divided into two similarly sized populations based on their axonal projections: direct pathway SPNs (dMSNs) and indirect pathway MSNs (iMSNs).
Two forms of plasticity in corticostriatal synapses on MSNs have been characterized extensively long-term potentiation (LTP) and long-term depression (LTD). LTP and LTD describe persistent changes in the efficacy of synaptic transmission that are induced by repetitive activation of cortical excitatory afferents. A unique characteristic of striatal MSNs is that DA plays a critical role in both the induction and maintenance of their neuroplasticity.
Activity-dependent LTD induction is associated with the postsynaptic generation of endocannabinoids (eCBs). This eCB-dependent form of LTD (eCB-LTD) is seen in D2R-expressing iMSNs, but not D1R-expressing dMSNs (Kreitzer and Malenka, 2007). Meanwhile, activation of D2Rs on iMSNs restrains local type 2a adenosine receptor (A2AR) signaling. Spike-timing-dependent plasticity experiments have suggested that local A2AR activation can inhibit both eCB synthesis and LTD induction (Shen et al., 2008). Signaling via D2Rs and A2ARs appears to be linked to eCB-LTD through cAMP/protein kinase A (PKA) and regulator of G protein signaling 4 (RGS4; Lerner and Kreitzer, 2012).
LTP can be induced in MSNs by high-frequency stimulation (HFS) of glutamatergic inputs that results in co-activation of D1Rs and N-methyl-D-aspartate type glutamate receptors (NMDARs; Calabresi et al., 2007; Surmeier et al., 2014). In striatal MSNs, D1Rs co-localize with NMDARs and form heteromeric complexes on dendritic spines (Fiorentini et al., 2006; Calabresi et al., 2010). These D1R/NMDAR complexes facilitate rapid trafficking of NMDAR subunits and modulate the potentiation of NMDAR responses, giving rise to activity-dependent synaptic plasticity changes that involve PKA and dopamine- and cAMP-regulated phosphoprotein (DARPP)-32-regulated phosphorylation of the NR1 subunits of NMDARs (Fiorentini et al., 2008; Murphy et al., 2014).
In experimental models of the LID, a form of synaptic plasticity known as depotentiation has been observed; depotentiation reverses LTP and may represent a homeostatic mechanism (Picconi et al., 2003, 2008). Although the specific mechanisms responsible for homeostatic depotentiation are not yet known, several recent studies have provided important pieces of information (Table 1). One of the most reproducible observations has been that positive allostericmodulator, which modulates M4Rs, enables depotentiation in dSPNs by suppressing RGS4 signaling (Shen et al., 2015). Additionally, extracellular signal-regulated kinase (ERK) signaling has been reported to facilitate depotentiation under normal (unaltered) conditions, but to oppose it under dyskinetic conditions (Cerovic et al., 2015). Striatal synaptic depotentiation could be restored in a subset of striatal MSNs by 5-HT1A/1B receptor agonism with eltoprazine via a mechanism that involved normalization of D1R-dependent cAMP/PKA and ERK/mTORC signaling pathways and recovery of NMDAR subunit balance (Ghiglieri et al., 2016). Mice lacking D-aspartate oxidase (Ddo−/−) display high levels of free D-aspartate and NMDA, which stimulate NMDAR transmission; a low-frequency stimulation protocol failed to depotentiate HFS-induced LTP in Ddo−/− mice (Errico et al., 2011). Finally, nociceptin/orphanin FQ, the endogenous agonist of the nociception receptor, has been shown to prevent D1R agonism-induced ERK phosphorylation and loss of depotentiation in MSNs (Marti et al., 2012). These findings provide insights into the mechanism of striatal neuron depotentiation and could, eventually, lead to novel therapeutic strategies for alleviating LID.
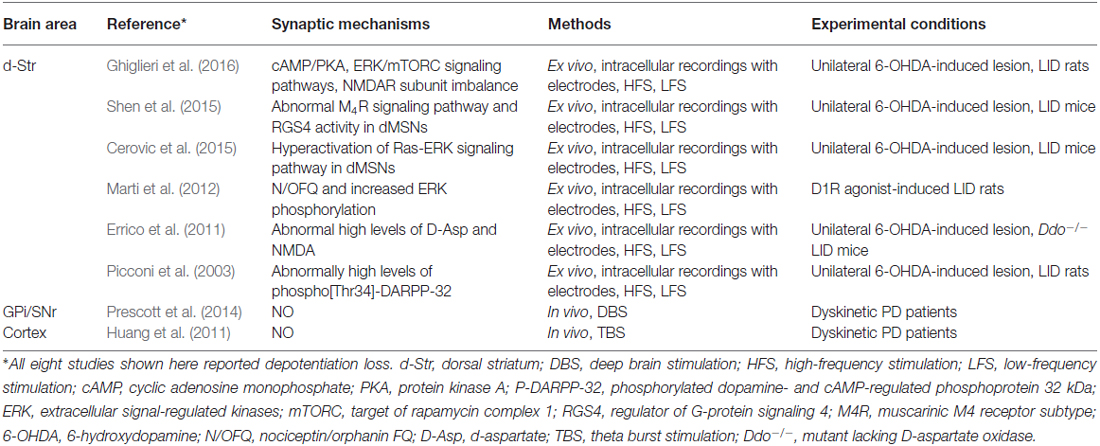
Table 1. Summary of maladaptive depotentiation plasticity in cortico-basal ganglionic circuitry in LID.
Chronic L-Dopa-Induced Corticostriatal-Synapse Adaptations in LID
In a DA-depleted striatum that is being supplemented with L-DOPA, DA striatal levels can be preserved through sprouting of DA terminals and decreased DA uptake by DA transporters (Lee et al., 2008). Distinct degrees of DA denervation affect the induction and maintenance of two distinct forms of corticostriatal synaptic plasticity differently. Initially, DA depletion affects NMDAR-dependent LTP exclusively; with further depletion, sufficient to produce clinical symptoms, LTD is also influenced (Paillé et al., 2010).
As PD degeneration advances, nigrostriatal neurons lose their DA storage capacity, resulting in unregulated DA release and large fluctuations in extracellular DA levels (Rylander et al., 2010). The consequent large fluctuations in extracellular DA concentrations contribute to the establishment of further morphological and functional changes at both pre- and postsynaptic levels. Considerable attention has been devoted to the participation of persistent sensitization of canonical signaling downstream of D1R in the development and manifestation of dyskinesia. In striatal neurons, D1R activates adenylyl-cyclase through G proteins (Gαolf). Increased Gαolf levels has been associated with LID both unilateral lesion mice model (Alcacer et al., 2012) and PD patients who had received a chronic L-dopa treatment (Corvol et al., 2004). Another signaling component leading to the abnormal D1R-mediated transmission involved in LID is the adenylyl cyclase type 5 (AC5), which is highly expressed in striatal MSNs (Glatt and Snyder, 1993; Mons and Cooper, 1994). Recently, Park et al. (2014) found that AC5 knock-out mice exhibits attenuated LID by inhibition of cAMP as well as ERK signaling. In addition, several lines of evidence indicate that abnormal activation of PKA-mediated phosphorylation of DARPP-32 at T34 (Picconi et al., 2003; Santini et al., 2007; Lebel et al., 2010) and PKA dependent phosphorylation of GluA1 at Ser845 (Santini et al., 2007, 2010) in dyskinesia may have profound repercussions on synaptic plasticity (Figure 1).
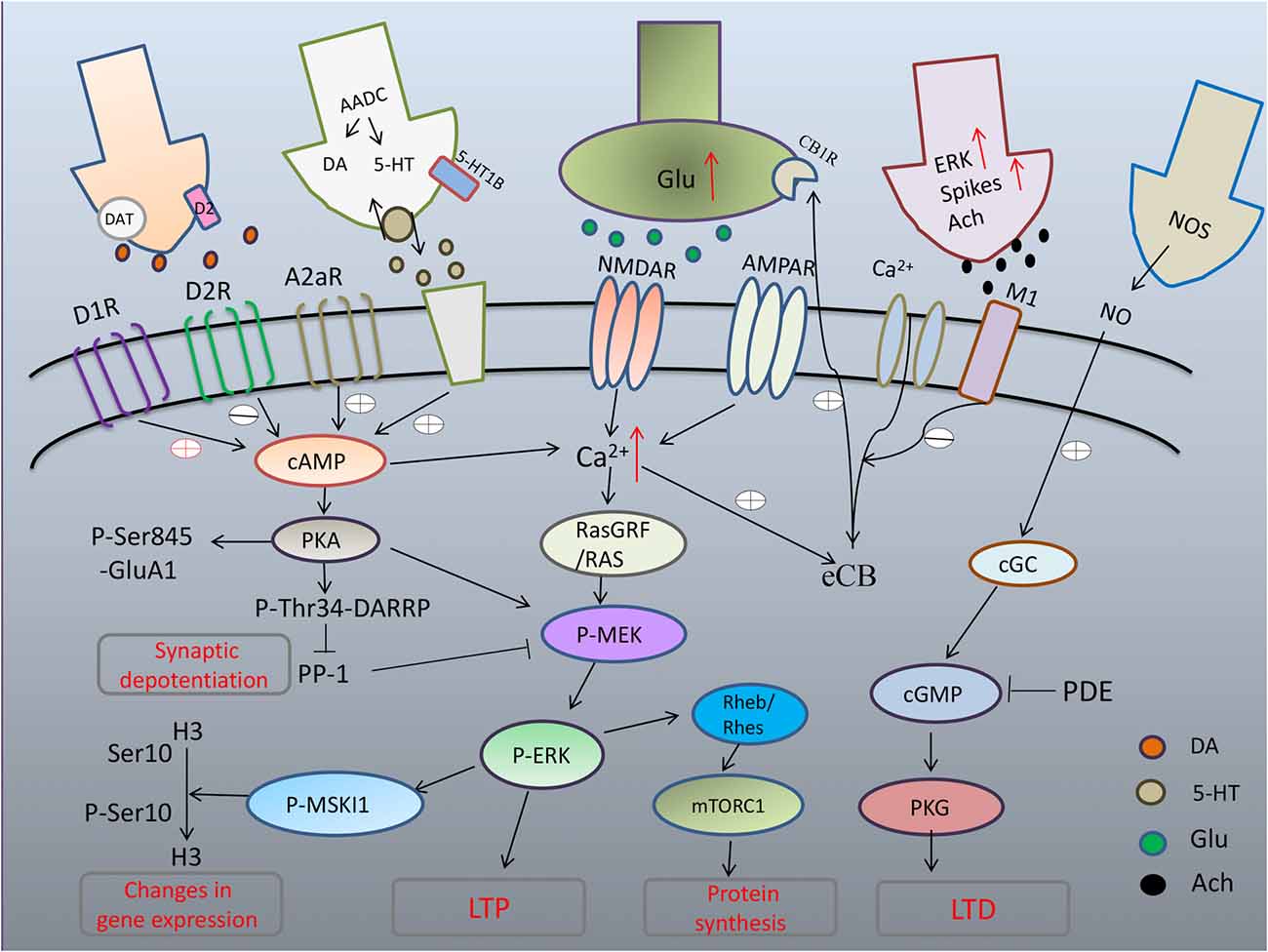
Figure 1. Maladaptive corticostriatal synaptic plasticity mechanism in L-DOPA-induced dyskinesia (LID). Excess of dopamine (DA) abnormally stimulates D1 pathway with hyperphosphorylation of extracellular signal-regulated kinase (ERK) and uncontrolled activation of protein kinase A (PKA) that leads to hyperphosphorylation of DARPP32, which blocks PP-1 causing loss of synaptic depotentiation. PKA/DARPP-32 and ERK/mitogen- and stress-activated kinase 1 (MSK1) signaling lead to phosphorylation of histone H3 in the nucleus, inducing changes in gene expression. Hyperactivation of ERK through convergent altered signaling pathways brings to increased inhibition of tuberous sclerosis complex (TSC)1/2, and consequent disinhibition of Rheb/Rhes, leading to excessive increase of signaling of mTORC1 that, in turn, exerts its long term effects through changes in protein synthesis. After chronic L-DOPA, cholinergic interneurons show increased phospho-ERK immunoreactivity and higher firing rates with increased release of acetylcholine (Ach). Striatal cGMP signaling is declined and activity-dependent LTD, which strictly relies on the nitric-oxide- (NO-) dependent activation of protein kinase G (PKG) is absent in LID.
Newer evidence indicates that D1R do crosstalk to glutamate signaling (mainly NMDA receptors), which are strictly correlated to abnormal synaptic plasticity and motor behavior in L-DOPA-treated dyskinetic rats. ERK dependent signaling and its downstream targets, including molecules involved in the regulation of protein translation and gene transcription, was shown to be apparently hyperactivated in DA-denervated striatum already by a single administration of L-dopa and chronic L-dopa administration (Pavón et al., 2006). L-DOPA produces pronounced activation of ERK1/2 signaling through D1 class of DA receptors. Phosphorylation of ERK1/2 and mitogen- and stress-activated kinase 1 (MSK1), a downstream target of ERK1/2, was dose-dependently blocked by the D1R antagonist, SCH23390 (Westin et al., 2007). Further to these results, (Darmopil et al., 2009) revealed that genetic inactivation of dopamine D1 but not D2 receptors inhibits LID and suppress ERK phosphorylation, phospho-acetylation of Histone H3 (pAcH3; a direct substrate of MSK-1) and FosB/ΔFosB accumulation. Recent evidence indicates that MSK1 could be involved in LID. Genetic inactivation of MSK1 attenuated LID and reduced the phosphorylation of histone H3 at Ser10 in the striatum (Feyder et al., 2016). Similarly to ERK, dopamine D1 receptor-mediated activation of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) occurs in mice that developed dyskinesia (Santini et al., 2009). Remarkably, an upstream component of the mTOR pathway, the Ras homolog enriched in striatum (Rhes), is critically involved in the pathological upregulation of mTORC1 during LID (Subramaniam et al., 2011).
In control condition, concomitant activation of DA D2 receptors and blockade of A2A adenosine receptors is able to reduce striatal glutamatergic transmission via a retrograde action of endocannabinoid-dependent mechanism (Tozzi et al., 2011). Alterations in A2A receptor expression and signaling have been observed in PD patients undergoing L-DOPA therapy (Calon et al., 2004; Ramlackhansingh et al., 2011) and in experimental models of LID (Pinna et al., 2002; Blandini and Armentero, 2012). Recently, G-protein-coupled adenosine A2A, cannabinoid CB1 and dopamine D2 receptors (A2A-CB1-D2 receptor heteromers) has been unraveled. This heteromer, present in normal and DA-depleted striatum, is however lost following acute or chronic treatment with L-dopa in rats and monkeys (Bonaventura et al., 2014; Pinna et al., 2014).
Early studies founded altered acetylcholine signaling in dopamine depletion striatum resulting in a loss of feedback control of acetylcholine release (Kayadjanian et al., 1999; Ding et al., 2006). Notably, striatal cholinergic interneurons, are involved in the D2/A2A and endocannabinoid-mediated retrograde effects. Concomitant activation of D2 DA receptors and blockade of A2A receptors reduces the firing rate of these interneurons and primary motor cortex (M1) receptor antagonism blocks the D2/A2A receptor-mediated modulation of excitatory transmission in both dMSN and iMSN (Tozzi et al., 2011). In 6-hydroxydopamine lesion mice, repeated L-DOPA treatment increases basal firing rate and stronger excitatory responses to dopamine in striatal cholinergic neurons with increased phospho-ERK immunoreactivity in this neuronal population (Ding et al., 2011). Taken together, these data suggest increased dopamine sensitivity of striatal cholinergic neurons contributes to the expression of LID.
An in vivo electrophysiological study demonstrated distinct effects of chronic L-DOPA administration on dMSNs vs. iMSNs in DA-depleted rats. The dMSNs had abnormally persistent cortically-evoked LTD, whereas the iMSNs exhibited LTP, rather than LTD, in response to the same stimulation (Belujon et al., 2010). Such findings suggest that LID might be caused by cell type-specific altered induction of plasticity in striatal MSNs (Calabresi et al., 2015). In a dyskinetic state, the direct pathway exhibits only LTP, while the indirect pathway exhibits only LTD. By contrast, in a parkinsonian state, the indirect pathway exhibits only LTP and the direct pathway exhibits only LTD. These pathophysiological changes are associated with a loss of bidirectional plasticity, such that only unidirectional changes in synaptic strength can occur (Thiele et al., 2014).
Apart from the major direct vs. indirect divisions of the striatal MSN population, the striatal circuitry can also be divided biochemically into two striatal compartments known as the striosomes (also known as striatal patches) and matrix. It has been proposed that neurodegenerative dysfunctions of the BG, such as LID, could involve a striosome-matrix imbalance (Crittenden and Graybiel, 2011). A recent article shows that electrically evoked dopamine release differs between the striosome and matrix compartments in a regionally-distinct manner. In the VS (ventral lateral striatum and nucleus accumbens), dopamine release in striosomes is greater than in the proximal matrix region, and in the DS (medial and lateral), the opposite is true (Salinas et al., 2016). A recent study reveals that neuromodulator substance P acting through neurokinin-1 receptor (NK1Rs) can boost DA release within the centers of striosomes, but diminish DA release in a border region where striosomes and matrix interface (Brimblecombe and Cragg, 2015). If this notion were to be confirmed with further studies, it could have explanatory implications for a range of motor and nonmotor symptoms associated with BG neurodegeneration.
Structural Plasticity of Striatal Dendritic Spines in LID
The spiny dendrites of MSNs—the smallest processing units of biochemical signals generated at corticostriatal synapses—integrate synaptic afferents from different origins (Chen and Sabatini, 2012; Yuste, 2013). PD patients exhibit morphological changes in their striatal dendritic spines, including spine loss (McNeill et al., 1988). Using two-photon laser scanning microscopy and BAC transgenic mice, Day et al. (2006) observed that DA depletion led to a selective loss of spines and glutamatergic synapses on iMSNs, but not dMSNs.
Accumulating evidence indicates that synaptic plasticity consequent to morphologic changes in dendritic spines may be key to resolving the mechanisms underlying both PD and dyskinesia (Fieblinger and Cenci, 2015). Zhang et al. (2013) observed that PD model rats have abnormally few synapses and corticostriatal multisynaptic boutons (contacting dendritic shafts or a shaft and a spine), whereas LID model rats show a restoration in the total number of corticostriatal synapses and high densities of mushroom spines (enlarged postsynaptic densities receiving multisynaptic excitatory input). It is unclear yet whether the newly sprouted spines in the rat model form functional synapses.
Interestingly, iMSNs appear to re-grow spines that had been lost due to loss of dopaminergic innervation (Fieblinger et al., 2014; Suarez et al., 2014). The behavior of dMSNs following DA depletion, however, is less clear. Both Fieblinger et al. (2014) and Nishijima et al. (2014) did not see changes in dMSN density following a DA-denervating lesion; rather, they observed decreases in dMSN density in response to long-term L-DOPA exposure. On the contrary, Suarez et al. (2014) suggested that dMSN spine loss occurs after DA depletion, with chronic L-DOPA having no effect. It is possible that these discrepancies could be related to methodological differences, such as differences in the models or neurotoxin injection sites employed.
In summary, MSN dendrite atrophy should be regarded as a potential therapeutic target for PD. Meanwhile, LID might be the result of an L-DOPA induced mis-rewiring of corticostriatal synapses (Zhang et al., 2013; Fieblinger et al., 2014). This hypothesis should be examined further, especially with respect to cell-type specificity and the corresponding changes in intrinsic excitability (Fieblinger et al., 2014; Surmeier et al., 2014).
Plastic Adaptations in Other Cortico-Basal Ganglion-Thalamic Circuitry in LID
The synaptic and molecular rearrangements that occur in LID have been relatively well studied in the striatum. However, plastic adaptations occurring in other parts of the cortico-basal ganglionic circuitry have received less attention. There is a growing appreciation for activity-dependent synaptic plasticity throughout the cortico-basal ganglionic loop.
The striatum is the major input station of glutamatergic innervation arising from the cortex and the thalamus. However, plastic adaptations occurring in thalamostriatal system have been poorly explored, but are likely to play an important role (Smith et al., 2014; Tritsch and Carter, 2016). Parker et al. (2016) find that dopamine depletion selectively reduces thalamostriatal drive in dMSNs mediated by AMPA rather than NMDA receptors. Combination of in vivo pharmacogenetics and optogenetics, the authors reveal that inhibition of thalamostriatal inputs rescues PD motor behavior, implicating maladaptive synaptic plasticity in the thalamus as playing a key role in dopamine depletion animal.
Prescott et al. (2009) reported that extrastriatal DA modulates activity-dependent synaptic plasticity in the BG output neurons of the SNr. HFS induced LTP-like potentiation of field-evoked potential amplitudes when delivered with, but not without, L-DOPA administration. Interestingly, in a more recent study, the same research group suggested that the ability of BG output nuclei to undergo depotentiation might be selectively lost in patients who develop LID (Prescott et al., 2014). Their work suggests that depotentiation in the SNr and GPi—the output n of the BG—may suppress nonessential synaptic information while integrating and normalizing signals that are to be relayed out to the thalamo-cortical network.
Ueno et al. (2014) observed that intratelencephalic-type pyramidal neurons (which project to dMSNs) in M1 of LID model rats had enlarged spines and elevated miniature excitatory postsynaptic current amplitudes. These morphological and electrophysiological changes in intratelencephalic-type pyramidal neurons in M1 could explain on a cellular level, at least in part, the loss of depotentiation-like plasticity that occurs in PD patients with LID.
Huang et al. (2011) found that depotentiation could not be induced following HFS-induced LTP-like plasticity in the M1 of PD patients with LID. Recent findings suggest that alterations in cerebellar sensory processing function may be an important contributor to maladaptive sensorimotor plasticity in M1 (Popa et al., 2013). Kishore et al. (2014) proposed that loss of M1 plasticity may reflect a loss of co-ordination among BG, cerebellar, and cortical inputs, resulting in abnormal plasticity of motor maps within M1 and, eventually, the involuntary movements characteristic of LID.
Concluding Remarks
L-DOPA-induced changes in the plasticity of corticostriatal synapses are key to understanding the pathophysiology of LID. LID-associated changes in the synaptic and molecular biology of the striatum have been well described, whereas our understanding of activity-dependent synaptic plasticity elsewhere in the BG-thalamo-cortical network in LID is relatively undeveloped. Notably promising research areas moving forward include cell type-specific structural plasticity of striatal MSN dendritic spines, the mis-rewiring hypothesis for LID, and homeostatic adaptations in the intrinsic excitability and synaptic connectivity of striatal MSNs.
Author Contributions
QW read related references and wrote this manuscript. WZ gave several guidance about this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81272150). Additionally, the authors are very grateful for assistance from The Guangdong Provincial Clinical Medical Center for Neurosurgery (No. 2013B020400005).
References
Alcacer, C., Santini, E., Valjent, E., Gaven, F., Girault, J. A., and Hervé, D. (2012). Gαolf mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J. Neurosci. 32, 5900–5910. doi: 10.1523/JNEUROSCI.0837-12.2012
Belujon, P., Lodge, D. J., and Grace, A. A. (2010). Aberrant striatal plasticity is specifically associated with dyskinesia following levodopa treatment. Mov. Disord. 25, 1568–1576. doi: 10.1002/mds.23245
Blandini, F., and Armentero, M. T. (2012). New pharmacological avenues for the treatment of L-DOPA-induced dyskinesias in Parkinson’s disease: targeting glutamate and adenosine receptors. Expert Opin. Investig. Drugs 21, 153–168. doi: 10.1517/13543784.2012.651457
Bonaventura, J., Rico, A. J., Moreno, E., Sierra, S., Sánchez, M., Luquin, N., et al. (2014). L-DOPA-treatment in primates disrupts the expression of A2A adenosine-CB1 cannabinoid-D2 dopamine receptor heteromers in the caudate nucleus. Neuropharmacology 79, 90–100. doi: 10.1016/j.neuropharm.2013.10.036
Brimblecombe, K. R., and Cragg, S. J. (2015). Substance P weights striatal dopamine transmission differently within the striosome-matrix axis. J. Neurosci. 35, 9017–9023. doi: 10.1523/JNEUROSCI.0870-15.2015
Calabresi, P., Di Filippo, M., Ghiglieri, V., Tambasco, N., and Picconi, B. (2010). Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 9, 1106–1117. doi: 10.1016/s1474-4422(10)70218-0
Calabresi, P., Ghiglieri, V., Mazzocchetti, P., Corbelli, I., and Picconi, B. (2015). Levodopa-induced plasticity: a double-edged sword in Parkinson’s disease? Philos. Trans. R. Soc. Lond. B Biol. Sci. 370:1672. doi: 10.1098/rstb.2014.0184
Calabresi, P., Picconi, B., Tozzi, A., and Di Filippo, M. (2007). Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219. doi: 10.1016/j.tins.2007.03.001
Calon, F., Dridi, M., Hornykiewicz, O., Bédard, P. J., Rajput, A. H., and Di Paolo, T. (2004). Increased adenosine A2A receptors in the brain of Parkinson’s disease patients with dyskinesias. Brain 127, 1075–1084. doi: 10.1093/brain/awh128
Cenci, M. A., and Lundblad, M. (2006). Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J. Neurochem. 99, 381–392. doi: 10.1111/j.1471-4159.2006.04124.x
Cenci, M. A., Ohlin, K. E., and Odin, P. (2011). Current options and future possibilities for the treatment of dyskinesia and motor fluctuations in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 10, 670–684. doi: 10.2174/187152711797247885
Cerovic, M., Bagetta, V., Pendolino, V., Ghiglieri, V., Fasano, S., Morella, I., et al. (2015). Derangement of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and extracellular signal-regulated kinase (ERK) dependent striatal plasticity in L-DOPA-induced dyskinesia. Biol. Psychiatry 77, 106–115. doi: 10.1016/j.biopsych.2014.04.002
Chen, Y., and Sabatini, B. L. (2012). Signaling in dendritic spines and spine microdomains. Curr. Opin. Neurobiol. 22, 389–396. doi: 10.1016/j.conb.2012.03.003
Corvol, J. C., Muriel, M. P., Valjent, E., Féger, J., Hanoun, N., Girault, J. A., et al. (2004). Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J. Neurosci. 24, 7007–7014. doi: 10.1523/JNEUROSCI.0676-04.2004
Crittenden, J. R., and Graybiel, A. M. (2011). Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5:59. doi: 10.3389/fnana.2011.00059
Cui, G., Jun, S. B., Jin, X., Pham, M. D., Vogel, S. S., Lovinger, D. M., et al. (2013). Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242. doi: 10.1038/nature11846
Darmopil, S., Martín, A. B., De Diego, I. R., Ares, S., and Moratalla, R. (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613. doi: 10.1016/j.biopsych.2009.04.025
Day, M., Wang, Z., Ding, J., An, X., Ingham, C. A., Shering, A. F., et al. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 9, 251–259. doi: 10.1038/nn1632
Ding, J., Guzman, J. N., Tkatch, T., Chen, S., Goldberg, J. A., Ebert, P. J., et al. (2006). RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat. Neurosci. 9, 832–842. doi: 10.1038/nn1700
Ding, Y., Won, L., Britt, J. P., Lim, S. A., McGehee, D. S., and Kang, U. J. (2011). Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proc. Natl. Acad. Sci. U S A 108, 840–845. doi: 10.1073/pnas.1006511108
Errico, F., Bonito-Oliva, A., Bagetta, V., Vitucci, D., Romano, R., Zianni, E., et al. (2011). Higher free D-aspartate and N-methyl-D-aspartate levels prevent striatal depotentiation and anticipate L-DOPA-induced dyskinesia. Exp. Neurol. 232, 240–250. doi: 10.1016/j.expneurol.2011.09.013
Feyder, M., Södersten, E., Santini, E., Vialou, V., LaPlant, Q., Watts, E. L., et al. (2016). A role for mitogen- and stress-activated kinase 1 in L-DOPA-induced dyskinesia and FosB expression. Biol. Psychiatry 79, 362–371. doi: 10.1016/j.biopsych.2014.07.019
Fieblinger, T., and Cenci, M. A. (2015). Zooming in on the small: the plasticity of striatal dendritic spines in L-DOPA-induced dyskinesia. Mov. Disord. 30, 484–493. doi: 10.1002/mds.26139
Fieblinger, T., Graves, S. M., Sebel, L. E., Alcacer, C., Plotkin, J. L., Gertler, T. S., et al. (2014). Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat. Commun. 5:5316. doi: 10.1038/ncomms6316
Fiorentini, C., Busi, C., Spano, P., and Missale, C. (2008). Role of receptor heterodimers in the development of L-dopa-induced dyskinesias in the 6-hydroxydopamine rat model of Parkinson’s disease. Parkinsonism Relat. Disord. 14, S159–S164. doi: 10.1016/j.parkreldis.2008.04.022
Fiorentini, C., Rizzetti, M. C., Busi, C., Bontempi, S., Collo, G., Spano, P., et al. (2006). Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol. Pharmacol. 69, 805–812. doi: 10.1124/mol.105.016667
Ghiglieri, V., Mineo, D., Vannelli, A., Cacace, F., Mancini, M., Pendolino, V., et al. (2016). Modulation of serotonergic transmission by eltoprazine in L-DOPA-induced dyskinesia: behavioral, molecular and synaptic mechanisms. Neurobiol. Dis. 86, 140–153. doi: 10.1016/j.nbd.2015.11.022
Glatt, C. E., and Snyder, S. H. (1993). Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature 361, 536–538. doi: 10.1038/361536a0
Huang, Y. Z., Rothwell, J. C., Lu, C. S., Chuang, W. L., and Chen, R. S. (2011). Abnormal bidirectional plasticity-like effects in Parkinson’s disease. Brain 134, 2312–2320. doi: 10.1093/brain/awr158
Kayadjanian, N., Schofield, W. N., Andren, J., Sirinathsinghji, D. J., and Besson, M. J. (1999). Cortical and nigral deafferentation and striatal cholinergic markers in the rat dorsal striatum: different effects on the expression of mRNAs encoding choline acetyltransferase and muscarinic m1 and m4 receptors. Eur. J. Neurosci. 11, 3659–3668. doi: 10.1046/j.1460-9568.1999.00788.x
Kishore, A., Popa, T., Balachandran, A., Chandran, S., Pradeep, S., Backer, F., et al. (2014). Cerebellar sensory processing alterations impact motor cortical plasticity in Parkinson’s disease: clues from dyskinetic patients. Cereb. Cortex 24, 2055–2067. doi: 10.1093/cercor/bht058
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647. doi: 10.1038/nature05506
Lebel, M., Chagniel, L., Bureau, G., and Cyr, M. (2010). Striatal inhibition of PKA prevents levodopa-induced behavioural and molecular changes in the hemiparkinsonian rat. Neurobiol. Dis. 38, 59–67. doi: 10.1016/j.nbd.2009.12.027
Lee, J., Zhu, W. M., Stanic, D., Finkelstein, D. I., Horne, M. H., Henderson, J., et al. (2008). Sprouting of dopamine terminals and altered dopamine release and uptake in Parkinsonian dyskinaesia. Brain 131, 1574–1587. doi: 10.1093/brain/awn085
Lerner, T. N., and Kreitzer, A. C. (2012). RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron 73, 347–359. doi: 10.1016/j.neuron.2011.11.015
Marti, M., Rodi, D., Li, Q., Guerrini, R., Fasano, S., Morella, I., et al. (2012). Nociceptin/orphanin FQ receptor agonists attenuate L-DOPA-induced dyskinesias. J. Neurosci. 32, 16106–16119. doi: 10.1523/JNEUROSCI.6408-11.2012
McNeill, T. H., Brown, S. A., Rafols, J. A., and Shoulson, I. (1988). Atrophy of medium spiny I striatal dendrites in advanced Parkinson’s disease. Brain Res. 455, 148–152. doi: 10.1016/0006-8993(88)90124-2
Mons, N., and Cooper, D. M. (1994). Selective expression of one Ca2+-inhibitable adenylyl cyclase in dopaminergically innervated rat brain regions. Mol. Brain Res. 22, 236–244. doi: 10.1016/0169-328x(94)90052-3
Murphy, J. A., Stein, I. S., Lau, C. G., Peixoto, R. T., Aman, T. K., Kaneko, N., et al. (2014). Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. J. Neurosci. 34, 869–879. doi: 10.1523/JNEUROSCI.4538-13.2014
Nishijima, H., Suzuki, S., Kon, T., Funamizu, Y., Ueno, T., Haga, R., et al. (2014). Morphologic changes of dendritic spines of striatal neurons in the levodopa-induced dyskinesia model. Mov. Disord. 29, 336–343. doi: 10.1002/mds.25826
Paillé, V., Picconi, B., Bagetta, V., Ghiglieri, V., Sgobio, C., Di Filippo, M., et al. (2010). Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J. Neurosci. 30, 14182–14193. doi: 10.1523/JNEUROSCI.2149-10.2010
Park, H. Y., Kang, Y. M., Kang, Y., Park, T. S., Ryu, Y. K., Hwang, J. H., et al. (2014). Inhibition of adenylyl cyclase type 5 prevents L-DOPA-induced dyskinesia in an animal model of Parkinson’s disease. J. Neurosci. 34, 11744–11753. doi: 10.1523/JNEUROSCI.0864-14.2014
Parker, P. R., Lalive, A. L., and Kreitzer, A. C. (2016). Pathway-specific remodeling of thalamostriatal synapses in parkinsonian mice. Neuron 89, 734–740. doi: 10.1016/j.neuron.2015.12.038
Pavón, N., Martín, A. B., Mendialdua, A., and Moratalla, R. (2006). ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 59, 64–74. doi: 10.1016/j.biopsych.2005.05.044
Picconi, B., Centonze, D., Håkansson, K., Bernardi, G., Greengard, P., Fisone, G., et al. (2003). Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 6, 501–506. doi: 10.1038/nn1040
Picconi, B., Paillé, V., Ghiglieri, V., Bagetta, V., Barone, I., Lindgren, H. S., et al. (2008). L-DOPA dosage is critically involved in dyskinesia via loss of synaptic depotentiation. Neurobiol. Dis. 29, 327–335. doi: 10.1016/j.nbd.2007.10.001
Pinna, A., Bonaventura, J., Farré, D., Sánchez, M., Simola, N., Mallol, J., et al. (2014). L-DOPA disrupts adenosine A2A-cannabinoid C1-dopamine D2 receptor heteromer cross-talk in the striatum of hemiparkinsonian rats: biochemical and behavioral studies. Exp. Neurol. 253, 180–191. doi: 10.1016/j.expneurol.2013.12.021
Pinna, A., Corsi, C., Carta, A. R., Valentini, V., Pedata, F., and Morelli, M. (2002). Modification of adenosine extracellular levels and adenosine A2A receptor mRNA by dopamine denervation. Eur. J. Pharmacol. 446, 75–82. doi: 10.1016/s0014-2999(02)01818-6
Popa, T., Velayudhan, B., Hubsch, C., Pradeep, S., Roze, E., Vidailhet, M., et al. (2013). Cerebellar processing of sensory inputs primes motor cortex plasticity. Cereb. Cortex 23, 305–314. doi: 10.1093/cercor/bhs016
Prescott, I. A., Dostrovsky, J. O., Moro, E., Hodaie, M., Lozano, A. M., and Hutchison, W. D. (2009). Levodopa enhances synaptic plasticity in the substantia nigra pars reticulata of Parkinson’s disease patients. Brain 132, 309–318. doi: 10.1093/brain/awn322
Prescott, I. A., Liu, L. D., Dostrovsky, J. O., Hodaie, M., Lozano, A. M., and Hutchison, W. D. (2014). Lack of depotentiation at basal ganglia output neurons in PD patients with levodopa-induced dyskinesia. Neurobiol. Dis. 71, 24–33. doi: 10.1016/j.nbd.2014.08.002
Ramlackhansingh, A. F., Bose, S. K., Ahmed, I., Turkheimer, F. E., Pavese, N., and Brooks, D. J. (2011). Adenosine 2A receptor availability in dyskinetic and nondyskinetic patients with Parkinson disease. Neurology 76, 1811–1816. doi: 10.1212/WNL.0b013e31821ccce4
Rylander, D., Parent, M., O’Sullivan, S. S., Dovero, S., Lees, A. J., Bezard, E., et al. (2010). Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann. Neurol. 68, 619–628. doi: 10.1002/ana.22097
Salinas, A. G., Davis, M. I., Lovinger, D. M., and Mateo, Y. (2016). Dopamine dynamics and cocaine sensitivity differ between striosome and matrix compartments of the striatum. Neuropharmacology 108, 275–283. doi: 10.1016/j.neuropharm.2016.03.049
Santini, E., Heiman, M., Greengard, P., Valjent, E., and Fisone, G. (2009). Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2:ra36. doi: 10.1126/scisignal.2000308
Santini, E., Sgambato-Faure, V., Li, Q., Savasta, M., Dovero, S., Fisone, G., et al. (2010). Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS One 5:e12322. doi: 10.1371/journal.pone.0012322
Santini, E., Valjent, E., Usiello, A., Carta, M., Borgkvist, A., Girault, J. A., et al. (2007). Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 27, 6995–7005. doi: 10.1523/JNEUROSCI.0852-07.2007
Shen, W., Flajolet, M., Greengard, P., and Surmeier, D. J. (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851. doi: 10.1126/science.1160575
Shen, W., Plotkin, J. L., Francardo, V., Ko, W. K., Xie, Z., Li, Q., et al. (2015). M4 muscarinic receptor signaling ameliorates striatal plasticity deficits in models of L-DOPA-induced dyskinesia. Neuron 88, 762–773. doi: 10.1016/j.neuron.2015.10.039
Smith, Y., Galvan, A., Ellender, T. J., Doig, N., Villalba, R. M., Huerta-Ocampo, I., et al. (2014). The thalamostriatal system in normal and diseased states. Front. Syst. Neurosci. 8:5. doi: 10.3389/fnsys.2014.00005
Suarez, L. M., Solis, O., Carames, J. M., Taravini, I. R., Solis, J. M., Murer, M. G., et al. (2014). L-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol. Psychiatry 75, 711–722. doi: 10.1016/j.biopsych.2013.05.006
Subramaniam, S., Napolitano, F., Mealer, R. G., Kim, S., Errico, F., Barrow, R., et al. (2011). Rhes, a striatal-enriched small G protein, mediates mTOR signaling and L-DOPA-induced dyskinesia. Nat. Neurosci. 15, 191–193. doi: 10.1038/nn.2994
Surmeier, D. J., Graves, S. M., and Shen, W. (2014). Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr. Opin. Neurobiol. 29, 109–117. doi: 10.1016/j.conb.2014.07.008
Thiele, S. L., Chen, B., Lo, C., Gertler, T. S., Warre, R., Surmeier, J. D., et al. (2014). Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and L-DOPA induced dyskinesia in mouse models. Neurobiol. Dis. 71, 334–344. doi: 10.1016/j.nbd.2014.08.006
Tozzi, A., de Iure, A., Di Filippo, M., Tantucci, M., Costa, C., Borsini, F., et al. (2011). The distinct role of medium spiny neurons and cholinergic interneurons in the D2/A2A receptor interaction in the striatum: implications for Parkinson’s disease. J. Neurosci. 31, 1850–1862. doi: 10.1523/JNEUROSCI.4082-10.2011
Tritsch, N. X., and Carter, A. G. (2016). Parkinson’s disease: a thalamostriatal rebalancing act? Neuron 89, 675–677. doi: 10.1016/j.neuron.2016.02.008
Ueno, T., Yamada, J., Nishijima, H., Arai, A., Migita, K., Baba, M., et al. (2014). Morphological and electrophysiological changes in intratelencephalic-type pyramidal neurons in the motor cortex of a rat model of levodopa-induced dyskinesia. Neurobiol. Dis. 64, 142–149. doi: 10.1016/j.nbd.2013.12.014
Voon, V., Fernagut, P. O., Wickens, J., Baunez, C., Rodriguez, M., Pavon, N., et al. (2009). Chronic dopaminergic stimulation in Parkinson’s disease: from dyskinesias to impulse control disorders. Lancet Neurol. 8, 1140–1149. doi: 10.1016/s1474-4422(09)70287-x
Westin, J. E., Vercammen, L., Strome, E. M., Konradi, C., and Cenci, M. A. (2007). Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol. Psychiatry 62, 800–810. doi: 10.1016/j.biopsych.2006.11.032
Yuste, R. (2013). Electrical compartmentalization in dendritic spines. Annu. Rev. Neurosci. 36, 429–449. doi: 10.1146/annurev-neuro-062111-150455
Zhang, Y., Meredith, G. E., Mendoza-Elias, N., Rademacher, D. J., Tseng, K. Y., and Steece-Collier, K. (2013). Aberrant restoration of spines and their synapses in L-DOPA-induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J. Neurosci. 33, 11655–11667. doi: 10.1523/JNEUROSCI.0288-13.2013
Keywords: Parkinson disease, synaptic plasticity, dopamine, basal ganglia, motor cortex
Citation: Wang Q and Zhang W (2016) Maladaptive Synaptic Plasticity in L-DOPA-Induced Dyskinesia. Front. Neural Circuits 10:105. doi: 10.3389/fncir.2016.00105
Received: 17 February 2016; Accepted: 05 December 2016;
Published: 20 December 2016.
Edited by:
Kuan Hong Wang, National Institute of Mental Health, NIH, USAReviewed by:
Xin Jin, Salk Institute for Biological Studies, USADivya Sitaraman, University of San Diego, USA
Copyright © 2016 Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wangming Zhang, d3poYW5nQHZpcC4xMjYuY29t