 Gabriela L. Carrillo
Gabriela L. Carrillo Jianmin Su
Jianmin Su Aboozar Monavarfeshani
Aboozar Monavarfeshani Michael A. Fox
Michael A. Fox- 1Developmental and Translational Neurobiology Center, Virginia Tech Carilion Research Institute, Roanoke, VA, United States
- 2Graduate Program in Translational Biology, Medicine and Health, Virginia Tech, Blacksburg, VA, United States
- 3Department of Biological Sciences, Virginia Tech, Blacksburg, VA, United States
- 4Department of Pediatrics, Virginia Tech Carilion School of Medicine, Roanoke, VA, United States
The suprachiasmatic nucleus (SCN) is the master pacemaker that drives circadian behaviors. SCN neurons have intrinsic, self-sustained rhythmicity that is governed by transcription-translation feedback loops. Intrinsic rhythms within the SCN do not match the day-night cycle and are therefore entrained by light-derived cues. Such cues are transmitted to the SCN by a class of intrinsically photosensitive retinal ganglion cells (ipRGCs). In the present study, we sought to identify how axons from ipRGCs target the SCN. While none of the potential targeting cues identified appeared necessary for retinohypothalamic innervation, we unexpectedly identified a novel role for the extracellular matrix protein F-spondin in circadian behavior. In the absence of F-spondin, mice lost their ability to maintain typical intrinsic rhythmicity. Moreover, F-spondin loss results in the displacement of vasoactive intestinal peptide (VIP)-expressing neurons, a class of neurons that are essential for maintaining rhythmicity among SCN neurons. Thus, this study highlights a novel role for F-spondin in maintaining circadian rhythms.
Introduction
Most light-sensitive organisms contain biological clocks that entrain biochemical, physiological and behavioral processes with daily fluctuations of the solar day-night cycle. In mammals, circadian rhythmicity results from the cell-autonomous cyclic transcription and translation of tightly regulated core circadian “clock” genes, including period genes (per1, per2, per3), cryptochrome genes (cry1 and cry2), brain and muscle arnt-like 1 (bmal1) and circadian locomotor output cycles kaput (clock, Bell-Pedersen et al., 2005; Dibner et al., 2010; Mohawk et al., 2012). The cyclic expression and activity of proteins encoded by these core “clock” genes do not precisely match the 24-h day-night cycle, but rather must be entrained to changes in this cycle by environmental cues. The principle cue that entrains mammalian circadian rhythms to the solar day-night cycle is light. Environmental lighting conditions are detected and transformed into neural activity by retinal photoreceptors and are relayed to a set of densely packed neurons in the suprachiasmatic nucleus (SCN) of the ventral hypothalamus by retinal ganglion cells (RGCs; Figures 1A,B; Fox and Guido, 2011). The SCN then functions as the master regulator of circadian clocks throughout the body and orchestrates the coordination of molecular and biochemical oscillations in all other tissues (Dibner et al., 2010; Mohawk et al., 2012). Neurons within the SCN generate self-sustained oscillations of the core “clock” machinery and intercellular signaling between these neurons, through both synaptic connections and gap junction coupling, allow for the rapid synchronization of light-induced gene expression and oscillations (Challet et al., 2003; Aton and Herzog, 2005). In the absence of light-derived signals, core “clock” machinery within SCN neurons maintain their oscillatory activity but these oscillations “free-run” and fail to entrain to changes in the solar day-night cycle (Foster, 1998; Brzezinski et al., 2005).
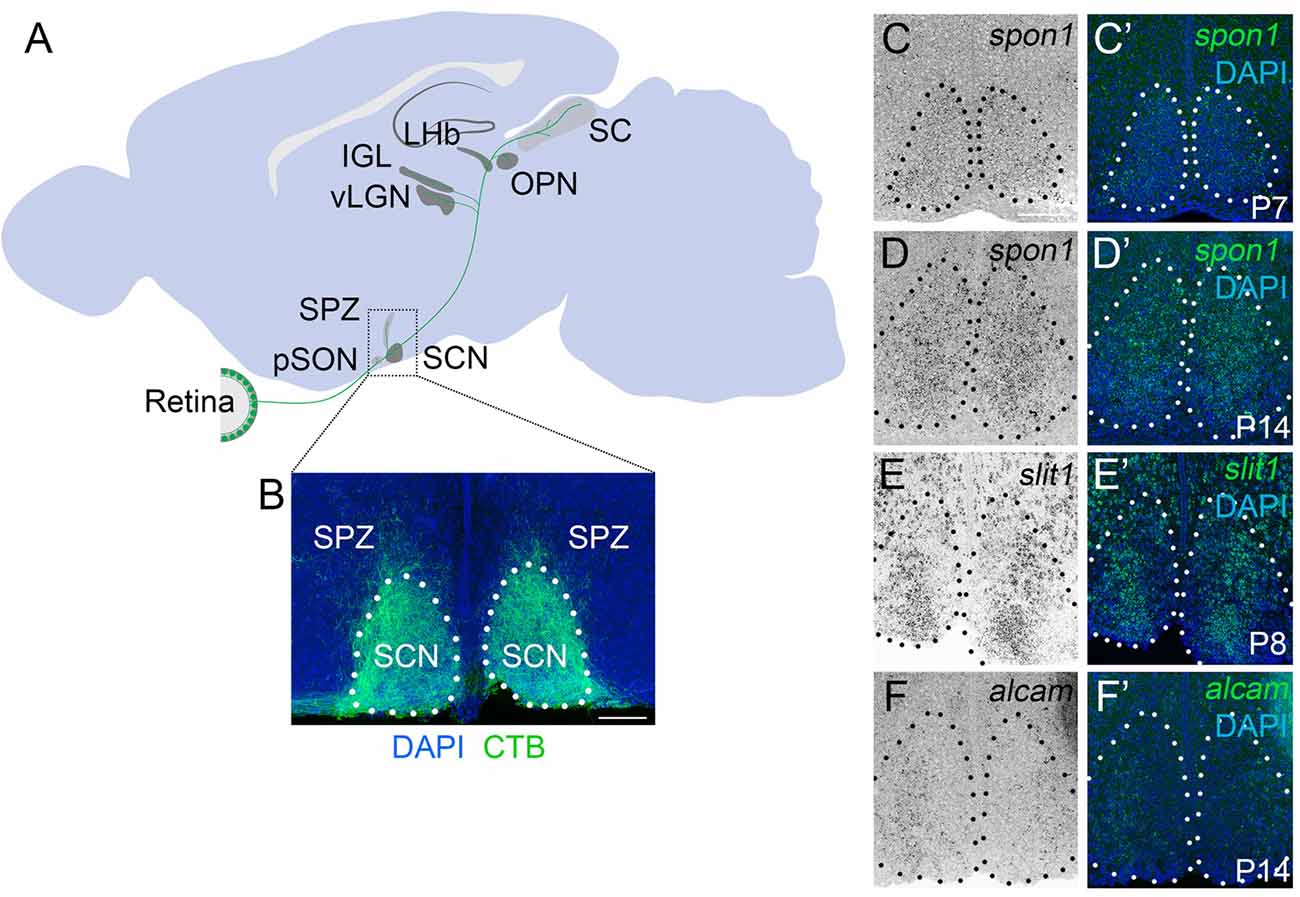
Figure 1. F-spondin,Slit1 and ALCAM are expressed in the developing SCN. (A) Schematic representation of the major retino-recipient targets of M1 intrinsically photosensitive retinal ganglion cell (ipRGC) axons (labeled green). SCN, suprachiasmatic nucleus; pSON, peri-supraoptic nucleus; SPZ, subparaventricular zone; vLGN, ventral lateral geniculate nucleus; IGL, intergeniculate leaflet; LHb, lateral habenula; OPN, olivary pretectal nucleus. (B) Coronal image of cholera toxin subunit B (CTB)-labeled retinal projections in SCN. Scale bar = 100 μm. (C–F) In situ hybridization (ISH) for spon1, slit1 and alcam mRNAs in P7–P14 mouse SCN.(C′–F′) Depict ISH and DAPI-labeling. Black (C–F) and white (C′–F′) dots encircle SCN.Scale bar = 100 μm.
Despite the importance of light-derived signals in entraining SCN neurons, only a small subset of RGCs supply photic information to the SCN (Morin and Studholme, 2014). This small set of RGCs express the photopigment melanopsin and are intrinsically photosensitive (Hattar et al., 2002, 2006). Loss of these intrinsically photosensitive RGCs (ipRGCs) leads to “free-running” circadian rhythms even in the presence of a normal solar day-night cycle (Guler et al., 2008). Despite having been under extreme scrutiny since their discovery almost two decades ago, the cellular and molecular mechanisms responsible for regulating ipRGC axon innervation of the SCN remain unclear (Fox and Guido, 2011). In the present study, we sought to answer this question by identifying and testing SCN-specific axonal targeting cues. Using a bio-informatics approach, we identified F-spondin and Slit1, two extracellular matrix proteins, and ALCAM, a cell adhesion molecule, that were all enriched in the adult SCN. The ability of these three cues to direct axonal growth, guidance and targeting in other regions of the developing brain has been well established (Ott et al., 1998; Burstyn-Cohen et al., 1999; Tzarfati-Majar et al., 2001; Plump et al., 2002; Diekmann and Stuermer, 2009). In fact, two of these cues (Slit1 and ALCAM) have established roles in the development of the retinofugal pathway in rodents (Ott et al., 1998; Weiner et al., 2004; Buhusi et al., 2009; Diekmann and Stuermer, 2009). While F-spondin acts as a guidance cue in developing motor circuits (Burstyn-Cohen et al., 1999; Tzarfati-Majar et al., 2001), its role in the developing visual system remains unexplored. It is noteworthy, however, that F-spondin shares homology with the extracellular matrix protein Reelin and can bind canonical Reelin receptors, both of which are important for the assembly of connections between ipRGCs and visual thalamus (Tzarfati-Majar et al., 2001; Hoe et al., 2005; Su et al., 2011, 2013).
Here, we tested whether these factors were necessary for the formation of connections between ipRGCs and the SCN. While our results demonstrate that each of these cues is dispensable for retinohypothalamic targeting, we found that F-spondin-deficient (spon1−/−) mutants (but not mutants lacking Slit1 or ALCAM) displayed severely disrupted “free-running” rhythmicity. Furthermore, F-spondin-deficient mutants displayed overtly normal circadian rhythms in normal day-night conditions, suggesting that the loss of intrinsic circadian rhythmicity was masked in the presence of light. The rapid ability of spon1−/− mutants to adapt to changes in lighting conditions and to entrain to ultradian photoperiods suggests that SCN neurons are weakly coupled in the absence of this extracellular matrix protein. Taken together, these results identify a novel role for F-spondin in maintaining intrinsic circadian rhythms.
Materials and Methods
Animals
C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA, USA). Spon1−/− mutant mice were purchased from Taconic Biosciences Inc. (Hudson, NY, USA) and slit1−/− were purchased from MMRRC1. The generation of alcam−/−, math5−/− and opn4taulacz/+ mice were described previously (Wang et al., 2001; Weiner et al., 2004; Hattar et al., 2006). Genomic DNA was isolated from tail using the HotSHOT method (Truett et al., 2000) and genotyping was performed with the following primers: spon1 (wildtype, WT) 5′-GAC CGG AGA TCT AGG AAC CCC TAG-3′ and 5′-CAC TCT CGC CAA CAG CTG GAG CG-3′, spon1 (mutant) 5′-CTC CGC TCA GAG CAG CGC AGC TC-3′ and 5′-CCC TAG GAA TGC TCG TCA AGA-3′; lacZ 5′-TTC ACT GGC CGT CGT TTT ACA ACGTCG TGA-3′ and 5′-ATG TGA GCG AGT AAC AAC CCG TCG GAT TCT-3′; math5, ATG GCG CTC AGC TAC ATC AT and GGG TCT ACC TGG AGC CTA GC; neomycin (neo), GCC GGC CAC AGT CGA TGA ATC and CAT TGA ACA AGA TGG ATT GCA; slit1 (WT) 5′-AAG ATG CCT CCT CTG ACT TC-3′ and 5′-ACC CTT AGC TTC TAC CAA CC-3′; slit1 (mutant) 5′-TCT CCT TTG ATC TGA GAC CG-3′ and 5′-AGG TTT CTC GAG CGT CAT AG-3′; alcam (common) 5′-AAA GTC GCT GTC CCC CTA AG-3′, alcam (mutant) 5′-GGT CTT GTA GTT GCC GTC GT-3′ and alcam (WT) 5′-GAG CAG ACC AGT CAA GCC TAA-3′. The following cycling conditions were used on an Eppendorf or Bio-Rad Mastercycler EP: spon1, 94°C for 5 min, followed by 33 cycles of amplification (94°C for 30 s, 62°C for 30 s, and 72°C for 45 s) and 10 min at 72°C; lacZ, 95°C for 5 min, followed by 35 cycles of amplification (95°C for 30 s, 52°C for 30 s, 72°C for 45 s), and 10 min at 72°C; math5, 95°C for 5 min, followed by 35 cycles of amplification (94°C for 30 s, 59°C for 30 s, and 72°C for 45 s) and 10 min at 72°C; neo, 94°C for 3 min, followed by 35 cycles of amplification (94°C for 30 s, 56°C for 30 s, and 72°C for 45 s) and 10 min at 72°C; slit1, 95°C for 5 min, followed by 30 cycles of amplification (95°C for 30 s, 60°C for 30 s, and 72°C for 30 s) and 10 min at 72°C; alcam, 94°C for 2 min, followed by 10 cycles of amplification (94°C for 20 s, 65°C for 15 s and decrease 0.5°C per cycle, 68°C for 10 s) and additional 28 cycles of amplification (94°C for 15 s, 60°C for 15 s, and 72°C for 10 s) and 1 min at 72°C. All analyses conformed to National Institutes of Health (NIH) guidelines and protocols, approved by the Virginia Polytechnic Institute and State University Institutional Animal Care and Use Committees.
Reagents
All chemicals and reagents were purchased from Fisher (Fairlawn, NJ, USA) or Sigma (St. Louis, MO, USA) unless otherwise stated.
Immunohistochemistry
Fluorescent immunohistochemistry (IHC) was performed on 16 μm cryosectioned paraformaldehyde (PFA)-fixed brain tissue as described previously (Su et al., 2010, 2012). Briefly, tissue slides were allowed to air dry for 15 min before being incubated with blocking buffer (2.5% normal goat serum, 2.5% bovine serum albumin and 0.1% Triton X-100 in PBS) for 30 min. Rabbit anti-Vasoactive Intestinal Peptide (VIP) antibodies (diluted 1:150 for IHC) were purchased from Immunostar and rabbit anti-vasopressin (AVP) antibodies (diluted 1:1000 for IHC) were purchased from Millipore (Cat# AB1565). Fluorescent secondary antibodies were from Life Technologies (diluted 1:1000). Primary antibodies were diluted in blocking buffer and incubated on tissue sections for overnight at 4°C. On the following day, tissue slides were washed in PBS and secondary antibodies diluted 1:1000 in blocking buffer were applied to slides for 1 h at room temperature. After thoroughly washing in PBS, tissue slides were coverslipped with VectaShield (Vector Laboratories, Burlingame, CA, USA). Images were acquired on a Zeiss Axio Imager A2 fluorescent microscope, a Zeiss Examiner Z1 LSM 710 confocal microscope, or a Zeiss LSM 700 confocal microscope (Oberkochen, Germany). Intensity and area of signal occupation of fluorescent, confocal images were measured in ImageJ as previously described (Singh et al., 2012; Su et al., 2016). A total of 4–6 animals (three sections per animal) were analyzed per genotype and Student’s T tests were used to assess statistical significance.
In Situ Hybridization
In situ hybridization (ISH) was performed on 16 μm coronal cryosectioned tissues as previously described. Antisense riboprobes were generated from full length cDNAs of slit1 (MMM1013-98685876), alcam (MMM1013-202762192), spon1 (MMM1013-202701079) and per1 (MMM1013-202764685; from Open Biosystems; Huntsville, AL, USA). Riboprobes were synthesized using digoxigenin (DIG)-labeled UTP (Roche, Mannheim, Germany) and the MAXIscript in vitro Transcription Kit (Ambion, Austin, TX, USA). Probes were hydrolyzed to 500 nt. Coronal brain sections were prepared and hybridized at 65°C as previously described (Su et al., 2010), and bound riboprobes were detected by horseradish peroxidase (POD)-conjugated anti-DIG antibodies and fluorescent staining with Tyramide Signal Amplification (TSA) systems (PerkinElmer, Shelton, CT, USA). Images were obtained on a Zeiss Axio Imager A2 fluorescent microscope or a Zeiss Examiner Z1 LSM 700 confocal microscope. A minimum of three animals per genotype, age and time were compared in ISH experiments.
Intraocular Injection of Anterograde Tracers
Intraocular injection of cholera toxin subunit B (CTB) conjugated to Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen) was performed in P13 mice as described previously (Su et al., 2011, 2013). After 2 days, mice were killed, and brains were fixed in 4% PFA. One hundred micrometer coronal sections were sectioned on a vibratome (Microm HM 650 V; Thermo Scientific, Waltham, MA, USA) and mounted in VectaShield (Vector Laboratories, Burlingame, CA, USA). Retinal projections in SCN were analyzed from at least three animals for each age and genotype. Images were acquired on a Zeiss Examiner Z1 LSM 710 confocal microscope or a Zeiss LSM 700 confocal microscope.
Quantitative Real-Time PCR
RNA was isolated from the SCN from both 12:12 LD and DD conditions. Samples from 12:12 light:dark (LD) were isolated at ZT4, ZT10, ZT16 and ZT22. Samples from constant darkness (DD) conditions were isolated at ZT4 5 days after mice were exposed to constant darkness. For samples obtained from the dark phase of ther standard 12:12 LD cycle or from anytime during a DD cycle, mice were euthanized and brain removed in the dark under dim red light illumination. In all cases, brains were removed, vibratomed (200 μm) in iced-cold DEPC-PBS and Suprachiasmatic nuclei (SCN) were dissected. For each sample (at each time point) SCN from a total of six mice was pooled. RNA from these SCN was isolated using the BioRad Total RNA Extraction from Fibrous and Fatty Tissue kit (BioRad). cDNAs were generated from 200 ng RNA with the Superscript II Reverse Transcription First Strand cDNA Synthesis kit (Invitrogen). Quantitative real-time PCR (qPCR) was performed on a Chromo4 Four Color Real-Time system (BioRad) using iQ SYBRGreen Supermix (BioRad; Su et al., 2010). qPCR was performed with the following primers Bmal1: 5′-TCCTTCCAGGCAGTCAACTT-3′ and 5′-CTGCAGTGAATGCTTTTGGA-3′; cry1: 5′-TGG CAT CAA GAT CCT CAA GA-3′ and 5′-TCC GCT GCG TCT ATA TCC TC-3′; gjd2: 5′-TGCTCATCATCGTACACCGT-3′ and 5′-GCAGCAGCACTCCACTATGA-3′; gapdh: 5′-CG TCCCGTAGACAAAATGGT-3′ and 5′-TTGATGGCAACA ATCTCCAC-3′; per1: 5′-AAC GCT TTG CTT TAG ATC GG-3′ and 5′-TCC TCA ACC GCT TCA GAG AT-3′; per2: 5′-GTA TCC ATT CAT GTC GGG CT-3′ and 5′-TAC TGG GAC TAG CGG CTC C-3′; vip: 5′-CGT GGT TGT TTT CCT TCG AG-3′ and 5′-GGA GCA GTG AGG GAG ATT CTG-3′. qPCR primers were designed over introns. The following cycling conditions were used with 10 ng RNA: 95°C for 30 s, followed by 40 cycles of amplification (95°C for 5 s, 60°C for 30 s, 55°C for 60 s, read plate) and a melting curve analysis. Relative quantities of RNA were determined using the ∆∆−CT method. A minimum of n = 3 experiments (each in triplicate) was run and examined for spon1−/− mutants and littermate controls at each time point (ZT4, ZT10, ZT16, ZT22) during 12:12 LD or DD (ZT4). Each individual run included separate gapdh control reactions at each time point.
Wheel Running Activity Assay
Wheel running activity was monitored in wheel cages from Lafayette Instruments which recorded each wheel revolution as an event and sent that information to a compatible computer in 5-min bins using ClockLab software R2011b. All mice were individually housed. Mice were habituated for 2 days before continually recording activity for 2 weeks of 12:12 LD and 2 weeks of DD. To test phase advance and phase delay, activity was recorded for 2 weeks in 12:12 LD, then the onset of light was advanced 6 h and activity was recorded in 12:12 LD. After 2 weeks, the onset of light was delayed 6 h to the original onset of light for 2 weeks of LD. A total of six spon1-mutant and six littermate WT mice were analyzed for the phase advance and delay conditions. To test activity in skeleton photoperiods, spon1−/− and littermate controls were exposed to 2 weeks of 12:12 LD then were exposed to 2 weeks of 1:11 LD cycle and finally 2 weeks of 12:12 LD. To assess spon1−/− and littermate control mice to an ultradian solar cycle, mice were exposed to more than 2 weeks of 12:12 LD circle, then were exposed to 1 week of 3.5 h light/3.5 h dark.
Results
Slit1, ALCAM, F-spondin Are Enriched in the Developing Suprachiasmatic Nucleus
To begin to identify potential retinohypothalamic targeting cues we screened available on-line expression databases for any extracellular matrix proteins, growth factors, morphogens or cell adhesion molecules enriched in adult mouse SCN (Lein et al., 20072). We identified two genes encoding extracellular matrix proteins (Slit1 and F-spondin) and one gene encoding a transmembrane cell adhesion molecule (ALCAM) all of which are enriched in adult SCN. Two of these candidates have previously been reported to contribute to the guidance and growth of retinal axons: members of the Slit family of extracellular matrix proteins mediate retinal pathfinding through binding to Robo receptors (Ringstedt et al., 2000; Plump et al., 2002; Thompson et al., 2006) and the immunoglobulin superfamily adhesion molecule ALCAM (also called BEN, SC1, DM-GRASP, Neurolin and CD166) contributes to the guidance, fasciculation, and topographic mapping of retinal axons (Ott et al., 1998; Weiner et al., 2004; Buhusi et al., 2009; Diekmann and Stuermer, 2009). While F-spondin (which is encoded by the gene spon1) has not been implicated in guiding retinal axon growth or subcortical visual circuit formation, it has been implicated in axonal outgrowth in the developing spinal cord (Burstyn-Cohen et al., 1999; Tzarfati-Majar et al., 2001). Importantly, RGCs express amyloid precursor protein (APP), a transmembrane receptor capable of binding to F-spondin (Ho and Südhof, 2004; Osterhout et al., 2015).
Although retinal axons begin to innervate targets in the mouse midbrain during embryogenesis, they do not fully innervate the SCN until the end of the first postnatal week of mouse development (Fox and Guido, 2011; McNeill et al., 2011). To assess whether the expression of these candidate targeting cues coincided with retinohypothalamic innervation, we generated riboprobes and tested whether their mRNAs were present in developing SCN. Not only were all three genes present at early postnatal ages, but each cue exhibited a unique expression pattern in the developing SCN (Figures 1C–F).
F-spondin, Slit1 and ALCAM Are Not Required for Retinohypothalamic Tract Formation
Since Slit1, ALCAM and F-spondin are generated in the developing SCN, we assessed their necessity for retinohypothalamic innervation in targeted mouse mutants lacking each cue (i.e., slit1−/−, alcam−/− and spon1−/− mutants). All three mutants are viable, fertile, and appear indistinguishable from littermate controls in regards to size and cage activity. To assess retinohypothalamic innervation in these mutants, we performed intraocular injections of fluorescently conjugated CTB, an anterograde tracer that efficiently labels all retinal projections into the brain (Muscat et al., 2003). In controls, retinal projections from each eye project bilaterally to the SCN in each hemisphere on the hypothalamus and these projections arborize to fill the entire SCN (Figure 2A). To our surprise CTB-labeled retinohypothalamic projections appear unaltered by the loss of Slit1, ALCAM or F-spondin (Figures 2B–D).
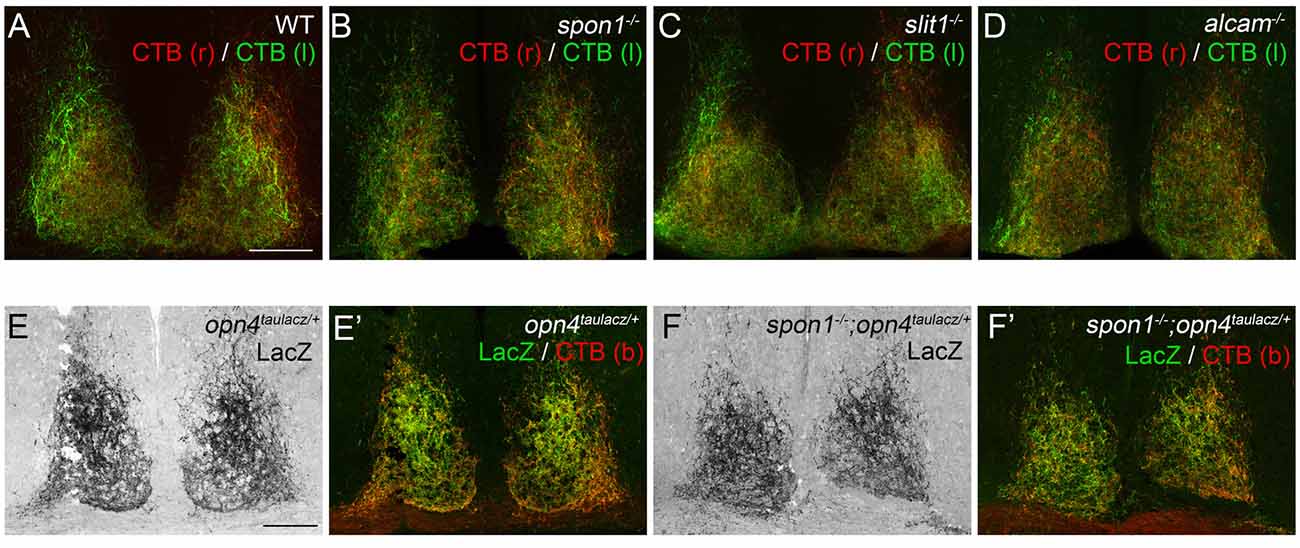
Figure 2. F-spondin, Slit1 and ALCAM are dispensable for retinal innervation of the SCN. (A–D) CTB-labeled retinal projections in the SCN of P15 wildtype (WT) (A), spon1−/− (B), slit1−/− (C) and alcam−/− (D) mice. CTB(r)—CTB labeled retinal projections from right eye; CTB(l)—CTB labeled retinal projections from left eye. Scale bar = 100 μm. (E,F) M1 ipRGC arbors in the SCN are labeled by immunohistochemistry (IHC) for LacZ in P24 opn4tauLacZ/+ reporter mice. Arbors of M1 ipRGCs correctly target SCN in spon1−/−; opn4tauLacZ/+ mutant mice (F). (E′,F′) show both LacZ-immunoreactivity and CTB-labeling in control (E′) and mutant (F′) SCN. CTB (b)—binocular CTB labeled retina projections. Scale bar = 100 μm.
Since CTB labels all retinal projections indiscriminately, despite only a very small population of RGCs actually projecting axons to the SCN, it remained possible that axons from ipRGCs were no longer innervating SCN in these mutants and instead other classes of RGC axons were present. Consequently, we tested whether ipRGC axons correctly innervated the SCN in mutants lacking F-spondin, the targeting candidate whose developmental expression pattern most closely resembled the distribution of ipRGC arbors in SCN (Figure 1D; Hattar et al., 2006). Spon1−/− mutant mice were crossed with opn4tau-lacz/+ transgenic mice, in which ipRGC projections are labeled with Tau-LacZ fusion protein (Hattar et al., 2002, 2006). The majority of retinal projections in the SCN of control opn4tau-laz/+ mice contained Tau-LacZ (Figure 2E). Patterns of ipRGC axon arbors in mutants were indistinguishable from controls (Figure 2F). Altogether, our results strongly suggest Slit1, ALCAM and F-spondin are each individually dispensable for the formation of the retinohypothalamic tract.
F-spondin Deficiency Leads to a Loss of Free Running Rhythmicity
Given that retinal innervation of the SCN appeared normal in slit1−/−, alcam−/− and spon1−/− mutants, we next explored whether these factors may play other roles in SCN function. Since the SCN is the master regulator of circadian rhythms and responsible for the entrainment of these rhythms to changes in light:dark (LD) cycles, we monitored control and mutant mouse activity in normal cycles of 12 h of light and 12 h of darkness (12:12 LD) and in constant darkness (DD). Control mice entrained their activity to 12:12 LD conditions, with a robust increase in activity at the onset of the dark period (Figures 3, 4A). When control mice were shifted to DD, they immediately commenced free-running rhythmicity with each circadian period being less than 24 h in the absence of light signals (period length = 23.63 ± 0.09 [SEM]; n = 21; Figures 3, 4A). Actograms of slit1−/− and alcam−/− mutant activity resembled that of control mice in three ways: (1) their activity was entrained to the dark periods of 12:12 LD (Figure 3); (2) they exhibited free-running rhythmicity in DD (period length for slit1−/− mutants = 23.61 ± 0.04 [SEM]; n = 8; period length for alcam−/− mutants = 23.71 ± 0.04 [SEM]; n = 15; Figure 3); (3) their levels of activity in either 12:12 LD or DD appeared comparable to WT controls (Figure 3).
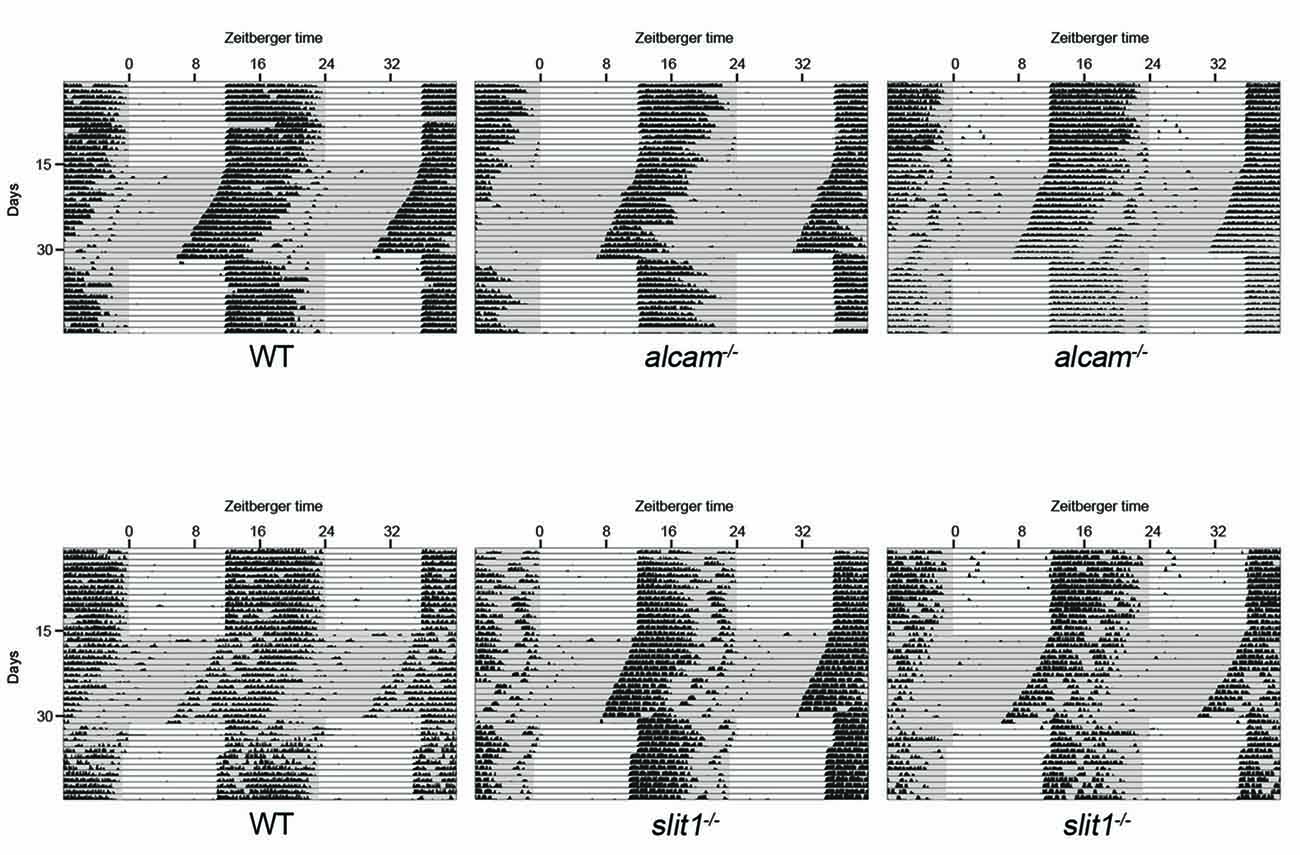
Figure 3. Mice lacking Slit1 and ALCAM exhibit normal photoentrainment and free-running rhythmicity. Representative actograms of wheel running activity of WT, slit1−/− and alcam−/− mice. Mice were housed in a 12:12 light:dark (LD) cycle and then shifted to constant darkness (DD) conditions. Gray background indicates periods of darkness; white background indicates periods of light. Black bars indicate relative level of wheel running activity. Actograms are double plotted.
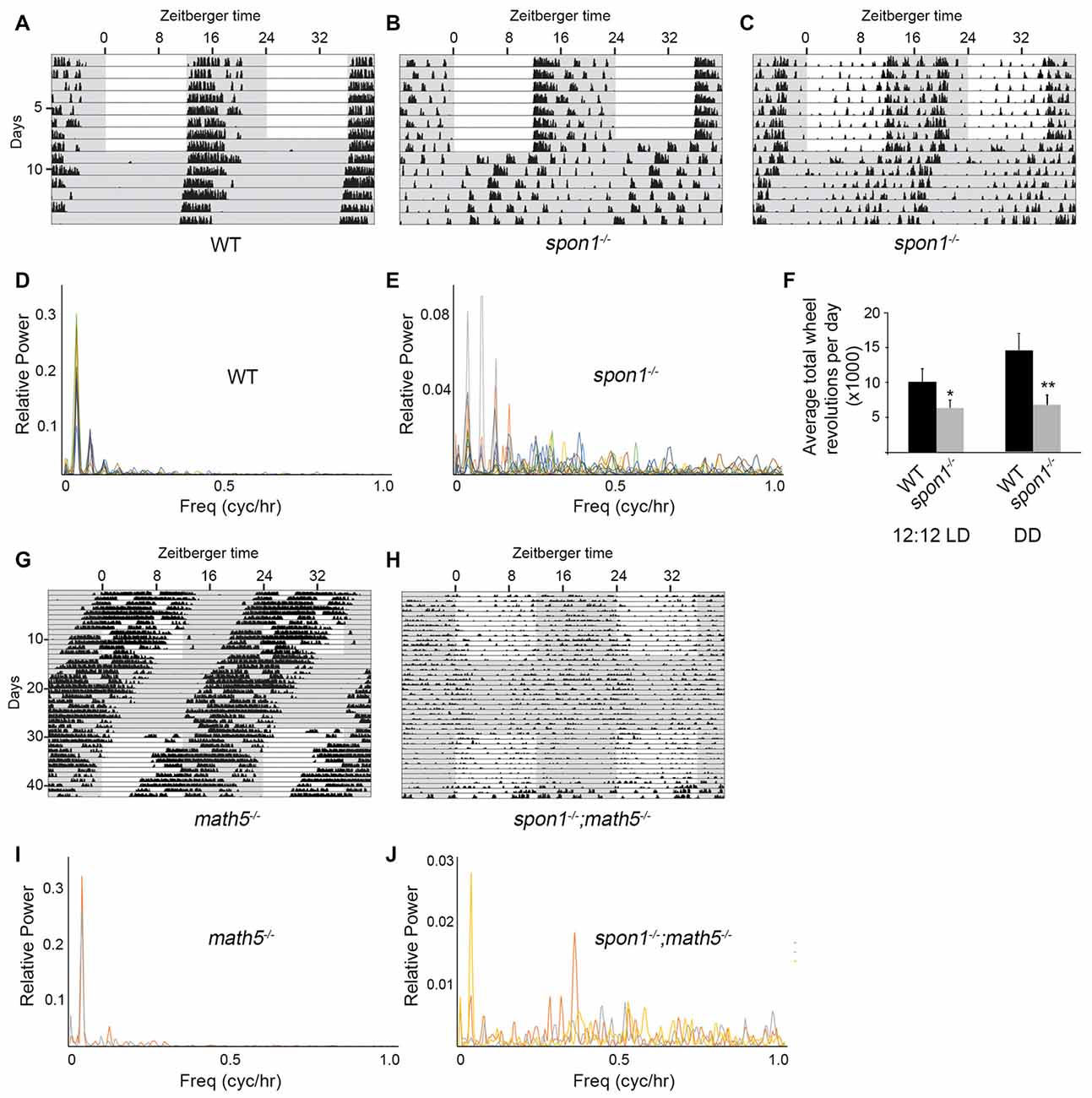
Figure 4. F-spondin deficiency leads to a loss of free running rhythmicity. (A–C) Representative actograms of wheel running activity of spon1−/− mutants (B,C) or littermate WT mice (A) housed in a 12:12 LD cycle and then shifted to DD conditions. Gray background indicates periods of darkness; white background indicates periods of light. Black bars indicate relative level of wheel running activity. Actograms are double plotted. (D,E) Fast Fourier Transform analysis of the activity patterns of 10 WT mice (D) and 10 spon1−/− mutants (E) in DD conditions. Analysis of each individual mouse is shown with a distinct color. (F) Levels of wheel running activity of spon1−/− mutants was reduced in both 12:12 LD and DD conditions compared with littermate controls. Data are shown as mean ± SEM. *Indicates p < 0.05 and **indicates p < 0.005 by Students t-test (n = 14 WT and 27 spon1−/− mutants). (G,H) Representative actograms of wheel running activity of math5−/− mutants (E) or spon1−/−; math5−/− mutants (F) housed in alternating 12:12 LD and DD cycles. (I,J) Fast Fourier Transform analysis of the activity patterns of two math5−/− mutant mice (I) and three math5−/− ; spon1−/− double mutants (J) in DD conditions. Analysis of each individual mouse is shown with a distinct color.
While mice lacking F-spondin also appeared to entrain the majority of their activity to the dark phase of the 12:12 LD cycle (Figures 4A,B), their behavior differed from controls (and from slit1−/− and alcam−/− mutants) in several ways. First, a cohort of spon1−/− mutants exhibited weak, spontaneous activity during the light phase of the 12:12 LD cycle (Figure 4C). Second, their level of activity was significantly reduced compared to controls (Figure 4F), a result that could be explained by previously identified roles for F-spondin in motoneuron and spinal cord development (Burstyn-Cohen et al., 1999; Tzarfati-Majar et al., 2001). Third, and most striking, the majority of mice lacking F-spondin exhibited disrupted free-running rhythmicity in DD conditions (61% of spon1−/− mutants exhibited severely disrupted or arrhythmic activity in DD; n = 13/21 mutants; Figures 4A–E). These results reveal that spon1−/− mutants have major impairment of internal rhythmicity and they suggest that the presence of light is sufficient to mask such impaired activity.
To confirm that spon1−/− mutants lack rhythmicity in the absence of visual stimuli, we genetically removed retinal inputs to SCN. To accomplish this, we crossed spon1−/− mutants to math5−/− mutants in which the deletion of the transcription factor Math5 impairs retinogenesis and leads to a failure in RGC development (Wang et al., 2001). Math5−/− mutants are viable but lack connections between the retina and retino-recipient nuclei within the brain (Wang et al., 2001; Brooks et al., 2013; Seabrook et al., 2013). For this reason, math5−/− mutants are blind and exhibit free-running rhythmicity regardless of the lighting conditions (Figures 4G,I; Brzezinski et al., 2005). Loss of retinal inputs to the SCN in F-spondin-deficient mutants (i.e., spon1−/−:math5−/− double mutants) resulted in arrhythmic locomotor activity regardless of lighting conditions (n = 3; all spon1−/−:math5−/− mutants exhibited arrhythmic activity; Figures 4H,J).
Light Plays a Dominant Influence in Masking the Lack of Internal Rhythms in Spon1−/− Mutants
Loss or dysfunction of several core “clock” proteins has been shown to result in arrhythmic behavior in DD, as shown here for a significant number of spon1−/− mutants (van der Horst et al., 1999; Vitaterna et al., 1999; Bunger et al., 2000; Bae et al., 2001; Zheng et al., 2001). In several cases, it has been demonstrated that the lack of internal rhythmicity and weak synchronization between SCN neurons in these “clock” mutants allow for more rapid adaptation of their activity patterns to changes in lighting conditions (Herzog, 2007; An et al., 2013; Hatori et al., 2014). Therefore, we sought to determine whether the same was true for mutants lacking F-spondin. We assessed behavioral responses to advances or delays in the onset of light in a 12:12 LD cycle in mutant and control mice entrained to a normal 12:12 LD cycle. After 10 days in 12:12 LD the onset of light was advanced 6 h and after an additional 10 days the onset of light was delayed 6 h. It took control mice more than 3 days to re-entrain to the LD cycle that was advanced or delayed by 6 h (Figure 5A). In sharp contrast, spon1−/− mutants immediately adapted to shifts in lighting conditions (whether that was an advance or delay in the LD cycle) and began wheel running at the onset of the new dark period (Figure 5B).
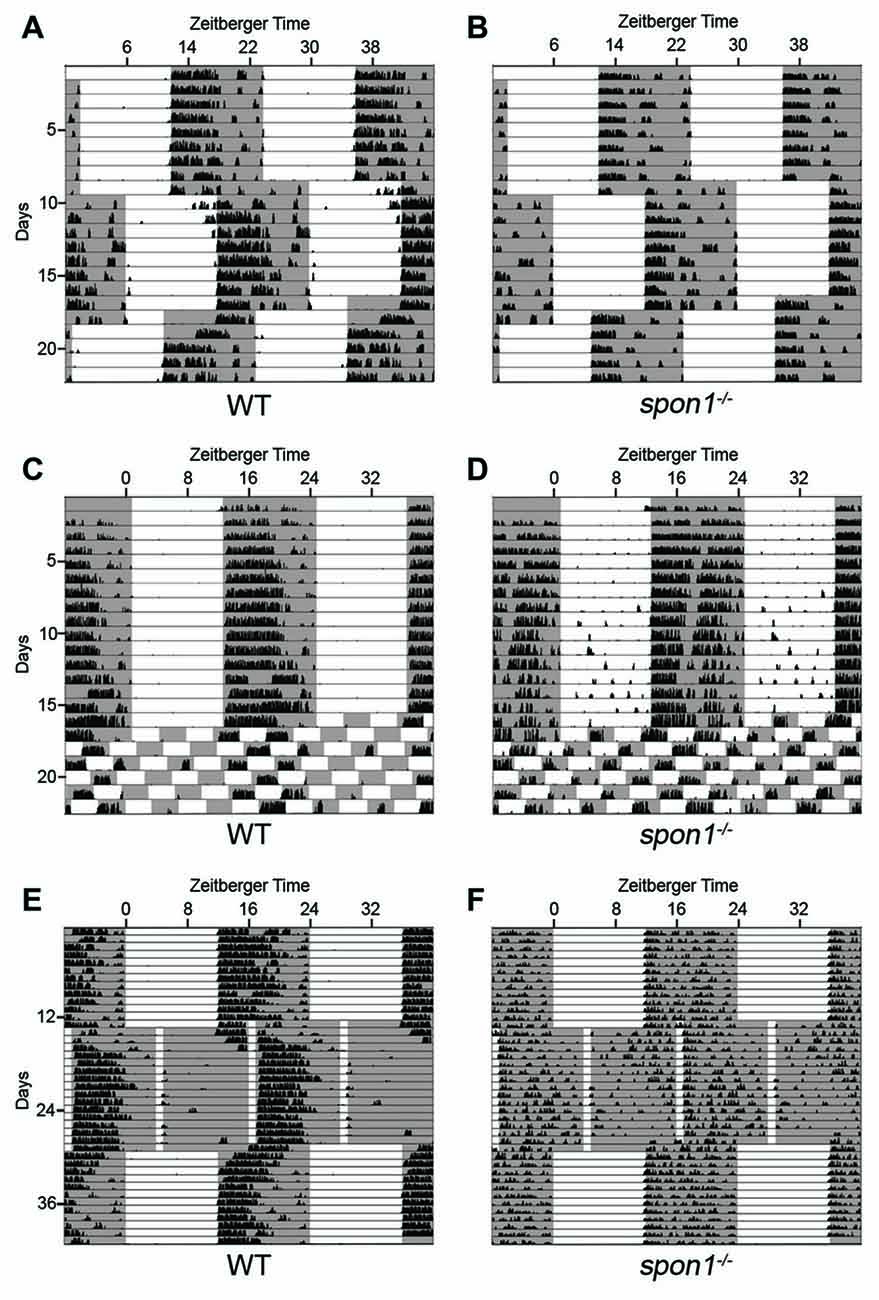
Figure 5. Light masks the lack of internal rhythms in spon1−/− mutants. (A,B) Representative actograms of wheel running activity of spon1−/− mutants (B) or littermate WT mice (A) housed in a 12:12 LD cycle, exposed to a 6 h advance in the light-dark cycle, and then exposed to a 6 h delay in the light-dark cycle. Gray background represents periods of darkness; white background indicates periods of light. Black bars indicate level of wheel running activity. Spon1−/− mutants immediately adjust to shifts in the light dark cycle. n = 22 WT and 22 spon1−/− mutants. (C,D) Representative actograms of wheel running activity of spon1−/− mutants (D) or littermate WT (C) housed in a 12:12 LD cycle before being exposed to 7 days of an ultradian light-dark cycle (3.5:3.5 LD). While WT mice cannot entrain to this light-dark cycle, spon1−/− mutants exhibit wheel running activity during each dark period of the 3.5:3.5 LD cycle. n = 4 WT and eight spon1−/− mutants. (E,F) Representative actograms of wheel running activity of spon1−/− mutants (D) or littermate WT (C) housed in a 12:12 LD cycle before being exposed to skeleton photoperiod (1:11 LD). While WT mice photoentrain in response to a 1:11 LD cycle, mutants do not. n = 5 WT and six spon1−/− mutants.
These results suggest that light has an immediate and dominant influence on the locomotor activity of spon1−/− mutants and masks the disrupted of internal rhythmicity in mutants. To further test this, we assessed responses to ultradian lighting conditions, in which the light:dark cycle was reduced to a 7 h cycle (3.5 h light; 3.5 h dark). When shifted from a 12:12 LD cycle to an ultradian LD cycle, WT-mice cannot entrain their circadian rhythmicity to each short photoperiod (Redlin and Mrosovsky, 1999; Redlin et al., 2005; Abraham et al., 2006), and instead display activity only during the dark periods that coincide with dark periods of the previous 12:12 LD cycle (Figure 5C). In contrast, spon1−/− mutants exhibited significant bouts of activity during all dark periods and activity was masked in each period of light (Figure 5D). These results support the notion that spon1−/− mutants do not actually entrain to 12:12 LD cycles but rather their activity is masked by the presence of light.
Previous studies have demonstrated that short pulses of light coinciding with dusk and dawn are sufficient to maintain circadian rhythms in nocturnal rodents (Schwartz et al., 1996; Aton et al., 2005). If spon1−/− mutants were not entraining to 12:12 LD cycles, but were merely masking activity in the presence of light, we hypothesized that skeleton photoperiods would be insufficient to entrain mutant activity. To test this hypothesis, we exposed mutant and control mice to a skeleton photoperiod of 1 h of light followed by 11 h of darkness (1:11 LD). In such skeleton photoperiods, WT mice consolidated more than 80% of their daily activity to a singular dark period (Figure 5E; average WT activity in photoperiod 1 = 9676 ± 1388 [SEM] revolutions per cycle and in photoperiod 2 = 2446 ± 725 revolutions per cycle; p < 0.01 by Students t-test; n = 5). In contrast, activity in spon1−/− mutants remained disrupted and was less confined to a single photoperiod in this paradigm (Figure 5F; average spon1−/− mutant activity in photoperiod 1 = 5893 ± 761 revolutions per cycle and in photoperiod 2 = 4233 ± 451 [SEM] revolutions per cycle; p = 0.27 by Students t-test; n = 6). Thus, short pulses of light appear insufficient to correct impaired internal rhythms in F-spondin-deficient mutants.
Loss of F-spondin Leads to an Altered Distribution of VIP-expressing Neurons in SCN
Loss of internal circadian rhythms and immediate arrhythmic activity in DD has been reported in mice lacking core “clock” genes (van der Horst et al., 1999; Vitaterna et al., 1999; Bunger et al., 2000; Bae et al., 2001; Zheng et al., 2001; Abraham et al., 2006), the gap junction protein connexin 36 (Cx36; encoded by the gjd2 gene; Long et al., 2005), and VIP signaling (Harmar et al., 2002; Aton et al., 2005). We, therefore, tested whether any of these pathways were altered in mutants lacking F-spondin. First, we assessed the expression of bmal1, cry1, per1 and per2 mRNAs in mutants and control SCN every 6 h during a normal 12:12 LD cycle. For all genes, the cyclical expression of these circadian genes appeared similar in control and spon1−/− SCN (Figure 6A). The only noteworthy difference in circadian gene expression in spon1−/− mutants was a significantly elevated expression level of per2 at the end of the light period (Figure 6A). We also assessed per1 and per2 mRNAs in spon1−/− mutant and control SCN exposed to DD conditions for 5 days. RNA was isolated at Zeitbeger time 4, a time at which controls were likely inactive after 5 days of DD and mutants were likely active (see Figures 2, 3). RNA analysis revealed no significant differences in per1 or per2 gene expression after 5 days of DD in the absence of F-spondin (Figure 6B). We confirmed this result by generating riboprobes against per1 mRNA and performing ISH in the SCN of mutant control mice (Figure 6C). Qualitatively, per1 mRNA levels and distribution in the SCN appeared similar in mutants and controls at ZT4 and ZT16 after 5 days in DD conditions (Figure 6C). These results were surprising given previous studies on these core clock genes. Based on these results, we interpret this to indicate that disrupted internal rhythms in spon1−/− mutants are not the result of loss of core “clock” gene expression in the absence of F-spondin.
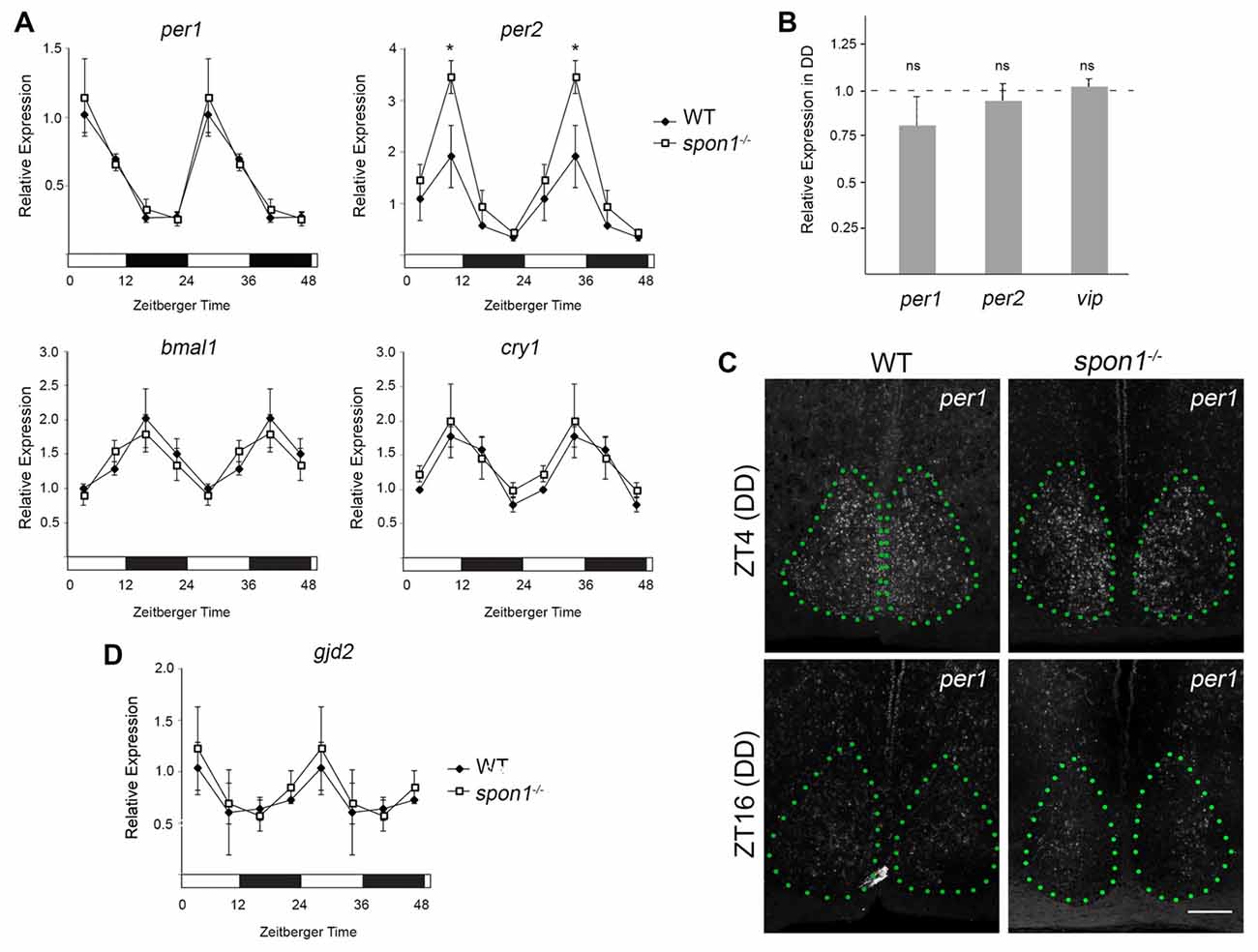
Figure 6. Core clock gene expression in spon1−/− mutant SCN. (A) Expression of bmal1, cry1, per1 and per2 mRNAs in the SCN of adult littermate WT mice and spon1−/− mutants exposed to 12:12 LD. SCNs were dissected at four time points and mRNA levels were assessed by quantitative real-time PCR (qPCR). Samples were normalized to gapdh levels. At ZT4, expression of per2 was significantly elevated in spon1−/− mutants. Data are shown as mean ± SEM. *Indicates p < 0.03 by Students t-test (n = 3 in triplicate per genotype). (B) Expression of per1, per2 and vasoactive intestinal peptide (vip) mRNAs in the SCN of littermate WT mice (dashed line) and spon1−/− mutants (gray bars) exposed to 5 days of DD. RNA was isolated at ZT4 and analyzed by qPCR. Samples were normalized to gapdh levels. Data are shown as mean ± SEM. ns—not statistically different. (C) ISH for per1 mRNA in SCN of adult control and spon1−/− exposed to 5 days of DD. Tissue was analyzed at ZT4 and ZT16. SCN depicted by green dots. Scale bar = 100 μm. (D) Expression of gjd2 mRNA in the SCN of WT and spon1−/− mutants exposed to 12:12 LD. RNA levels were assessed by qPCR and were normalized to gapdh levels. Data are shown as mean ± SEM.
Next, we applied similar approaches to address whether gjd2 mRNA levels were altered in spon1−/− SCN. In control mice exposed to 12:12 LD conditions, gjd2 mRNA levels appeared to be regulated in a rhythmic pattern in SCN, with peak expression coinciding with early phases of the light cycle (similar to per1 expression patterns; Figure 6D). Similar levels and temporal patterns of gjd2 were observed in spon1−/− mutant SCN (Figure 6D). Taken together, these results suggest that the expression of Cx36 was not perturbed by the loss of F-spondin.
Finally, we addressed whether VIP levels or VIP-expressing neurons were altered by the loss of F-spondin. In WT mice, VIP-containing fibers densely arborize throughout the entire SCN however the majority of VIP-expressing neurons (which receive retinal input) are confined to the ventral most region of SCN that overlies the optic chiasm (OC; Abrahamson and Moore, 2001; Bedont et al., 2014; Fernandez et al., 2016; Figure 7A). In spon1−/− mutants, we observed similar levels of VIP-immunoreactivity (Figures 7A–C,E) and similar levels of vip mRNA (Figure 6B) within the SCN compared to littermate controls. However, an increased number of VIP-expressing neurons were displaced into the OC and optic tract in mutants (Figure 7). In contrast, we detected no difference in the distribution of vasopressin (AVP)-expressing neurons in control or spon1−/− mice (Figures 7F,G). Thus, F-spondin appears necessary for the proper migration of VIP+ neurons into the SCN.
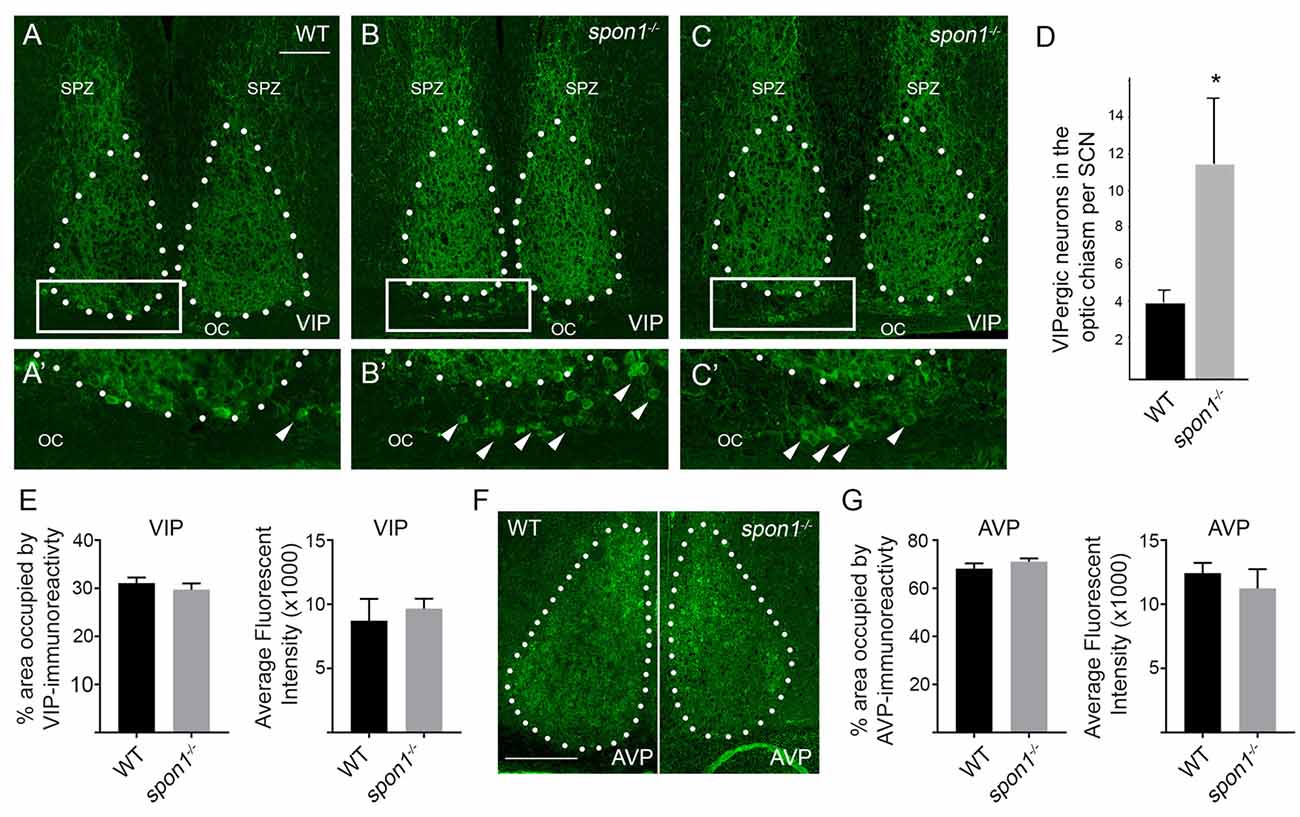
Figure 7. Loss of F-spondin leads to an altered distribution of VIP-expressing neurons in SCN. (A–C) IHC for VIP in adult WT and spon1−/− mutants. Tissue was isolated at ZT4. White dots encircle SCN. Boxed areas are shown in higher magnification in (A′–C′). Arrowheads highlight VIP+ neurons in the optic chiasm (OC). SPZ, subparaventricular zone. Scale bar in (A) = 100 μm. (D) Quantification of VIP+ neurons mislocalized in the OC of adult WT and spon1−/− mutants. Data are shown as mean ± SEM. *Indicates p < 0.03 by Students t-test (n = 4 per genotype). (E) Quantification of the area occupied by VIP-immunoreactivity and the average fluorescent intensity of this signal in the SCN in adult WT and spon1−/− mutants. Data are shown as mean ± SEM. (F) IHC for anti-vasopressin (AVP) in adult WT and spon1−/− mutants. White dots encircle SCN. Scale bar in (F) = 100 μm. (G) Quantification of the area occupied by AVP-immunoreactivity and the average fluorescent intensity of this signal in the SCN in adult WT and spon1−/− mutants. Data are shown as mean ± SEM.
Discussion
Although these studies failed to identify targeting cues necessary for guiding ipRGC axons into the SCN, they unexpectedly discovered a novel role for F-spondin in maintaining circadian rhythms in the absence of visual stimuli. Mice lacking spon1 not only lose intrinsic rhythmicity, but are also more quickly able to modify their activity to changes in the light:dark cycle—results suggesting a loss of neuronal synchrony in the SCN. Although similar results have been observed in mutants lacking core “clock” genes (such as in cry1−/−:cry2−/− double mutants, cry1−/−:per2−/− double mutants, bmal1−/− mutants, per1−/−:per2−/− double mutants; van der Horst et al., 1999; Bunger et al., 2000; Bae et al., 2001; Zheng et al., 2001; Oster et al., 2002; Abraham et al., 2006) and gap junctional coupling (gjd2−/−; Long et al., 2005), we failed to detect significant alterations in the expression or distribution of many of these elements in spon1−/−. We did detect alterations in the expression of per2 mRNA in spon1−/− mutants in 12:12 LD conditions. Constitutive and dramatic overexpression of Per2 has been shown to abolish intrinsic rhythmicity by increasing nuclear Per2:Cry complexes which inhibits the negative feedback loop of circadian gene transcriptions (Chen et al., 2013; Wang et al., 2016). It seems unlikely that elevated per2 levels underlie the loss of rhythmicity in spon1−/− mutants since the levels are not constitutively upregulated (in either 12:12 LD or DD conditions) and since expression is only modestly upregulated during exposure to light (Figures 4A,B). More striking than these modest increases in per2 expression, we observed alterations in the distribution of VIPergic neurons in spon1−/− mutant SCN. Disruption of VIP signaling (by either the loss of VIP itself, the loss of its receptors, or the loss of transcription factors necessary for the differentiation of VIP neurons) reduces synchrony among SCN neurons and leads to immediate arrhythmic behavior in the absence of visual stimuli (Harmar et al., 2002; Aton et al., 2005; Maywood et al., 2006, 2011; Bedont et al., 2014; Hatori et al., 2014), just as we show here for spon1−/− mutants.
VIP neurons comprise 10%–20% of the neurons in SCN (Abrahamson and Moore, 2001). Importantly, VIPergic neurons in the SCN receive direct input from the retina (Fernandez et al., 2016), display induction of activity-dependent immediate early genes (such as cFos) upon delivery of visual stimuli to the retina (Aioun et al., 1998), and are essential for photoentrainment. Although they represent a minority of cells in the SCN, the influence of VIPergic neurons on SCN function is profound with projections that densely arborize throughout the SCN (and adjacent subparaventricular zone [SPZ], a downstream synaptic target of SCN neurons), with the ability to alter the firing rates of more than half the entire population of SCN neurons by releasing VIP (Reed et al., 2001), and with the ability to regulate the coupling of a network of cellular oscillators in the SCN (Aton and Herzog, 2005; Aton et al., 2005). In regards to the studies presented here, it remains unclear whether (and if so, how) mislocalization of VIP neurons in spon1−/− mutants alters their upstream connectivity with RGCs or downstream connections with other SCN neurons or other targets. Moreover, although F-spondin has known roles in directing cell migration (Debby-Brafman et al., 1999) and the mRNAs of F-spondin receptors are highly expressed in mouse SCN (Allen Brain Atlas; Ho and Südhof, 2004; Hoe et al., 2005; Oka et al., 2015), it remains unclear whether and how F-spondin influences VIPergic neuronal migration in the SCN. Moreover, while we identified F-spondin as being dramatically enriched in SCN vs. other brain regions, it is generated in other brain regions (Allen Brain Atlas). It remains possible that its expression and function in other brain regions (especially those associated with circadian behaviors, such as the intergeniculate nucleus) contribute to alterations in circadian behaviors observed here.
While this study is the first to demonstrate a role for F-spondin in maintaining intrinsic rhythmicity and circadian function, there are in fact few known roles for this ECM molecule in the brain. A recent series of studies, however, has identified spon1 gene variants and risk alleles in influencing dementia severity and cognitive decline (Jahanshad et al., 2013; Sherva et al., 2014; Varol et al., 2017). Loss or mutation of F-spondin may affect dementia severity via its role in binding APP and clearing Aβ peptides (Ho and Südhof, 2004; Habisch et al., 2010). However, in light of results presented here, it is also possible that F-spondin’s role in maintaining circadian rhythms contribute to limiting AB pathology and pathogenesis of Alzheimer’s disease (AD). Disruption of circadian rhythms have well established roles in the pathogenesis of AD (Musiek, 2015)—roles that are both consequences of the disease and potentially causative of the disease. In support of the latter, mutations in core “clock” genes have been identified (like those in spon1) that are associated with increased risk for patients developing AD (Chen et al., 2013). With such studies in mind, it is possible that disruption of intrinsic rhythms in F-spondin-deficient mammals may have significant impact on the development and progression of cognitive and neurodegenerative diseases.
Author Contributions
GLC, JS and AM carried out all of the collection, preparation and analysis of data for these studies. Specifically, GLC performed bioinformatics screening, behavioral analysis and analysis in spon1−/− mutants. JS and AM performed ISH and analysis of slit1−/− and alcam−/− mutants. GLC and JS helped draft portions of the manuscript. MAF helped conceive, design and coordinate the study and drafted the manuscript. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by the National Institutes of Health—National Eye Institute grants EY021222 and EY024712 (MAF) and a fellowship from the VTCRI Medical Research Scholars Program (AM).
Footnotes
References
Abraham, D., Dallmann, R., Steinlechner, S., Albrecht, U., Eichele, G., and Oster, H. (2006). Restoration of circadian rhythmicity in circadian clock-deficient mice in constant light. J. Biol. Rhythms 21, 169–176. doi: 10.1177/0748730406288040
Abrahamson, E. E., and Moore, R. Y. (2001). Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res. 916, 172–191. doi: 10.1016/s0006-8993(01)02890-6
Aioun, J., Chambille, I., Peytevin, J., and Martinet, L. (1998). Neurons containing gastrin-releasing peptide and vasoactive intestinal polypeptide are involved in the reception of the photic signal in the suprachiasmatic nucleus of the Syrian hamster: an immunocytochemical ultrastructural study. Cell Tissue Res. 291, 239–253. doi: 10.1007/s004410050994
An, S., Harang, R., Meeker, K., Granados-Fuentes, D., Tsai, C. A., Mazuski, C., et al. (2013). A neuropeptide speeds circadian entrainment by reducing intercellular synchrony. Proc. Natl. Acad. Sci. U S A 110, E4355–E4361. doi: 10.1073/pnas.1307088110
Aton, S. J., Colwell, C. S., Harmar, A. J., Waschek, J., and Herzog, E. D. (2005). Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat. Neurosci. 8, 476–483. doi: 10.1038/nn1419
Aton, S. J., and Herzog, E. D. (2005). Come together, right‥.now: synchronization of rhythms in a mammalian circadian clock. Neuron 48, 531–534. doi: 10.1016/j.neuron.2005.11.001
Bae, K., Jin, X., Maywood, E. S., Hastings, M. H., Reppert, S. M., and Weaver, D. R. (2001). Differential functions of mPer1, mPer2 and mPer3 in the SCN circadian clock. Neuron 30, 525–536. doi: 10.1016/s0896-6273(01)00302-6
Bedont, J. L., LeGates, T. A., Slat, E. A., Byerly, M. S., Wang, H., Hu, J., et al. (2014). Lhx1 controls terminal differentiation and circadian function of the suprachiasmatic nucleus. Cell Rep. 7, 609–622. doi: 10.1016/j.celrep.2014.03.060
Bell-Pedersen, D., Cassone, V. M., Earnest, D. J., Golden, S. S., Hardin, P. E., Thomas, T. L., et al. (2005). Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat. Rev. Genet. 6, 544–556. doi: 10.1038/nrg1633
Brooks, J. M., Su, J., Levy, C., Wang, J. S., Seabrook, T. A., Guido, W., et al. (2013). A molecular mechanism regulating the timing of corticogeniculate innervation. Cell Rep. 5, 573–581. doi: 10.1016/j.celrep.2013.09.041
Brzezinski, J. A. IV., Brown, N. L., Tanikawa, A., Bush, R. A., Sieving, P. A., Vitaterna, M. H., et al. (2005). Loss of circadian photoentrainment and abnormal retinal electrophysiology in Math5 mutant mice. Invest. Ophthalmol. Vis. Sci. 46, 2540–2551. doi: 10.1167/iovs.04-1123
Buhusi, M., Demyanenko, G. P., Jannie, K. M., Dalal, J., Darnell, E. P., Weiner, J. A., et al. (2009). ALCAM regulates mediolateral retinotopic mapping in the superior colliculus. J. Neurosci. 29, 15630–15641. doi: 10.1523/JNEUROSCI.2215-09.2009
Bunger, M. K., Wilsbacher, L. D., Moran, S. M., Clendenin, C., Radcliffe, L. A., Hogenesch, J. B., et al. (2000). Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017. doi: 10.1016/s0092-8674(00)00205-1
Burstyn-Cohen, T., Tzarfaty, V., Frumkin, A., Feinstein, Y., Stoeckli, E., and Klar, A. (1999). F-Spondin is required for accurate pathfinding of commissural axons at the floor plate. Neuron 23, 233–246. doi: 10.1016/s0896-6273(00)80776-x
Challet, E., Caldelas, I., Graff, C., and Pévet, P. (2003). Synchronization of the molecular clockwork by light- and food-related cues in mammals. Biol. Chem. 384, 711–719. doi: 10.1515/bc.2003.079
Chen, Q., Huang, C. Q., Hu, X. Y., Li, S. B., and Zhang, X. M. (2013). Functional CLOCK gene rs1554483 G/C polymorphism is associated with susceptibility to Alzheimer’s disease in the Chinese population. J. Int. Med. Res. 41, 340–346. doi: 10.1177/0300060513476430
Debby-Brafman, A., Burstyn-Cohen, T., Klar, A., and Kalcheim, C. (1999). F-Spondin, expressed in somite regions avoided by neural crest cells, mediates inhibition of distinct somite domains to neural crest migration. Neuron 22, 475–488. doi: 10.1016/s0896-6273(00)80703-5
Dibner, C., Schibler, U., and Albrecht, U. (2010). The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 72, 517–549. doi: 10.1146/annurev-physiol-021909-135821
Diekmann, H., and Stuermer, C. A. (2009). Zebrafish neurolin-a and -b, orthologs of ALCAM, are involved in retinal ganglion cell differentiation and retinal axon pathfinding. J. Comp. Neurol. 513, 38–50. doi: 10.1002/cne.21928
Fernandez, D. C., Chang, Y. T., Hattar, S., and Chen, S. K. (2016). Architecture of retinal projections to the central circadian pacemaker. Proc. Natl. Acad. Sci. U S A 113, 6047–6052. doi: 10.1073/pnas.1523629113
Foster, R. G. (1998). Shedding light on the biological clock. Neuron 20, 829–832. doi: 10.1016/s0896-6273(00)80464-x
Fox, M. A., and Guido, W. (2011). Shedding light on class-specific wiring: development of intrinsically photosensitive retinal ganglion cell circuitry. Mol. Neurobiol. 44, 321–329. doi: 10.1007/s12035-011-8199-8
Guler, A. D., Ecker, J. L., Lall, G. S., Haq, S., Altimus, C. M., Liao, H. W., et al. (2008). Melanopsin cells are the principal conduits for rod-cone input to non-image-forming vision. Nature 453, 102–105. doi: 10.1038/nature06829
Habisch, H. J., Schmid, B., von Arnim, C. A., Ludolph, A. C., Brenner, R., and Storch, A. (2010). Efficient processing of Alzheimer’s disease amyloid-β peptides by neuroectodermally converted mesenchymal stem cells. Stem Cells Dev. 19, 629–633. doi: 10.1089/scd.2009.0045
Harmar, A. J., Marston, H. M., Shen, S., Spratt, C., West, K. M., Sheward, W. J., et al. (2002). The VPAC2 receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 109, 497–508. doi: 10.1016/s0092-8674(02)00736-5
Hatori, M., Gill, S., Mure, L. S., Goulding, M., O’Leary, D. D., and Panda, S. (2014). Lhx1 maintains synchrony among circadian oscillator neurons of the SCN. Elife 3:e03357. doi: 10.7554/elife.03357
Hattar, S., Kumar, M., Park, A., Tong, P., Tung, J., Yau, K. W., et al. (2006). Central projections of melanopsin-expressing retinal ganglion cells in the mouse. J. Comp. Neurol. 497, 326–349. doi: 10.1002/cne.20970
Hattar, S., Liao, H. W., Takao, M., Berson, D. M., and Yau, K. W. (2002). Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science 295, 1065–1070. doi: 10.1126/science.1069609
Herzog, E. D. (2007). Neurons and networks in daily rhythms. Nat. Rev. Neurosci. 8, 790–802. doi: 10.1038/nrn2215
Ho, A., and Südhof, T. C. (2004). Binding of F-spondin to amyloid-β precursor protein: a candidate amyloid-β precursor protein ligand that modulates amyloid-β precursor protein cleavage. Proc. Natl. Acad. Sci. U S A 101, 2548–2553. doi: 10.1073/pnas.0308655100
Hoe, H. S., Wessner, D., Beffert, U., Becker, A. G., Matsuoka, Y., and Rebeck, G. W. (2005). F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol. Cell. Biol. 25, 9259–9268. doi: 10.1128/mcb.25.21.9259-9268.2005
Jahanshad, N., Rajagopalan, P., Hua, X., Hibar, D. P., Nir, T. M., Toga, A. W., et al. (2013). Genome-wide scan of healthy human connectome discovers SPON1 gene variant influencing dementia severity. Proc. Natl. Acad. Sci. U S A 110, 4768–4773. doi: 10.1073/pnas.1216206110
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. doi: 10.1038/nature05453
Long, M. A., Jutras, M. J., Connors, B. W., and Burwell, R. D. (2005). Electrical synapses coordinate activity in the suprachiasmatic nucleus. Nat. Neurosci. 8, 61–66. doi: 10.1038/nn1361
Maywood, E. S., Chesham, J. E., O’Brien, J. A., and Hastings, M. H. (2011). A diversity of paracrine signals sustains molecular circadian cycling in suprachiasmatic nucleus circuits. Proc. Natl. Acad. Sci. U S A 108, 14306–14311. doi: 10.1073/pnas.1101767108
Maywood, E. S., Reddy, A. B., Wong, G. K., O’Neill, J. S., O’Brien, J. A., McMahon, D. G., et al. (2006). Synchronization and maintenance of timekeeping in suprachiasmatic circadian clock cells by neuropeptidergic signaling. Curr. Biol. 16, 599–605. doi: 10.1016/j.cub.2006.02.023
McNeill, D. S., Sheely, C. J., Ecker, J. L., Badea, T. C., Morhardt, D., Guido, W., et al. (2011). Development of melanopsin-based irradiance detecting circuitry. Neural Dev. 6:8. doi: 10.1186/1749-8104-6-8
Mohawk, J. A., Green, C. B., and Takahashi, J. S. (2012). Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 35, 445–462. doi: 10.1146/annurev-neuro-060909-153128
Morin, L. P., and Studholme, K. M. (2014). Retinofugal projections in the mouse. J. Comp. Neurol. 522, 3733–3753. doi: 10.1002/cne.23635
Muscat, L., Huberman, A. D., Jordan, C. L., and Morin, L. P. (2003). Crossed and uncrossed retinal projections to the hamster circadian system. J. Comp. Neurol. 466, 513–524. doi: 10.1002/cne.10894
Musiek, E. S. (2015). Circadian clock disruption in neurodegenerative diseases: cause and effect? Front. Pharmacol. 6:29. doi: 10.3389/fphar.2015.00029
Oka, H., Kitagawa, M., and Takata, T. (2015). F-spondin inhibits differentiation of clastic precursors via lipoprotein receptor-related protein 8 (LRP8). J. Periodontol. 86, 465–472. doi: 10.1902/jop.2014.140419
Oster, H., Yasui, A., van der Horst, G. T., and Albrecht, U. (2002). Disruption of mCry2 restores circadian rhythmicity in mPer2 mutant mice. Genes Dev. 16, 2633–2638. doi: 10.1101/gad.233702
Osterhout, J. A., Stafford, B. K., Nguyen, P. L., Yoshihara, Y., and Huberman, A. D. (2015). Contactin-4 mediates axon-target specificity and functional development of the accessory optic system. Neuron 86, 985–999. doi: 10.1016/j.neuron.2015.04.005
Ott, H., Bastmeyer, M., and Stuermer, C. A. (1998). Neurolin, the goldfish homolog of DM-GRASP, is involved in retinal axon pathfinding to the optic disk. J. Neurosci. 18, 3363–3372.
Plump, A. S., Erskine, L., Sabatier, C., Brose, K., Epstein, C. J., Goodman, C. S., et al. (2002). Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron 33, 219–232. doi: 10.1016/s0896-6273(01)00586-4
Redlin, U., Hattar, S., and Mrosovsky, N. (2005). The circadian Clock mutant mouse: impaired masking response to light. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 191, 51–59. doi: 10.1007/s00359-004-0570-z
Redlin, U., and Mrosovsky, N. (1999). Masking of locomotor activity in hamsters. J. Comp. Physiol. A 184, 429–437. doi: 10.1007/s003590050342
Reed, H. E., Meyer-Spasche, A., Cutler, D. J., Coen, C. W., and Piggins, H. D. (2001). Vasoactive intestinal polypeptide (VIP) phase-shifts the rat suprachiasmatic nucleus clock in vitro. Eur. J. Neurosci. 13, 839–843. doi: 10.1046/j.0953-816x.2000.01437.x
Ringstedt, T., Braisted, J. E., Brose, K., Kidd, T., Goodman, C., Tessier-Lavigne, M., et al. (2000). Slit inhibition of retinal axon growth and its role in retinal axon pathfinding and innervation patterns in the diencephalon. J. Neurosci. 20, 4983–4991.
Schwartz, W. J., Peters, R. V., Aronin, N., and Bennett, M. R. (1996). Unexpected c-fos gene expression in the suprachiasmatic nucleus of mice entrained to a skeleton photoperiod. J. Biol. Rhythms 11, 35–44. doi: 10.1177/074873049601100104
Seabrook, T. A., El-Danaf, R. N., Krahe, T. E., Fox, M. A., and Guido, W. (2013). Retinal input regulates the timing of corticogeniculate innervation. J. Neurosci. 33, 10085–10097. doi: 10.1523/JNEUROSCI.5271-12.2013
Sherva, R., Tripodis, Y., Bennett, D. A., Chibnik, L. B., Crane, P. K., de Jager, P. L., et al. (2014). Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimers Dement. 10, 45–52. doi: 10.1016/j.jalz.2013.01.008
Singh, R., Su, J., Brooks, J. M., Terauchi, A., Umemori, H., and Fox, M. A. (2012). Fibroblast growth factor 22 contributes to the development of retinal nerve terminals in the dorsal lateral geniculate nucleus. Front. Mol. Neurosci. 4:61. doi: 10.3389/fnmol.2011.00061
Su, J., Chen, J., Lippold, K., Monavarfeshani, A., Carrillo, G. L., Jenkins, R., et al. (2016). Collagen-derived matricryptins promote inhibitory nerve terminal formation in the developing neocortex. J. Cell Biol. 212, 721–736. doi: 10.1083/jcb.201509085
Su, J., Gorse, K., Ramirez, F., and Fox, M. A. (2010). Collagen XIX is expressed by interneurons and contributes to the formation of hippocampal synapses. J. Comp. Neurol. 518, 229–253. doi: 10.1002/cne.22228
Su, J., Haner, C. V., Imbery, T. E., Brooks, J. M., Morhardt, D. R., Gorse, K., et al. (2011). Reelin is required for class-specific retinogeniculate targeting. J. Neurosci. 31, 575–586. doi: 10.1523/JNEUROSCI.4227-10.2011
Su, J., Klemm, M. A., Josephson, A. M., and Fox, M. A. (2013). Contributions of VLDLR and LRP8 in the establishment of retinogeniculate projections. Neural Dev. 8:11. doi: 10.1186/1749-8104-8-11
Su, J., Stenbjorn, R. S., Gorse, K., Su, K., Hauser, K. F., Ricard-Blum, S., et al. (2012). Target-derived matricryptins organize cerebellar synapse formation through α3β1 integrins. Cell Rep. 2, 223–230. doi: 10.1016/j.celrep.2012.07.001
Thompson, H., Camand, O., Barker, D., and Erskine, L. (2006). Slit proteins regulate distinct aspects of retinal ganglion cell axon guidance within dorsal and ventral retina. J. Neurosci. 26, 8082–8091. doi: 10.1523/JNEUROSCI.1342-06.2006
Truett, G. E., Heeger, P., Mynatt, R. L., Truett, A. A., Walker, J. A., and Warman, M. L. (2000). Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 29, 52–54.
Tzarfati-Majar, V., Burstyn-Cohen, T., and Klar, A. (2001). F-spondin is a contact-repellent molecule for embryonic motor neurons. Proc. Natl. Acad. Sci. U S A 98, 4722–4727. doi: 10.1073/pnas.081062398
van der Horst, G. T., Muijtjens, M., Kobayashi, K., Takano, R., Kanno, S., Takao, M., et al. (1999). Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature 398, 627–630. doi: 10.1038/19323
Varol, E., Sotiras, A., Davatzikos, C., and Alzheimer’s Disease Neuroimaging Initiative. (2017). HYDRA: revealing heterogeneity of imaging and genetic patterns through a multiple max-margin discriminative analysis framework. Neuroimage 145, 346–364. doi: 10.1016/j.neuroimage.2016.02.041
Vitaterna, M. H., Selby, C. P., Todo, T., Niwa, H., Thompson, C., Fruechte, E. M., et al. (1999). Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl. Acad. Sci. U S A 96, 12114–12119. doi: 10.1073/pnas.96.21.12114
Wang, S. W., Kim, B. S., Ding, K., Wang, H., Sun, D., Johnson, R. L., et al. (2001). Requirement for math5 in the development of retinal ganglion cells. Genes Dev. 15, 24–29. doi: 10.1101/gad.855301
Wang, R. H., Zhao, T., Cui, K., Hu, G., Chen, Q., Chen, W., et al. (2016). Negative reciprocal regulation between Sirt1 and Per2 modulates the circadian clock and aging. Sci. Rep. 6:28633. doi: 10.1038/srep28633
Weiner, J. A., Koo, S. J., Nicolas, S., Fraboulet, S., Pfaff, S. L., Pourquié, O., et al. (2004). Axon fasciculation defects and retinal dysplasias in mice lacking the immunoglobulin superfamily adhesion molecule BEN/ALCAM/SC1. Mol. Cell. Neurosci. 27, 59–69. doi: 10.1016/j.mcn.2004.06.005
Keywords: suprachiasmatic nucleus, extracellular matrix, circadian rhythm, photoentrainment, intrinsically photosensitive retinal ganglion cells
Citation: Carrillo GL, Su J, Monavarfeshani A and Fox MA (2018) F-spondin Is Essential for Maintaining Circadian Rhythms. Front. Neural Circuits 12:13. doi: 10.3389/fncir.2018.00013
Received: 06 September 2017; Accepted: 25 January 2018;
Published: 08 February 2018.
Edited by:
Carol Mason, Columbia University, United StatesReviewed by:
Sujata Rao, Cleveland Clinic, United StatesAlexandra Rebsam, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Samer Hattar, Section on Light and Circadian Rhythms (SLCR), National Institute of Mental Health, United States
Copyright © 2018 Carrillo, Su, Monavarfeshani and Fox. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael A. Fox, bWFmb3gxQHZ0Yy52dC5lZHU=