 Ruwani Wijeyekoon
Ruwani Wijeyekoon Roger A. Barker*
Roger A. Barker*- Cambridge Centre for Brain Repair, Department of Clinical Neurosciences, University of Cambridge, Cambridge, UK
Huntington’s disease (HD) is a devastating, fatal, autosomal dominant condition in which the abnormal gene codes for a mutant form of huntingtin that causes widespread neuronal dysfunction and death. This leads to a clinical presentation, typically in midlife, with a combination of motor, psychiatric, cognitive, metabolic, and sleep abnormalities, for which there are some effective symptomatic therapies that can produce some transient benefits. The disease, though, runs a progressive course over a 20-year period ultimately leading to death, and there are currently no proven disease modifying therapies. However whilst the neuronal dysfunction and loss affects much of the central nervous system, the striatum is affected early on in the disease and is one of the areas most affected by the pathogenic process. As a result the prospect of treating HD using neural transplants of striatal tissue has been explored and to date the clinical data is inconclusive. In this review we discuss the rationale for treating HD using this approach, before discussing the clinical trial data and what we have learnt to date using this therapeutic strategy.
Introduction
Huntington’s disease (HD) is a genetic, autosomal dominant, trinucleotide repeat disorder caused by an expansion of CAG repeats in exon 1 of the huntingtin gene and the formation of abnormal huntingtin protein with N-terminal polyglutamine tracts (The Huntington’s Disease Collaborative Research Group, 1993). It is characterized by progressive neurodegeneration within the central nervous system (CNS), with areas such as the striatum and the cerebral cortex traditionally being affected at a relatively early stage, although this is having to be revised in the light of the recent TRACK-HD data (Tabrizi et al., 2009, 2011; Ross and Tabrizi, 2011). Clinically, the disease manifests with the development of movement abnormalities and cognitive and psychiatric symptoms, which gradually deteriorate and invariably lead to severe disability, along with early metabolic, and sleep abnormalities (Goodman et al., 2008, 2011; Wood et al., 2008; Goodman and Barker, 2010).
Although knowledge has advanced since the gene for HD was identified, the biochemical and cellular pathophysiology of the mutant huntingtin gene and protein and their wider effects are complex and still not well understood, leading to difficulties in the development of curative, or disease modifying therapies (Ross and Tabrizi, 2011). Thus, current treatments for HD remain mainly symptomatic and alternative strategies, including cell replacement by fetal striatal tissue transplantation have continued to be explored.
Before the identification of the genetic basis of HD and the research that has stemmed from this, HD was thought of as a disease which primarily involved significant atrophy and degeneration of the striatum-particularly the medium spiny neurons. Whilst it is now recognized that the condition has a much more diffuse pathology from disease onset, it is nevertheless clear that the striatum is a major site of pathology and thus could be a target for cell replacement therapies. As such the rationale developed that replacing the lost neurons through striatal grafting could work for this condition, if the cells could be shown to survive and connect appropriately with the host brain and by so doing recreate the striatal circuitry. This was originally shown experimentally in various older types of animal models of HD.
The Experimental Evidence for Successful Striatal Allografting in Animal Models of HD
Prior to the discovery of the gene for HD and the subsequent generation of transgenic models, the main animal model of HD involved excitotoxic lesions of the striatum (Coyle and Schwarcz, 1976; DiFiglia, 1990; Ferrante et al., 1993). Using this model it was shown that allografts of fetal striatal tissue could survive the transplantation process and become integrated in the host brain (Wictorin, 1992). This integration was shown using an array of labeling techniques, and clearly showed that the striatal projection neurons that developed out of the transplant received cortical and nigral afferents, whilst also projecting to the pallidum and nigra (Clarke et al., 1988; Campbell et al., 1993; Clarke and Dunnett, 1993; Dunnett, 1999). This “circuit reconstruction” led to behavioral improvements in these animals, and provided evidence that such an approach could, in theory, work in patients with HD (Isacson et al., 1984; Dunnett et al., 1988; Reading and Dunnett, 1995).
These early rodent studies were important in showing a proof of concept that striatal allografts could replace and restore function in a “diseased striatum.” However it must be realized that the tissue grafted contains more than just striatal tissue, as typically the whole ganglionic eminence (WGE) is dissected and implanted and this structure gives rise to a range of non-striatal structures such as the pallidum and cortex (Graybiel et al., 1989; Watts et al., 2000a). As such grafts of WGE tissue produced heterogeneous transplants of which only a part are striatal in structure and at least 40% of the total transplant needs to be “striatal” in nature in order for an effect to be seen (Nakao et al., 1996; Watts et al., 2000b). The contribution of this non-striatal tissue to the efficacy, or not, of the graft is unknown, but attempts to circumvent this with more selective dissections [e.g., dissection of the lateral part of the lateral ganglionic eminence (LGE)], whilst increasing the percentage of striatal tissue within the grafts, has failed to enhance or even reproduce the behavioral effects of WGE transplants (Freeman et al., 1995; Peschanski et al., 1995; Brundin et al., 1996; Brasted et al., 1999). In addition these models work on the premise that striatal pathology lies at the heart of HD, and that excitotoxic lesions of this structure, accurately or at least adequately models disease. This is a big assumption given what we now know about HD, and indeed similar allotransplants into a commonly used transgenic mouse model of HD (the R6/2 mouse) did not show any benefits (Dunnett et al., 1998). This is important because ultimately for striatal allografts to work they must form connections to and from the host brain and this process may be compromised if those structures to which they connect are themselves affected by the HD disease process (see below Post-mortem Studies).
Nevertheless by the mid-1990s, when the first transgenic models of HD were emerging, sufficient data had been acquired from the rodent excitotoxic models, to move toward non-human primate studies. In 1998 two groups reported their findings in this area, showing that fetal striatal allografts placed into the excitotoxic lesioned striatum of marmosets and macaques behaved in an analogous fashion to that seen in similar rodent studies (Kendall et al., 1998; Palfi et al., 1998). It was against this background that clinical studies commenced, even though the negative transgenic model transplant study had yet to be done and published and some of these early clinical studies were done at centers not previously involved with the experimental animal transplantation work.
Clinical Studies of Striatal Allografting in Huntington’s Disease
The positive results from animal models led to a number of small open-label studies of fetal striatal transplantation in HD patients at several centers around the world with the expectation that the grafted tissue would work by recreating local striatal circuits, rather than through any neurotrophic effects or an enhancement of endogenous repair processes. These studies are presented in terms of where they were done, as each center tended to adopt their own techniques for tissue preparation and implantation as well as trial design.
Sramka et al. (1992) in Bratislava published an early report on fetal neural transplantation into the caudate nucleus of HD patients using stereotactic neurosurgery. Four HD patients with relatively severe disease received transplants of “mesencephalic” tissue from two to three embryos into the caudate nucleus bilaterally. Immunosuppression was provided with cyclosporine and postoperatively the authors observed a reduction in the amplitude and frequency of hyperkinesia.
This was followed by Madrazo et al. (1995) in Mexico. They published case reports of fetal striatal transplantation in two patients with HD. This study involved a 39-year old female with disease duration of 9 years and a 29-year old male with disease duration of 5 years. Fetal striatal tissue (aged 13 weeks for the first patient and 12 weeks for the second) was transplanted into the right caudate nucleus followed by immunosuppression with cyclosporine A and prednisolone. No surgical complications were reported at 33 and 16 months postoperatively for the two patients who appeared to have a slower or more stable disease progression as compared to their preoperative state.
Quinn et al. (1996) established a protocol by which to assess the safety and efficacy of neural transplantation in HD across studies and centers, – the Core Assessment Program for Intracerebral Transplantation in HD – the CAPIT-HD protocol. This has been used by many centers and has been adopted by most of those trials that we will now discuss (Table 1).
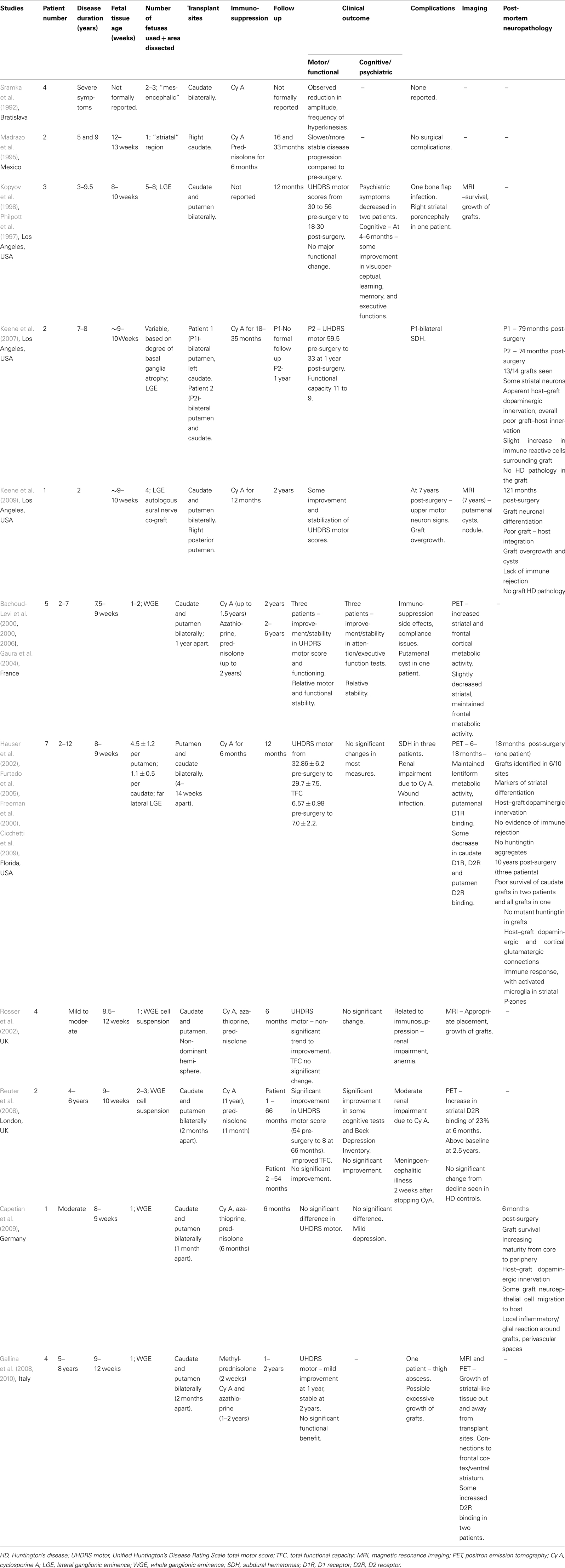
Table 1. Summary of reported clinical studies on fetal striatal transplantation in Huntington’s disease.
Los Angeles, USA
Philpott et al. (1997) and Kopyov et al. (1998) reported a safety study on three HD patients in Los Angeles. The HD patients were non-demented males, age 25–48, with disease duration of 3–9.5 years, and total Unified Huntington’s Disease Rating Scale (UHDRS) motor scores of 30–56. Fetal striatal precursor tissue from the LGE of five to eight donors aged 8–10 weeks was transplanted bilaterally into the caudate and putamen. At 12 months follow up post-surgery there was one bone flap infection, but no major unexpected adverse effects. Overall, UHDRS motor scores improved slightly to 18–30, but in one patient their preoperative myoclonus continued to worsen. Psychiatric symptoms decreased in frequency and intensity in 2/3 cases and at 4–6 months, some scores improved on measures of cognitive functioning.
Magnetic resonance imaging (MRI) scans showed survival and growth of grafts, but at 12 months one patient developed a hypodense area in the right striatum which was thought to represent porencephaly secondary to delayed graft necrosis or an asymptomatic infarct. Overall, the trial concluded that fetal striatal transplantation in HD was possible without major unexpected complications. (See also Post-mortem Studies below).
France
Bachoud-Levi et al. (2000a,b, 2006) reported on fetal striatal transplantation in five HD patients in France in 2000. Pre-transplant, the patients had a disease duration of 2–7 years, UHDRS motor scores of 20–91, total functional capacity (TFC) of 11–12 and all but one had little cognitive impairment. Interestingly, their rate of progression was quite rapid coming up to the grafting itself (Lazic and Barker, 2004). WGE tissue from 7.5- to 9-week-old fetuses was transplanted into the right and left (1 year apart) caudate and putamen. Immunosuppression was with cyclosporine (stopped 6 months post second graft), azathioprine, and prednisolone (gradually stopped up to 1 year post second graft). Two years after receiving the first transplant, three patients showed improvement or stability in the UHDRS motor score, TFC, and neuropsychological tests of attention and executive function along with some neurophysiological tests, compared to control HD patients. 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) imaging showed increased striatal and frontal cortical metabolic activity at 2 years post grafting in these three patients, while the other two patients had no evidence of improvement both clinically and on PET imaging (Gaura et al., 2004). The authors concluded that metabolically active grafts were surviving in the striatum and could underlie the clinical benefits seen in the three patients. At 2–6 year follow up times (Bachoud-Levi et al., 2006) these three patients were seen to demonstrate relative stability in motor features (apart from dystonia), non-timed cognitive, and functional assessments, with slightly decreased striatal and maintained frontal metabolic activity on PET imaging. One other patient improved initially, but then deteriorated following the appearance of a putamenal cyst on MRI. The remaining patient continued to decline throughout.
This group are currently involved in an on-going European Phase II trial of fetal striatal transplantation in HD1. An ancillary study to the main trial has reported on 13 patients aged 39–55, disease duration 4–11 years, who have received fetal striatal transplants (bilaterally in 12 patients, 3 weeks–3 months apart) (Krystkowiak et al., 2007). Immunosuppression involved cyclosporine A (stopped within the first year post bilateral grafting) and azathioprine and prednisolone (gradually decreased and stopped in the following months). Two patients developed subdural hematomas (SDHs) postoperatively and one had a cerebral infection. In addition, one patient showed evidence of an on-going rejection process clinically, immunologically, and on imaging at 14 months post-transplant, which reversed with further immunosuppressive therapy. Four other patients developed evidence of alloimmunization without rejection. The extent to which this will be a significant complication of this approach is not known at the present time, but may become more obvious in the on-going larger trial.
Florida, USA
Hauser et al. (2002) reported on fetal striatal transplantation in HD patients in Florida, USA. Seven non-demented patients, aged 28–64, 2–12 years from diagnosis, with a mean UHDRS total motor score of 32.9 and a TFC of 6.6, received far lateral LGE grafts from 8- to 9-week-old fetuses into the caudate and putamen bilaterally. Immunosuppression with cyclosporine A was given for 6 months. Three patients developed SDHs post operatively, with two requiring surgical evacuation. This was thought to be due to the increased cortical atrophy seen in HD and intraoperative CSF loss, highlighting the risks associated with surgery on more advanced HD patients. At 12 months follow up, mean UHDRS motor scores (29.7 ± 7.54) and TFC (7.0 ± 2.16), indicated no significant improvement or worsening, which may reflect some clinical benefit in the context of the natural progression of HD. Post hoc analysis excluding patient 5 (who deteriorated after the development of the SDHs) indicated a significant improvement in UHDRS motor scores. FDG and (11C)-Raclopride PET imaging demonstrated maintenance of lentiform metabolic activity and putamenal D1 receptor binding. Some decrease in caudate D1 and D2 receptor binding and putamenal D2 binding was seen at 6–18 months, with a significant loss of D2 binding at 2 years (Hauser et al., 2002; Furtado et al., 2005). Several of these cases have now come to post-mortem (see below) with findings that fit well with the PET scanning data.
UK
Rosser et al. (2002) reported on a UK trial of fetal striatal transplantation in HD. Four patients with mild to moderate HD each had WGE cell suspensions from one 8.5- to 12-week-old fetus transplanted into the caudate and putamen of the non-dominant hemisphere. Triple immunosuppression with prednisolone, cyclosporine, and azathioprine was used. At 6 months post-transplant, there was a non-significant trend toward improvement in the UHDRS motor score, but no significant changes in hand tapping, functional capacity, or neuropsychological test scores. MRI showed appropriate positioning and growth of grafts. Up to 6 months, predicted complications from the immunosuppression were seen and included renal impairment and anemia.
These four patients went onto have striatal transplants on the contralateral side on average 22.5 months after the first transplant. One other patient was transplanted bilaterally simultaneously. The patients were followed up with the CAPIT-HD protocol and compared to non-transplanted HD patients. At 2 years post-transplant they were observed to show no significant improvement (Unpublished data; Barker and Swain, 2010).
London, UK
Reuter et al. (2008) reported on two patients receiving fetal striatal allografts in HD. One male and one female patient with disease duration 4–6 years, received WGE cell suspension grafts from two to three 9- to 10-week-old fetuses into the caudate and putamen bilaterally (2 months between sides). Immunosuppression was planned to involve prednisolone for 1 month and cyclosporine for 1 year. Patient 1 was followed up with the CAPIT-HD protocol for 66 months and displayed a gradual but marked improvement which was also reported subjectively. His UHDRS score improved from 54 preoperatively, to 8 at 66 months, with significant improvements in his TFC, Beck Depression score, and some cognitive task scores. (11C)-Raclopride PET imaging showed evidence for increased striatal D2 receptor binding at 6 months, which then decreased at a rate similar to that seen in non-grafted HD patients, but remained above baseline at 2.5 years. This was thought to relate to graft survival within a degenerating striatal complex. Cyclosporine was discontinued at 3 months due to renal complications. Patient 2 did not demonstrate any significant change clinically or on imaging post-transplantation and unfortunately developed an acute meningoencephalitic illness following a fall 2 weeks after stopping the cyclosporine.
Germany
Capetian et al. (2009) reported on the case of a 42-year old male HD patient who underwent fetal striatal transplantation as part of the German component of the European HD phase II study. At baseline he had moderate chorea, with a normal psychopathological status. WGE tissue from one 8- to 9-week-old fetus was transplanted into the caudate and putamen bilaterally, 1 month apart and standard triple immunosuppression was used. At 6 months post-transplantation, there was no significant difference in the UHDRS or cognitive score, but he showed evidence of slight depression. He died of unrelated causes at 6 months post-transplantation and the histology of the transplant is discussed below.
Italy
Recently Gallina et al. (2008, 2010) reported on four cases of fetal striatal transplantation in HD. The patients aged 33–53 were 5–8 years into their illness and each had WGE tissue from one 9- to 12-week-old fetus transplanted into the caudate and putamen bilaterally, 2 months apart. Prednisolone, cyclosporine, and azathioprine immunosuppression was used. The patients had combinations of motor dysfunction (chorea, dystonia), cognitive working memory problems, and mild psychiatric symptoms pre-transplant. On follow up, the patients generally showed mild improvements in UHDRS scores at 1 year post-transplant, which remained relatively stable at 2 years. However, no significant functional benefit was evident and one patient’s postoperative course was complicated by the development of a thigh abscess.
Magnetic resonance imaging and PET imaging showed macroscopic growth of metabolically active, striatal – like tissue with extension beyond the deposition site in six of eight grafts and additional development of new tissue connecting the grafts with the frontal cortex and the ventral striatum. There was some reported increase in D2 receptor binding in three patients as observed with (123)IBZM-SPECT imaging. The authors report that they found no clinical or imaging evidence of uncontrolled graft growth. However in relation to this study, Freeman et al. (2011) have discussed the concern of graft overgrowth, the importance of optimizing donor, and grafting conditions and other key technical issues involved in ensuring the safety of fetal neural transplantation using this approach in HD.
Clinical Studies of Striatal Xenografting in Huntington’s Disease
The use of fetal striatal tissue for transplantation brings with it significant ethical issues and limitations in the amount of tissue available at any one time. Xenotransplantation, with the use of porcine neural tissue may avoid some of these issues and one clinical study has been reported so far (Fink et al., 2000). In this study 12 HD patients received 24 million fetal porcine striatal cells into the striatum unilaterally. One year post-transplantation, they exhibited a favorable safety profile, but there was no significant change in the TFC score. This is not surprising given what we know about the survival of porcine tissue in the much less immunologically challenging situation in the laboratory (Barker and Widner, 2004).
Post-Mortem Studies of Striatal Allografting in Huntington’s Disease
Several centers have now reported post-mortem analyses on HD patients with fetal striatal transplants.
Florida, USA
The first post-mortem report (Freeman et al., 2000) related to a patient who received bilateral LGE transplants in Hauser et al.’s (2002) study in Florida, USA. Three grafts were placed in the right putamen, with one in the right caudate, while the left putamen and caudate received four and two grafts respectively. He showed benefit in chorea and gait scores post-transplant, but died at 18 months. Post-mortem analysis identified three graft sites in the right putamen, two in the left putamen, and one in the left anterior limb of the internal capsule, indicating survival of grafts 12 months after stopping immunosuppression. There was evidence of markers of graft striatal differentiation and host dopaminergic innervation of the graft but no evidence of immune rejection or aggregated huntingtin in grafted tissue.
Three other patients transplanted at the same center (patients 1, 3, and 5; Hauser et al., 2002) came to post-mortem 10 years post-surgery (Cicchetti et al., 2009). They had received four to six grafts into each putamen and one to two grafts into each caudate (Hauser et al., 2002). Graft survival was very poor in patient 3, with only 1 (putamenal graft) of 16 transplants identified at post-mortem. In the other two patients, there was complete loss of all caudate grafts, but surviving putamenal grafts displayed areas with striatal markers (P-zones). These regions contained typical striatal projection neurons and interneurons, with the projection neurons appearing less healthy compared to the interneurons. This pattern of neuronal degeneration was noted to mimic that seen in HD and the authors concluded that this suggested disease-like neuronal degeneration was occurring in genetically unrelated striatal grafts. Host cortical glutamatergic projections were found to be closely apposed to striatal projection neurons and there appeared to be an on-going immune response. Although the astroglial response respected transplant boundaries, activated microglia were seen within the striatal P-zones, with some indication of potential phagocytosis of neuronal elements. There was no evidence of abnormal huntingtin aggregation within the graft (Cicchetti et al., 2009, 2011).
Los Angeles, USA
Keene et al. (2007) reported on the post-mortem results of two patients transplanted at the Los Angeles center. Both patients appeared moderately impaired at transplantation. Patient 1 received two grafts in the right putamen, three in the left putamen, and one in the left caudate. He developed bilateral SDHs 2 months post operatively, but was lost to follow up and died 79 months post-transplantation. Patient 2 received three grafts per putamen and one graft per caudate. She initially displayed apparent slower progression at 3 months post-transplant, before continuing to progress at a rate typical for HD and died 74 months after surgery. Post-mortem studies showed similar findings in both patients. All grafts, except the left caudate graft in patient 2, were identified and consisted of neurons and dense neuropil, with few astrocytes, and microglia. Neurons expressed markers consistent with striatal neuronal differentiation and although there was some apparent host dopaminergic innervation of the graft, evidence of graft to host innervation was scant. Neuronal intranuclear polyubiquitinated inclusions, characteristic of HD pathology, were absent in the grafts.
Keene et al. (2009) also reported a case of fetal striatal transplantation at the same center which showed evidence of graft overgrowth at post-mortem. A 29-year old female HD patient with mild motor, cognitive, and psychiatric symptoms received 9- to 10-week-old fetal LGE transplants bilaterally (four in each putamen and one in each caudate). In addition, based on some data suggesting the possible neurotrophic benefits of peripheral nerve co-grafts (Watts et al., 1995, 1997), autologous sural nerve was also implanted into the most posterior right putamenal graft site. She received cyclosporine immunosuppression for 12 months. She showed improvement and stabilization of the UHDRS motor and behavior scales and in some other indices up to 2 years post-transplant, but then developed upper motor neuron signs, with MRI evidence of a cyst and a nodular mass in the left and right putamen respectively. HD disease features also then continued to progress and she died around 121 months post-transplantation. Post-mortem revealed graft overgrowth, with five mass lesions and two cysts in the right caudate and putamen and three mass lesions and one large cyst in the left caudate and putamen. The masses contained unorganized neuropil and a diffuse population of neurons, with islands of reactive astrocytes and scattered oligodendrocytes. The authors reported no evidence of on-going mitotic activity and interpreted the graft overgrowth as an excess of “normal” graft tissue, rather then neoplastic proliferation. Factors influencing this outcome were considered to include original graft composition, host factors, and possibly effects of the sural nerve graft. Generally poor graft–host integration, lack of immune rejection of allografts and absence of graft inclusion pathology were also reported.
Germany
Post-mortem results of the case described by Capetian et al. (2009) in Germany have also been reported. The patient received two fetal WGE grafts with two trajectories into the head of the caudate, two trajectories into the pre-commissural putamen and one trajectory into the post-commissural putamen, bilaterally. The patient died of unrelated causes at 6 months post-transplant and post-mortem showed evidence of surviving grafts along all trajectories, with increasing maturity from the core to the periphery. There was host dopaminergic innervation of the graft and some migration of graft neuroepithelial cells into the host. Local inflammatory infiltrates and an increased surrounding glial reaction were seen with perivascular cuffing of lymphocytes in response to grafted cells.
Based on the post-mortem studies so far, fetal striatal transplants in HD appear to display poor long-term survival. The reasons for this are not known, but allograft immunoreactivity, excitotoxicity, and targeted microglial responses have been suggested as potential factors in this poor survival (Cicchetti et al., 2011). This has obvious implications for the long-term therapeutic efficacy of striatal transplantation in HD, as it would suggest that any cell placed into the HD brain is likely to succumb to the disease process in the tissue into which it is placed as the grafts try to integrate.
Clinical Challenges for Striatal Allografting in Huntington’s Disease
Unlike ventral mesencephalic allografting for Parkinson’s disease (PD), the case for neural grafting with fetal striatal tissue for HD remains unproven. There are case reports of benefits that can last for years, but there is also much data suggesting that it does not work in the long term and post-mortem data suggesting that the grafts eventually succumb to the disease process. This raises a number of questions and challenges in using this approach for HD, including:
• Why graft only the striatum when the disease affects many areas of the CNS from disease onset?
• How can the graft survive and integrate when it is being placed into a hostile environment where the disease process is well established?
• What contribution does the non-striatal tissue in the graft make to the efficacy or otherwise of the transplant?
• If neural grafting is to be attempted in HD, when in the disease process should it be done?
• Why bother using such an approach when a more logical one would be to target the pathogenic cascade downstream of the mutant gene and the protein it codes for?
• How can the benefits of cell therapy be properly assessed in small numbers of HD patients, each of whom may progress rather differently from each other?
We will now briefly deal with each of these issues in turn.
Why graft only the striatum when the disease affects many areas of the CNS from disease onset?
It is now well recognized that HD has a diffuse pathology from the onset and that whilst the striatum is affected early on and significantly, it is not the only site of pathology. However, the fact that it is a majorly affected site does at least give some credence to targeting it with replacement therapies, especially given some of the experimental data in animal models of HD. Indeed in the field of PD, it is now recognized that the disease process involves many system outside the dopaminergic nigrostriatal pathway, but this does not mean that targeting this latter pathway is without effect (Brundin et al., 2010). So the same could be true of HD, neural striatal allografting is not trying to cure patients of HD, it is simply trying to restore one part of a more global CNS disorder that does have major striatal pathology as part of that process. The question then arises as to how much of the features of HD are dependent on striatal pathology, as opposed to pathology at other CNS sites, and how significant are those features to the patients quality of life (Tippett et al., 2007; Ho et al., 2009; Thu et al., 2010). This is not known, but at least some of the problems seen in this disorder do have a striatal origin and thus targeting this structure for cell replacement is not in itself a bad idea, it just has limited potential to help the patient.
How can the graft survive and integrate when it is being placed into a hostile environment where the disease process is well established?
This has only recently become an issue as the post-mortem studies emerge, and does represent a major problem to the field. Experimentally grafts of fetal striatal tissue have been shown to survive and integrate well in the excitotoxic model of HD (Clarke and Dunnett, 1993), but this is very different from the genetic disorder where all cells are affected to a degree by the mutant gene and its product. Studies in HD transgenic models have been very limited, but at least clinically in those grafts that have been looked at years after implantation, there is evidence that they are not surviving. The reasons for this are not known, although a number of theories have been proposed (Cicchetti et al., 2011). Nevertheless this suggests that striatal allografts for HD patients will always have only limited benefits. The question then arises as to whether these transient benefits are worthwhile, given there are no disease modifying therapies for this condition.
What contribution does the non-striatal tissue in the graft make to the efficacy or otherwise of the transplant?
This is unknown, as attempts to increase the yield of striatal tissue in the transplant have failed to translate into behavioral benefits, probably because the selective dissections undertaken to do this remove a critical population of cells that form a vital functional component of the transplant. Nevertheless it is clearly important to know what contribution these non-striatal populations of cells make to the efficacy or otherwise of the graft (Freeman et al., 2011), and this will become more of an issue as we move toward the production of striatal neurons from stem cells sources (Dunnett and Rosser, 2007). For the moment, all we can say is that the role of the non-striatal tissue to the survival, integration, and efficacy of WGE striatal transplants is unknown.
If neural grafting is to be attempted in HD, when in the disease process should it be done?
This raises a number of important issues to do with how novel therapeutics of an invasive nature can be done in chronic neurodegenerative disorders of the CNS. The rationale for neural allografting is to replace the striatal tissue lost to the disease process and so one could argue that the tissue should be grafted when the striatum has significant atrophy in patients with moderately advanced disease. This though brings with it a number of issues; (a) how do you neurosurgically place tissue into a significantly atrophied structure; (b) how can that tissue be expected to recreate circuits when most of the components of that circuit have been lost; (c) is it sensible to place tissue in an environment where the disease process is active; finally (d) how can the patient be consented at a stage of disease when they have significant cognitive deficits. Conversely if you go for very early stage disease then you run the risk of; (a) causing or exacerbating striatal damage with sequelae to the patient; and (b) treating a stage of disease that is not that problematic to the patient. Thus, it would seem logical to pick some intermediate stage between these two stages and although exactly which stage this would be is unclear, it would probably represent early-mild disease stages.
Why bother using such an approach when a more logical one would be to target the pathogenic cascade downstream of the mutant gene and the protein it codes for?
Ultimately the cure for HD will come from better understanding the pathogenic pathway and blocking the disease process using a cocktail of agents. However, there are currently no disease modifying agents for HD, despite several attempts to find one including studies with Co-enzyme Q10; remacemide; riluzole, EPA, creatine, lamotrigine, baclofen, idebenone and D-alpha-tocopherol (Shoulson et al., 1989; Peyser et al., 1995; Ranen et al., 1996; Kremer et al., 1999; Huntington Study Group, 2001; Verbessem et al., 2003; Landwehrmeyer et al., 2007; Huntington Study Group TREND-HD Investigators, 2008; Mestre et al., 2009). As such neural grafting must still be seen as a possible therapy, especially since it seeks to replace lost cells, not stop a disease process and thus could be used together with disease modifying therapies in some instances. Whether it has any real benefits in HD is unproven, and until such time as this is resolved, it should still be considered as one therapeutic strategy.
How can the benefits of cell therapy be properly assessed in small numbers of HD patients, each of whom may progress rather differently from each other?
The optimal way to do any cell therapy trial for any neurodegenerative disorder of the CNS is not known, as the diseases are not easy to monitor; vary from patient to patient and over time; studies involve small numbers of patients; rely on data from imperfect animal models and often seek to find quick solutions in complex diffuse disease processes. Fetal striatal allografts and HD are no exception to this, as although the disease can be diagnosed with absolute confidence based on a genetic test in the right clinical context, its natural history is not properly known. Thus individual patients with HD follow different trajectories for reasons that are not clear, although work is being done to identify disease modifiers – from both a genetic and non-genetic perspective. The best controls in small trials may be the patient themselves but recently it has been suggested that disease progression varies as a function of disease stage, and is not linear (Tabrizi et al., 2011). This inability to understand what controls and predicts disease progression is a major issue, especially as it is now becoming clear that environmental factors may improve graft function (Brasted et al., 2000) as well as have effects on the disease course (van Dellen et al., 2000, 2008; Nithianantharajah and Hannan, 2006) in experimental models of disease and repair in HD.
Ultimately the best way to do cell therapy studies in HD is either to:
(a) use larger numbers with a delayed start design as has been done in the on-going transplant study in France (see text Footnote 1). Whilst an obvious solution, this is not straightforward given the variability of disease onset and progression, for which a large cohort is not immune, along with the ethical concerns of undertaking irreversible experimental therapies in large numbers of patients;
(b) use intensive multi-modal assessments in small numbers of patients and ascertain whether they all point in the same direction (Reuter et al., 2008); This adoption of multiple end points is useful, although if one has discordant results it will then be important to know which is the more meaningful change and this may prove difficult. In addition, unlike the use of disease modifying therapies, neural grafts in HD currently are used to repair the brain and not change the underlying neuropathological process and as such changes in any biomarker measures may not be informative;
(c) use very long follow up times (Reuter et al., 2008; Politis et al., 2010). This is useful but is complicated by the fact that long-term follow up brings with it the problems of avoiding the introduction of any symptomatic drugs which will alter some disease measures.
Which of these approaches one adopts is dependent to some extent on what is being trialed, as safety is paramount in these studies and the amount of data that speaks to that, or is already known, determines the risk one can take with patient cohorts. However the challenge of doing experimental therapeutics in neurodegenerative disorders of the CNS such as HD remains controversial, but indefinite follow up of small numbers of intensely studied patients currently represents the best approach, as has been seen in trials of ventral mesencephalic grafting in PD (Politis et al., 2010).
Conclusion
The use of fetal striatal tissue as a therapeutic strategy for HD has a long history experimentally, but clinically the data is lacking as to whether it really works. The experimental work goes back 30 years and has mainly been based on lesion models of HD, with relatively little work having been done in the newer transgenic model of HD. These newer transgenic models suffer from an absence of cell loss, but many may regard these as being more akin to the clinical scenario. Thus the failure to show benefits from grafting in the R6/2 mouse model (Dunnett et al., 1998) may be a more realistic approach, although the rate at the disease progresses in these animals makes the interpretation more difficult. Indeed further studies need to be done in the more slowly progressing newer transgenic animals before any conclusions can be drawn on the efficacy of fetal striatal allografting in this model of disease. What the experimental studies in the striatal lesion models of HD have shown, is that fetal striatal tissue can survive, differentiate appropriately, make connections, and circuits and restore behavior when grafted into such a lesion. What remains unresolved is whether this is possible when other structures in these circuits are also involved in the disease process.
In this review we have laid out the rationale and basis for the clinical studies that have adopted this approach, and highlighted that much of this work was done in the pre-gene era when the disease was thought to have a much more exclusive striatal pathology. The clinical studies that have subsequently been undertaken have been done with differing degrees of rigor and produced mixed results, but with little evidence to date that it produces long-term benefits in the majority of patients. This, coupled to the emerging post-mortem data in some of these patients, suggests that this approach may not be the optimal therapeutic strategy for treating HD. However, until clear evidence emerges of disease modifying therapies for HD, it remains one possible strategy and could even be used in conjunction with such treatments as a way of repairing and retarding the disease. Ultimately, though, neural striatal allografting is only treating one part of a much more diffuse disease process and as such will never be a cure for this devastating disorder of the CNS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Some of the authors’ work is supported by a NIHR Biomedical Research Centre award to the University of Cambridge and Addenbrooke’s Hospital.
Footnote
References
Bachoud-Levi, A., Bourdet, C., Brugieres, P., Nguyen, J. P., Grandmougin, T., Haddad, B., Jeny, R., Bartolomeo, P., Boisse, M. F., Barba, G. D., Degos, J. D., Ergis, A. M., Lefaucheur, J. P., Lisovoski, F., Pailhous, E., Rémy, P., Palfi, S., Defer, G. L., Cesaro, P., Hantraye, P., and Peschanski, M. (2000a). Safety and tolerability assessment of intrastriatal neural allografts in five patients with Huntington’s disease. Exp. Neurol. 161, 194–202.
Bachoud-Levi, A. C., Remy, P., Nguyen, J. P., Brugieres, P., Lefaucheur, J. P., Bourdet, C., Baudic, S., Gaura, V., Maison, P., Haddad, B., Boissé, M. F., Grandmougin, T., Jény, R., Bartolomeo, P., Dalla Barba, G., Degos, J. D., Lisovoski, F., Ergis, A. M., Pailhous, E., Cesaro, P., Hantraye, P., and Peschanski, M. (2000b). Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet 356, 1975–1979.
Bachoud-Levi, A. C., Gaura, V., Brugieres, P., Lefaucheur, J. P., Boisse, M. F., Maison, P., Baudic, S., Ribeiro, M. J., Bourdet, C., Remy, P., Cesaro, P., Hantraye, P., and Peschanski, M. (2006). Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: a long-term follow-up study. Lancet Neurol. 5, 303–309.
Barker, R. A., and Swain, R. (2010). Neural transplantation for the treatment of Huntington’s Disease. Eur. Neurol. Rev. 5, 41–45.
Barker, R. A., and Widner, H. (2004). Immune problems in central nervous system cell therapy. NeuroRx 1, 472–481.
Brasted, P. J., Watts, C., Torres, E. M., Robbins, T. W., and Dunnett, S. B. (1999). Behavioural recovery following striatal transplantation: effects of postoperative training and P-zone volume. Exp. Brain Res. 128, 535–538.
Brasted, P. J., Watts, C., Torres, E. M., Robbins, T. W., and Dunnett, S. B. (2000). Behavioral recovery after transplantation into a rat model of Huntington’s disease: dependence on anatomical connectivity and extensive postoperative training. Behav. Neurosci. 114, 431–436.
Brundin, P., Barker, R. A., and Parmar, M. (2010). Neural grafting in Parkinson’s disease problems and possibilities. Prog. Brain Res. 184, 265–294.
Brundin, P., Fricker, R. A., and Nakao, N. (1996). Paucity of P-zones in striatal grafts prohibit commencement of clinical trials in Huntington’s disease. Neuroscience 71, 895–897.
Campbell, K., Kalen, P., Wictorin, K., Lundberg, C., Mandel, R. J., and Bjorklund, A. (1993). Characterization of GABA release from intrastriatal striatal transplants: dependence on host-derived afferents. Neuroscience 53, 403–415.
Capetian, P., Knoth, R., Maciaczyk, J., Pantazis, G., Ditter, M., Bokla, L., Landwehrmeyer, G. B., Volk, B., and Nikkhah, G. (2009). Histological findings on fetal striatal grafts in a Huntington’s disease patient early after transplantation. Neuroscience 160, 661–675.
Cicchetti, F., Saporta, S., Hauser, R. A., Parent, M., Saint-Pierre, M., Sanberg, P. R., Li, X. J., Parker, J. R., Chu, Y., Mufson, E. J., Kordower, J. H., and Freeman, T. B. (2009). Neural transplants in patients with Huntington’s disease undergo disease-like neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 12483–12488.
Cicchetti, F., Soulet, D., and Freeman, T. B. (2011). Neuronal degeneration in striatal transplants and Huntington’s disease: potential mechanisms and clinical implications. Brain 134(Pt 3), 641–652.
Clarke, D. J., and Dunnett, S. B. (1993). Synaptic relationships between cortical and dopaminergic inputs and intrinsic GABAergic systems within intrastriatal striatal grafts. J. Chem. Neuroanat. 6, 147–158.
Clarke, D. J., Dunnett, S. B., Isacson, O., Sirinathsinghji, D. J., and Bjorklund, A. (1988). Striatal grafts in rats with unilateral neostriatal lesions – I. Ultrastructural evidence of afferent synaptic inputs from the host nigrostriatal pathway. Neuroscience 24, 791–801.
Coyle, J. T., and Schwarcz, R. (1976). Lesion of striatal neurones with kainic acid provides a model for Huntington’s chorea. Nature 263, 244–246.
DiFiglia, M. (1990). Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 13, 286–289.
Dunnett, S. B. (1999). Striatal reconstruction by striatal grafts. J. Neural Transm. Suppl. 55, 115–129.
Dunnett, S. B., Carter, R. J., Watts, C., Torres, E. M., Mahal, A., Mangiarini, L., Bates, G., and Morton, A. J. (1998). Striatal transplantation in a transgenic mouse model of Huntington’s disease. Exp. Neurol. 154, 31–40.
Dunnett, S. B., Isacson, O., Sirinathsinghji, D. J., Clarke, D. J., and Bjorklund, A. (1988). Striatal grafts in rats with unilateral neostriatal lesions – III. Recovery from dopamine-dependent motor asymmetry and deficits in skilled paw reaching. Neuroscience 24, 813–820.
Dunnett, S. B., and Rosser, A. E. (2007). Stem cell transplantation for Huntington’s disease. Exp. Neurol. 203, 279–292.
Ferrante, R. J., Kowall, N. W., Cipolloni, P. B., Storey, E., and Beal, M. F. (1993). Excitotoxin lesions in primates as a model for Huntington’s disease: histopathologic and neurochemical characterization. Exp. Neurol. 119, 46–71.
Fink, J. S., Schumacher, J. M., Ellias, S. L., Palmer, E. P., Saint-Hilaire, M., Shannon, K., Penn, R., Starr, P., VanHorne, C., Kott, H. S., Dempsey, P. K., Fischman, A. J., Raineri, R., Manhart, C., Dinsmore, J., and Isacson, O. (2000). Porcine xenografts in Parkinson’s disease and Huntington’s disease patients: preliminary results. Cell Transplant. 9, 273–278.
Freeman, T. B., Cicchetti, F., Bachoud-Levi, A. C., and Dunnett, S. B. (2011). Technical factors that influence neural transplant safety in Huntington’s disease. Exp. Neurol. 227, 1–9.
Freeman, T. B., Cicchetti, F., Hauser, R. A., Deacon, T. W., Li, X. J., Hersch, S. M., Nauert, G. M., Sanberg, P. R., Kordower, J. H., Saporta, S., and Isacson, O. (2000). Transplanted fetal striatum in Huntington’s disease: phenotypic development and lack of pathology. Proc. Natl. Acad. Sci. U.S.A. 97, 13877–13882.
Freeman, T. B., Sanberg, P. R., and Isacson, O. (1995). Development of the human striatum: implications for fetal striatal transplantation in the treatment of Huntington’s disease. Cell Transplant. 4, 539–545.
Furtado, S., Sossi, V., Hauser, R. A., Samii, A., Schulzer, M., Murphy, C. B., Freeman, T. B., and Stoessl, A. J. (2005). Positron emission tomography after fetal transplantation in Huntington’s disease. Ann. Neurol. 58, 331–337.
Gallina, P., Paganini, M., Lombardini, L., Mascalchi, M., Porfirio, B., Gadda, D., Marini, M., Pinzani, P., Salvianti, F., Crescioli, C., Bucciantini, S., Mechi, C., Sarchielli, E., Romoli, A. M., Bertini, E., Urbani, S., Bartolozzi, B., De Cristofaro, M. T., Piacentini, S., Saccardi, R., Pupi, A., Vannelli, G. B., and Di Lorenzo, N. (2010). Human striatal neuroblasts develop and build a striatal-like structure into the brain of Huntington’s disease patients after transplantation. Exp. Neurol. 222, 30–41.
Gallina, P., Paganini, M., Lombardini, L., Saccardi, R., Marini, M., De Cristofaro, M. T., Pinzani, P., Salvianti, F., Crescioli, C., Di Rita, A., Bucciantini, S., Mechi, C., Sarchielli, E., Moretti, M., Piacentini, S., Gritti, G., Bosi, A., Sorbi, S., Orlandini, G., Vannelli, G. B., and Di Lorenzo, N. (2008). Development of human striatal anlagen after transplantation in a patient with Huntington’s disease. Exp. Neurol. 213, 241–244.
Gaura, V., Bachoud-Levi, A. C., Ribeiro, M. J., Nguyen, J. P., Frouin, V., Baudic, S., Brugieres, P., Mangin, J. F., Boisse, M. F., Palfi, S., Cesaro, P., Samson, Y., Hantraye, P., Peschanski, M., and Remy, P. (2004). Striatal neural grafting improves cortical metabolism in Huntington’s disease patients. Brain 127(Pt 1), 65–72.
Goodman, A. O., and Barker, R. A. (2010). How vital is sleep in Huntington’s disease? J. Neurol. 257, 882–897.
Goodman, A. O., Murgatroyd, P. R., Medina-Gomez, G., Wood, N. I., Finer, N., Vidal-Puig, A. J., Morton, A. J., and Barker, R. A. (2008). The metabolic profile of early Huntington’s disease – a combined human and transgenic mouse study. Exp. Neurol. 210, 691–698.
Goodman, A. O., Rogers, L., Pilsworth, S., McAllister, C. J., Shneerson, J. M., Morton, A. J., and Barker, R. A. (2011). Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease. Curr. Neurol. Neurosci. Rep. 11, 821–827.
Graybiel, A. M., Liu, F. C., and Dunnett, S. B. (1989). Intrastriatal grafts derived from fetal striatal primordia. I. Phenotypy and modular organization. J. Neurosci. 9, 3250–3271.
Hauser, R. A., Furtado, S., Cimino, C. R., Delgado, H., Eichler, S., Schwartz, S., Scott, D., Nauert, G. M., Soety, E., Sossi, V., Holt, D. A., Sanberg, P. R., Stoessl, A. J., and Freeman, T. B. (2002). Bilateral human fetal striatal transplantation in Huntington’s disease. Neurology 58, 687–695.
Ho, A. K., Gilbert, A. S., Mason, S. L., Goodman, A. O., and Barker, R. A. (2009). Health-related quality of life in Huntington’s disease: which factors matter most? Mov. Disord. 24, 574–578.
Huntington Study Group. (2001). A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology 57, 397–404.
Huntington Study Group TREND-HD Investigators. (2008). Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: the TREND-HD study. Arch. Neurol. 65, 1582–1589.
Isacson, O., Brundin, P., Kelly, P. A., Gage, F. H., and Bjorklund, A. (1984). Functional neuronal replacement by grafted striatal neurones in the ibotenic acid-lesioned rat striatum. Nature 311, 458–460.
Keene, C. D., Chang, R. C., Leverenz, J. B., Kopyov, O., Perlman, S., Hevner, R. F., Born, D. E., Bird, T. D., and Montine, T. J. (2009). A patient with Huntington’s disease and long-surviving fetal neural transplants that developed mass lesions. Acta Neuropathol. 117, 329–338.
Keene, C. D., Sonnen, J. A., Swanson, P. D., Kopyov, O., Leverenz, J. B., Bird, T. D., and Montine, T. J. (2007). Neural transplantation in Huntington disease: long-term grafts in two patients. Neurology 68, 2093–2098.
Kendall, A. L., Rayment, F. D., Torres, E. M., Baker, H. F., Ridley, R. M., and Dunnett, S. B. (1998). Functional integration of striatal allografts in a primate model of Huntington’s disease. Nat. Med. 4, 727–729.
Kopyov, O. V., Jacques, S., Lieberman, A., Duma, C. M., and Eagle, K. S. (1998). Safety of intrastriatal neurotransplantation for Huntington’s disease patients. Exp. Neurol. 149, 97–108.
Kremer, B., Clark, C. M., Almqvist, E. W., Raymond, L. A., Graf, P., Jacova, C., Mezei, M., Hardy, M. A., Snow, B., Martin, W., and Hayden, M. R. (1999). Influence of lamotrigine on progression of early Huntington disease: a randomized clinical trial. Neurology 53, 1000–1011.
Krystkowiak, P., Gaura, V., Labalette, M., Rialland, A., Remy, P., Peschanski, M., and Bachoud-Levi, A. C. (2007). Alloimmunisation to donor antigens and immune rejection following foetal neural grafts to the brain in patients with Huntington’s disease. PLoS ONE 2, e166. doi:10.1371/journal.pone.0000166
Landwehrmeyer, G. B., Dubois, B., de Yebenes, J. G., Kremer, B., Gaus, W., Kraus, P. H., Przuntek, H., Dib, M., Doble, A., Fischer, W., Ludolph, A. C., and European Huntington’s Disease Initiative Study Group. (2007). Riluzole in Huntington’s disease: a 3-year, randomized controlled study. Ann. Neurol. 62, 262–272.
Lazic, S. E., and Barker, R. A. (2004). Rapid decline in motor symptoms in HD neural transplant patients prior to surgery. Brain Res. Bull. 63, 83–84.
Madrazo, I., Franco-Bourland, R. E., Castrejon, H., Cuevas, C., and Ostrosky-Solis, F. (1995). Fetal striatal homotransplantation for Huntington’s disease: first two case reports. Neurol. Res. 17, 312–315.
Mestre, T., Ferreira, J., Coelho, M. M., Rosa, M., and Sampaio, C. (2009). Therapeutic interventions for disease progression in Huntington’s disease. Cochrane Database Syst. Rev. CD006455.
Nakao, N., Grasbon-Frodl, E. M., Widner, H., and Brundin, P. (1996). DARPP-32-rich zones in grafts of lateral ganglionic eminence govern the extent of functional recovery in skilled paw reaching in an animal model of Huntington’s disease. Neuroscience 74, 959–970.
Nithianantharajah, J., and Hannan, A. J. (2006). Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat. Rev. Neurosci. 7, 697–709.
Palfi, S., Conde, F., Riche, D., Brouillet, E., Dautry, C., Mittoux, V., Chibois, A., Peschanski, M., and Hantraye, P. (1998). Fetal striatal allografts reverse cognitive deficits in a primate model of Huntington disease. Nat. Med. 4, 963–966.
Peschanski, M., Cesaro, P., and Hantraye, P. (1995). Rationale for intrastriatal grafting of striatal neuroblasts in patients with Huntington’s disease. Neuroscience 68, 273–285.
Peyser, C. E., Folstein, M., Chase, G. A., Starkstein, S., Brandt, J., Cockrell, J. R., Bylsma, F., Coyle, J. T., McHugh, P. R., and Folstein, S. E. (1995). Trial of D-alpha-tocopherol in Huntington’s disease. Am. J. Psychiatry 152, 1771–1775.
Philpott, L. M., Kopyov, O. V., Lee, A. J., Jacques, S., Duma, C. M., Caine, S., Yang, M., and Eagle, K. S. (1997). Neuropsychological functioning following fetal striatal transplantation in Huntington’s chorea: three case presentations. Cell Transplant. 6, 203–212.
Politis, M., Wu, K., Loane, C., Quinn, N. P., Brooks, D. J., Rehncrona, S., Bjorklund, A., Lindvall, O., and Piccini, P. (2010). Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci. Transl. Med. 2, 38ra46.
Quinn, N., Brown, R., Craufurd, D., Goldman, S., Hodges, J., Kieburtz, K., Lindvall, O., MacMillan, J., and Roos, R. (1996). Core Assessment Program for Intracerebral Transplantation in Huntington’s Disease (CAPIT-HD). Mov. Disord. 11, 143–150.
Ranen, N. G., Peyser, C. E., Coyle, J. T., Bylsma, F. W., Sherr, M., Day, L., Folstein, M. F., Brandt, J., Ross, C. A., and Folstein, S. E. (1996). A controlled trial of idebenone in Huntington’s disease. Mov. Disord. 11, 549–554.
Reading, P. J., and Dunnett, S. B. (1995). Embryonic striatal grafts reverse the disinhibitory effects of ibotenic acid lesions of the ventral striatum. Exp. Brain Res. 105, 76–86.
Reuter, I., Tai, Y. F., Pavese, N., Chaudhuri, K. R., Mason, S., Polkey, C. E., Clough, C., Brooks, D. J., Barker, R. A., and Piccini, P. (2008). Long-term clinical and positron emission tomography outcome of fetal striatal transplantation in Huntington’s disease. J. Neurol. Neurosurg. Psychiatr. 79, 948–951.
Ross, C. A., and Tabrizi, S. J. (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98.
Rosser, A. E., Barker, R. A., Harrower, T., Watts, C., Farrington, M., Ho, A. K., Burnstein, R. M., Menon, D. K., Gillard, J. H., Pickard, J., Dunnett, S. B., and NEST-UK. (2002). Unilateral transplantation of human primary fetal tissue in four patients with Huntington’s disease: NEST-UK safety report ISRCTN no 36485475. J. Neurol. Neurosurg. Psychiatr. 73, 678–685.
Shoulson, I., Odoroff, C., Oakes, D., Behr, J., Goldblatt, D., Caine, E., Kennedy, J., Miller, C., Bamford, K., Rubin, A., Plumb, S., and Kurlan, R. (1989). A controlled clinical trial of baclofen as protective therapy in early Huntington’s disease. Ann. Neurol. 25, 252–259.
Sramka, M., Rattaj, M., Molina, H., Vojtassak, J., Belan, V., and Ruzicky, E. (1992). Stereotactic technique and pathophysiological mechanisms of neurotransplantation in Huntington’s chorea. Stereotact. Funct. Neurosurg. 58, 79–83.
Tabrizi, S. J., Langbehn, D. R., Leavitt, B. R., Roos, R. A., Durr, A., Craufurd, D., Kennard, C., Hicks, S. L., Fox, N. C., Scahill, R. I., Borowsky, B., Tobin, A. J., Rosas, H. D., Johnson, H., Reilmann, R., Landwehrmeyer, B., Stout, J. C., and TRACK-HD investigators. (2009). Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 8, 791–801.
Tabrizi, S. J., Scahill, R. I., Durr, A., Roos, R. A., Leavitt, B. R., Jones, R., Landwehrmeyer, G. B., Fox, N. C., Johnson, H., Hicks, S. L., Kennard, C., Craufurd, D., Frost, C., Langbehn, D. R., Reilmann, R., Stout, J. C., and TRACK-HD Investigators. (2011). Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 10, 31–42.
The Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983.
Thu, D. C., Oorschot, D. E., Tippett, L. J., Nana, A. L., Hogg, V. M., Synek, B. J., Luthi-Carter, R., Waldvogel, H. J., and Faull, R. L. (2010). Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain 133(Pt 4), 1094–1110.
Tippett, L. J., Waldvogel, H. J., Thomas, S. J., Hogg, V. M., van Roon-Mom, W., Synek, B. J., Graybiel, A. M., and Faull, R. L. (2007). Striosomes and mood dysfunction in Huntington’s disease. Brain 130(Pt 1), 206–221.
van Dellen, A., Blakemore, C., Deacon, R., York, D., and Hannan, A. J. (2000). Delaying the onset of Huntington’s in mice. Nature 404, 721–722.
van Dellen, A., Cordery, P. M., Spires, T. L., Blakemore, C., and Hannan, A. J. (2008). Wheel running from a juvenile age delays onset of specific motor deficits but does not alter protein aggregate density in a mouse model of Huntington’s disease. BMC Neurosci. 9, 34. doi:10.1186/1471-2202-9-34
Verbessem, P., Lemiere, J., Eijnde, B. O., Swinnen, S., Vanhees, L., Van Leemputte, M., Hespel, P., and Dom, R. (2003). Creatine supplementation in Huntington’s disease: a placebo-controlled pilot trial. Neurology 61, 925–930.
Watts, C., Brasted, P. J., and Dunnett, S. B. (2000a). Embryonic donor age and dissection influences striatal graft development and functional integration in a rodent model of Huntington’s disease. Exp. Neurol. 163, 85–97.
Watts, C., McNamara, I. R., and Dunnett, S. B. (2000b). Volume and differentiation of striatal grafts in rats: relationship to the number of cells implanted. Cell Transplant. 9, 65–72.
Watts, R. L., Mandir, A. S., and Bakay, R. A. (1995). Intrastriatal cografts of autologous adrenal medulla and sural nerve in MPTP-induced parkinsonian macaques: behavioral and anatomical assessment. Cell Transplant. 4, 27–38.
Watts, R. L., Subramanian, T., Freeman, A., Goetz, C. G., Penn, R. D., Stebbins, G. T., Kordower, J. H., and Bakay, R. A. (1997). Effect of stereotaxic intrastriatal cografts of autologous adrenal medulla and peripheral nerve in Parkinson’s disease: two-year follow-up study. Exp. Neurol. 147, 510–517.
Wictorin, K. (1992). Anatomy and connectivity of intrastriatal striatal transplants. Prog. Neurobiol. 38, 611–639.
Keywords: Huntington’s disease, neural transplantation, clinical studies
Citation: Wijeyekoon R and Barker RA (2011) The current status of neural grafting in the treatment of Huntington’s disease. A review. Front. Integr. Neurosci. 5:78. doi: 10.3389/fnint.2011.00078
Received: 22 July 2011; Paper pending published: 14 August 2011;
Accepted: 15 November 2011; Published online: 07 December 2011.
Edited by:
Hongyun Huang, Neuroscience Institute of Taishan Medical University, ChinaReviewed by:
Marcos R. Costa, Federal University of Rio Grande do Norte, BrazilMarcelo F. Costa, Universidade de São Paulo, Brazil
Copyright: © 2011 Wijeyekoon and Barker. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Roger A. Barker, Cambridge Centre for Brain Repair, Department of Clinical Neurosciences, University of Cambridge, Forvie Site, Robinson Way, Cambridge CB2 2PY, UK. e-mail:cmFiNDZAY2FtLmFjLnVr