 Eva R. Zamora-Martinez
Eva R. Zamora-Martinez Scott Edwards
Scott Edwards- 1Department of Molecular and Cellular Neuroscience, The Scripps Research Institute, La Jolla, CA, USA
- 2Department of Physiology and Alcohol and Drug Abuse Center of Excellence, Louisiana State University Health Sciences Center, New Orleans, LA, USA
Early pioneering work in the field of biochemistry identified phosphorylation as a crucial post-translational modification of proteins with the ability to both indicate and arbitrate complex physiological processes. More recent investigations have functionally linked phosphorylation of extracellular signal-regulated kinase (ERK) to a variety of neurophysiological mechanisms ranging from acute neurotransmitter action to long-term gene expression. ERK phosphorylation serves as an intracellular bridging mechanism that facilitates neuronal communication and plasticity. Drugs of abuse, including alcohol and opioids, act as artificial yet powerful rewards that impinge upon natural reinforcement processes critical for survival. The graded progression from initial exposure to addiction (or substance dependence) is believed to result from drug- and drug context-induced adaptations in neuronal signaling processes across brain reward and stress circuits following excessive drug use. In this regard, commonly abused drugs as well as drug-associated experiences are capable of modifying the phosphorylation of ERK within central reinforcement systems. In addition, chronic drug and alcohol exposure may drive ERK-regulated epigenetic and structural alterations that underlie a long-term propensity for escalating drug use. Under the influence of such a neurobiological vulnerability, encountering drug-associated cues and contexts can produce subsequent alterations in ERK signaling that drive relapse to drug and alcohol seeking. Current studies are determining precisely which molecular and regional ERK phosphorylation-associated events contribute to the addiction process, as well as which neuroadaptations need to be targeted in order to return dependent individuals to a healthy state.
Introduction
Drugs of abuse act upon a variety of brain centers to alter neurotransmission and intracellular signaling cascades. With the perplexing abundance of drug-induced neuroadaptations, the question remains which alterations result from simple pharmacological mechanisms following drug exposure, and which changes go on to drive the aberrant behavioral mechanisms responsible for addiction. This dilemma has led to multiple conceptualizations of the neurophysiological mechanisms underlying the graded transition to addiction. Some have noted the remarkable persistence of relapse propensity during periods of attempted abstinence, characterizing addiction as a disorder of learning and memory (Kelley, 2004; Hyman, 2005; Hyman et al., 2006). Within this model, drugs of abuse act as unconditioned stimuli that usurp brain reinforcement circuitry to focus reward-related pursuits away from natural or adaptive rewards toward pursuit of the drug of choice (Kalivas and Volkow, 2005). Such a narrowing of behavioral repertoire is driven by highly incentivized drug-related cues and contexts that have been repeatedly paired with drug experiences (Self and Nestler, 1998; Kalivas and O’brien, 2008). Others have highlighted the engagement and potentiation of brain stress (or anti-reward) circuitry that follows particularly excessive or escalated drug intake observed in addicted populations (Ahmed and Koob, 1998). The gradual transition from limited drug sampling to compulsive intake may actually represent a substantial interaction of both within- and between-system neuroadaptations across brain reinforcement circuitry that contributes to the persistence of the addicted phenotype (Edwards and Koob, 2010).
With the overwhelming plasticity that has been observed in preclinical models of reward and addiction (Edwards and Koob, 2012), neuroscientists have long-searched for the most important molecular substrates for purposes of disease conceptualization and therapeutic targeting (Kalivas and Volkow, 2005; Nestler, 2005). In 2007, Girault and colleagues proposed that extracellular signal-regulated kinase (ERK1/2), specifically the ERK2 isoform, acts as an intracellular interface for signaling the intersection of drug reward and drug-related contextual information (Girault et al., 2007). Their conceptualization focused on the convergence of dopamine and glutamate transmission within medium spiny neurons of the striatum (Valjent et al., 2005; Philibin et al., 2011), although similar signaling convergences could involve a variety of neurotransmitters at a number of critical limbic centers involved in various stages of the addiction process. The two major ERK isoforms are present in multiple brain regions, yet exhibit a noticeable rostral-caudal gradient whereby ERK1 levels increase and ERK2 levels decrease from rostral (frontal cortex ERK1/ERK2 ratio of 0.16) to caudal (pons/medulla ERK1/ERK2 ratio of 1.5) regions (Ortiz et al., 1995). ERK1/2 activation is predicated on phosphorylation of neighboring tyrosine and threonine residues (Robbins et al., 1993) by an upstream ERK kinase (or MEK; Payne et al., 1991) in response to a wide array of extracellular signals. Pharmacological inhibition of MEK has implicated central ERK activity in a range of neurobiological phenomenon, including fear conditioning (Sananbenesi et al., 2003), affective behavior (Einat et al., 2003), and central pain processing (Fu et al., 2008; Cao et al., 2009). Moreover, recent data have implicated ERK phosphorylation and activity across multiple brain regions throughout various stages of the addiction timeline. The current review focuses on the critical role of ERK signaling in producing dependence on alcohol and opioids, two widely abused substance classes (Substance Abuse and Mental Health Services Administration, 2008). For an excellent overview of the role of ERK in cocaine addition, see Lu et al. (2006).
As to how transient ERK phosphorylation might mediate the chronic, relapsing disorder of addiction, it has been demonstrated that ERK/mitogen-activated protein kinase (MAPK) activation often leads to more stable changes in gene expression via modification of the post-translational properties of histone proteins (Day and Sweatt, 2011). Histones are nuclear proteins tightly and ionically linked with DNA, normally serving to silence gene transcription. Modifications of histone proteins alter their association with DNA, rendering genetic material more or less accessible to transcriptional machinery (Cheung et al., 2000). As one example of this signaling cascade, in striatal neurons, ERK is capable of activating mitogen- and stress-activated kinase 1 (MSK1), which phosphorylates histone H3, leading to the induction of the immediate early genes c-Fos and c-Jun (Brami-Cherrier et al., 2007). Consequently, repeated activation of the ERK cascade by drugs of abuse or environmental stimuli may promote long-term gene expression and neuronal plasticity associated with the persistence of addiction via such epigenetic modifications (Figure 1). In the following, we provide an overview of the role of ERK activity in the addiction process, from the initial actions of acute alcohol and opioid exposure to the effects of cues and contexts that characterize addiction as a chronic, relapsing disease.
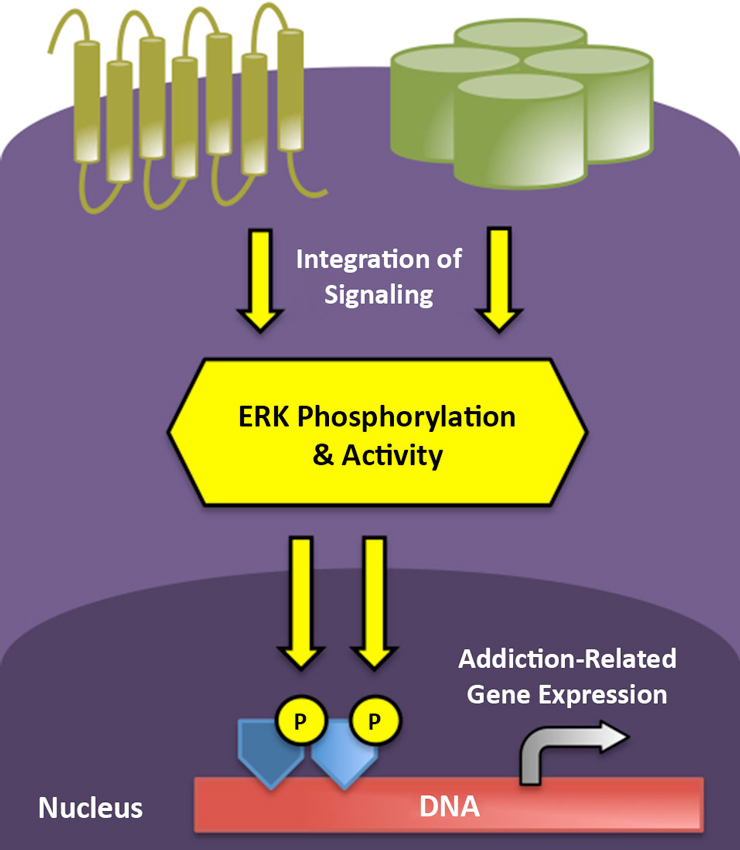
Figure 1. ERK phosphorylation integrates and impacts multiple levels of intracellular signaling to mediate short- and long-term neuroplasticity associated with the transition to addicted states. G protein-coupled receptors (gold) and ion channels (green) represent two major receptor classes that modify ERK phosphorylation in stress and reward circuitry following neurotransmitter binding. Active ERK can translocate to the nucleus to drive addiction-related patterns of gene expression following the subsequent phosphorylation of transcription factors (dark blue) and core histones (light blue).
Effects of Alcohol Exposure on Neuronal Extracellular Signal-Regulated Kinase (ERK) Phosphorylation
The role of the ERK signaling pathway in mediating ethanol sensitivity has been demonstrated in a variety of species, including zebrafish (Peng et al., 2009) and drosophila (Corl et al., 2009; Eddison et al., 2011). For example, ethanol acts through the ERK pathway to promote the adaptive camouflage response in zebrafish via corticotropin-releasing factor (CRF; Wagle et al., 2011). Ethanol impacts ERK/MAPK signaling across multiple organ systems (Aroor and Shukla, 2004), highlighting the central role of alcohol in human health and disease processes (National Institute on Alcohol Abuse and Alcoholism, 2010). Soon (15 min) after a 1 g/kg intragastric or intraperitoneal administration of alcohol to rats, neuronal ERK phosphorylation is increased in extended amygdala regions (Ibba et al., 2009; Thorsell et al., 2013), including the nucleus accumbens, bed nucleus of the stria terminalis, and central amygdala. This time point corresponds to peak ethanol (Quertemont et al., 2003) and dopamine (Melis et al., 2007) levels after administration of this dose, and also produces a conditioned place preference (CPP) in male (Bahi, 2013) and female (Torres et al., 2013) rats. In concert with these acute rewarding effects, Ibba et al. (2009) blocked ethanol-induced ERK phosphorylation (1 g/kg, 15 min) with a dopamine D1 receptor antagonist. The association between the rewarding effects of ethanol and accumbens dopamine release has also been linked to Ras-GRF2, an upstream effector of ERK signaling (Stacey et al., 2012). Increases in ERK phosphorylation post-alcohol appear to be maintained for a longer period of time in the amygdala (60–90 min) vs. accumbens and may mediate the anxiolytic and discriminative stimulus properties of alcohol exposure (Pandey et al., 2008; Besheer et al., 2012). In concert with these effects, Pandey and colleagues have elegantly described a functional role for a compromised BDNF-ERK-CREB signaling axis in the amygdala in the promotion of alcohol drinking (Pandey et al., 2006). They were able to demonstrate a reduction in ERK phosphorylation following antisense-mediated knockdown of the neuropeptide BDNF in three primary amygdala nuclei (basolateral, medial, and central). However, only a reduction of BDNF/pERK/cAMP response element-binding protein (pCREB) in the medial and central amygdala led to increased drinking, possibly as a means to restore functional signaling along this axis. This study demonstrates the ability of alcohol to alter nuclear and gene transcription systems downstream of ERK (also see Hansson et al., 2008). Indeed, Pandey and colleagues have hypothesized that compromised CREB function in the amygdala engenders an anxiety-like state that encourages drinking, and this theory has been supported in both rats and mice (Pandey et al., 2003, 2004, 2005; Zhang et al., 2011). Signaling along this axis has even been shown to mediate structural plasticity in the central and medial amygdala in response to alcohol drinking (Pandey et al., 2008). In comparison to rewarding doses of alcohol (~1.0–1.5 g/kg), acute administration of higher doses (2.5–4.5 g/kg) in rats produce reductions in neuronal ERK phosphorylation, and this may correspond to the aversive effects of higher doses (Anderson et al., 2010; Torres et al., 2013; Zhu et al., 2013) that tend to limit alcohol consumption in non-dependent drinkers. Interestingly, Hodge and colleagues reported that a lower dose of a MEK-ERK inhibitor given systemically increased alcohol drinking in mice while higher doses decreased drinking (Faccidomo et al., 2009), further suggesting that alcohol preference is directly related to neuronal ERK levels.
In addition to D1 dopamine receptors, a recent study described the role of brain peptide signaling, specifically the neuropeptide S (NPS)/NPS receptor system, in mediating the reinforcing effects of alcohol via ERK phosphorylation in the central amygdala (Thorsell et al., 2013). Typically, neuropeptides act through G protein-coupled receptors to activate several signal transduction cascades at once. However, Heilig and colleagues took advantage of the selective NPS receptor antagonist NCGC00185684, which preferentially blocks ERK phosphorylation over cAMP and calcium signaling. They demonstrated that NCGC00185684 reduced alcohol drinking and progressive ratio responding (a measure of motivation) for alcohol in non-dependent animals. Although this drug did not block reinstatement to alcohol-seeking behavior in rats, relapse may be more of a concern in dependent subjects seeking to continue along an abstinence regimen. In this regard, Ruggeri et al. (2010) found that amygdala NPS receptor expression is increased in withdrawn, alcohol-dependent rats, wherein NCGC00185684 might be expected to prove efficacious against relapse-like behaviors.
Neuronal Extracellular Signal-Regulated Kinase (ERK) Phosphorylation in the Alcohol-Dependent State
Indeed, mounting evidence suggests that ERK phosphorylation may play a unique role in alcohol dependence in addition to its effects in mediating the positive reinforcing effects of acute alcohol exposure. Importantly, the transition from recreational alcohol use to dependence is associated with an emergence of negative motivational symptomatology that promotes excessive drinking (Hansson et al., 2007; Gilpin and Koob, 2008). Such a condition is modeled via repeated cycles of alcohol intoxication and abstinence in rodents (Roberts et al., 1996, 2000), whereby intermittent exposure both represents the human alcoholic condition and is thought to expedite alcoholism-related plasticity (O’Dell et al., 2004; Breese et al., 2005). To measure the effects of excessive and repeated alcohol exposures on neuronal ERK phosphorylation, Sanna et al. (2002) employed a model of alcohol dependence that exposed rats to intermittent cycles (14 h or alcohol exposure, 10 h of abstinence) of dependence-inducing alcohol vapor (Gilpin et al., 2008). Chronic (14 d) vapor exposure reduced pERK levels at the end of the alcohol exposure period (i.e., at peak blood alcohol levels) in several brain regions including the accumbens and hippocampus (also see Roberto et al., 2003). In contrast, withdrawal (7–13 h) from this regimen significantly elevated ERK phosphorylation in the amygdala, at a time point commensurate with diminished blood alcohol levels and elevated operant alcohol self-administration (Edwards et al., 2012a). This neuroadaptation suggests that ERK-mediated signaling may mark and mediate plasticity induced by alcohol withdrawal in addition to the effects produced by drinking. Withdrawal-induced ERK phosphorylation is also in line with opponent process theories of drug addiction (Koob and Le Moal, 2008), whereby brain stress systems are hypothesized to be recruited and potentiated during abstinence from excessive drug exposure.
A remaining question is which neurotransmitters contribute to excessive amygdala ERK phosphorylation during withdrawal in alcohol-dependent animals. The neurobiological basis for the negative reinforcement mechanisms important for the development and maintenance of alcoholism may include neuroadaptations in central neuropeptide systems associated with stress and anxiety, including the amygdala CRF system (Timpl et al., 1998; Shekhar et al., 2005). Indeed, previous studies have demonstrated the recruitment of CRF in the amygdala as a critical element driving the excessive ethanol intake observed during both acute and protracted abstinence (Heilig and Koob, 2007). These studies implicated enhanced CRF signaling in discrete amygdala nuclei, including the central (Lack et al., 2005; Funk and Koob, 2007; Funk et al., 2007) and basolateral (Sommer et al., 2008) nuclei as neuroanatomical substrates driving ethanol dependence. Interestingly, intracerebroventricular administration of CRF in vivo increases ERK phosphorylation in the basolateral amygdala, but not central nucleus (Refojo et al., 2005).
In addition, given the remarkable persistence of the addicted state, some investigators have sought to focus on neuronal changes that last well into abstinence. Such a “post-dependent” state is associated with a stable elevation of forebrain MAPK signaling elements, including ERK kinases (Rimondini et al., 2002). As an extension of their preliminary studies, Heilig and colleagues (Hansson et al., 2008) sought to understand ERK cascade responsiveness to alcohol in the post-dependent state. Interestingly, ethanol-induced activation of MEK/ERK-associated pathways in the medial amygdala is lost in dependent animals. Moreover, ERK signaling is potentiated and suppresses the neuronal response to alcohol in the frontal cortex and nucleus accumbens. Both of these changes may possibly correspond to tolerance to the rewarding effects of alcohol in dependence, leading to an escalation of drinking if relapse occurs.
Extracellular Signal-Regulated Kinase (ERK) Modulation of Alcohol Withdrawal and Compulsive Alcohol Seeking
Cue-induced reinstatement of alcohol seeking activates ERK phosphorylation in the basolateral amygdala (Radwanska et al., 2008) and nucleus accumbens shell, and this is dependent on mGluR5 activation in both regions (Schroeder et al., 2008; Sinclair et al., 2012). These studies suggest that excessive ERK activity may correspond with potentiated glutamatergic transmission in alcohol dependence and relapse (Ron, 2004; Chandler et al., 2006; Szumlinski et al., 2007; Mason et al., 2009; Holmes et al., 2013).
ERK signaling would also appear to play a connecting role in the critical interplay between negative affective states (including anxiety and stress) and ethanol drinking (De Witte et al., 2003). Alcohol withdrawal potentiates anxiety-like behavior (Valdez et al., 2002) and sensitizes contextual fear memories (Bertotto et al., 2006) that in turn increase alcohol consumption (Bertotto et al., 2010). Interestingly, fear conditioning potentiates ERK phosphorylation in the basolateral amygdala during withdrawal in alcohol dependent animals (Bertotto et al., 2011). This effect was blocked by administration of MK-801, an antagonist of NMDA receptor channels, which are known to be dysregulated in the amygdala of dependent rats (Roberto et al., 2004, 2006). In a similar investigation, Gilpin and colleagues assessed neuronal activations patterns by measuring ERK phosphorylation following exposure to a discrete context previously paired with traumatic stress (bobcat urine), mimicking symptom provocation in human post-traumatic stress disorder (PTSD) patients (Edwards et al., 2013). In this study, a subgroup of rats that displayed a persistently high avoidance of trauma-related stimuli also exhibited a long-lasting post-stress escalation of alcohol drinking. High-avoidance, high-drinking rats also exhibited greater synchronicity between the dorsal prefrontal cortex and basolateral amygdala, as measured by individual within-subject and between-region correlations in ERK phosphorylation upon re-exposure to the traumatic stress-paired context. This finding corresponds to the regional synchronization found in PTSD patients upon symptom provocation (Gilboa et al., 2004), and may underlie the increased propensity for alcohol abuse in this population (Engdahl et al., 1998; Jacobsen et al., 2001). In addition, a heightened proclivity for high-risk prescription opioid use in PTSD sufferers (Seal et al., 2012) is one piece of evidence suggesting a common neurobiological substrate underlying alcohol and opioid use disorders in vulnerable populations.
Effects of Opioid Exposure on Neuronal Extracellular Signal-Regulated Kinase (ERK) Phosphorylation
Endogenous opioids represent one of the body’s primary natural reward systems and play a central role in health and disease states (Bodnar, 2013), including drug addiction (Trigo et al., 2010). Exogenous opioids, ranging from opiate derivatives (e.g., morphine) to prescription opioid analgesics (e.g., oxycodone), interact with opioid receptors in the body to produce effects ranging from therapeutic to pathological (Shurman et al., 2010). Similar to alcohol, morphine (5 mg/kg, 20 min pretreatment) acutely elicits ERK phosphorylation in the rodent extended amygdala through a dopamine D1 receptor-dependent process (Valjent et al., 2004), while higher doses (10–50 mg/kg) produce no changes or even decreases in neuronal pERK levels, depending on brain region (Eitan et al., 2003; Muller and Unterwald, 2004; Valjent et al., 2004; Moron et al., 2010). In striatal neurons, mu-opioid receptor activation of the ERK cascade also appears to require receptor phosphorylation by G protein-coupled receptor kinase (GRK3) and arrestin3 recruitment (Macey et al., 2006). The rewarding effects of opioids, including oxycodone (Liu et al., 2009), are associated with ERK activity in the nucleus accumbens (Xu et al., 2012). With repeated exposure, psychomotor sensitization to morphine requires ERK phosphorylation in striatal D1 neurons (Borgkvist et al., 2008). Interestingly, genetic deletion of the ERK1 isoform leads to enhanced D1 signaling, greater phosphorylation of the ERK2 isoform, and potentiation of morphine reward (Mazzucchelli et al., 2002). In addition to the ventral striatum, Li et al. (2011) linked morphine reward to increases in central amygdala ERK phosphorylation downstream of NMDA receptor activation. A D1 receptor-dependent, ERK mediated process in the basolateral amygdala was also found to be required for opioid-related memory formation (Lyons et al., 2013). Interestingly, Laviolette and colleagues have provided evidence that opioid reward memory consolidation transitions from an ERK- and basolateral amygdala-dependent process to a CaMKII-dependent mechanism in the prefrontal cortex (Gholizadeh et al., 2013).
Neuronal Extracellular Signal-Regulated Kinase (ERK) Phosphorylation in the Opioid-Dependent State
Chronic heroin self-administration in rats produces a multitude of symptoms of dependence including elevated brain reward thresholds (Kenny et al., 2006) and a marked hyperalgesia that correlates with heroin intake (Edwards et al., 2012b). With extended access to heroin self-administration (≥6 h/day), animals will escalate their intake over time and begin to display signs of physical withdrawal either spontaneously or following mu-opioid receptor antagonism (Vendruscolo et al., 2011). Intake escalation represents a critical Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria for dependence and is associated with multiple behavioral and neurochemical adaptations that persist or become manifest in protracted abstinence (Edwards and Koob, 2013).
In an attempt to shed light on brain regional neuroadaptations in ERK signaling following escalation of heroin self-administration, Self and colleagues (Edwards et al., 2009) examined ERK phosphorylation across eleven mesolimbic brain regions. Interestingly, naltrexone by itself significantly reduced ERK phosphorylation by 24–30% in heroin-naïve animals in both the ventral tegmental area and substantia nigra, suggesting a tonic activation of the ERK signaling cascade via endogenous mu-opioid receptor stimulation in these regions. This blunting of the ERK pathway by naltrexone was normalized in heroin self-administering animals receiving a naltrexone challenge, suggesting a relative potentiation of midbrain ERK signaling in heroin dependence. These data are in harmony with increases in total ERK1/2 levels found in the midbrain locus coeruleus in morphine-dependent rats (Ortiz et al., 1995). In forebrain regions, increases in pERK in the prefrontal cortex and hippocampus predominated following 24 h withdrawal either in the presence or absence of a naltrexone challenge. In comparison, naltrexone-precipitated withdrawal (vs. spontaneous withdrawal) facilitated greater increases in pERK in the nucleus accumbens and central amygdala. A similar pattern of accumbens ERK activation was found in morphine-dependent mice receiving a naloxone challenge, and did not generalize to the other MAPK-family members p38MAPK or c-Jun N-terminal kinase (JNK; Li et al., 2010). Extending these findings, Ciccarelli et al. (2013) delineated a role for ERK signaling in the regulation of epigenetic marking during opioid withdrawal. They found that naloxone-precipitated morphine withdrawal increased histone H3 phosphorylation through an ERK-dependent process in the nucleus accumbens. Such epigenetic changes at the histone level likely represent stable, long-term alterations in gene expression capacity (Robison and Nestler, 2011) that may underlie the persistence of addiction (Maze and Nestler, 2011). Recruitment of ERK signaling in the central amygdala was also found in morphine-dependent mice (Hofford et al., 2009). The central amygdala is critically involved in the expression of dysphoria and hyperalgesia that occurs during opioid dependence, a phenomenon mediated by CRF signaling (Heinrichs et al., 1995; McNally and Akil, 2002). Potentiated ERK signaling may enhance central amygdala CRF levels via CREB activity, which is increased in CRF neurons in morphine-dependent animals (Shaw-Lutchman et al., 2002).
Extracellular Signal-Regulated Kinase (ERK) Signaling in the Context of Opioid Withdrawal and Relapse Behaviors
Opioid withdrawal produces a long-lasting conditioned place aversion (CPA) in dependent animals (Stinus et al., 2000), indicative of the negative motivational state made manifest following dependence induction (Koob and Le Moal, 2008). Wang et al. (2012) recently demonstrated the importance of ERK-mediated epigenetic regulation within the ventromedial prefrontal cortex (vmPFC) in the extinction of aversive memories within the context of morphine withdrawal. In this study, re-exposure to a withdrawal-paired context elicited ERK phosphorylation and histone H3 acetylation in the vmPFC, while MEK-ERK inhibition prevented both induction of H3 acetylation and extinction of CPA. Thus, disruption of vmPFC ERK signaling may play a role in opioid craving and relapse under aversive conditions. In concert with this hypothesis, reduced levels of mu-opioid receptors and ERK signaling components were found in the prefrontal cortex of humans who suffered an opioid overdose (Ferrer-Alcón et al., 2004a,b).
A heightened propensity to relapse is one hallmark of opioid addiction that persists well into abstinence and is typically preceded by re-exposure to drug-associated cues and contexts (O’Brien, 1997). The motivating properties of such contextual cues to elicit relapse-like behavior has been modeled in rodents (Neisewander et al., 2000; Grimm et al., 2001), whereby alcohol (Bienkowski et al., 2004) and drug seeking actually increases with time in the abstinent state. For example, Shaham and colleagues demonstrated withdrawal time-dependent increases in baseline heroin seeking as well as footshock stress-induced reinstatement of heroin seeking (Shalev et al., 2001). This phenomenon has also been observed in classical (Pavlovian) conditioning models such as CPP. Expression of morphine CPP gradually increases over 2 weeks after the last drug-environment pairing (Li et al., 2008). Interestingly, re-exposure of animals to the morphine-paired environment at the latest withdrawal time point (14 days) increased ERK phosphorylation in the central but not basolateral amygdala. This was matched by similar increases in CREB phosphorylation, providing additional evidence linking this signaling axis in drug reward and addiction liability. Further, inhibition of central amygdala MEK-ERK activity with U0126 also prevented the expression of the magnified CPP in protracted abstinence.
Conclusion and Future Challenges
Valjent et al. (2004) hypothesized that a strong activation of pERK across all three extended amygdala regions distinguishes drugs most likely to be abused from other psychoactive substances that are not considered to produce addiction (e.g., antidepressants, antipsychotics), in accordance with the suggested central role of these regions in dependence (Koob et al., 2014). Such a molecular litmus test may be useful to screen the potential addiction liability of trending recreational substances to complement traditional behavioral assays (Huang et al., 2012; Aarde et al., 2013). On a similar note, it is important to mention that the effect of ERK signaling in mediating compulsive drug seeking may generalize to natural rewards as well. Grimm et al. (2002, 2005) found that operant seeking of sucrose increases with withdrawal time, similar to artificial rewards. Following 3 weeks withdrawal, both cocaine- and sucrose-seeking behavior elicit striatal ERK phosphorylation, while only cocaine seeking increases protein kinase A (PKA) phosphorylation of AMPA GluR1 subunits (Edwards et al., 2011). Given the role of striatal endogenous opioids in signaling food palatability (Kelley et al., 2002), ERK activity in central reinforcement systems may play a critical neurobiological role in the global obesity epidemic (Potenza, 2014). With the extensive overlap in the neural circuitry engaged by both drug and food cues (Kelley et al., 2005; Volkow et al., 2013), further studies into the shared role of ERK signaling in addiction and obesity are warranted.
Ultimately, neuroscientists hope to produce medical breakthroughs from the basic science knowledge gleaned from a better understanding of altered ERK signaling in disease processes. Upon initial examination, the demonstrated role of ERK phosphorylation in mediating drug and alcohol dependence makes MEK/ERK blockade an attractive therapeutic strategy. Unfortunately, the ubiquity of MAPK/ERK activity in virtually all physiological processes makes systemic inhibition more poison than therapy (Balmanno and Cook, 2009). However, as discussed above in the study of Thorsell et al. (2013), directed activation or blockade of ERK signaling via target receptor ligands that are capable of selectively influencing this pathway represent one solution to this dilemma. In addition to NPS receptors, other promising targets of interest couple to ERK pathways, including CRF1 receptors (Punn et al., 2006; Kageyama et al., 2007; Hauger et al., 2009; Meng et al., 2011). In addition, Bruchas and Chavkin (2010) have proposed that targeting ligand-directed signaling at kappa-opioid receptors (KORs) may prove useful in the treatment of pain and addiction by selectively altering distinct members of the MAPK family (ERK, p38, and JNK). For example, while arrestin-dependent p38 MAPK activation appears to mediate aversion in animals, recruitment of JNK signaling in the absence of arrestin results in an inactivation of KOR activity. The same group demonstrated that repeated forced swim stress induces striatal ERK phosphorylation (Bruchas et al., 2008), similar to heroin withdrawal (Edwards et al., 2009). Stress-induced ERK activation was blocked by the long-lasting KOR antagonist norbinaltorphimine, a drug that has also been demonstrated to block escalation of heroin self-administration in rodents when injected systemically or into the ventral striatum (Schlosburg et al., 2013). These remarkable findings have propelled the design and development of new small molecule, functionally selective KOR antagonists (Zhou et al., 2013) that may prove efficacious in managing the hedonic dysregulation associated with drug and alcohol dependence (Koob and Le Moal, 1997; Kenny et al., 2006; Potter et al., 2011; Walker et al., 2012).
Finally, it will be critical to understand how ERK signaling interfaces with the multitude of other molecular signaling processes influenced by alcohol and opioids, ranging from classical kinase pathways to systemic inflammatory responses (Crews et al., 2011; Ting and Van Der Kooy, 2012; Ron and Messing, 2013). In addition, the interactive nature of and co-dependence on alcohol and opioids is a subject that has not been investigated at the level of neuronal ERK signaling. Alcohol and opioids activate a shared subset of neurobiological systems (Koob and Bloom, 1988; Herz, 1997; Koob et al., 2003; Siggins et al., 2003; Gianoulakis, 2009) and exhibit substantial similarity in terms of withdrawal symptomatology (West and Gossop, 1994; Edwards et al., 2012b). Thus, it is not surprising that existing studies have revealed a parallel recruitment of ERK signaling within stress and reward circuitry across important stages of the addiction timeline. The addiction field is currently challenged with the task of translating this basic knowledge at the signal transduction level into more effective treatments to curb excessive opioid and alcohol use. Continuing collaboration between addiction scientists and clinicians in the context of an important functional integration of addiction-related research aims at the National Institutes of Health is expected to drive these efforts in the coming decades.
Authors and Contributors
Eva R. Zamora-Martinez and Scott Edwards contributed to the conceptualization and writing of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health grant AA020839 from the National Institute on Alcohol Abuse and Alcoholism (to Scott Edwards).
References
Aarde, S. M., Angrish, D., Barlow, D. J., Wright, M. J. Jr., Vandewater, S. A., Creehan, K. M., et al. (2013). Mephedrone (4-methylmethcathinone) supports intravenous self-administration in Sprague-Dawley and Wistar rats. Addict. Biol. 18, 786–799. doi: 10.1111/adb.12038
Ahmed, S. H., and Koob, G. F. (1998). Transition from moderate to excessive drug intake: change in hedonic set point. Science 282, 298–300. doi: 10.1126/science.282.5387.298
Anderson, R. I., Varlinskaya, E. I., and Spear, L. P. (2010). Ethanol-induced conditioned taste aversion in male sprague-dawley rats: impact of age and stress. Alcohol. Clin. Exp. Res. 34, 2106–2115. doi: 10.1111/j.1530-0277.2010.01307.x
Aroor, A. R., and Shukla, S. D. (2004). MAP kinase signaling in diverse effects of ethanol. Life Sci. 74, 2339–2364. doi: 10.1016/j.lfs.2003.11.001
Bahi, A. (2013). Viral-mediated knockdown of mGluR7 in the nucleus accumbens mediates excessive alcohol drinking and increased ethanol-elicited conditioned place preference in rats. Neuropsychopharmacology 38, 2109–2119. doi: 10.1038/npp.2012.122
Balmanno, K., and Cook, S. J. (2009). Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 16, 368–377. doi: 10.1038/cdd.2008.148
Bertotto, M. E., Bussolino, D. F., Molina, V. A., and Martijena, I. D. (2010). Increased voluntary ethanol consumption and c-Fos expression in selected brain areas induced by fear memory retrieval in ethanol withdrawn rats. Eur. Neuropsychopharmacol. 20, 568–581. doi: 10.1016/j.euroneuro.2010.02.014
Bertotto, M. E., Bustos, S. G., Molina, V. A., and Martijena, I. D. (2006). Influence of ethanol withdrawal on fear memory: effect of D-cycloserine. Neuroscience 142, 979–990. doi: 10.1016/j.neuroscience.2006.07.013
Bertotto, M. E., Maldonado, N. M., Bignante, E. A., Gorosito, S. V., Cambiasso, M. J., Molina, V. A., et al. (2011). ERK activation in the amygdala and hippocampus induced by fear conditioning in ethanol withdrawn rats: modulation by MK-801. Eur. Neuropsychopharmacol. 21, 892–904. doi: 10.1016/j.euroneuro.2011.01.001
Besheer, J., Fisher, K. R., Cannady, R., Grondin, J. J., and Hodge, C. W. (2012). Intra-amygdala inhibition of ERK(1/2) potentiates the discriminative stimulus effects of alcohol. Behav. Brain Res. 228, 398–405. doi: 10.1016/j.bbr.2011.12.023
Bienkowski, P., Rogowski, A., Korkosz, A., Mierzejewski, P., Radwanska, K., Kaczmarek, L., et al. (2004). Time-dependent changes in alcohol-seeking behaviour during abstinence. Eur. Neuropsychopharmacol. 14, 355–360. doi: 10.1016/j.euroneuro.2003.10.005
Bodnar, R. J. (2013). Endogenous opiates and behavior: 2012. Peptides 50, 55–95. doi: 10.1016/j.peptides.2013.10.001
Borgkvist, A., Valjent, E., Santini, E., Herve, D., Girault, J. A., and Fisone, G. (2008). Delayed, context- and dopamine D1 receptor-dependent activation of ERK in morphine-sensitized mice. Neuropharmacology 55, 230–237. doi: 10.1016/j.neuropharm.2008.05.028
Brami-Cherrier, K., Lavaur, J., Pages, C., Arthur, J. S., and Caboche, J. (2007). Glutamate induces histone H3 phosphorylation but not acetylation in striatal neurons: role of mitogen- and stress-activated kinase-1. J. Neurochem. 101, 697–708. doi: 10.1111/j.1471-4159.2006.04352.x
Breese, G. R., Overstreet, D. H., Knapp, D. J., and Navarro, M. (2005). Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1- and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology 30, 1662–1669. doi: 10.1038/sj.npp.1300706
Bruchas, M. R., and Chavkin, C. (2010). Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 210, 137–147. doi: 10.1007/s00213-010-1806-y
Bruchas, M. R., Xu, M., and Chavkin, C. (2008). Repeated swim stress induces kappa opioid-mediated activation of extracellular signal-regulated kinase 1/2. Neuroreport 19, 1417–1422. doi: 10.1097/WNR.0b013e32830dd655
Cao, H., Gao, Y. J., Ren, W. H., Li, T. T., Duan, K. Z., Cui, Y. H., et al. (2009). Activation of extracellular signal-regulated kinase in the anterior cingulate cortex contributes to the induction and expression of affective pain. J. Neurosci. 29, 3307–3321. doi: 10.1523/JNEUROSCI.4300-08.2009
Chandler, L. J., Carpenter-Hyland, E., Hendricson, A. W., Maldve, R. E., Morrisett, R. A., Zhou, F. C., et al. (2006). Structural and functional modifications in glutamateric synapses following prolonged ethanol exposure. Alcohol. Clin. Exp. Res. 30, 368–376. doi: 10.1111/j.1530-0277.2006.00041.x
Cheung, P., Allis, C. D., and Sassone-Corsi, P. (2000). Signaling to chromatin through histone modifications. Cell 103, 263–271. doi: 10.1016/s0092-8674(00)00118-5
Ciccarelli, A., Calza, A., Santoru, F., Grasso, F., Concas, A., Sassoe-Pognetto, M., et al. (2013). Morphine withdrawal produces ERK-dependent and ERK-independent epigenetic marks in neurons of the nucleus accumbens and lateral septum. Neuropharmacology 70, 168–179. doi: 10.1016/j.neuropharm.2012.12.010
Corl, A. B., Berger, K. H., Ophir-Shohat, G., Gesch, J., Simms, J. A., Bartlett, S. E., et al. (2009). Happyhour, a Ste20 family kinase, implicates EGFR signaling in ethanol-induced behaviors. Cell 137, 949–960. doi: 10.1016/j.cell.2009.03.020
Crews, F. T., Zou, J., and Qin, L. (2011). Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav. Immun. 25(Suppl. 1), S4–S12. doi: 10.1016/j.bbi.2011.03.003
Day, J. J., and Sweatt, J. D. (2011). Epigenetic mechanisms in cognition. Neuron 70, 813–829. doi: 10.1016/j.neuron.2011.05.019
De Witte, P., Pinto, E., Ansseau, M., and Verbanck, P. (2003). Alcohol and withdrawal: from animal research to clinical issues. Neurosci. Biobehav. Rev. 27, 189–197. doi: 10.1016/s0149-7634(03)00030-7
Eddison, M., Guarnieri, D. J., Cheng, L., Liu, C. H., Moffat, K. G., Davis, G., et al. (2011). Arouser reveals a role for synapse number in the regulation of ethanol sensitivity. Neuron 70, 979–990. doi: 10.1016/j.neuron.2011.03.030
Edwards, S., and Koob, G. F. (2010). Neurobiology of dysregulated motivational systems in drug addiction. Future Neurol. 5, 393–401. doi: 10.2217/fnl.10.14
Edwards, S., and Koob, G. F. (2012). Experimental psychiatric illness and drug abuse models: from human to animal, an overview. Methods Mol. Biol. 829, 31–48. doi: 10.1007/978-1-61779-458-2_2
Edwards, S., and Koob, G. F. (2013). Escalation of drug self-administration as a hallmark of persistent addiction liability. Behav. Pharmacol. 24, 356–362. doi: 10.1097/FBP.0b013e3283644d15
Edwards, S., Bachtell, R. K., Guzman, D., Whisler, K. N., and Self, D. W. (2011). Emergence of context-associated GluR(1) and ERK phosphorylation in the nucleus accumbens core during withdrawal from cocaine self-administration. Addict. Biol. 16, 450–457. doi: 10.1111/j.1369-1600.2010.00296.x
Edwards, S., Baynes, B. B., Carmichael, C. Y., Zamora-Martinez, E. R., Barrus, M., Koob, G. F., et al. (2013). Traumatic stress reactivity promotes excessive alcohol drinking and alters the balance of prefrontal cortex-amygdala activity. Transl. Psychiatry 3:e296. doi: 10.1038/tp.2013.70
Edwards, S., Graham, D. L., Whisler, K. N., and Self, D. W. (2009). Phosphorylation of GluR1, ERK and CREB during spontaneous withdrawal from chronic heroin self-administration. Synapse 63, 224–235. doi: 10.1002/syn.20601
Edwards, S., Guerrero, M., Ghoneim, O. M., Roberts, E., and Koob, G. F. (2012a). Evidence that vasopressin V1b receptors mediate the transition to excessive drinking in ethanol-dependent rats. Addict. Biol. 17, 76–85. doi: 10.1111/j.1369-1600.2010.00291.x
Edwards, S., Vendruscolo, L. F., Schlosburg, J. E., Misra, K. K., Wee, S., Park, P. E., et al. (2012b). Development of mechanical hypersensitivity in rats during heroin and ethanol dependence: alleviation by CRF(1) receptor antagonism. Neuropharmacology 62, 1142–1151. doi: 10.1016/j.neuropharm.2011.11.006
Eitan, S., Bryant, C. D., Saliminejad, N., Yang, Y. C., Vojdani, E., Keith, D. Jr., et al. (2003). Brain region-specific mechanisms for acute morphine-induced mitogen-activated protein kinase modulation and distinct patterns of activation during analgesic tolerance and locomotor sensitization. J. Neurosci. 23, 8360–8369.
Einat, H., Yuan, P., Gould, T. D., Li, J., Du, J., Zhang, L., et al. (2003). The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J. Neurosci. 23, 7311–7316.
Engdahl, B., Dikel, T. N., Eberly, R., and Blank, A. Jr. (1998). Comorbidity and course of psychiatric disorders in a community sample of former prisoners of war. Am. J. Psychiatry 155, 1740–1745.
Faccidomo, S., Besheer, J., Stanford, P. C., and Hodge, C. W. (2009). Increased operant responding for ethanol in male C57BL/6J mice: specific regulation by the ERK1/2, but not JNK, MAP kinase pathway. Psychopharmacology (Berl) 204, 135–147. doi: 10.1007/s00213-008-1444-9
Ferrer-Alcón, M., Garcia-Fuster, M. J., La Harpe, R., and Garcia-Sevilla, J. A. (2004a). Long-term regulation of signalling components of adenylyl cyclase and mitogen-activated protein kinase in the pre-frontal cortex of human opiate addicts. J. Neurochem. 90, 220–230. doi: 10.1111/j.1471-4159.2004.02473.x
Ferrer-Alcón, M., La Harpe, R., and Garcia-Sevilla, J. A. (2004b). Decreased immunodensities of micro-opioid receptors, receptor kinases GRK 2/6 and beta-arrestin-2 in postmortem brains of opiate addicts. Brain Res. Mol. Brain Res. 121, 114–122. doi: 10.1016/j.molbrainres.2003.11.009
Fu, Y., Han, J., Ishola, T., Scerbo, M., Adwanikar, H., Ramsey, C., et al. (2008). PKA and ERK, but not PKC, in the amygdala contribute to pain-related synaptic plasticity and behavior. Mol. Pain 4:26. doi: 10.1186/1744-8069-4-26
Funk, C. K., and Koob, G. F. (2007). A CRF(2) agonist administered into the central nucleus of the amygdala decreases ethanol self-administration in ethanol-dependent rats. Brain Res. 1155, 172–178. doi: 10.1016/j.brainres.2007.04.009
Funk, C. K., Zorrilla, E. P., Lee, M. J., Rice, K. C., and Koob, G. F. (2007). Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol. Psychiatry 61, 78–86. doi: 10.1016/j.biopsych.2006.03.063
Gholizadeh, S., Sun, N., De Jaeger, X., Bechard, M., Coolen, L., and Laviolette, S. R. (2013). Early versus late-phase consolidation of opiate reward memories requires distinct molecular and temporal mechanisms in the amygdala-prefrontal cortical pathway. PLoS One 8:e63612. doi: 10.1371/journal.pone.0063612
Gianoulakis, C. (2009). Endogenous opioids and addiction to alcohol and other drugs of abuse. Curr. Top. Med. Chem. 9, 999–1015. doi: 10.2174/156802609789630956
Gilboa, A., Shalev, A. Y., Laor, L., Lester, H., Louzoun, Y., Chisin, R., et al. (2004). Functional connectivity of the prefrontal cortex and the amygdala in posttraumatic stress disorder. Biol. Psychiatry 55, 263–272. doi: 10.1016/j.biopsych.2003.08.004
Gilpin, N. W., and Koob, G. F. (2008). Neurobiology of alcohol dependence: focus on motivational mechanisms. Alcohol Res. Health 31, 185–195.
Gilpin, N. W., Richardson, H. N., Cole, M., and Koob, G. F. (2008). Vapor inhalation of alcohol in rats. Curr. Protoc. Neurosci. Chapter 9, Unit 9.29. doi: 10.1002/0471142301.ns0929s44
Girault, J. A., Valjent, E., Caboche, J., and Herve, D. (2007). ERK2: a logical AND gate critical for drug-induced plasticity? Curr. Opin. Pharmacol. 7, 77–85. doi: 10.1016/j.coph.2006.08.012
Grimm, J. W., Fyall, A. M., and Osincup, D. P. (2005). Incubation of sucrose craving: effects of reduced training and sucrose pre-loading. Physiol. Behav. 84, 73–79. doi: 10.1016/j.physbeh.2004.10.011
Grimm, J. W., Hope, B. T., Wise, R. A., and Shaham, Y. (2001). Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature 412, 141–142. doi: 10.1038/35084134
Grimm, J. W., Shaham, Y., and Hope, B. T. (2002). Effect of cocaine and sucrose withdrawal period on extinction behavior, cue-induced reinstatement and protein levels of the dopamine transporter and tyrosine hydroxylase in limbic and cortical areas in rats. Behav. Pharmacol. 13, 379–388. doi: 10.1097/00008877-200209000-00011
Hansson, A. C., Bermudez-Silva, F. J., Malinen, H., Hyytia, P., Sanchez-Vera, I., Rimondini, R., et al. (2007). Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacology 32, 117–126. doi: 10.1038/sj.npp.1301034
Hansson, A. C., Rimondini, R., Neznanova, O., Sommer, W. H., and Heilig, M. (2008). Neuroplasticity in brain reward circuitry following a history of ethanol dependence. Eur. J. Neurosci. 27, 1912–1922. doi: 10.1111/j.1460-9568.2008.06159.x
Hauger, R. L., Risbrough, V., Oakley, R. H., Olivares-Reyes, J. A., and Dautzenberg, F. M. (2009). Role of CRF receptor signaling in stress vulnerability, anxiety and depression. Ann. N Y Acad. Sci. 1179, 120–143. doi: 10.1111/j.1749-6632.2009.05011.x
Heilig, M., and Koob, G. F. (2007). A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 30, 399–406. doi: 10.1016/j.tins.2007.06.006
Heinrichs, S. C., Menzaghi, F., Schulteis, G., Koob, G. F., and Stinus, L. (1995). Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav. Pharmacol. 6, 74–80. doi: 10.1097/00008877-199501000-00011
Herz, A. (1997). Endogenous opioid systems and alcohol addiction. Psychopharmacology (Berl) 129, 99–111. doi: 10.1007/s002130050169
Hofford, R. S., Hodgson, S. R., Roberts, K. W., Bryant, C. D., Evans, C. J., and Eitan, S. (2009). Extracellular signal-regulated kinase activation in the amygdala mediates elevated plus maze behavior during opioid withdrawal. Behav. Pharmacol. 20, 576–583. doi: 10.1097/FBP.0b013e32832ec57e
Holmes, A., Spanagel, R., and Krystal, J. H. (2013). Glutamatergic targets for new alcohol medications. Psychopharmacology (Berl) 229, 539–554. doi: 10.1007/s00213-013-3226-2
Huang, P. K., Aarde, S. M., Angrish, D., Houseknecht, K. L., Dickerson, T. J., and Taffe, M. A. (2012). Contrasting effects of d-methamphetamine, 3,4-methylenedioxymethamphetamine, 3,4-methylenedioxypyrovalerone and 4-methylmethcathinone on wheel activity in rats. Drug Alcohol Depend. 126, 168–175. doi: 10.1016/j.drugalcdep.2012.05.011
Hyman, S. E. (2005). Addiction: a disease of learning and memory. Am. J. Psychiatry 162, 1414–1422. doi: 10.1176/appi.ajp.162.8.1414
Hyman, S. E., Malenka, R. C., and Nestler, E. J. (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 29, 565–598. doi: 10.1146/annurev.neuro.29.051605.113009
Ibba, F., Vinci, S., Spiga, S., Peana, A. T., Assaretti, A. R., Spina, L., et al. (2009). Ethanol-induced extracellular signal regulated kinase: role of dopamine D1 receptors. Alcohol. Clin. Exp. Res. 33, 858–867. doi: 10.1111/j.1530-0277.2009.00907.x
Jacobsen, L. K., Southwick, S. M., and Kosten, T. R. (2001). Substance use disorders in patients with posttraumatic stress disorder: a review of the literature. Am. J. Psychiatry 158, 1184–1190. doi: 10.1176/appi.ajp.158.8.1184
Kageyama, K., Hanada, K., Moriyama, T., Imaizumi, T., Satoh, K., and Suda, T. (2007). Differential regulation of CREB and ERK phosphorylation through corticotropin-releasing factor receptors type 1 and 2 in AtT-20 and A7r5 cells. Mol. Cell. Endocrinol. 263, 90–102. doi: 10.1016/j.mce.2006.08.011
Kalivas, P. W., and O’brien, C. (2008). Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 33, 166–180. doi: 10.1038/sj.npp.1301564
Kalivas, P. W., and Volkow, N. D. (2005). The neural basis of addiction: a pathology of motivation and choice. Am. J. Psychiatry 162, 1403–1413. doi: 10.1176/appi.ajp.162.8.1403
Kelley, A. E. (2004). Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron 44, 161–179. doi: 10.1016/j.neuron.2004.09.016
Kelley, A. E., Bakshi, V. P., Haber, S. N., Steininger, T. L., Will, M. J., and Zhang, M. (2002). Opioid modulation of taste hedonics within the ventral striatum. Physiol. Behav. 76, 365–377. doi: 10.1016/s0031-9384(02)00751-5
Kelley, A. E., Schiltz, C. A., and Landry, C. F. (2005). Neural systems recruited by drug- and food-related cues: studies of gene activation in corticolimbic regions. Physiol. Behav. 86, 11–14. doi: 10.1016/j.physbeh.2005.06.018
Kenny, P. J., Chen, S. A., Kitamura, O., Markou, A., and Koob, G. F. (2006). Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J. Neurosci. 26, 5894–5900. doi: 10.1523/jneurosci.0740-06.2006
Koob, G. F., and Bloom, F. E. (1988). Cellular and molecular mechanisms of drug dependence. Science 242, 715–723. doi: 10.1126/science.2903550
Koob, G. F., and Le Moal, M. (1997). Drug abuse: hedonic homeostatic dysregulation. Science 278, 52–58. doi: 10.1126/science.278.5335.52
Koob, G. F., and Le Moal, M. (2008). Addiction and the brain antireward system. Annu. Rev. Psychol. 59, 29–53. doi: 10.1146/annurev.psych.59.103006.093548
Koob, G. F., Buck, C. L., Cohen, A., Edwards, S., Park, P. E., Schlosburg, J. E., et al. (2014). Addiction as a stress surfeit disorder. Neuropharmacology 76, 370–382. doi: 10.1016/j.neuropharm.2013.05.024
Koob, G. F., Roberts, A. J., Kieffer, B. L., Heyser, C. J., Katner, S. N., Ciccocioppo, R., et al. (2003). Animal models of motivation for drinking in rodents with a focus on opioid receptor neuropharmacology. Recent Dev. Alcohol. 16, 263–281. doi: 10.1007/0-306-47939-7_19
Lack, A. K., Floyd, D. W., and Mccool, B. A. (2005). Chronic ethanol ingestion modulates proanxiety factors expressed in rat central amygdala. Alcohol 36, 83–90. doi: 10.1016/j.alcohol.2005.07.004
Li, T., Hou, Y., Cao, W., Yan, C. X., Chen, T., and Li, S. B. (2010). Naloxone-precipitated withdrawal enhances ERK phosphorylation in prefrontal association cortex and accumbens nucleus of morphine-dependent mice. Neurosci. Lett. 468, 348–352. doi: 10.1016/j.neulet.2009.11.030
Li, Y. Q., Li, F. Q., Wang, X. Y., Wu, P., Zhao, M., Xu, C. M., et al. (2008). Central amygdala extracellular signal-regulated kinase signaling pathway is critical to incubation of opiate craving. J. Neurosci. 28, 13248–13257. doi: 10.1523/JNEUROSCI.3027-08.2008
Li, F., Wang, X. S., Dai, R. P., Zhang, J. Y., Zhou, X. F., Hao, W., et al. (2011). The activation of NMDA receptor-ERK pathway in the central amygdala is required for the expression of morphine-conditioned place preference in the rat. Neurotox. Res. 20, 362–371. doi: 10.1007/s12640-011-9250-2
Liu, Y. L., Yan, L. D., Zhou, P. L., Wu, C. F., and Gong, Z. H. (2009). Levo-tetrahydropalmatine attenuates oxycodone-induced conditioned place preference in rats. Eur. J. Pharmacol. 602, 321–327. doi: 10.1016/j.ejphar.2008.11.031
Lu, L., Koya, E., Zhai, H., Hope, B. T., and Shaham, Y. (2006). Role of ERK in cocaine addiction. Trends Neurosci. 29, 695–703. doi: 10.1016/j.tins.2006.10.005
Lyons, D., De Jaeger, X., Rosen, L. G., Ahmad, T., Lauzon, N. M., Zunder, J., et al. (2013). Opiate exposure and withdrawal induces a molecular memory switch in the basolateral amygdala between ERK1/2 and CaMKIIalpha-dependent signaling substrates. J. Neurosci. 33, 14693–14704. doi: 10.1523/JNEUROSCI.1226-13.2013
Macey, T. A., Lowe, J. D., and Chavkin, C. (2006). Mu opioid receptor activation of ERK1/2 is GRK3 and arrestin dependent in striatal neurons. J. Biol. Chem. 281, 34515–34524. doi: 10.1074/jbc.m604278200
Mason, B. J., Shaham, Y., Weiss, F., and Le, A. D. (2009). Stress, alcohol craving and relapse risk: mechanisms and viable treatment targets. Alcohol 43, 541–543. doi: 10.1016/j.alcohol.2009.09.024
Maze, I., and Nestler, E. J. (2011). The epigenetic landscape of addiction. Ann. N Y Acad. Sci. 1216, 99–113. doi: 10.1111/j.1749-6632.2010.05893.x
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W., et al. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34, 807–820. doi: 10.1016/s0896-6273(02)00716-x
McNally, G. P., and Akil, H. (2002). Role of corticotropin-releasing hormone in the amygdala and bed nucleus of the stria terminalis in the behavioral, pain modulatory and endocrine consequences of opiate withdrawal. Neuroscience 112, 605–617. doi: 10.1016/s0306-4522(02)00105-7
Melis, M., Enrico, P., Peana, A. T., and Diana, M. (2007). Acetaldehyde mediates alcohol activation of the mesolimbic dopamine system. Eur. J. Neurosci. 26, 2824–2833. doi: 10.1111/j.1460-9568.2007.05887.x
Meng, Q. Y., Chen, X. N., Tong, D. L., and Zhou, J. N. (2011). Stress and glucocorticoids regulated corticotropin releasing factor in rat prefrontal cortex. Mol. Cell. Endocrinol. 342, 54–63. doi: 10.1016/j.mce.2011.05.035
Moron, J. A., Gullapalli, S., Taylor, C., Gupta, A., Gomes, I., and Devi, L. A. (2010). Modulation of opiate-related signaling molecules in morphine-dependent conditioned behavior: conditioned place preference to morphine induces CREB phosphorylation. Neuropsychopharmacology 35, 955–966. doi: 10.1038/npp.2009.199
Muller, D. L., and Unterwald, E. M. (2004). In vivo regulation of extracellular signal-regulated protein kinase (ERK) and protein kinase B (Akt) phosphorylation by acute and chronic morphine. J. Pharmacol. Exp. Ther. 310, 774–782. doi: 10.1124/jpet.104.066548
National Institute on Alcohol Abuse and Alcoholism. (2010). Beyond hangovers: understanding alcohol’s impact on your health. NIH Publication No. 13-7604. http://pubs.niaaa.nih.gov/publications/Hangovers/beyondHangovers.htm
Neisewander, J. L., Baker, D. A., Fuchs, R. A., Tran-Nguyen, L. T., Palmer, A., and Marshall, J. F. (2000). Fos protein expression and cocaine-seeking behavior in rats after exposure to a cocaine self-administration environment. J. Neurosci. 20, 798–805.
Nestler, E. J. (2005). Is there a common molecular pathway for addiction? Nat. Neurosci. 8, 1445–1449. doi: 10.1038/nn1578
O’Brien, C. P. (1997). A range of research-based pharmacotherapies for addiction. Science 278, 66–70. doi: 10.1126/science.278.5335.66
O’Dell, L. E., Roberts, A. J., Smith, R. T., and Koob, G. F. (2004). Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol. Clin. Exp. Res. 28, 1676–1682. doi: 10.1097/01.alc.0000145781.11923.4e
Ortiz, J., Harris, H. W., Guitart, X., Terwilliger, R. Z., Haycock, J. W., and Nestler, E. J. (1995). Extracellular signal-regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: regional distribution and regulation by chronic morphine. J. Neurosci. 15, 1285–1297.
Pandey, S. C., Roy, A., and Zhang, H. (2003). The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol. Clin. Exp. Res. 27, 396–409. doi: 10.1097/01.alc.0000056616.81971.49
Pandey, S. C., Roy, A., Zhang, H., and Xu, T. (2004). Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J. Neurosci. 24, 5022–5030. doi: 10.1523/jneurosci.5557-03.2004
Pandey, S. C., Zhang, H., Roy, A., and Misra, K. (2006). Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J. Neurosci. 26, 8320–8331. doi: 10.1523/jneurosci.4988-05.2006
Pandey, S. C., Zhang, H., Roy, A., and Xu, T. (2005). Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J. Clin. Invest. 115, 2762–2773. doi: 10.1172/jci24381
Pandey, S. C., Zhang, H., Ugale, R., Prakash, A., Xu, T., and Misra, K. (2008). Effector immediate-early gene arc in the amygdala plays a critical role in alcoholism. J. Neurosci. 28, 2589–2600. doi: 10.1523/JNEUROSCI.4752-07.2008
Payne, D. M., Rossomando, A. J., Martino, P., Erickson, A. K., Her, J. H., Shabanowitz, J., et al. (1991). Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 10, 885–892.
Peng, J., Wagle, M., Mueller, T., Mathur, P., Lockwood, B. L., Bretaud, S., et al. (2009). Ethanol-modulated camouflage response screen in zebrafish uncovers a novel role for cAMP and extracellular signal-regulated kinase signaling in behavioral sensitivity to ethanol. J. Neurosci. 29, 8408–8418. doi: 10.1523/JNEUROSCI.0714-09.2009
Philibin, S. D., Hernandez, A., Self, D. W., and Bibb, J. A. (2011). Striatal signal transduction and drug addiction. Front. Neuroanat. 5:60. doi: 10.3389/fnana.2011.00060
Potenza, M. N. (2014). Obesity, food and addiction: emerging neuroscience and clinical and public health implications. Neuropsychopharmacology 39, 249–250. doi: 10.1038/npp.2013.198
Potter, D. N., Damez-Werno, D., Carlezon, W. A. Jr., Cohen, B. M., and Chartoff, E. H. (2011). Repeated exposure to the kappa-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol. Psychiatry 70, 744–753. doi: 10.1016/j.biopsych.2011.05.021
Punn, A., Levine, M. A., and Grammatopoulos, D. K. (2006). Identification of signaling molecules mediating corticotropin-releasing hormone-R1alpha-mitogen-activated protein kinase (MAPK) interactions: the critical role of phosphatidylinositol 3-kinase in regulating ERK1/2 but not p38 MAPK activation. Mol. Endocrinol. 20, 3179–3195. doi: 10.1210/me.2006-0255
Quertemont, E., Green, H. L., and Grant, K. A. (2003). Brain ethanol concentrations and ethanol discrimination in rats: effects of dose and time. Psychopharmacology (Berl) 168, 262–270. doi: 10.1007/s00213-003-1437-7
Radwanska, K., Wrobel, E., Korkosz, A., Rogowski, A., Kostowski, W., Bienkowski, P., et al. (2008). Alcohol relapse induced by discrete cues activates components of AP-1 transcription factor and ERK pathway in the rat basolateral and central amygdala. Neuropsychopharmacology 33, 1835–1846. doi: 10.1038/sj.npp.1301567
Refojo, D., Echenique, C., Muller, M. B., Reul, J. M., Deussing, J. M., Wurst, W., et al. (2005). Corticotropin-releasing hormone activates ERK1/2 MAPK in specific brain areas. Proc. Natl. Acad. Sci. U S A 102, 6183–6188. doi: 10.1073/pnas.0502070102
Rimondini, R., Arlinde, C., Sommer, W., and Heilig, M. (2002). Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J. 16, 27–35. doi: 10.1096/fj.01-0593com
Robbins, D. J., Zhen, E., Owaki, H., Vanderbilt, C. A., Ebert, D., Geppert, T. D., et al. (1993). Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 268, 5097–5106.
Roberto, M., Bajo, M., Crawford, E., Madamba, S. G., and Siggins, G. R. (2006). Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology 31, 988–996. doi: 10.1038/sj.npp.1300840
Roberts, A. J., Cole, M., and Koob, G. F. (1996). Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol. Clin. Exp. Res. 20, 1289–1298. doi: 10.1111/j.1530-0277.1996.tb01125.x
Roberto, M., Nelson, T. E., Ur, C. L., Brunelli, M., Sanna, P. P., and Gruol, D. L. (2003). The transient depression of hippocampal CA1 LTP induced by chronic intermittent ethanol exposure is associated with an inhibition of the MAP kinase pathway. Eur. J. Neurosci. 17, 1646–1654. doi: 10.1046/j.1460-9568.2003.02614.x
Roberto, M., Schweitzer, P., Madamba, S. G., Stouffer, D. G., Parsons, L. H., and Siggins, G. R. (2004). Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J. Neurosci. 24, 1594–1603. doi: 10.1523/jneurosci.5077-03.2004
Roberts, A. J., Heyser, C. J., Cole, M., Griffin, P., and Koob, G. F. (2000). Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology 22, 581–594. doi: 10.1016/s0893-133x(99)00167-0
Robison, A. J., and Nestler, E. J. (2011). Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 12, 623–637. doi: 10.1038/nrn3111
Ron, D. (2004). Signaling cascades regulating NMDA receptor sensitivity to ethanol. Neuroscientist 10, 325–336. doi: 10.1177/1073858404263516
Ron, D., and Messing, R. O. (2013). Signaling pathways mediating alcohol effects. Curr. Top. Behav. Neurosci. 13, 87–126. doi: 10.1007/978-3-642-28720-6_161
Ruggeri, B., Braconi, S., Cannella, N., Kallupi, M., Soverchia, L., Ciccocioppo, R., et al. (2010). Neuropeptide S receptor gene expression in alcohol withdrawal and protracted abstinence in postdependent rats. Alcohol. Clin. Exp. Res. 34, 90–97. doi: 10.1111/j.1530-0277.2009.01070.x
Sananbenesi, F., Fischer, A., Schrick, C., Spiess, J., and Radulovic, J. (2003). Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. J. Neurosci. 23, 11436–11443.
Sanna, P. P., Simpson, C., Lutjens, R., and Koob, G. (2002). ERK regulation in chronic ethanol exposure and withdrawal. Brain Res. 948, 186–191. doi: 10.1016/s0006-8993(02)03191-8
Schlosburg, J. E., Whitfield, T. W. Jr., Park, P. E., Crawford, E. F., George, O., Vendruscolo, L. F., et al. (2013). Long-term antagonism of kappa opioid receptors prevents escalation of and increased motivation for heroin intake. J. Neurosci. 33, 19384–19392. doi: 10.1523/JNEUROSCI.1979-13.2013
Schroeder, J. P., Spanos, M., Stevenson, J. R., Besheer, J., Salling, M., and Hodge, C. W. (2008). Cue-induced reinstatement of alcohol-seeking behavior is associated with increased ERK1/2 phosphorylation in specific limbic brain regions: blockade by the mGluR5 antagonist MPEP. Neuropharmacology 55, 546–554. doi: 10.1016/j.neuropharm.2008.06.057
Seal, K. H., Shi, Y., Cohen, G., Cohen, B. E., Maguen, S., Krebs, E. E., et al. (2012). Association of mental health disorders with prescription opioids and high-risk opioid use in US veterans of Iraq and Afghanistan. JAMA 307, 940–947. doi: 10.1001/jama.2012.5573
Self, D. W., and Nestler, E. J. (1998). Relapse to drug-seeking: neural and molecular mechanisms. Drug Alcohol Depend. 51, 49–60. doi: 10.1016/s0376-8716(98)00065-9
Shalev, U., Morales, M., Hope, B., Yap, J., and Shaham, Y. (2001). Time-dependent changes in extinction behavior and stress-induced reinstatement of drug seeking following withdrawal from heroin in rats. Psychopharmacology (Berl) 156, 98–107. doi: 10.1007/s002130100748
Shaw-Lutchman, T. Z., Barrot, M., Wallace, T., Gilden, L., Zachariou, V., Impey, S., et al. (2002). Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J. Neurosci. 22, 3663–3672.
Shekhar, A., Truitt, W., Rainnie, D., and Sajdyk, T. (2005). Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress 8, 209–219. doi: 10.1080/10253890500504557
Shurman, J., Koob, G. F., and Gutstein, H. B. (2010). Opioids, pain, the brain and hyperkatifeia: a framework for the rational use of opioids for pain. Pain Med. 11, 1092–1098. doi: 10.1111/j.1526-4637.2010.00881.x
Siggins, G. R., Martin, G., Roberto, M., Nie, Z., Madamba, S., and De Lecea, L. (2003). Glutamatergic transmission in opiate and alcohol dependence. Ann. N Y Acad. Sci. 1003, 196–211. doi: 10.1196/annals.1300.012
Sinclair, C. M., Cleva, R. M., Hood, L. E., Olive, M. F., and Gass, J. T. (2012). mGluR5 receptors in the basolateral amygdala and nucleus accumbens regulate cue-induced reinstatement of ethanol-seeking behavior. Pharmacol. Biochem. Behav. 101, 329–335. doi: 10.1016/j.pbb.2012.01.014
Sommer, W. H., Rimondini, R., Hansson, A. C., Hipskind, P. A., Gehlert, D. R., Barr, C. S., et al. (2008). Upregulation of voluntary alcohol intake, behavioral sensitivity to stress and amygdala crhr1 expression following a history of dependence. Biol. Psychiatry 63, 139–145. doi: 10.1016/j.biopsych.2007.01.010
Stacey, D., Bilbao, A., Maroteaux, M., Jia, T., Easton, A. C., Longueville, S., et al. (2012). RASGRF2 regulates alcohol-induced reinforcement by influencing mesolimbic dopamine neuron activity and dopamine release. Proc. Natl. Acad. Sci. U S A 109, 21128–21133. doi: 10.1073/pnas.1211844110
Stinus, L., Caille, S., and Koob, G. F. (2000). Opiate withdrawal-induced place aversion lasts for up to 16 weeks. Psychopharmacology (Berl) 149, 115–120. doi: 10.1007/s002139900358
Substance Abuse and Mental Health Services Administration. (2008). Results from the 2007 National Survey on Drug Use and Health: National Findings, MD, USA: Office of Applied Statistics.
Szumlinski, K. K., Diab, M. E., Friedman, R., Henze, L. M., Lominac, K. D., and Bowers, M. S. (2007). Accumbens neurochemical adaptations produced by binge-like alcohol consumption. Psychopharmacology (Berl) 190, 415–431. doi: 10.1007/s00213-006-0641-7
Thorsell, A., Tapocik, J. D., Liu, K., Zook, M., Bell, L., Flanigan, M., et al. (2013). A novel brain penetrant NPS receptor antagonist, NCGC00185684, blocks alcohol-induced ERK-phosphorylation in the central amygdala and decreases operant alcohol self-administration in rats. J. Neurosci. 33, 10132–10142. doi: 10.1523/JNEUROSCI.4742-12.2013
Timpl, P., Spanagel, R., Sillaber, I., Kresse, A., Reul, J. M., Stalla, G. K., et al. (1998). Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat. Genet. 19, 162–166. doi: 10.1038/520
Ting, A. K. R., and Van Der Kooy, D. (2012). The neurobiology of opiate motivation. Cold Spring Harb. Perspect. Med. 2:a012096. doi: 10.1101/cshperspect.a012096
Torres, O. V., Walker, E. M., Beas, B. S., and O’dell, L. E. (2013). Female rats display enhanced rewarding effects of ethanol that are hormone dependent. Alcohol. Clin. Exp. Res. 38, 108–115. doi: 10.1111/acer.12213
Trigo, J. M., Martin-Garcia, E., Berrendero, F., Robledo, P., and Maldonado, R. (2010). The endogenous opioid system: a common substrate in drug addiction. Drug Alcohol Depend. 108, 183–194. doi: 10.1016/j.drugalcdep.2009.10.011
Valdez, G. R., Roberts, A. J., Chan, K., Davis, H., Brennan, M., Zorrilla, E. P., et al. (2002). Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol. Clin. Exp. Res. 26, 1494–1501. doi: 10.1111/j.1530-0277.2002.tb02448.x
Valjent, E., Pages, C., Herve, D., Girault, J. A., and Caboche, J. (2004). Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur. J. Neurosci. 19, 1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x
Valjent, E., Pascoli, V., Svenningsson, P., Paul, S., Enslen, H., Corvol, J. C., et al. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U S A 102, 491–496. doi: 10.1073/pnas.0408305102
Vendruscolo, L. F., Schlosburg, J. E., Misra, K. K., Chen, S. A., Greenwell, T. N., and Koob, G. F. (2011). Escalation patterns of varying periods of heroin access. Pharmacol. Biochem. Behav. 98, 570–574. doi: 10.1016/j.pbb.2011.03.004
Volkow, N. D., Wang, G. J., Tomasi, D., and Baler, R. D. (2013). Obesity and addiction: neurobiological overlaps. Obes. Rev. 14, 2–18. doi: 10.1111/j.1467-789X.2012.01031.x
Wagle, M., Mathur, P., and Guo, S. (2011). Corticotropin-releasing factor critical for zebrafish camouflage behavior is regulated by light and sensitive to ethanol. J. Neurosci. 31, 214–224. doi: 10.1523/JNEUROSCI.3339-10.2011
Walker, B. M., Valdez, G. R., Mclaughlin, J. P., and Bakalkin, G. (2012). Targeting dynorphin/kappa opioid receptor systems to treat alcohol abuse and dependence. Alcohol 46, 359–370. doi: 10.1016/j.alcohol.2011.10.006
Wang, W. S., Kang, S., Liu, W. T., Li, M., Liu, Y., Yu, C., et al. (2012). Extinction of aversive memories associated with morphine withdrawal requires ERK-mediated epigenetic regulation of brain-derived neurotrophic factor transcription in the rat ventromedial prefrontal cortex. J. Neurosci. 32, 13763–13775. doi: 10.1523/JNEUROSCI.1991-12.2012
West, R., and Gossop, M. (1994). Overview: a comparison of withdrawal symptoms from different drug classes. Addiction 89, 1483–1489. doi: 10.1111/j.1360-0443.1994.tb03747.x
Xu, Y., Lv, X. F., Cui, C. L., Ge, F. F., Li, Y. J., and Zhang, H. L. (2012). Essential role of NR2B-containing NMDA receptor-ERK pathway in nucleus accumbens shell in morphine-associated contextual memory. Brain Res. Bull. 89, 22–30. doi: 10.1016/j.brainresbull.2012.06.012
Zhang, L. L., Wang, J. J., Liu, Y., Lu, X. B., Kuang, Y., Wan, Y. H., et al. (2011). GPR26-deficient mice display increased anxiety- and depression-like behaviors accompanied by reduced phosphorylated cyclic AMP responsive element-binding protein level in central amygdala. Neuroscience 196, 203–214. doi: 10.1016/j.neuroscience.2011.08.069
Zhou, L., Lovell, K. M., Frankowski, K. J., Slauson, S. R., Phillips, A. M., Streicher, J. M., et al. (2013). Development of functionally selective, small molecule agonists at kappa opioid receptors. J. Biol. Chem. 288, 36703–36716. doi: 10.1074/jbc.m113.504381
Keywords: addiction, amygdala, drug dependence, extracellular signal-regulated kinase, nucleus accumbens, protein phosphorylation, reward, withdrawal
Citation: Zamora-Martinez ER and Edwards S (2014) Neuronal extracellular signal-regulated kinase (ERK) activity as marker and mediator of alcohol and opioid dependence. Front. Integr. Neurosci. 8:24. doi: 10.3389/fnint.2014.00024
Received: 02 January 2014; Accepted: 19 February 2014;
Published online: 11 March 2014.
Edited by:
Melissa A. Herman, The Scripps Research Institute, USAReviewed by:
Jean-Antoine Girault, INSERM, FranceKaren K. Szumlinski, University of California, USA
Copyright © 2014 Zamora-Martinez and Edwards. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott Edwards, Department of Physiology and Alcohol and Drug Abuse Center of Excellence, Louisiana State University Health Sciences Center, 1901 Perdido Street, New Orleans, LA 70112, USA e-mail:c2Vkd2E1QGxzdWhzYy5lZHU=