 Jessica M. D'Amico1,2
Jessica M. D'Amico1,2 Elizabeth G. Condliffe1,2,3,4
Elizabeth G. Condliffe1,2,3,4 Karen J. B. Martins1,5
Karen J. B. Martins1,5 David J. Bennett1,6
David J. Bennett1,6 Monica A. Gorassini1,2,3*
Monica A. Gorassini1,2,3*- 1Centre for Neuroscience, University of Alberta, Edmonton, AB, Canada
- 2Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
- 3Department of Biomedical Engineering, University of Alberta, Edmonton, AB, Canada
- 4Division of Physical Medicine and Rehabilitation, University of Alberta, Edmonton, AB, Canada
- 5Faculty of Physical Education and Recreation, University of Alberta, Edmonton, AB, Canada
- 6Faculty of Rehabilitation Medicine, University of Alberta, Edmonton, AB, Canada
The state of areflexia and muscle weakness that immediately follows a spinal cord injury (SCI) is gradually replaced by the recovery of neuronal and network excitability, leading to both improvements in residual motor function and the development of spasticity. In this review we summarize recent animal and human studies that describe how motoneurons and their activation by sensory pathways become hyperexcitable to compensate for the reduction of functional activation of the spinal cord and the eventual impact on the muscle. Specifically, decreases in the inhibitory control of sensory transmission and increases in intrinsic motoneuron excitability are described. We present the idea that replacing lost patterned activation of the spinal cord by activating synaptic inputs via assisted movements, pharmacology or electrical stimulation may help to recover lost spinal inhibition. This may lead to a reduction of uncontrolled activation of the spinal cord and thus, improve its controlled activation by synaptic inputs to ultimately normalize circuit function. Increasing the excitation of the spinal cord with spared descending and/or peripheral inputs by facilitating movement, instead of suppressing it pharmacologically, may provide the best avenue to improve residual motor function and manage spasticity after SCI.
Introduction
In the months following a spinal cord injury (SCI), 70–80% of individuals develop spasticity (Maynard et al., 1990; Skold et al., 1999), which is characterized by involuntary muscle activity such as spasms, hyperreflexia, clonus and co-contraction (Pandyan et al., 2005; Nielsen et al., 2007). There is also an increased resistance to passive stretch and the development of joint contractures, the latter characterized by reduced joint range of motion and deformity mediated, in part, by changes to tissues in the muscle and joint (Diong et al., 2012). Although spasticity can interfere with residual motor function and produce pain, it can also be useful. For example, involuntary muscle spasms (i.e., prolonged contractions) and tone in extensor muscles can facilitate walking (Skold, 2000), standing and transfers (Satkunam, 2003; Adams and Hicks, 2005). In this review, we compare evidence from both animal and human studies to discuss how muscle and non-muscle tissues respond to the different phases of SCI and how neurons and neuronal circuits increase their excitability and decrease their inhibitory capacity to compensate for the loss of descending and movement-related sensory inputs, ultimately leading to the development of spasticity. In many cases, there is good agreement with animal and human studies but also some important differences. We propose that instead of further suppressing spinal cord activity with antispastic medications, perhaps we should be doing just the opposite, by finding ways to functionally reactivate the spinal cord to restore lost inhibition and normalize neuronal and circuit function. In this way, improvements in both residual motor control and/or reductions in spasticity can occur without the unwanted side effects of antispastic medications.
Assessments of Spasticity in Humans
Throughout this review, several clinical measures of spasticity are discussed as they are used to evaluate the effectiveness of different interventions (Hsieh et al., 2008). The frequently used Ashworth (Ashworth, 1964) and modified Ashworth (Bohannon and Smith, 1987) scales rate, from 0 to 4, the resistance of a relaxed, single joint to movement imposed by the evaluator throughout its full available range. The pendulum test, which also measures resistance to passive motion, records changes in knee joint angle after the relaxed leg is allowed to swing freely following its release from an extended position (Wartenberg, 1951; Bajd and Vodovnik, 1984). To measure spasticity evoked by various stimuli, the Spinal Cord Assessment Tool for Spasticity (SCATS) rates, on a scale of 0–3, the severity of low frequency (4–12 Hz) rhythmic movements of the ankle (i.e., clonus, Wallace et al., 2005) and flexor and extensor spasms in response to pinprick or rapid leg extension, respectively (Benz et al., 2005). The self-reported Penn spasm frequency scale asks patients to assess the average frequency and source of spasms from 0 (no spasms on most days) to 4 (>10 spontaneous spasms/hour) (Penn et al., 1989). However, like most of the other assessment methods, the scale is ordinal with incompletely explored reliability, validity, and responsiveness (Biering-Sorensen et al., 2006; Hsieh et al., 2008). Moreover, these assessments are limited to a single point in time during the assessment, even though spasticity varies by time of day, recent activity level, temperature and emotional status (Biering-Sorensen et al., 2006). Thus, more objective measures of spasticity, based on actual muscle activation patterns recorded over many hours/days, are needed.
Changes in Tissue Properties After SCI
Muscle
As with neurons and neuronal circuits, muscle and connective tissue also adapt to SCI. With a reduced activity and unloading after SCI in humans, muscles begin to atrophy within the first months as measured by decreased fiber diameter, cross-sectional area and volume (Scelsi et al., 1982; Lotta et al., 1991; Biering-Sorensen et al., 2009). There are also changes in the joint angle-torque relationship of some muscles that may accompany reductions in the range of joint motion. For example, in plantarflexors (but not dorsiflexors) peak twitch torque occurs at more plantarflexed joint angles in SCI participants compared to non-injured controls (McDonald et al., 2005; Pelletier and Hicks, 2010). The shift in the joint angle-torque relationship has been attributed to shortening of the muscle as a result of sarcomere loss and increases in connective tissue, but this has not been directly proven. Changes in the composition of the muscle fiber can also occur after SCI, although different results have been obtained when examining non-spastic vs. spastic muscles. Several animal models of SCI, where no clear spasticity develops, and several human studies have a reported a decrease in the proportion of the fatigue-resistant type I and less-fatigable type IIa fibers coupled with an increase in the most-fatigable type IIb or IId/x fibers [abbreviated together as IIb(x)](Scelsi et al., 1982; Lotta et al., 1991; Roy et al., 1992, 1999; Harris et al., 2007; Malisoux et al., 2007). In humans with complete SCI, the slower-to-faster fiber type transitions begin ~4–7 months post-SCI and continue until almost all fibers are type IIb(x), which can occur 2–6 years post-SCI (reviewed in Biering-Sorensen et al., 2009).
Importantly, in the studies described above, the amount of muscle activity present from day-to-day was not well described. It is reasonable to assume that muscle activity has an effect on skeletal muscle properties. As a model of this, acute transection of the sacral spinal cord in adult rats initially results in tail muscles that are inactive, but then a clear spasticity syndrome develops 2 weeks later that mimics what happens after SCI in humans (Bennett et al., 1999, 2004). Acutely, when muscles of the tail were inactive, as measured by 24-h EMG recordings, there was a transition from slow, fatigue-resistant type I to predominantly fast, fatigable type IIb(x) fibers compared to uninjured control animals (Harris et al., 2007). However, in spastic muscles with enhanced EMG activity compared to controls, the amount of atrophy measured in the myofibers was substantially reduced. Moreover, the proportion of type I to type II fibers was similar to age-matched control rats (Harris et al., 2007), although the muscles were more fatigable (Harris et al., 2006). In agreement with the rat results, participants with motor incomplete [ASIA Impairment Scale (AIS) C&D] or motor complete (AIS A&B) injuries having marked spasticity exhibited stimulus-torque responses characteristic of plantarflexor muscles with slow contractile properties (Hidler et al., 2002; Pelletier and Hicks, 2010). Additionally, in participants with motor complete and incomplete SCI (AIS B&C), muscle cross-sectional area was positively correlated to modified Ashworth scores (Gorgey and Dudley, 2008). In contrast, muscles of individuals with low measures of spasticity displayed faster contractile properties compared to control muscles, in line with the human studies described earlier (reviewed in Biering-Sorensen et al., 2009).
Collectively, these results indicate that similar to exercise (Roy et al., 1999), the involuntary activity present in spastic muscles allows some retention of normal muscle fiber type composition and contractile properties; however, the muscles still remain more fatigable. Increased fatigability after SCI in humans appears to be related to changes in the muscle (Klein et al., 2006). For example, the metabolic capacity of muscle, as measured by oxidative enzyme activity (Shields, 1995; Wang et al., 1999) and concentrations of Na+/K+-ATPase (Ditor et al., 2004), is correlated to the amount of fatigue resistance in individuals with SCI (Shields, 1995). In summary, although spastic muscle activity promotes slow contractile and fiber-type properties of muscle, it is not enough to preserve fatigue resistance. Treatments such as intensive exercise (Roy et al., 1999) or electrical stimulation (Rochester et al., 1995; Gerrits et al., 2002, 2003) are likely also needed to improve muscle endurance via increases in oxidative capacities of the muscle (Gerrits et al., 2003).
Muscle Connective Tissue
Similar to muscle, remodeling of non-muscle tissue also occurs after SCI. For example, after SCI in humans atrophic myofibers become replaced by adipocytes, collagen, and other amorphous substances (Scelsi et al., 1982; Olsson et al., 2006). It is thought that these morphological changes increase the intrinsic stiffness of the muscle (Mirbagheri et al., 2001; Schleip et al., 2006). However, in individuals with increased passive tension in the vastus lateralis at the whole muscle and muscle fiber level, specifically in type IIb(x) fibers, there were no changes in passive tension at the myofibril level (Olsson et al., 2006; Malisoux et al., 2007). Likewise, the muscle protein titin, a main contributor to passive tension (Horowits et al., 1986; Labeit and Kolmerer, 1995), remained unchanged in spastic muscles when compared to control muscles, as did the properties of the intermediate filaments (Olsson et al., 2006). Thus, as muscle atrophy occurs, the increased passive tension evident at the whole muscle and muscle fiber level is likely not due to structural changes of the myofibril, but rather due, in part, to the replacement of myofibrils by amorphous substances such as collagen and connective tissue (Scelsi et al., 1982; Olsson et al., 2006). Moreover, adaptations to extracellular and joint tissues may also contribute to joint stiffness after SCI, but these changes have only been demonstrated in immobilization models (Gracies, 2005).
In summary, changes in muscle and non-muscle tissue, combined with adaptations to preserved spinal circuitry as described next, likely contributes to spastic motor behaviors after SCI. For example, increases in muscle stiffness and changes in the joint angles that produce optimal torque may contribute to the decreased threshold and increased gain of the stretch reflex pathway that mediate, in part, the oscillatory activation of muscles during clonus (de Vlugt et al., 2012).
Changes in Motoneuron Properties After SCI
Spinal Shock
Immediately after injury, the spinal cord enters a state of “spinal shock” (Ditunno et al., 2004; Dietz, 2010) that is characterized by severe muscle paralysis, flaccid muscle tone (Bastian, 1890; Sherrington, 1899, 1909) and an initial loss of reflexes and sensation caudal to the lesion (Leis et al., 1996; Little et al., 1999; Ditunno et al., 2004; Dietz, 2010). The duration and severity of spinal shock differs between species, lasting only minutes to hours in the frog (Hall, 1850), cat (Sherrington, 1899; Hunt et al., 1963; Chambers et al., 1966) and dog (Sherrington, 1899; Fulton and Sherrington, 1932) but up to 2 weeks in the monkey (Sherrington, 1899; Fulton and Sherrington, 1932; Hunt et al., 1963; McCouch et al., 1966), rat (Bennett et al., 1999) and several weeks in humans (Ko et al., 1999). In humans, all reflexes are absent only for the first 24 h after SCI (Phase 1) (Ditunno et al., 2004), which coincides with the first 2 weeks of areflexia in the rat tail model of SCI (Bennett et al., 1999). By 1–3 days post-injury, human H-reflexes and cutaneous reflexes begin to return (Diamantopoulos and Olsen, 1967; Ashby et al., 1974; Ko et al., 1999; Hiersemenzel et al., 2000) while a delayed plantar response also develops (Phase 2: Ko et al., 1999; Ditunno et al., 2004). Phase 3, lasting 4 days to 1 month, is characterized by a gradual increase in all reflexes with the disappearance of the abnormal plantar response. Spinal shock in humans is considered resolved by 1–6 months post-injury with eventual development of hyperreflexia, clonus and muscle spasms (Ko et al., 1999; Ditunno et al., 2004). Similarly in the rat, hypertonus in flexor and extensor muscles, clonus and hyperreflexia (cutaneous tail flick) develops by 2 months post-injury (Bennett et al., 1999).
Several factors contribute to the initial suppression of spinal cord excitability after SCI. For example, there is a slight hyperpolarization (2–6 mV) of the resting membrane potential in cat (Cope et al., 1980; Schadt and Barnes, 1980) and rat motoneurons (Li et al., 2007), which may explain why antidromic activation of human motoneurons, as measured by F-waves, is difficult acutely after SCI (Ashby et al., 1974). In contrast, H-reflexes, but not tendon tap reflexes, recover during spinal shock, as shown in both the cat (Hunt et al., 1963; Zapata, 1966) and human (Weaver et al., 1963; Hiersemenzel et al., 2000). This suggests that fusimotor drive is also reduced acutely after SCI given that tendon taps rely on muscle spindle excitability whereas H-reflexes do not. Pre-synaptic inhibition of primary afferents is also increased in spinal shock, as measured by increases in primary afferent depolarization in cats (Quevedo et al., 1993).
One of the more prominent contributors to spinal shock is likely the disappearance of dendritic, voltage-activated sodium and calcium persistent inward currents (PICs) in motoneurons acutely after SCI. PICs contain a TTX-sensitive persistent Na current and a low-voltage activated, slowly inactivating L-type Ca current (CaV1.3) that is sensitive to nimodipine (Li and Bennett, 2003). PICs act to amplify synaptic inputs (Lee and Heckman, 2000) and because they are activated sub-threshold to cell firing (Bennett et al., 1998), PICs aid in the secure and rapid recruitment of motoneurons (Lee and Heckman, 2000). After recruitment, PICs produce a sustained depolarization, or plateau potential, which can last for many seconds to produce self-sustained firing of the motoneuron in the presence of reduced synaptic inputs (Hounsgaard et al., 1984, 1986; Crill and Schwindt, 1986; Carlin et al., 2000; Powers and Binder, 2003; reviewed in Heckman et al., 2005). For example, a 1-s long, sub-threshold depolarization of the motoneuron by a sensory-evoked, excitatory post-synaptic potential (EPSP) activates a plateau potential (Figure 1A), which then keeps the motoneuron discharging for many seconds even though the synaptic input (EPSP, see hyperpolarized trace) has subsided. During spinal shock, PICs are no longer activated in response to sensory stimulation. In motoneurons of acutely injured rats (Figure 1B), the same 1-s long EPSP does not activate a plateau potential and subsequently, self-sustained firing does not occur. Thus, the acute disappearance of PICs dramatically reduces the excitability of the motoneuron as the amplification and prolongation of synaptic inputs does not occur. This, coupled with motoneuron hyperpolarization, increases in pre-synaptic inhibition and decreases in background synaptic and gamma motoneuron drive, renders the motoneuron and spinal circuits unexcitable in the first days and weeks after SCI.
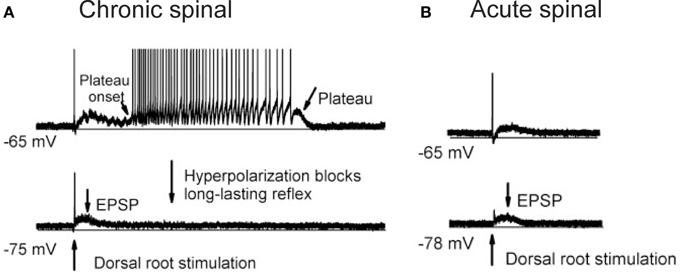
Figure 1. Re-emergence of PICs after chronic SCI. (A) Re-emergence of PICs in a completely transected (S2) rat after chronic (>50 days post-injury) SCI. Motoneuron recording in response to single pulse (3 × 's sensory threshold) dorsal root stimulation. At resting membrane potential (−65 mV, top trace), PIC activation produces a plateau potential and self-sustained firing. Hyperpolarization of the motoneuron (−75 mV, bottom trace) deactivates the PIC, eliminating the long-lasting reflex (LLR or spasm) and reveals the long (1 s) EPSP. (B) Spinal shock in acutely-spinalized rat (hours after injury), same preparation and experimental set-up as in (A). Note absence of long-lasting reflex response at resting membrane potential (−65 mV, top trace) due to elimination of PICs acutely after injury. The long-duration motoneuron EPSP (~1 s) appears acutely after injury as revealed in hyperpolarized motoneuron (−78 mV, bottom trace). Taken from Li et al. (2004a).
Recovery of Motoneuron PICs
In the weeks and months that follow a SCI, neurons and neuronal circuits below the injury adapt to inactivity by increasing intrinsic excitability. This recovered excitability is marked in human SCI by increases in residual muscle strength, H-reflexes, F-wave persistence and the return of flexor reflex responses (Leis et al., 1996; Little et al., 1999; Hiersemenzel et al., 2000). In addition to changes in the transmission of sensory inputs (as discussed later on), motoneuron PICs also re-emerge in the weeks following a SCI to contribute to both the recovery of motor function and the development of spasticity. For example, the motoneuron described previously in Figure 1A actually comes from a rat whose sacral spinal cord was completely severed 2 months previously and whose tail muscles developed prolonged muscle spasms. Thus, the self-sustained activity in this motoneuron, which is mediated by the PIC and subsequent plateau potential, reflects the involuntary muscle spasms that develop in these animals. The long time course of the plateau potential, which can last for several seconds, is mainly produced by the slowly inactivating CaPIC given that the NaPIC inactivates over a few seconds (Lee and Heckman, 2001; Miles et al., 2005; Harvey et al., 2006) and blocking CaPICs greatly reduces long-lasting reflexes (LLR), i.e., spasms (Murray et al., 2011b).
Instead of producing prolonged plateau potentials, the persistent NaPIC facilitates repetitive discharge of the motoneuron and mediates the slow, but steady firing that is characteristic of spontaneous motor unit discharge in individuals with chronic SCI (e.g., 5 Hz discharge in Figure 2C; Gorassini et al., 2004; see also Zijdewind and Thomas, 2012). For example, in motoneurons from chronically spinalized rats, the NaPIC is activated shortly after the upswing on the afterhyperpolarization (AHP) to produce a slow ramp and then acceleration in membrane potential (Vm) which then triggers an action potential (see bottom trace in Figure 2A). Following this, during the fast depolarizing phase of the action potential (spike), the NaPIC deactivates but then re-activates on the upsweep of the next AHP to slowly ramp up the Vm and trigger another spike. In this way, the NaPIC helps to regeneratively fire the motoneuron at interspike intervals that are much longer than the duration of the AHP (marked by bar), especially under conditions of weak or absent depolarizing synaptic drive (Li and Bennett, 2007). Thus, repetitive activation of the NaPIC produces the slow but steady motoneuron firing often observed after SCI (e.g., 1.8 Hz firing in Figure 2A). In motoneurons from acutely injured rats, there is little to no NaPIC as demonstrated by the absence of a slow acceleration in Vm after the AHP (first inset in Figure 2B, bottom trace). Because of this, repetitive firing only occurs during a strong depolarizing drive, as occurs during a large current injection (see third current pulse in Figure 2B). The large current injection provides sufficient acceleration of the Vm after the AHP to bring the motoneuron to threshold (second inset, bottom trace). Thus, without the contribution of the NaPIC, the motoneuron can only fire under a strong depolarizing drive, with interspike intervals that are close to the duration of the AHP, resulting in faster firing rates (e.g., 7.7 Hz firing in Figure 2B).
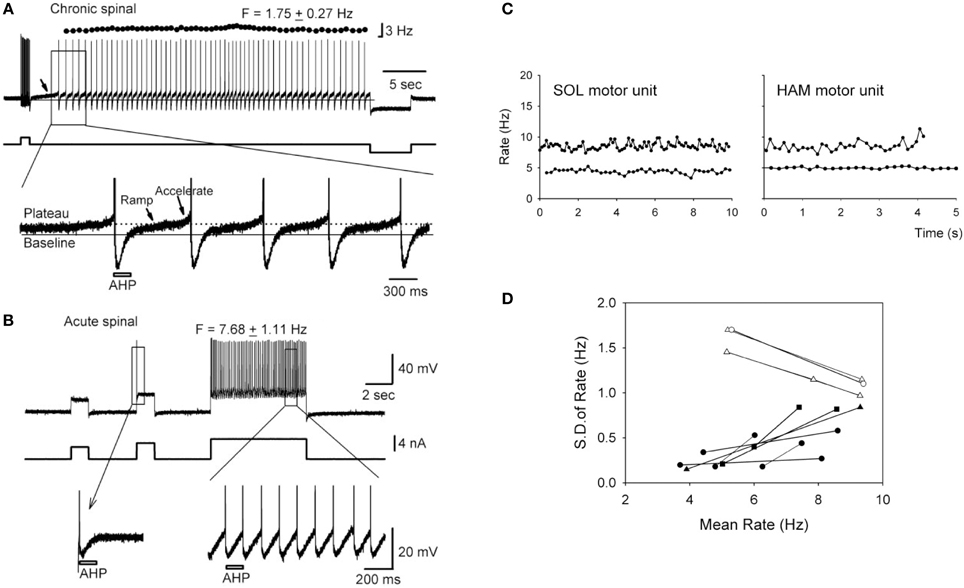
Figure 2. Slow, steady firing in rat and human motoneurons after chronic SCI. (A) Top trace: Firing response of a motoneuron from a chronic spinal rat in response to a brief depolarizing current pulse (~2 nA). Note low mean rate (1.75 Hz) and small standard deviation (±0.27 Hz) in firing. Bottom trace: Magnification of top trace indicated by rectangle. Plateau activation due to NaPIC is indicated by dotted line, and baseline membrane potential (Vm) is represented by solid black line. Note the ramp and acceleration (arrows) in Vm after each AHP due to NaPIC, producing interspike intervals much longer than the duration of the AHP (gray bar). (B) Top trace: Firing response of motoneuron from an acute spinal rat without plateau potentials in response to large (~4–5 nA) depolarizing current injections. Note increased firing rate (7.68 Hz) during large current pulse. Bottom trace: Magnification of top trace as indicated by rectangle. Interspike intervals are now closer to the duration of the AHP (gray bar) and lack NaPIC-mediated ramp in Vm. (A,B) taken from Li et al. (2004a). (C) Slow, steady firing rate of spontaneously-active soleus (SOL) and hamstrings (HAM) motor units from two participants with incomplete SCI (5 Hz firing). Note increase in firing rate and variability during superimposed voluntary activation of the muscles (10 Hz firing). (D) Relationship between the firing rate variability [standard deviation (SD)] and mean rate of motor units from SCI participants (black symbols) and control, uninjured participants (white symbols). (C,D) taken from Gorassini et al. (2004).
Evidence for the activation of NaPICs in mediating the slow, but steady firing in human motoneurons after SCI is demonstrated by the increase in firing rate variability when volitional drive is superimposed on a spontaneously active motor unit (Figure 2C). Here, the steady but slow discharge of the motor unit at 5 Hz is thought to be mediated by the regenerative activation of a strong NaPIC under conditions of low, involuntary depolarizing synaptic drive, similar to rat motoneurons. With the introduction of volitional drive, synaptic noise to the motoneuron increases and subsequently, both the firing rate of the motor unit (e.g., 10 Hz in Figure 2C), along with its spike-to-spike variability, increase (black symbols in Figure 2D). This suggests that the faster, more variable firing is produced by the noisy depolarization from the voluntary synaptic drive that recruits the motoneuron at random times during the slow, NaPIC-mediated ramp in Vm. The relationship between firing rate and firing variability is opposite in motor units from uninjured control participants during increasing levels of voluntary drive (white symbols in Figure 2D). Under low levels of voluntary drive, firing rates are low (~5 Hz) but variable due to low levels of synaptic noise (EPSPs) that sporadically brings the Vm to threshold after the AHP (Matthews, 1996). However, when voluntary drive increases, so does the mean depolarizing drive to the motoneuron and this helps to more consistently accelerate the Vm to threshold after the AHP (as in Figure 2B). Thus, during strong voluntary drive in uninjured individuals, firing rate variability at high rates decreases because the interspike interval (ISI) is more dependent upon the stable AHP rather than the synaptic noise (Miles et al., 2005). For example, firing rate variability is low and plateaus beyond 10 Hz (ISI's ≤ 100 ms, Person and Kudina, 1971), likely because the ~100 ms AHP occupies a larger proportion of the ISI compared to at lower rates (ISI's > 100 ms) where random synaptic noise has more opportunity to recruit the motoneuron during the NaPIC-mediated, slow ramp in Vm. Interestingly, when the firing rate in the SCI and uninjured motor units both reach ~10 Hz, the firing rate variability in the two groups become similar, suggesting that at rates >10 Hz, the AHP is the main determinant of the ISI.
In summary, after SCI both CaPIC-mediated plateau potentials and NaPIC-mediated regenerative firing allow motoneurons to exhibit prolonged, involuntary firing in response to brief or low-levels of depolarizing synaptic drive.
Estimating PICs from Human Motor Unit Recordings
The use of paired motor unit recordings in humans has provided indirect evidence that PICs contribute to the involuntary activation of motoneurons during muscle spasms in chronic SCI. The basis of estimating PIC activation by using the firing profiles of two motor units (motoneurons) is demonstrated from direct intracellular recordings in the rat (Figure 3A). For example, the amplitude of the PIC can be estimated indirectly during current clamp when the motoneuron is activated by a triangular-shaped current injection (Bennett et al., 2001). Once the PICs are activated, the added depolarization provided by the PIC allows the motoneuron to fire at levels of injected current (input) below that needed to recruit the motoneuron (below top horizontal line: Figure 3Ai), with cell firing stopping only when the injected current is substantially reduced (at second horizontal line). The difference in injected current between derecruitment and recruitment of the motoneuron (ΔI) is then used as a measure of the PIC amplitude because it is the amount of current that needs to be removed to counteract the added depolarization provided by the PIC and stop the motoneuron from firing.
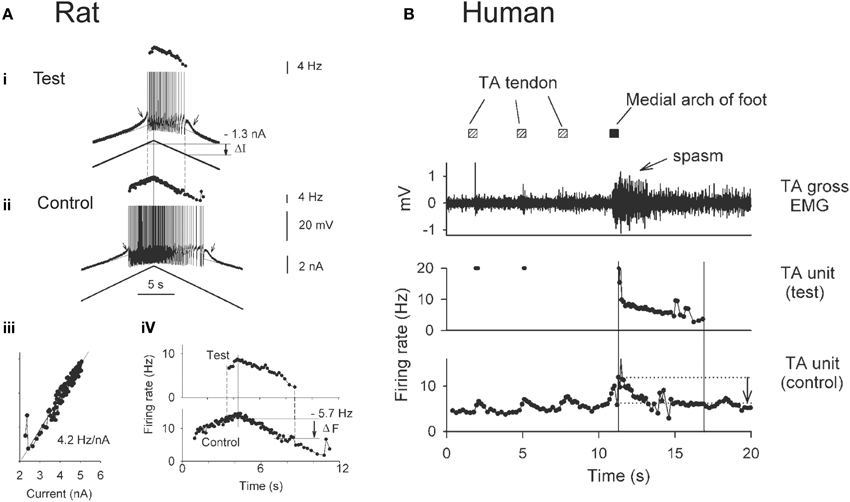
Figure 3. Estimating PICs in rat and human motoneurons. (A) Rat motoneurons: Paired motor unit analysis (ΔF) technique in rat motoneurons. (i,ii) Firing rate profiles of a control and test motoneuron in response to the same triangular current injection. Measurement of PIC as the difference in current input at recruitment and derecruitment of the test motoneuron (dashed lines): ΔI value. (iii) Linear relationship between the firing rate and injected current in the control motoneuron. (iv) The control motoneuron is serving as a measure of input to the test motoneuron. Measurement of the PIC as the difference in firing rate of the control motoneuron at recruitment and derecruitment (dashed lines) of the test motoneuron: ΔF value. (B) Human motoneurons: ΔF during muscle spasm in human SCI. Top trace: Tibialis anterior (TA) surface EMG during sustained dorsiflexion and application of transient vibration to TA tendon (gray squares) and medial arch of the foot (black square). Middle trace: Firing rate of a higher-threshold TA test unit in response to the vibrations. Bottom trace: Firing rate profile of tonically active lower-threshold control motor unit. ΔF is measured as the difference in control unit rate at derecruitment and recruitment of test motor unit (vertical lines and arrow). (A,B) taken from Gorassini et al. (2004).
Using the firing rate profiles of two motoneurons to estimate the amplitude of the PIC requires that both cells receive a common input and that the firing rate of one of the motoneurons (e.g., control motoneuron in Figure 3Aii) serves as an estimate of the input to the second motoneuron (e.g., test motoneuron). This is possible because the firing rate of a motoneuron is linearly related to the amount of current injection (or synaptic input) it receives once the PIC is fully activated, as shown in Figure 3Aiii. Thus, when using the firing rate (F) of the control motoneuron as a measure of input to the test motoneuron (Figure 3Aiv), rather than the injected current, the difference in firing rate of the control motoneuron at derecruitment and recruitment of the test motoneuron (dashed lines) corresponds to the reduction in input that is required to counteract the added depolarization from the PIC (Fderecruitment-Frecruitment = ΔF). Dividing the ΔF value (5.7 Hz) by the slope of the F-I relationship measured in Figure 3Aiii (4.2 Hz/nA) results in an estimation of current (1.4 nA) that is close to the amplitude of the PIC measured from the injected current profile (1.3 nA in Figure 3Ai).
By using the firing rate profiles of two motoneurons (motor units) as described above, the contribution of PICs to the activation of motoneurons during involuntary muscles spasms has been estimated in participants with SCI (Figure 3B). Here the firing rate of a lower-threshold control motor unit from the tibialis anterior (TA) muscle that is activated by vibration to the TA tendon (Figure 3B, bottom trace) is used as a measure of input to the TA motoneuron pool and to a higher threshold test motor unit. As evidence of this, vibration of the TA tendon (hatched squares) produced a transient increase in the firing rate of the control unit and a transient recruitment of a higher-threshold test unit (Figure 3B: middle trace). Vibration of the tendon did not produce a strong enough afferent input to evoke a muscle spasm. However, vibration of the medial arch of the foot (Figure 3B: black square) produced a greater increase in the firing rate of the tonically-active control unit, signifying greater synaptic input to the motor units, and consequently resulted in stable recruitment of the higher-threshold test unit. In fact, the newly recruited test unit continued to fire well after the removal of the vibratory input, and exhibited sustained firing even at levels of synaptic input (as measured by the firing rate of the control unit) well below the level needed to recruit the test unit (difference between horizontal lines, arrow indicates ΔF). This difference indicates the activation of a PIC. By comparing the estimated PIC amplitude (ΔF = 5 Hz) to the total amount of rate modulation in the test unit during a moderate contraction or involuntary spasm (~12 Hz), it is estimated that the PIC provides ~40% of the depolarizing drive to the motoneuron during self-sustained firing (Gorassini et al., 2004) and therefore, plays a major role in driving involuntary muscle spasms after SCI in humans. During stronger muscle spasms, peak firing rates of motor units can reach as high as 35 Hz (Thomas and Ross, 1997) where the relative contribution of PIC and synaptic activation of the motoneuron is unknown.
Using the paired motor unit technique to estimate PIC amplitude (ΔF) in human participants relies on the important assumptions that: (1) the firing rate of the control unit is an accurate representation of its synaptic input and (2) the control unit receives the same synaptic drive as the test unit (Gorassini et al., 2004). Thus, it is important to measure the firing rate of the control unit during periods of linear input-output which likely occurs during moderate rates of discharge (5–20 Hz) and durations of spike trains (10–20 s), to avoid firing rate saturation and adaptation, respectively (Sawczuk et al., 1997; Brownstone, 2006). These values are based on firing rate properties of sacral rat motoneurons that are similar to firing properties of human motor units in the lower limb (Li et al., 2004a). Likewise, firing rates of the control unit during the first and last 1–2 s of activity should be avoided as initial activation of PICs and AHP conductances, respectively, could affect the firing rate of the motoneuron (Bennett et al., 1998; Wienecke et al., 2009). Finally, the correlation between the smoothed firing rate of a control and test motor unit should be high (correlation coefficient > 0.8) to ensure that both units are receiving a common synaptic drive (De Luca and Erim, 2002). Thus, ΔF measures can provide a reasonable estimate of PIC amplitude when the above conditions are followed. However, one must keep in mind that it is not possible to directly measure the linearity of the input-output relation of human motoneurons.
Modulation of Motoneuron PICs by Monoamine Receptors
In addition to being voltage-sensitive, PICs require the concomitant activation of serotonergic (5-HT) or noradrenergic (NA) receptors located on the motoneurons. Serotonin is synthesized from the amino acid, L-tryptophan, via the enzymes L-tryptophan hydroxylase and amino acid decarboxylase (AADC) (Feldman et al., 1997). Neuronal excitability is modified by 5-HT via seven main types of receptors, 5-HT1−7, which are all G-protein coupled receptors (GPCRs) except for the ligand-activated 5-HT3 receptor (Boess and Martin, 1994; Hoyer et al., 2002; Nichols and Nichols, 2008). PICs on motoneurons are modulated by 5-HT2B/C receptors specifically (Miller et al., 1996; Murray et al., 2011a). Noradrenaline, like dopamine and adrenaline, is a catecholamine synthesized by adrenal chromaffin cells and sympathetic nerves and is converted from the amino acid L-tyrosine into L-Dopa via tyrosine hydroxylase (Kopin, 1968). L-Dopa is then converted into dopamine via AADC and subsequently into noradrenaline via the enzyme dopamine beta hydroxylase (DβH). Noradrenaline exerts its actions via three main different types of GPCRs: α1, α2, and β subtypes (Ahlquist, 1980; Bylund, 1992), where α1 receptors are coupled to downstream Gq pathways and affect motoneuron PICs, similar to 5-HT2 receptors (Gershengorn, 1989; Minneman and Esbenshade, 1994; Lee and Heckman, 1996) and α2 receptors are coupled to downstream Gi pathways to affect sensory transmission, similar to 5-HT1 receptors (Ruffolo et al., 1991; Murray et al., 2011b). The major source of serotonin to the spinal cord, as identified in rodents, comes from 5-HT containing neurons in the medullary raphe pallidus (B1), raphe obscuris (B2), raphe magnus (B3) and from adjacent parts of the reticular formation (reviewed in Schmidt and Jordan, 2000). The dorsal horn is mainly innervated by neurons from the raphe magnus, whereas motoneurons in the ventral horn receive 5-HT inputs mainly from the raphe pallidus and obscuris. The A5, A6 (locus coeruleus) and A7 NA cell groups in the pontine region provide the major source of NA fibers to the spinal cord, with a predominant innervation of the dorsal horn by A6 axons and of the ventral horn by A7 axons (Bruinstroop et al., 2012).
The role of 5-HT and NA receptors in facilitating motoneuron PICs was first demonstrated in the decerebrate cat where PICs and sustained reflex responses were blocked by methysergide; a 5-HT and NA receptor blocker (Hounsgaard et al., 1986, 1988). Likewise, immediately following a complete spinal transection where descending sources of 5-HT and NA were abolished (i.e., acute injury), PICs were reduced or eliminated but could be subsequently restored with an intravenous application of 5-HTP, a 5-HT precursor (Hounsgaard et al., 1988) or by L-DOPA (Conway et al., 1988), indicating loss of 5-HT2 and NAα1 receptor activity. In contrast to acute SCI, serotonergic and NA receptors are tonically activated on motoneurons below a complete, chronic transection, as evidenced by the reduction of PIC activation and spasms by cyproheptadine, a 5-HT2 and NAα1 receptor blocker (Figure 4A). Interestingly, the activation of these monoaminergic receptors occurs even though there is a marked disappearance of both 5-HT and NA fibers and their monoamines below the lesion within 7 and 9 days after injury (Carlsson et al., 1963; Anden et al., 1964). For example, in rat spinal cords there is a reduction in immunolabeling for 5-HT below the lesion 2 months after a complete transection (Figure 4B). As will be described below, the reactivation of 5-HT2 and NAα1 receptors on the motoneuron, and the recovery of PICs in chronic SCI, results from an increase in monoamine receptors that can be activated without the binding of 5-HT and NA. This “self-activation” of 5-HT2 and NAα1 receptors is one strategy the spinal cord uses to regain its lost excitability.
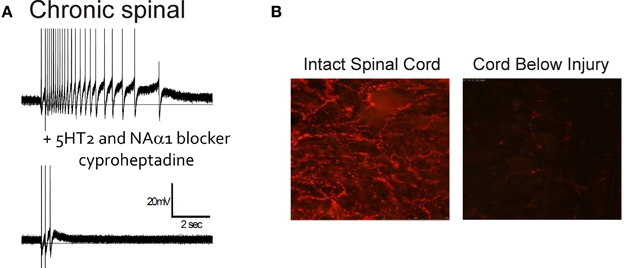
Figure 4. Endogenous activation of 5HT2/NAα1 receptors with little monoamines below injury. (A) Top trace: Intracellular recording from motoneuron of chronic spinal rat. Single-pulse, dorsal root stimulation evokes long-lasting plateau potential and self-sustained firing. Bottom trace: Blocking of 5-HT2 and NAα1 receptors with cyproheptadine eliminates PIC-mediated plateau potential and self-sustained firing, leaving only sensory-mediated activation of motoneuron. (B) Immunofluorescence imaging of residual serotonergic fibers (beaded red) labeled with Texas-Red in spinal cords above (left) and below (right) a complete spinal transection from a rat spinalized 2 months previously. (B) was taken from Murray et al. (2010).
Constitutively-Active 5-HT2 and NAα1 Receptors
5-HT2 and NAα1 receptors are coupled to a hetero-trimeric G protein which consists of an α, β, and γ subunit (Raymond et al., 2001; Heckman et al., 2003). Activation of GPCRs induces a conformational change in the receptor complex which results in the release of the α-subunit by facilitating the exchange of GDP for GTP. The dissociated α-subunit then acts to stimulate the β isoform of the enzyme phospholipase C (PLC), resulting in the hydrolysis of phosphatidylinositol bisphosphate into the secondary messengers inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 increases calcium mobilization via release of intracellular, IP3-regulated calcium stores and DAG activates the downstream protein kinase C (PKC) (Mizuno and Itoh, 2009) which subsequently phosphorylates and activates voltage-gated channels such as the Na and Ca channels mediating the PICs. An interesting characteristic of GPCRs, like the 5-HT2 and NAα1 receptors, is their ability to display constitutive receptor activity (Gether et al., 1997; Seifert and Wenzel-Seifert, 2002; Berg et al., 2005; Navailles et al., 2006). GPCRs exist in equilibrium between inactive (R) and active (R*) receptor states (Figure 5). In the active state, the receptor is coupled to its G-protein and can activate downstream signaling pathways, such as those which facilitate the motoneuron PIC. Typically receptors enter their active state only when the appropriate ligand binds to the receptor complex (ligand-activation). However, different isoforms of a receptor can also spontaneously enter their active state without activation by a ligand and this is termed constitutive receptor activity.
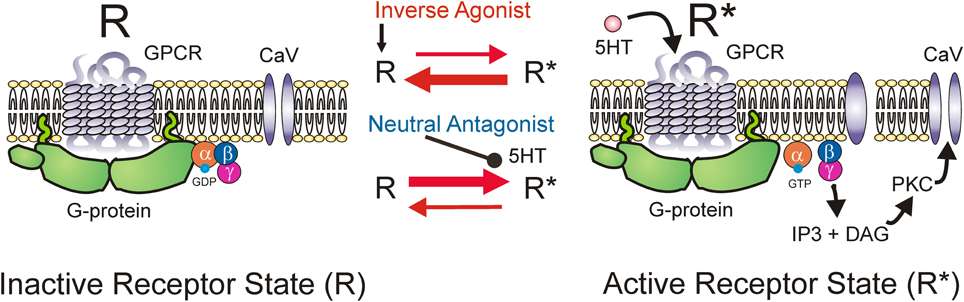
Figure 5. Mechanism of inverse agonists and neutral antagonists. Receptor state equilibrium: Inactive state (R, left panel) of G-protein coupled receptor (GPCR) where α, β, and γ subunits of the G-protein and downstream pathways are not activated. Active receptor state (R*, right panel) where the α-subunit is released by facilitating exchange of GDP for GTP. Dissociated α-subunit activates phospholipase C (PLC, not shown) resulting in the hydrolysis of phosphatidylinositol bisphosphate into the secondary messengers inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 increases calcium mobilization via release of intracellular, IP3-regulated calcium stores and DAG activates the downstream protein kinase C (PKC) which subsequently phosphorylates and activates voltage-gated channels such as the Na and Ca channels mediating the PICs.
5-HT2C and NAα1 receptors demonstrate constitutive receptor activity, as measured by basal IP3 levels and through pharmacological methods in human (HEK -293) and monkey (COS-7) kidney cells and in cultured mouse cortical neurons (Herrick-Davis et al., 2000; Rauser et al., 2001; Berg et al., 2005; Navailles et al., 2006; Chanrion et al., 2008). To examine if constitutive activity in 5-HT2 and NAα1 receptors mediates the recovery of motoneuron PICs in chronic injury, the effects of two types of antagonists, an inverse agonist and a neutral antagonist, on PIC-mediated LLR (spasms) has been examined, first in completely transected rats (Murray et al., 2010) and then in humans with SCI (D'Amico et al., 2013b). An inverse agonist stabilizes the receptor in its inactive resting state (R), preventing it from entering into its active state (R*). It also blocks other agonists from binding to the receptor and thus, inverse agonists block both constitutive receptor activity and conventional ligand-activation of the receptor (Figure 5; Westphal and Sanders-Bush, 1994; Herrick-Davis et al., 2000; Chanrion et al., 2008). In contrast, a neutral antagonist works by only blocking the ligand activation of the receptor. The 5-HT2 and NAα1 receptor inverse agonist, cyproheptadine, and the selective 5-HT2 receptor inverse agonist SB206553, both reduced PIC-mediated, LLR in chronically-spinalized rats (Figures 6C,D: red traces/bars) and had no effect on sensory-mediated, short-lasting reflexes (SLR). In contrast, the neutral antagonists methysergide and SB242084, which only block the ligand from activating the 5-HT2 and NAα1 receptors, had no effect on PIC-mediated responses (Figures 6B,D: blue traces/bars). Because the inverse agonist reduced 5-HT2 and NAα1 receptor activity and its downstream effects (e.g., reduction of PIC-mediated LLR) and the neutral antagonist had no effect, then the effects of the inverse agonists can be attributed solely to blocking receptors that were constitutively active. Furthermore, mRNA analysis revealed an upregulation in the INI isoform of the 5-HT2C receptor which displays the highest degree of constitutive receptor activity (Murray et al., 2010). Consistent with the mRNA findings, 5HT2C immunoreactivity is increased by 60% in motoneurons below a complete transection (Ren et al., 2013). Evidence for constitutively active NAα1 receptors was also found whereby the inverse agonists prazosin and WB4101 significantly reduced PIC-mediated responses in chronic spinal rats whereas the neutral antagonists methysergide (5-HT2/NAα1) and REC (NAα1) had no effect (Rank et al., 2011). Thus, following complete SCI in the chronic animal, the recovery of PIC activation in the motoneuron occurs as a result of the emergence of constitutive 5-HT2 and NAα1 receptor activity.
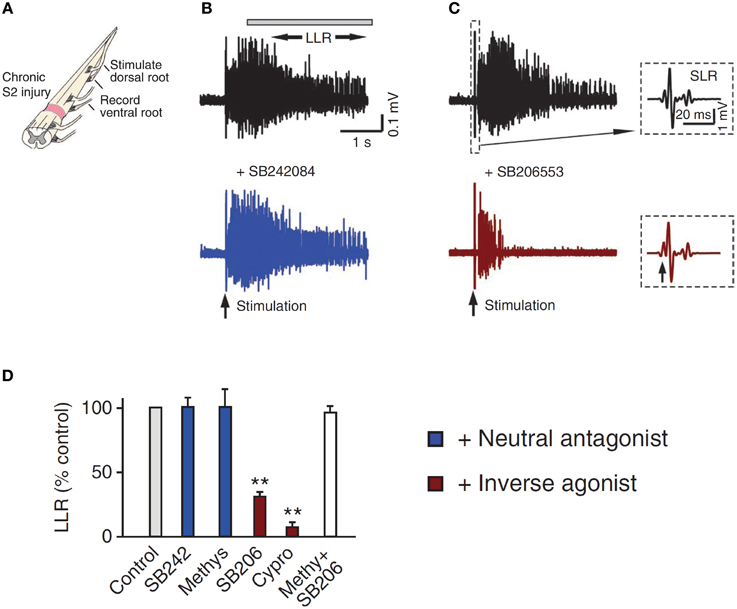
Figure 6. Constitutive receptor activity in chronic SCI rat. (A) In vitro ventral root recordings in response to single-pulse (3 × T) dorsal root stimulation in chronically spinalized (S2) rats. (B) PIC-mediated long-lasting reflex (LLR: 500–4000 ms after stimulation) before (black trace) and after (blue trace) application of the neutral antagonist SB242084. (C) Evoked LLR before (black trace) and after (red trace) application of the inverse agonist SB206553. Inset shows the sensory evoked, short-latency reflex (SLR: 10–40 ms) that is not mediated by the slow activating PICs or affected by the inverse agonist. (D) Group means of the LLR (expressed as a % of pre-drug values) after application of the neutral antagonists (blue), inverse agonists (red), and application of inverse agonist after the receptor is first blocked with the neutral antagonist (white) to render the inverse agonist ineffective. Taken from Murray et al. (2010). **p < 0.01.
To examine the emergence of constitutively-active monoaminergic receptors after SCI in humans, similar pharmacological methods have been employed in individuals with both motor incomplete (iSCI: AIS C&D) and motor complete (cSCI: AIS A&B) injury (D'Amico et al., 2013b). Oral administration of cyproheptadine, the inverse agonist to 5-HT2 and NAα1 receptors, also reduced PIC-mediated LLR in participants with incomplete SCI (Figure 7A) and had no effect on the sensory-mediated SLRs similar to the rat (D'Amico et al., 2013b). In these same participants oral intake of citalopram, a selective serotonin reuptake inhibitor, increased the LLR (Figure 7B), indicating that residual levels of serotonin were present below the injury and likely facilitated motoneuron PICs via ligand activation of the 5-HT2 receptors (see also Thompson and Hornby, 2013). Similar to participants with incomplete SCI, participants with motor complete injuries also displayed tonic activation of 5-HT2 and NAα1 receptors given that cyproheptadine also reduced PIC-mediated LLR (Figure 7C). However, oral intake of the neutral antagonist, chlorpromazine, had no effect on LLR (Figure 7D), even though it was effective in reducing PIC-mediated ΔF measures in uninjured controls having intact 5-HT and NA fibers (D'Amico et al., 2013b). Based on these two findings, participants with motor complete SCI appeared not to have residual levels of 5-HT and NA below the lesion so that PICs were solely facilitated by constitutive activity in 5-HT2 and NAα1 receptors, similar to the complete SCI model in rats. How the facilitation or control of these monoamine receptors may be used to promote motor recovery or control spasticity, respectively, will be discussed at the end of this review.
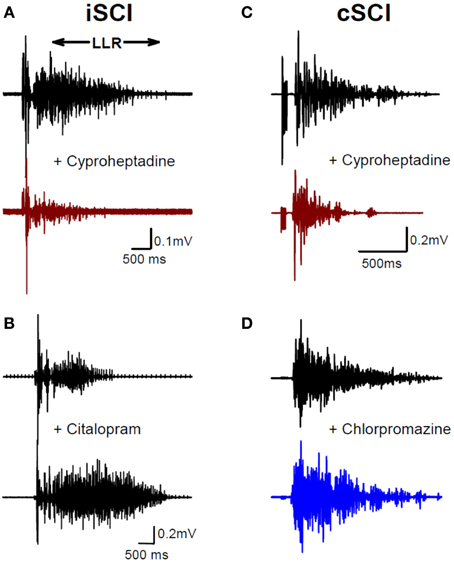
Figure 7. Constitutive receptor activity in chronic human SCI. (A) Inverse agonist cyproheptadine in a motor incomplete SCI participant (iSCI, AIS C). Unrectified surface EMG during long-lasting reflex (LLR) evoked in the TA muscle from medial arch stimulation before (top trace) and after (bottom trace) 8 mg of oral cyproheptadine. Note reduction in the presumably PIC-mediated long-lasting reflex (LLR: 500 ms after stimulation onwards). (B) Serotonin re-uptake inhibitor citalopram in iSCI (AIS C) participant. Long-lasting reflex (LLR) recorded in TA muscle before (top trace) and after (bottom trace) 20 mg of oral citalopram. (C) Same as in (A) but for 12 mg oral dose of cyproheptadine in motor and sensory complete SCI participant (cSCI, AIS A). (D) Effect of neutral antagonist chlorpromazine (12.5 mg) on PIC-mediated LLR in same cSCI participant. (A–C) taken from D'Amico et al. (2013b).
Changes in Sensory Transmission to Motoneurons After SCI
Emergence of Long-Duration EPSPs
In addition to adaptive changes in the motoneuron, the transmission of sensory inputs to the motoneuron is also affected following SCI. One of the main changes is the loss or reduction of early, inhibitory post-synaptic potentials (IPSPs) and the conversion to pure, prolonged EPSPs in response to brief, sensory stimulation (Baker and Chandler, 1987). As demonstrated in Figure 1, following acute or chronic SCI, a single pulse of electrical stimulation to a sacral dorsal root, which contains predominantly cutaneous afferents, produced a very long (~1 s) EPSP that was dependent upon the activation of NMDA channels (Bennett et al., 2001). The profile of this EPSP was revealed by blocking voltage-dependent conductances with cell hyperpolarization so that the synaptic activation profile of the motoneuron predominated (Li et al., 2004a). This 1-s EPSP provided a long enough depolarization of the motoneuron to activate the CaPIC, the latter requiring at least 500 ms of synaptic depolarization to activate (Moritz et al., 2007; Murray et al., 2011b). Thus, when the motoneuron PIC recovers after chronic SCI, the long EPSP can trigger plateau potentials and self-sustained firing that is unchecked due to the loss of descending and intrinsic inhibition of the motoneuron, resulting in the activation of involuntary muscle spasms.
The loss of IPSPs and conversion to pure EPSPs in response to cutaneomuscular stimulation is also evident in the activation profile of motoneurons (motor units) recorded in participants with chronic, incomplete SCI (Norton et al., 2008) using the peristimulus frequencygram (PSF) technique (Awiszus et al., 1991; Turker and Powers, 1999, 2003). To construct a PSF, the instantaneous firing rate of a tonically activated motor unit is plotted time-locked to a sensory stimulus (Figure 8). Because the firing rate of a motoneuron mainly reflects the net current reaching the soma (Baldissera et al., 1982; Powers et al., 1992), any modulation in firing rate from a sensory stimulus should reflect the underlying shape of the EPSP or IPSP (Turker and Powers, 1999, 2003). For example, the firing rate of a tonically active motor unit recorded from the TA muscle of an incomplete SCI participant increased above baseline for a duration of ~1 s in response to stimulating cutaneomuscular afferents from the medial arch of the foot. This was most likely due to an EPSP lasting for ~1 s, similar to that measured in motoneurons from chronically spinalized rats (Figure 1A). In a control participant (Figure 8B), the PSF only lasted for ~300 ms, with a pause in firing after the initial acceleration in rate and an interposed cluster of action potentials at firing rates near or below the mean background rate (gray circle: Figure 8B). The shape of this PSF was likely mediated by a 300 ms long EPSP that contained a brief, interposed IPSP. To demonstrate this, a current profile was injected into a hyperpolarized motoneuron from a chronic spinal rat (inset in Figure 8C) to produce the hypothesized PSP (gray trace in Figure 8C). When a steady current was injected into the motoneuron to produce tonic firing and the simulated current profile was then added, the resulting PSF (black dots in Figure 8C) was similar to the PSF recorded in the control participant (Figure 8B). Thus, it has been proposed that, unlike uninjured control participants, robust IPSPs from cutaneomuscular afferent stimulation are not activated in motoneurons of SCI participants and are replaced by prolonged EPSPs. These 1 s-long EPSPs produce a long enough depolarization of the motoneuron to activate CaPICs and trigger self-sustained firing, resulting in the activation of unchecked, involuntary muscle spasms.
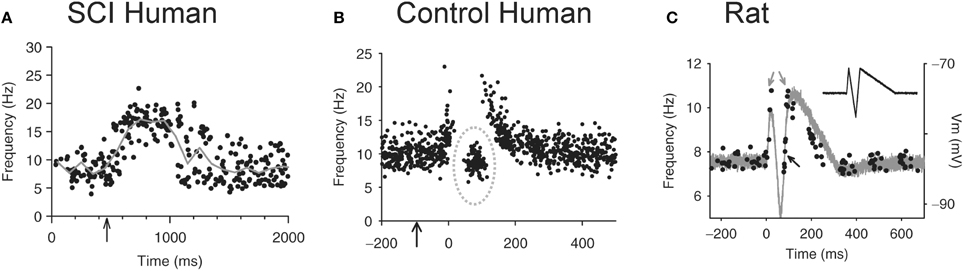
Figure 8. Peristimulus frequencygrams (PSF) in SCI and uninjured participants and in rat. (A) PSF of TA motor units recorded in incomplete SCI participant (AIS C) in response to stimulation of the medial arch of the foot (time marked by arrow, 0.2 ms pulse width, 3 pulses, 300 Hz, ~50 mA) while participant maintained tonic dorsiflexion. Mean rate indicated by solid gray line. (B) PSF recorded from uninjured control participant in response to same medial arch stimulation. Note shorter duration of response, pause and cluster of motor unit action potentials as marked by the dashed gray circle. (C) PSF (black dots) recorded in rat motoneuron that mimics the PSF obtained in the uninjured control participant from (B). The intracellular current injection profile required to obtain this PSF is shown in inset and the resulting Vm response (gray trace) to this injected profile in a hyperpolarized motoneuron. Taken from Norton et al. (2008).
The animal and human studies described above mainly examined changes in the transmission of cutaneous reflex pathways. As described next, transmission in other sensory pathways is also altered after SCI. Generally, transmission is increased in excitatory reflex pathways and reduced in inhibitory reflex pathways.
Pre-Synaptic Inhibition
Pre-synaptic inhibition effectively modulates the efficacy of afferent transmission to the motoneuron via primary afferent depolarizing interneurons (PAD INs, gray neurons in Figure 9A). Although pre-synaptic inhibition can occur at all afferent terminals, studies in humans have mainly focused on pre-synaptic inhibition of Ia afferents. The mechanisms of pre-synaptic inhibition involve the activation of fast, ionotropic γ-aminobutyric acidA (GABAA) receptors and slow, metabotropic GABAB receptors. Activation of GABAA receptors on primary afferent terminals produces an efflux of Cl− resulting in primary afferent depolarization and subsequently, reducing the size of the propagating action potential. This ultimately results in less calcium influx and less neurotransmitter release (Rudomin and Schmidt, 1999; Seki et al., 2003; Rudomin, 2009). In contrast, G-protein coupled GABAB receptors directly modulate the calcium channels in afferent terminals, resulting in less calcium influx and subsequently, less neurotransmitter release (Rudomin and Schmidt, 1999; Castro et al., 2006; Soto et al., 2006).
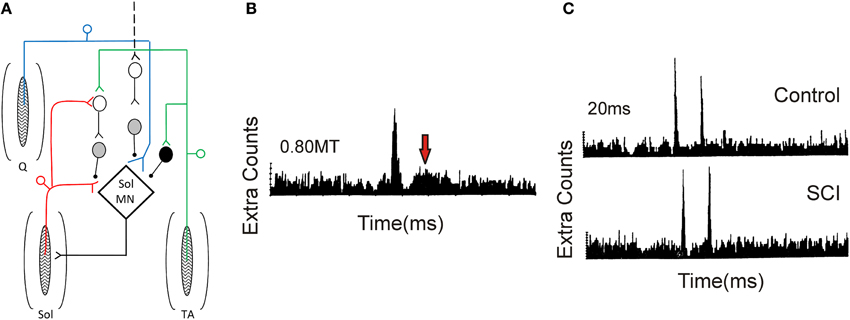
Figure 9. Pre-synaptic and reciprocal inhibition in humans. (A) Pre-synaptic and reciprocal inhibitory pathways. Red trace: Monosynaptic Ia pathway to soleus (Sol, lower pathway) and homonymous activation of PAD interneurons (upper pathway). PAD interneurons marked by gray circles. Blue trace: Heteronymous, monosynaptic facilitation of soleus motoneurons by quadriceps (Q) Ia afferents that are tonically inhibited by PAD Ins, the latter activated by descending pathways (dashed line). Green trace: Reciprocal inhibitory pathway from tibialis anterior (TA) afferents onto soleus Ia reciprocal inhibitory interneuron (black circle) and onto soleus PAD interneuron via second order interneuron (D1/D2 inhibition). (B) PSTH obtained from a SCI participant in response to low-threshold electrical stimulation of the posterior tibial nerve (bottom red pathway in A). Note second peak in PSTH (arrow). (C) Decrease of second PSTH peak in response to paired activation (20 ms ISI) of posterior tibial nerve in uninjured control (top trace) but not SCI (bottom trace) participant indicative of reduced pre-synaptic inhibition (top red pathway in A). (B,C) were modified from Mailis and Ashby (1990).
Mailis and Ashby (1990) examined transmission from Ia afferents to motoneurons after SCI by recording motor unit action potentials in the soleus muscle in response to low-intensity (~0.8 xMT) posterior tibial nerve stimulation (lower red path in Figure 9). In only the most spastic SCI participants, the peak amplitude of the post-stimulus time histogram (PSTH) at the Ia monosynaptic latency was augmented (first peak in Figure 9B). In addition, there was the emergence of a second peak 11–15 ms later (at arrow in Figure 9B), suggesting the emergence of an oligosynaptic, group I excitatory pathway. To examine if this enhanced response was due to a reduction in pre-synaptic inhibition on the Ia afferent terminals, paired electrical stimulation of homonymous afferents from the soleus muscle was used (Birnbaum and Ashby, 1982; Lev-Tov et al., 1983). A prior conditioning stimulus (0.8 xMT) was applied to the posterior tibial nerve to activate PAD INs (upper red pathway in Figure 9), which in turn pre-synaptically inhibited Ia afferents with monosynaptic connections onto soleus motoneurons (lower red pathway). Here, the first peak of the PSTH was produced by the arrival of Ia inputs from the first conditioning volley and the second peak from the test volley (Figure 9C). In controls, the second peak in the PSTH was reduced in comparison to the first peak at interstimulus intervals (ISIs) of 20 and 50 ms, likely due to pre-synaptic inhibition of Ia afferents from the first conditioning volley (upper red pathway). Reduction of the second peak was smaller or absent in the SCI participants (Figure 9C), indicating a reduction in homonymous, pre-synaptic inhibition (Mailis and Ashby, 1990).
Reduction of pre-synaptic inhibition from heteronymous nerves, using the techniques of Hultborn et al. (1987) and Meunier and Pierrot-Deseilligny (1989), was also examined after SCI. Excitatory monosynaptic inputs from quadriceps Ia afferents (blue pathway in Figure 9A) can facilitate the soleus H-reflex (lower red pathway) but the amount of this facilitation is modulated by the amount of pre-synaptic inhibition that is tonically present on the quadriceps Ia afferent terminals, likely from descending pathways (marked as black dashed line, Figure 9A). Heteronymous facilitation of the soleus H-reflex by quadriceps Ia afferents (4–5 xMT, ~80 ms ISI) was greater in SCI participants compared to uninjured controls, indicating reduced levels of background pre-synaptic inhibition after SCI (Faist et al., 1994). Pre-synaptic inhibition of soleus Ia afferent terminals through PAD interneurons can also be activated by afferents supplying the antagonist tibialis anterior muscle (TA, Figure 9A, green pathway). In uninjured controls, conditioning the TA afferents produces two phases of inhibition onto soleus motoneurons: an early D1 inhibition at ISIs of 10–25 ms and a late, longer-lasting D2 inhibition at ISIs of 70–200 ms (Mizuno et al., 1971). A pre-synaptic site for the inhibition was proposed since the same conditioning volleys did not suppress cortical motor evoked potentials (MEPs) in the soleus, thereby excluding post-synaptic inhibitory mechanisms (Capaday et al., 1995; Faist et al., 1996). When an H-reflex in the flexor carpi radialis (FCR) muscle was conditioned by a prior stimulation to the antagonist radial nerve (0.95 xMT, 13 ms ISI), there was no early D1 inhibition in the agonist H-reflex in tetraplegic participants, indicating reduced pre-synaptic inhibition of Ia afferents in the median nerve (Aymard et al., 2000). In summary, pre-synaptic inhibition appears to be significantly reduced after SCI, likely resulting, in part, from decreased descending facilitation of PAD INs (Figure 9A). However, there is no correlation between the reduction in pre-synaptic inhibition and Ashworth scores of spasticity (Faist et al., 1994).
Reciprocal Inhibition
Reciprocal Ia inhibition is a disynaptic pathway through Ia interneurons (black neuron, Figure 9A) that is activated by Ia afferents from the agonist (e.g., TA) muscle and inhibits motoneurons of the antagonist (e.g., soleus) muscle. Activation of Ia inhibitory interneurons ensures that antagonist muscles remain relaxed during contraction of the agonist muscle, preventing undesired co-contractions. Reciprocal inhibition can be measured by conditioning a test H-reflex evoked in the antagonist muscle with a low-threshold stimulation (0.8–1.0 xMT, ISIs 2–4 ms) to the nerve supplying the agonist muscle (Kots and Zhukov, 1971; Mizuno et al., 1971; Crone et al., 1987). In SCI participants, suppression of the soleus H-reflex by a prior conditioning stimulation of the common peroneal nerve supplying the TA muscle was greatly reduced compared to control participants and, in some cases, replaced by reciprocal facilitation (Crone et al., 2003). Although it is apparent that reduced reciprocal inhibition may contribute to increased motoneuron excitability after SCI, there is no correlation between the strength of reciprocal inhibition and the degree of spasticity, as measured by the Ashworth scale (Crone et al., 1994; Nielsen et al., 2007).
Post-Activation Depression
Post-activation (or rate-dependant) depression (PAD2) of H-reflexes occurs when the same (homonymous) Ia afferent pathway is repetitively activated (Crone and Nielsen, 1989). PAD2 produces a strong, long-lasting (~10 s) inhibition of H-reflexes and motoneuron activity (Magladery et al., 1952; Rothwell et al., 1986; Crone and Nielsen, 1989; Hultborn et al., 1996). The mechanism of PAD2 is unknown and may include decreases in neurotransmitter release at the Ia-motoneuron synapse (Hultborn et al., 1996; Kohn et al., 1997) or frequency-dependent facilitation of inhibitory spinal interneurons (Boulenguez et al., 2010). The level of PAD2 can be measured by examining the gradual decrease in H-reflex size when trains of reflexes are elicited at incrementing frequencies between 1 and 10 Hz (Lloyd and Wilson, 1957). PAD2 is reduced in individuals with SCI (Schindler-Ivens and Shields, 2000) and can be increased by a single bout of treadmill training (Trimble et al., 1998). Additionally, PAD2 can be measured by examining the reduction of H-reflexes by homonymous tendon vibration because the vibration repeatedly activates the same Ia afferents recruited during the H-reflex. Tendon vibration failed to attenuate H-reflexes in participants with chronic SCI compared to uninjured controls, again indicating reduced PAD2 (and/or classical pre-synaptic inhibition) as a possible contributor to enhanced sensory transmission and the triggering of muscle spasms after SCI (Calancie et al., 1993).
Flexor Withdrawal Reflexes
Flexor reflex afferent (FRA) pathways include group II, joint and cutaneous afferents that activate ipsilateral flexors and contralateral extensors and inhibit ipsilateral extensors. In uninjured participants, noxious cutaneous stimulation produces an excitatory, short-latency (~50–70 ms) flexor withdrawal response with a duration of ~50 ms (Pedersen, 1954; Shahani and Young, 1971). In participants with motor complete SCI, this short latency response is not present and is replaced by a long-latency (>100 ms) excitation of flexors at both high and low intensities of stimulation (Roby-Brami and Bussel, 1987). A long-latency FRA response can also be obtained in uninjured participants but only at high stimulation intensities (Shahani and Young, 1971). In spinalized cats the early and late FRA responses are produced from distinct pathways whereby the early FRA pathway tonically inhibits the late FRA pathway. This inhibition can be released by the application of L-DOPA and NA, resulting in the emergence of long FRA responses that are similar to those present in individuals with complete SCI (Anden et al., 1964, 1966a,b; Jankowska et al., 1967; Lundberg, 1979). The development of constitutive NAα1 receptor activity on interneurons in the FRA pathway, which has been shown to occur on motoneurons (Rank et al., 2011), may contribute to the unmasking of long FRA reflexes after motor complete SCI in humans.
Mechanisms for Reduced Spinal Inhibition After SCI
The excitability of several inhibitory spinal pathways is reduced after SCI and likely contributes to the prolonged EPSPs that readily trigger PICs and spasms. As discussed below, inhibition and the regulation of excitatory sensory pathways is influenced by several factors.
Depolarization of Chloride Reversal Potential
The strength of IPSP activation in neurons is modulated by the activity of a potassium-chloride cotransporter (KCC2, Figure 10A) which removes chloride from the neuron in exchange for potassium to maintain the chloride equilibrium potential below rest at ~75 mV (Price et al., 2005). When GABAA and glycine receptors are activated, Cl− flows into the neuron to produce IPSPs due to the low intracellular concentration of Cl−. Following SCI, there is a downregulation and decreased membrane insertion of KCC2 (Boulenguez et al., 2010), resulting in an increase in intracellular Cl− concentration and an ~10 mV depolarization of the Cl− reversal potential in 6–8 day old rats (Figure 10B). In adult rats, the depolarization of the Cl− reversal potential is less at ~5 mV (Murray et al., 2011b). The 10 mV depolarization of the Cl− reversal potential to above the resting membrane potential in neonates subsequently results in a switch from GABAA and glycine-mediated inhibition to excitation because chloride moves out of the cell rather than into the cell during activation of the receptors (Boulenguez et al., 2010). Such a reversal was not seen in adult rats but the 5 mV depolarization of the Cl− reversal potential to near rest likely shifts the balance of EPSP activation over IPSP activation after SCI, as shown for the PSF profiles in the human (Figure 8; Norton et al., 2008). Functionally, the reduction of KCC2 activity results in a decrease of rate-dependent depression (or PAD2) of the H-reflex in rats (Boulenguez et al., 2010) and may mediate the reduction of other spinal inhibitory networks such as those activated during reciprocal inhibition and cutaneomuscular reflexes. Interestingly, rate-dependent depression (or PAD2) of the H-reflex can be restored via activation of TrkB receptors with BDNF after SCI (but not in intact animals), which also produces an upregulation of the KCC2 cotransporter (Boulenguez et al., 2010). Activation of 5-HT2A receptors on the motoneuron, via a calcium-independent PKC pathway, also restores KCC2 expression and rate-dependent depression of the H-reflex after SCI in rodents (Bos et al., 2013). Thus, strategies which enhance KCC2 activity may restore intrinsic inhibition to the spinal cord to reduce spasticity after SCI. The specific 5-HT2A receptor agonist used in this study (TCB-2) is not yet available for human use and it is likely that other 5-HT2A receptor agonists (e.g., DOI) would have hallucinogenic effects (Sadzot et al., 1989).
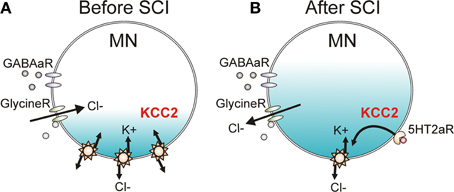
Figure 10. KCC2 cotransporter and chloride equilibrium before and after SCI. (A) A potassium chloride cotransporter (KCC2) removes chloride (Cl−) from the motoneuron (MN) in exchange for potassium (K+) to maintain Cl− equilibrium potential below resting membrane potential, allowing Cl− influx and MN hyperpolarization during activation of GABA and Glycine receptors (R). (B) Downregulation of KCC2 expression in motoneurons after SCI increases intracellular Cl− concentration, depolarizing Cl− equilibrium potential to above rest. This produces efflux of Cl− and depolarization of MN during activation of GABA and Glycine receptors. Activation of 5-HT2A receptors increases cell membrane expression of KCC2 after SCI to restore endogenous inhibition.
Disinhibition from Decreased Monoamine Receptor Activation
In addition to facilitating motoneuron PICs, serotonin and noradrenaline also regulate sensory transmission pre- and post-synaptically via the activation of Gq and Gi-coupled receptors. For example, activation of Gq-coupled 5-HT2 and NAα1 receptors on Ia and Ib inhibitory interneurons facilitates Ia reciprocal and Ia/Ib non-reciprocal inhibition (Jankowska et al., 2000; Hammar and Jankowska, 2003). Activation of Gi-coupled 5-HT1 and NAα2 receptors on the terminals of group I/II muscle, skin and high threshold pain afferents and on excitatory interneurons, such as propriospinal and group II interneurons, reduces transmission in these pathways (Jankowska et al., 1993, 1994; Rekling et al., 2000; Millan, 2002; Yoshimura and Furue, 2006; Jordan et al., 2008). Because there is a dramatic loss of serotonin and noradrenaline below a SCI, recent experiments have examined the effects of administering specific 5-HT1 and NAα2 receptor agonists to restore control over sensory afferent transmission to the motoneuron. As mentioned previously, a single-shock stimulation to a dorsal root produces a 1-s long EPSP as revealed during cell hyperpolarization (bottom trace, Figure 11Ai). This EPSP contains both a short-latency, short-duration component (short EPSP) and a longer-latency, longer-lasting component (long EPSP). At rest (−72 mV), the long EPSP produced by the dorsal root stimulation activates a CaPIC to produce a plateau potential and self-sustained firing or long-lasting reflex response (LLR, top trace Figure 11Ai). When the specific agonist to the 5-HT1B/D receptor, zolmitriptan, was applied the long (but not short) EPSP was eliminated (bottom trace, Figure 11Aii) so that at rest, a plateau potential and LLR was not triggered (top trace, Figure 11Aii). Testing with multiple 5-HT1 receptor agonists/antagonists showed that sensory transmission is mainly produced by the specific activation of 5-HT1B and 5-HT1F receptors (Murray et al., 2011b). Application of an agonist to the NAα2 receptor, clonidine, also reduced the long sensory-evoked EPSP to prevent triggering of plateaus/LLRs (Rank et al., 2011). Both of these findings indicate that facilitation of Gi-coupled 5-HT1 and NAα2 receptors can reduce transmission of excitatory sensory pathways after SCI in rats.
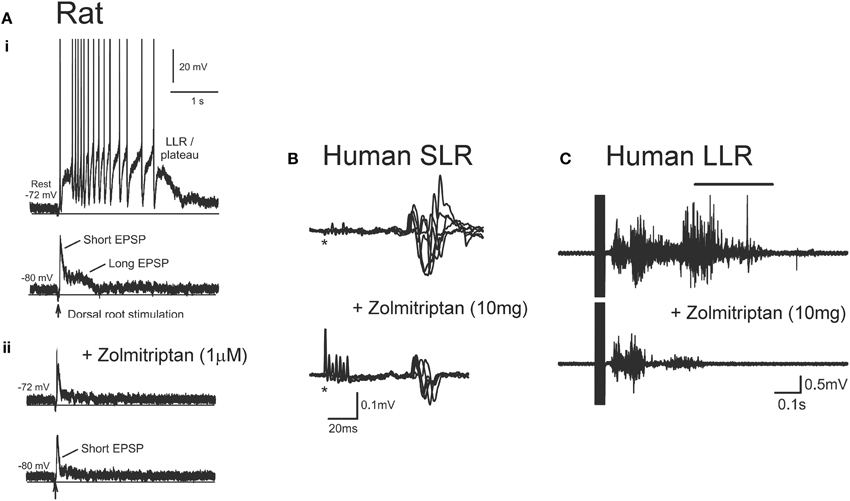
Figure 11. Effect of zolmitriptan on sensory transmission in SCI rat and human. (A) Effects of zolmitriptan in chronically injured rat motoneurons. (i)Top trace: Intracellular recording from chronic spinal rat motoneuron at rest (−72 mV) where single-pulse dorsal root stimulation produces PIC-mediated long-lasting reflex (LLR). Bottom trace: same motoneuron hyperpolarized to −80 mV to reveal short and long EPSP in response to dorsal root stimulation. (ii) Recording from same motoneuron in (i) at rest (top trace) and during hyperpolarization (bottom trace) after application of the 5HT1B/D receptor agonist zolmitriptan. Note elimination of the long-duration EPSP which prevents the triggering of the PIC-mediated long-lasting reflex. Taken from Murray et al. (2011b). (B) Unrectified EMG showing short- lasting reflex (SLR: first 50 ms of reflex response) recorded from the TA muscle in a single SCI participantbefore (top trace) and after (bottom trace) 10 mg oral zolmitriptan in response to medial arch stimulation. *Marks time of stimulation. (C) Unrectified TA EMG before (top trace) and after (bottom trace) 10 mg zolmitriptan in a single SCI participant in response to medial arch stimulation. Black bar denotes long-lasting reflex response (LLR: 500 ms after stimulation and beyond). (B,C) were taken from D'Amico et al. (2013a).
Similar findings have been obtained in humans with motor complete SCI where oral intake of zolmitriptan (10 mg dose) suppresses the activation of (SLR, Figure 11B). The reduction of these SLRs resulted in reduced triggering of the LLR responses or spasms (Figure 11C), similar to the findings in the rat. The reduction in spasms was likely not mediated by a reduction in plateau potentials given that zolmitriptan does not affect the Na or CaPIC (Murray et al., 2011b). The mainly monosynaptic H-reflex was also reduced by zolmitriptan (D'Amico et al., 2013a). The same dose of zolmitriptan had similar effects on the H-reflex in uninjured control participants indicating that despite the decreased levels of serotonin below the motor-complete injury, the 5-HT1 receptors were not supersensitive. A tonic elevation in the transmission of sensory inputs after SCI suggests there is a low or absent level of basal 5-HT1 and NAα2 receptor activity below an injury. As such, it is unlikely that Gi-coupled 5-HT1 and NAα2 receptors display functional levels of constitutive receptor activity, unlike the Gq-coupled 5-HT2 and NAα1 receptors on motoneurons. Understanding why 5-HT1 and NAα2 receptors do not become constitutively active may help devise new strategies to control sensory transmission and spasticity after SCI.
Additional mechanisms producing enhanced sensory transmission to motoneurons after SCI are reviewed in Roy and Edgerton (2012) and include the suggestion by Kitzman (2005) that a decrease in dendritic number, without a subsequent increase in voltage-gated channels, may also increase the excitability of motoneurons after SCI. Moreover, elevations in motoneuron vesicular glutamate transporter 2 (2-IR), but not the 1-IR transporter on myelinated primary afferents, suggests that polysynaptic glutamatergic inputs from interneurons (but not from monosynaptic pathways) are elevated after SCI (Kitzman, 2006, 2007). In addition to neuronal changes, overexpression of glutamate receptor 1 (GluR1) on astrocytes has also been implicated in exacerbating sensory-evoked spasms in an ischemic model of rat SCI (Hefferan et al., 2007).
Treatments for Spasticity and Recovery of Motoneuron Function
When involuntary muscle activity leads to impairments in function, difficulties in hygiene, skin breakdown and/or pain, treatment is warranted. As discussed above, spasticity is caused by adaptations to the muscle and spinal circuits as a result of a disruption in descending activation of the spinal cord. Most treatment strategies for spasticity target these secondary adaptations with limited success. Strategies that aim to fix the primary cause of spasticity by enhancing functional descending and/or peripheral activation of the spinal cord, in both motor complete and incomplete SCI, may prove to be more efficacious.
Current Pharmacological Treatments
Current pharmacological treatments for spasticity aim to suppress spinal cord or muscle activity. Three main sites are targeted to reduce: (1) sensory transmission to motoneurons, (2) motoneuron excitability and (3) muscle contractions (Figure 12). Unfortunately when taken orally, these medications are not specific to the spinal cord or muscle and the majority produce some generalized depression of the CNS, resulting in altered mental states such as drowsiness and dizziness and in generalized weakness (Satkunam, 2003). For many of these agents long-term, high-dose use can also result in physiologic dependence (Simon and Yelnik, 2010) such that sudden withdrawal can result in serious consequences such as seizures (Grosset and Grosset, 2004). The most commonly prescribed agent for spasticity is baclofen, a GABAB agonist which acts pre-synaptically (site 1, Figure 12) to reduce the amount of excitatory neurotransmitter released into synapses (Curtis et al., 1997; Li et al., 2004b). Although oral baclofen can reduce spasticity, it has detrimental effects on walking in patients with incomplete SCI, where other antispastics such as clonidine and cyproheptadine do not (Wainberg et al., 1986, 1990; Norman et al., 1998; Barbeau and Norman, 2003). Long-term use of oral baclofen also exacerbates the weakness and fatigability of motor units (Thomas et al., 2010). In cases of severe spasticity where patients are refractory to other treatments, baclofen is applied to the cerebrospinal fluid surrounding the spinal cord via an intrathecal pump (Lazorthes et al., 1990; Stempien and Tsai, 2000; Krach, 2001). Although intrathecal delivery reduces systemic side effects, it has a high rate (1%/month) of serious complications such as catheter obstruction which results in sudden withdrawal of baclofen and requires surgical intervention to repair (Draulans et al., 2013). Similar to baclofen, tizanidine (and clonidine) enhances pre-synaptic inhibition but via activation of NAα2 receptors on primary afferents and excitatory interneurons (site 1, Figure 12) (Davidoff, 1985; Krach, 2001). In one of the largest studies to date (78 SCI individuals), tizanidine reduced Ashworth scores by 37% in comparison to placebo (4%), with 81% of individuals reporting side effects (54% placebo) and no change in activities of daily living (Nance et al., 1994). Zolmitriptan also reduces sensory transmission by binding to 5-HT1B receptors (site 1, Figure 12) (Murray et al., 2011b) but is not used as an oral anti-spastic given that regular use can induce headaches (Limmroth et al., 1999) and it is contraindicated in people with cardio-, cerebro-, or peripheral vascular disease (Peterlin and Rapoport, 2007). Diazepam, an allosteric modulator of the GABAA receptor, both pre and post-synaptically (site 4) (Polc et al., 1974; Sieghart, 1994), and gabapentin, a GABA analog with unknown mechanism of action (Taylor, 1994; Sills, 2006) are also used as anti-spastics in SCI, but as with most of the under-powered studies in SCI, there is no quantitative evidence for a significant clinical effect (Taricco et al., 2006).
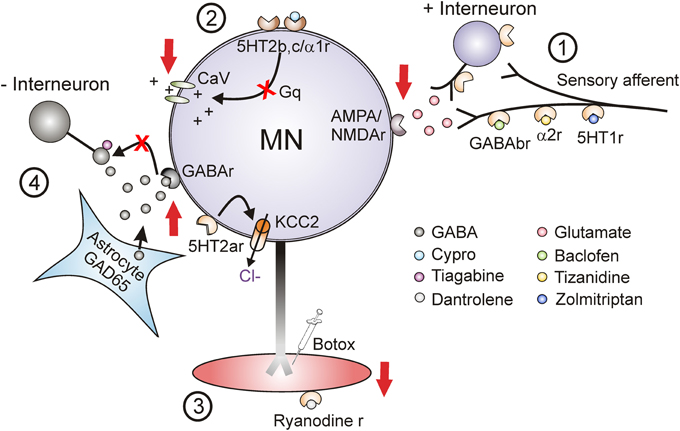
Figure 12. Target sites for current and future antispastic treatments. Pre-synaptic (1), motoneuron (2), muscle (3), and novel (4) sites for antispastic treatments. Site 1: GABAB, NAα2, and 5-HT1 receptors (r) located on pre-synaptic terminals or on interposed excitatory interneurons can be activated by baclofen, tizanidine, and zolmitriptan, respectively, to reduce excitatory glutamatergic release and activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors located on the motoneurons. Site 2: 5-HT2 and NAα1 receptors on the motoneuron with constitutive or ligand activation facilitate downstream voltage-gated calcium channels (CaV) mediating PICs via Gq protein-coupled pathways. Inverse agonists, such as cyproheptadine, switch the 5HT2 and NAα1 receptors to their inactive state, reducing activity in the Gq pathway and lessening activation of PICs and consequently muscle spasms. Site 3: Botulinum toxin (Botox®) injection directly into spastic muscles or activation of ryanodine receptors by dantrolene sodium (dantrolene) reduces spasticity by targeting the muscle site directly. Site 4: Spinal injection of the HIV1-CMV-GAD65 lentivirus increases GAD65 gene expression and GABA release from astrocytes. Systemic administration of tiagabine, a GABA reuptake inhibitor, increases amount of enhanced GABA in synapse to activate pre- and post-synaptic GABA receptors to reduce motoneuron excitability. Activation of 5-HT2A receptors increases cell expression of KCC2 cotransporter to reduce intracellular concentration of Cl− and increase endogenous motoneuron inhibition. Adapted from D'Amico et al. (2013a).
The only currently used antispastic that directly affects the excitability of the motoneuron (site 2, Figure 12) is cyproheptadine (Nance, 1994; Gracies et al., 1997); however, it lacks regulatory approval for this use. Cyproheptadine is as effective in reducing excessive muscle activity (17% reduction in Ashworth scores) compared to clonidine (22%) and baclofen (22%) (Nance, 1994), although this was only tested in 25 participants. Associated with the anti-histamine and anti-cholinergic properties of cyproheptadine were side effects such as CNS depression, dry mouth, appetite stimulation and thus, the potential for weight gain (Gracies et al., 1997). Despite these side effects, at low doses (12 mg/day) cyproheptadine decreased involuntary muscle spasms and clonus to improve residual walking function in participants with incomplete SCI (Wainberg et al., 1990). The development of more specific inverse agonists to 5-HT2/NAα1-receptors with fewer side effects than cyproheptadine may lead to better oral control of involuntary muscle spasms and clonus in SCI.
Reduction of spasticity by directly targeting the muscle (site 3, Figure 12) is achieved with botulinum toxin injected into specific muscles and with oral dantrolene sodium (Satkunam, 2003). Botulinum toxin, which blocks the release of acetylcholine at the neuromuscular junction, allows a more localized reduction in spasticity to injected muscles, but comes at the cost of further reducing muscle strength (Das and Park, 1989a,b; Simpson, 2004). Botulinum toxin produced stable improvements in upper-extremity function, hygiene and decreased pain in ~65% of individuals with SCI (Marciniak et al., 2008). However, the duration of effect is only 2–4 months, requiring repeated injections which may further increase the amount of non-contractile tissue within the target muscle (Fortuna et al., 2013). Dantrolene sodium is an oral antispastic that affects the excitation-contraction coupling in skeletal muscles by decreasing the release of calcium from the sarcoplasmic reticulum through blocking the ryanodine receptor (Britt, 1984; Krause et al., 2004; Lanner et al., 2010). In individuals with SCI, dantrolene sodium effectively reduced torque in response to passive stretch of the muscle (Herman et al., 1972; Glass, 1974; Glass and Hannah, 1974; Meyler et al., 1981), decreased reflex hyperexcitability and clonus and improvements in activities of daily life (Meyler et al., 1981). While CNS depression may occur with dantrolene sodium, it is less severe than other oral antispastics (Watanabe, 2009; Lapeyre et al., 2010). Despite the reduction in reflexes, dantrolene sodium is not commonly prescribed for SCI spasticity likely because it produces generalized muscle weakness (Adams and Hicks, 2005) and can result in hepatotoxicity (Gracies et al., 1997).
New Avenues for Pharmacological and Gene Therapy Treatments
A common disadvantage of all oral treatments is their action on supraspinal centers. Therefore, the ability to direct drug therapies specifically to the spinal cord is very desirable. Recently, a spinally targeted strategy was proposed by Marsala's group (site 4, Figure 12) where, in a rat model of ischemic SCI, the efficacy of a GABA uptake inhibitor tiagabine, applied systemically, was increased while localizing its effects to the lumbar spinal cord (Kakinohana et al., 2012). To do this, a lentivirus applied to the lumbar spinal cord locally upregulated GAD65 gene expression in astrocytes. Because of the enhanced production of GABA, the previously ineffective dose of tiagabine was now able to reduce hindlimb stretch reflexes without producing side effects such as CNS depression (Kakinohana et al., 2012). The use of spinally-targeted gene therapies opens up several different avenues for anti-spastic treatments with numerous potential targets, such as the constitutively-active isoforms of the 5-HT2C receptor or the KCC2 transporter. Despite the promise of this strategy and its success in treating other disorders such as Parkinson's disease (Bjorklund et al., 2000; Kaplitt et al., 2007) and blindness (Dinculescu et al., 2005; Buch et al., 2008; Stieger et al., 2011), there are potential drawbacks to the use of gene therapies including small packaging capacity, the risk of insertional mutagenesis, immune responses against a vector and difficulty in supplying sufficient concentrations of the selected genes needed for human clinical trials (Monahan and Samulski, 2000). Thus, further research into this promising application is needed.
As discussed above, novel targets for reducing spasticity, such as the application of agonists to the 5-HT1B/F receptors, like zolmitriptan, to reduce sensory transmission (site 1, Figure 12) and 5-HT2A receptors to enhance KCC2 activity (site 4, Figure 12) are not suitable as oral antispastics due to their potentially harmful effects at supraspinal and cardiovascular centers. However, if these agonists were delivered intrathecally to the spinal cord, reductions in spasticity may be achieved over the long-term. It would be worthwhile examining in animal models of SCI if long-term use of intrathecal 5-HT1/2A agonists is effective and if their symptoms of sudden withdrawal are less severe than with baclofen. Additionally, it would be worth testing the efficacy of intrathecal application of cyproheptadine which exerts its effects directly on the motoneuron by reducing 5-HT2 and NAα1 receptor activity. Because cyproheptadine reduces the excitability of the motoneuron directly, it may be used to treat spasticity for CNS disorders of varying etiology, such as amyolateral sclerosis, multiple sclerosis, stroke, traumatic brain injury and cerebral palsy. Better technologies for intrathecal or spinal drug delivery will open possibilities for the application of these newly discovered antispastics. However, as discussed above, any agents that suppress spinal cord excitability may further promote the compensatory changes within the spinal cord that increases its excitability, such as increases in constitutive monoamine receptor activity and sensory transmission and decreases in KCC2 activity. In fact, the development of tolerance to antispastic drugs may, in part, be produced by increasing these compensatory mechanisms by pharmacological suppression of the spinal cord and muscle. Thus, strategies which enhance functional synaptic activation of the spinal cord, such as the physical treatments described below, may be the best solution to curb hyperexcitabilty of the spinal cord after injury.
Physical Treatments
Physical treatments can successfully reduce spasticity and they avoid the negative side effects associated with pharmacological treatments. The first line of prevention and treatment of spasticity by physical treatments targets the maintenance of joint range of motion through strategies such as stretching (Bovend'Eerdt et al., 2008), range of motion exercises (Harvey et al., 2009a), static weight bearing (Odeen and Knutsson, 1981), and passive cycling (Rayegani et al., 2011). All of these techniques have produced significant reductions in measures of spasticity such as the Ashworth score and the pendulum test. However, these techniques have produced inconsistent results on range of motion and there is sparse evidence regarding improvements in motor function (Harvey et al., 2009b), though reductions in co-contraction and improved gait from stretching have been reported (Bovend'Eerdt et al., 2008). Physical treatments produced a reduction in atrophy-related changes in muscle architecture (Willoughby et al., 2000), reduced passive stiffness (Guissard and Duchateau, 2004), reduced Ia transmission as assessed by Hmax/Mmax ratios (Guissard and Duchateau, 2004; Rayegani et al., 2011) and normalization of motoneuronal excitability as assessed by F-wave/M-wave ratios (Rayegani et al., 2011) to potentially contribute to the observed reductions in spasticity.
In participants with incomplete SCI who can perform voluntary contractions, therapies initiated to improve motor function can also reduce spasticity. For example, body-weight supported treadmill training consistently reduces ankle clonus across various studies, in addition to reducing flexor and extensor spasms and co-contraction and increasing PAD2 (Gorassini et al., 2009; Adams and Hicks, 2011; Manella and Field-Fote, 2013; Knikou and Mummidisetty, 2014). These measures of spasticity are also associated with improvements in motor control such as increases in voluntary motor strength, walking speed, and self-reported mobility. In a recent pilot study, resistance training at maximal intensity also improved walking and balance with a trend to decrease the modified Ashworth score (Jayaraman et al., 2013). Other forms of therapy involving voluntary contractions include neural facilitation, hippotherapy and hydrotherapy, and all have resulted in reduced measures of spasticity (Hsieh et al., 2012). For example, 10 weeks of hydrotherapy (3 × 's per week), in addition to passive range of motion exercises, significantly decreased the Ashworth and Penn spasm frequency scores and resulted in a significant decrease in the dosage of oral baclofen taken compared to passive exercises alone (Kesiktas et al., 2004). Increases in the strength of spared corticospinal pathways have also been produced from physical treatments that incorporate voluntary contractions, such as treadmill training (Thomas and Gorassini, 2005). Thus, reductions in spasticity from these active physical treatments that provide functional, patterned synaptic activation of the spinal cord may result from enhanced activation of spinal inhibitory circuitry by strengthened descending motor pathways (Zewdie et al., 2011) and/or from decreases in sensory transmission by enhanced activation of the dorsal horn by descending monoaminergic pathways (Murray et al., 2011b; Rank et al., 2011). In addition, the increased activation of the spinal cord by synaptic inputs during active treatments may increase spinal BDNF to facilitate KCC2 activity and subsequently, intrinsic motoneuron inhibition (Gomez-Pinilla et al., 2007; Boulenguez et al., 2010).
Electrical stimulation of the spinal cord, peripheral nerves and muscles has also been used to treat spasticity and/or motor impairment. A single session of stimulation that results in muscle contractions consistently demonstrates acute reductions in spasticity, which is thought to be mediated by muscle fatigue induced from electrically-elicited contractions (Marsolais and Kobetic, 1988). For example, stimulation of the triceps surae generating isometric contractions decreased the modified Ashworth score for the plantarflexors (Van Der Salm et al., 2006). Similarly, functional electrical stimulation (FES)-assisted cycling also decreased the modified Ashworth score and pendulum test in people with complete SCI (Krause et al., 2008). It has been theorized that long-term stimulation which increases muscle hypertrophy (Skold et al., 2002) and strength (Granat et al., 1993) could be associated with increases in spasm strength. However, research has failed to consistently confirm that long-term stimulation protocols adversely impact spasticity. In contrast, an 8 week program of stimulation of the quadriceps and hamstrings that produced subtetanic isometric contractions reduced spasms as measured by SCATs in people with motor complete SCI (Carty et al., 2013). Similarly people with incomplete SCI who used FES to augment gait training for at least 16 months had decreased intrinsic and reflex stiffness (Mirbagheri et al., 2002). Moreover, 15 days of tibial nerve stimulation that did not result in perceptible contractions reduced Ashworth scores, spasm frequency, tendon and H-reflex to a similar extent as oral baclofen (Aydin et al., 2005). Although most studies show improvements in spasticity with long-term electrical stimulation, some individuals did rank their spasticity as worse following 6 months of FES cycling, even though there were no changes in their modified Ashworth Scores (Skold et al., 2002). More invasive modes of stimulation, such as spinal cord stimulation with epidural electrodes, improved resistance to passive stretch and reduced the use of antispastic medications (Pinter et al., 2000). However, some negative side effects with a previous epidural stimulator included loss of effectiveness and stimulator failure over ~10 year period (Midha and Schmitt, 1998) and suggest, like intrathecal drug delivery, some technical issues need improvement before more invasive methods of treating spasticity become mainstream.
Combined Excitatory Pharmacological and Physical Treatments
An alternative method to reduce spasticity may be to first increase the functional activation of the spinal cord by residual descending and intact peripheral synaptic inputs. This would then increase control over excitatory circuits and enhance the activation of intrinsic inhibitory mechanisms (KCC2) to curb involuntary, intrinsic (PIC) activation of motoneurons. Recently, combined excitatory pharmacological and physical treatments have been used in both animals and humans to increase spinal cord excitability and promote the recovery of walking function. In participants with incomplete SCI, treadmill training was combined with the oral administration of escitalopram, a selective serotonin reuptake blocker, to increase the amount of serotonin released in the spinal cord (Hornby et al., 2009). Elevation in serotonin levels would potentially increase the excitability of motoneurons via 5-HT2B/C receptors (Murray et al., 2011a), increase the excitability of locomotor interneurons via 5-HT1A, 5-HT7, and 5-HT2A/C receptors (Courtine et al., 2009; Jordan and Slawinska, 2011; Slawinska et al., 2013) and decrease transmission of sensory inputs via 5-HT1B/F receptors (Murray et al., 2011b). When combined with 8 weeks of treadmill training, escitalopram produced a greater improvement in walking function (maximum speed) when compared to participants receiving treadmill training alone (Hornby et al., 2009). Likewise, a combination of spinal cord stimulation and intrathecal application of 5-HT1A, 5-HT7, and 5-HT2A receptor agonists improved volitional control of walking in a rat model of incomplete SCI (Van Den Brand et al., 2012). In the human study, although walking performance improved with increases in 5-HT receptor activation, measures of spasticity also increased (Thompson et al., 2011; Thompson and Hornby, 2013), likely due to the facilitation of 5-HT2B/C receptors. It remains to be seen if, over time, motor training can better shape the enhanced activation of the spinal cord by descending and peripheral inputs to improve control of spasticity when excitability of the spinal cord is increased pharmacologically. Perhaps application of 5-HT receptor agonists that specifically target locomotor-related interneurons (e.g., 5-HT1A, 5-HT7, and 5-HT2A) over motoneurons (5-HT2B/C) may produce greater improvements in walking function with fewer increases in spastic motor behaviors.
Conclusions
Following the reduction of synaptic excitation by descending and movement-related sensory inputs, the spinal cord below the injury adapts by increasing the intrinsic excitability of motoneurons via increases in constitutive monoamine receptors and downregulation of KCC2, resulting in uncontrolled activation of muscles below the injury. Strategies to help improve controlled and functional activation of motoneurons via synaptic inputs may help to strengthen the purposeful activation of muscles and reduce the involuntary activation of motoneurons by suppressing some of these maladaptive changes. For example, enhanced descending and/or sensory activation of motoneurons during facilitated motor tasks, such as treadmill or exoskeleton walking, FES-assisted cycling or rowing, which can be applied to participants with motor complete or incomplete injuries, may produce enough pattered activation of motoneurons to help curb, in an activity dependent manner, increases in constitutive monoamine receptor activity and/or promote BDNF-facilitation of KCC2 activity (Boulenguez et al., 2010). Thus, rather than suppressing the involuntary activation of motoneurons and muscles with suppressive pharmacology, perhaps we should be exciting the spinal cord with functional, patterned activity that occurs, or mimics, inputs produced during natural movement to keep intrinsic, inhibitory mechanisms viable. Despite the numerous and effective physical strategies to improve motor function and treat spasticity, individuals with SCI are typically prescribed pharmacological interventions that reduce spinal cord excitability as the first line of treatment (Rekand et al., 2012). Further studies comparing the effectiveness of physical, vs. suppressive pharmacological, treatments over the long-term are needed to determine the best approach for treating and managing spasticity after SCI. The best approach to control spasticity and improve residual motor function may be to increase the activation of the spinal cord with functional, patterned inputs akin to what happens during natural movements such as walking and reaching. This may improve both descending and peripheral control over excitatory spinal circuits and enhance intrinsic inhibition in the spinal cord to reduce spasticity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Jaynie F. Yang for helpful comments on the review. This work was supported by the Canadian Institute of Health Research (MOP-106549) and National Institute of Health Research (R01NS048170) grants to Monica A. Gorassini and Canadian Institute of Health Research (MOP 14697) and National Institute of Health Research (R01NS047567) grants to David J. Bennett. Salary support was provided by Alberta Innovates: Health Solutions to Monica A. Gorassini, David J. Bennett, Jessica M. D'Amico, and Elizabeth G. Condliffe and the Alberta Paraplegic Foundation to Jessica M. D'Amico.
References
Adams, M. M., and Hicks, A. L. (2005). Spasticity after spinal cord injury. Spinal Cord 43, 577–586. doi: 10.1038/sj.sc.3101757
Adams, M. M., and Hicks, A. L. (2011). Comparison of the effects of body-weight-supported treadmill training and tilt-table standing on spasticity in individuals with chronic spinal cord injury. J. Spinal Cord Med. 34, 488–494. doi: 10.1179/2045772311Y.0000000028
Ahlquist, R. P. (1980). Historical perspective. Classification of adrenoreceptors. J. Auton. Pharmacol. 1, 101–106. doi: 10.1111/j.1474-8673.1980.tb00445.x
Anden, N. E., Haeggendal, J., Magnusson, T., and Rosengren, E. (1964). The time course of the disappearance of noradrenaline and 5-hydroxytryptamine in the spinal cord after transection. Acta Physiol. Scand. 62, 115–118. doi: 10.1111/j.1748-1716.1964.tb03958.x
Anden, N. E., Jukes, M. G., and Lundberg, A. (1966a). The effect of DOPA on the spinal cord. 2. A pharmacological analysis. Acta Physiol. Scand. 67, 387–397. doi: 10.1111/j.1748-1716.1966.tb03325.x
Anden, N. E., Jukes, M. G., Lundberg, A., and Vyklicky, L. (1966b). The effect of DOPA on the spinal cord. 1. Influence on transmission from primary afferents. Acta Physiol. Scand. 67, 373–386. doi: 10.1111/j.1748-1716.1966.tb03324.x
Ashby, P., Verrier, M., and Lightfoot, E. (1974). Segmental reflex pathways in spinal shock and spinal spasticity in man. J. Neurol. Neurosurg. Psychiatry 37, 1352–1360. doi: 10.1136/jnnp.37.12.1352
Ashworth, B. (1964). Preliminary trial of carisoprodol in multiple sclerosis. Practitioner 192, 540–542.
Awiszus, F., Feistner, H., and Schafer, S. S. (1991). On a method to detect long-latency excitations and inhibitions of single hand muscle motoneurones in man. Exp. Brain Res. 86, 440–446. doi: 10.1007/BF00228970
Aydin, G., Tomruk, S., Keles, I., Demir, S. O., and Orkun, S. (2005). Transcutaneous electrical nerve stimulation versus baclofen in spasticity: clinical and electrophysiologic comparison. Am. J. Phys. Med. Rehabil. 84, 584–592. doi: 10.1097/01.phm.0000171173.86312.69
Aymard, C., Katz, R., Lafitte, C., Lo, E., Penicaud, A., Pradat-Diehl, P., et al. (2000). Presynaptic inhibition and homosynaptic depression: a comparison between lower and upper limbs in normal human subjects and patients with hemiplegia. Brain 123, 1688–1702. doi: 10.1093/brain/123.8.1688
Bajd, T., and Vodovnik, L. (1984). Pendulum testing of spasticity. J. Biomed. Eng. 6, 9–16. doi: 10.1016/0141-5425(84)90003-7
Baker, L. L., and Chandler, S. H. (1987). Characterization of postsynaptic potentials evoked by sural nerve stimulation in hindlimb motoneurons from acute and chronic spinal cats. Brain Res. 420, 340–350. doi: 10.1016/0006-8993(87)91255-8
Baldissera, F., Campadelli, P., and Piccinelli, L. (1982). Impulse coding of ramp currents intracellularly injected into pyramidal tract neurones. Exp. Brain Res. 48, 455–458. doi: 10.1007/BF00238623
Barbeau, H., and Norman, K. E. (2003). The effect of noradrenergic drugs on the recovery of walking after spinal cord injury. Spinal Cord 41, 137–143. doi: 10.1038/sj.sc.3101374
Bastian, H. C. (1890). On the symptomatology of total transverse lesions of the spinal cord; with special reference to the condition of the various reflexes. Med. Chir. Trans. 73, 151–217.
Bennett, D. J., Gorassini, M., Fouad, K., Sanelli, L., Han, Y., and Cheng, J. (1999). Spasticity in rats with sacral spinal cord injury. J. Neurotrauma 16, 69–84. doi: 10.1089/neu.1999.16.69
Bennett, D. J., Hultborn, H., Fedirchuk, B., and Gorassini, M. (1998). Synaptic activation of plateaus in hindlimb motoneurons of decerebrate cats. J. Neurophysiol. 80, 2023–2037.
Bennett, D. J., Li, Y., and Siu, M. (2001). Plateau potentials in sacrocaudal motoneurons of chronic spinal rats, recorded in vitro. J. Neurophysiol. 86, 1955–1971.
Bennett, D. J., Sanelli, L., Cooke, C. L., Harvey, P. J., and Gorassini, M. A. (2004). Spastic long-lasting reflexes in the awake rat after sacral spinal cord injury. J. Neurophysiol. 91, 2247–2258. doi: 10.1152/jn.00946.2003
Benz, E. N., Hornby, T. G., Bode, R. K., Scheidt, R. A., and Schmit, B. D. (2005). A physiologically based clinical measure for spastic reflexes in spinal cord injury. Arch. Phys. Med. Rehabil. 86, 52–59. doi: 10.1016/j.apmr.2004.01.033
Berg, K. A., Harvey, J. A., Spampinato, U., and Clarke, W. P. (2005). Physiological relevance of constitutive activity of 5-HT2A and 5-HT2C receptors. Trends Pharmacol. Sci. 26, 625–630. doi: 10.1016/j.tips.2005.10.008
Biering-Sorensen, B., Kristensen, I. B., Kjaer, M., and Biering-Sorensen, F. (2009). Muscle after spinal cord injury. Muscle Nerve 40, 499–519. doi: 10.1002/mus.21391
Biering-Sorensen, B., Nielsen, J. B., and Klinge, K. (2006). Spasticity assessment: a review. Spinal Cord 44, 708–722. doi: 10.1038/sj.sc.3101928
Birnbaum, A., and Ashby, P. (1982). Postsynaptic potentials in individual soleus motoneurons in man produced by achilles tendon taps and electrical stimulation of tibial nerve. Electroencephalogr. Clin. Neurophysiol. 54, 469–471. doi: 10.1016/0013-4694(82)90211-5
Bjorklund, A., Kirik, D., Rosenblad, C., Georgievska, B., Lundberg, C., and Mandel, R. J. (2000). Towards a neuroprotective gene therapy for Parkinson's disease: use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res. 886, 82–98. doi: 10.1016/S0006-8993(00)02915-2
Boess, F. G., and Martin, I. L. (1994). Molecular biology of 5-HT receptors. Neuropharmacology 33, 275–317. doi: 10.1016/0028-3908(94)90059-0
Bohannon, R. W., and Smith, M. B. (1987). Interrater reliability of a modified Ashworth Scale of muscle spasticity. Phys. Ther. 67, 206–207.
Bos, R., Sadlaoud, K., Boulenguez, P., Buttigieg, D., Liabeuf, S., Brocard, C., et al. (2013). Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 110, 348–353. doi: 10.1073/pnas.1213680110
Boulenguez, P., Liabeuf, S., Bos, R., Bras, H., Jean-Xavier, C., Brocard, C., et al. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307. doi: 10.1038/nm.2107
Bovend'Eerdt, T. J., Newman, M., Barker, K., Dawes, H., Minelli, C., and Wade, D. T. (2008). The effects of stretching in spasticity: a systematic review. Arch. Phys. Med. Rehabil. 89, 1395–1406. doi: 10.1016/j.apmr.2008.02.015
Brownstone, R. M. (2006). Beginning at the end: repetitive firing properties in the final common pathway. Prog Neurobiol. 78, 156–172. doi: 10.1016/j.pneurobio.2006.04.002
Bruinstroop, E., Cano, G., Vanderhorst, V. G., Cavalcante, J. C., Wirth, J., Sena-Esteves, M., et al. (2012). Spinal projections of the A5, A6 (locus coeruleus), and A7 noradrenergic cell groups in rats. J. Comp. Neurol. 520, 1985–2001. doi: 10.1002/cne.23024
Buch, P. K., Bainbridge, J. W., and Ali, R. R. (2008). AAV-mediated gene therapy for retinal disorders: from mouse to man. Gene Ther. 15, 849–857. doi: 10.1038/gt.2008.66
Calancie, B., Broton, J. G., Klose, K. J., Traad, M., Difini, J., and Ayyar, D. R. (1993). Evidence that alterations in presynaptic inhibition contribute to segmental hypo- and hyperexcitability after spinal cord injury in man. Electroencephalogr. Clin. Neurophysiol. 89, 177–186. doi: 10.1016/0168-5597(93)90131-8
Capaday, C., Lavoie, B. A., and Comeau, F. (1995). Differential effects of a flexor nerve input on the human soleus H-reflex during standing versus walking. Can. J. Physiol. Pharmacol. 73, 436–449. doi: 10.1139/y95-056
Carlin, K. P., Jones, K. M., Jiang, Z., Jordan, L. M., and Brownstone, R. M. (2000). Dendritic L-type calciume currents in mouse spinal motoneurons: implications for bistability. Eur. J. Neurosci. 12, 1635–1646. doi: 10.1046/j.1460-9568.2000.00055.x
Carlsson, A., Magnusson, T., and Ronsengren, E. (1963). 5-Hydroxytryptamine of the spinal cord normally and after transection. Experientia 19, 359. doi: 10.1007/BF02152316
Carty, A., McCormack, K., Coughlan, G. F., Crowe, L., and Caulfield, B. (2013). Alterations in body composition and spasticity following subtetanic neuromuscular electrical stimulation training in spinal cord injury. J. Rehabil. Res. Dev. 50, 193–202. doi: 10.1682/JRRD.2011.11.0220
Castro, A. R., Pinto, M., Lima, D., and Tavares, I. (2006). Secondary hyperalgesia in the monoarthritic rat is mediated by GABAB and NK1 receptors of spinal dorsal horn neurons: a behavior and c-fos study. Neuroscience 141, 2087–2095. doi: 10.1016/j.neuroscience.2006.05.048
Chambers, W. W., Liu, C. N., McCouch, G. P., and D'Aquili, E. (1966). Descehding tracts and spinal shock in the cat. Brain 89, 377–390. doi: 10.1093/brain/89.2.377
Chanrion, B., Manourry La Cour, C., Gavarini, S., Seimandi, M., Vincent, L., Pujol, J. F., et al. (2008). Inverse agonist and neutral antagonist actions of antidepressants at recombinant and native 5-Ht2C receptors: differential modulation of cell surface expression and signal transduction. Mol. Pharmacol. 73, 748–757. doi: 10.1124/mol.107.041574
Conway, B. A., Hultborn, H., Kiehn, O., and Mintz, I. (1988). Plateau potentials in alpha-motoneurones induced by intravenous injection of L-DOPA and clonidine in the spinal cat. J. Physiol. 405, 369–384.
Cope, T. C., Nelson, S. G., and Mendell, L. M. (1980). Factors outside neuraxis mediate “acute” increase in EPSP amplitude caudal to spinal cord transection. J. Neurophysiol. 44, 174–183.
Courtine, G., Gerasimenko, Y., Van Den Brand, R., Yew, A., Musienko, P., Zhong, H., et al. (2009). Transformation of nonfunctional spinal circuits into functional states after the loss of brain input. Nat. Neurosci. 12, 1333–1342. doi: 10.1038/nn.2401
Crill, W. E., and Schwindt, P. C. (1986). Role of persistent inward and outward membrane currents in epileptiform bursting in mammalian neurons. Adv. Neurol. 44, 225–233.
Crone, C., Hultborn, H., Jespersen, B., and Nielsen, J. (1987). Reciprocal Ia inhibition between ankle flexors and extensors in man. J. Physiol. 389, 163–185.
Crone, C., Johnsen, L. L., Biering-Sorensen, F., and Nielsen, J. B. (2003). Appearance of reciprocal facilitation of ankle extensors from ankle flexors in patients with stroke or spinal cord injury. Brain 126, 495–507. doi: 10.1093/brain/awg036
Crone, C., and Nielsen, J. (1989). Methodological implications of the post activation depression of the soleus H-reflex in man. Exp. Brain Res. 78, 28–32. doi: 10.1007/BF00230683
Crone, C., Nielsen, J., Petersen, N., Ballegaard, M., and Hultborn, H. (1994). Disynaptic reciprocal inhibition of ankle extensors in spastic patients. Brain 117, 1161–1168. doi: 10.1093/brain/117.5.1161
Curtis, D. R., Gynther, B. D., Lacey, G., and Beattie, D. T. (1997). Baclofen: reduction of presynaptic calcium influx in the cat spinal cord in vivo. Exp. Brain Res. 113, 520–533. doi: 10.1007/PL00005604
D'Amico, J. M., Li, Y., Bennett, D. J., and Gorassini, M. A. (2013a). Reduction of spinal sensory transmission by facilitation of 5-HT1B/D receptors in noninjured and spinal cord-injured humans. J. Neurophysiol. 109, 1485–1493. doi: 10.1152/jn.00822.2012
D'Amico, J. M., Murray, K. C., Li, Y., Chan, K. M., Finlay, M. G., Bennett, D. J., et al. (2013b). Constitutively active 5-HT2/alpha1 receptors facilitate muscle spasms after human spinal cord injury. J. Neurophysiol. 109, 1473–1484. doi: 10.1152/jn.00821.2012
Das, T. K., and Park, D. M. (1989a). Botulinum toxin in treating spasticity. Br. J. Clin. Pract. 43, 401–403.
Das, T. K., and Park, D. M. (1989b). Effect of treatment with botulinum toxin on spasticity. Postgrad. Med. J. 65, 208–210. doi: 10.1136/pgmj.65.762.208
Davidoff, R. A. (1985). Antispasticity drugs: mechanisms of action. Ann. Neurol. 17, 107–116. doi: 10.1002/ana.410170202
De Luca, C. J., and Erim, Z. (2002). Common drive in motor units of a synergistic muscle pair. J. Neurophysiol. 87, 2200–2204. doi: 10.1152/jn.00793.2001
de Vlugt, E., De Groot, J. H., Wisman, W. H., and Meskers, C. G. (2012). Clonus is explained from increased reflex gain and enlarged tissue visoelasticity. J. Biomech. 45, 148–155. doi: 10.1016/j.jbiomech.2011.09.023
Diamantopoulos, E., and Olsen, P. Z. (1967). Excitability of motor neurones in spinal shock in man. J. Neurol. Neurosurg. Psychiatry 30, 427–431. doi: 10.1136/jnnp.30.5.427
Dietz, V. (2010). Behavior of spinal neurons deprived of supraspinal input. Nat. Rev. Neurol. 6, 167–174. doi: 10.1038/nrneurol.2009.227
Dinculescu, A., Glushakova, L., Min, S. H., and Hauswirth, W. W. (2005). Adeno-associated virus-vectored gene therapy for retinal disease. Hum. Gene Ther. 16, 649–663. doi: 10.1089/hum.2005.16.649
Diong, J., Harvey, L. A., Kwah, L. K., Eyles, J., Ling, M. J., Ben, M., et al. (2012). Incidence and predictors of contracture after spinal cord injury–a prospective cohort study. Spinal Cord 50, 579–584. doi: 10.1038/sc.2012.25
Ditor, D. S., Hamilton, S., Tarnopolsky, M. A., Green, H. J., Craven, B. C., Parise, G., et al. (2004). Na+,K+-ATPase concentration and fiber type distribution after spinal cord injury. Muscle Nerve 29, 38–45. doi: 10.1002/mus.10534
Ditunno, J. F., Little, J. W., Tessler, A., and Burns, A. S. (2004). Spinal shock revisited: a four-phase model. Spinal Cord 42, 383–395. doi: 10.1038/sj.sc.3101603
Draulans, N., Vermeersch, K., Degraeuwe, B., Meurrens, T., Peers, K., Nuttin, B., et al. (2013). Intrathecal baclofen in multiple sclerosis and spinal cord injury: complications and long-term dosage evolution. Clin. Rehabil. 27, 1137–1143. doi: 10.1177/0269215513488607
Faist, M., Dietz, V., and Pierrot-Deseilligny, E. (1996). Modulation, probably presynaptic in origin, of monosynaptic Ia excitation during human gait. Exp. Brain Res. 109, 441–449. doi: 10.1007/BF00229628
Faist, M., Mazevet, D., Dietz, V., and Pierrot-Deseilligny, E. (1994). A quantitative assessment of presynaptic inhibition of Ia afferents in spastics. Differences in hemiplegics and paraplegics. Brain 117, 1449–1455. doi: 10.1093/brain/117.6.1449
Feldman, R. S., Meyer, J. S., and Quenzer, L. F. (1997). “Serotonin,” in Principles of Neuropsychopharmacology, eds R. S. Feldman, J. S. Meyer, and L. F. Quenzer (Sunderland, MA: Sinauer Associates Inc.), 345–389.
Fortuna, R., Horisberger, M., Vaz, M. A., and Herzog, W. (2013). Do skeletal muscle properties recover following repeat onabotulinum toxin A injections? J. Biomech. 46, 2426–2433. doi: 10.1016/j.jbiomech.2013.07.028
Fulton, J. F., and Sherrington, C. S. (1932). State of the flexor reflex in paraplegic dog and monkey respectively. J. Physiol. 75, 17–22.
Gerrits, H. L., Hopman, M. T., Offringa, C., Engelen, B. G., Sargeant, A. J., Jones, D. A., et al. (2003). Variability in fibre properties in paralysed human quadriceps muscles and effects of training. Pflugers Arch. 445, 734–740. doi: 10.1007/s00424-002-0997-4
Gerrits, H. L., Hopman, M. T., Sargeant, A. J., Jones, D. A., and De Haan, A. (2002). Effects of training on contractile properties of paralyzed quadriceps muscle. Muscle Nerve 25, 559–567. doi: 10.1002/mus.10071
Gershengorn, M. C. (1989). Mechanism of signal transduction by TRH. Ann. N.Y. Acad. Sci. 553, 191–196. doi: 10.1111/j.1749-6632.1989.tb46641.x
Gether, U., Ballesteros, J. A., Seifert, R., Sanders-Bush, E., Weinstein, H., and Kobilka, B. K. (1997). Structural instability of a constitutively-active G protein-coupled receptor. J. Biol. Chem. 272, 2587–2590. doi: 10.1074/jbc.272.5.2587
Glass, A., and Hannah, A. (1974). A comparison of dantrolene sodium and diazepam in the treatment of spasticity. Paraplegia 12, 170–174. doi: 10.1038/sc.1974.27
Gomez-Pinilla, F., Huie, J. R., Ying, Z., Ferguson, A. R., Crown, E. D., Baumbauer, K. M., et al. (2007). BDNF and learning: evidence that instrumental training promotes learning within the spinal cord by up-regulating BDNF expression. Neuroscience 148, 893–906. doi: 10.1016/j.neuroscience.2007.05.051
Gorassini, M. A., Knash, M. E., Harvey, P. J., Bennett, D. J., and Yang, J. F. (2004). Role of motoneurons in the generation of muscle spasms after spinal cord injury. Brain 127, 2247–2258. doi: 10.1093/brain/awh243
Gorassini, M. A., Norton, J. A., Nevett-Duchcherer, J., Roy, F. D., and Yang, J. F. (2009). Changes in locomotor muscle activity after treadmill training in subjects with incomplete spinal cord injury. J. Neurophysiol. 101, 969–979. doi: 10.1152/jn.91131.2008
Gorgey, A. S., and Dudley, G. A. (2008). Spasticity may defend skeletal muscle composition after incomplete spinal cord injury. Spinal Cord 46, 96–102. doi: 10.1038/sj.sc.3102087
Gracies, J. M. (2005). Pathophysiology of spastic paresis. I: paresis and soft tissue changes. Muscle Nerve 31, 535–551. doi: 10.1002/mus.20284
Gracies, J. M., Nance, P., Elovic, E., McGuire, J., and Simpson, D. M. (1997). Traditional pharmacological treatments for spasticity. Part II: general and regional treatments. Muscle Nerve Suppl. 6, S92–S120.
Granat, M. H., Ferguson, A. C., Andrews, B. J., and Delargy, M. (1993). The role of functional electrical stimulation in the rehabilitation of patients with incomplete spinal cord injury–observed benefits during gait studies. Paraplegia 31, 207–215. doi: 10.1038/sc.1993.39
Grosset, K. A., and Grosset, D. G. (2004). Prescribed drugs and neurological complications. J. Neurol. Neurosurg. Psychiatry 75, iii2–iii8. doi: 10.1136/jnnp.2004.045757
Guissard, N., and Duchateau, J. (2004). Effect of static stretch training on neural and mechanical properties of the human plantar-flexor muscles. Muscle Nerve 29, 248–255. doi: 10.1002/mus.10549
Hall, M. (1850). Synopsis of the Diastaltic Nervous System: or The System of the Spinal Marrow, and its Reflex Arcs; as the Nervous Agent in all the Functions of Ingestion and of Egestion in the Animal Oeconomy. London: Mallett, J.
Hammar, I., and Jankowska, E. (2003). Modulatory effects of alpha1-,alpha2-, and beta -receptor agonists on feline spinal interneurons with monosynaptic input from group I muscle afferents. J. Neurosci. 23, 332–338.
Harris, R. L., Bobet, J., Sanelli, L., and Bennett, D. J. (2006). Tail muscles become slow but fatigable in chronic sacral spinal rats with spasticity. J. Neurophysiol. 95, 1124–1133. doi: 10.1152/jn.00456.2005
Harris, R. L., Putman, C. T., Rank, M., Sanelli, L., and Bennett, D. J. (2007). Spastic tail muscles recover from myofiber atrophy and myosin heavy chain transformations in chronic spinal rats. J. Neurophysiol. 97, 1040–1051. doi: 10.1152/jn.00622.2006
Harvey, L. A., Herbert, R. D., Glinsky, J., Moseley, A. M., and Bowden, J. (2009a). Effects of 6 months of regular passive movements on ankle joint mobility in people with spinal cord injury: a randomized controlled trial. Spinal Cord 47, 62–66. doi: 10.1038/sc.2008.71
Harvey, L. A., Lin, C. W., Glinsky, J. V., and De Wolf, A. (2009b). The effectiveness of physical interventions for people with spinal cord injuries: a systematic review. Spinal Cord 47, 184–195. doi: 10.1038/sc.2008.100
Harvey, P. J., Li, Y., Li, X., and Bennett, D. J. (2006). Persistent sodium currents and repetitive firing in motoneurons of the sacrocaudal spinal cord of adult rats. J. Neurophysiol. 96, 1141–1157. doi: 10.1152/jn.00335.2005
Heckman, C. J., Gorassini, M. A., and Bennett, D. J. (2005). Persistent inward currents in motoneuron dendrites: implications for motor output. Muscle Nerve 31, 135–156. doi: 10.1002/mus.20261
Heckman, C. J., Lee, R. H., and Brownstone, R. M. (2003). Hyperexcitable dendrites in motoneurons and their neuromodulatory control during motor behaviour. Trends Neurosci. 26, 688–695. doi: 10.1016/j.tins.2003.10.002
Hefferan, M. P., Kucharova, K., Kinjo, K., Kakinohama, O., Sekerkova, G., Nakamura, S., et al. (2007). Spinal astrocyte glutamate receptor 1 over-expression after ischemic insult facilitates behavioural signs of spasticity and rigidity. J. Neurosci. 27, 11179–11191. doi: 10.1523/JNEUROSCI.0989-07.2007
Herman, R., Mayer, N., and Mecomber, S. A. (1972). Clinical pharmaco-physiology of dantrolene sodium. Am. J. Phys. Med. 51, 296–311.
Herrick-Davis, K., Grinde, E., and Teitler, M. (2000). Inverse agonist activity of atypical antipsychotic drugs at human 5HT2C receptors. J. Pharmacol. Exp. Ther. 295, 226–232.
Hidler, J. M., Harvey, R. L., and Rymer, W. Z. (2002). Frequency response characteristics of ankle plantar flexors in humans following spinal cord injury: relation to degree of spasticity. Ann. Biomed. Eng. 30, 969–981. doi: 10.1114/1.1500409
Hiersemenzel, L. P., Curt, A., and Dietz, V. (2000). From spinal shock to spasticity: neuronal adaptations to a spinal cord injury. Neurology 54, 1574–1582. doi: 10.1212/WNL.54.8.1574
Hornby, T. G., Kahn, J. H., Moore, J. L., Kinnaird, C., and Echauz, A. (2009). Combined selective serotonin reuptake inhibitors and locomotor training improves treadmill walking vs locomotor training alone in individuals with motor incomplete spinal cord injury. Soc. Neurosci. Abstract 176.03.
Horowits, R., Kempner, E. S., Bisher, M. E., and Podolsky, R. J. (1986). A physiological role for titin and nebulin in skeletal muscle. Nature 323, 160–164. doi: 10.1038/323160a0
Hounsgaard, J., Hultborn, H., Jespersen, B., and Kiehn, O. (1984). Intrinsic membrane properties causing a bistable behaviour of alpha-motoneurones. Exp. Brain Res. 55, 391–394. doi: 10.1007/BF00237290
Hounsgaard, J., Hultborn, H., Jespersen, B., and Kiehn, O. (1988). Bistability of alpha-motoneurons in the decerebrate cat and in the acute spinal cat after intravenous 5-hydroxytryptophan. J. Physiol. 405, 345–367.
Hounsgaard, J., Hultborn, H., and Kiehn, O. (1986). Transmitter-controlled properties of alpha-motoneurons causing long-lasting motor discharge to brief excitatory inputs. Brain Res. 64, 39–49. doi: 10.1016/S0079-6123(08)63398-1
Hoyer, D., Hannon, J. P., and Martin, G. R. (2002). Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 71, 533–554. doi: 10.1016/S0091-3057(01)00746-8
Hsieh, J. T. C., Wolfe, D. L., Townson, A. F., Short, C., Connolly, S. J., Mehta, S., et al. (2012). “Spasticity following spinal cord injury,” in Spinal Cord Injury Rehabilitation Evidence, Version 4.0, eds J. J. Eng, R. W. Teasell, W. C. Miller, D. L. Wolfe, A. F. Townson, J. T. C. Hsieh, et al. Available online at: http://www.scireproject.com/rehabilitation-evidence/spasticity
Hsieh, J. T., Wolfe, D. L., Miller, W. C., and Curt, A. (2008). Spasticity outcome measures in spinal cord injury: psychometric properties and clinical utility. Spinal Cord 46, 86–95. doi: 10.1038/sj.sc.3102125
Hultborn, H., Illert, M., Nielsen, J., Paul, A., Ballegaard, M., and Wiese, H. (1996). On the mechanism of the post-activation depression of the H-reflex in human subjects. Exp. Brain Res. 108, 450–462. doi: 10.1007/BF00227268
Hultborn, H., Meunier, S., Morin, C., and Pierrot-Deseilligny, E. (1987). Assessing changes in presynaptic inhibition of I a fibres: a study in man and the cat. J. Physiol. 389, 729–756.
Hunt, R. S., Meltzer, G. E., and Landau, W. M. (1963). Fusimotor function. I. Spinal shock of the cat and the monkey. Arch. Neurol. 9, 120–126. doi: 10.1001/archneur.1963.00460080030003
Jankowska, E., Hammar, I., Chojnicka, B., and Heden, C. H. (2000). Effects of monoamines on interneurons in four spinal reflex pathways from group I and/or group II muscle afferents. Eur. J. Neurosci. 12, 701–714. doi: 10.1046/j.1460-9568.2000.00955.x
Jankowska, E., Jukes, M. G., Lund, S., and Lundberg, A. (1967). The effect of DOPA on the spinal cord. 6. Half-centre organization of interneurones transmitting effects from the flexor reflex afferents. Acta Physiol. Scand. 70, 389–402. doi: 10.1111/j.1748-1716.1967.tb03637.x
Jankowska, E., Lackberg, Z. S., and Dyrehag, L. E. (1994). Effects of monoamines on transmission from group II muscle afferents in sacral segments in the cat. Eur. J. Neurosci. 6, 1058–1061. doi: 10.1111/j.1460-9568.1994.tb00601.x
Jankowska, E., Riddell, J. S., Skoog, B., and Noga, B. R. (1993). Gating of transmission to motoneurones by stimuli applied in the locus coeruleus and raphe nuclei of the cat. J. Physiol. 461, 705–722.
Jayaraman, A., Thompson, C. K., Rymer, W. Z., and Hornby, T. G. (2013). Short-term maximal-intensity resistance training increases volitional function and strength in chronic incomplete spinal cord injury: a pilot study. J. Neurol. Phys. Ther. 37, 112–117. doi: 10.1097/NPT.0b013e31828390a1
Jordan, L. M., Liu, J., Hedlund, P. B., Akay, T., and Pearson, K. G. (2008). Descending command systems for the initiation of locomotion in mammals. Brain Res. Rev. 57, 183–191. doi: 10.1016/j.brainresrev.2007.07.019
Jordan, L. M., and Slawinska, U. (2011). Chapter 12-modulation of rhythmic movement: control of coordination. Prog. Brain Res. 188, 181–195. doi: 10.1016/B978-0-444-53825-3.00017-6
Kakinohana, O., Hefferan, M. P., Miyanohara, A., Nejime, T., Marsala, S., Juhas, S., et al. (2012). Combinational spinal GAD65 gene delivery and systemic GABA-mimetic treatment for modulation of spasticity. PLoS ONE 7:e30561. doi: 10.1371/journal.pone.0030561
Kaplitt, M. G., Feigin, A., Tang, C., Fitzsimons, H. L., Mattis, P., Lawlor, P. A., et al. (2007). Safety and tolerability of gene therapy with an adeno-assicated virus (AAV) borne GAD gene for Parkinson's disease: an open label phase I trial. Lancet 369, 2097–2105. doi: 10.1016/S0140-6736(07)60982-9
Kesiktas, N., Paker, N., Erdogan, N., Gulsen, G., Bicki, D., and Yilmaz, H. (2004). The use of hydrotherapy for the management of spasticity. Neurorehabil. Neural Repair 18, 268–273. doi: 10.1177/1545968304270002
Kitzman, P. (2005). Alteration in axial motoneuronal morphology in the spinal cord injured spastic rat. Exp. Neurol. 192, 100–108. doi: 10.1016/j.expneurol.2004.10.021
Kitzman, P. (2006). Changes in vesicular glutamate transporter 2, vesicular GABA transporter and vesicular acetylcholine transporter labeling of sacrocaudal motoneurons in the spastic rat. Exp. Neurol. 197, 407–419. doi: 10.1016/j.expneurol.2005.10.005
Kitzman, P. (2007). VGLUT1 and GLYT2 labeling of the sacrocaudal motoneurons in the spinal cord injured spastic rat. Exp. Neurol. 204, 195–204. doi: 10.1016/j.expneurol.2006.10.008
Klein, C. S., Hager-Ross, C. K., and Thomas, C. K. (2006). Fatigue properties of human thenar motor units paralysed by chronic spinal cord injury. J. Physiol. 573(pt 1), 161–171. doi: 10.1113/jphysiol.2005.103044
Knikou, M., and Mummidisetty, C. K. (2014). Locomotor training improves premotoneuronal control after chronic spinal cord injury. J. Neurophysiol. doi: 10.1152/jn.00871.2013. [Epub ahead of print].
Ko, H. Y., Ditunno, J. F., Graziani, V., and Little, J. W. (1999). The pattern of reflex recovery during spinal shock. Spinal Cord 37, 402–409. doi: 10.1038/sj.sc.3100840
Kohn, A. F., Floeter, M. K., and Hallett, M. (1997). Presynaptic inhibition compared with homosynaptic depression as an explanation for soleus H-reflex depression in humans. Exp. Brain Res. 116, 375–380. doi: 10.1007/PL00005765
Kopin, I. J. (1968). Biosynthesis and metabolism of catecholamines. Anesthesiology 29, 654–660. doi: 10.1097/00000542-196807000-00007
Kots, I., and Zhukov, V. I. (1971). Superspinal control of segmentary centers of muscle-antagonists in man. 3. “Tuning” of a spinal apparatus of reciprocal inhibition during organization of voluntary movement. Biofizika 16, 1085–1092.
Krach, L. E. (2001). Pharmacotherapy of spasticity: oral medications and intrathecal baclofen. J. Child Neurol. 16, 31–36. doi: 10.1177/088307380101600106
Krause, P., Szecsi, J., and Straube, A. (2008). Changes in spastic muscle tone increase in patients with spinal cord injury using functional electrical stimulation and passive leg movements. Clin. Rehabil. 22, 627–634. doi: 10.1177/0269215507084648
Krause, T., Gerbershagen, M. U., Fiege, M., Weisshorn, R., and Wappler, F. (2004). Dantrolene–a review of its pharmacology, therapeutic use and new developments. Anaesthesia 59, 364–373. doi: 10.1111/j.1365-2044.2004.03658.x
Labeit, S., and Kolmerer, B. (1995). Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270, 293–296. doi: 10.1126/science.270.5234.293
Lanner, J. T., Georgiou, D. K., Joshi, A. D., and Hamilton, S. L. (2010). Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2:a003996. doi: 10.1101/cshperspect.a003996
Lapeyre, E., Kuks, J. B., and Meijler, W. J. (2010). Spasticity: revisiting the role and the individual value of several pharmacological treatments. NeuroRehabilitation 27, 193–200. doi: 10.3233/NRE-2010-0596
Lazorthes, Y., Sallerin-Caute, B., Verdie, J. C., Bastide, R., and Carillo, J. P. (1990). Chronic intrathecal baclofen administration for control of severe spasticity. J. Neurosurg. 72, 393–402. doi: 10.3171/jns.1990.72.3.0393
Lee, R. H., and Heckman, C. J. (1996). Influence of voltage-sensitive dendritic conductances on bistable firing and effective synaptic current in cat spinal motoneurons in vivo. J. Neurophysiol. 76, 2107–2110.
Lee, R. H., and Heckman, C. J. (2000). Adjustable amplification of synaptic input in the dendrites of spinal motoneurons in vivo. J. Neurosci. 20, 6734–6740.
Lee, R. H., and Heckman, C. J. (2001). Essential role of a fast persistent inward current in action potential initiation and control of rhythmic firing. J. Neurophysiol. 85, 472–475.
Leis, A. A., Kronenberg, M. F., Stetkarova, I., Paske, W. C., and Stokic, D. S. (1996). Spinal motoneuron excitability after acute spinal cord injury in humans. Neurology 47, 231–237. doi: 10.1212/WNL.47.1.231
Lev-Tov, A., Fleshman, J. W., and Burke, R. E. (1983). Primary afferent depolarization and presynaptic inhibition of monosynaptic group Ia EPSPs during posttetanic potentiation. J. Neurophysiol. 50, 413–427.
Li, X., and Bennett, D. J. (2007). Apamin-sensitive calcium-activated potassium currents (SK) are activated by persistent calcium currents in rat motoneurons. J. Neurophysiol. 97, 3314–3330. doi: 10.1152/jn.01068.2006
Li, X., Murray, K., Harvey, P. J., Ballou, E. W., and Bennett, D. J. (2007). Serotonin facilitates a persistent calcium current in motoneurons of rats with and without chronic spinal cord injury. J. Neurophysiol. 97, 1236–1246. doi: 10.1152/jn.00995.2006
Li, Y., and Bennett, D. J. (2003). Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J. Neurophysiol. 90, 857–869. doi: 10.1152/jn.00236.2003
Li, Y., Gorassini, M. A., and Bennett, D. J. (2004a). Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J. Neurophysiol. 91, 767–783. doi: 10.1152/jn.00788.2003
Li, Y., Li, X., Harvey, P. J., and Bennett, D. J. (2004b). Effects of baclofen on spinal reflexes and persistent inward currents in motoneurons of chronic spinal rats with spasticity. J. Neurophysiol. 92, 2694–2703. doi: 10.1152/jn.00164.2004
Limmroth, V., Kazarawa, Z., Fritsche, G., and Diener, H. C. (1999). Headache after frequent use of serotonin agonists zolmitriptan and naratriptan. Lancet 353, 378. doi: 10.1016/S0140-6736(05)74950-3
Little, J. W., Ditunno, J. F. Jr., Stiens, S. A., and Harris, R. M. (1999). Incomplete spinal cord injury: neuronal mechanisms of motor recovery and hyperreflexia. Arch. Phys. Med. Rehabil. 80, 587–599. doi: 10.1016/S0003-9993(99)90204-6
Lloyd, D. P., and Wilson, V. J. (1957). Reflex depression in rhythmically active monosynaptic reflex pathways. J. Gen. Physiol. 40, 409–426. doi: 10.1085/jgp.40.3.409
Lotta, S., Scelsi, R., Alfonsi, E., Saitta, A., Nicolotti, D., Epifani, P., et al. (1991). Morphometric and neurophysiological analysis of skeletal muscle in paraplegic patients with traumatic cord lesion. Paraplegia 29, 247–252. doi: 10.1038/sc.1991.35
Lundberg, A. (1979). Multisensory control of spinal reflex pathways. Prog. Brain Res. 50, 11–28. doi: 10.1016/S0079-6123(08)60803-1
Magladery, J. W., Teasdall, R. D., Park, A. M., and Languth, H. W. (1952). Electrophysiological studies of reflex activity in patients with lesions of the nervous system. I. A comparison of spinal motoneurone excitability following afferent nerve volleys in normal persons and patients with upper motor neurone lesions. Bull. Johns Hopkins Hosp. 91, 219–244.
Mailis, A., and Ashby, P. (1990). Alterations in group Ia projections to motoneurons following spinal lesions in humans. J. Neurophysiol. 64, 637–647.
Malisoux, L., Jamart, C., Delplace, K., Nielens, H., Francaux, M., and Theisen, D. (2007). Effect of long-term muscle paralysis on human single fiber mechanics. J. Appl. Physiol. 102, 340–349. doi: 10.1152/japplphysiol.00609.2006
Manella, K. J., and Field-Fote, E. C. (2013). Modulatory effects of locomotor training on extensor spasticity in individuals with motor-incomplete spinal cord injury. Restor. Neurol. Neurosci. 31, 633–646. doi: 10.3233/RNN-120255
Marciniak, C., Rader, L., and Gagnon, C. (2008). The use of botulinum toxin for spasticity after spinal cord injury. Am. J. Phys. Med. Rehabil. 87, 312–317. doi: 10.1097/PHM.0b013e318168ceaf
Marsolais, E. B., and Kobetic, R. (1988). Development of a practical electrical stimulation system for restoring gait in the paralyzed patient. Clin. Orthop. Relat. Res. 233, 64–74.
Matthews, P. B. (1996). Relationship of firing intervals of human motor units to the trajectory of post-spike after-hyperpolarization and synaptic noise. J. Physiol. 492, 597–628.
Maynard, F. M., Karunas, R. S., and Waring, W. P. 3rd. (1990). Epidemiology of spasticity following traumatic spinal cord injury. Arch. Phys. Med. Rehabil. 71, 566–569.
McCouch, G. P., Liu, C. N., and Chambers, W. W. (1966). Descending tracts and spinal shock in the monkey (Macaca mulatta). Brain 89, 359–376. doi: 10.1093/brain/89.2.359
McDonald, M. F., Kevin Garrison, M., and Schmit, B. D. (2005). Length-tension properties of ankle muscles in chronic human spinal cord injury. J. Biomech. 38, 2344–2353. doi: 10.1016/j.jbiomech.2004.10.024
Meunier, S., and Pierrot-Deseilligny, E. (1989). Gating of the afferent volley of the monosynaptic stretch reflex during movement in man. J. Physiol. 419, 753–763.
Meyler, W. J., Bakker, H., Kok, J. J., Agoston, S., and Wesseling, H. (1981). The effect of dantrolene sodium in relation to blood levels in spastic patients after prolonged administration. J. Neurol. Neurosurg. Psychiatry 44, 334–339. doi: 10.1136/jnnp.44.4.334
Midha, M., and Schmitt, J. K. (1998). Epidural spinal cord stimulation for the control of spasticity in spinal cord injury patients lacks long-term efficacy and is not cost-effective. Spinal Cord 36, 190–192. doi: 10.1038/sj.sc.3100532
Miles, G. B., Dai, Y., and Brownstone, R. M. (2005). Mechanisms underlying the early phase of spike frequency adaptation in mouse spinal motoneurones. J. Physiol. 566, 519–532. doi: 10.1113/jphysiol.2005.086033
Millan, M. J. (2002). Descending control of pain. Prog. Neurobiol. 66, 355–474. doi: 10.1016/S0301-0082(02)00009-6
Miller, J. F., Paul, K. D., Lee, R. H., Rymer, W. Z., and Heckman, C. J. (1996). Restoration of extensor excitability in the acute spinal cat by the 5-HT2 agonist DOI. J. Neurophysiol. 75, 620–628.
Minneman, K. P., and Esbenshade, T. A. (1994). Alpha 1-adrenergic receptor subtypes. Annu. Rev. Pharmacol. Toxicol. 34, 117–133. doi: 10.1146/annurev.pa.34.040194.001001
Mirbagheri, M. M., Barbeau, H., Ladouceur, M., and Kearney, R. E. (2001). Intrinsic and reflex stiffness in normal and spastic, spinal cord injured subjects. Exp. Brain Res. 141, 446–459. doi: 10.1007/s00221-001-0901-z
Mirbagheri, M. M., Ladouceur, M., Barbeau, H., and Kearney, R. E. (2002). The effects of long-term FES-assisted walking on intrinsic and reflex dynamic stiffness in spastic spinal-cord-injured subjects. IEEE Trans. Neural Syst. Rehabil. Eng. 10, 280–289. doi: 10.1109/TNSRE.2002.806838
Mizuno, N., and Itoh, H. (2009). Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals 17, 42–54. doi: 10.1159/000186689
Mizuno, Y., Tanaka, R., and Yanagisawa, N. (1971). Reciprocal group I inhibition on triceps surae motoneurons in man. J. Neurophysiol. 34, 1010–1017.
Monahan, P. E., and Samulski, R. J. (2000). Adeno-associated virus vectors for gene therapy: more pros than cons? Mol. Med. Today 6, 433–440. doi: 10.1016/S1357-4310(00)01810-4
Moritz, A. T., Newkirk, G., Powers, R. K., and Binder, M. D. (2007). Facilitation of somatic calcium channels can evoke prolonged tail currents in rat hypoglossal motoneurons. J. Neurophysiol. 98, 1042–1047. doi: 10.1152/jn.01294.2006
Murray, K. C., Nakae, A., Stephens, M. J., Rank, M., D'amico, J. M., Harvey, P. J., et al. (2010). Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5HT2C receptors. Nat. Med. 16, 694–700. doi: 10.1038/nm.2160
Murray, K. C., Stephens, M. J., Ballou, E. W., Heckman, C. J., and Bennett, D. J. (2011a). Motoneuron excitability and muscle spasms are regulated by 5-HT2B and 5-HT2C receptor activity. J. Neurophysiol. 105, 731–748. doi: 10.1152/jn.00774.2010
Murray, K. C., Stephens, M. J., Rank, M., D'amico, J., Gorassini, M. A., and Bennett, D. J. (2011b). Polysynaptic excitatory postsynaptic potentials that trigger spasms after spinal cord injury in rats are inhibited by 5-HT1B and 5-HT1F receptors. J. Neurophysiol. 106, 925–943. doi: 10.1152/jn.01011.2010
Nance, P. W. (1994). A comparison of clonidine, cyproheptadine and baclofen in spastic spinal cord injured patients. J. Am. Paraplegia Soc. 17, 150–156.
Nance, P. W., Bugaresti, J., Shellenberger, K., Sheremata, W., and Martinez-Arizala, A. (1994). Efficacy and safety of tizanidine in the treatment of spasticity in patients with spinal cord injury. North American Tizanidine Study Group. Neurology 44(11 Suppl. 9), S44–S51.
Navailles, S., De Deurwaerdere, P., and Spampinato, U. (2006). Clozapine and haloperidol differentially alter the constitutive activity of central serotonin2C receptors in vivo. Biol. Psychiatry 59, 568–575. doi: 10.1016/j.biopsych.2005.07.035
Nichols, D. E., and Nichols, C. D. (2008). Serotonin receptors. Chem. Rev. 108, 1614–1641. doi: 10.1021/cr078224o
Nielsen, J. B., Crone, C., and Hultborn, H. (2007). The spinal pathophysiology of spasticity-from a basic science point of view. Acta Physiol. 189, 171–180. doi: 10.1111/j.1748-1716.2006.01652.x
Norman, K. E., Pepin, A., and Barbeau, H. (1998). Effects of drugs on walking after spinal cord injury. Spinal Cord 36, 699–715. doi: 10.1038/sj.sc.3100674
Norton, J. A., Bennett, D. J., Knash, M. E., Murray, K. C., and Gorassini, M. A. (2008). Changes in sensory-evoked synaptic activation of motoneurons after spinal cord injury in man. Brain 131, 1478–1491. doi: 10.1093/brain/awn050
Odeen, I., and Knutsson, E. (1981). Evaluation of the effects of muscle stretch and weight load in patients with spastic paraplegia. Scand. J. Rehabil. Med. 13, 117–121.
Olsson, M. C., Kruger, M., Meyer, L. H., Ahnlund, L., Gransberg, L., Linke, W. A., et al. (2006). Fibre type-specific increase in passive muscle tension in spinal cord-injured subjects with spasticity. J. Physiol. 577, 339–352. doi: 10.1113/jphysiol.2006.116749
Pandyan, A. D., Gregoric, M., Barnes, M. P., Wood, D., Van Wijck, F., Burridge, J., et al. (2005). Spasticity: clinical perceptions, neurological realities and meaningful measurement. Disabil. Rehabil. 27, 2–6. doi: 10.1080/09638280400014576
Pedersen, E. (1954). Studies on the central pathway of the flexion reflex in man and animal. Acta Psychiatr. Neurol. Scand. Suppl. 88, 1–81.
Pelletier, C. A., and Hicks, A. L. (2010). The length-tension relationship of human dorsiflexor and plantarflexor muscles after spinal cord injury. Spinal Cord 48, 202–206. doi: 10.1038/sc.2009.106
Penn, R. D., Savoy, S. M., Corcos, D., Latash, M., Gottlieb, G., Parke, B., et al. (1989). Intrathecal baclofen for severe spinal spasticity. N. Engl. J. Med. 320, 1517–1521. doi: 10.1056/NEJM198906083202303
Person, R. S., and Kudina, L. P. (1971). Discharge frequency and discharge pattern of human motor units during voluntary contraction of muscle. Electroencephalogr. Clin. Neurophysiol. 32, 471–483. doi: 10.1016/0013-4694(72)90058-2
Peterlin, B. L., and Rapoport, A. M. (2007). Clinical pharmacology of the serotonin receptor agonist, zolmitriptan. Expert Opin. Drug Metab. Toxicol. 3, 899–911. doi: 10.1517/17425255.3.6.899
Pinter, M. M., Gerstenbrand, F., and Dimitrijevic, M. R. (2000). Epidural electrical stimulation of posterior structures of the human lumbosacral cord: 3. control of spasticity. Spinal Cord 38, 524–531. doi: 10.1038/sj.sc.3101040
Polc, P., Mohler, H., and Haefely, W. (1974). The effect of diazepam on spinal cord activities: possible sites and mechanisms of action. Naunyn Schmiedebergs Arch. Pharmacol. 284, 319–337. doi: 10.1007/BF00504702
Powers, R. K., and Binder, M. D. (2003). Persistent sodium and calcium currents in rat hypoglossal motoneurons. J. Neurophysiol. 89, 615–624. doi: 10.1152/jn.00241.2002
Powers, R. K., Robinson, F. R., Konodi, M. A., and Binder, M. D. (1992). Effective synaptic current can be estimated from measurements of neuronal discharge. J. Neurophysiol. 68, 964–968.
Price, T. J., Cervero, F., and De Koninck, Y. (2005). Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr. Top. Med. Chem. 5, 547–555. doi: 10.2174/1568026054367629
Quevedo, J., Eguibar, J. R., Jimenez, I., Schmidt, R. F., and Rudomin, P. (1993). Primary afferent depolarization of muscle afferents elicited by stimulation of joint afferents in cats with intact neuraxis and during reversible spinalization. J. Neurophysiol. 70, 1899–1910.
Rank, M. M., Murray, K. C., Stephens, M. J., D'amico, J., Gorassini, M. A., and Bennett, D. J. (2011). Adrenergic receptors modulate motoneuron excitability, sensory synaptic transmission and muscle spasms after chronic spinal cord injury. J. Neurophysiol. 105, 410–422. doi: 10.1152/jn.00775.2010
Rauser, L., Savage, J. E., Meltzer, H. Y., and Roth, B. L. (2001). Inverse agonist actions of typical and atypical antipsychotic drugs at the human 5HT2C receptor. J. Pharmacol. Exp. Ther. 299, 83–89.
Rayegani, S. M., Shojaee, H., Sedighipour, L., Soroush, M. R., Baghbani, M., and Amirani, O. B. (2011). The effect of electrical passive cycling on spasticity in war veterans with spinal cord injury. Front. Neurol. 2:39. doi: 10.3389/fneur.2011.00039
Raymond, J. R., Mukhin, Y. V., Gelasco, A., Turner, J., Collinsworth, G., Gettys, T. W., et al. (2001). Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol. Ther. 92, 179–212. doi: 10.1016/S0163-7258(01)00169-3
Rekand, T., Hagen, E. M., and Gronning, M. (2012). Spasticity following spinal cord injury. Tidsskr. Nor. Legeforen. 132, 970–973. doi: 10.4045/tidsskr.10.0872
Rekling, J. C., Funk, G. D., Bayliss, D. A., Dong, X. W., and Feldman, J. L. (2000). Synaptic control of motoneuronal excitability. Physiol. Rev. 80, 767–852.
Ren, L. Q., Wienecke, J., Chen, M., Møller, M., Hultborn, H., and Zhang, M. (2013). The time course of serotonin 2C receptor expression after spinal transection of rats: an immunohistochemical study. Neuroscience 236, 31–46. doi: 10.1016/j.neuroscience.2012.12.063
Roby-Brami, A., and Bussel, B. (1987). Long-latency spinal reflex in man after flexor reflex afferent stimulation. Brain 110, 707–725. doi: 10.1093/brain/110.3.707
Rochester, L., Chandler, C. S., Johnson, M. A., Sutton, R. A., and Miller, S. (1995). Influence of electrical stimulation of the tibialis anterior muscle in paraplegic subjects. 1. contractile properties. Paraplegia 33, 437–449. doi: 10.1038/sc.1995.97
Rothwell, J. C., Day, B. L., Berardelli, A., and Marsden, C. D. (1986). Habituation and conditioning of the human long latency stretch reflex. Exp. Brain Res. 63, 197–204. doi: 10.1007/BF00235664
Roy, R. R., and Edgerton, V. R. (2012). Neurobiological perspective of spasticity as occurs after a spinal cord injury. Exp. Neurol. 235, 116–122. doi: 10.1016/j.expneurol.2012.01.017
Roy, R. R., Pierotti, D. J., Flores, V., Rudolph, W., and Edgerton, V. R. (1992). Fibre size and type adaptations to spinal isolation and cyclical passive stretch in cat hindlimb. J. Anat. 180, 491–499.
Roy, R. R., Talmadge, R. J., Hodgson, J. A., Oishi, Y., Baldwin, K. M., and Edgerton, V. R. (1999). Differential response of fast hindlimb extensor and flexor muscles to exercise in adult spinalized cats. Muscle Nerve 22, 230–241.
Rudomin, P. (2009). In search of lost presynaptic inhibition. Exp. Brain Res. 196, 139–151. doi: 10.1007/s00221-009-1758-9
Rudomin, P., and Schmidt, R. F. (1999). Presynaptic inhibition in the vertebrate spinal cord revisited. Exp. Brain Res. 129, 1–37. doi: 10.1007/s002210050933
Ruffolo, R. R. Jr., Nichols, A. J., Stadel, J. M., and Hieble, J. P. (1991). Structure and function of alpha-adrenoceptors. Pharmacol. Rev. 43, 475–505.
Sadzot, B., Baraban, J. M., Glennon, R. A., Lyon, R. A., Leonhardt, S., Jan, C. R., et al. (1989). Hallucinogenic drug interactions at human brain 5-HT2 receptors: implications for treating LSD-induced hallucinogenesis. Psychopharmacology (Berl) 98, 495–499. doi: 10.1007/BF00441948
Satkunam, L. E. (2003). Rehabilitation medicine: 3. Management of adult spasticity. CMAJ 169, 1173–1179.
Sawczuk, A., Powers, R. K., and Binder, M. D. (1997). Contribution of outward currents to spike-frequency adaptation in the hypoglossal motoneurons of the rat. J. Neurophysiol. 78, 2246–2253.
Scelsi, R., Marchetti, C., Poggi, P., Lotta, S., and Lommi, G. (1982). Muscle fiber type morphology and distribution in paraplegic patients with traumatic cord lesion. Histochemical and ultrastructural aspects of rectus femoris muscle. Acta. Neuropathol. 57, 243–248. doi: 10.1007/BF00692178
Schadt, J. C., and Barnes, C. D. (1980). Motoneuron membrane changes associated with spinal shock and the Schiff-Sherrington phenomenon. Brain Res. 201, 373–383. doi: 10.1016/0006-8993(80)91041-0
Schindler-Ivens, S., and Shields, R. K. (2000). Low frequency depression of H-reflexes in humans with acute and chronic spinal-cord injury. Exp. Brain Res. 133, 233–241. doi: 10.1007/s002210000377
Schleip, R., Naylor, I. L., Ursu, D., Melzer, W., Zorn, A., Wilke, H. J., et al. (2006). Passive muscle stiffness may be influenced by active contractility of intramuscular connective tissue. Med. Hypotheses 66, 66–71. doi: 10.1016/j.mehy.2005.08.025
Schmidt, B. J., and Jordan, L. M. (2000). The role of serotonin in reflex modulation and locomotor rhythm production in the mammalian spinal cord. Brain Res. Bull. 53, 689–710. doi: 10.1016/S0361-9230(00)00402-0
Seifert, R., and Wenzel-Seifert, K. (2002). Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch. Pharmacol. 366, 381–416. doi: 10.1007/s00210-002-0588-0
Seki, K., Perlmutter, S. I., and Fetz, E. E. (2003). Sensory input to primate spinal cord is presynaptically inhibited during voluntary movement. Nat. Neurosci. 6, 1309–1316. doi: 10.1038/nn1154
Shahani, B. T., and Young, R. R. (1971). Human flexor reflexes. J. Neurol. Neurosurg. Psychiatry 34, 616–627. doi: 10.1136/jnnp.34.5.616
Shields, R. K. (1995). Fatigability, relaxation properties and electromyographic responses of the human paralyzed soleus muscle. J. Neurophysiol. 73, 2195–2206.
Sieghart, W. (1994). Pharmacology of benzodiazepine receptors: an update. J. Psychiatry Neurosci. 19, 24–29.
Sills, G. J. (2006). The mechanisms of action of gabapentin and pregabalin. Curr. Opin. Pharmacol. 6, 108–113. doi: 10.1016/j.coph.2005.11.003
Simon, O., and Yelnik, A. P. (2010). Managing spasticity with drugs. Eur. J. Phys. Rehabil. Med. 46, 401–410.
Simpson, L. L. (2004). Identification of the major steps in botulinum toxin action. Annu. Rev. Pharmacol. Toxicol. 44, 167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554
Skold, C. (2000). Spasticity in spinal cord injury: self- and clinically rated intrinsic fluctuations and intervention-induced changes. Arch. Phys. Med. Rehabil. 81, 144–149. doi: 10.1016/S0003-9993(00)90132-1
Skold, C., Levi, R., and Seiger, A. (1999). Spasticity after traumatic spinal cord injury: nature, severity, and location. Arch. Phys. Med. Rehabil. 80, 1548–1557. doi: 10.1016/S0003-9993(99)90329-5
Skold, C., Lonn, L., Harms-Ringdahl, K., Hultling, C., Levi, R., Nash, M., et al. (2002). Effects of functional electrical stimulation training for six months on body composition and spasticity in motor complete tetraplegic spinal cord-injured individuals. J. Rehabil. Med. 34, 25–32. doi: 10.1080/165019702317242677
Slawinska, U., Miazga, K., Cabaj, A. M., Leszczynska, A. N., Majczynski, H., Nagy, J. I., et al. (2013). Grafting of fetal brainstem 5-HT neurons into the sublesional spinal cord of paraplegic rats restores coordinated hindlimb locomotion. Exp. Neurol. 247, 572–581. doi: 10.1016/j.expneurol.2013.02.008
Soto, C., Martin-Cora, F., Leiras, R., Velo, P., and Canedo, A. (2006). GABA(B) receptor-mediated modulation of cutaneous input at the cuneate nucleus in anesthetized cats. Neuroscience 137, 1015–1030. doi: 10.1016/j.neuroscience.2005.09.026
Stempien, L., and Tsai, T. (2000). Intrathecal baclofen pump use for spasticity: a clinical survey. Am. J. Phys. Med. Rehabil. 79, 536–541. doi: 10.1097/00002060-200011000-00010
Stieger, K., Cronin, T., Bennett, J., and Rolling, F. (2011). Adeno-associated virus mediated gene therapy for retinal degenerative diseases. Methods Mol. Biol. 807, 179–218. doi: 10.1007/978-1-61779-370-7_8
Taricco, M., Pagliacci, M. C., Telaro, E., and Adone, R. (2006). Pharmacological interventions for spasticity following spinal cord injury: results of a cochrane systematic review. Eura. Medicophys. 42, 5–15.
Taylor, C. P. (1994). Emerging perspectives on the mechanism of action of gabapentin. Neurology 44, S10–S16. discussion: S31–S32.
Thomas, C. K., Hager-Ross, C. K., and Klein, C. S. (2010). Effects of baclofen on motor units paralysed by chronic cervical spinal cord injury. Brain 133, 117–125. doi: 10.1093/brain/awp285
Thomas, C. K., and Ross, B. H. (1997). Distinct patterns of motor unit behaviour during muscle spasms in spinal cord injured subjects. J. Neurophysiol. 77, 2847–2850.
Thomas, S. L., and Gorassini, M. A. (2005). Increases in corticospinal tract function by treadmill training after incomplete spinal cord injury. J. Neurophysiol. 94, 2844–2855. doi: 10.1152/jn.00532.2005
Thompson, C. K., and Hornby, T. G. (2013). Divergent modulation of clinical measures of volitional and reflexive motor behaviors following serotonergic medications in human incomplete spinal cord injury. J. Neurotrauma 30, 498–502. doi: 10.1089/neu.2012.2515
Thompson, C. K., Jayaraman, A., Kinnaird, C., and Hornby, T. G. (2011). Methods to quantify pharmacologically induced alterations in motor function in human incomplete SCI. J. Vis. Exp. 50:2148. doi: 10.3791/2148
Trimble, M. H., Kukulka, C. G., and Behrman, A. L. (1998). The effect of treadmill gait training on low-frequency depression of the soleus H-reflex: comparison of a spinal cord injured man to normal subjects. Neurosci. Lett. 246, 186–188. doi: 10.1016/S0304-3940(98)00259-6
Turker, K. S., and Powers, R. K. (1999). Effects of large excitatory and inhibitory inputs on motoneuron discharge rate and probability. J. Neurophysiol. 82, 829–840.
Turker, K. S., and Powers, R. K. (2003). Estimation of postsynaptic potentials in rat hypoglossal motoneurones: insights for human work. J. Physiol. 551, 419–431. doi: 10.1113/jphysiol.2003.044982
Van Den Brand, R., Heutschi, J., Barraud, Q., Digiovanna, J., Bartholdi, K., Huerlimann, M., et al. (2012). Restoring voluntary control of locomotion after paralyzing spinal cord injury. Science 336, 1182–1185. doi: 10.1126/science.1217416
Van Der Salm, A., Veltink, P. H., Ijzerman, M. J., Groothuis-Oudshoorn, K. C., Nene, A. V., and Hermens, H. J. (2006). Comparison of electric stimulation methods for reduction of triceps surae spasticity in spinal cord injury. Arch. Phys. Med. Rehabil. 87, 222–228. doi: 10.1016/j.apmr.2005.09.024
Wainberg, M., Barbeau, H., and Gautheir, S. (1986). Quantitative assessment of the effect of cyproheptadine on spastic paretic gait: a preliminary study. J. Neurol. 233, 311–314. doi: 10.1007/BF00314166
Wainberg, M., Barbeau, H., and Gauthier, S. (1990). The effects of cyproheptadine on locomotion and on spasticity in patients with spinal cord injuries. J. Neurol. Neurosurg. Psychiatry 53, 754–763. doi: 10.1136/jnnp.53.9.754
Wallace, D. M., Ross, B. H., and Thomas, C. K. (2005). Motor unit behavior during clonus. J. Appl. Physiol. 99, 2166–2172. doi: 10.1152/japplphysiol.00649.2005
Wang, H., Hiatt, W. R., Barstow, T. J., and Brass, E. P. (1999). Relationships between muscle mitochondrial DNA content, mitochondrial enzyme activity and oxidative capacity in man: alterations with disease. Eur. J. Appl. Physiol. Occup. Physiol. 80, 22–27. doi: 10.1007/s004210050553
Wartenberg, R. (1951). Pendulousness of the legs as a diagnostic test. Neurology 1, 18–24. doi: 10.1212/WNL.1.1.18
Watanabe, T. K. (2009). Role of oral medications in spasticity management. PM R. 1, 839–841. doi: 10.1016/j.pmrj.2009.07.014
Weaver, R. A., Landau, W. M., and Higgins, J. F. (1963). Fusimotor function II. Evidence of fusimotor depression in human spinal shock. Arch. Neurol. 9, 127–132. doi: 10.1001/archneur.1963.00460080037004
Westphal, R. S., and Sanders-Bush, E. (1994). Reciprocal binding properties of 5HT2C receptor agonists and inverse agonists. Mol. Pharmacol. 46, 937–942.
Wienecke, J., Zhang, M., and Hultborn, H. (2009). A prolongation of the postspike afterhyperpolarization following spike trains can partly explain the lower firing rates at derecruitment than those at recruitment. J. Neurophysiol. 102, 3698–3710. doi: 10.1152/jn.90995.2008
Willoughby, D. S., Priest, J. W., and Jennings, R. A. (2000). Myosin heavy chain isoform and ubiquitin protease mRNA expression after passive leg cycling in persons with spinal cord injury. Arch. Phys. Med. Rehabil. 81, 157–163. doi: 10.1016/S0003-9993(00)90134-5
Yoshimura, M., and Furue, H. (2006). Mechanisms for the anti-nociceptive actions of the descending noradrenergic and serotonergic systems in the spinal cord. J. Pharmacol. Sci. 101, 107–117. doi: 10.1254/jphs.CRJ06008X
Zapata, P. (1966). Peripheral and central factors in the pathophysiology of spinal shock. Acta. Physiol. Lat. Am. 16, 266–277.
Zewdie, E. T., Roy, F. D., Yang, J., and Gorassini, M. (2011). Increase in the excitability of spinal inhibitory pathways from intensive locomotor training after incomplete spinal cord injury. Clin. Neurophysiol. 122:S177. doi: 10.1016/S1388-2457(11)60641-X
Keywords: serotonin, noradrenaline, motoneuron, persistent inward currents, reflexes, spinal cord injuries
Citation: D'Amico JM, Condliffe EG, Martins KJB, Bennett DJ and Gorassini MA (2014) Recovery of neuronal and network excitability after spinal cord injury and implications for spasticity. Front. Integr. Neurosci. 8:36. doi: 10.3389/fnint.2014.00036
Received: 05 November 2013; Accepted: 17 April 2014;
Published online: 12 May 2014.
Edited by:
Martin Oudega, University of Pittsburgh, USAReviewed by:
Robert M. Brownstone, Dalhousie University, CanadaChristine Thomas, University of Miami, USA
Copyright © 2014 D'Amico, Condliffe, Martins, Bennett and Gorassini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monica A. Gorassini, Department of Biomedical Engineering, Faculty of Medicine and Dentistry, Centre for Neuroscience, University of Alberta, 5-005 Katz Group—Rexall Centre, Edmonton, AB T6G 2E1, Canada e-mail:bW9uaWNhLmdvcmFzc2luaUB1YWxiZXJ0YS5jYQ==