

 Scott J. Barton
Scott J. Barton- Program in Neuroscience and Department of Psychological and Brain Sciences, Indiana University, Bloomington, IN, USA
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder that targets the corticostriatal system and results in progressive deterioration of cognitive, emotional, and motor skills. Although cortical and striatal neurons are widely studied in animal models of HD, there is little information on neuronal function during expression of the HD behavioral phenotype. To address this knowledge gap, we used chronically implanted micro-wire bundles to record extracellular spikes and local field potentials (LFPs) in truncated (R6/1 and R6/2) and full-length (knock-in, KI) mouse models as well as in transgenic HD rats (tgHD rats) behaving in an open-field arena. Spike activity was recorded in the striatum of all models and in prefrontal cortex (PFC) of R6/2 and KI mice, and in primary motor cortex (M1) of R6/2 mice. We also recorded LFP activity in R6/2 striatum. All HD models exhibited altered neuronal activity relative to wild-type (WT) controls. Although there was no consistent effect on firing rate across models and brain areas, burst firing was reduced in striatum, PFC, and M1 of R6/2 mice, and in striatum of KI mice. Consistent with a decline in bursting, the inter-spike-interval coefficient of variation was reduced in all regions of all models, except PFC of KI mice and striatum of tgHD rats. Among simultaneously recorded neuron pairs, correlated firing was reduced in all brain regions of all models, while coincident bursting, which measures the temporal overlap between bursting pairs, was reduced in striatum of all models as well as in M1 of R6/2s. Preliminary analysis of striatal LFPs revealed aberrant behavior-related oscillations in the delta to theta range and in gamma activity. Collectively, our results indicate that disrupted corticostriatal processing occurs across multiple HD models despite differences in the severity of the behavioral phenotype. Efforts aimed at normalizing corticostriatal activity may hold the key to developing new HD therapeutics.
Introduction
The striatum receives input from all areas of cerebral cortex and uses that information to guide behavior. Corticostriatal processing plays a critical role in decision making, habit formation, movement selection, and reward expectancy (Alvarez and Eichenbaum, 2002; Costa et al., 2004; Graybiel, 2008; Israel and Bergman, 2008). In fact, cortical neurons are the primary driver of striatal neuronal activity. Without cortical input, striatal neurons are silent owing to an inwardly rectifying K+ current that keeps neuronal membranes hyperpolarized (Wilson and Kawaguchi, 1996). Glutamate released from cortical afferents increases striatal excitability, and when this input is coordinated across large numbers of afferents, the resulting activation of striatal circuits drives downstream processing through the rest of the basal ganglia (Wickens and Wilson, 1998). Thus, the striatum selects and refines the cortical signals that shape behavioral output.
Huntington’s disease (HD), an autosomal dominant disorder caused by expansion of a translated CAG (glutamine) repeat in the N-terminal domain of the Huntingtin (HTT) protein (Huntington’s Disease Collaborative Research Group, 1993), results in widespread pathology of the corticostriatal system (Vonsattel and Figlia, 1998). Autopsy of end-stage HD patients reveals substantial degeneration and loss of medium spiny neurons, which account for more than 90% of the striatal neuronal population (Groves, 1983). Medium spiny neurons are the sole output system of the striatum, and they receive massive glutamate input from cortical pyramidal cells, which also undergo substantial degeneration and loss. Although damage occurs in other brain regions, corticostriatal pathology appears to be the primary cause of the cognitive and motor abnormalities that characterize HD (Lawrence et al., 1998).
Various rodent models of HD have been developed with the goal of identifying pathogenic mechanisms that can speed the search for safe and effective treatments. Different approaches have been used to model the HD phenotype. One approach, exemplified by the R6 line of mice, involves expression of the N-terminal fragment consisting of either the first exon or the first 171 amino acids (Mangiarini et al., 1996; Schilling et al., 1999). The result is expression of the truncated mutant HTT protein and an early and robust motor phenotype. The emergence of this phenotype soon after weaning has made the R6/2 model one of the most widely used in the study of HD (Heng et al., 2008). R6/1 mice also show a robust phenotype but, because of a smaller polyglutamine expansion, onset occurs several weeks later than in R6/2 mice. In these and other truncated models, the polyglutamine expansion occurs independently of its natural genomic and protein context (Ehrnhoefer et al., 2009). An alternative approach is to express the full-length human mutant HTT as exemplified by the knock-in (KI) model, which has the polyglutamine expansion inserted or “knocked in” to the endogenous mouse gene (Shelbourne et al., 1999). In this case, the KI model contains a chimeric mouse–human HD gene, and thus the natural genomic and protein context of the polyglutamine expansion is largely preserved. Relative to the R6 line, KI mice display a less intense and later onset phenotype (Dorner et al., 2007; Menalled et al., 2009). Interestingly, assessments of both truncated and full-length mouse models indicate that the phenotype emerges well before substantial neuronal loss (Hickey et al., 2008). In fact, neuronal loss is not a primary feature of these models, suggesting that neuronal dysfunction, rather than loss per se, plays a critical role in the HD behavioral phenotype – an idea supported by clinical evaluations (Gutekunst et al., 2002; Palop et al., 2006).
To determine if functional deficits in corticostriatal neuronal processing is a common feature of HD models, we previously assessed the activity of individual striatal and prefrontal cortical (PFC) neurons in both R6/2 and KI mice (Miller et al., 2008b; Walker et al., 2008) as they behaved in an open-field arena where they could engage in naturally occurring episodes of spontaneous behavior. To assess the generality of our findings, we also recorded open-field striatal firing patterns in a transgenic HD rat (tgHD rat) model (Miller et al., 2010), which expresses a truncated fragment of the HD gene (von Hörsten et al., 2003). Overall, corticostriatal activity patterns are markedly altered in the HD animals relative to wild-type (WT) controls. To extend and update our previous findings to other HD models and other brain regions, we present here preliminary data indicating altered neuronal activity from motor cortex (M1) of R6/2 mice, and striatum of R6/1 mice. We also present early evidence from R6/2 striatum that local field potentials (LFPs), which represent the peri-synaptic activity of a large number of neurons, are altered in HD.
Materials and Methods
Detailed methods can be found in Miller et al. (2008b, 2010), and Walker et al. (2008).
Animals
Transgenic R6/1 and R6/2 mice (B6CBA–TgN[HDexon1]62Gpb) contain exon 1 of the human HD gene and are based on the C57BL/6 and CBA background strains (Mangiarini et al., 1996). The R6/2 mouse is characterized by a rapidly progressive phenotype with onset at ∼4 weeks and death in ∼16 weeks (Mangiarini et al., 1996; Carter et al., 1999; Levine et al., 2004). R6/1 mice are similar to R6/2s, albeit with a shorter repeat length and a later onset phenotype (Mangiarini et al., 1996). Homozygous KI (CAG 140) mice express a chimeric mouse/human exon 1 of the HD gene inserted into the mouse gene via homologous targeting of W9.5 ES cells from a 129sv background strain (Menalled et al., 2003). Homozygous tgHD rat have a Sprague-Dawley background and express a truncated HTT cDNA fragment with 51 CAG (polyglutamine) repeats under control of the native rat HTT promoter (von Hörsten et al., 2003). Both the KI mouse and tgHD rat express a late onset HD phenotype, relative to R6/1 and R6/2 mice.
All animals, including corresponding WTs for each HD model, were housed individually under standard conditions (12-h light/dark cycle with lights on at 07:30 h) with access to food and water ad libitum. All housing and animal-use procedures followed NIH guidelines and were approved by the Indiana University Institutional Animal Care and Use Committee.
Surgical Procedures
All animals were anesthetized as appropriate (see Miller et al., 2008b, 2010) and mounted in a stereotaxic frame. The scalp was shaved, swabbed with betadine, and after lidocaine (20 mg/ml) was injected subcutaneously, an incision was made at the midline to expose the skull. Trepanations were made over the appropriate brain area according to mouse and rat brain atlases (Paxinos and Watson, 1998; Franklin and Paxinos, 2008). Multi-wire electrode bundles were lowered into each region. Additional holes were drilled for stainless steel anchor screws. Electrode assemblies were permanently attached to the skull with dental acrylic. Antibiotics were applied to the surgical site to prevent infection. Lactated Ringer’s solution was administered subcutaneously to counteract dehydration. All animals were allowed 1 week of post-surgical recovery.
In vivo Electrophysiology
Electrode assemblies were made in-house and consisted of either four or eight, 25–50 μm Formvar-insulated stainless steel recording wires (California Fine Wire Company, Grover Beach, CA, USA) and one, 50 μm uninsulated stainless steel ground wire assembled in a custom fabricated hub. Electrode impedance was consistently ∼1 MΩ. The electrode assembly was small, light-weight, and well-tolerated by all animals so that they could behave freely.
All experiments were conducted during the light phase of the diurnal cycle and were 30 min to 1 h in duration. Animals were placed in an open-field arena located in a sound-attenuated and electrically shielded recording chamber. Animals explored freely during the entire recording session. For recording, the electrode assembly was connected to a light-weight flexible harness equipped with field-effect transistors that provide unity-gain current amplification for each of the micro-wires. Extracellular neuronal action potentials and LFPs were routed through preamplifiers with 500× gain and 154 Hz to 8.8 kHz band-pass filters for spikes, and 1,000× gain and 0.7–170 Hz filters for LFPs. All signals were digitized at 40 kHz and acquired by the Multichannel Acquisition Processor system (Plexon, Dallas, TX, USA). Spikes were sorted online prior to the recording session. Voltage thresholds ≥2.5 times background noise were established, and waveform samples (∼1,000) were collected to define a template via principal component analysis. The raw signal for each spike was routed to an oscilloscope and audio monitor to facilitate action potential discrimination by matching the analog signal with the digitized template. To maximize the probability that spikes consisted of only one signal (i.e., one neuron source), autocorrelation and inter-spike-interval (ISI) analyses were used to detect the presence of the absolute refractory period. In some cases, Spike 2 software (Cambridge Electronic Design, Cambridge, England) was used to confirm signal isolations offline.
Behavioral Analysis
Mouse behaviors for all electrophysiological sessions were recorded by videotape and coded by observers who were blind to genotype. We coded open-field behavioral activity (e.g., ambulation, grooming, rearing, sniffing), and quiet rest, which was defined as absence of these and other overt behaviors. Rat behaviors were recorded using an open-field force-plate actometer, which provides a multivariate behavioral sensing arena (Fowler et al., 2001, 2009; Miller et al., 2010).
Histology
Electrode placement in each brain area was verified before analysis. Animals were deeply anesthetized and a current pulse (30 μA for 5 s) was passed through each micro-wire to mark recording sites. Animals were transcardially perfused with saline followed by 10% potassium ferrocyanide [K4Fe(CN)6] in 10% formalin to produce blue deposits at the site of the recording electrode (“Prussian blue” reaction). Brains were removed and cryoprotected in 30% sucrose dissolved in 10% formalin. The brains were then frozen, and coronal sections (50 μM) were mounted on gelatin subbed slides to confirm micro-wire location.
Data Analysis
Neuronal data were analyzed by NeuroExplorer (Nex Technologies, Littleton, MA, USA) and custom written Matlab scripts (Mathworks, Natick, MA, USA). Statistical analyses used GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) and SigmaPlot 11 (Systat Software, San Jose, CA, USA), and the alpha level of significance was p < 0.05. Timestamps of all waveforms obtained from recording sessions were included for analysis. Firing rate was calculated by dividing the spike-trains into 1 s bins (spikes/s). To assess spike-train variability, the coefficient of variation of inter-spike-intervals (CV ISI) was calculated by dividing the SD of all ISIs in a train by the mean ISI of the train. Note that a CV = 1 indicates a Poisson and a CV = 0 is a completely regular spike-train. A CV > 1 indicates a complex spike-train pattern and often indicates bursting activity.
To quantify burst activity in spike-trains, the Poisson surprise algorithm was used (Legendy and Salcman, 1985). We used a minimum burst surprise value of five, which estimates that bursts occur ∼150 times (p < 0.007) more frequently than would be expected in a Poisson spike-train with the same mean firing rate. The surprise value provides an estimate of the statistical significance of each burst in the spike-train. Therefore, the surprise value is an index of how intense or “surprising” the ISIs of a particular burst is compared with other ISIs in the same train. This method is a rigorous detector of bursts because it is not sensitive to fluctuations in average firing rate and treats each spike-train as an independent source (Legendy and Salcman, 1985). The method, moreover, is well-established for detecting bursts in striatum and cortical structures (Aldridge and Gilman, 1991; Homayoun et al., 2005; Wichmann and Soares, 2006). We used it to measure various indices of bursts (e.g., burst rate, burst duration, ISI in bursts, percent of spikes in the train that participate in burst activity, and the burst surprise value).
Coincident bursts were defined as the number of bursts from two neurons that overlap in time (Lisman, 1997, Miller et al., 2008b, 2010). The mean time that bursts were coincident (coincidence duration), also was calculated. Coincident bursting and coincidence duration were determined for each pair-wise comparison in each session.
To assess correlated and synchronous firing (coherent firing) between two spike-trains, cross-correlation histograms (CCHs) were constructed for each pair-wise comparison (Perkel et al., 1967) in each recording session. All CCHs were constructed based on 1 ms bins and either a ±1-s (striatum) or ±0.5 s (cortex) time lag from the zero bin. The CCHs were smoothed using a Gaussian filter with a bin width of three. Significant peaks, which indicate correlated and synchronous firing, were identified using a 95% confidence interval by assuming the null hypothesis that each spike-train is a Poisson process and that firing between neuronal pairs is independent (i.e., flat cross-correlogram; Abeles 1982).
Local field potential oscillations were assessed by analyzing power spectral densities (Fourier transforms) and spectrograms (frequency through time plots) generated in NeuroExplorer. LFP data were collected during discrete behaviors of quiet rest (lack of behavioral activation; see above), rearing and grooming. Each behavioral episode lasted a minimum of 3 s and LFP data were time-locked to the epoch. Power spectral densities were constructed by averaging LFP data for three different mice (three trials per mouse) for each behavior.
Results
Spike Electrophysiology
Table 1 provides information on all the HD models used in our recording experiments (for comprehensive reviews of various HD rodent models see Levine et al., 2004; Heng et al., 2008). All our electrophysiological data are based on comparisons between HD animals and their respective WTs. All mouse models were recorded at varying stages of the disease from periods of early phenotype expression through later stages. Because of a longer period of phenotype expression, both the R6/1 and KI models are especially useful for this type of analysis, but in all cases, the electrophysiological changes that were evident early on persisted through later stages of disease progression. Thus, data for each model were combined across recording sessions. Assessment of the tgHD rat cohort occurred when animals expressed a mild, but stable phenotype.

Table 1. Characteristics of genetic rodent models of HD.
Table 2 summarizes our spike electrophysiology results. Neurons were putatively labeled as either medium spiny in striatum or pyramidal in cortex based on well-established waveform and firing properties (see Miller et al., 2008b; Walker et al., 2008, and Discussion for details). We assessed the rate and pattern of spike activity of individual neurons as well as the correlated activity of simultaneously recorded neuronal pairs.
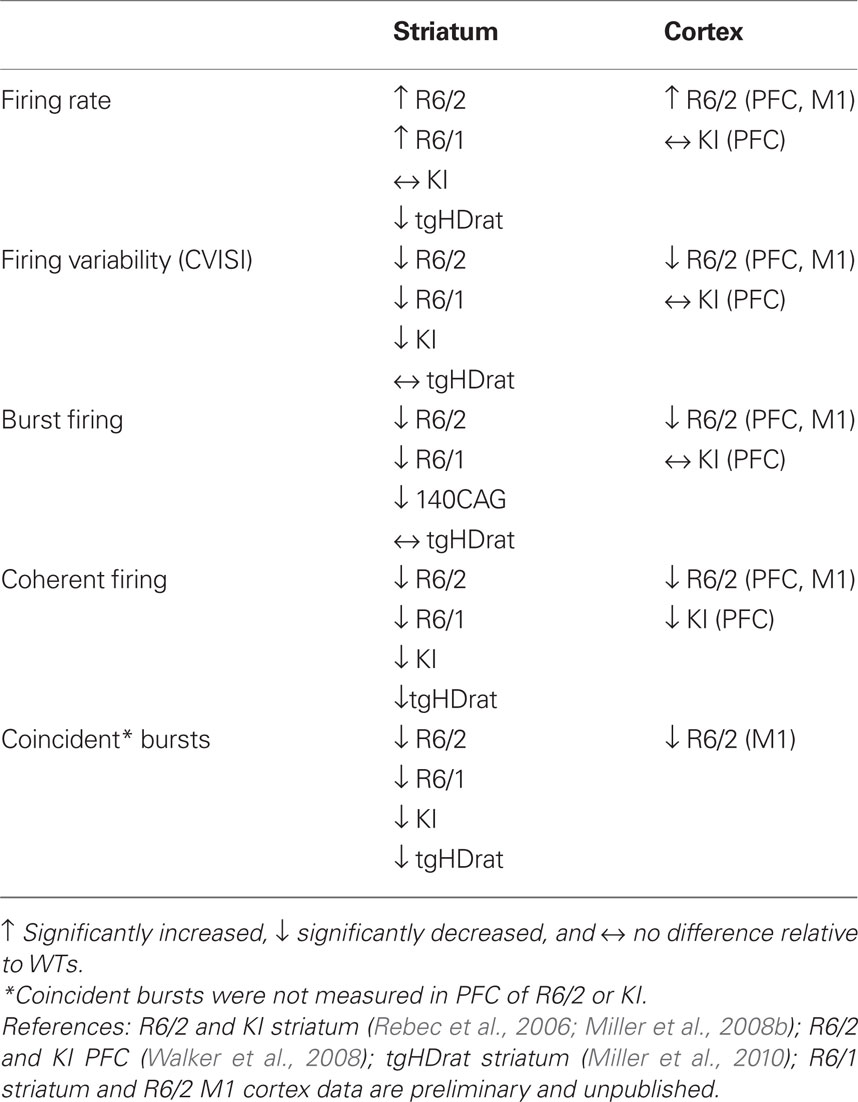
Table 2. Summary of neuronal activity altered in HD models in vivo.
Changes in rate
Neuronal firing rate provides a metric of the overall level of neuronal activity. Increased levels of firing were found in striatum of both R6 models, but not KI mice or tgHD rats, relative to WTs. In fact, rate was decreased in tgHD rats. Increased firing rate also was found in PFC of R6/2, but not KI mice, nor M1 cortex of R6/2s. It appears, therefore, that the R6 truncated mouse models, which show a robust phenotype, are prone to neuronal hyper-activity.
Changes in pattern
Spike pattern, reflected in the CV ISI and burst activity, represents a more detailed aspect of neuronal processing. The CV ISI is commonly used to assess spike-train variability, indicating complex firing patterns of activity (i.e., CV ISI > 1; see Materials and Methods). To quantify individual burst parameters, we used the Poisson burst surprise method to measure the percent of spikes that participate in bursts within each train as well as the burst surprise value (the higher the value the more “surprising” or prominent the burst). All our HD mouse models showed reductions in both CV ISI values and various aspects of bursting relative to WT. The only exceptions were tgHD striatum and the PFC of KI mice. Interestingly, the structure of individual bursts, which includes burst duration and ISI within bursts, was not altered in any model or brain area, suggesting that HD neurons have the capacity to burst, but lack the ability to generate a proper bursting pattern.
Both the CV ISI and the Poisson surprise methods rely on the deviation of the ISI from a Poisson distribution (Robin et al., 2009). In fact, as shown in Figure 1, a strong correlation exists between CV ISI and the percent of spikes in bursts. Note that bursts cluster near the origin in R6/2 mice indicating a reduction in the overall level of burst activity relative to WT.
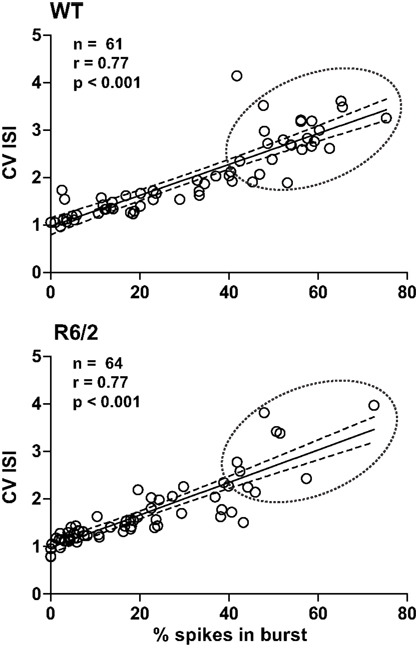
Figure 1. Metrics of burst firing. Plots illustrate the strong correlation between the percentage of spikes in bursts and CV ISI. Note that this correlation is true for both WT and R6/2 mice. Thus, CV ISI and % spikes in bursts are complimentary indicators of the pattern of burst firing. It is clear, however, that data in the R6/2 plot are clustered at the origin, indicating a reduction in a prominent burst pattern relative to WT. The solid line indicates the mean and the broken lines represent the 95% confidence intervals. The broken circle emphasizes the difference in distributions.
Reductions in coherent firing among neuron pairs
We also assessed the temporal relationship in spiking between simultaneously recorded pairs of neurons. For this analysis, we constructed cross-correlograms, and in each case, peaks in the histogram that exceeded the confidence interval were defined as significantly coherent neuronal pairs. Interestingly, as shown in Figure 2, the temporal dynamics between coherent spiking differs between cortical and striatal neurons. For example, cortical neuron pairs tend to spike in precise temporal or “synchronized” coherence (Sakurai and Takahashi, 2006; Walker et al., 2008), whereas striatal neuron pairs discharge with much broader temporal coherence (Miller et al., 2008b; Ponzi and Wickens, 2010). In all cases, however, we found a marked reduction in temporally coherent neuronal activity from all brain areas and all HD models recorded.
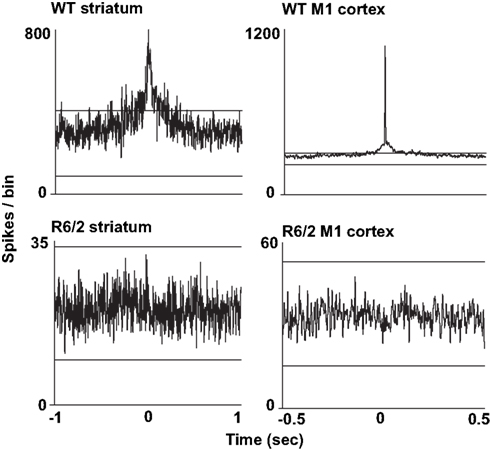
Figure 2. Representative cross-correlograms constructed from a pair of WT and R6/2 striatal neurons (left column) and a pair of WT and R6/2 M1 cortical neurons (right column). The two horizontal lines in each plot indicate the 95% confidence intervals. The two WT peaks exceed the upper confidence interval, indicating significance. Note the different time scales between striatum and cortical cross-correlograms indicating either correlated or synchronous firing, respectively. Striatal and M1 neuron pairs show markedly reduced coherent spike activity (flat and non-significant peak). We note that we have yet to find repetitive peaks and troughs in the cross-correlograms, which indicates oscillatory firing activity between neuronal pairs.
Reductions in coincident bursts among neuron pairs
We explored the temporal relationship in burst firing between neuron pairs by measuring the number of bursts that overlap in time (Lisman, 1997). Similar to coherent spikes, coincident bursts were reduced in all brain areas and HD models tested. Moreover, in striatum of R6/2 and KI mice and their respective WTs, a higher percentage of coincident bursts occurs in correlated than non-correlated neuronal pairs, suggesting that coherent bursting contributes to coherent spike activity (Miller et al., 2008b).
Figure 3 shows a sample of spike rasters recorded from individual WT and R6/2 mice. Altered firing properties are apparent at the single-neuron level (firing rate, bursts) and at the pair-wise level (coherent firing, coincident bursts). These activity patterns are comparable to what we have reported for striatum and PFC of other HD models (Miller et al., 2008b, 2010; Walker et al., 2008).

Figure 3. Dysregulated neuronal firing patterns among populations of neurons. Long-timescale (60 s) spike rasters of four simultaneously recorded striatal neurons from WT and R6/2 mice. The horizontal lines above each raster define bursts. Arrows represent representative instances of coincident bursts in the WT raster. It is evident that population activity in WT mice is more phasic (more complex) and bursts are often temporally coherent across neurons relative to R6/2 mice.
Striatal LFPs
We have begun to investigate large ensembles of neurons by recording LFPs, which capture the sum of pre- and post-synaptic neuronal activity. In striatum, ample evidence indicates that LFPs represent a form of local processing, rather than volume conduction from other brain regions (Berke et al., 2004; DeCoteau et al., 2007). To date, we have recorded LFPs from dorsal striatum of three WT and three R6/2 mice during discrete, behaviorally matched epochs (minimum of three epochs per animal) in the open-field. During epochs of quiet rest, striatal LFP oscillations from WT mice were characterized by prominent delta to low theta oscillations (<5–7 Hz); the same was true for R6/2 mice, although a second prominent gamma band (30–40 Hz) oscillation also appeared (Figure 4). Behavioral episodes of rearing, which were strictly defined as upper forelimb rears on the side of the open-field for a minimum of 3 s, revealed delta oscillations in WT and R6/2 mice, but with an additional strong theta band (7–14 Hz) in the R6/2 model. In contrast, episodes of grooming showed no difference in LFP oscillations between R6/2 and WT.
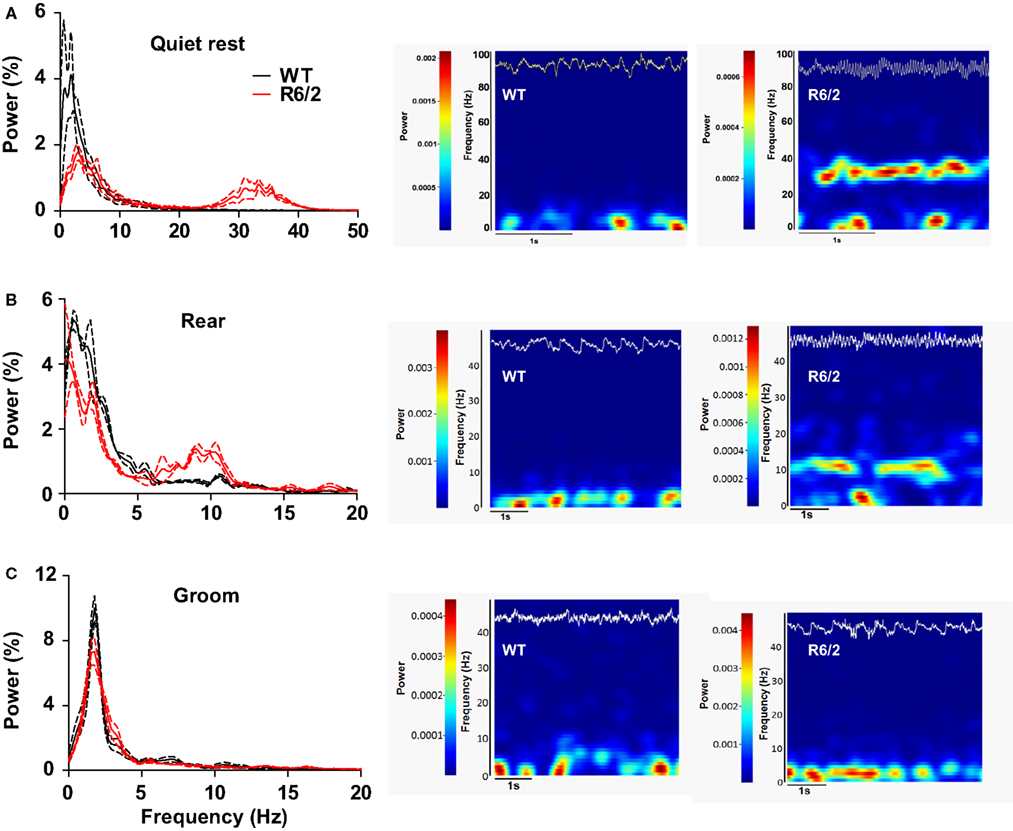
Figure 4. Dysregulated striatal LFP oscillations in R6/2 mice. [(A), left] Power spectral densities of LFPs in striatum during quiet rest. Prominent 30–40 Hz (gamma) oscillations are present in R6/2 and absent in WT. Solid lines represent the mean density and broken lines indicate ±SEM. [(A), right] Representative spectrograms (frequency through time) of LFP activity during a single epoch of quite rest from a WT and R6/2 mouse. Frequency and power are on the y axis; hotter colors represent greater power. The top trace (white) on each spectrogram is the corresponding raw LFP record (to scale). [(B), left and right] Same as (A), but striatal LFPs recorded during rearing. Note the prominent 10 Hz (theta) oscillation in R6/2 and its absence in WTs. [(C), left and right] Striatal LFPs during epochs of grooming reveal no difference in LFP oscillations between the genotypes.
Discussion
Our results indicate that corticostriatal processing is altered in behaving, symptomatic HD rodent models compared to WT controls. This effect is evident in all the models we tested, albeit with some differences that may be related to behavioral phenotype or genetic background of the HD model. At the single-neuron level, for example, firing rate is elevated in strongly symptomatic R6/2 mice, but not in mildly symptomatic KI mice. Similarly, PFC burst firing is altered in R6/2s, but not KIs. In striatum, however, burst firing is attenuated in all mouse models. Interestingly, deficits in population-level neuronal processing occur in all brain regions. For example, analysis of cross-correlograms indicates coherent spiking is reduced in all HD models. Coincident bursting (i.e., temporally correlated burst activity between simultaneously recorded neurons) and oscillations of LFPs also show alterations in HD. Thus, neuronal firing patterns and their temporal dynamics are impaired in the HD corticostriatal system during spontaneous open-field behavior.
Our assessments focused on open-field behavior for two reasons. First, open-field testing is conducive to the long-duration (30 min to 1 h) recording sessions typically required for in vivo electrophysiology. For example, analysis of bursting, CV ISI, coincident bursting, and construction of reliable cross-correlograms (a measure of coherent firing between neurons; see Materials and Methods) require records of many hundreds of spikes. Without sufficient spike data, the fidelity of these metrics is diminished. Second, coherent spike activity in both cortex (Constantinidis et al., 2002; Putrino et al., 2010) and striatum (Kimura et al., 2003), as well as striatal LFPs (Barnes et al., 2005), have been shown to be differentially driven by task performance, but rodent HD models show robust deficiencies in performing skilled behavioral tasks (Brooks and Dunnett, 2009). Such tasks, moreover, promote synaptic plasticity, which is compromised in cortex (Cummings et al., 2006, 2007) and striatum (Milnerwood et al., 2006) of HD models. In contrast, natural spontaneous open-field behaviors do not require prolonged learning periods and are unlikely to depend on overt changes in synaptic plasticity. Thus, animals were studied in an open-field arena, which allowed for analysis of multiple, ongoing patterns of neuronal activity.
All HD mouse models showed a progressive behavioral phenotype relative to WT controls (Miller et al., 2008b; Walker et al., 2008). Although tgHD rats lose fine motor skills and develop prominent cognitive deficits at the ages used in our analysis (von Hörsten et al., 2003; Cao et al., 2006; Nguyen, 2006), we found no overt phenotype in the open-field (Miller et al., 2010). In fact, there was no substantial difference in spontaneous behavior (motor activity vs. quiet rest) between the genotypes of each HD model. Thus, it is unlikely that any differences that emerged in our electrophysiological data could be explained simply by differences in behavioral activity in the open-field, but instead are likely to reflect neuronal circuit differences between genotypes (e.g., WT vs. HD). Individual HD models, however, showed some electrophysiological differences (see Table 2 for summary data; for HD phenotype references see Levine et al., 2004; Heng et al., 2008). These differences could emerge from the genetic background (i.e., strain and species) of each model, which in turn could influence firing activity of corticostriatal circuits. Interestingly, none of the tested models showed a clear trend for corticostriatal activity to worsen through HD progression. This outcome, however, is not entirely unexpected since the disruption in neuronal activity is prominent early on, even in the first recording session. It is likely, therefore, that corticostriatal activity is an early event in HD and persists throughout HD progression. Indeed, similar to our in vivo results, ex vivo cortical culture networks grown on microelectrode arrays and infected with mutant HTT fragments show reduced spontaneous collective burst firing (Gambazzi et al., 2010). Thus, the mutant HTT protein dramatically changes spontaneous network activity.
In WT striatum, neuronal activity is relatively slow but highly irregular as reflected in a CV ISI > 1 along with prominent episodes of spike bursting. In fact, irregular firing is a common feature of striatal medium spiny neurons (Wilson, 1993). That attenuation of this irregular pattern of activity occurs in most HD models that we have studied to date underscores the point that a simple change in rate (i.e., hyper-excitability) is not the fingerprint of striatal neuropathology. In fact, the change in pattern coupled with reductions in coordinated activity between simultaneously recorded neuronal pairs provides strong evidence for widespread dysfunction in cortical and striatal circuits. Our preliminary LFP data, which represents circuit rather than single-neuron activity, supports this view.
Circuit dysfunction indicates fundamental problems in neuronal communication, and evidence now suggests that a change in the extracellular dynamics of glutamate, an excitatory amino acid that shapes the pattern of both cortical and striatal activity, plays a critical role in HD (Miller and Bezprozvanny, 2010). In striatum, for example, the clearance of extracellular glutamate is significantly decreased in R6/2 relative to WT mice (Miller et al., 2008a). This effect, moreover, is evident early in the progression of the HD behavioral phenotype and can be reversed by increased expression of GLT1 (Miller et al., 2008a), the astroglial glutamate transporter responsible for the uptake of most extracellular glutamate (Danbolt, 2001). Increasing GLT1 expression also ameliorates the HD behavioral phenotype (Miller et al., 2008a). Failure of glutamate uptake likely contributes to the neurodegeneration that occurs over the course of HD, which is driven in part by activation of extrasynaptic glutamate receptors (Milnerwood et al., 2010). Consistent with this view, deficits in glutamate uptake have been identified in post-mortem striatal tissue obtained from HD patients (Hassel et al., 2008). Dysregulation of glutamate dynamics, therefore, could be a key mechanism underlying our electrophysiological results in HD models. Striatal neurons also receive glutamate input from midline thalamus (Smith and Bolam, 1990), which may comprise another source of glutamate dysfunction in HD (Carroll et al., 2011).
Interneurons also may play a role. In both cortex and striatum, interneurons control the timing and synchrony of spikes generated by output neurons (Hestrin and Galarreta, 2005; Tiesinga et al., 2008). Interestingly, mutant HTT disrupts the mechanism by which receptors for GABA, an inhibitory amino acid released by cortical and striatal interneurons, are trafficked to synapses (Twelvetrees et al., 2010). Thus, loss of GABA-mediated inhibition may interact with deficits in glutamate clearance to disrupt corticostriatal processing in HD.
Striatal medium spiny neurons can be identified apart from other striatal neuron types (e.g., interneurons) by a combination of firing rate and spike waveform shape (Berke et al., 2004; Sharott et al., 2009). But because of the altered firing patterns recorded in a portion of our HD samples, we took a conservative approach and pooled all neurons for analysis. Note, however, that we ensured that all waveform shapes were consistent with putative medium spiny neurons (Berke et al., 2004; Miller et al., 2008b, 2010). It also is relevant that medium spiny neurons comprise >90% of neurons in striatum (Groves, 1983), making other neuron types less likely to be sampled. Moreover, we recognize that medium spiny neurons are not homogeneous. In fact, there is evidence that striato-pallidal neurons, which represent the so-called indirect pathway, degenerate earlier than the striato-nigral or direct pathway in HD patients (Deng et al., 2004; Starr et al., 2008). It is possible, therefore, that medium spiny neurons in our HD mice comprise two distinct populations. Nevertheless, a sizable number of striatal output neurons contribute axon collaterals to both the direct and indirect pathways (Levesque and Parent, 2005), making it difficult to speculate on the implications of possible differences between striato-pallidal and striato-nigral neurons in HD.
Like medium spiny neurons in striatum, cortical neurons can be classified by firing rate and waveform into fast-spiking (>10 spikes/s with narrow after hypolarizations, AHPs) and regular-spiking (<10 spikes/s with wide AHPs) neurons, which are thought to correspond to interneurons and pyramidal cells, respectively (McCormick et al., 1985; Connors and Gutnick, 1990; Homayoun and Moghaddam, 2007). In our PFC investigation (Walker et al., 2008), to determine if >10 spikes/s represents a separate class of neurons, we plotted the average of 50 waveforms and measured the AHPs. These data were compared to a random sample of AHPs from those classified as regular-spiking cells based on firing rate. Less than 10% of the cortical neurons (∼500) recorded in our PFC investigation could be identified as fast-spiking neurons. Although some of these may be interneurons, our subsequent analysis of AHPs revealed no difference from regular-spiking AHPs, suggesting that our cortical neurons comprise a homogeneous population of presumed pyramidal cells.
Early studies of LFPs suggested that they arose mainly from laminar structures, while detectable oscillations in non-laminar areas (e.g., striatum) were thought to result from simple volume conduction. Ample evidence now argues against this view (Berke et al., 2004; Gatev et al., 2006; DeCoteau et al., 2007; Boraud et al., 2008). For example, entrainment of striatal neurons to LFP oscillations occurs at both beta and theta frequencies (Courtemanche et al., 2003; Berke et al., 2004; DeCoteau et al., 2007). It also is relevant that during periods of quiet rest our WT mice show a prominent LFP oscillation in the theta band (Figure 4), a finding in line with what has been reported in striatum of resting rats (DeCoteau et al., 2007). In fact, data obtained from M1 cortex and striatum indicate that the mechanism underlying LFP oscillations is similar in rat and mouse (Costa et al., 2006). LFPs are key features of behaviorally relevant neuronal processing (Buzsaki, 2006) and thus can make an important contribution to understanding the neural basis of the HD behavioral phenotype, apart from spike pattern analysis. It also appears that LFPs are strongly influenced by glutamate (Buzsaki, 2006), making their assessment in HD models especially appropriate.
Numerous in vitro studies of HD models report corticostriatal synaptic dysfunction (for reviews see Cepeda et al., 2007, 2010; Miller and Bezprozvanny, 2010), which may underlie the altered activity patterns observed in our HD animals. Although no one synaptic problem can be pinpointed, it is clear that striatal medium spiny neurons are hyperactive, which is in part due to a progressive increase in input resistance, reduction in cell capacitance, exaggerated glutamate-dependent responses, and increased intracellular calcium levels (see above, and Cepeda et al., 2001; Bezprozvanny and Hayden, 2004). On the other hand, subpopulations of striatal neurons show a decrease in spontaneous excitatory post-synaptic currents and an increase in spontaneous inhibitory post-synaptic currents (Cummings et al., 2010). Recordings from cortical neurons in multiple HD models has revealed marked changes in synaptic events, including increased frequencies of spontaneous excitatory post-synaptic currents, a higher magnitude of evoked excitatory post-synaptic currents, and biphasic changes in inhibitory post-synaptic currents (Cummings et al., 2009). Interestingly, changes in striatal and cortical synaptic transmission are consistent across various genetic models of HD (Cummings et al., 2009, 2010). Therefore, it will be important to distinguish which facets of synaptic transmission in HD underscore the changes in neuronal activity patterns.
Conclusion
Our results point to corticostriatal processing deficits as an underlying feature of the HD behavioral phenotype. Because these deficits are present early in the progression of the disease, they likely set the stage for the increase in symptom severity that emerges later when neuronal degeneration and loss become prominent. Thus, it would be useful to assess HD models even before the behavioral phenotype emerges not only to gage the onset of neuronal dysfunction but also to establish a target for testing new, more effective therapeutic strategies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by R01 NS 035663, F31 NS 064791, F31 066603, and by the Indiana METACyt Initiative of Indiana University, funded in part through a major grant from the Lilly Endowment, Inc. Jason Ummel, Lauren Walker, Prianka Chilukuri, Wajeeha Hussain, and Desiree Cossyleon assisted with data collection. We also acknowledge the technical support of Paul Langley and the editorial assistance of Faye Caylor.
References
Abeles, M. (1982). Quantification, smoothing, and confidence limits for single-units’ histograms. J. Neurosci. Methods 5, 317–325.
Aldridge, J. W., and Gilman, S. (1991). The temporal structure of spike trains in the primate basal ganglia: afferent regulation of bursting demonstrated with precentral cerebral cortical ablation. Brain Res. 543, 123–138.
Alvarez, P., and Eichenbaum, H. (2002). Representations of odors in the rat orbitofrontal cortex change during and after learning. Behav. Neurosci. 116, 421–433.
Barnes, B. W., Liljeholm, M., and Ostlund, S. B. (2005). Activity of striatal neurons reflects dynamic encoding and recoding of procedural memories. Nature 437, 1158–1161.
Berke, J. D., Okatan, M., Skurski, J., and Eichenbaum, H. B. (2004). Oscillatory entrainment of striatal neurons in freely moving rats. Neuron 43, 883–896.
Bezprozvanny, I., and Hayden, M. R. (2004). Deranged calcium signaling and Huntington’s disease. Biochem. Biophys. Res. Commun. 322, 1310–1317.
Boraud, T., Brown, P., Goldberg, J. A., Graybiel, A. M., and Magill, P. J. (2008). “Oscillations in the basal ganglia: the good, the bad, and the unexpected,” in The Basal Ganglia VIII, eds J. P. Bolam, C. A. Ingham, and P. J. Magill (New York: Springer Science and Business Media), 3–22.
Brooks, S. P., and Dunnett, S. B. (2009). Tests to assess motor phenotype in mice: a user’s guide. Nat. Rev. Neurosci. 10, 519–529.
Cao, C., Temel, Y., Blokland, A., Ozen, H., Steinbusch, H. W., Vlamings, R., Nguyen, H. P., von Horsten, S., Schmitz, C., and Visser-Vandewalle, V. (2006). Progressive deterioration of reaction time performance and choreiform symptoms in a new Huntington’s disease transgenic ratmodel. Behav. Brain Res. 170, 257–261.
Carroll, J. B., Lerch, J. P., Franciosi, S., Spreeuw, A., Bissada, N., Henkelman, R. M., and Hayden, M. R. (2011). Natural history of disease in the YAC128 mouse reveals a discrete signature of pathology in Huntington’s disease. Neurobiol. Dis. PMID: 21458571. [Epub ahead of print].
Carter, R. J., Lione, L. A., Humby, T., Mangiarini, L., Mahal, A., Bates, G. P., Dunnett, S. B., and Morton, A. J. (1999). Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. 19, 3248–3257.
Cepeda, C., Ariano, M. A., Calvert, C. R., Flores-Hernandez, J., Chandler, S. H., Leavitt, B. R., Hayden, M. R., and Levine, M. S. (2001). NMDA receptor function in mouse models of Huntington disease. J. Neurosci. Res. 66, 525–539.
Cepeda, C., Cummings, D. M., André, V. M., Holley, S. M., and Levine, M. S. (2010). Genetic mouse models of Huntington’s disease: focus on electrophysiological mechanisms. ASN Neuro 2, e00033.
Cepeda, C., Wu, N., Andre, V. M., Cummings, D. M., and Levine, M. S. (2007). The corticostriatal pathway in Huntington’s disease. Prog. Neurobiol. 81, 253–271.
Connors, B. W., and Gutnick, M. J. (1990). Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci. 13, 99–104.
Constantinidis, C., Williams, G. V., and Goldman-Rakic, P. S. (2002). A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat. Neurosci. 5, 175–180.
Costa, R. M., Cohen, D., and Nicolelis, M. A. (2004). Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr. Biol. 14, 1124–1134.
Costa, R. M., Lin, S. C., Sotnikova, T. D., Cyr, M., Gainetdinov, R. R., Caron, M. G., and Nicolelis, M. A. (2006). Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron 52, 359–369.
Courtemanche, R., Fujii, N., and Graybiel, A. M. (2003). Synchronous, focally modulated beta-band oscillations characterize local field potential activity in the striatum of awake behaving monkeys. J. Neurosci. 23, 11741–11752.
Cummings, D. M., Andre, V. M., Uzgil, B. O., Gee, S. M., Fisher, Y. E., Cepeda, C., and Levine, M. S. (2009). Alterations in cortical excitation and inhibition in genetic mouse models of Huntington’s disease. J. Neurosci. 29, 10371–10386.
Cummings, D. M., Cepeda, C., and Levine, M. S. (2010). Alterations in striatal synaptic transmission are consistent across genetic mouse models of Huntington’s disease. ASN Neuro 2, e00036.
Cummings, D. M., Milnerwood, A. J., Dallerac, G. M., Vatsavayai, S. C., Hirst, M. C., and Murphy, K. P. (2007). Abnormal cortical synaptic plasticity in a mouse model of Huntington’s disease. Brain Res. Bull. 72, 103–107.
Cummings, D. M., Milnerwood, A. J., Dallérac, G. M., Waights, V., Brown, J. Y., Vatsavayai, S. C., Hirst, M. C., and Murphy, K. P. (2006). Aberrant cortical synaptic plasticity and dopaminergic dysfunction in a mouse model of Huntington’s disease. Hum. Mol. Genet. 15, 2856–2868.
DeCoteau, W. E., Thorn, C., Gibson, D. J., Courtemanche, R., Mitra, P., Kubota, Y., and Graybiel, A. M. (2007). Oscillations of local field potentials in the rat dorsal striatum during spontaneous and instructed behaviors. J. Neurophysiol. 97, 3800–3805.
Deng, Y. P., Albin, R. L., Penney, J. B., Young, A. B., Anderson, K. D., and Reiner, A. (2004). Differential loss of striatal projection systems in Huntington’s disease: a quantitative immunohistochemical study. J. Chem. Neuroanat. 27, 143–164.
Dorner, J. L., Miller, B. R., Barton, S. J., Brock, T. J., and Rebec, G. V. (2007). Sex differences in behavior and striatal ascorbate release in the 140 CAG knock-in mouse model of Huntington’s disease. Behav. Brain Res. 178, 90–97.
Ehrnhoefer, D. E., Butland, S. L., Pouladi, M. A., and Hayden, M. R. (2009). Mouse models of Huntington disease: variations on a theme. Dis. Model Mech. 2, 123–129.
Fowler, S. C., Birkestrand, B. R., Chen, R., Moss, S. J., Vorontsova, E., Wang, G., and Zarcone, T. J. (2001). A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial patterning, stereotypies and tremor. J. Neurosci. Methods 107, 107–124.
Fowler, S. C., Miller, B. R., Gaither, T. W., Johnson, M. A., and Rebec, G. V. (2009). Force-plate quantification of progressive behavioral deficits in the R6/2 mouse model of Huntington’s disease. Behav. Brain Res. 202, 130–137.
Franklin, K., and Paxinos, G. (2008). The Mouse Brain in Stereotaxic Coordinates, 3rd Edn. New York: Academic Press.
Gambazzi, L., Gokce, O., Seredenina, T., Katsyuba, E., Runne, H., Markram, H., Giugliano, M., and Luthi-Carter, R. (2010). Factor expression underlies cortical neuron microcircuit hypoconnectivity resulting from exposure to mutant Huntingtin fragments. J. Pharmacol. Exp. Ther. 335, 13–22.
Gatev, P., Darbin, O., and Wichmann, T. (2006). Oscillations in the basal ganglia under normal conditions and in movement disorders. Mov. Disord. 21, 1566–1577.
Graybiel, A. M. (2008). Habits, rituals, and the evaluative brain. Annu. Rev. Neurosci. 31, 359–387.
Groves, P. M. (1983). A theory of the functional organization of the neostriatum and the neostriatal control of voluntary movement. Brain Res. 286, 109–132.
Gutekunst, C.-A., Norflus, F., and Hersch, S. M. (2002). “The neuropathology of Huntington’s disease,” in Huntington’s Disease, 3rd Edn, eds G. Bates, P. Harper, and L. Jones (New York: Oxford University Press), 251–275.
Hassel, B., Tessler, S., Faull, R. L., and Emson, P. C. (2008). Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 33, 232–237.
Heng, M. Y., Detloff, P. J., and Albin, R. L. (2008). Rodent genetic models of Huntington disease. Neurobiol. Dis. 32, 1–9.
Hestrin, S., and Galarreta, M. (2005). Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci. 28, 304–309.
Hickey, M. A., Kosmalska, A., Enayati, J., Cohen, R., Zeitlin, S., Levine, M. S., and Chesselet, M. F. (2008). Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington’s disease mice. Neuroscience 157, 280–295.
Homayoun, H., Jackson, M. E., and Moghaddam, B. (2005). Activation of metabotropic glutamate 2/3 receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats. J. Neurophysiol. 93, 1989–2001.
Homayoun, H., and Moghaddam, B. (2007). NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 27, 11496–11500.
Huntington’s Disease Collaborative Research Group. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983.
Israel, Z., and Bergman, H. (2008). Pathophysiology of the basal ganglia and movement disorders: from animal models to human clinical applications. Neurosci. Biobehav. Rev. 32, 367–377.
Kimura, M., Matsumoto, N., Okahashi, K., Ueda, Y., Satoh, T., Minamimoto, T., Sakamoto, M., and Yamada, H. (2003). Goal-directed, serial and synchronous activation of neurons in the primate striatum. Neuroreport 14, 799–802.
Lawrence, A. D., Sahakian, B. J., and Robbins, T. W. (1998). Cognitive functions and corticostriatal circuits: insights from Huntington’s disease. Trends Cogn. Sci. 2, 379–388.
Legendy, C. R., and Salcman, M. (1985). Bursts and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. J. Neurophysiol. 53, 926–939.
Levesque, M., and Parent, A. (2005). The striatofugal fiber system in primates: a reevaluation of its organization based on single-axon tracing studies. Proc. Natl. Acad. Sci. U.S.A. 102, 11888–11893.
Levine, M. S., Cepeda, C., Hickey, M. A., Fleming, S. M., and Chesselet, M. F. (2004). Genetic mouse models of Huntington’s and Parkinson’s diseases: illuminating but imperfect. Trends Neurosci. 27, 691–697.
Lisman, J. E. (1997). Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 20, 38–43.
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S. W., and Bates, G. P. (1996). Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506.
McCormick, D. A., Connors, B. W., Lighthall, J. W., and Prince, D. A. (1985). Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J. Neurophysiol. 54, 782–806.
Menalled, L., El-Khodor, B. F., Patry, M., Suárez-Fariñas, M., Orenstein, S. J., Zahasky, B., Leahy, C., Wheeler, V., Yang, X. W., MacDonald, M., Morton, A. J., Bates, G., Leeds, J., Park, L., Howland, D., Signer, E., Tobin, A., and Brunner, D. (2009). Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 35, 319–336.
Menalled, L. B., Sison, J. D., Dragatsis, I., Zeitlin, S., and Chesselet, M. F. (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J. Comp. Neurol. 465, 11–26.
Miller, B. R., and Bezprozvanny, I. (2010). Corticostriatal circuit dysfunction in Huntington’s disease: intersection of glutamate, dopamine and calcium. Future Neurol. 5, 735–756.
Miller, B. R., Dorner, J. L., Shou, M., Sari, Y., Barton, S. J., Sengelaub, D. R., Kennedy, R. T., and Rebec, G. V. (2008a). Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience 153, 329–337.
Miller, B. R., Walker, A. G., Shah, A. S., Barton, S. J., and Rebec, G. V. (2008b). Dysregulated information processing by medium-spiny neurons in striatum of freely behaving mouse models of Huntington’s disease. J. Neurophysiol. 100, 2205–2216.
Miller, B. R., Walker, A. G., Fowler, S. C., von Hörsten, S., Riess, O., Johnson, M. A., and Rebec, G. V. (2010). Dysregulation of coordinated neuronal firing patterns in striatum of freely behaving transgenic rats that model Huntington’s disease. Neurobiol. Dis. 37, 106–113.
Milnerwood, A. J., Cummings, D. M., Dallérac, G. M., Brown, J. Y., Vatsavayai, S. C., Hirst, M. C., Rezaie, P., and Murphy, K. P. (2006). Early development of aberrant synaptic plasticity in a mouse model of Huntington’s disease. Hum. Mol. Genet. 15, 1690–1703.
Milnerwood, A. J., Gladding, C. M., Pouladi, M. A., Kaufman, A. M., Hines, R. M., Boyd, J. D., Ko, R. W., Vasuta, O. C., Graham, R. K., Hayden, M. R., Murphy, T. H., and Raymond, L. A. (2010). Early increase in extrasynaptic nmda receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 65, 178–190.
Nguyen, P. V. (2006). Comparative plasticity of brain synapses in inbred mouse strains. J. Exp. Biol. 209, 2293–2303.
Palop, J. J., Chin, J., and Mucke, L. (2006). A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773.
Paxinos, G., and Watson, C. (1998). The Rat Brain in Stereotaxic Coordinates. New York: Academic Press.
Perkel, D. H., Gerstein, G. L., and Moore, G. P. (1967). Neuronal spike trains and stochastic point processes. II. Simultaneous spike trains. Biophys. J. 7, 419–440.
Ponzi, A., and Wickens, J. (2010). Sequentially switching cell assemblies in random inhibitory networks of spiking neurons in the striatum. J. Neurosci. 30, 5894–5911.
Putrino, D., Brown, E. N., Mastaglia, F. L., and Ghosh, S. (2010). Differential involvement of excitatory and inhibitory neurons of cat motor cortex in coincident spike activity related to behavioral context. J. Neurosci. 30, 8048–8056.
Rebec, G. V., Conroy, S. K., and Barton, S. J. (2006). Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience 137, 327–336.
Robin, K., Maurice, N., Degos, B., Deniau, J. M., Martinerie, J., and Pezard, L. (2009). Assessment of bursting activity and interspike intervals variability: a case study for methodological comparison. J. Neurosci. Methods 179, 142–149.
Sakurai, Y., and Takahashi, S. (2006). Dynamic synchrony of firing in the monkey prefrontal cortex during working-memory tasks. J. Neurosci. 26, 10141–10153.
Schilling, G., Becher, M. W., Sharp, A. H., Jinnah, H. A., Duan, K., Kotzuk, J. A., Slunt, H. H., Ratovitski, T., Cooper, J. K., Jenkins, N. A., Copeland, N. G., Price, D. L., Ross, C. A., and Borchelt, D. R. (1999). Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of Huntingtin. Hum. Mol. Genet. 8, 397–407.
Sharott, A., Moll, C. K., Engler, G., Denker, M., Grun, S., and Engel, A. K. (2009). Different subtypes of striatal neurons are selectively modulated by cortical oscillations. J. Neurosci. 29, 4571–4585.
Shelbourne, P. F., Killeen, N., Hevner, R. F., Johnston, H. M., Tecott, L., Lewandoski, M., Ennis, M., Ramirez, L., Li, Z., Iannicola, C., Littman, D. R., and Myers, R. M. (1999). A Huntington’s disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum. Mol. Genet. 8, 763–774.
Smith, A. D., and Bolam, J. P. (1990). The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 13, 259–265.
Starr, P. A., Kang, G. A., Heath, S., Shimamoto, S., and Turner, R. S. (2008). Pallidal neuronal discharge in Huntington’s disease: support for selective loss of striatal cells originating the indirect pathway. Exp. Neurol. 211, 227–233.
Tiesinga, P., Fellous, J. M., and Sejnowski, T. J. (2008). Regulation of spike timing in visual cortical circuits. Nat. Rev. Neurosci. 9, 97–107.
Twelvetrees, A. E., Yuen, E. Y., Arancibia-Carcamo, I. L., MacAskill, A. F., Rostaing, P., Lumb, M. J., Humbert, S., Triller, A., Saudou, F., Yan, Z., and Kittler, J. T. (2010). Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant Huntingtin. Neuron 65, 53–65.
von Hörsten, S., Schmitt, I., Nguyen, H. P., Holzmann, C., Schmidt, T., Walther, T., Bader, M., Pabst, R., Kobbe, P., Krotova, J., Stiller, D., Kask, A., Vaarmann, A., Rathke-Hartlieb, S., Schulz, J. B., Grasshoff, U., Bauer, I., Vieira-Saecker, A. M., Paul, M., Jones, L., Lindenberg, K. S., Landwehrmeyer, B., Bauer, A., Li, X. J., and Riess, O. (2003). Transgenic rat model of Huntington’s disease. Hum. Mol. Genet. 12, 617–624.
Vonsattel, J. P., and Figlia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 360–384.
Walker, A. G., Miller, B. R., Fritsch, J. N., Barton, S. J., and Rebec, G. V. (2008). Altered information processing in the prefrontal cortex of Huntington’s disease mouse models. J. Neurosci. 28, 8973–8982.
Wichmann, T., and Soares, J. (2006). Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J. Neurophysiol. 95, 2120–2133.
Wickens, J. R., and Wilson, C. J. (1998). Regulation of action-potential firing in spiny neurons of the rat neostriatum in vivo. J. Neurophysiol. 79, 2358–2364.
Wilson, C. J. (1993). The generation of natural firing patterns in neostriatal neurons. Prog. Brain Res. 99, 277–297.
Keywords: mouse models of Huntington’s disease, behavioral electrophysiology, striatal local field potentials, spike synchrony, bursting
Citation: Miller BR, Walker AG, Barton SJ and Rebec GV (2011) Dysregulated neuronal activity patterns implicate corticostriatal circuit dysfunction in multiple rodent models of huntington’s disease. Front. Syst. Neurosci. 5:26. doi: 10.3389/fnsys.2011.00026
Received: 04 January 2011; Accepted: 27 April 2011;
Published online: 09 May 2011.
Edited by:
Elizabeth Abercrombie, Rutgers-Newark: The State University of New Jersey, USAReviewed by:
John A. Wolf, University of Pennsylvania, USAMichael S. Levine, Brain Research Institute, USA
Copyright: © 2011 Miller, Walker, Barton and Rebec. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: George V. Rebec, Program in Neuroscience and Department of Psychological and Brain Sciences, Indiana University, 1101 East 10th Street, Bloomington, IN 47405, USA. e-mail:cmViZWNAaW5kaWFuYS5lZHU=