 Emily L. Jocoy1
Emily L. Jocoy1 Véronique M. André1
Véronique M. André1 Damian M. Cummings1
Damian M. Cummings1 Shilpa P. Rao1 Nanping Wu1
Shilpa P. Rao1 Nanping Wu1 Amy J. Ramsey2
Amy J. Ramsey2 Marc G. Caron2
Marc G. Caron2 Carlos Cepeda1
Carlos Cepeda1 Michael S. Levine1*
Michael S. Levine1*- 1 Intellectual and Developmental Disabilities Research Center, Semel Institute, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USA
- 2 Department of Cell Biology, Duke University, Durham, NC, USA
Dopamine, via activation of D1 receptors, enhances N-methyl-D-aspartate (NMDA) receptor-mediated responses in striatal medium-sized spiny neurons. However, the role of specific NMDA receptor subunits in this enhancement remains unknown. Here we used genetic and pharmacological tools to dissect the contribution of NR1 and NR2A/B subunits to NMDA responses and their modulation by dopamine receptors. We demonstrate that D1 enhancement of NMDA responses does not occur or is significantly reduced in mice with genetic knock-down of NR1 subunits, indicating a critical role of these subunits. Interestingly, spontaneous and evoked α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid (AMPA) receptor-mediated responses were significantly enhanced in NR1 knock-down animals, probably as a compensatory mechanism for the marked reduction in NMDA receptor function. The NMDA receptor subunits NR2A and NR2B played differential roles in D1 modulation. Whereas genetic deletion or pharmacological blockade of NR2A subunits enhanced D1 potentiation of NMDA responses, blockade of NR2B subunits reduced this potentiation, suggesting that these regulatory subunits of the NMDA receptor counterbalance their respective functions. In addition, using D1 and D2 receptor EGFP-expressing mice, we demonstrate that NR2A subunits contribute more to NMDA responses in D1-MSSNs, whereas NR2B subunits contribute more to NMDA responses in D2 cells. The differential contribution of discrete receptor subunits to NMDA responses and dopamine modulation in the striatum has important implications for synaptic plasticity and selective neuronal vulnerability in disease states.
Introduction
Glutamate and dopamine (DA) receptor interactions are vital to numerous functions including learning and memory, motor coordination, and reward mechanisms (Calabresi et al., 2000; Surmeier et al., 2007; Schultz, 2010). When dysfunctional, these receptor interactions contribute to the manifestation of psychiatric and neurodegenerative disorders (Andre et al., 2010b). Glutamatergic and DA inputs converge on the spines of striatal medium-sized spiny neurons (MSSNs) to regulate neuronal excitability (Bouyer et al., 1984; Freund et al., 1984). DA differentially modulates excitatory inputs. Specifically, N-methyl-D-aspartate receptor (NMDAR)-mediated responses are potentiated by D1 receptor (D1R) and attenuated by D2R activation (Cepeda et al., 1993; Snyder et al., 1998; Chergui and Lacey, 1999; Levine et al., 1996b; Brady and O’Donnell, 2004; Tseng and O’Donnell, 2004). Modulation can occur through a variety of mechanisms including multiple transduction pathways, voltage-gated Ca2+ channels, receptor subunit phosphorylation and mobilization, changes in receptor surface expression, and direct protein–protein coupling (Cepeda et al., 1998; Snyder et al., 1998; Lee et al., 2002; Surmeier et al., 2007; Pascoli et al., 2011).
NMDA receptors are heteromers constituted by different NR1, NR2, and/or NR3 subunits. The most common receptor combination consists of two NR1 subunits, which are obligatory for a functional receptor, and two NR2 subunits which confer different physiological properties (Laube et al., 1998). NMDA receptors expressed by MSSNs consist of NR1, NR2A, and/or NR2B subunits, thus allowing for diheteromeric (NR1/NR2A or NR1/NR2B) and triheteromeric (NR1/NR2A/NR2B) receptors (Dunah and Standaert, 2003).
Mice with genetic deletion of specific NMDA receptor subunits have been created (Ikeda et al., 1995; Sakimura et al., 1995; Kutsuwada et al., 1996; Mohn et al., 1999). Although deletion of some subunits (e.g., NR1 or NR2B) can be lethal, other genetic strategies can be used to examine the role of these subunits. This, in conjunction with the availability of relatively selective pharmacological antagonists, constitutes a powerful tool to begin dissecting the role of NMDA receptor subunits. The present study examined how specific NMDA receptor subunits sculpt NMDAR-mediated responses and how they affect DA modulation in MSSNs, particularly the enhancement of NMDA currents by D1 receptor activation. In addition, we used mice expressing the reporter gene, enhanced green fluorescent protein (EGFP) in direct (D1R-containing) and indirect (D2R-containing) pathway MSSNs to examine the relative contribution of NR2A/B subunits to NMDA responses and their modulation by dopamine receptor agonists.
Materials and Methods
All experimental procedures were performed in accordance with the United States Public Health Service Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of California, Los Angeles. Every effort was made to minimize pain and discomfort. Experiments were conducted on four groups of mice: (1) NR1 knock-down (KD; WT n = 4, age 98 ± 7 days; KD n = 6, age 100 ± 6 days), generated using homologous recombination in embryonic stem cells (Mohn et al., 1999). This mouse line expresses only 5% of the subunit and is viable. (2) Mice with the NR2A subunit genetically removed (NR2AKO; slice: WT n = 31, age 37 ± 3 days; NR2AKO n = 28, 36 ± 3 days; acute dissociation: WT n = 34, age 37 ± 3 days; NR2AKO n = 44, age 37 ± 2 days). (3) Mice with the NR2A subunit genetically removed and expressing EGFP under the control of the D1R promoter (D1 WT n = 21, age 38 ± 3 days; D1 NR2AKO n = 15, age 38 ± 2 days), and (4) Mice with the NR2A subunit genetically removed and expressing EGFP under the control of the D2R promoter (D2 WT n = 15, age 38 ± 4 days; D2 NR2AKO n = 21, age 38 ± 2 days). EGFP mice were heterozygous. This is important as it was recently shown that D2-EGFP homozygous mice have significant increases in D2R expression (Kramer et al., 2011). Electrophysiological properties were examined in slices and acutely isolated MSSNs using whole-cell patch clamp recordings in voltage clamp mode.
Brain Slice Preparation
Mice were deeply anesthetized with halothane, killed by decapitation, and the brains dissected and immediately placed in oxygenated ice cold low Ca2+ and high Mg2+ artificial cerebrospinal fluid (ACSF) containing (in mM) NaCl, 130; NaH2PO4, 1.25; KCl, 3; NaHCO3, 26; MgCl2, 5; CaCl2, 1, and glucose, 10. Coronal slices (350 μm) were cut and transferred to an incubating chamber containing ACSF (with 2 mM CaCl2 and 2 mM MgCl2) oxygenated with 95% O2–5% CO2 (pH 7.2–7.4, 290–310 mOsm, 25 ± 2°C). Following recovery, slices were placed on the stage of an upright microscope (Olympus BX51), submerged in continuously flowing ACSF (4 ml/min). All experiments were performed at room temperature.
Whole-cell patch clamp recordings in voltage clamp mode were obtained from MSSNs in the dorsolateral striatum visualized with the aid of infrared videomicroscopy (Cepeda et al., 1998). MSSNs were identified by somatic size and typical basic membrane properties (input resistance, membrane capacitance, and time constant). Series resistance was <25 MΩ. The patch pipette (4–6 MΩ) contained the following solution (in mM): Cs–methanesulfonate 125, KCl 3, NaCl 4, MgCl2 1, MgATP 5, EGTA 9, HEPES 8, GTP 1, phosphocreatine 10, leupeptin 0.1, QX-314 4 (pH 7.25–7.3, osmolarity 280–290 mOsm). Passive membrane properties of MSSNs were determined by applying a depolarizing step voltage command (10 mV) and using the membrane test function integrated in the pClamp8 software (Axon Instruments, Foster City, CA, USA). This function reports membrane capacitance (in pF), input resistance (in MΩ) and time constant (in ms or μs).
Spontaneous EPSCs were isolated by holding the membrane at −70 mV and blocking GABAA receptors with bicuculline methiodide (BIC, 20 μM). The amplitude, frequency, and kinetic properties of spontaneous EPSCs were measured using MiniAnalysis software (Synaptosoft Inc, Fort Lee, NJ, USA). Evoked EPSCs were recorded by stimulating the corpus callosum (0.01–1 mA square pulses, 0.1 ms duration) using a monopolar glass electrode placed 150–250 μm dorsal from the recording pipette. To isolate synaptic responses mediated by non-NMDA receptors, the GABAA receptor blockers BIC (20 μM) or picrotoxin (100 μM) were applied, while CNQX or NBQX (10 μM) was applied to block non-NMDARs and isolate NMDAR responses. NMDAR–EPSCs were recorded either at +40 or −70 mV depending on the presence or absence of Mg2+ in the ACSF solution. Three responses (15 s between stimulations) were recorded and averaged at each intensity to construct input-output relationships.
Acutely Isolated Neurons
Detailed procedures have been published (Flores-Hernandez et al., 2002; Cepeda et al., 2008b). Briefly, slices were obtained as described above. After at least 1 h of incubation in oxygenated ACSF, the dorsal striatum was dissected, placed in an oxygenated cell-stir chamber (Wheaton, Millville, NJ, USA) and enzymatically treated for 20 min with papain (0.5 mg/ml, Calbiochem) at 35°C in a N-[2-hydroxyethyl] piperazine-N-[2-ethanesulfonic acid] (HEPES)-buffered Hank’s balanced salts solution (HBSS, Sigma Chemical) supplemented with (in mM) 1 pyruvic acid, 0.005 glutathione, 0.1 NG-nitro-L-arginine, and 1 kynurenic acid (pH 7.4, 300–310 mOsm). After enzymatic digestion, the tissue was rinsed with a low Ca2+ HEPES-buffered Na-isethionate solution containing (in mM) 140 Na-isethionate, 2 KCl, 2MgCl2, 0.1 CaCl2, 23 glucose, and 15 HEPES (pH 7.4, 300–310 mOsm/l). Striatal slices were mechanically dissociated with fire-polished Pasteur pipettes. The cell suspension was then plated into a 35-mm nunclon Petri dish mounted on the stage of an upright microscope (Zeiss Axioscope, Thornwood, NY, USA) containing a HEPES-buffered salt solution.
Standard whole-cell patch clamp techniques were used to obtain voltage clamp recordings (Bargas et al., 1994). The internal solution consisted of (in mM) 175 N-methyl-D-glucamine (NMDG), 40 HEPES, 2 MgCl2, 10 ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 12 phosphocreatine, 2 Na2 ATP, 0.2 Na2 GTP, and 0.1 leupeptin (pH 7.25, 265–270 mOsm/l). The external solution consisted of (in mM) 135 NaCl, 20 CsCl, 3 BaCl2, 2 CaCl2, 10 glucose, 10 HEPES, and 0.0003 tetrodotoxin (TTX; pH 7.4, 300–310 mOsm/l). Recordings were obtained with an Axon Instruments 200A patch clamp amplifier and controlled by a computer running pClamp (v. 8.1; Axon Instruments, Foster City, CA, USA).
Drugs were applied through a pressure-driven fast perfusion system (SF-77B, Warner Instruments, Hamden, CT) synchronized by pClamp. Values for peak currents and peak current densities were calculated for all neurons. NMDA currents (3 s duration every 20 s) were evoked by applying different concentrations of the agonist while holding the cell at −70 mV.
Drugs
The selective NR2A antagonist NVP-AAM077 (NVP, 0.001–1 μM) was a gift from Novartis; zinc (10–300 nM), another NR2A antagonist, was applied in the presence of 10 mM tricine to remove ambient zinc. NR2B antagonists Ifenprodil (0.01–20 μM) was from Sigma; and Conantokin G (1 μM) from Peptides International. Stocks of NMDA (100 mM), NVP (1 mM), Ifenprodil (1 mM), 6-Chloro PB (SKF 81297, 1 mM), and quinpirole (10 mM) were all dissolved in H2O and stored in the freezer until use.
Data Analysis and Statistics
In the text, values are presented as means ± SEMs. Group means for all measures were compared using Student’s t-tests (for two group comparisons) and appropriately designed ANOVAs followed by suitable post hoc comparison tests (for multiple group comparisons) using SigmaStat software (SPSS, Chicago, IL, USA). Differences were considered statistically significant when p < 0.05.
Results
Role of NR1 Subunits
As deletion of NR1 subunits is lethal, the contribution of these subunits to NMDAR-mediated responses was examined in NR1-KD mice (Mohn et al., 1999). MSSNs in slices from WT and NR1-KD mice displayed similar passive membrane properties including capacitance, input resistance and time constant (Table 1). As expected, NMDAR-mediated currents evoked by electrical stimulation of cortical inputs were severely reduced in NR1-KD MSSNs (Figure 1A, left). Interestingly, evoked AMPAR-mediated currents were significantly increased, suggesting compensatory mechanisms (Figure 1A right). Similarly, the frequency of spontaneous EPSCs at a holding potential of −70 mV in the presence of BIC was significantly increased in MSSNs from NR1-KD mice (Figure 1B). In addition, the kinetics of spontaneous synaptic events was changed. EPSCs displayed significantly faster rise and decay times, as well as reduced half-amplitude durations in cells from NR1-KD compared to control mice (Table 2).
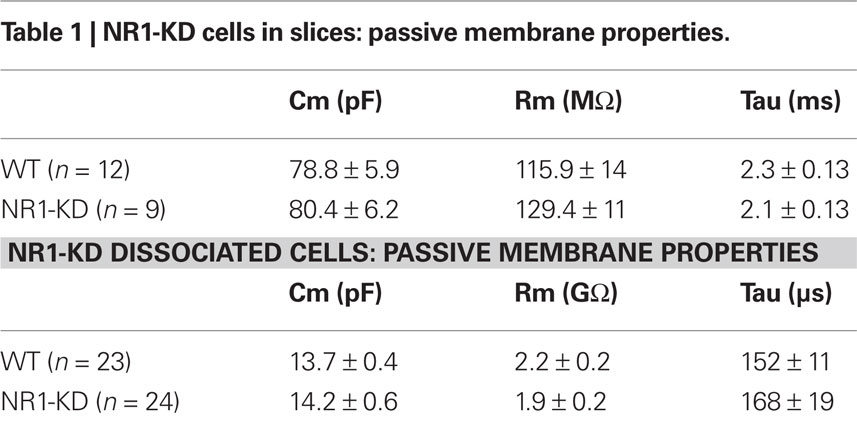
Table 1. NR1-KD cells in slices: passive membrane properties.
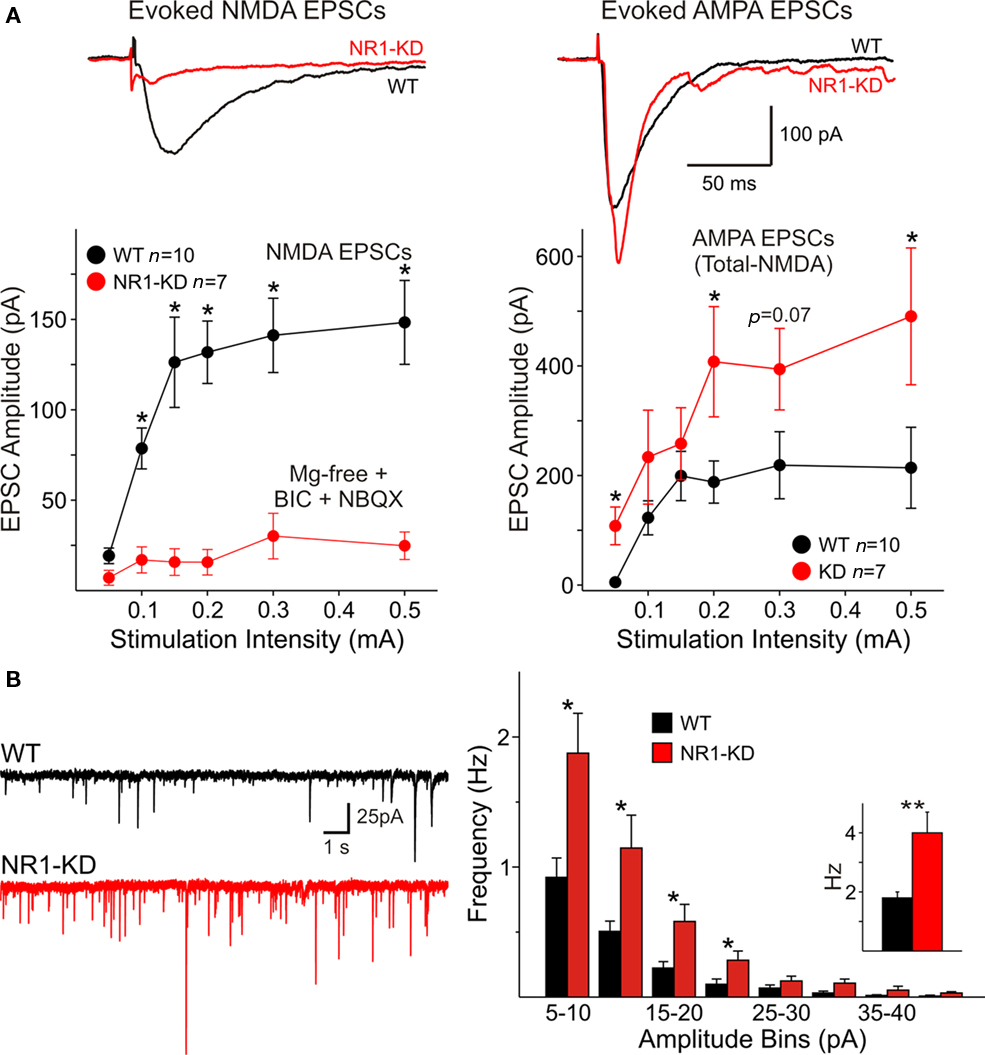
Figure 1. (A) Traces show average NMDA and AMPAR-mediated responses evoked by cortical stimulation in Mg2+-free conditions and in the presence of non-NMDA (NBQX) and GABAA receptor blockers (BIC). Currents evoked by activation of NMDA receptors (left) were severely reduced in MSSNs from NR1-KD mice. In contrast, AMPA responses (right) were significantly increased. Cells were voltage clamped at −70 mV. Graphs indicate that at different stimulation intensities the NMDA currents were almost non-existent in NR1-KD cells (left), whereas the amplitude of AMPA currents was increased (right). Amplitude of AMPA currents was determined by subtraction of NMDA currents from the total current in the absence of glutamatergic antagonists. (B) Spontaneous excitatory postsynaptic currents (sEPSCs) recorded in MSSNs of ∼3 month-old NR1-KD and control mice. Traces show sEPSCs recorded in the presence of a GABAA receptor antagonist (BIC, 20 μM) with membranes voltage clamped at −70 mV. Amplitude–frequency histograms for sEPSCs. Mean frequency of sEPSCs is indicated in the inset. *p < 0.05; **p < 0.01; ***p < 0.001 in this and subsequent figures.

Table 2. Kinetics of sEPSCs in NR1-KD.
In acutely isolated MSSNs, passive membrane properties were also similar (Table 1). Bath application of increasing concentrations of NMDA-induced inward currents but the amplitudes of these responses were significantly smaller in cells from NR1-KD mice (Figure 2A). The peak and current density amplitudes of AMPAR-induced currents were also significantly increased in NR1-KD mice (Figure 2B), suggesting changes in postsynaptic AMPA receptors.
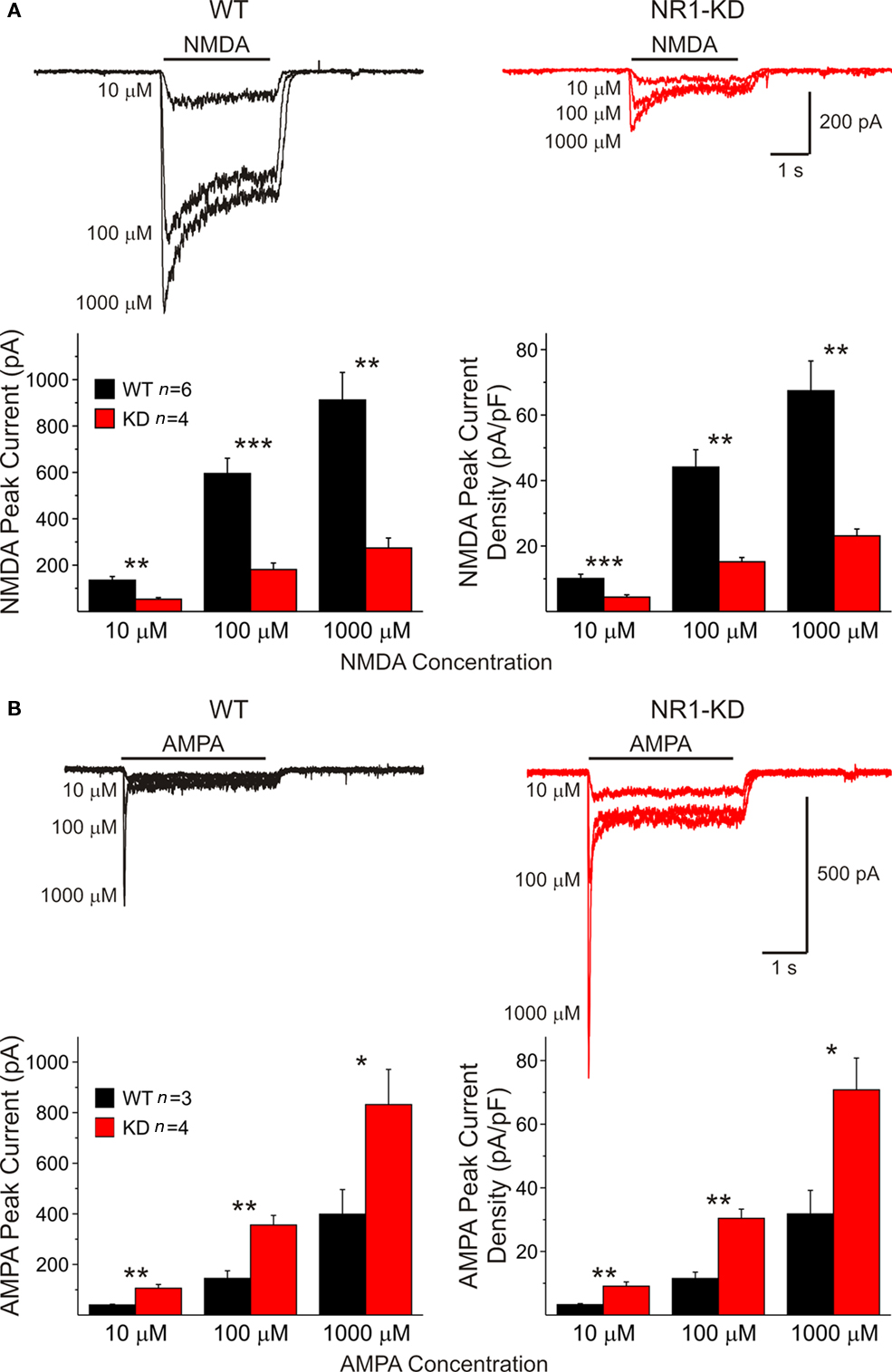
Figure 2. (A) In isolated MSSNs, bath application of increasing concentrations of NMDA-induced inward currents. As shown in the graphs, the amplitude of NMDAR-mediated peak and current densities was significantly smaller in cells from NR1-KD mice. (B) In contrast, peak and current densities induced by AMPA were significantly increased, suggesting compensatory mechanisms.
Dopamine D1 Receptor Modulation of NMDA Currents is Altered in NR1-KD Mice
We have shown previously that activation of D1 receptors enhances synaptically evoked NMDA responses (Levine et al., 1996a). In cells from WT mice the D1 agonist SKF 81297 (1 μM) increased peak current amplitude whereas in cells from NR1-KD mice the agonist produced little effects or even decreased peak current amplitude (Figure 3A).
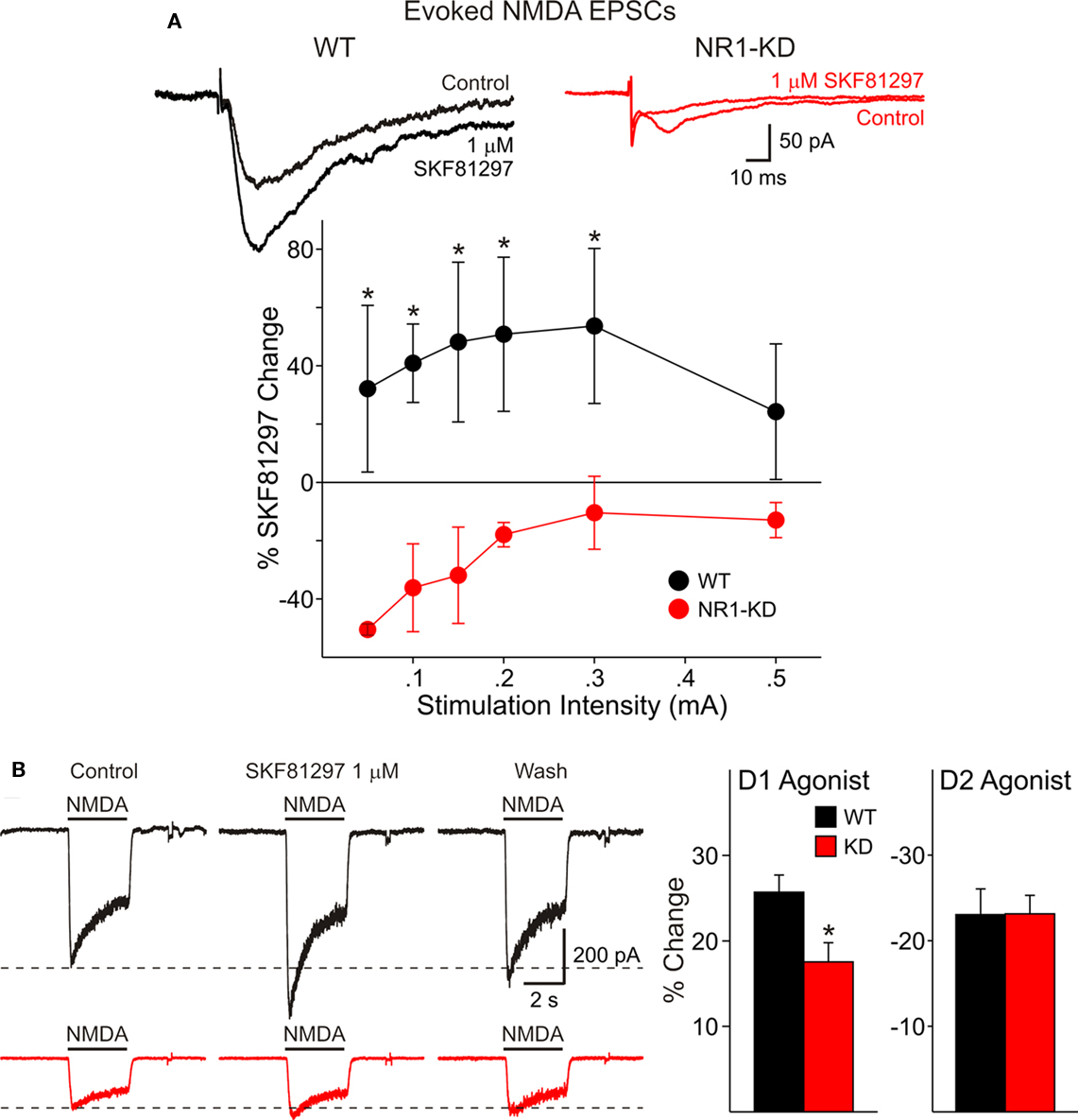
Figure 3. (A) In cells from WT mice (left) the D1 agonist SKF 81297 increased peak current amplitude of evoked NMDA responses, whereas in cells from NR1-KD mice (right) the agonist produced no change or decreased peak current amplitude. (B) There also was decreased modulation of NMDA responses by the dopamine D1 receptor agonist in isolated MSSNs. The percent increase in NMDA responses was significantly reduced in NR1-KD animals (left bar graph) whereas the decrease by a D2 agonist was unaffected (right bar graph).
There also was decreased modulation of NMDAR-mediated responses by the dopamine D1 receptor agonist in isolated MSSNs (Figure 3B). The percent increase in NMDA responses was significantly reduced in NR1-KD animals. No differences were found in D2 receptor modulation (Figure 3B, right bar graph).
Role of NR2A Subunits
The membrane properties of MSSNs in slice or after acute dissociation were practically identical in control and NR2AKO mice. NMDAR-mediated EPSCs evoked by electrical stimulation were compared in slices. The membrane potential was held at +40 mV to remove the Mg2+ block of NMDARs and GABAA and non-NMDA glutamate receptors were blocked with picrotoxin and CNQX, respectively. Responses from NR2AKOs (n = 18) had smaller peak amplitudes than those of WTs (n = 14) and statistically significant differences occurred at stimulation intensities between 0.8 and 1.0 mA (p < 0.05; Figure 4A). Decay times were slower in cells from NR2AKOs than those of WTs at higher intensities as well (p < 0.05; Figure 4A right graph).
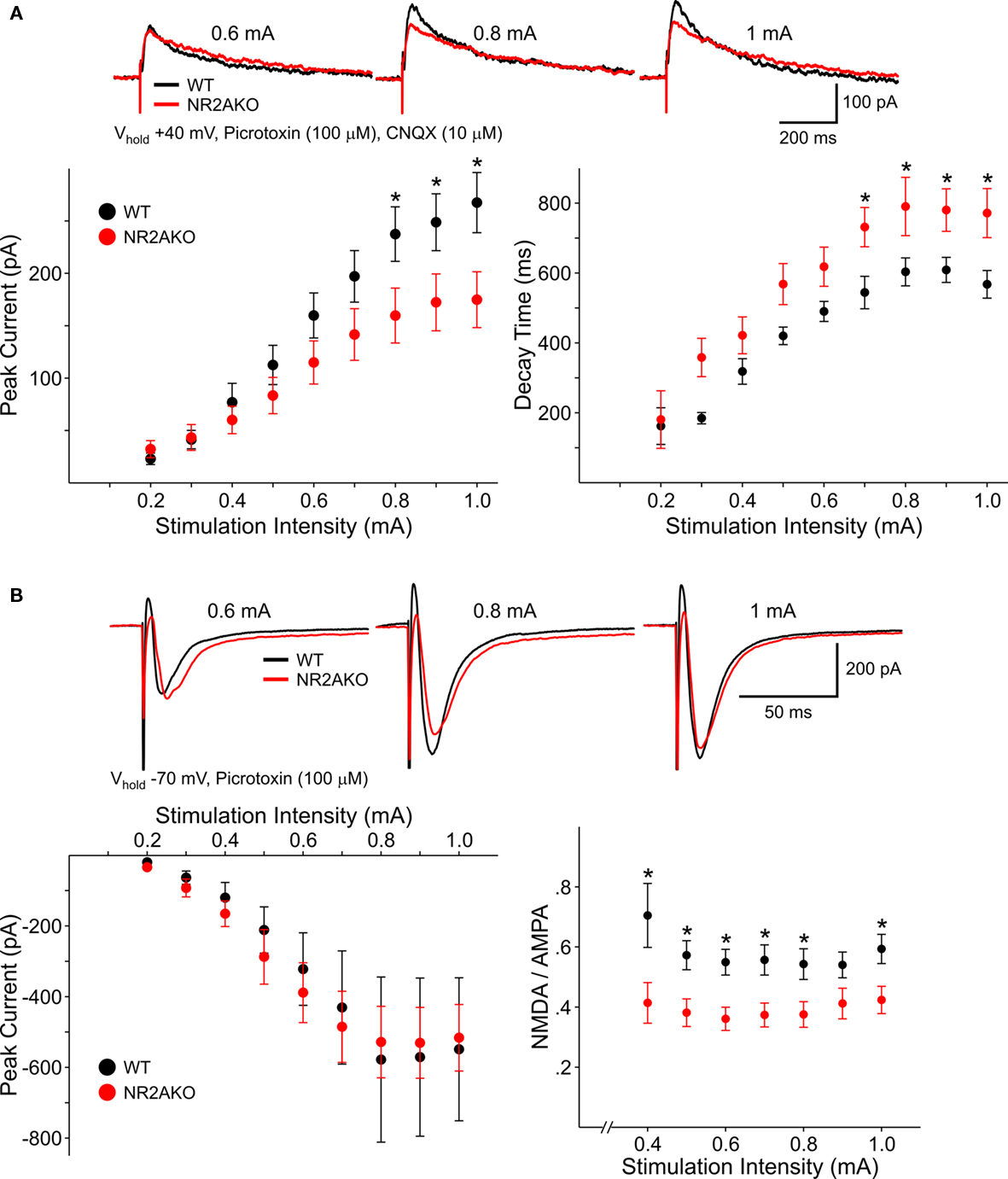
Figure 4. (A) Representative traces of NMDAR-mediated evoked EPSCs from WT and NR2AKO MSSNs at different stimulation current intensities. Graphs are summary data illustrating that mean peak amplitudes of NMDAR–EPSCs are significantly smaller in MSSNs from NR2AKOs compared to those of WTs at higher stimulation intensities. In addition, deletion of NR2A results in slower mean decay times. (B) In contrast, genetic deletion of NR2A does not alter AMPAR-mediated EPSCs. Representative traces of evoked AMPAR–EPSCs from WT and NR2AKO MSSNs at different stimulation current intensities. Mean peak amplitudes of AMPAR–EPSCs are similar between genotypes at all intensities. Ratios of mean peak amplitudes of NMDAR- to AMPAR–EPSCs are significantly smaller in NR2AKOs than WTs.
To isolate AMPAR-mediated responses, EPSCs were evoked at a holding potential of −70 mV in the presence of picrotoxin. Peak amplitudes were similar between cells from NR2AKOs (n = 18) and WTs (n = 14; Figure 4B). Moreover, ratios of NMDAR- to AMPAR-mediated currents were consistently smaller (p < 0.01) in NR2AKOs, indicating that smaller NMDAR–EPSC amplitudes were due to alterations in NMDARs and not due to differences in activation of excitatory inputs to MSSNs.
The effects of acutely blocking NR2A-containing NMDARs were also examined using a pharmacological approach. Following 5 min perfusion of NVP (0.1 μM), peak amplitudes of evoked NMDAR-mediated EPSCs in WTs were decreased significantly compared to baseline responses (34.6 ± 3.8%, n = 12, p < 0.01) and decay times were increased (31.1 ± 13.9%, p < 0.05). In contrast, NVP had practically no effect on the amplitude of evoked responses in cells from NR2AKO mice (Figure 5). Taken together, these findings suggest that the NR2A subunit contributes significantly to synaptic NMDAR-mediated responses. Genetic deletion or acute pharmacological block of NR2A-containing NMDARs caused a reduction in peak amplitudes and prolonged the decay time of the response.
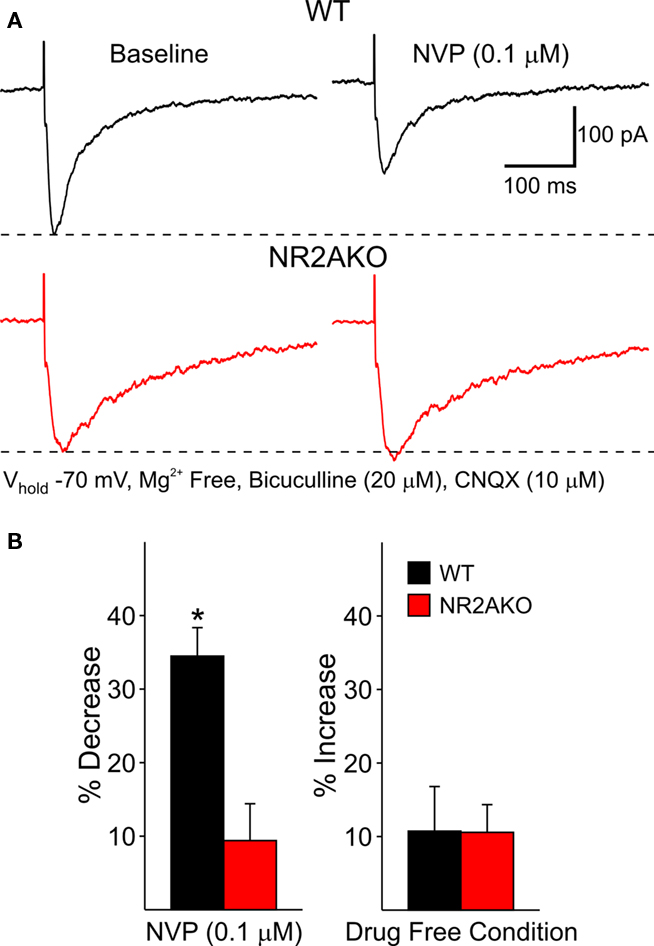
Figure 5. (A) Representative evoked NMDAR-EPSCs from WT and NR2AKO MSSNs before (left) and after (right) 5 min application of NVP. (B) Graph on the left illustrates that NVP (0.1 μM) significantly attenuates mean peak amplitudes of NMDAR-EPSCs from WT cells only. Bar graph on the right demonstrates that mean peak amplitudes remain stable during a 5 min recording period in the absence of the NR2A antagonist.
In dissociated WT cells increasing concentrations of NVP (0.001, 0.01, 0.1, and 1 μM) or zinc (10, 30, 100, 300 nM) attenuated both peak and steady-state currents in a dose-dependent manner (Figures 6A,B). The peak component of the current was attenuated by NVP more than the steady-state component (p < 0.05) whereas zinc attenuated steady-state currents more than peak currents (p < 0.05; Figure 6A graphs). This demonstrates that NR2A subunits contribute not only to synaptic NMDA responses but also to responses induced in the soma and proximal dendrites, where excitatory synaptic inputs are less abundant.
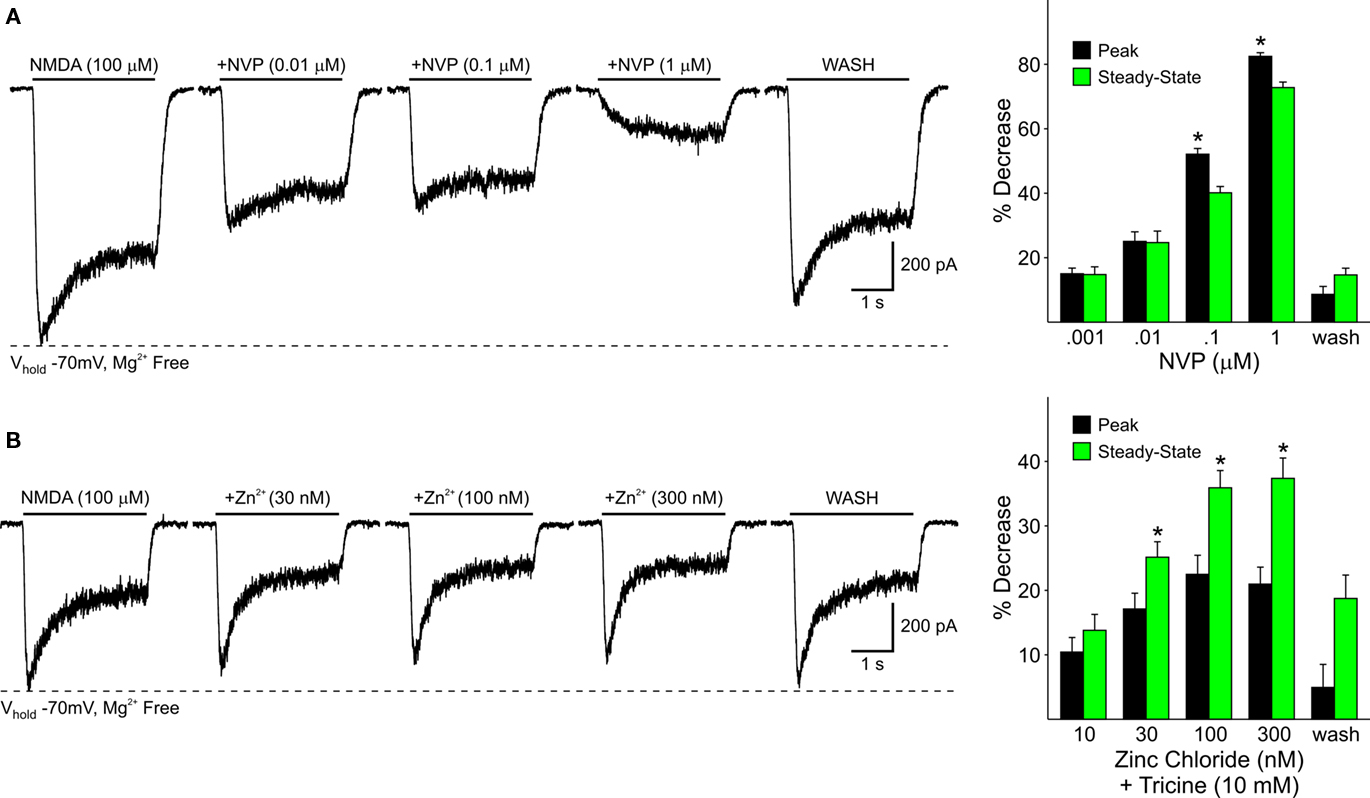
Figure 6. (A) Representative traces from a WT MSSN of NMDAR-mediated currents in the presence of increasing concentrations of NVP. Bar graphs summarize the decrease in peak and steady-state average NMDAR currents in WT MSSNs in the presence of increasing concentrations of NVP. Peak currents were attenuated more than steady-state currents at higher concentrations. (B) Representative traces of NMDAR-mediated currents from WT cells in the presence of increasing concentrations of zinc. Bar graphs illustrate the decrease in peak and steady-state average NMDAR-mediated currents in the presence of increasing concentrations of zinc. Steady-state currents were attenuated more than peak currents.
The effect of genetic deletion of NR2A subunits on NMDA responses induced by activation of mostly extrasynaptic receptors located at or near the soma was then studied in dissociated MSSNs from WT and NR2AKO mice. Peak current and current density amplitudes evoked by increasing concentrations of NMDA (1, 10, 100, and 1000 μM) were similar in NR2AKO and WT mice (not shown). Moreover, EC50 values for mean peak current amplitudes and current densities were also similar [45.3 ± 8.4 μM for WTs (n = 8) and 39.1 ± 3.4 μM for NR2AKOs (n = 16)]. However, in the presence of increasing concentrations of NVP or zinc, the attenuation of NMDAR-mediated currents was significantly reduced in NR2AKOs (Figures 7A,B). Because synaptic inputs are mostly eliminated in dissociated MSSNs, these results suggest that either the NR2A subunit does not contribute significantly to mostly extrasynaptic NMDA receptors, or that compensatory effects result from genetic ablation of this subunit. In support of the second alternative, application of Ifenprodil (1 and 10 μM), a NR2B antagonist, attenuated peak currents from NR2AKOs significantly more in MSSNs from NR2AKO compared to WT mice (p < 0.05; Figure 7C).
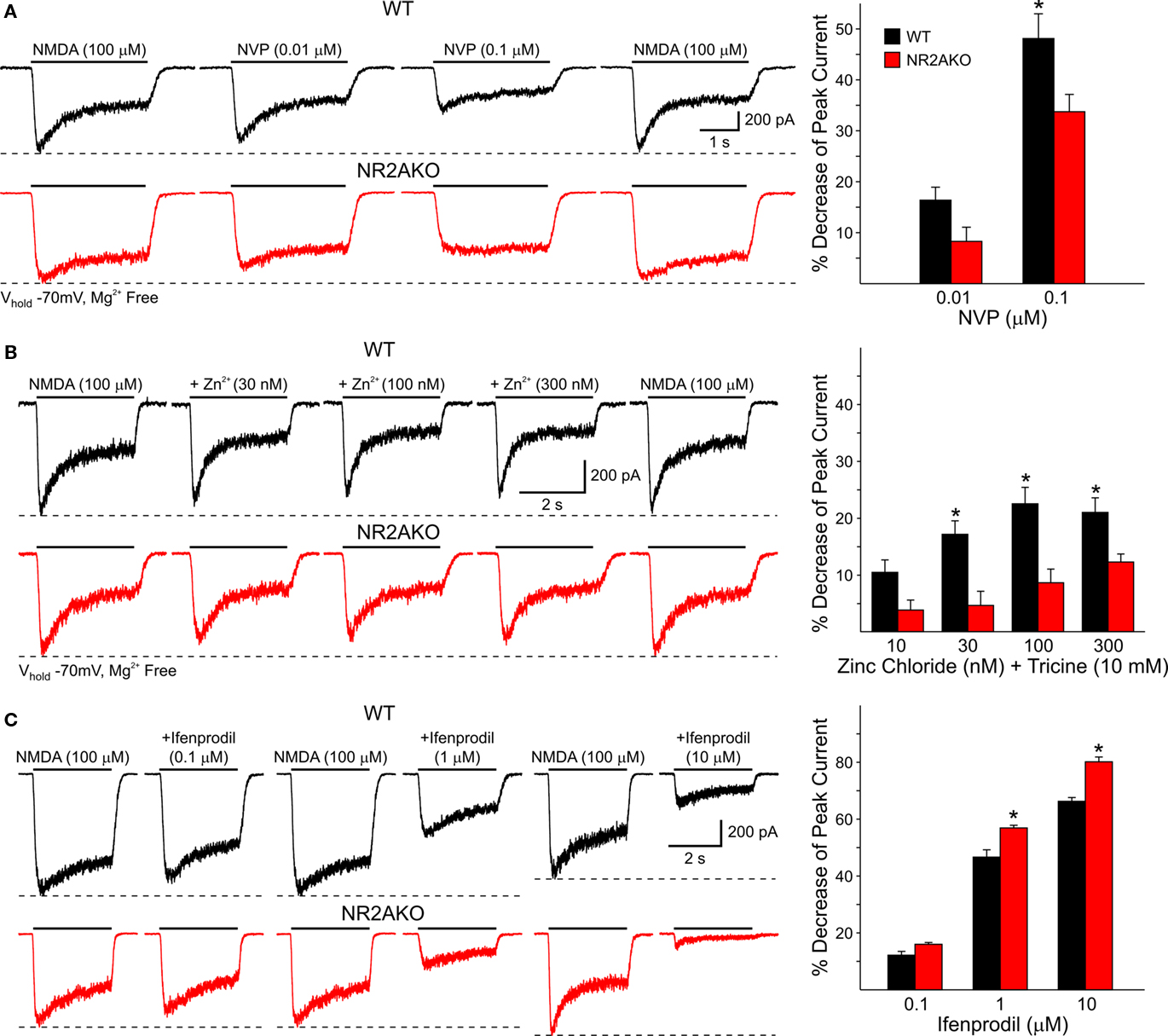
Figure 7. (A) Traces of NMDAR-mediated currents from WT and NR2AKO isolated MSSNs in the presence of increasing concentrations of NVP demonstrate that attenuation of NMDAR-mediated currents by the antagonist is reduced in NR2AKOs. Graph shows that NVP decreases mean peak NMDAR-mediated currents more in WT than in NR2AKOs. (B) Zinc attenuation of NMDAR-mediated currents is also reduced in NR2AKOs at most concentrations. (C) In NR2AKO MSSNs, the functional contribution of NR2B to NMDAR-mediated currents is increased. Traces of NMDAR-mediated currents from WT (top) and NR2AKO (bottom) MSSNs in the presence of Ifenprodil. Graph illustrates increased Ifenprodil sensitivity in NR2AKO compared to WT MSSNs.
To examine if there is a differential contribution of NR2A subunits in D1R- and D2R-expressing MSSNs, D1R-, or D2R EGFP mice were crossed to the NR2AKO mice, thereby creating double transgenic mice with EGFP labeling of D1R- or D2R-expressing cells with and without NR2A. In acutely isolated D1R-expressing MSSNs, NMDAR-mediated peak current densities were consistently smaller in NR2AKOs than WTs across all concentrations (100, 500, and 1000 μM NMDA, p < 0.05; Figure 8A). In contrast, NMDAR-mediated current densities were similar between D2-NR2AKO and D2-WT MSSNs at all NMDA concentrations (Figure 8B). These data suggest a greater contribution of NR2A to D1R- compared to D2R-expressing MSSNs.
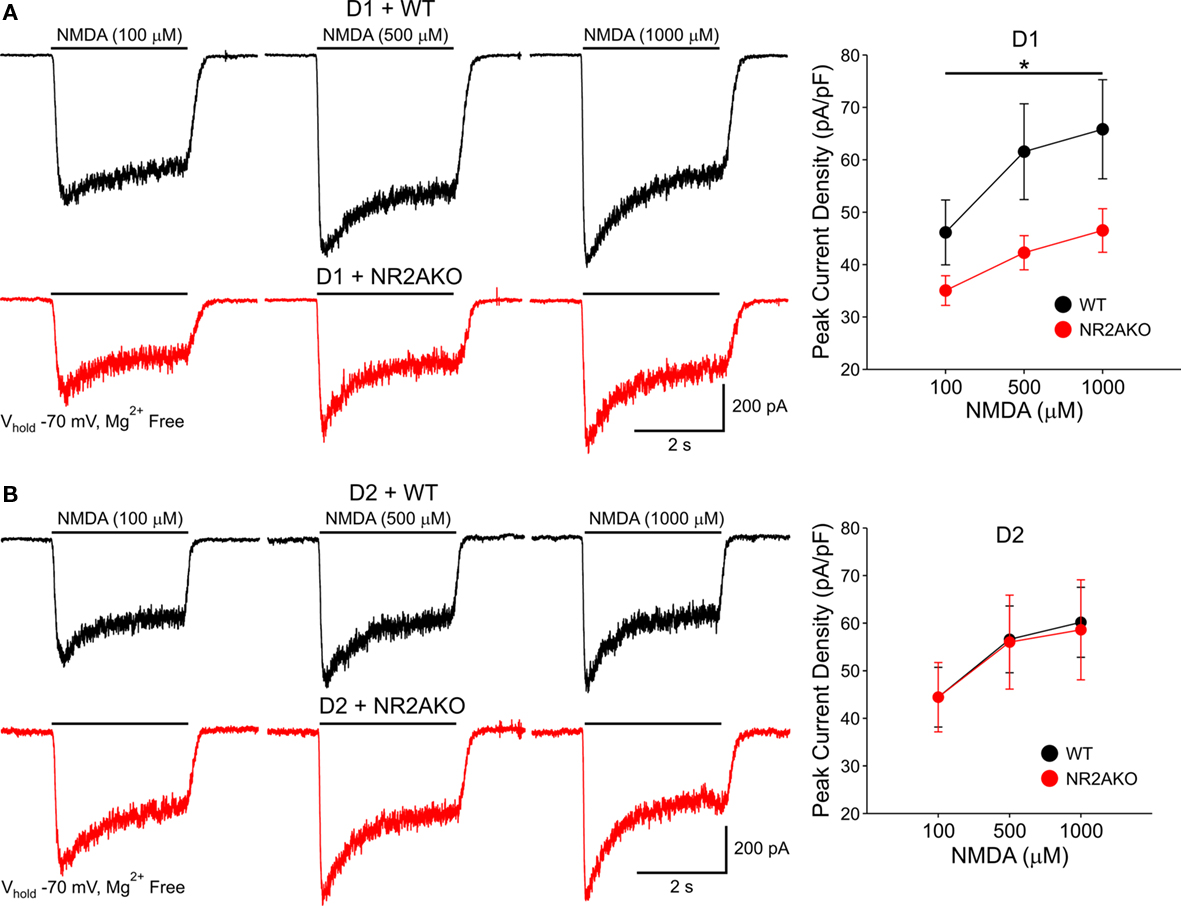
Figure 8. (A) Traces of NMDAR-mediated currents from acutely isolated D1-WT and D1-NR2AKO MSSNs demonstrate a larger contribution of NR2A subunits to NMDA responses in D1R-expressing MSSNs. Line graph shows that genetic deletion of NR2A significantly decreases mean peak NMDAR-mediated current densities in D1R-expressing MSSNs. (B) In contrast, NR2A subunits contribute less to NMDA responses in D2R-expressing MSSNs. Representative traces of NMDAR-mediated currents from acutely isolated D2-WT and D2-NR2AKO MSSNs. Line graph illustrates that genetic deletion of NR2A does not significantly alter mean peak NMDAR-mediated current densities in D2R-expressing MSSNs.
Genetic Deletion of NR2A Enhances D1R Potentiation of NMDA Responses
The selective D1R agonist SKF 81297 (1 μM) was used to examine the potentiation of NMDAR-mediated responses in isolated MSSNs from NR2AKO and control animals. In the entire population of MSSNs, without distinction of DA receptor-subtype expression, the percent increase was greater in NR2AKO mice than in WTs but the difference was not statistically significant (Figure 9A graph). However, after separating MSSNs by subtype using EGFP, NMDA currents in D1R-expressing neurons (n = 24) from NR2AKO mice displayed significant increases in potentiation compared to D1R-expressing cells (n = 21) from WTs (p < 0.05; Figure 9A traces and Figure 9B left). In contrast, no significant differences in D2R attenuation of NMDAR-mediated currents occurred between D2-WT (n = 13) and D2-NR2AKO (n = 10) MSSNs (Figure 10 left). This indicates that D1R potentiation of NMDA responses is enhanced in the absence of NR2A subunits in D1R-expressing MSSNs.
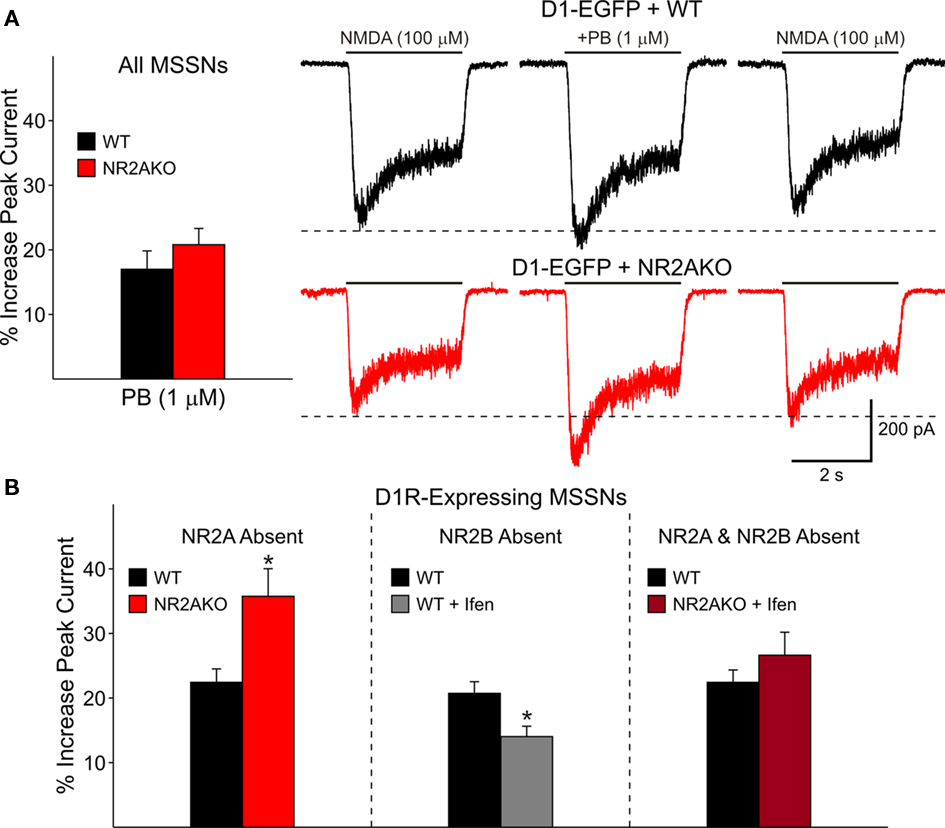
Figure 9. (A) Bar graph shows that genetic deletion of NR2A does not significantly alter D1R-modulation of NMDAR-mediated currents in both D1R- and D2R-expressing cells. Traces on the right illustrate NMDAR-mediated currents in D1-WT and D1-NR2AKO MSSNs. The D1 agonist (PB) significantly potentiates NMDAR-mediated currents more in D1R-expressing cells from NR2AKOs. (B) The bar graph on the left illustrates that genetic deletion of NR2A significantly increases D1R potentiation of NMDA currents. The bar graph in the middle demonstrates that pharmacological block of NR2B-expressing NMDARs results in a significant decrease in D1R potentiation. The bar graph on the right illustrates that the combined genetic deletion of NR2A and pharmacological block of NR2B-expressing NMDARs does not significantly alter D1R modulation.
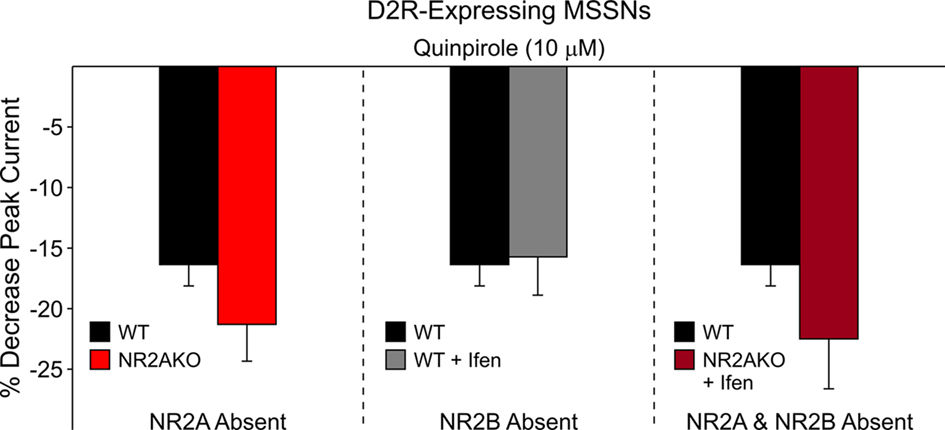
Figure 10. The bar graph on the left illustrates that genetic deletion of NR2A does not significantly alter D2R attenuation of NMDAR-mediated currents. The bar graph in the middle demonstrates that pharmacological block of NR2B-containing NMDARs does not significantly affect D2R modulation. The bar graph on the right shows that the combined genetic deletion of NR2A and pharmacological block of NR2B-expressing NMDARs does not significantly alter D2R modulation of NMDAR-mediated currents.
Role of NR2B Subunits
As NR2BKO mice do not survive postnatally (Tovar et al., 2000), two NR2B subunit antagonists, Ifenprodil and Conantokin G were used to examine the contribution of NR2B subunits to NMDA responses and DA receptor modulation: NMDAR–EPSCs were isolated pharmacologically (Mg2+-free ACSF in the presence of BIC and CNQX). Ifenprodil (5 μM), a non-competitive NR2B antagonist, only slightly decreased peak NMDAR–EPSC amplitudes 14.2 ± 4.2% (p < 0.05; Figure 11A). The competitive antagonist, Conantokin G (1 μM) significantly decreased peak NMDAR-mediated EPSCs in MSSNs (36.9 ± 8.1%, p < 0.05; Figure 11B). Faster decay times tended to occur after either Ifenprodil or Conantokin application, although the difference did not reach statistical significance. Taken together, these findings indicate that the NR2B subunit also contributes to synaptic NMDA responses.
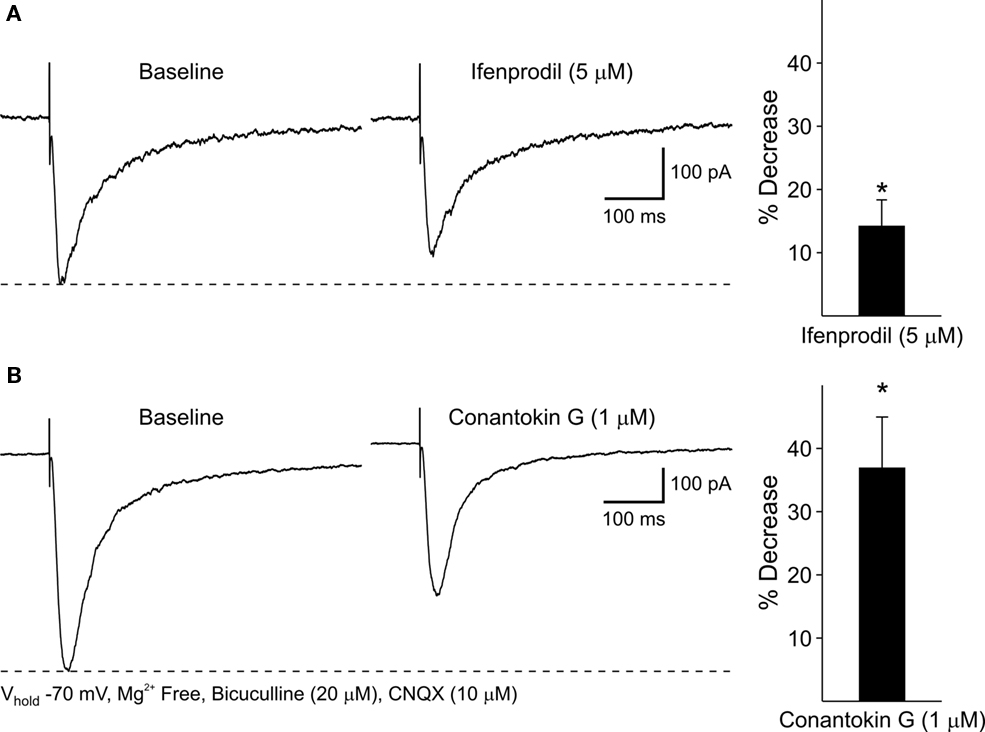
Figure 11. (A) Representative traces of evoked NMDAR–EPSCs before and after 5 min application of the NR2B antagonist, Ifenprodil. Bar graph illustrates that Ifenprodil decreases mean peak amplitudes of NMDAR–EPSCs compared to baseline responses. (B) Representative traces of NMDAR–EPSCs before and after 5 min application of the NR2B antagonist, Conantokin G. Bar graph shows that Conantokin G significantly decreases mean peak amplitudes of NMDAR–EPSCs in WT MSSNs compared to baseline responses.
In isolated MSSNs increasing concentrations of Ifenprodil (0.1, 1, and 10 μM) significantly attenuated both peak and steady-state NMDA currents. Steady-state currents were attenuated more that peak currents (p < 0.05; Figure 12A). The role of NR2B within MSSN subpopulations was examined by applying Ifenprodil to D1-WT and D2-WT cells. NMDAR-mediated peak current densities were blocked in both groups, however current densities from D2-WTs (n = 8) were slightly, but consistently, more sensitive to Ifenprodil than D1-WTs (n = 8, p < 0.05; Figure 12B). This difference in Ifenprodil sensitivity suggests that, functionally, levels of NR2B-containing NMDARs are greater in D2-WT than D1-WT cells.
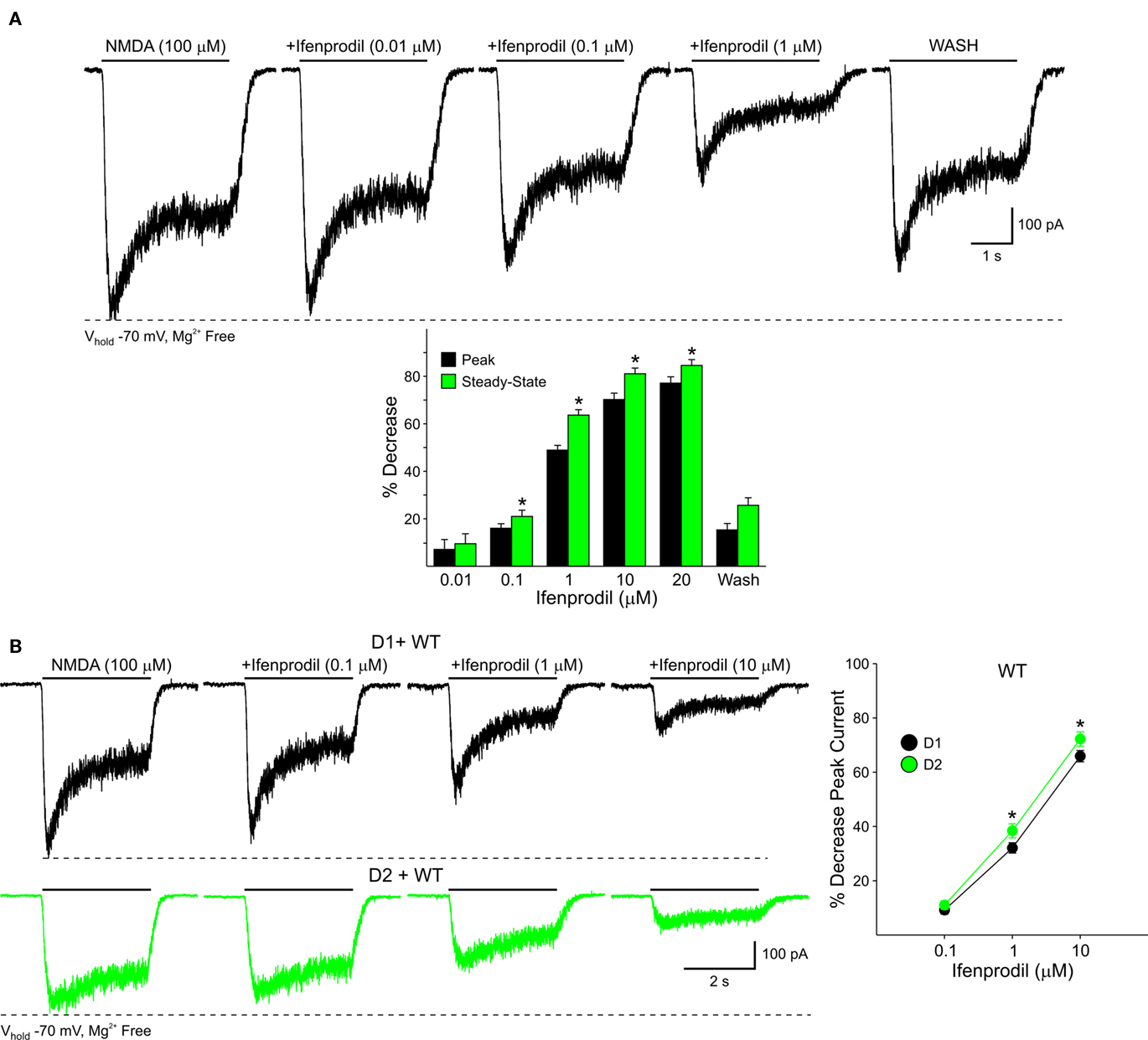
Figure 12. (A) Pharmacological blockade of NR2B-expressing NMDARs with increasing concentrations of Ifenprodil reveals a contribution of these subunits to NMDAR-mediated currents in acutely isolated MSSNs. Bar graph illustrates the decrease in peak and steady-state mean NMDAR currents. Steady-state currents are attenuated more than peak currents. (B) NR2B subunits contribute more to D2R-than D1R-expressing MSSNs. Representative traces of NMDAR-mediated currents from D1-WT and D2-WT cells in the presence of increasing concentrations of Ifenprodil. Line graph illustrates that in both D1-WT and D2-WT MSSNs, NMDAR-mediated currents are blocked by Ifenprodil, but the sensitivity is greater in D2 cells.
Pharmacological Blockade of NR2B Subunits Reduces D1R Potentiation of NMDA Responses
To determine if D1R modulation of NMDAR-mediated currents is affected by the NR2B subunit, recordings from D1-WT MSSNs were made with and without Ifenprodil (1 μM). NMDAR-mediated currents were potentiated regardless of the presence or absence of Ifenprodil. However, a significant decrease in potentiation by SKF 81297 (1 μM) was observed when Ifenprodil was present (20.8 ± 1.8%, n = 28 in the absence, and 14.1 ± 1.6%, n = 23 in the presence of Ifenprodil, p = 0.01; Figure 9B middle). These data suggest that the NR2B subunit finely tunes D1R modulation; without NR2B, D1R modulation of NMDAR-mediated currents is dampened. In contrast, pharmacological blockade of NR2B subunits did not significantly affect D2R-mediated attenuation of NMDAR-mediated responses with quinpirole (10 μM; Figure 10 middle).
Effects of Combined Genetic and Pharmacological Block of NR2A and NR2B Subunits on D1R Modulation
The combined effect of NR2A and NR2B subunits on D1R modulation was analyzed by genetically deleting NR2A and pharmacologically blocking NR2B-expressing NMDARs. Modulation was compared between D1R-expressing WT cells (n = 24), in which NR2A and NR2B subunits remained intact, and D1R-expressing cells in which NR2A was genetically deleted and NR2B subunits were blocked by Ifenprodil (n = 11). Interestingly, potentiation levels of NMDAR-mediated currents were similar for both groups (Figure 9B right). These data suggest that in the absence of NR2 subunits, the presence of NR1 is enough to maintain normal D1R potentiation levels.
The combined role of NR2 subunits in D2R attenuation of NMDAR-mediated currents also was examined. D2R modulation was compared between MSSNs with NR2 subunits intact (D2-WTs, n = 9) and MSSNs in which NR2A was genetically deleted and NR2B was blocked by Ifenprodil (D2-NR2AKO + Ifenprodil, n = 9). NMDAR-mediated currents from both groups were attenuated similarly by quinpirole, suggesting that the combined presence or absence of NR2A and NR2B subunits does not play a significant role in D2R modulation of NMDA currents (Figure 10 right).
Discussion
The major findings of this study are that NR1, NR2A, and NR2B subunits differentially sculpt and modulate glutamatergic inputs in striatal MSSNs. Down-regulation of NR1 subunits in NR1-KD mice produces a significant reduction of NMDAR-mediated responses and removes the ability of D1 receptors to enhance these responses. Associated with these changes, AMPA receptor function is increased in striatal slices and dissociated neurons. Absence or pharmacological blockade of NR2A and NR2B subunits affect the amplitude and kinetics of NMDA responses and produce contrasting effects on D1 modulation. Whereas absence of NR2A subunits enhances NMDAR-mediated potentiation, pharmacological blockade of NR2B subunits decreases this potentiation, suggesting that these subunits have different roles and counterbalance each other. In contrast, D2R modulation of NMDA responses is largely unaffected by NR2A/B subunits. Finally, we also demonstrate that the relative proportion of NR2 subunits is different in subpopulations of MSSNs, with D1 cells (direct pathway) functionally expressing relatively more NR2A-containing NMDARs and D2 cells (indirect pathway) functionally expressing more NR2B-containing NMDARs. The fact that many of the findings could be demonstrated in slices and in isolated cell preparations is one of the strengths of the present study.
The NR1 subunit is obligatory for a functional NMDA receptor. Thus, the reduction in NMDA receptor-mediated currents in MSSNs from NR1-KD mice was expected. However, the increase in AMPAR-mediated responses was unexpected and suggested compensatory mechanisms to counter deficits in NMDA receptor function. While this effect could be the result of homeostatic mechanisms to balance such deficits, it could also reflect a limitation of the use of genetic knock-out or knock-down animals to examine the function of specific receptor subunits. The very low expression of NR1 subunits during development could have contributed to the changes in AMPA receptor function observed in the present study.
Spontaneous EPSCs displayed faster kinetics indicating that AMPA receptor subunit composition or glutamate transporters may also have changed. However, it cannot be ruled out that the near absence of NR1 subunits contributed to faster EPSCs as NMDAR-mediated responses have slower kinetics than AMPA responses. Finally, in dissociated MSSNs responses to AMPA were increased in NR1-KD animals. Alterations in frequency and kinetics of spontaneous EPSCs, as well as increased responsiveness to AMPA, suggest changes at both the circuit and receptor levels after know-down of NR1 subunits.
Dopamine D1 receptor modulation of NMDAR-mediated currents in NR1-KD mice showed significant deficits. Enhancement of NMDAR-mediated responses by D1 receptors in striatum is enacted through a number of intracellular signaling cascades including cAMP–PKA–DARPP32, Ca2+ conductances, and phosphorylation of NMDA receptor subunits (Snyder et al., 1998; Fiorentini and Missale, 2004; Cepeda et al., 2008a). Reduced expression of NR1 subunits completely obliterated D1 receptor modulation, suggesting that this subunit may represent a final common path for these signaling cascades.
The NR2A/B regulatory subunits confer different sensitivity to zinc and polyamines, open probability, deactivation time, and channel conductance, to name a few (Kohr, 2006). Although still controversial, it appears that NR2 subunit expression differs depending on receptor location with the most prominent contribution of NR2A-containing NMDARs at synaptic sites and NR2B-containing NMDARs at both synaptic and extrasynaptic receptor locations. The present study provides some support for this idea, as genetic KO of NR2A subunits changed the kinetics of evoked synaptic responses.
D1 receptor potentiation of NMDA currents is altered by NR2 subunit composition, but D2R attenuation of these currents is not. Genetic deletion of the NR2A subunit leads to enhanced D1R potentiation of NMDAR-mediated currents, suggesting that the presence of the NR2A subunit normally counters D1R potentiation. Direct protein coupling is known to occur between the C-terminus of the D1R and the C-terminus of the NR2A subunit, and in the presence of PKA and PKC antagonists, this direct coupling causes a decrease in NMDAR-mediated currents (Lee et al., 2002; Lee and Liu, 2004). This attenuation of currents occurs because fewer NMDARs are expressed on the membrane. In NR2AKO mice, the NR2A subunit is missing, thus direct coupling between D1Rs and NR2A-containing NMDARs cannot occur.
In contrast, the presence of the NR2B subunit enhances D1R potentiation. Genetic deletion of the NR2A subunit at NMDARs located at the soma and proximal dendrites leads to increased Ifenprodil sensitivity and pharmacological blockade of NR2B-containing NMDARs reduces D1R potentiation. Interestingly, studies in the striatum and prefrontal cortex have demonstrated that activation of D1Rs leads to increased surface expression of the NR2B subunit, potentially explaining the observed enhancement of NMDAR-mediated currents (Dunah and Standaert, 2001; Hallett et al., 2006). The mechanism responsible for this transfer of NMDARs from the intracellular membrane to the plasma membrane surface involves phosphorylation of NR2B subunits by tyrosine kinases (Dunah and Standaert, 2001; Dunah et al., 2004; Hallett et al., 2006; Gao and Wolf, 2008; Pascoli et al., 2011). Therefore, pharmacological blockade of the NR2B subunit will prevent increased surface expression by D1R activation and the level of potentiation of NMDAR-mediated currents will decrease. Heteromeric complexes consisting of NMDA and D1Rs are known to exist and these complexes increase the density of D1Rs at the postsynaptic density (Fiorentini and Missale, 2004).
It appears that NR2A and NR2B subunits maintain a balance, and when one of these subunits is missing or antagonized, the other predominates and the degree of D1R modulation becomes affected. However, when both NR2A and NR2B subunits are blocked, D1R modulation levels of NMDAR-mediated currents are similar to WT levels. Perhaps phosphorylation of NR1 subunits is sufficient to allow renormalization of D1R potentiation (Snyder et al., 1998).
Interestingly, changes in NR2 subunit composition do not alter D2R modulation of NMDA currents. Direct protein–protein coupling does occur between NMDARs and D2Rs, although only in the presence of cocaine (Liu et al., 2006). It is striking that despite the existence of D2-NR2B heteroreceptors, pharmacological block of NR2B-expressing receptors does not alter D2R modulation. Because these heteroreceptors appear to occur only under particular conditions, they may not play an important role in normal D2R modulation of NMDAR-mediated currents.
Prior studies examining NR2 subunit expression in striatum observed the presence of NR2A and NR2B subunits in MSSNs. However, conclusive data regarding the preferred expression of NR2 subunits between MSSNs of the direct (D1) and indirect (D2) pathways was not observed (Landwehrmeyer et al., 1995; Standaert et al., 1999). In the present study, significant functional differences in NR2 subunit composition of NMDARs were observed with D1R-containing cells expressing relatively greater NR2A subunit levels while D2R-containing cells express relatively greater NR2B subunit levels. Opposite results were observed by Fantin et al. (2007), concluding that NR2A subunits regulate D2 while NR2B subunits regulate D1 MSSNs. Differences in methodology could explain these opposing findings. One caveat of the present study is that only acutely isolated MSSNs were used for the NR2 subunit experiments. Thus, observed differences in NR2 subunit expression between D1R- and D2R-expressing MSSNs are applicable only to NMDARs located at the soma and proximal dendrites. These data do not provide information about the distribution of NR2A- and NR2B-containing NMDARs at distal dendrite locations on MSSN subpopulations.
Differences in NR2 subunit expression may play a key role in excitotoxicity and, therefore, could increase the overall vulnerability of subpopulations of MSSNs. Several pieces of evidence suggest that D1 MSSNs are less vulnerable to excitotoxicity than D2 MSSNs. D1 cells express greater levels of NR2A-containing NMDARs than D2 cells and these NMDARs have faster decay times (Monyer et al., 1994; Flint et al., 1997). As a result, Ca2+ flux is more restricted in D1 cells, potentially providing more protection for these cells. The combination of increased excitability (Kreitzer and Malenka, 2007; Cepeda et al., 2008b; Gertler et al., 2008) and greater functional expression of NR2B-containing NMDARs in D2 cells may increase their vulnerability to excitotoxicity.
The functional importance of NR2 subunit composition of NMDARs is highlighted by pathological conditions in which changes in subunit composition occur, including Huntington’s disease (HD). Alterations in NR2 subunit composition are observed in a mouse model of HD (Ali and Levine, 2006). Additionally, shifts in the NR2 subunit ratio of another HD model leads to increased striatal neurodegeneration by overexpression of NR2B subunits, further emphasizing that changes in NR2 composition can exacerbate pathological conditions (Heng et al., 2009). The location of NR2 subunits also plays an important role. NR2A subunits are predominantly found at the synapse, whereas NR2B subunits occur at both synaptic and extrasynaptic sites. It has been shown that stimulation of synaptic NMDA receptors confers cell protection in mouse models of HD, whereas stimulation of NR2B-containing NMDA receptors at extrasynaptic locations is pro-apoptotic and detrimental for cell survival (Okamoto et al., 2009; Milnerwood et al., 2010).
NMDA receptors play an essential role in cognition and mood regulation. Blockade or deficits in NMDA receptor-function have been associated with the negative symptoms of schizophrenia (Olney et al., 1999). However, individual subunits appear to play different roles. In the NR1-KD mouse used in the present study, behavioral alterations similar to those observed in pharmacologically induced animal models of schizophrenia have been reported (Mohn et al., 1999; Ramsey, 2009). Cognitive deficits in NR1-KD mice could be compounded by the lack of D1 receptor potentiation of NMDA responses. In contrast, NR2A subunit KO mice demonstrate a selective reduction in anxiety and depression, supporting a role of NR2A subunits in the modulation of emotional behaviors (Boyce-Rustay and Holmes, 2006). Absence of the NR2A subunits in mice also leads to deficits in discrimination learning (Brigman et al., 2008), suggesting this subunit plays a role in synaptic plasticity. In contrast, NR2B antagonists appear to improve cognitive performance in attentional tasks (Kos et al., 2011). The idea that NR2A and NR2B subunits play differential roles in synaptic plasticity remains controversial, and most studies indicate that both are required for long-term synaptic changes (Erreger et al., 2005; Fox et al., 2006; Schotanus and Chergui, 2008; Muller et al., 2009). The fact that their interactions with D1 receptors are bidirectional lends support to the idea of specific functions of each subunit in synaptic plasticity, at least in striatum.
Conclusion
In conclusion, using genetic and pharmacological tools we provide evidence that NMDA receptor subunits differentially contribute to the sculpting of NMDA responses and their modulation by D1 dopamine receptors in striatal MSSNs. The NR1 subunit is critical for both functions as down-regulation of this subunit almost completely eliminates NMDA responses and their modulation. NR2A/B subunits can affect amplitude and kinetics of the response and play contrasting roles in D1 receptor modulation. In addition, we demonstrate that in D1 receptor-containing MSSNs the NR2A subunit has a more prominent functional role, whereas in D2 receptor-containing MSSNs the NR2B subunit predominates. This could have important implications for differential synaptic plasticity and neuronal vulnerability of MSSNs of the direct and indirect pathways. We have demonstrated that D1 receptor activation enhances NMDA responses (Cepeda et al., 1993; Andre et al., 2010a). If NR2A subunits are more concentrated around synapses, it is possible that D1 receptor enhancement of synaptic NMDA responses would support plasticity in direct pathway MSSNs. In contrast, the presence of NR2B subunits at both synaptic and extrasynaptic loci, could make neurons of the indirect pathway more vulnerable to excess glutamate release and would be detrimental for cell survival (Hardingham et al., 2002). Additional studies will be necessary to unravel the role of second messenger cascades and protein–protein interactions in these contrasting roles of NR2A/B subunits.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by USPHS Grant NS33538. We would like to thank Amaal J. Starling, Yvette E. Fisher, and Steven M. Gee for data collection and Donna Crandall for help with the illustrations. Damian M. Cummings present address is Department of Neuroscience, Physiology and Pharmacology, University College London, UK. Amy J. Ramsey present address is University of Toronto, Department of Pharmacology and Toxicology, Toronto, ON, Canada.
References
Ali, N. J., and Levine, M. S. (2006). Changes in expression of N-methyl-D-aspartate receptor subunits occur early in the R6/2 mouse model of Huntington’s disease. Dev. Neurosci. 28, 230–238.
Andre, V. M., Cepeda, C., Cummings, D. M., Jocoy, E. L., Fisher, Y. E., William Yang, X., and Levine, M. S. (2010a). Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur. J. Neurosci. 31, 14–28.
Andre, V. M., Cepeda, C., and Levine, M. S. (2010b). Dopamine and glutamate in Huntington’s disease: a balancing act. CNS Neurosci. Ther. 16, 163–178.
Bargas, J., Howe, A., Eberwine, J., Cao, Y., and Surmeier, D. J. (1994). Cellular and molecular characterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. J. Neurosci. 14, 6667–6686.
Bouyer, J. J., Park, D. H., Joh, T. H., and Pickel, V. M. (1984). Chemical and structural analysis of the relation between cortical inputs and tyrosine hydroxylase-containing terminals in rat neostriatum. Brain Res. 302, 267–275.
Boyce-Rustay, J. M., and Holmes, A. (2006). Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology 31, 2405–2414.
Brady, A. M., and O’Donnell, P. (2004). Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J. Neurosci. 24, 1040–1049.
Brigman, J. L., Feyder, M., Saksida, L. M., Bussey, T. J., Mishina, M., and Holmes, A. (2008). Impaired discrimination learning in mice lacking the NMDA receptor NR2A subunit. Learn Mem. 15, 50–54.
Calabresi, P., Gubellini, P., Centonze, D., Picconi, B., Bernardi, G., Chergui, K., Svenningsson, P., Fienberg, A. A., and Greengard, P. (2000). Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J. Neurosci. 20, 8443–8451.
Cepeda, C., Andre, V. M., Jocoy, E. L., and Levine, M. S. (2008a). “NMDA and dopamine: diverse mechanisms applied to interacting receptor systems,” in The Biology of the NMDA Receptor, ed. A. M. Vandongen (Boca Raton, FL: Taylor & Francis Group, LLC), 41–57.
Cepeda, C., André, V. M., Yamazaki, I., Wu, N., Kleiman-Weiner, M., and Levine, M. S. (2008b). Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur. J. Neurosci. 27, 671–682.
Cepeda, C., Buchwald, N. A., and Levine, M. S. (1993). Neuromodulatory actions of dopamine in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc. Natl. Acad. Sci. U.S.A. 90, 9576–9580.
Cepeda, C., Colwell, C. S., Itri, J. N., Chandler, S. H., and Levine, M. S. (1998). Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J. Neurophysiol. 79, 82–94.
Chergui, K., and Lacey, M. G. (1999). Modulation by dopamine D1-like receptors of synaptic transmission and NMDA receptors in rat nucleus accumbens is attenuated by the protein kinase C inhibitor Ro 32-0432. Neuropharmacology 38, 223–231.
Dunah, A. W., Sirianni, A. C., Fienberg, A. A., Bastia, E., Schwarzschild, M. A., and Standaert, D. G. (2004). Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol. 65, 121–129.
Dunah, A. W., and Standaert, D. G. (2001). Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J. Neurosci. 21, 5546–5558.
Dunah, A. W., and Standaert, D. G. (2003). Subcellular segregation of distinct heteromeric NMDA glutamate receptors in the striatum. J. Neurochem. 85, 935–943.
Erreger, K., Dravid, S. M., Banke, T. G., Wyllie, D. J., and Traynelis, S. F. (2005). Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J. Physiol. 563, 345–358.
Fantin, M., Marti, M., Auberson, Y. P., and Morari, M. (2007). NR2A and NR2B subunit containing NMDA receptors differentially regulate striatal output pathways. J. Neurochem. 103, 2200–2211.
Fiorentini, C., and Missale, C. (2004). Oligomeric assembly of dopamine D1 and glutamate NMDA receptors: molecular mechanisms and functional implications. Biochem. Soc. Trans. 32, 1025–1028.
Flint, A. C., Maisch, U. S., Weishaupt, J. H., Kriegstein, A. R., and Monyer, H. (1997). NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J. Neurosci. 17, 2469–2476.
Flores-Hernandez, J., Cepeda, C., Hernandez-Echeagaray, E., Calvert, C. R., Jokel, E. S., Fienberg, A. A., Greengard, P., and Levine, M. S. (2002). Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J. Neurophysiol. 88, 3010–3020.
Fox, C. J., Russell, K. I., Wang, Y. T., and Christie, B. R. (2006). Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus 16, 907–915.
Freund, T. F., Powell, J. F., and Smith, A. D. (1984). Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience 13, 1189–1215.
Gao, C., and Wolf, M. E. (2008). Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J. Neurochem. 106, 2489–2501.
Gertler, T. S., Chan, C. S., and Surmeier, D. J. (2008). Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci. 28, 10814–10824.
Hallett, P. J., Spoelgen, R., Hyman, B. T., Standaert, D. G., and Dunah, A. W. (2006). Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J. Neurosci. 26, 4690–4700.
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414.
Heng, M. Y., Detloff, P. J., Wang, P. L., Tsien, J. Z., and Albin, R. L. (2009). In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J. Neurosci. 29, 3200–3205.
Ikeda, K., Araki, K., Takayama, C., Inoue, Y., Yagi, T., Aizawa, S., and Mishina, M. (1995). Reduced spontaneous activity of mice defective in the epsilon 4 subunit of the NMDA receptor channel. Brain Res. Mol. Brain Res. 33, 61–71.
Kohr, G. (2006). NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 326, 439–446.
Kos, T., Nikiforuk, A., Rafa, D., and Popik, P. (2011). The effects of NMDA receptor antagonists on attentional set-shifting task performance in mice. Psychopharmacology (Berl) 214, 911–921.
Kramer, P. F., Christensen, C. H., Hazelwood, L. A., Dobi, A., Bock, R., Sibley, D. R., Mateo, Y., and Alvarez, V. A. (2011). Dopamine D2 receptor overexpression alters behavior and physiology in Drd2-EGFP mice. J. Neurosci. 31, 126–132.
Kreitzer, A. C., and Malenka, R. C. (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647.
Kutsuwada, T., Sakimura, K., Manabe, T., Takayama, C., Katakura, N., Kushiya, E., Natsume, R., Watanabe, M., Inoue, Y., Yagi, T., Aizawa, S., Arakawa, M., Takahashi, T., Nakamura, Y., Mori, H., and Mishina, M. (1996). Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16, 333–344.
Landwehrmeyer, G. B., Standaert, D. G., Testa, C. M., Penney, J. B. Jr., and Young, A. B. (1995). NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J. Neurosci. 15, 5297–5307.
Laube, B., Kuhse, J., and Betz, H. (1998). Evidence for a tetrameric structure of recombinant NMDA receptors. J. Neurosci. 18, 2954–2961.
Lee, F. J., and Liu, F. (2004). Direct interactions between NMDA and D1 receptors: a tale of tails. Biochem. Soc. Trans. 32, 1032–1036.
Lee, F. J., Xue, S., Pei, L., Vukusic, B., Chery, N., Wang, Y., Wang, Y. T., Niznik, H. B., Yu, X. M., and Liu, F. (2002). Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 111, 219–230.
Levine, M. S., Altemus, K. L., Cepeda, C., Cromwell, H. C., Crawford, C., Ariano, M. A., Drago, J., Sibley, D. R., and Westphal, H. (1996a). Modulatory actions of dopamine on NMDA receptor-mediated responses are reduced in D1A-deficient mutant mice. J. Neurosci. 16, 5870–5882.
Levine, M. S., Li, Z., Cepeda, C., Cromwell, H. C., and Altemus, K. L. (1996b). Neuromodulatory actions of dopamine on synaptically-evoked neostriatal responses in slices. Synapse 24, 65–78.
Liu, X. Y., Chu, X. P., Mao, L. M., Wang, M., Lan, H. X., Li, M. H., Zhang, G. C., Parelkar, N. K., Fibuch, E. E., Haines, M., Neve, K. A., Liu, F., Xiong, Z. G., and Wang, J. Q. (2006). Modulation of D2R-NR2B interactions in response to cocaine. Neuron 52, 897–909.
Milnerwood, A. J., Gladding, C. M., Pouladi, M. A., Kaufman, A. M., Hines, R. M., Boyd, J. D., Ko, R. W., Vasuta, O. C., Graham, R. K., Hayden, M. R., Murphy, T. H., and Raymond, L. A. (2010). Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 65, 178–190.
Mohn, A. R., Gainetdinov, R. R., Caron, M. G., and Koller, B. H. (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436.
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540.
Muller, T., Albrecht, D., and Gebhardt, C. (2009). Both NR2A and NR2B subunits of the NMDA receptor are critical for long-term potentiation and long-term depression in the lateral amygdala of horizontal slices of adult mice. Learn Mem. 16, 395–405.
Okamoto, S., Pouladi, M. A., Talantova, M., Yao, D., Xia, P., Ehrnhoefer, D. E., Zaidi, R., Clemente, A., Kaul, M., Graham, R. K., Zhang, D., Vincent Chen, H. S., Tong, G., Hayden, M. R., and Lipton, S. A. (2009). Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 15, 1407–1413.
Olney, J. W., Newcomer, J. W., and Farber, N. B. (1999). NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 33, 523–533.
Pascoli, V., Besnard, A., Herve, D., Pages, C., Heck, N., Girault, J. A., Caboche, J., and Vanhoutte, P. (2011). Cyclic adenosine monophosphate-independent tyrosine phosphorylation of NR2B mediates cocaine-induced extracellular signal-regulated kinase activation. Biol. Psychiatry 69, 218–227.
Ramsey, A. J. (2009). NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog. Brain Res. 179, 51–58.
Sakimura, K., Kutsuwada, T., Ito, I., Manabe, T., Takayama, C., Kushiya, E., Yagi, T., Aizawa, S., Inoue, Y., Sugiyama, H., and Mishina, M. (1995). Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 373, 151–155.
Schotanus, S. M., and Chergui, K. (2008). NR2A-containing NMDA receptors depress glutamatergic synaptic transmission and evoked-dopamine release in the mouse striatum. J. Neurochem. 106, 1758–1765.
Schultz, W. (2010). Dopamine signals for reward value and risk: basic and recent data. Behav. Brain Funct. 6, 24.
Snyder, G. L., Fienberg, A. A., Huganir, R. L., and Greengard, P. (1998). A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J. Neurosci. 18, 10297–10303.
Standaert, D. G., Friberg, I. K., Landwehrmeyer, G. B., Young, A. B., and Penney, J. B. Jr. (1999). Expression of NMDA glutamate receptor subunit mRNAs in neurochemically identified projection and interneurons in the striatum of the rat. Brain Res. Mol. Brain Res. 64, 11–23.
Surmeier, D. J., Ding, J., Day, M., Wang, Z., and Shen, W. (2007). D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235.
Tovar, K. R., Sprouffske, K., and Westbrook, G. L. (2000). Fast NMDA receptor-mediated synaptic currents in neurons from mice lacking the epsilon2 (NR2B) subunit. J. Neurophysiol. 83, 616–620.
Keywords: NMDA, dopamine, receptor subunits, modulation, striatum
Citation: Jocoy EL, André VM, Cummings DM, Rao SP, Wu N, Ramsey AJ, Caron MG, Cepeda C and Levine MS (2011) Dissecting the contribution of individual receptor subunits to the enhancement of N-methyl-D-aspartate currents by dopamine D1 receptor activation in striatum. Front. Syst. Neurosci. 5:28. doi: 10.3389/fnsys.2011.00028
Received: 26 January 2011;
Paper pending published: 26 March 2011;
Accepted: 28 April 2011;
Published online: 11 May 2011.
Edited by:
Elizabeth Abercrombie, Rutgers-Newark: The State University of New Jersey, USAReviewed by:
Kuei Y. Tseng, Rosalind Franklin University of Medicine and Science, USAStephen Rayport, Columbia University, USA
Copyright: © 2011 Jocoy, André, Cummings, Rao, Wu, Ramsey, Caron, Cepeda and Levine. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Michael S. Levine, Intellectual and Developmental Disabilities Research Center, Semel Institute, Room 58-258, David Geffen School of Medicine, University of California Los Angeles, 760 Westwood Plaza, Los Angeles, CA 90024, USA. e-mail:bWxldmluZUBtZWRuZXQudWNsYS5lZHU=