
 Arthur Riegel1
Arthur Riegel1
- 1 Department of Neurosciences, Medical University of South Carolina, Charleston, SC, USA
- 2 Department of Electrical and Computer Engineering, University of Missouri, Columbia, MO, USA
Extracellular glutamate of glial origin modulates glial and neuronal glutamate release and synaptic plasticity. Estimates of the tonic basal concentration of extracellular glutamate range over three orders of magnitude (0.02–20 μM) depending on the technology employed to make the measurement. Based upon binding constants for glutamate receptors and transporters, this range of concentrations translates into distinct physiological and pathophysiological roles for extracellular glutamate. Here we speculate that the difference in glutamate measurements can be explained if there is patterned membrane surface expression of glutamate release and transporter sites creating extracellular subcompartments that vary in glutamate concentration and are preferentially sampled by different technologies.
The concentration of glutamate varies dramatically depending upon the biological compartment being measured. For example, plasma glutamate levels are estimated in the range of 150 μM, cerebrospinal fluid levels around 10 μM, and intracellular glutamate concentrations in the brain are approximately 10 mM (Danbolt, 2001; Featherstone and Shippy, 2008). The concentration of glutamate in the synaptic cleft following action potential mediated release exceeds 1 mM for <10 ms, and rapidly returns to <20 nM between release events due high affinity glutamate uptake by neurons and glia (Dzubay and Jahr, 1999). In contrast to the agreed upon glutamate concentrations in these biological compartments, estimates of the tonic basal concentration of glutamate within the extracellular space outside of the synaptic cleft varies over three orders of magnitude, ranging from 0.02 to 30 μM (Herman and Jahr, 2007; Chefer et al., 2009). The variance in estimated extracellular glutamate concentration results from electrophysiological estimates made in vitro from tissue slices (0.02–0.1 μM) versus in vivo measurements using microdialysis or voltammetry (1–30 μM). Discerning the reasons behind these discrepancies and ascertaining the correct level of extracellular glutamate outside the synaptic cleft is critical to understanding the physiology of how glutamate receptors, transporters, and antiporters regulate synaptic and glial glutamate release. Also, the concentration of extracellular glutamate will determine its role in metabolic processes such as cellular redox potential and neurometabolic coupling between synaptic activity, glial metabolism, and blood flow (Aoyama et al., 2008; Magistretti, 2009). Knowledge of how these glial and neuronal glutamatergic processes homeostatically regulate extracellular glutamate concentrations is central to understanding the physiology of excitatory neuroplasticity and its emerging role in neuropsychiatric diseases. For example, extracellular levels outside the synaptic cleft in the low micromolar range place glutamate in the range of the Kd for glutamate binding to extrasynaptic metabotropic glutamate receptors (mGluR), glutamate transporters, and extrasynaptic NMDA ionotropic glutamate receptors, while levels in low nanomolar range would not significantly occupy these binding sites (Conn and Pin, 1997; Danbolt, 2001; Herman and Jahr, 2007). Here we explore possible reasons why the in vitro electrophysiology and in vivo experiments deliver such disparate values. Further, we propose that both measurements are correctly assessing extracellular subcompartments that differentially contain nanomolar and micromolar extracellular glutamate, and that the concentration difference arises from the patterned expression of release and uptake sites on the membrane surface. The multicompartment proposal is illustrated in Figure 1, where we outline a working definition of extracellular glutamate into three subcompartments, including synaptic, perisynaptic, and nonsynaptic subcompartments.
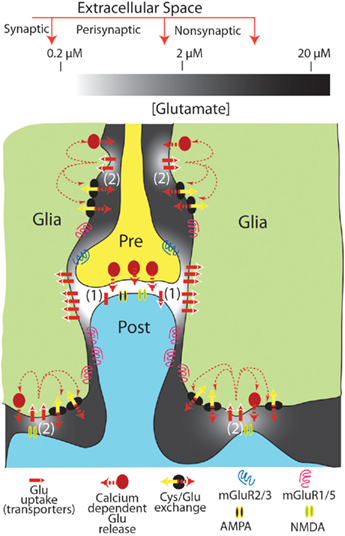
Figure 1. Tonic levels of extracellular glutamate measured in the synaptic, perisynaptic, and nonsynaptic compartments of the extracellular space. The levels of glutamate shown are approximated for resting conditions (i.e., in the absence of action potential mediated synaptic release). It is proposed that extracellular glutamate in compartmentalized morphologically by close appositions between neuronal and glial processes (note that the distances between glia–neuron distances are exaggerated for illustrative purposes), and by the patterned distribution of glutamate uptake sites and neuronal or glial release sites. This creates 2 regions of nanomolar glutamate high in iGluR, uptake sites and either synaptic (1) or glial (2) glutamate release sites. A second region surrounding the nanomolar region contains low micromolar levels of glutamate that are maintained largely from glial release and relatively lower glutamate uptake via high affinity transporters. This latter compartment is in part populated by mGluR that receive tonic glutamate stimulation and thereby modulate synaptic (and probably glial; D’Ascenzo et al., 2007) release and neuroplasticity.
In vivo (μM) Versus in vitro (nM) Measures of Extracellular Glutamate in Brain
Estimates of the in vivo concentration of extracellular glutamate using microdialysis have been made in a variety of mammalian species and in many brain regions (Chefer et al., 2009). The most accepted approach is the no-net-flux method where increasing concentrations of exogenous glutamate are passed through the probe and extracellular glutamate is inferred as the point where in vivo glutamate and exogenously applied glutamate are equal (i.e., no-net-flux of glutamate across the dialysis membrane). These studies measure 1–30 μM extracellular glutamate in different brain regions between different mammalian species, with the majority of studies finding levels between 1 and 5 μM (Jacobson et al., 1985; Lerma et al., 1986; Miele et al., 1996; Baker et al., 2003; Galvan et al., 2003; Szumlinski et al., 2004, 2005; Melendez et al., 2005a; Miller et al., 2008; Berglind et al., 2009). Microdialysis typically provides measurements at ≥1 min intervals, with most samples obtained in the range of 10–30 min intervals, and the measurement surface area (active dialysis membrane) is 103–104 μm2 (conventional microdialysis probes range from 150 to 250 μm diameter and 500 to 2000 μm length). More recently, in vivo voltammetry using biosensors coated with glutamate oxidase to enzymatically reduce glutamate have been employed to make measurements of glutamate over 0.2–30 s intervals, and employ a measurement surface area ranging from 30 to 250 μm2 (Oldenziel et al., 2004; Rahman et al., 2005). To estimate extracellular concentrations, voltammetry typically uses both a background electrode to subtract non-specific oxidative currents and an external glutamate calibration curve. Although the technology and probe characteristics differ, the measures of extracellular glutamate with biosensors are similar to microdialysis estimates in the range of 1–30 μM (Day et al., 2006; Oldenziel et al., 2007; Hascup et al., 2008; McLamore et al., 2010).
The size of the measurement surface for either microdialysis or biosensors obviates measuring release from single synapses. Moreover, the >1-min measurement interval in microdialysis studies contributes to an inability of dialysis to quantify synaptic glutamate release. Thus, the pool of glutamate being sampled by microdialysis under resting conditions in awake animals is not derived from action potential mediated release, as revealed by the fact that blocking voltage-dependent sodium channels with tetrodotoxin (TTX) does not significantly reduce extracellular levels of glutamate (Moghaddam, 1993; Morari et al., 1993; Timmerman and Westerink, 1997; Timmerman et al., 1999; Baker et al., 2002; Del Arco et al., 2003; Chefer et al., 2009). However, during certain behavioral tasks, TTX-sensitive increases in extracellular glutamate are measured using microdialysis (LaLumiere and Kalivas, 2008; Madayag et al., 2010). Some microdialysis studies find that voltage-dependent calcium channel antagonists partially (15–30%) decrease extracellular glutamate, supporting contribution by calcium-dependent glial glutamate release (D’Ascenzo et al., 2007). While microdialysis provides a measure of tonic extracellular glutamate that is integrated over time and is largely not of synaptic origin, glutamate biosensors measure basal levels of extracellular glutamate that are ∼30–50% inhibited by TTX (Hascup et al., 2008; van der Zeyden et al., 2008). Taken together, microdialysis and glutamate biosensors identify a tonic pool of 1–5 μM extracellular glutamate, the majority of which is derived from non-synaptic sources.
The sources of non-synaptic glutamate measured in vivo include Ca++-dependent glial release and cystine–glutamate exchange, and the basal non-synaptic concentration of extracellular glutamate results from a balance between these sources and glutamate uptake via high affinity glutamate transporters (Baker et al., 2002; Haydon et al., 2009). The cystine–glutamate antiporter exchanges one extracellular cystine molecule for one glutamate molecule and is a rate-limiting step in glutathione synthesis. Glia provide the dominant contribution to dialysis measurements of both cystine–glutamate exchange and glutamate transporter activity (Pow, 2001; Sato et al., 2002). In some brain regions inhibiting cystine–glutamate exchange reduces the basal extracellular concentration of glutamate by >50%, and inhibiting glutamate uptake can elevate levels fivefold (Baker et al., 2002). Confirming the role of cystine–glutamate exchange mediated release versus synaptic release in regulating the extracellular pool of glutamate measured by microdialysis, the rise in glutamate produced by blocking glutamate transporters is entirely prevented by blocking cystine–glutamate exchange while TTX has no effect (Baker et al., 2002; Melendez et al., 2005b). Also, deleting the gene transcribing the catalytic subunit of the cystine–glutamate exchanger (xCT) causes ∼50% reduction in extracellular glutamate measured by microdialysis (Massie et al., 2011). Finally, the chronic administration of addictive drugs such as cocaine and nicotine reduce xCT expression (Knackstedt et al., 2009), and this is reflected by reduced basal extracellular glutamate (Baker et al., 2003).
In contrast to the in vivo microdialysis and biosensor estimates of low micromolar extracellular glutamate in the hippocampus (Oldenziel et al., 2006a; Chefer et al., 2011; De Bundel et al., 2011), two studies using hippocampal slices find extracellular glutamate levels in the low nanomolar range (Cavelier and Attwell, 2005; Herman and Jahr, 2007). The authors measured tonic NMDA receptor (NMDAr) currents mediated by non-synaptic extracellular glutamate. These studies compared this tonic NMDAr current to the NMDAr current induced by saturating agonist (NMDA) concentration, and used an NMDAr dose–response curve for glutamate activation (Hill coefficient = 1.5, EC50 = 1.8–2.3 μM) generated in cultured hippocampal neurons (Patneau and Mayer, 1990) or a dose–response curve determined in a nucleated patch of neuronal membrane from the hippocampal slice (Herman and Jahr, 2007). Through these measurements they conclude that extracellular glutamate concentrations in hippocampal slices are between 23 and 89 nM (Cavelier and Attwell, 2005; Herman and Jahr, 2007). A low nanomolar concentration of glutamate is also consistent with estimates based upon the capacity of glutamate transporters to scavenge extracellular glutamate at concentrations up to 103 below the transporter Kd (1–10 μM; Zerangue and Kavanaugh, 1996; Danbolt, 2001).
Methodological Caveats for in vivo and in vitro Estimates of Glutamate
Below we outline various concerns that can be raised regarding the veracity the measures of extracellular glutamate by each technology. However, our primary thesis is that the methodological caveats only marginally contribute to the three-order of magnitude distinction between the in vitro and in vivo studies, and that this difference largely reflects the physiology of glutamate regulation in the extracellular space.
Damage Produced by in vivo Microdialysis Probes or Biosensors
The presence of non-synaptic, glial glutamate measured by dialysis and biosensors has created concern that these techniques may be over-estimating the level of extracellular glutamate, since 1–5 μM glutamate is in the vicinity of the Kd for NMDA receptors and would desensitize a portion of these receptors. Accordingly, damage to neuropil induced by inserting the dialysis probe or a large diameter (>100 μm) biosensor may cause non-physiological elevations of glutamate either directly from acute damage or due to the glial infiltration that occurs when the probes are in the brain for many hours, as is typical of in vivo experiments. Acute damage is likely not a concern because most experiments are conducted hours after probe insertion, and the levels of glutamate decline to a steady-state value by the time of measurement (van der Zeyden et al., 2008). However, the potential for glial infiltration and creation of an artificial extracellular compartment where glutamate can accumulate over the course of an experiment lasting many hours needs to be considered. Since both cystine–glutamate exchange and glutamate transport would be expected to accompany glial infiltration, creating a privileged environment with elevated glutamate concentrations is a reasonable concern. To some extent, this concern may be obviated by the fact that small diameter (10 μm) carbon fiber glutamate biosensors in the striatum find that tonic glutamate concentrations are higher (18–29 μM glutamate) than is typical for microdialysis or standard (>100 μm diameter) biosensors (1–5 μM; Kulagina et al., 1999; Oldenziel et al., 2006b); although, one carbon fiber voltammetry study reported 2 μM glutamate (Rahman et al., 2005). Interestingly, the higher levels of basal glutamate measured using carbon fiber biosensors were up to 90% TTX-dependent (van der Zeyden et al., 2008), and if one considers that the total basal levels of glutamate measured in this carbon fiber voltammetry study were 18 μM, the level of TTX-insensitive glutamate remaining is ∼1.8 μM glutamate, which is in the range measured by microdialysis (Oldenziel et al., 2006b).
Larger basal levels with carbon fiber voltammetry compared with dialysis have also been observed in studies of extracellular dopamine where voltammetry measures ∼10-fold more dopamine than is measured with microdialysis. Thus, mathematical modeling of microdialysis in brain has interposed a trauma layer between the neuropil and probe in an effort to understand the potential impact of damage on no-net-flux microdialysis estimates of extracellular neurotransmitters (Bungay et al., 2003; Tang et al., 2003; Chen, 2006). The conclusion from these analyses is that damage from a microdialysis probe produces a glial barrier separating the probe from neuropil where active dopamine uptake and synaptic release occurs, causing dialysis to underestimate extracellular dopamine levels. Thus, akin to the dopamine measurements the trauma layer created by microdialysis probes may constitute a barrier to synaptically released glutamate. This possibility is consistent with the analysis above that subtracting the TTX-sensitive glutamate from carbon fiber voltammetry measures of glutamate results in the 1- to 5-μM TTX-insensitive glutamate measured by microdialysis.
In spite of data indicating that dialysis could underestimate tonic extracellular levels of glutamate by eliminating synaptically released glutamate, it is possible that oxidative stress associated with a trauma layer could promote glutamate levels. For example, oxidative stress triggers transcriptional increases in xCT to support glutathione synthesis (Shih et al., 2003; Lewerenz et al., 2009), and thereby increases cystine/glutamate exchange. However, oxidative stress also increases glutamate uptake, in part to provide a source of intracellular glutamate for cystine–glutamate exchange to more effectively scavenge cystine from the extracellular environment for glutathione synthesis (Lewerenz et al., 2006). Also, while microglia invading an area of neuronal damage express GLT-1, these microglia also cause a reduction in glutamate transporter expression on astrocytes (Domercq et al., 2007; Nakajima et al., 2008; Tilleux and Hermans, 2008). Given these inferential and contradictory data, conclusions on how oxidative stress induced by in vivo probe damage to neuropil may contribute to basal glutamate measurements remains unclear.
Technical Caveats with in vitro Slice Experiments that May Affect Basal Glutamate
Three technical caveats with in vitro tissue slice physiology that could influence the concentration of extracellular glutamate measured are worth brief consideration. First, in contrast to in vivo experiments conducted largely in adult animals, the electrophysiological estimates of glutamate used P15–P19 mice or P11–P13 rats (Cavelier and Attwell, 2005; Herman and Jahr, 2007), and neither glutamate transporters nor cystine–glutamate exchange are fully expressed at this age (Kugler and Schleyer, 2004; La Bella et al., 2007). Also, glutamatergic tone on mGluR2/3 increases during development, such that the mGluR2/3 antagonist LY341495 is without effect on release probability at P13–17, but tone is demonstrable by P28–42 (Chen and Roper, 2004).
A second caveat is that the open diffusion perimeter across the slice surfaces can cause partial depletion of extracellular constituents unless they are actively replaced in the incubation medium. For example, depletion of ascorbic acid and glutathione normally found in vivo results in significant oxidative stress in tissue slices (Rice, 1999; Brahma et al., 2000); accordingly ascorbate is added to the buffer in most slice experiments. Interestingly, the reduction in glutathione may be related in part to depletion of the glutathione precursor cystine, which can directly affect extracellular glutamate levels in tissue slices via activating cystine–glutamate exchange. For example, cystine loss and a corresponding decrease in extracellular glutamate is indicated by the fact that restoring extracellular cystine to physiological levels of 100–300 nM in cortical or striatal slices activates cystine/glutamate exchange to increase extracellular glutamate levels measured by slice microdialysis, and restores tone onto mGluR2/3 release regulating autoreceptors (Moran et al., 2005). Contrary to this observation, when glutamate transporters are blocked in hippocampal slices NMDAr currents do not plateau, indicating that significant washout of glutamate may not be occurring (Herman and Jahr, 2007).
A final consideration is also related to the impact of making tissue slices on measures of synaptic transmission. A number of changes are induced by making hippocampal tissue slices on dendritic spine morphology, glial coverage of synapses and spines, and on the intracellular structure of dendrites (Witcher et al., 2007). For example, spine density is higher in hippocampal pyramidal neurons in recovered tissue slices than in hippocampal tissue fixed in vivo, and the extent of difference depends on different incubations conditions such as temperature and buffer constituents (Fiala et al., 2003; Bourne and Harris, 2008). As well, synapses in recovered hippocampal slices contain less glial coverage (Witcher et al., 2007). While all of these factors could affect the extracellular levels of glutamate in tissue slices, it remains to be determined how influential they are on synaptic physiology and extracellular glutamate levels.
Can Extracellular Subcompartments Explain the in vitro Estimate of Tonic Glutamate and the μM Levels Measured in vivo?
One explanation for the difference between the in vivo and in vitro measures of glutamate may lie in the presence of subcompartments of extracellular glutamate. The synaptic cleft is one subcompartment where glutamate levels are tightly regulated by both synaptic and glial glutamate uptake, and the concentration of glutamate is in the low nanomolar range to prevent desensitization of iGluR. The standing NMDAr current used to estimate extracellular glutamate levels in vitro are located in both the synaptic cleft and extrasynaptic membrane, with extrasynaptic NMDAr constituting ∼36% of total NMDAr in P14–21 acute hippocampal slices (Harris and Pettit, 2007); the age of animals used to estimate glutamate levels from NMDAr currents in slice studies (see discussion above). Electron microscopy reveals that akin to synaptic NMDAr, approximately 80% of the extrasynaptic NMDAr are localized to points of near contact between adjacent processes (Hardingham and Bading, 2010; Petralia et al., 2010). This is similar to estimates for the proportion of glutamate transporters also clustered adjacent to the synaptic cleft and at points of near contact by neuron–glial processes (Chaudhry et al., 1995; Cholet et al., 2002). Although never experimentally tested, if one assumes that the patterned expression of NMDAr and glutamate transporters is fairly widespread in the brain and not just the hippocampus or cerebellum (Bourne and Harris, 2008), the NMDAr may contact lower concentrations of tonic extracellular glutamate due to being localized in the vicinity of glutamate transporters. If indeed NMDAr both inside and outside the synaptic cleft are localized to areas of relatively high glutamate uptake, and thereby in regions of low basal glutamate tone, the electrophysiological estimates of extracellular glutamate made using whole cell patch measures of standing NMDAr currents would reflect extracellular subcompartment(s) that contain nanomolar glutamate. In contrast, outside these areas of relatively high glutamate uptake and NMDAr, glutamate would have a tendency to accumulate. The localization of the majority of NMDAr to such specialized compartments also offers an explanation for the observation that in contrast to applying NMDA, glutamate application to tissue slices in concentrations akin to those measured in vivo (2 μM) does not readily activate NMDAr (Herman and Jahr, 2007). Thus, a high density of glutamate transporters in the vicinity of the synapse would protect synaptic NMDAr from both endogenous non-synaptic glutamate released from glia and experimenter delivered non-synaptic glutamate. Together these data are consistent with a perspective that the standing NMDAr current created by endogenous tonic glutamate is in the low nanomolar range because the majority of NMDAr are localized to extracellular compartments where glutamate transporters are also enriched and basal glutamate levels are low (i.e., in the synapse and extrasynaptic specializations with high capacity for glutamate uptake).
The possible compartmentalization of different concentrations of extracellular glutamate is buttressed by measuring glutamatergic tone on mGluRs that are largely localized to extrasynaptic sites, and may therefore be exposed to higher basal levels of glutamate (Whittington et al., 1995; Schrader and Tasker, 1997; Marek et al., 2000; Oliet et al., 2001; Losonczy et al., 2003; Acuna-Goycolea et al., 2004; Wang et al., 2005; Ren et al., 2007). Although not intended to quantify extracellular glutamate concentrations in tissue slices, by considering EC50s of glutamate at mGluR and mGluR antagonist dose–response curves, these studies indicate extracellular glutamatergic tone on mGluRs in the low micromolar range. For example, in hippocampal slices the mGluR2/3 selective antagonist LY341495 potentiates the amplitude of evoked EPSCs by 36%, and a 15.5% potentiation of spontaneous EPSC (sEPSC) frequency was produced by the group III mGluR (4/6/7/8) antagonist CPPG in hypothalamic orexin neurons (Acuna-Goycolea et al., 2004). Equivalent findings exist from studies using mGluR2/3 or mGluR4/6/7/8 antagonists in the medial PFC, substantia nigra, and supraoptic nucleus (Marek et al., 2000; Oliet et al., 2001; Wang et al., 2005). Since mGluR2/3 and mGluR4/5/6/7 are largely extrasynaptic and have an EC50 for glutamate in the range of 1–10 μM (Tanabe et al., 1992; Conn and Pin, 1997; Pin et al., 1999), these results suggest that extracellular, extrasynaptic glutamate in brain slices is in the low micromolar range. Finally, studies measuring presynaptic AMPA/kainite ionotropic receptors [glutamate EC50 = 10.2 μM (Li et al., 1995)] indicate low micromolar glutamate tone since the AMPA antagonist NBQX reduces glutamate release by 36% (Ren et al., 2007). Finally, in vivo studies using local application of mGluR2/3 antagonist reveal glutamatergic tone on mGluR2/3s regulating the size of excitatory field potentials. By passing increasing concentrations of an mGluR2/3 antagonist through a dialysis probe adjacent to a recording electrode in the nucleus accumbens, it was shown that the amplitude of field potentials elicited by stimulating prefrontal glutamatergic afferents to the accumbens were under tonic inhibition by glutamate activation of mGluR2/3 (Moussawi et al., 2011). While these in vitro and in vivo data are consistent with glutamate tone on mGluRs, it is possible that constitutive activity of the mGluRs could account for some of the observed tone.
In summary, the use of standing NMDAr currents to estimate tonic extracellular glutamate may reflect glutamate concentrations in subcompartments of the extracellular space that are particularly dense in both NMDAr and glutamate transporters. Accordingly, this measure may not represent levels of glutamate that can accumulate in extracellular compartments where relatively less glutamate uptake and tonic glial release may create levels closer to the 1- to 10-μM measured by biosensors or microdialysis. Thus, when extrasynaptic effects of glutamate are measured at mGluR or presynaptic iGluR, the electrophysiological data are largely consistent with in vivo estimates of extracellular glutamate in the low micromolar range. A resolution to the marked differences in estimated concentrations of extracellular glutamate is depicted in Figure 1. Thus, at least two compartments of extracellular glutamate could exist; one compartment high in iGluR and glutamate transport and release processes (e.g., the synapse and perhaps non-synaptic neuron–neuron and neuron–glia appositions) that contains low nanomolar glutamate. Another non-synaptic compartment containing low micromolar glutamate and mGluRs is maintained largely by glial release and maintains a relatively lower density of glutamate transporters.
Subcompartments of Extracellular Glutamate Differentially Regulate Excitatory Transmission and Plasticity
A computational model recently suggested that extracellular glutamate exists at different concentrations in distinctly regulated but continuous subcompartments, termed synaptic, perisynaptic, and nonsynaptic (Pendyam et al., 2009). This model accommodates the discrepancy between in vitro electrophysiological estimates of glutamate based on NMDAr currents (synaptic), the in vitro electrophysiological estimates based on mGluR pharmacology (perisynaptic), and the dialysis/biosensor in vivo studies that directly measure glutamate levels (nonsynaptic). Thus, the synaptic compartment contains low nanomolar glutamate that is regulated by synaptic release and neuronal/glial glutamate transporters, while the nonsynaptic space contains low micromolar glutamate that is regulated by glial glutamate release and glial transporters, and the perisynaptic compartment containing mGluRs was modeled as a transition zone influenced by both tonic glial glutamate release and synaptic glutamate release (Figure 1; Pendyam et al., 2009).
An important aspect of this model is the patterned membrane surface expression of glutamate transporters and glial release sites that creates the equivalent of a semipermeable barrier between the synaptic and non-synaptic extracellular environments. The high affinity Na+-dependent glutamate transporter family includes five subtypes known as excitatory amino acid transporters (EAAT1–5; Danbolt, 2001). EAAT1 (GLAST) and EAAT2 (GLT-1) are expressed mainly in astrocytes, and EAAT3 and EAAT4 in neurons, whereas EAAT5 expression is limited to retina (Danbolt, 2001). GLAST and GLT-1 are non-uniformly expressed on glial membranes, with 73% of labeled transporters ensheathing excitatory synapses, dendritic spines, and other neuronal process contact sites (Cholet et al., 2002). Similarly, high expression of neuronal EAAT4 in the synaptic compartment strongly regulates the access of synaptic glutamate to mGluRs in the perisynaptic environment (Wadiche and Jahr, 2005). In addition to the patterned expression of transporters, the activity of transporters is non-linear in their contribution to reducing extracellular glutamate (Herman and Jahr, 2007), posing the possibility that different affinity transporters may be selectively localized to maintain different levels of extracellular glutamate. An additional subtlety arising from glia–neuron morphological specializations and patterned expression of glutamate transporters is that synaptic glutamate may have greater access to the perisynaptic environment at the pre-synapse than at the post-synapse, indicating greater influence on release regulating mGluRs (Lehre and Rusakov, 2002). It is also noteworthy that non-synaptic neuronal release may occur in synapse-like extracellular compartments that maintain nanomolar concentrations of glutamate. Thus, the rapid/transient stimulation of AMPAr on cerebellar Bergman glial cells by neuronal glutamate arises from a non-synaptic specialization containing high glial AMPAr density and rapid glutamate elimination (Matsui and Jahr, 2004; Matsui et al., 2005).
In contrast to neuronal release in the synaptic compartment, both in vitro electrophysiological and in vivo dialysis/biosensor studies indicate the presence of tonic extracellular glutamate derived from glia (Cavelier and Attwell, 2005; Chefer et al., 2009). Three sources of glutamate release from glia into the extrasynaptic space under physiological conditions have obtained significant experimental evidence, including Ca++-dependent (Halassa and Haydon, 2010), cystine–glutamate exchange (Baker et al., 2002), and passive diffusion (perhaps regulated by pH; Cavelier and Attwell, 2005). As with disparities in the concentration of extracellular glutamate, which glial source of glutamate contributes to the extracellular environment depends on the method of measurement. While in vitro electrophysiological studies using NMDAr currents to estimate the source of tonic glutamate in brain slices find no influence by Ca++-dependent or cystine–glutamate exchange (Jabaudon et al., 1999; Cavelier and Attwell, 2005), microdialysis/biosensors find contributions by both of these mechanisms (Baker et al., 2002; Halassa and Haydon, 2010). Conversely, NMDAr current estimates of tonic extracellular glutamate identified only passive glutamate release, perhaps regulated by pH (Cavelier and Attwell, 2005), indicating an estimate reflecting levels of glutamate in an extracellular compartment that largely excludes glutamate derived from Ca++-dependent glial release and cystine–glutamate exchange, such as the synaptic cleft (Figure 1).
As with synaptic release, the accumulation of extracellular glutamate derived from glial release is regulated by glutamate transporters (Melendez et al., 2005b); although the density of transporters is lower, thereby permitting extracellular glutamate to accumulate into a low micromolar range. While patterned expression of glutamate transporters relative to the location of glial Ca++-dependent release sites or cystine–glutamate exchangers has not been studied, it is noteworthy that the preferred source of intracellular glutamate released by cystine–glutamate exchange is glutamate recently taken-up by GLT-1, implying physical proximity of GLT-1 and cystine–glutamate exchange (Lewerenz et al., 2006), and the surface expression of GLT-1 and xCT may be co-regulated (Knackstedt et al., 2010). Thus, akin to the synaptic cleft where patterned expression of glutamate transport adjacent to the cleft tightly regulates tonic glutamate at low nanomolar levels, co-expression and regulation of glutamate transport and cystine–glutamate exchange may also regulate tonic micromolar extracellular glutamate.
Finally it is important to caveat the model illustrated in Figure 1. First, most research characterizing patterned transporter localization and glial apposition to excitatory synapses has been conducted at hippocampal, and to a lesser extent cerebellar and cortical excitatory synapses (Diamond and Jahr, 2000; Bourne and Harris, 2008). Thus, it remains unknown to what extent these morphological and attending physiological implications apply outside these areas. Second, even in the hippocampus serial electron microscopy and three-dimensional reconstruction shows that only ∼60% of the synapses have adjacent glial projections (Ventura and Harris, 1999; Witcher et al., 2007). While it has been speculated that these synapses may be the most active synapses based on having larger synapse diameters (Witcher et al., 2007), this poses the possibility that not all excitatory synapses in the brain contain the configuration illustrated in Figure 1, and that some synaptic glutamate will have access to extrasynaptic space, and perhaps even to adjacent synapses (see Rusakov and Kullmann, 1998; Bergles et al., 1999; Witcher et al., 2007 for detailed discussion of these issues).
Conclusion
The disparate estimates of the concentration and glial sources of tonic extracellular glutamate depend upon the technologies applied and the possibility that the different approaches may be measuring distinct extracellular compartments (Table 1 for summary). Thus, low nanomolar estimates of tonic glutamate are made in vitro using NMDAr currents, while low micromolar estimates are derived both in vivo or in vitro using microdialysis or glutamate biosensors. The latter direct measures of glutamate also identify Ca++-dependent release and cystine–glutamate exchange as the important sources of glial glutamate release, while using NMDAr currents eliminates these sources and finds only passive diffusion to provide tonic non-synaptic glutamate. Based upon glutamate transporters being in highest density adjacent to the synaptic cleft, and perhaps at non-synaptic contacts containing NMDAr (Figure 1), we propose that tonic NMDAr currents are estimating glutamate largely at points of cellular contact where glutamate values are in the low nanomolar range and contributions from Ca++-dependent glial release and cystine–glutamate exchange are largely absent. Conversely, direct measurement of glutamate in vivo identifies micromolar concentrations of glutamate that exist outside of the synaptic environment, are derived in part from glial Ca++-dependent mechanisms and cystine–glutamate exchange, and provide tone onto perisynaptic mGluR. In this way, the influence of synaptic glutamate release is largely limited in space to the synaptic and immediate perisynaptic environment, thereby providing temporally coded excitatory neuronal communication in the msec range. In contrast, glutamate of glial origin in the non-synaptic extracellular compartment provides tonic influence on synaptic release and postsynaptic responsiveness through pre- and postsynaptic mGluR. The role of mGluR stimulation in regulating synaptic plasticity is well-characterized and permits glial release to contribute to the long-term regulation of synaptic strength (Losonczy et al., 2003; Gladding et al., 2009). Importantly, neuropsychiatric diseases have been linked to neuropathologies in mGluRs and glial glutamate release and uptake that in turn alter how mGluRs regulate the potentiation and depotentiation of excitatory transmission (Coyle, 2006; Luscher and Huber, 2009; Moussawi et al., 2009; Yang et al., 2009). Thus, clear understanding of the physiological concentrations and regulation of synaptic and non-synaptic glutamate and how these two compartments of extracellular glutamate interact is necessary to assess the significance of disease-associated changes in proteins regulating glutamate receptors and glutamate release and uptake mechanisms. Unfortunately, experiments to directly evaluate the possibility of the extracellular subcompartments outlined in Figure 1 remain to be conducted. Such future studies include quantitative morphological measurements to determine how pervasive clustering of relevant proteins is at points of glia–neuron apposition (e.g., NMDAr, glutamate transporters, cystine–glutamate antiporter). Also, simultaneous measurements with carbon fiber voltammetry and whole cell patch measurements in tissue slices would reveal if both compartments can be simultaneously measured.
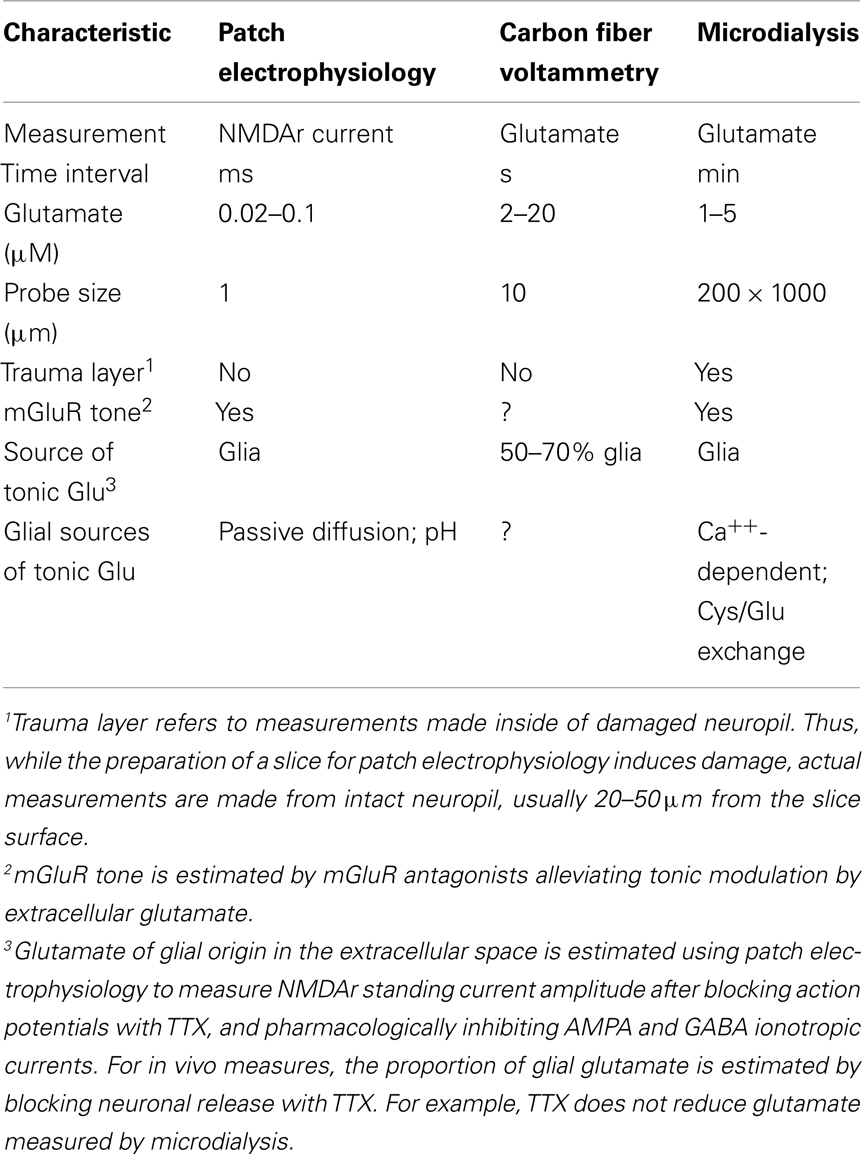
Table 1. Characteristics of tonic extracellular glutamate measured using different technologies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The concepts in this manuscript arose in part from work supported by NIH grants DA03906, DA12513, and DA015369.
References
Acuna-Goycolea, C., Li, Y., and van den Pol, A. N. (2004). Group III metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J. Neurosci. 24, 3013–3022.
Aoyama, K., Watabe, M., and Nakaki, T. (2008). Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 108, 227–238.
Baker, D. A., McFarland, K., Lake, R. W., Shen, H., Tang, X. C., Toda, S., and Kalivas, P. W. (2003). Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 6, 743–749.
Baker, D. A., Xi, Z. X., Shen, H., Swanson, C. J., and Kalivas, P. W. (2002). The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 22, 9134–9141.
Bergles, D. E., Diamond, J. S., and Jahr, C. E. (1999). Clearance of glutamate inside the synapse and beyond. Curr. Opin. Neurobiol. 9, 293–298.
Berglind, W. J., Whitfield, T. W. Jr., LaLumiere, R. T., Kalivas, P. W., and McGinty, J. F. (2009). A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J. Neurosci. 29, 3715–3719.
Bourne, J. N., and Harris, K. M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47–67.
Brahma, B., Forman, R. E., Stewart, E. E., Nicholson, C., and Rice, M. E. (2000). Ascorbate inhibits edema in brain slices. J. Neurochem. 74, 1263–1270.
Bungay, P. M., Newton-Vinson, P., Isele, W., Garris, P. A., and Justice, J. B. (2003). Microdialysis of dopamine interpreted with quantitative model incorporating probe implantation trauma. J. Neurochem. 86, 932–946.
Cavelier, P., and Attwell, D. (2005). Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J. Physiol. (Lond.) 564, 397–410.
Chaudhry, F. A., Lehre, K. P., van Lookeren Campagne, M., Ottersen, O. P., Danbolt, N. C., and Storm-Mathisen, J. (1995). Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15, 711–720.
Chefer, V., Meis, J., Wang, G., Kuzmin, A., Bakalkin, G., and Shippenberg, T. (2011). Repeated exposure to moderate doses of ethanol augments hippocampal glutamate neurotransmission by increasing release. Addict. Biol. 16, 229–237.
Chefer, V. I., Thompson, A. C., Zapata, A., and Shippenberg, T. S. (2009). Overview of brain microdialysis. Curr. Protoc. Neurosci. Chapter 7, Unit7.1.
Chen, H.-X., and Roper, S. N. (2004). Tonic activity of metabotropic glutamate receptors is involved in developmental modification of short-term plasticity in the neocortex. J. Neurophysiol. 92, 838–844.
Chen, K. C. (2006). Effects of tissue trauma on the characteristics of microdialysis zero-net-flux method sampling neurotransmitters. J. Theor. Biol. 238, 863–881.
Cholet, N., Pellerin, L., Magistretti, P. J., and Hamel, E. (2002). Similar perisynaptic glial localization for the Na+,K+-ATPase alpha 2 subunit and the glutamate transporters GLAST and GLT-1 in the rat somatosensory cortex. Cereb. Cortex 12, 515–525.
Conn, P. J., and Pin, J. P. (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37, 205–237.
Coyle, J. T. (2006). Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 26, 365–384.
D’Ascenzo, M., Fellin, T., Terunuma, M., Revilla-Sanchez, R., Meaney, D. F., Auberson, Y. P., Moss, S. J., and Haydon, P. G. (2007). mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 104, 1995–2000.
Day, B. K., Pomerleau, F., Burmeister, J. J., Huettl, P., and Gerhardt, G. A. (2006). Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J. Neurochem. 96, 1626–1635.
De Bundel, D., Schallier, A., Loyens, E., Fernando, R., Miyashita, H., Van Liefferinge, J., Vermoesen, K., Bannai, S., Sato, H., Michotte, Y., Smolders, I., and Massie, A. (2011). Loss of system x(c)- formula does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J. Neurosci. 31, 5792–5803.
Del Arco, A., Segovia, G., Fuxe, K., and Mora, F. (2003). Changes in dialysate concentrations of glutamate and GABA in the brain: an index of volume transmission mediated actions? J. Neurochem. 85, 23–33.
Diamond, J. S., and Jahr, C. E. (2000). Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J. Neurophysiol. 83, 2835–2843.
Domercq, M., Sanchez-Gomez, M. V., Sherwin, C., Etxebarria, E., Fern, R., and Matute, C. (2007). System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J. Immunol. 178, 6549–6556.
Dzubay, J. A., and Jahr, C. E. (1999). The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J. Neurosci. 19, 5265–5274.
Featherstone, D. E., and Shippy, S. A. (2008). Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist 14, 171–181.
Fiala, J. C., Kirov, S. A., Feinberg, M. D., Petrak, L. J., George, P., Goddard, C. A., and Harris, K. M. (2003). Timing of neuronal and glial ultrastructure disruption during brain slice preparation and recovery in vitro. J. Comp. Neurol. 465, 90–103.
Galvan, A., Smith, Y., and Wichmann, T. (2003). Continuous monitoring of intracerebral glutamate levels in awake monkeys using microdialysis and enzyme fluorometric detection. J. Neurosci. Methods 126, 175–185.
Gladding, C. M., Fitzjohn, S. M., and Molnar, E. (2009). Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol. Rev. 61, 395–412.
Halassa, M. M., and Haydon, P. G. (2010). Integrated brain circuits astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 72, 335–355.
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696.
Harris, A. Z., and Pettit, D. L. (2007). Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J. Physiol. (Lond.) 584, 509–519.
Hascup, K. N., Hascup, E. R., Pomerleau, F., Huettl, P., and Gerhardt, G. A. (2008). Second-by-second measures of L-glutamate in the prefrontal cortex and striatum of freely moving mice. J. Pharmacol. Exp. Ther. 324, 725–731.
Haydon, P. G., Blendy, J., Moss, S. J., and Rob Jackson, F. (2009). Astrocytic control of synaptic transmission and plasticity: a target for drugs of abuse? Neuropharmacology 56(Suppl. 1), 83–90.
Herman, M. A., and Jahr, C. E. (2007). Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 27, 9736–9741.
Jabaudon, D., Shimamoto, K., Yasuda-Kamatani, Y., Scanziani, M., Gahwiler, B., and Gerber, U. (1999). Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc. Natl. Acad. Sci. U.S.A. 96, 8733–8738.
Jacobson, I., Sandberg, M., and Hamberger, A. (1985). Mass transfer in brain dialysis devices – a new method for the estimation of extracellular amino acids concentration. J. Neurosci. Methods 15, 263–268.
Knackstedt, L. A., LaRowe, S., Mardikian, P., Malcolm, R., Upadhyaya, H., Hedden, S., Markou, A., and Kalivas, P. W. (2009). The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol. Psychiatry 65, 841–845.
Knackstedt, L. A., Melendez, R. I., and Kalivas, P. W. (2010). Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol. Psychiatry 67, 81–84.
Kugler, P., and Schleyer, V. (2004). Developmental expression of glutamate transporters and glutamate dehydrogenase in astrocytes of the postnatal rat hippocampus. Hippocampus 14, 975–985.
Kulagina, N. V., Shankar, L., and Michael, A. C. (1999). Monitoring glutamate and ascorbate in the extracellular space of brain tissue with electrochemical microsensors. Anal. Chem. 71, 5093–5100.
La Bella, V., Valentino, F., Piccoli, T., and Piccoli, F. (2007). Expression and developmental regulation of the cystine/glutamate exchanger (xc-) in the rat. Neurochem. Res. 32, 1081–1090.
LaLumiere, R. T., and Kalivas, P. W. (2008). Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 28, 3170–3177.
Lehre, K. P., and Rusakov, D. A. (2002). Asymmetry of glia near central synapses favors presynaptically directed glutamate escape. Biophys. J. 83, 125–134.
Lerma, J., Herranz, A. S., Herreras, O., Abraira, V., and Martin del Rio, R. (1986) In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 384, 145–155.
Lewerenz, J., Albrecht, P., Tien, M. L., Henke, N., Karumbayaram, S., Kornblum, H. I., Wiedau-Pazos, M., Schubert, D., Maher, P., and Methner, A. (2009). Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J. Neurochem. 111, 332–334.
Lewerenz, J., Klein, M., and Methner, A. (2006). Cooperative action of glutamate transporters and cystine/glutamate antiporter system X protects from oxidative glutamate toxicity. J. Neurochem. 98, 916–925.
Li, F., Owens, N., and Verdoorn, T. A. (1995). Functional effects of mutations in the putative agonist binding region of recombinant alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Mol. Pharmacol. 47, 148–154.
Losonczy, A., Somogyi, P., and Nusser, Z. (2003). Reduction of excitatory postsynaptic responses by persistently active metabotropic glutamate receptors in the hippocampus. J. Neurophysiol. 89, 1910–1919.
Luscher, C., and Huber, K. M. (2009). Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459.
Madayag, A., Kau, K. S., Lobner, D., Mantsch, J. R., Wisniewski, S., and Baker, D. A. (2010). Drug-induced plasticity contributing to heightened relapse susceptibility: neurochemical changes and augmented reinstatement in high-intake rats. J. Neurosci. 30, 210–217.
Magistretti, P. J. (2009). Role of glutamate in neuron-glia metabolic coupling. Am. J. Clin. Nutr. 90, 875S–880S.
Marek, G. J., Wright, R. A., Schoepp, D. D., Monn, J. A., and Aghajanian, G. K. (2000). Physiological antagonism between 5-hydroxytryptamine(2A) and group II metabotropic glutamate receptors in prefrontal cortex. J. Pharmacol. Exp. Ther. 292, 76–87.
Massie, A., Schallier, A., Kim, S. W., Fernando, R., Kobayashi, S., Beck, H., De Bundel, D., Vermoesen, K., Bannai, S., Smolders, I., Conrad, M., Plesnila, N., Sato, H., and Michotte, Y. (2011). Dopaminergic neurons of system x(c)-deficient mice are highly protected against 6-hydroxydopamine-induced toxicity. FASEB J. 25, 1359–1369.
Matsui, K., and Jahr, C. E. (2004). Differential control of synaptic and ectopic vesicular release of glutamate. J. Neurosci. 24, 8932–8939.
Matsui, K., Jahr, C. E., and Rubio, M. E. (2005). High-concentration rapid transients of glutamate mediate neural-glial communication via ectopic release. J. Neurosci. 25, 7538–7547.
McLamore, E. S., Mohanty, S., Shi, J., Claussen, J., Jedlicka, S. S., Rickus, J. L., and Porterfield, D. M. (2010). A self-referencing glutamate biosensor for measuring real time neuronal glutamate flux. J. Neurosci. Methods 189, 14–22.
Melendez, R. I., Hicks, M. P., Cagle, S. S., and Kalivas, P. W. (2005a). Ethanol exposure decreases glutamate uptake in the nucleus accumbens. Alcohol. Clin. Exp. Res. 29, 326–333.
Melendez, R. I., Vuthiganon, J., and Kalivas, P. W. (2005b). Regulation of extracellular glutamate in the prefrontal cortex: focus on the cystine glutamate exchanger and group I metabotropic glutamate receptors. J. Pharmacol. Exp. Ther. 314, 139–147.
Miele, M., Berners, M., Boutelle, M. G., Kusakabe, H., and Fillenz, M. (1996). The determination of the extracellular concentration of brain glutamate using quantitative microdialysis. Brain Res. 707, 131–133.
Miller, B. R., Dorner, J. L., Shou, M., Sari, Y., Barton, S. J., Sengelaub, D. R., Kennedy, R. T., and Rebec, G. V. (2008). Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience 153, 329–337.
Moghaddam, B. (1993). Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J. Neurochem. 60, 1650–1657.
Moran, M. M., McFarland, K., Melendez, R. I., Kalivas, P. W., and Seamans, J. K. (2005). Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J. Neurosci. 25, 6389–6393.
Morari, M., O’Connor, W. T., Ungerstedt, U., and Fuxe, K. (1993). N-methyl-D-aspartic acid differentially regulates extracellular dopamine, GABA, and glutamate levels in the dorsolateral neostriatum of the halothane-anesthetized rat: an in vivo microdialysis study. J. Neurochem. 60, 1884–1893.
Moussawi, K., Pacchioni, A., Moran, M., Olive, M. F., Gass, J. T., Lavin, A., and Kalivas, P. W. (2009). N-acetylcysteine reverses cocaine-induced metaplasticity. Nat. Neurosci. 12, 182–189.
Moussawi, K., Zhou, W., Shen, H., Reichel, C. M., See, R. E., Carr, D. B., and Kalivas, P. W. (2011). Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc. Natl. Acad. Sci. U.S.A. 108, 385–390.
Nakajima, K., Yamamoto, S., Kohsaka, S., and Kurihara, T. (2008). Neuronal stimulation leading to upregulation of glutamate transporter-1 (GLT-1) in rat microglia in vitro. Neurosci. Lett. 436, 331–334.
Oldenziel, W. H., Beukema, W., and Westerink, B. H. (2004). Improving the reproducibility of hydrogel-coated glutamate microsensors by using an automated dipcoater. J. Neurosci. Methods 140, 117–126.
Oldenziel, W. H., Dijkstra, G., Cremers, T. I., and Westerink, B. H. (2006a). Evaluation of hydrogel-coated glutamate microsensors. Anal. Chem. 78, 3366–3378.
Oldenziel, W. H., Dijkstra, G., Cremers, T. I., and Westerink, B. H. (2006b) In vivo monitoring of extracellular glutamate in the brain with a microsensor. Brain Res. 1118, 34–42.
Oldenziel, W. H., van der Zeyden, M., Dijkstra, G., Ghijsen, W. E., Karst, H., Cremers, T. I., and Westerink, B. H. (2007). Monitoring extracellular glutamate in hippocampal slices with a microsensor. J. Neurosci. Methods 160, 37–44.
Oliet, S., Piet, R., and Poulain, D. (2001). Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292, 923–925.
Patneau, D. K., and Mayer, M. L. (1990). Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J. Neurosci. 10, 2385–2399.
Pendyam, S., Mohan, A., Kalivas, P. W., and Nair, S. S. (2009). Computational model of extracellular glutamate in the nucleus accumbens incorporates neuroadaptations by chronic cocaine. Neuroscience 158, 1266–1276.
Petralia, R. S., Wang, Y. X., Hua, F., Yi, Z., Zhou, A., Ge, L., Stephenson, F. A., and Wenthold, R. J. (2010). Organization of NMDA receptors at extrasynaptic locations. Neuroscience 167, 68–87.
Pin, J. P., De Colle, C., Bessis, A. S., and Acher, F. (1999). New perspectives for the development of selective metabotropic glutamate receptor ligands. Eur. J. Pharmacol. 375, 277–294.
Pow, D. V. (2001). Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia 34, 27–38.
Rahman, M. A., Kwon, N. H., Won, M. S., Choe, E. S., and Shim, Y. B. (2005). Functionalized conducting polymer as an enzyme-immobilizing substrate: an amperometric glutamate microbiosensor for in vivo measurements. Anal. Chem. 77, 4854–4860.
Ren, M., Yoshimura, Y., Takada, N., Horibe, S., and Komatsu, Y. (2007). Specialized inhibitory synaptic actions between nearby neocortical pyramidal neurons. Science 316, 758–761.
Rice, M. E. (1999). Use of ascorbate in the preparation and maintenance of brain slices. Methods 18, 144–149.
Rusakov, D. A., and Kullmann, D. M. (1998). Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J. Neurosci. 18, 3158–3170.
Sato, H., Tamba, M., Okuno, S., Sato, K., Keino-Masu, K., Masu, M., and Bannai, S. (2002). Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J. Neurosci. 22, 8028–8033.
Schrader, L. A., and Tasker, J. G. (1997). Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J. Neurophysiol. 77, 527–536.
Shih, A. Y., Johnson, D. A., Wong, G., Kraft, A. D., Jiang, L., Erb, H., Johnson, J. A., and Murphy, T. H. (2003). Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 23, 3394–3406.
Szumlinski, K. K., Dehoff, M. H., Kang, S. H., Frys, K. A., Lominac, K. D., Klugmann, M., Rohrer, J., Griffin, W. III, Toda, S., Champtiaux, N. P., Berry, T., Tu, J. C., Shealy, S. E., During, M. J., Middaugh, L. D., Worley, P. F., and Kalivas, P. W. (2004). Homer proteins regulate sensitivity to cocaine. Neuron 43, 401–413.
Szumlinski, K. K., Lominac, K. D., Kleschen, M. J., Oleson, E. B., Dehoff, M. H., Schwarz, M. K., Seeburg, P. H., Worley, P. F., and Kalivas, P. W. (2005). Behavioral and neurochemical phenotyping of Homer1 mutant mice: possible relevance to schizophrenia. Genes Brain Behav. 4, 273–288.
Tanabe, Y., Masu, M., Ishii, T., Shigemoto, R., and Nakanishi, S. (1992). A family of metabotropic glutamate receptors. Neuron 8, 169–179.
Tang, A., Bungay, P. M., and Gonzales, R. A. (2003). Characterization of probe and tissue factors that influence interpretation of quantitative microdialysis experiments for dopamine. J. Neurosci. Methods 126, 1–11.
Tilleux, S., and Hermans, E. (2008). Down-regulation of astrocytic GLAST by microglia-related inflammation is abrogated in dibutyryl cAMP-differentiated cultures. J. Neurochem. 105, 2224–2236.
Timmerman, W., Cisci, G., Nap, A., de Vries, J. B., and Westerink, B. H. (1999). Effects of handling on extracellular levels of glutamate and other amino acids in various areas of the brain measured by microdialysis. Brain Res. 833, 150–160.
Timmerman, W., and Westerink, B. H. (1997). Brain microdialysis of GABA and glutamate: what does it signify? Synapse 27, 242–261.
van der Zeyden, M., Oldenziel, W. H., Rea, K., Cremers, T. I., and Westerink, B. H. (2008). Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol. Biochem. Behav. 90, 135–147.
Ventura, R., and Harris, K. M. (1999). Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 19, 6897–6906.
Wadiche, J. I., and Jahr, C. E. (2005). Patterned expression of Purkinje cell glutamate transporters controls synaptic plasticity. Nat. Neurosci. 8, 1329–1334.
Wang, L., Kitai, S. T., and Xiang, Z. (2005). Modulation of excitatory synaptic transmission by endogenous glutamate acting on presynaptic group II mGluRs in rat substantia nigra compacta. J. Neurosci. Res. 82, 778–787.
Whittington, M. A., Traub, R. D., and Jefferys, J. G. (1995). Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature 373, 612–615.
Witcher, M. R., Kirov, S. A., and Harris, K. M. (2007). Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia 55, 13–23.
Yang, Y., Gozen, O., Watkins, A., Lorenzini, I., Lepore, A., Gao, Y., Vidensky, S., Brennan, J., Poulsen, D., Won Park, J., Li Jeon, N., Robinson, M. B., and Rothstein, J. D. (2009). Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron 61, 880–894.
Keywords: glutamate, synapse, glia, glutamate uptake, cystine–glutamate exchange, mGluR
Citation: Moussawi K, Riegel A, Nair S and Kalivas PW (2011) Extracellular glutamate: functional compartments operate in different concentration ranges. Front. Syst. Neurosci. 5:94. doi: 10.3389/fnsys.2011.00094
Received: 02 April 2011; Accepted: 31 October 2011;
Published online: 24 November 2011.
Edited by:
Raphael Pinaud, University of Oklahoma Health Sciences Center, USAReviewed by:
Stephen Rayport, Columbia University, USAJose Bargas, Universidad Nacional Autónoma de México, Mexico
Copyright: © 2011 Moussawi, Riegel, Nair and Kalivas. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Peter W. Kalivas, Medical Unversity of South Carolina, 173 Ashley Avenue, DD205, Charleston, SC 29425, USA. e-mail:a2FsaXZhc3BAbXVzYy5lZHU=