
- 1 Department of Biological Science, The Florida State University, Tallahassee, FL, USA
- 2 Department of Physics, The Florida State University, Tallahassee, FL, USA
- 3 Institute of Molecular Biophysics, The Florida State University, Tallahassee, FL, USA
Striated muscle contraction is regulated by dynamic and cooperative interactions among Ca2+, troponin, and tropomyosin on the thin filament. While Ca2+ regulation has been extensively studied, little is known about the dynamics of individual regulatory units and structural changes of individual tropomyosin molecules in relation to their mechanical properties, and how these factors are altered by cardiomyopathy mutations in the Ca2+ regulatory proteins. In this hypothesis paper, we explore how various experimental and analytical approaches could broaden our understanding of the cooperative regulation of cardiac contraction in health and disease.
Cardiac muscle contraction is regulated by a Ca2+-dependent switch mechanism. Each basic contractile unit, or sarcomere, consists of a staggered array of thin and thick filaments. Contraction takes place when myosin heads in thick filaments engage in the cross-bridge cycle to generate isometric force or to slide adjacent thin filaments toward a sarcomere’s center, thereby shortening the sarcomere. Ca2+ regulation of striated muscle contraction is achieved primarily by the orchestrated action of troponin (Tn) and tropomyosin (Tm) on thin filaments. Tn is a three-subunit complex consisting of TnC, TnI, and TnT, where TnC is the Ca2+ sensor, TnI is the inhibitory subunit that holds the other two Tn subunits in a ternary complex and also binds actin, and TnT anchors the Tn complex to Tm. Tm is a dimeric, α-helical coiled-coil protein that binds along both grooves of an actin filament’s helix (O’Brien et al., 1971; Milligan et al., 1990; Perry, 2001). αTm is the predominant Tm isoform in adult human hearts. The molecular contour length (Lc) of αTm is about 40 nm (Smillie, 1996; Perry, 2001). All native Tm molecules polymerize into a long strand through head-to-tail overlap that involves about nine amino acids from the carboxy- and the amino-termini of adjacent molecules (McLachlan and Stewart, 1975). Systolic contraction is initiated by elevation of cytoplasmic Ca2+ that binds to Tn, which undergoes a conformational change that induces azimuthal movement of Tm on the thin filament to uncover myosin binding sites, thereby permitting cross-bridge cycling and concomitant ATP hydrolysis. As demonstrated by various biochemical (Lehrer et al., 1997; Maytum et al., 1999) and physiological (Metzger, 1995; Iwamoto, 1998; Dobesh et al., 2002) studies, cooperativity is a central feature of striated muscle activation and contraction. In the following discussion, unless otherwise specified, we focus on the Ca2+-induced cooperative activation of cardiac thin filaments in which Tm plays a central role through the end-to-end overlap between adjacent molecules.
Thin Filament Activation: Two State Versus Three State Model
Several models have been proposed depicting the mechanism by which thin filaments are activated. Hill’s model assumes two states that correspond to the thin filament being in an “on” or “off” conformation (Hill et al., 1980). The entire regulatory unit switches between states where myosin can bind either weakly or strongly. This model is appealing because it is based on well-established structural and thermodynamic details, but aspects of it are not entirely consistent with subsequent observations of thin filament structural data which show Ca2+ binding causes a major movement of Tm (Vibert et al., 1997).
A widely accepted model for striated muscle regulation based on kinetic studies was proposed by McKillop and Geeves (1993); it describes thin filament regulatory units in equilibrium between three states, termed Blocked, Closed, and Open. In each state, Tm occupies a different position on the thin filament that correlates with three states identified by structural studies. In the absence of Ca2+, the thin filament is said to be in the “blocked” state in which Tm sterically blocks myosin binding sites on actin; this state predominates in relaxed muscle and cardiac diastole. When Ca2+ binds to TnC, the equilibrium is shifted toward the “closed” state in which conformational changes in the Tn complex cause an azimuthal shift of Tm by 25° relative to the actin filament (Lehman et al., 1994; Xu et al., 1999). This partially uncovers myosin binding sites on actin and allows weak cross-bridge formation. These changes are followed by a structural transition of the thin filament to a fully active “open” state where Tm shifts further toward actin’s inner domain. Myosin binding sites on actin become fully exposed, thus allowing formation of additional strong crossbridges. Thus the three state model implies both Ca2+ and strong crossbridges have roles in thin filament activation; the relative importance of Ca2+ versus crossbridges appears to differ between cardiac and skeletal muscle, with cardiac muscle activation being more affected by strong crossbridge binding (Regnier et al., 2002; Gillis et al., 2007).
Cooperative Mechanism of Striated Muscle Activation: The Central Role of Tropomyosin
Cooperativity is an essential feature of striated muscle activation, especially in cardiac muscle because the heart functions in a highly coordinated manner. In addition to local cooperativity within a regulatory unit implied above, three models have been proposed for longer range cooperativity between regulatory units along a thin filament. An initial model was proposed by Hill et al. (1980). Hill suggested that each Tm–Tn and associated seven actin monomers act as a single unit that changes state individually, but the state of one unit affects the probability of activation of the adjacent, nearest-neighbor units; this can result in positive or negative cooperativity between nearest-neighbor units. In a later model of Geeves and Lehrer (1994), the concept of cooperative unit size n was introduced which represents the number of adjacent actin monomers that become available for myosin binding when a single regulatory unit turns on. This model incorporated Tm as a continuous, flexible filament allowing signal propagation along the surface of thin filaments. A third model was developed by Tobacman and Butters (2000) and involves actin–actin cooperativity which allows long range propagation of actin structural changes along a thin filament.
All three models have been shown to apply for a particular set of in vitro data, but currently none can fully explain the range of experimental data available (Boussouf and Geeves, 2007). This may be due, in part, to the complexity of the myofilament lattice in striated muscle. Each thin filament is comprised of several hundred molecules, and can interact with a similarly large number (tens to hundreds) of calcium ions and myosin molecules, with binding sites distributed along its length. Thus not only are there several different types of cooperative mechanisms in striated muscle, there are an even larger number of possible cooperative interactions (Gordon et al., 2000) that could affect muscle function. In Table 1, we summarize various cooperative mechanisms that may be involved in striated muscle regulation: Ca2+ binding to one regulatory unit may induce Ca2+ binding to an adjacent unit; formation of strong actomyosin crossbridges in one regulatory unit may induce Ca2+ binding to and/or crossbridge formation in the same and/or an adjacent unit; displacement of one tropomyosin (e.g., by Ca2+ binding to troponin and/or crossbridge formation) leads to the spread of activation which allows further crossbridge binding not only within that regulatory unit but also, through end-to-end contacts between adjacent tropomyosin molecules, in adjacent units; alternatively, it was proposed that activation might spread along the thin filament through actin monomers. All of these mechanisms might involve tropomyosin directly or indirectly. For instance, binding of Ca2+ to a regulatory unit leads to displacement of tropomyosin which affects the neighboring subunit, possibly through the Tm end-to-end overlap, and may induce Ca2+ binding to the neighboring Tn. Tm does in fact play a central role in cooperativity and is involved in the different mechanisms proposed above; it is indispensable for a coordinated activation of the muscle. There is an additional mechanism specific to skeletal muscle regulation, i.e., Ca2+ binding to one trigger site in the N-lobe of skeletal troponin C may induce Ca2+ binding to the second trigger site in the N-lobe of that troponin molecule; this only applies to skeletal muscle because the N-lobe of skeletal troponin C has two physiologically active EF-hand Ca2+-binding sites, while cardiac troponin C only has one. Furthermore, the mechanical properties – particularly myofilament compliance – of the myofilament assemblies could modulate both the actual cooperative interactions and the apparent cooperativity of force generation in the sarcomere (Chase et al., 2004; Kataoka et al., 2007).

Table 1. Cooperative mechanisms in muscle regulation where tropomyosin might play a central role.
A quantitative model described by Hill treats Tm as a rigid rod having end-to-end overlap with adjacent molecules of Tm (Hill et al., 1980). If only a single regulatory unit was activated, however, this simplification would mean that the Tm strand would be broken as one Tm molecule changes its structural state. It was later demonstrated experimentally that Tm is a semi-flexible molecule and Tn increases communication between neighboring structural units (Geeves and Lehrer, 1994). It was further shown that skeletal Tm induces cooperative binding of S1-ADP to actin in solution (Hill et al., 1980), and cooperative activation of actomyosin subfragment 1 (acto-S1) solution ATPase activity which is manifested by a sigmoidal ATPase versus (S1) relationship in the presence of skeletal Tm (Lehrer and Morris, 1982). Using a variety of complementary assays at different levels of biological organization, cardiac Tm confers statistically similar degrees of apparent cooperativity when compared with skeletal Tm (Clemmens et al., 2005; Boussouf et al., 2007; Jagatheesan et al., 2010); interestingly, these studies demonstrate that additional aspects of Ca2+-activated actomyosin function – such as the maximum isometric force – may be influenced by which isoform of Tm is present, and also that the isoform of troponin is important. While this work is in accord with the idea that both Ca2+ and crossbridges cooperatively activate the thin filament regardless of Tm (and Tn) isoform, the experiments do not address their relative importance for cardiac versus skeletal muscle, as dissected by Gillis et al. (2007) and Regnier et al. (2002). Regardless of whether thin filament activation depends more on Ca2+ or crossbridges (Table 1), these data indicate that cooperative spread of activation from one regulatory unit to the next depends critically on both the presence of, and the molecular composition of tropomyosin.
Does Ca2+ Activate the Thin Filament as a Single Unit?
The primary structural regulatory unit (SRU) responsible for the Ca2+ switch, consisting of one Tm molecule, one Tn complex, and seven actin monomers, is approximately the length of one single Tm molecule. The functional regulatory unit (FRU) is defined by the length of the thin filament activated by Ca2+ binding to a single Tn. In case of negative cooperativity the FRU would be shorter than the SRU, whereas for positive cooperativity, the FRU would be longer than the SRU. It was suggested from isometric force measurements that Ca2+ binding to just one or a very small number of SRUs is sufficient to activate an entire thin filament (Brandt et al., 1980) which would indicate that the FRU is much longer than an SRU. In vitro motility studies by Fraser and Marston (1995) also showed that the thin filament is switched on with an “all or none” response acting as a single cooperative unit, again suggesting that thin filament activation is a highly cooperative process in which the FRU is much longer than an SRU. The role of cooperative interactions between individual SRUs was studied in cardiac and skeletal muscles (Regnier et al., 2002; Gillis et al., 2007). In contrast to the conclusion that an entire thin filament activates all at once, the FRU in skeletal muscle was determined from isometric force measurements to span 12–14 actin monomers, i.e., only two SRUs (Regnier et al., 2002); cooperative spread of activation in this study was most likely through Tm end-to-end overlap. Again using isometric force measurements, the length of a FRU in cardiac muscle was found to be even shorter, about seven actin monomers, which is the same as a single SRU (Gillis et al., 2007).
In further contrast to the idea that thin filaments are fully activated by only a few Ca2+ ions, the rate of isometric tension redevelopment exhibits little or no dependence on cooperative interactions between regulatory units, and instead is controlled by dynamics within a regulatory unit in both cardiac and skeletal muscle (Chase et al., 1994; Gillis et al., 2007; Moreno-Gonzalez et al., 2007). The relationship between steady-state isometric tension (P) and the rate of isometric tension redevelopment (kTR) has been measured in permeabilized (“skinned”) muscle preparations. Thin filament activation level, and thus P, has typically been varied by applying a series of steady Ca2+ concentrations; measurements using myofibrils, where diffusional gradients are minimal, have confirmed that kTR at a steady Ca2+ level accurately reflects activation kinetics when a rapid step change in Ca2+ is applied (Stehle et al., 2009). At a given level of activating Ca2+, P is measured prior to, and kTR is measured following mechanical maneuvers that consist of a rapid length release which unloads the muscle preparation, and a sudden re-stretch to the original length (Brenner, 1988; Sweeney and Stull, 1990). At saturating Ca2+ levels, P is primarily determined by the number of strong crossbridges whereas kTR reports the rates of crossbridge transitions between weak, non-force states and strongly bound, force generating states (Brenner and Eisenberg, 1986). When Ca2+ concentration is varied over the activating range, the relationship between kTR and P is such that at low P (P < 50% Pmax), kTR is slow and unchanging. When the level of Ca2+ activation is increased and P approaches Pmax, kTR increases 10- to 15-fold in skeletal muscle preparations (Metzger and Moss, 1992; Chase et al., 1994; Regnier et al., 1996, 1998).
To explain the activation dependence of kTR, a simple model was evaluated (Landesberg and Sideman, 1994; Hancock et al., 1997; Regnier et al., 1999). A version of the model with four states (Figure 1) was necessary to describe the relationship between kTR and P in skeletal muscle, while only three states (no state 4 in Figure 1) were required to describe the less-steep relationship in cardiac muscle (Hancock et al., 1997). In addition to the variable [Ca2+], the skeletal and cardiac versions of the model have two pairs of kinetic rate parameters: f and g which reflect the processes associated with strong crossbridge formation and dissociation, respectively, and kon 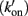 and koff
and koff 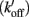 which reflect the processes associated with thin filament regulatory unit activation by Ca2+ and deactivation, respectively. The four-state version of the model for skeletal muscle has additional parameters, f ′ and g′: g′ reflects strong cross-bridge dissociation from regulatory units that have lost Ca2+; the rate parameter f ′ reflects the low probability of strong crossbridge formation at regulatory units in the blocked state, and is included for completeness. There is no inherent cooperative mechanism taken into consideration in the model, whether at the level of Ca2+ regulation or crossbridge formation. While this does not allow simulation of the steep dependence of P on Ca2+ concentration, the model can readily predict the thin filament activation dependence of P and kTR under a variety of conditions. Modeling and experiments, taken together, indicate that kTR at submaximal Ca2+ activation typically reflects the kinetics of individual thin filament regulatory units without requiring cooperative interactions between regulatory units, unlike steady-state tension where cooperativity plays a central role and simulations are not possible without it (Regnier et al., 1999; Moreno-Gonzalez et al., 2007). This shows that under physiologically relevant conditions, kTR is regulated by the dynamics of thin filament activation at submaximal Ca2+ levels and Ca2+ controls the rate limiting step in tension development, whereas at maximal Ca2+ the increase of kTR with force is governed by the rate of crossbridge cycling (Chase et al., 1994; Regnier et al., 1996, 1998).
which reflect the processes associated with thin filament regulatory unit activation by Ca2+ and deactivation, respectively. The four-state version of the model for skeletal muscle has additional parameters, f ′ and g′: g′ reflects strong cross-bridge dissociation from regulatory units that have lost Ca2+; the rate parameter f ′ reflects the low probability of strong crossbridge formation at regulatory units in the blocked state, and is included for completeness. There is no inherent cooperative mechanism taken into consideration in the model, whether at the level of Ca2+ regulation or crossbridge formation. While this does not allow simulation of the steep dependence of P on Ca2+ concentration, the model can readily predict the thin filament activation dependence of P and kTR under a variety of conditions. Modeling and experiments, taken together, indicate that kTR at submaximal Ca2+ activation typically reflects the kinetics of individual thin filament regulatory units without requiring cooperative interactions between regulatory units, unlike steady-state tension where cooperativity plays a central role and simulations are not possible without it (Regnier et al., 1999; Moreno-Gonzalez et al., 2007). This shows that under physiologically relevant conditions, kTR is regulated by the dynamics of thin filament activation at submaximal Ca2+ levels and Ca2+ controls the rate limiting step in tension development, whereas at maximal Ca2+ the increase of kTR with force is governed by the rate of crossbridge cycling (Chase et al., 1994; Regnier et al., 1996, 1998).
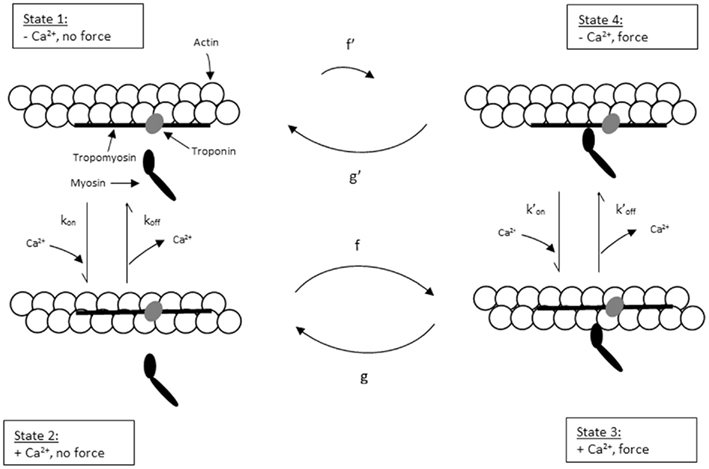
Figure 1. Schematic diagram of four-state model. On the left hand side are states where myosin forms weak, non-force-producing crossbridges. On the right hand side are force generating states where myosin binds strongly to actin. The upper states represent Ca2+ free states whereas the lower states represent Ca2+ bound. Ca2+ binds to TnC and activates regulatory units with rate constants kon (left) and 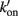 (right), while koff (left) and
(right), while koff (left) and 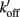 (right) reflect the kinetics of processes associated with regulatory units returning to the blocked state. Strong crossbridge formation and dissociation from thin filaments are governed by rate constants f and g, respectively. g′ Describes the rate of crossbridge detachment from regulatory units without Ca2+, as may occur during relaxation of skeletal muscle; the rate of strong crossbridge formation (f′) is practically non-existent in this condition. State 4 (upper right) is needed to model P–kTR relationships in skeletal muscle, but can be omitted when modeling cardiac muscle (Hancock et al., 1997).
(right) reflect the kinetics of processes associated with regulatory units returning to the blocked state. Strong crossbridge formation and dissociation from thin filaments are governed by rate constants f and g, respectively. g′ Describes the rate of crossbridge detachment from regulatory units without Ca2+, as may occur during relaxation of skeletal muscle; the rate of strong crossbridge formation (f′) is practically non-existent in this condition. State 4 (upper right) is needed to model P–kTR relationships in skeletal muscle, but can be omitted when modeling cardiac muscle (Hancock et al., 1997).
While the experiments described above provide information about the dynamics of individual regulatory units within the sarcomere, all biochemical and skinned-fiber experiments performed thus far to characterize cooperativity involved ensemble measurements. There is no direct measurement of cooperativity between individual neighboring SRUs, which is necessary to determine whether a thin filament activates as a single unit or activation involves numerous cooperative units.
Hypothesizing that we are able to incorporate a small number of reporters, each at an individual regulatory subunit along an individual thin filament, we could study the dynamics of activation of single regulatory units, as well as cooperative interactions between regulatory subunits. The reporter changes state when the regulatory unit turns on or off, reflecting the state of that regulatory unit (Figure 2). Statistical analysis of the signal from a single reporter would yield the dynamics of activation of the associated regulatory unit. We could also examine the cooperativity along the thin filament as a function of distance between two reporters. As shown in Figure 2A, two regulatory units separated by a short distance could show a highly correlated signal. Reporter signals from two regulatory units that are far apart, however, might not be correlated (Figure 2B). Introduction of such a novel technique would allow us to study cooperativity directly, and test the hypothesis of long range cooperativity along a thin filament.
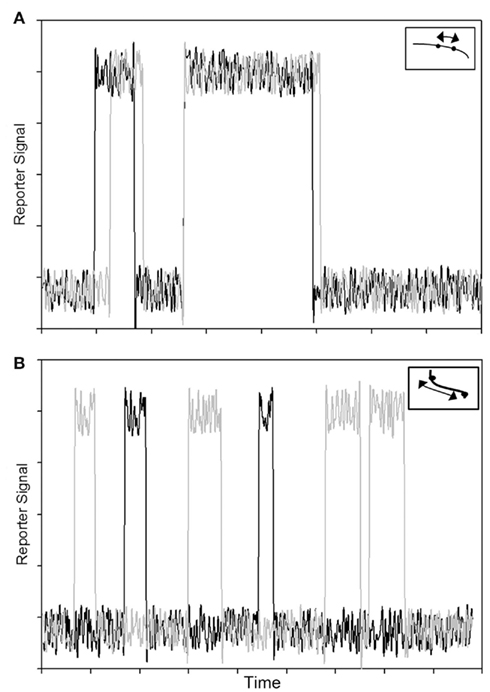
Figure 2. Overlay of signals (traces in black and gray) from two reporter probes corresponding to two regulatory subunits on a single thin filament. (A) Correlated signals from two reporter probes separated by a short distance demonstrate predicted result for cooperative interactions along a thin filament. (B) Uncorrelated signals from two reporter probes separated by a longer distance. The insets depict single thin filaments where the dots correspond to reporter probes on individual regulatory subunits; arrows represent distances between the reporter probes.
Familial Hypertrophic Cardiomyopathy Alters Cooperative Ca2+-Activation of Cardiac Thin Filaments
Independent of whether a thin filament activates as a single unit, familial hypertrophic cardiomyopathy (FHC)-related mutations have been found to alter Ca2+-sensitivity using in vitro functional assays. FHC is an inherited disease characterized by thickening of the myocardium. The disease affects an estimated 0.2% of the population and may be relatively benign, or may lead to heart failure or sudden cardiac death (Maron, 1997; Maron et al., 1998). A number of FHC-related mutations have been found in human cardiac α-tropomyosin (αTm), along with many other mutations primarily in cardiac cytoskeletal proteins (Bing et al., 2000; Fatkin and Graham, 2002; Roberts, 2002; Towbin and Bowles, 2002; Takeda, 2003; Wolska and Wieczorek, 2003). FHC-related αTm mutants have been linked to decreased thermal stability (Hilario et al., 2004; Kremneva et al., 2004; Wang et al., 2011) and a lower binding affinity for actin (Bing et al., 1997; Kremneva et al., 2004) compared to WT. In vitro studies with mutant αTm using myofibrillar ATPase activity, motility assays, and isometric force generation show significantly enhanced Ca2+-sensitivity and/or reduced cooperativity (Bing et al., 2000; Chang et al., 2005; Bai et al., 2011; Mathur et al., 2011; Wang et al., 2011). Cardiomyopathy mutations in αTm also affect skeletal muscle physiology (Bottinelli et al., 1998) because both α- and β-tropomyosins (WT and, in patients mutant αTm) are expressed in skeletal muscles, while cardiac muscle contains predominantly αTm; because the major pathology is in the heart, however, the consensus has been to look at the cardiac muscle and its proteins when studying the effect of FHC mutations on muscle contraction. Although the mechanistic relationship between the mutations and these observations is not yet clearly established (Tardiff, 2011), reduced rigidity of human cardiac αTm due to the mutations might be expected to affect the regulatory function of the molecule, leading to observed changes in Ca2+-sensitivity and/or cooperativity.
Is Tropomyosin a Semi-Flexible Molecule?
The flexural rigidity of a semi-flexible linear molecule can be characterized by its persistence length (Lp) which is the length over which the molecule loses directional correlation. Lp of tropomyosin from chicken and turkey gizzard smooth muscle, rabbit skeletal muscle, or rabbit and bovine cardiac muscle has been estimated to be 55–170 nm by various techniques at different temperatures (Swenson and Stellwagen, 1989; Phillips and Chacko, 1996; Li et al., 2010; Sousa et al., 2010), consistent with measurements obtained for other α-helical coiled-coil proteins (Hvidt et al., 1982; Howard and Spudich, 1996). Among these studies, Li et al. (2010) suggested that previous experimental estimates of Lp (i.e., apparent Lp), including that obtained for bovine cardiac tropomyosin in the same study, measured a combined effect of the inherent curved molecular structure and the true mechanical flexibility of tropomyosin. The apparent Lp is related reciprocally to the Lp due to true mechanical flexibility (i.e., dynamic Lp) and the Lp due to inherent molecular curvature (i.e., intrinsic Lp; Eq. 1):

The two effects were decoupled in a molecular dynamics simulation, which determined the dynamic Lp of Tm to be on the order of 500 nm, or ∼12 times the length of a single Tm molecule. We note that an Lp of ∼500 nm would imply that Tm behaves as a rigid body over the span of ∼12 SRU and thus the length of a FRU would be equivalent to approximately half the length of a thin filament within a sarcomere. This implication is consistent with the hypothesis that a thin filament activates as a single unit (Figure 3A, lower left). In contrast, however, previous studies showed the length of a FRU is no more than 12–14 actin monomers, which is approximately equivalent to the length of two SRUs, or more simply two Tm molecules (Regnier et al., 2002; Figure 3A, lower right). It is particularly noteworthy here that, assuming the intrinsic Lp of 135 nm determined by Li et al. (2010), Eq. 1 suggests the correction to apparent Lp due to intrinsic Lp diminishes drastically and non-linearly as apparent Lp approaches Lc (Figure 3): in case of a rigid Tm with apparent Lp > 2.5 Lc, the dynamic Lp is above 400 nm, or ∼10 SRU, consistent with the hypothesis that a thin filament activates as a single unit (Brandt et al., 1980; Fraser and Marston, 1995); on the other hand, in case of a semi-flexible Tm with apparent Lp ∼ Lc, dynamic Lp of the molecule is in the order of 60 nm, or ∼1.5 Lc, consistent with the hypothesis that the length of a FRU approximately equals 1–2 SRUs (Regnier et al., 2002; Gillis et al., 2007). It remains to be determined whether differences in experimentally determined values of apparent Lp over this crucial range (Figure 4) reflect differences inherent in the proteins or in experimental methodologies. Due to the non-linearity evident in Figure 4, a slight reduction in the experimentally measurable apparent Lp, depending on its exact value, may potentially imply a profound change in the true mechanical flexibility of Tm, as represented by its dynamic Lp.
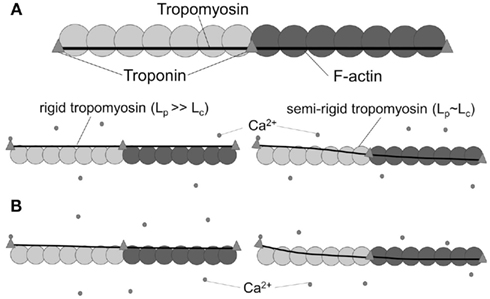
Figure 3. Tropomyosin rigidity is an important factor in the propagation of activation signal along the thin filament. (A) In the absence of Ca2+, tropomyosin sterically blocks myosin binding sites on actin (top). In the presence of Ca2+, a rigid (Lp ≫ Lc) tropomyosin strand (middle left) moves azimuthally nearly as a single unit, uncovering most of the myosin binding sites on actin along multiple SRUs (two SRUs, denoted by shading, are shown from each thin filament). In contrast, for semi-flexible (Lp ∼ Lc) tropomyosin (middle right), only a portion of the strand moves azimuthally and just 1–2 SRUs are activated. (B) Reduction in rigidity of an initially rigid tropomyosin has little impact on thin filament activation (bottom left), as activation signal still propagates along multiple SRUs. In contrast, a reduction in rigidity of a semi-flexible tropomyosin (bottom right) will significantly reduce the effective propagation length of activation signal, but also increases the likelihood of activation at lower Ca2+.
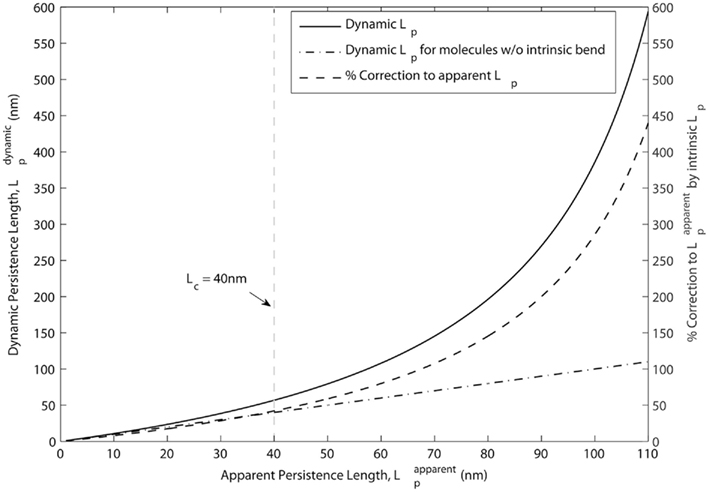
Figure 4. Correction due to intrinsic curvature of tropomyosin is less significant when apparent Lp approaches the contour length. According to Eq. 1, the percentage correction to apparent Lp due to intrinsic Lp (dashed line, right axis) is non-linear and is much smaller at values of apparent Lp ≤ Lc than at longer values. Dynamic Lp (solid line, left axis), measurement of the true mechanical flexibility obtained after the correction, is also close to apparent Lp when apparent Lp ≤ Lc. For reference, the linear relation obtained for molecules with no intrinsic bend is also shown (dot-dashed line, left axis). Also for reference, Lc (=40 nm) for Tm molecules is highlighted by the vertical, gray dashed line.
Reduction in Tropomyosin Rigidity Decreases Mechanical Correlation Along the Molecule
Transmission of a mechanical perturbation along the length of a Tm molecule can be modeled as an exponential decay according to the cosine correlation function (CCF; Howard, 2001). Since a Tm molecule follows the helical structure of a thin filament along its length, and each end of a Tm molecule is associated with a distinct Tn complex (at the head-to-tail overlap regions with neighboring molecules of Tm), the middle of a Tm molecule is expected to be the least perturbed region during activation at high Ca2+ levels (i.e., when the Tn complexes at both ends of a Tm molecule have Ca2+ bound). Assuming that configurations sampled by Tm on the thin filament are mainly determined by its mechanical properties, as illustrated in Figure 3, CCF can predict the propagation of activation. Figure 5A depicts the variation of perturbation signal transmitted from the ends to the middle of a Tm molecule at decreasing flexural rigidity, with initial Lp = 65 or 170 nm (as the lower and upper limit in crystallographic and solution studies of skeletal Tm). The effect of reduced signal transmission is noticeable in both cases, but is more significant for a more flexible Tm (Lp = 65 nm). On the other hand, since cytoplasmic Ca2+ does not normally achieve levels that fully saturate thin filament during systolic activation of cardiac muscle, only some but not all SRUs will have Ca2+ bound to the Tn complexes at both ends. Thus we also have to consider the case where Ca2+ binds to the Tn complex at only one end of a Tm molecule. In that situation, the opposite, Ca2+-free end would be the least perturbed region, and Figure 5B shows the variation of transmitted signal at the Ca2+-free end when Tm rigidity is altered. Compared to the case where Ca2+ binds to the Tn complexes at both ends of a Tm molecule, activation signal transmitted to the least perturbed region is significantly decreased at any given reduction in rigidity of Tm. Readers should note especially the case of a more flexible Tm (initial Lp = 65 nm), where signal transmission is nearly halved over the considered range of decrease in rigidity. It is clear that in most cases during systolic activation, when Ca2+ may be bound to the Tn complex only at one end of a Tm molecule, the transmission of mechanical perturbation is significantly impeded by reduction in rigidity of Tm, especially when the initial Lp is comparable to Lc. It is evident from this simple model that a drop in rigidity of a Tm molecule (e.g., associated with a disease-related mutation) can lead to a significant difference in mechanical correlation along its length, the extent of which depends on both the initial flexural rigidity of the molecule and where along the length this effect is measured. This effect is likely to have important functional consequences which are discussed further below. It can also underlie mechanical tuning at the molecular level through evolution, where sensitivity of Tm as a key component of the regulatory switch for thin filament activation may have been optimized within different physiological situations.
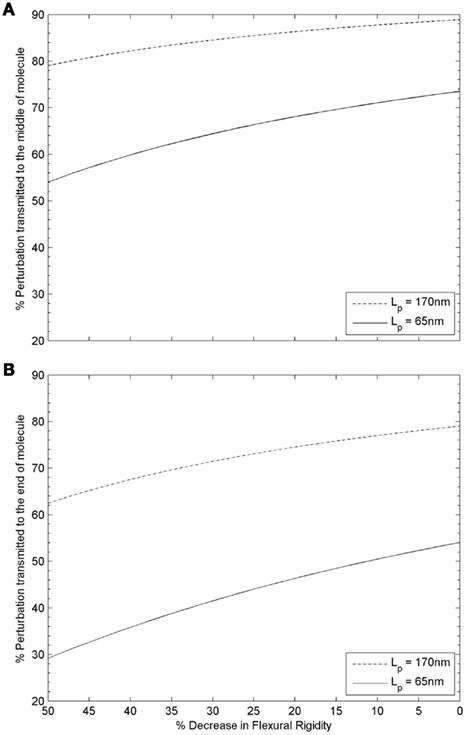
Figure 5. Variation of structural perturbation signal transmitted from (A) the ends to the middle of a tropomyosin molecule, or (B) from the Ca2+ bound end to the Ca2+ free end of a tropomyosin molecule as a function of the flexural rigidity of tropomyosin. (A) When the two ends of a Tm molecule are either both Ca2+ free, or both have Ca2+ bound, the structural change in the middle of the molecule, relative to that at either end, is greater for a rigid Tm (Lp > Lc, dotted-dash line) than for a semi-flexible Tm (Lp ∼ Lc, solid line) as expected. The effect of a reduction in flexural rigidity as is hypothesized to be found in some cases of FHC (compare left side of graph with initial value at right) is substantially more significant for the initially semi-flexible Tm (Lp ∼ Lc, solid line) than the rigid Tm (Lp > Lc, dotted-dash line); for the case of semi-flexible Tm (Lp ∼ Lc, solid line), an additional ∼25% drop is predicted when the initial rigidity is halved. (B) Thin filaments are not fully saturated by Ca2+ during systolic activation of cardiac muscle. When only one end of a Tm molecule has Ca2+ bound, the structural change at the Ca2+ free end relative to that at the end with Ca2+ bound is greater for a rigid Tm (Lp > Lc, dotted-dash line) than for a semi-flexible Tm (Lp ∼ Lc, solid line) as expected. As in panel A, the effect of a reduction in flexural rigidity as is hypothesized to be found in some cases of FHC (compare left side of graph with initial value at right) is much more significant for the initially semi-flexible Tm (Lp ∼ Lc, solid line) than the rigid Tm (Lp > Lc, dotted-dash line). For the case of a rigid Tm (Lp > Lc, dotted-dash line), 63% of the signal is transmitted when the initial rigidity is halved. In comparison, only 29% of the signal is transmitted in the case of a semi-flexible Tm (Lp ∼ Lc, solid line).
Implications of Reduced Correlation on Cooperative Thin Filament Regulation
We expect that a reduction in mechanical correlation would correspond to reduced cooperativity in transmission of activation along thin filaments, and could have major impacts on thin filament regulation.
First, decreased correlation along the length of a Tm molecule could influence ordered assembly of thin filaments by reducing the overall affinity of αTm strands for F-actin. It is therefore easier for αTm to move away from myosin binding sites during transition from the “blocked” to “closed” state during Ca2+-induced activation. Reduced mechanical correlation also implies a smaller turning moment and thus a lesser extent of conformational change of Tn is required to initiate azimuthal movement of αTm on the actin filament. Taken together, these can affect the functional Ca2+ sensitivity (i.e., pCa50) of regulation. Secondly, reduced correlation also means the mechanical turning moment due to Tn conformational change does not propagate as effectively along the length of Tm (Figure 3B, right). This may in turn affect the number of myosin binding sites uncovered and thus the force generated by each SRU at a given Ca2+ level. The exact functional implication and significance of these effects depend heavily on the innate flexibility of the wildtype αTm, which should be considered in three regimes: very flexible (Lp ≪ Lc), very rigid (Lp ≫ Lc), and semi-flexible (Lp ∼ Lc).
In the case of αTm that is already very flexible (Lp ≪ Lc), little or no intrinsic cooperative activation would exist between SRUs through end-to-end overlap. A further increase in flexibility (reduction in Lp) would result in even less mechanical correlation along the molecule, such that activation within each SRU would be reduced at all levels of Ca2+. In other words, an increase in flexibility of a highly flexible αTm leads to predicted reductions in both Ca2+ sensitivity and maximum force. The condition Lp ≪ Lc, however, is outside the range of existing measurements on α-helical coiled-coil proteins (see Is Tropomyosin a Semi-Flexible Molecule?), and thus this regime is not considered in detail.
Conversely, if αTm is inherently very rigid (Lp ≫ Lc), the reduction in mechanical correlation due to increased flexibility may be relatively insignificant (Figures 5A,B, dotted-dash line), such that the probability of uncovering any number of myosin binding sites does not change at all levels of Ca2+. Therefore, in this case, an increase in flexibility leads to the prediction of no or minimal change in Ca2+ sensitivity, cooperativity, and maximum force.
Lastly, for a semi-flexible αTm (Lp ∼ Lc), an increase in flexibility will result in a significant loss in correlation (Figure 5B, solid line), while the activation signal would still be able to propagate along a limited span of the molecule that is longer than an actin monomer. In this case where cooperativity is reduced, it would be mechanically more favorable for a Ca2+ bound Tn to induce sufficient movement of αTm to uncover at least some myosin binding sites, leading to increased functional Ca2+ sensitivity. As a result, the thin filament can be activated at a lower level of Ca2+, such as in early and late stages of systole or, in extreme cases that might be associated with cardiac disease, diastole. At saturating Ca2+, however, the reduced correlation implies that Ca2+ dependent azimuthal displacement at each end of Tm may not be fully transmitted to the middle of the molecule (Figure 5A, solid line). If Lp is near the low end of this regime and the increase in flexibility is sufficiently large, then some myosin binding sites would be more likely to remain blocked at high Ca2+ levels. On the other hand, if Lp is near the high end of this regime, there could be a tolerance in the system such that cardiac sarcomeres might maintain maximum force at systolic Ca2+ level despite a moderate increase in flexibility of αTm. Taken together, increased flexibility of a semi-flexible αTm enhances the probability to uncover at least some myosin binding sites at low Ca2+, but may slightly reduce the probability to uncover all myosin binding sites at high Ca2+.
Possible Link to Cardiac Hypertrophy
We reason by considering the mechanical correlation along the tropomyosin molecule that reduced Tm rigidity will contribute to higher Ca2+-sensitivity in cooperative thin filament regulation. This implies that human cardiac thin filaments harboring less rigid αTm mutants will undergo a prolonged systolic activation and perhaps diastolic dysfunction. Cardiac thin filaments will become activated earlier during systole, remain activated longer during the relaxation phase, and in extreme cases could possibly stay partially activated during diastole (Ho et al., 2009; Bai et al., 2011; Campbell and McCulloch, 2011). These effects would be expected to markedly impact the overall mechanics of the heart and cardiac output. Therefore, we hypothesize that at least some FHC-related mutations of human cardiac Tm exert their major influence on sarcomere mechanics through altered flexural rigidity of the molecule. This may in turn lead to functional changes in cooperative Ca2+ induced activation of cardiac thin filaments, such as previously observed in in vitro studies (Bing et al., 2000; Chang et al., 2005; Boussouf et al., 2007; Bai et al., 2011; Mathur et al., 2011; Wang et al., 2011). Techniques such as that illustrated in Figure 2 (described above) can directly decipher at the molecular level the effects FHC-related mutations impose on the dynamics of activation and cooperative interactions of individual and multiple regulatory units, respectively. While it is likely that additional factors will need to be considered to fully understand the complex phenotype observed in patients with these inherited cardiomyopathies, we fully anticipate that measurements of Tm’s flexural rigidity will provide an improved molecular understanding (Figures 3–5) of an important mechanistic link between FHC-related mutations of the molecule and hypertrophy of the heart due to increased workload.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Aya Katayoka Takeda, Dr. Brenda Schoffstall, and Dr. Nicolas Brunet for helpful discussions.
References
Bai, F., Weis, A., Takeda, A. K., Chase, P. B., and Kawai, M. (2011). Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin’s HCM mutations. Biophys. J. 100, 1014–1023.
Bing, W., Knott, A., Redwood, C., Esposito, G., Purcell, I., Watkins, H., and Marston, S. (2000). Effect of hypertrophic cardiomyopathy mutations in human cardiac muscle α-tropomyosin (Asp175Asn and Glu180Gly) on the regulatory properties of human cardiac troponin determined by in vitro motility assay. J. Mol. Cell. Cardiol. 32, 1489–1498.
Bing, W., Redwood, C. S., Purcell, I. F., Esposito, G., Watkins, H., and Marston, S. B. (1997). Effects of two hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glu180Gly, on Ca2+ regulation of thin filament motility. Biochem. Biophys. Res. Commun. 236, 760–764.
Bottinelli, R., Coviello, D. A., Redwood, C. S., Pellegrino, M. A., Maron, B. J., Spirito, P., Watkins, H., and Reggiani, C. (1998). A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ. Res. 82, 106–115.
Boussouf, S. E., and Geeves, M. A. (2007). Tropomyosin and troponin cooperativity on the thin filament. Adv. Exp. Med. Biol. 592, 99–109.
Boussouf, S. E., Maytum, R., Jaquet, K., and Geeves, M. A. (2007). Role of tropomyosin isoforms in the calcium sensitivity of striated muscle thin filaments. J. Muscle Res. Cell Motil. 28, 49–58.
Brandt, P. W., Cox, R. N., and Kawai, M. (1980). Can the binding of Ca2+ to two regulatory sites on troponin C determine the steep pCa/tension relationship of skeletal muscle? Proc. Natl. Acad. Sci. U.S.A. 77, 4717–4720.
Brenner, B. (1988). Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 85, 3265–3269.
Brenner, B., and Eisenberg, E. (1986). Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. U.S.A. 83, 3542–3546.
Campbell, S. G., and McCulloch, A. D. (2011). Multi-scale computational models of familial hypertrophic cardiomyopathy: genotype to phenotype. J. R. Soc. Interface 8, 1550–1561.
Chang, A. N., Harada, K., Ackerman, M. J., and Potter, J. D. (2005). Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in α-tropomyosin. J. Biol. Chem. 280, 34343–34349.
Chase, P. B., Macpherson, J. M., and Daniel, T. L. (2004). A spatially explicit nanomechanical model of the half-sarcomere: myofilament compliance affects Ca2+-activation. Ann. Biomed. Eng. 32, 1559–1568.
Chase, P. B., Martyn, D. A., and Hannon, J. D. (1994). Isometric force redevelopment of skinned muscle fibers from rabbit activated with and without Ca2+. Biophys. J. 67, 1994–2001.
Clemmens, E. W., Entezari, M., Martyn, D. A., and Regnier, M. (2005). Different effects of cardiac versus skeletal muscle regulatory proteins on in vitro measures of actin filament speed and force. J. Physiol. 566, 737–746.
Dobesh, D. P., Konhilas, J. P., and de Tombe, P. P. (2002). Cooperative activation in cardiac muscle: impact of sarcomere length. Am. J. Physiol. Heart Circ. Physiol. 282, H1055–H1062.
Fatkin, D., and Graham, R. M. (2002). Molecular mechanisms of inherited cardiomyopathies. Physiol. Rev. 82, 945–980.
Fraser, I. D., and Marston, S. B. (1995). In vitro motility analysis of actin-tropomyosin regulation by troponin and calcium. The thin filament is switched as a single cooperative unit. J. Biol. Chem. 270, 7836–7841.
Geeves, M. A., and Lehrer, S. S. (1994). Dynamics of the muscle thin filament regulatory switch: the size of the cooperative unit. Biophys. J. 67, 273–282.
Gillis, T. E., Martyn, D. A., Rivera, A. J., and Regnier, M. (2007). Investigation of thin filament near-neighbour regulatory unit interactions during force development in skinned cardiac and skeletal muscle. J. Physiol. 580, 561–576.
Gordon, A. M., Homsher, E., and Regnier, M. (2000). Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924.
Hancock, W. O., Huntsman, L. L., and Gordon, A. M. (1997). Models of calcium activation account for differences between skeletal and cardiac force redevelopment kinetics. J. Muscle Res. Cell Motil. 18, 671–681.
Hilario, E., da Silva, S. L., Ramos, C. H., and Bertolini, M. C. (2004). Effects of cardiomyopathic mutations on the biochemical and biophysical properties of the human α-tropomyosin. Eur. J. Biochem. 271, 4132–4140.
Hill, T., Eisenberg, E., and Greene, L. (1980). Theoretical model for the cooperative equilibrium binding of myosin subfragment 1 to the actin-troponin-tropomyosin complex. Proc. Natl. Acad. Sci. U.S.A. 77, 3186–3190.
Ho, C. Y., Carlsen, C., Thune, J. J., Havndrup, O., Bundgaard, H., Farrohi, F., Rivero, J., Cirino, A. L., Andersen, P. S., Christiansen, M., Maron, B. J., Orav, E. J., and Køber, L. (2009). Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2, 314–321.
Howard, J. (2001). Mechanics of Motor Proteins and the Cytoskeleton. Sunderland, MA: Sinauer Associates.
Howard, J., and Spudich, J. A. (1996). Is the lever arm of myosin a molecular elastic element? Proc. Natl. Acad. Sci. U.S.A. 93, 4462–4464.
Hvidt, S., Nestler, F. H., Greaser, M. L., and Ferry, J. D. (1982). Flexibility of myosin rod determined from dilute solution viscoelastic measurements. Biochemistry 21, 4064–4073.
Iwamoto, H. (1998). Thin filament cooperativity as a major determinant of shortening velocity in skeletal muscle fibers. Biophys. J. 74, 1452–1464.
Jagatheesan, G., Rajan, S., Ahmed, R. P., Petrashevskaya, N., Boivin, G., Arteaga, G. M., Tae, H. J., Liggett, S. B., Solaro, R. J., and Wieczorek, D. F. (2010). Striated muscle tropomyosin isoforms differentially regulate cardiac performance and myofilament calcium sensitivity. J. Muscle Res. Cell Motil. 31, 227–239.
Kataoka, A., Hemmer, C., and Chase, P. B. (2007). Computational simulation of hypertrophic cardiomyopathy mutations in troponin I: influence of increased myofilament calcium sensitivity on isometric force, ATPase and [Ca2+]i. J. Biomech. 40, 2044–2052.
Kremneva, E., Boussouf, S., Nikolaeva, O., Maytum, R., Geeves, M. A., and Levitsky, D. I. (2004). Effects of two familial hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys. J. 87, 3922–3933.
Landesberg, A., and Sideman, S. (1994). Coupling calcium binding to troponin C and cross-bridge cycling in skinned cardiac cells. Am. J. Physiol. 266, H1260–H1271.
Lehman, W., Craig, R., and Vibert, P. (1994). Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature 368, 65–67.
Lehrer, S., Golitsina, N., and Geeves, M. (1997). Actin-tropomyosin activation of myosin subfragment 1 ATPase and thin filament cooperativity. The role of tropomyosin flexibility and end-to-end interactions. Biochemistry 36, 13449–13454.
Lehrer, S. S., and Morris, E. P. (1982). Dual effects of tropomyosin and troponin-tropomyosin on actomyosin subfragment 1 ATPase. J. Biol. Chem. 257, 8073–8080.
Li, X., Holmes, K. C., Lehman, W., Jung, H., and Fischer, S. (2010). The shape and flexibility of tropomyosin coiled coils: implications for actin filament assembly and regulation. J. Mol. Biol. 395, 327–339.
Maron, B. J., Moller, J. H., Seidman, C. E., Vincent, G. M., Dietz, H. C., Moss, A. J., Towbin, J. A., Sondheimer, H. M., Pyeritz, R. E., McGee, G., and Epstein, A. E. (1998). Impact of laboratory molecular diagnosis on contemporary diagnostic criteria for genetically transmitted cardiovascular diseases: hypertrophic cardiomyopathy, long-QT syndrome, and Marfan syndrome: a statement for healthcare professionals from the councils on clinical cardiology, cardiovascular disease in the young, and basic science, American Heart Association. Circulation 98, 1460–1471.
Mathur, M. C., Chase, P. B., and Chalovich, J. M. (2011). Several cardiomyopathy causing mutations on tropomyosin either destabilize the active state of actomyosin or alter the binding properties of tropomyosin. Biochem. Biophys. Res. Commun. 406, 74–78.
Maytum, R., Lehrer, S., and Geeves, M. (1999). Cooperativity and switching within the three-state model of muscle regulation. Biochemistry 38, 1102–1110.
McKillop, D. F., and Geeves, M. A. (1993). Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 65, 693–701.
McLachlan, A. D., and Stewart, M. (1975). Tropomyosin coiled-coil interactions: evidence for an unstaggered structure. J. Mol. Biol. 98, 293–304.
Metzger, J. (1995). Myosin binding-induced cooperative activation of the thin filament in cardiac myocytes and skeletal muscle fibers. Biophys. J. 68, 1430–1442.
Metzger, J. M., and Moss, R. L. (1992). Myosin light chain 2 modulates calcium-sensitive cross-bridge transitions in vertebrate skeletal muscle. Biophys. J. 63, 460–468.
Milligan, R. A., Whittaker, M., and Safer, D. (1990). Molecular structure of F-actin and location of surface binding sites. Nature 348, 217–221.
Moreno-Gonzalez, A., Gillis, T. E., Rivera, A. J., Chase, P. B., Martyn, D. A., and Regnier, M. (2007). Thin-filament regulation of force redevelopment kinetics in rabbit skeletal muscle fibres. J. Physiol. 579, 313–326.
O’Brien, E. J., Bennett, P. M., and Hanson, J. (1971). Optical diffraction studies of myofibrillar structure. Philos. Trans. R. Soc. Lond. B Biol. Sci. 261, 201–208.
Perry, S. V. (2001). Vertebrate tropomyosin: distribution, properties and function. J. Muscle Res. Cell Motil. 22, 5–49.
Phillips, G. N. Jr., and Chacko, S. (1996). Mechanical properties of tropomyosin and implications for muscle regulation. Biopolymers 38, 89–95.
Regnier, M., Martyn, D. A., and Chase, P. B. (1996). Calmidazolium alters Ca2+ regulation of tension redevelopment rate in skinned skeletal muscle. Biophys. J. 71, 2786–2794.
Regnier, M., Martyn, D. A., and Chase, P. B. (1998). Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [ATP] in rabbit skeletal muscle. Biophys. J. 74, 2005–2015.
Regnier, M., Rivera, A., Chase, P., Smillie, L., and Sorenson, M. (1999). Regulation of skeletal muscle tension redevelopment by troponin C constructs with different Ca2+ affinities. Biophys. J. 76, 2664–2672.
Regnier, M., Rivera, A. J., Wang, C. K., Bates, M. A., Chase, P. B., and Gordon, A. M. (2002). Thin filament near-neighbour regulatory unit interactions affect rabbit skeletal muscle steady-state force-Ca2+ relations. J. Physiol. 540, 485–497.
Smillie, L. B. (1996). “Tropomyosin,” in Biochemistry of Smooth Muscle Contraction, ed. M. Bárány (San Diego: Academic Press), 63–75.
Sousa, D., Cammarato, A., Jang, K., Graceffa, P., Tobacman, L. S., Li, X., and Lehman, W. (2010). Electron microscopy and persistence length analysis of semi-rigid smooth muscle tropomyosin strands. Biophys. J. 99, 862–868.
Stehle, R., Solzin, J., Iorga, B., and Poggesi, C. (2009). Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflügers Arch. 458, 337–357.
Sweeney, H. L., and Stull, J. T. (1990). Alteration of cross-bridge kinetics by myosin light chain phosphorylation in rabbit skeletal muscle: implications for regulation of actin-myosin interaction. Proc. Natl. Acad. Sci. U.S.A. 87, 414–418.
Swenson, C. A., and Stellwagen, N. C. (1989). Flexibility of smooth and skeletal tropomyosins. Biopolymers 28, 955–963.
Takeda, N. (2003). Cardiomyopathy: molecular and immunological aspects (review). Int. J. Mol. Med. 11, 13–16.
Tardiff, J. C. (2011). Thin filament mutations: developing an integrative approach to a complex disorder. Circ. Res. 108, 765–782.
Tobacman, L. S., and Butters, C. A. (2000). A new model of cooperative myosin-thin filament binding. J. Biol. Chem. 275, 27587–27593.
Vibert, P., Craig, R., and Lehman, W. (1997). Steric-model for activation of muscle thin filaments. J. Mol. Biol. 266, 8–14.
Wang, F., Brunet, N. M., Grubich, J. R., Bienkiewicz, E. A., Asbury, T. M., Compton, L. A., Mihajlović, G., Miller, V. F., and Chase, P. B. (2011). Facilitated cross-bridge interactions with thin filaments by familial hypertrophic cardiomyopathy mutations in α-tropomyosin. J. Biomed. Biotechnol. 2011, 435271.
Wolska, B. M., and Wieczorek, D. M. F. (2003). The role of tropomyosin in the regulation of myocardial contraction and relaxation. Pflügers Arch. 446, 1–8.
Keywords: tropomyosin, thin filament, calcium activation, persistence length, cooperativity, heart, sarcomere, cardiomyopathy
Citation: Loong CKP, Badr MA and Chase PB (2012) Tropomyosin flexural rigidity and single Ca2+ regulatory unit dynamics: implications for cooperative regulation of cardiac muscle contraction and cardiomyocyte hypertrophy. Front. Physio. 3:80. doi: 10.3389/fphys.2012.00080
Received: 29 December 2011; Accepted: 18 March 2012;
Published online: 04 April 2012.
Edited by:
Kenneth S. Campbell, University of Kentucky, USAReviewed by:
Jonathan P. Davis, The Ohio State University, USAMargaret Westfall, University of Michigan, USA
Copyright: © 2012 Loong, Badr and Chase. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: P. Bryant Chase, Department of Biological Science, The Florida State University, Biology Unit One Building, Room 206, 81 Chieftain Way, Box 3064370, Tallahassee, FL 32306-4370, USA. e-mail: chase@bio.fsu.edu
†Campion K. P. Loong and Myriam A. Badr have contributed equally to this work.