 Sabzali Javadov
Sabzali JavadovA commentary on
Differential effects of buffer pH on Ca2+-induced ROS emission with inhibited mitochondrial complex I and III
by Lindsay, D. P., Camara, A. K., Stowe, D. F., Lubbe, R., and Aldakkak, M. (2015). Front. Physiol. 6:58. doi: 10.3389/fphys.2015.00058
Reperfusion of the heart following sustained ischemia is associated with enhanced reactive oxygen species (ROS) production, Ca2+ accumulation, and pHi normalization that are the major inducers of mitochondrial permeability transition (mPT). Despite intensive studies, a cause-and-effect relationship between the ROS-Ca2+-pHi triangle and mPT has not yet been established (Halestrap et al., 2004; Bernardi, 2013). Initially, several proteins such as VDAC, ANT, and phosphate carrier (PiC) have been suggested as the essential structural components of the mPTP. However, genetic studies from different groups demonstrated that pore opening can occur in the absence of these proteins indicating that they are not involved in the mPTP structure (reviewed in Bernardi, 2013; Halestrap and Richardson, 2015). Emerging studies suggest that the mitochondrial F0F1-ATP synthase or electron transport chain (ETC) complex V is involved in pore formation and may actually play an important role as a structural component of the mPTP (Giorgio et al., 2013; Alavian et al., 2014; Azarashvili et al., 2014; Carraro et al., 2014). In addition to the unknown molecular identity of the mPT pore (mPTP), a lack of in vitro models mimicking cardiac ischemia-reperfusion (IR) makes it difficult to elucidate the precise role of mitochondrial ROS, Ca2+, and pHi in response to oxidative stress. Mitochondrial ETC complexes I, II, and III are the main sites of ROS (superoxide anion) production (Figure 1). Dysfunction of the complexes induced by cardiac IR enhances ROS which are not efficiently eliminated by the mitochondrial antioxidant system due to high ROS generation and low ROS scavenging. Activity of ETC complexes may be diminished by a number of factors including cardiolipin oxidation, degradation of supercomplexes, alteration of the ion homeostasis/redox potential, etc.
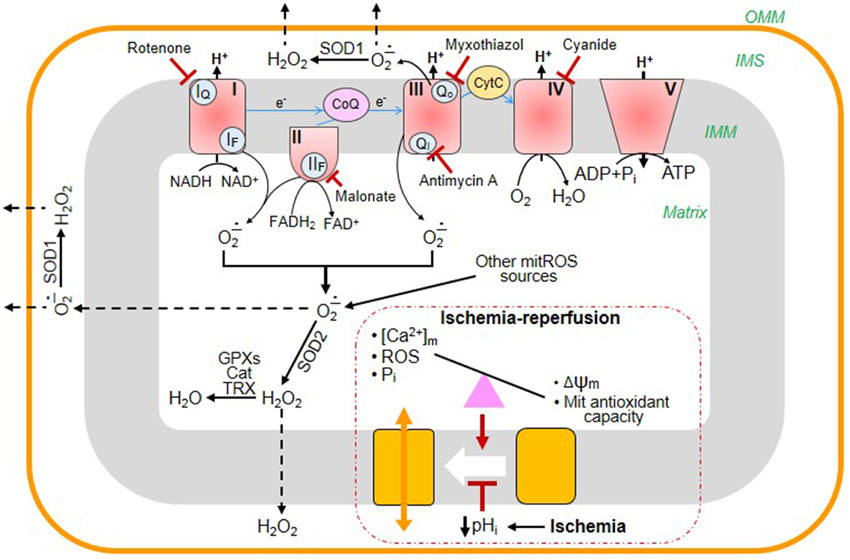
Figure 1. Main sites of ROS generation in mitochondria and their elimination by the antioxidant system: the effects of cardiac ischemia-reperfusion (IR). The mitochondrial electron transport chain (ETC) consists of four multi-subunit complexes (I, II, III, and IV), coenzyme Q (CoQ), and cytochrome c (Cyt C). Electrons (e-) are transferred through the ETC from the reducing NADH/FADH2 to O2, finally generating H2O at complex IV. The electron transfer through the ETC is accompanied by ROS generation at complexes I, II, and III. Complex I oxidizes NADH and produces superoxide (O−.2) at the flavin site (IF) and ubiquinone-binding site (IQ) that is released to the matrix. Complex II oxidizes succinate to fumarate and generates O−.2/H2O2 both in the reverse and forward reactions through the flavin site (IIF). Complex III is the main source of ROS that produces O−.2 to both cytoplasm and matrix. Under normal physiological conditions, the level of mitochondrial ROS is regulated by mitochondrial and cytoplasmic antioxidant systems. Superoxide dismutases (SOD1, SOD2) convert O−.2 into H2O2 which is then eliminated by catalase (Cat), glutathione peroxidases (GPXs), and reduced thioredoxin (TRX). During cardiac IR, the balance between generation and scavenging of ROS is altered due to high ROS production and low antioxidant capacity of mitochondria. Mitochondrial permeability transition pores (mPTP) remain closed during ischemia due to low pHi. However, at reperfusion, excessive ROS generation together with increased [Ca2+] and [Pi]m and diminished membrane potential (Δψm) cause mPTP opening. The blunted arrows indicate the sites where the inhibitors rotenone, malonate, myxothiazol, antimycin A, and cyanide bind to ETC complexes.
The article published by Lindsay et al. (2015) studies pH-dependence of Ca2+-induced swelling (a marker of mPTP opening), ROS generation and respiratory function of isolated guinea pig cardiac mitochondria using substrates and inhibitors for ETC complexes I and III. Results of the study demonstrated that pH and Ca2+-induced mPTP opening have different effects on ROS production at complexes I and III. The authors attempted to mimic cardiac IR by blocking complexes I and III with rotenone and antimycin in the presence of pyruvate and succinate, respectively. Although this is the only approach to assess the contribution of individual ETC complexes to ROS production in isolated mitochondria, it is rather different from the in vivo condition observed in cardiac IR. Each of complexes I and III contain two sites of ROS generation, and rotenone and antimycin inhibit only one site at complex I (the ubiquinone-binding site, IQ) and complex III (the quinone-reducing center, Qi), respectively. Complete chemical blocking of these sites and the use of only one substrate (pyruvate or succinate) for each complex are the major limitations of the study. On the other hand, more recent studies revealed that succinate is a general metabolic marker of ischemia in a variety of tissues including the heart, and that it is responsible for mitochondrial ROS production during reperfusion by reverse electron transport at complex I Inhibition of ischemic succinate accumulation and its oxidation after subsequent reperfusion was sufficient to ameliorate in vivo cardiac IR injury in rodents (Chouchani et al., 2014). Indeed, in the study by Lindsay et al. (2015), the authors measured ROS levels at complex I-III, but not complex III alone. Most of the ROS signal observed during succinate oxidation is rotenone-sensitive and this is associated with the IQ site of complex I due to the backflow of electrons from the reduced Q-pool. Accordingly, reverse electron transfer from the reduced QH2 pool at site IQ should be blocked to measure ROS generation solely at complex III.
Oxidative stress induces a complex of biochemical, biophysical and topographical changes of the inner mitochondrial membrane that ultimately result in malfunction of the ETC. The latter, when accompanied by membrane depolarization, ROS generation, matrix Ca2+ and Pi overload, can induce reversible (low conductance, physiological) or irreversible (high conductance, pathological) mPT depending on the severity of IR. Moreover, opening of the mPTP can further enhance the aforementioned alterations. Since low pHi in the ischemic myocardium blocks the mPTP, pore opening occurs only upon reperfusion with normalization of pHi (Griffiths and Halestrap, 1995). The contribution of each ETC complex may be different throughout the ischemic period due to changes in the redox potential, ion homeostasis, and antioxidant system of mitochondria. Mitochondrial respiration, ROS generation and mPTP opening were pH-dependent, which indicates that interactions between these parameters are complex (Lindsay et al., 2015). ROS production at pH 6.5 was significantly lower than that at pH 6.9 and pH 7.15 for complexes I and I-III in mitochondria with Ca2+ swelling. Notably, significant mitochondrial swelling associated increased ROS generation was observed in the presence of succinate and antimycin A at all pH (6.5; 6.9, and 7.15), and both swelling and ROS production were significantly reduced by cyclosporin A to basic levels. These data confirm previous studies that mPTP opening induces mitochondrial ROS production (Batandier et al., 2004) and lowering pH inhibits pore opening (Javadov et al., 2008). However some findings of the study remain unanswered. For instance, Ca2+-induced swelling was accompanied by a great increase in ROS release from complex I at pH 6.9 but not at pHs 7.15 or 6.5. Respiration rates (state three and RCI) were markedly affected at pHs 6.5 or 6.9 for complex I but not for complex I-III during Ca2+ swelling. Remarkably, inhibition of complex I by rotenone blocks PTP opening in tissues that express low levels of cyclophilin D, and, conversely rotenone does not affect the PTP in tissues which are characterized by high levels of expression of cyclophilin D and sensitivity to cyclosporin A (Li et al., 2012). The inhibitory effect of rotenone on PTP can complicate the interpretation of the results reported by Lindsay et al. (2015). In addition, it is very difficult to assess the effect of matrix pH on mPTP in vitro to mimic conditions observed in situ. First of all, the relevant parameter is matrix pH (Nicolli et al., 1993); in deenergized mitochondria the probability of pore opening has an optimum at matrix pH 7.4, although is changes both below and above this level. During IR, matrix pH is influenced by reenergization, and by the secondary events that follow transport of species that depend on the delta pH. This is critical for the PTP, because reenergization may offset the protective effects of an initially acidic matrix pH because of increased Pi uptake, and as shown in isolated brain mitochondria, ischemic and postischemic acidosis may worsen rather than relieve PTP-dependent tissue damage (Kristian et al., 2001).
In conclusion, despite certain limitations, the elucidation of the contribution of ROS and pHi to mPTP opening via chemical inhibition of complexes I and III by Lindsay et al. (2015) opens new directions for further studies.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health through Research Grant SC1HL118669 (SJ).
References
Alavian, K. N., Beutner, G., Lazrove, E., Sacchetti, S., Park, H. A., Licznerski, P., et al. (2014). An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 111, 10580–10585. doi: 10.1073/pnas.1401591111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Azarashvili, T., Odinokova, I., Bakunts, A., Ternovsky, V., Krestinina, O., Tyynelä, J., et al. (2014). Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 55, 69–77. doi: 10.1016/j.ceca.2013.12.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Batandier, C., Leverve, X., and Fontaine, E. (2004). Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 279, 17197–17204. doi: 10.1074/jbc.M310329200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bernardi, P. (2013). The mitochondrial permeability transition pore: a mystery solved. Front. Physiol. 4:95. doi: 10.3389/fphys.2013.00095
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carraro, M., Giorgio, V., Šileikyte, J., Sartori, G., Forte, M., Lippe, G., et al. (2014). Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J. Biol. Chem. 289, 15980–15985. doi: 10.1074/jbc.C114.559633
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijević, D., Sundier, S. Y., Robb, E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi: 10.1038/nature13909
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 110, 5887–5892. doi: 10.1073/pnas.1217823110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Griffiths, E. J., and Halestrap, A. P. (1995). Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 307, 93–98.
Halestrap, A. P., and Richardson, A. P. (2015). The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 78, 1291–1241. doi: 10.1016/j.yjmcc.2014.08.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Halestrap, A. P., Clarke, S. J., and Javadov, S. A. (2004). Mitochondrial permeability transition pore opening during myocardial reperfusion - a target for cardioprotection. Cardiovasc. Res. 61, 372–385. doi: 10.1016/S0008-6363(03)00533-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Javadov, S., Choi, A., Rajapurohitam, V., Zeidan, A., Basnakian, A. G., and Karmazyn, M. (2008). NHE-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc. Res. 77, 416–424. doi: 10.1093/cvr/cvm039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kristian, T., Bernardi, P., and Siesjö, B. K. (2001). Acidosis promotes the permeability transition in energized mitochondria: implications for reperfusion injury. J. Neurotrauma 18, 1059–1074. doi: 10.1089/08977150152693755
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, B., Chauvin, C., De Paulis, D., De Oliveira, F., Gharib, A., Vial, G., et al. (2012). Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin D. Biochim. Biophys. Acta 1817, 1628–1634. doi: 10.1016/j.bbabio.2012.05.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lindsay, D. P., Camara, A. K. S., Stowe, D. F., Lubbe, R., and Aldakkak, M. (2015). Differential effects of buffer pH on Ca2+-induced ROS emission with inhibited mitochondrial complexes I and III. Front. Physiol. 6:58. doi: 10.3389/fphys.2015.00058
Nicolli, A., Petronilli, V., and Bernardi, P. (1993). Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry 32, 4461–4465. doi: 10.1021/bi00067a039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: cardiac ischemia-reperfusion, mitochondria, permeability transition pore, calcium, ROS, pH
Citation: Javadov S (2015) The calcium-ROS-pH triangle and mitochondrial permeability transition: challenges to mimic cardiac ischemia-reperfusion. Front. Physiol. 6:83. doi: 10.3389/fphys.2015.00083
Received: 19 February 2015; Accepted: 03 March 2015;
Published: 18 March 2015.
Edited by:
Miguel A. Aon, Johns Hopkins University School of Medicine, USACopyright © 2015 Javadov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabzali Javadov,c2FiemFsaS5qYXZhZG92QHVwci5lZHU=