Department of Biological Sciences, Kennedy Center for Research on Human Development, Vanderbilt University, Nashville, TN, USA
Loss of fragile X mental retardation 1 (FMR1) gene function is the most common cause of inherited mental retardation and autism spectrum disorders, characterized by attention disorder, hyperactivity and disruption of circadian activity cycles. Pursuit of effective intervention strategies requires determining when the FMR1 product (FMRP) is required in the regulation of neuronal circuitry controlling these behaviors. In the well-characterized Drosophila disease model, loss of the highly conserved dFMRP causes circadian arrhythmicity and conspicuous abnormalities in the circadian clock circuitry. Here, a novel Sholl Analysis was used to quantify over-elaborated synaptic architecture in dfmr1-null small ventrolateral neurons (sLNvs), a key subset of clock neurons. The transgenic Gene-Switch system was employed to drive conditional neuronal dFMRP expression in the dfmr1-null mutant background in order to dissect temporal requirements within the clock circuit. Introduction of dFMRP during early brain development, including the stages of neurogenesis, neuronal fate specification and early pathfinding, provided no rescue of dfmr1 mutant phenotypes. Similarly, restoring normal dFMRP expression in the adult failed to restore circadian circuit architecture. In sharp contrast, supplying dFMRP during a transient window of very late brain development, wherein synaptogenesis and substantial subsequent synaptic reorganization (e.g. use-dependent pruning) occur, provided strong morphological rescue to reestablish normal sLNvs synaptic arbors. We conclude that dFMRP plays a developmentally restricted role in sculpting synaptic architecture in these neurons that cannot be compensated for by later reintroduction of the protein at maturity.
Fragile X Syndrome (FraX) is a common heritable neurological disease manifesting mental retardation, autism, attention deficit disorder and disrupted activity patterns (Bassell and Warren, 2008
; Gatto and Broadie, 2009
; Hagerman et al., 2009
). FraX patients display characteristic hyperactivity and disordered sleep associated with altered melatonin levels (Elia et al., 2000
; Gould et al., 2000
; Miano et al., 2008
). Similarly, FraX genetic disease models also show dramatically impaired circadian activity profiles. Drosophila mutants lacking their sole fragile X mental retardation 1 (FMR1) family member (dfmr1) display significantly altered motor activity, disrupted sleep and striking circadian rhythm phenotypes, hallmarked by arrhythmicity and erratic locomotion (Dockendorff et al., 2002
; Inoue et al., 2002
; Morales et al., 2002
; Bushey et al., 2009
; Gatto and Broadie, 2009
). In mammals, a tripartite gene family (FMR1, FXR1 and FXR2) encodes proteins with >60% amino acid identity and high functional domain conservation (Siomi et al., 1995
; Zhang et al., 1995
). Mice deficient for either Fmr1 or Fxr2 alone maintain fairly normal rhythmicity, albeit with free-run periodicity shortening (Zhang et al., 2008
). However, combinatorial Fmr1/Fxr2 double null animals are entirely arrhythmic and further lack basic entrainment capacity (Zhang et al., 2008
). Thus, the circadian regulation of behavior is clearly dependent on the FMR1 gene family across a wide range of species.
The Drosophila circadian clock circuitry is particularly well characterized (Helfrich-Forster, 2005
; Chang, 2006
; Nitabach and Taghert, 2008
). Within this circuit, the small ventrolateral neurons (sLNvs) are a key subset of clock neurons that possess sufficient circadian pacemaker capacity to modulate morning activity peaks in light:dark cycles and drive rhythmicity in constant darkness (Renn et al., 1999
; Grima et al., 2004
; Stoleru et al., 2004
). These critical clock neurons express the neuropeptide pigment dispersing factor (PDF) (Helfrich-Forster, 1995
, 2005
), which has long been proposed to serve as the clock output mediating coordination of downstream neurons. Characterization of pdf-null animals reveals impaired light cycle entrainment with advanced evening activity peaks and disrupted anticipation, while free-running behavior in constant darkness shows progressive deterioration of rhythmicity (Renn et al., 1999
). Further, disrupted cycling of PDF within the terminal sLNv projections induced by transgenically-mediated electrical hyperexcitability causes altered behavioral rhythms with multiple periods (Nitabach et al., 2006
), and work using a membrane-tethered toxin to influence neuronal ion channel properties revealed a 4-h phase advance in the PDF peak accumulation in the sLNvs, which coordinately shifts morning anticipatory behavior (Wu et al., 2008
). The PDF neuropeptide facilitates circadian oscillations in individual neurons, while driving synchrony in neuronal subgroups (Sheeba et al., 2008
; Yoshii et al., 2009
).
Null dfmr1 sLNvs exhibit striking architectural defects, including overgrowth, overextension and mistargeting beyond the normal branching and defasiculation point in the protocerebrum, projecting a more expansive terminal synaptic arborization (Dockendorff et al., 2002
; Morales et al., 2002
; Reeve et al., 2005
, 2008
; Sekine et al., 2008
; Gatto and Broadie, 2009
). Conversely, targeted dfmr1 overexpression in sLNvs results in a dramatic collapse of their dorsal synaptic arbor (Reeve et al., 2005
), indicating that the levels of the dFMR1 product (dFMRP) protein alone can bidirectionally modulate synaptic complexity. Thus, dFMRP plays a critical role in controlling the synaptic connectivity within the clock circuit that drives circadian activity patterns.
A fundamental need is to determine the critical period(s) of FMRP function. Treatment design, implementation and efficacy depend upon determining whether FraX is primarily a disease of development or maintained function. FMRP has been proposed to be required very early, during neurogenesis and pathfinding, later during stages of synaptogenesis and neuronal circuitry refinement (e.g. activity-dependent synaptic pruning), or at maturity for use-dependent circuit remodeling (Nimchinsky et al., 2001
; Galvez and Greenough, 2005
; Larson et al., 2005
; Yun et al., 2006
; Bureau et al., 2008
; Gatto and Broadie, 2008
; Tessier and Broadie, 2008
). Clearly, these potential roles may not be mutually exclusive. To distinguish these possibilities, we have used the conditional, transgenic Gene-Switch (GS) method (Osterwalder et al., 2001
; Roman et al., 2001
) to neuronally target wild-type dFMRP in null dfmr1 mutants (Gatto and Broadie, 2008
). Conditional expression was driven during windows throughout development and at maturity, followed by examination of the resultant sLNv synaptic architecture using a PDF-adapted modified Sholl Analysis. Neither early developmental nor adult-onset dFMRP expression was sufficient to prevent or reverse, respectively, the sLNv synaptic defects. Surprisingly, inappropriate dFMRP expression during early pupal metamorphosis actually exacerbated dfmr1-null phenotypes. Successful and striking structural resolution of synaptic connectivity defects was obtained only via targeted reintroduction of dFMRP during late pupal brain development. These results reveal a highly specific, dFMRP-dependent window of sLNv clock neuron development during the late stages of synaptogenesis and/or synaptic refinement.
Drosophila Genetics
Drosophila stocks were maintained at 25°C on standard cornmeal/agar/molasses medium supplemented with yeast. The w1118 line served as the genetic background control for the null dfmr1 allele (dfmr150M) (Zhang et al., 2001
; Gatto and Broadie, 2008
). Stocks carrying PDF-Gal4 and UAS-mCD8::GFP, both from the Drosophila Stock Center (Bloomington, IN, USA), as well as UAS-synaptotagmin-GFP (Zhang et al., 2002
), were introduced into the dfmr1-null background using standard genetic techniques. As previously described (Gatto and Broadie, 2008
), recombinant parental lines harboring the null dfmr1 allele (Zhang et al., 2001
) and either a wild-type dfmr1 transgene under UAS control (UAS-9557-3) (Zhang et al., 2001
) or the neuronal-specific driver elav-Gene-Switch construct (GSG-301) (Osterwalder et al., 2001
) were generated using standard genetic techniques. Accordingly, “eGS” as a genotype descriptor refers to the following: dfmr150M, elav-GSG-301/dfmr150M, UAS-9557-3. For RU486 (mifepristone; Sigma, St Louis, MO, USA) dosing (eGS + RU), the drug was dissolved at 10 mM in 80% ethanol (EtOH) and either mixed with food to the desired concentration for larval (0.5 μg/mL) or adult (30 μg/mL) feeding, or it was used directly for immersion treatment of both larvae and pupae as indicated. For pupal day 1 (P1) induction, late-stage wandering third instar larvae were washed briefly with EtOH, soaked in RU486 for 2 min, and then transferred to fresh vials for puparium formation. Induction from P2–P4 required an anterior incision in the pupal case with a 30-gauge needle and subsequent submersion in RU486 for 2 min. Pupae were then transferred to humid chambers to prevent desiccation. For vehicle control, equivalent volumes of 80% EtOH were used to identically treat the eGS line (eGS + E).
Western Blot Analyses
The central nervous system, including the brain and the ventral nerve cord, was dissected free from staged and treated larvae in Ca2+-free modified standard saline, while the brain proper was dissected from both pupae and adults (Gatto and Broadie, 2008
). Dissected samples were homogenized and boiled in 1X NuPage sample buffer (Invitrogen, Carlsbad, CA, USA) supplemented with 40 mM DTT. The total protein from four brains per sample was loaded onto 4–12% Bis-Tris gels and electrophoresed in NuPage MES Buffer (Invitrogen) for 1 h at 200 V. Transfer to nitrocellulose was carried out for 1 h at 100 V in NuPage transfer buffer (Invitrogen) with 10% MeOH. Processing was completed using the Odyssey near infrared fluorescence detection system (Li-COR, Lincoln, NE) to enable quantitative Western blot analysis. Antibodies used included: anti-dFMRP (1:2000; 6A15 monoclonal, Sigma), anti-α-tubulin (1:200,000; B512 monoclonal, Sigma), and Alexa-goat-anti-mouse-680 (1:10,000; Invitrogen-Molecular Probes). Raw integrated intensities were calculated for the lower molecular weight band of the dFMRP doublet and for the α-tubulin band. The ratio of dFMRP:α-tubulin normalized for loading and allowed comparison of eGS neuronally induced dFMRP relative to endogenous dFMRP in age matched wild-type (w1118) animals.
Immunohistochemistry
Immunohistochemistry was performed essentially as previously described (Gatto and Broadie, 2008
). Brains from staged and treated animals (0–3 days old) were dissected in standard saline and then fixed for 40 min with 4% paraformaldehyde/4% sucrose in phosphate buffered saline (PBS), pH 7.4. Preparations were rinsed with PBS, then blocked and permeablized with 0.2% triton X-100 in PBS (PBST) supplemented with 1% bovine serum albumin (BSA) and 0.5% normal goat serum (NGS) for 1 h at room temperature. Primary and secondary antibodies were diluted in PBST with 0.2% BSA and 0.1% NGS and incubated overnight at 4°C and 2 h at room temperature, respectively. Antibodies employed include: anti-GFP (1:2000; ab290 polyclonal, Abcam Inc., Cambridge, MA), anti-PDF [1:5; C7 monoclonal, Developmental Studies Hybridoma Bank (DSHB), University of Iowa], anti-ELAV (1:100; 9F8A9 monoclonal, DSHB), anti-dFMRP (1:500; 6A15, Sigma) and Alexa-Fluor secondaries (1:250; Invitrogen-Molecular Probes). All fluorescent images were collected using a ZEISS LSM 510 META laser scanning confocal microscope.
PDF-Adapted Modified Sholl Analysis
A PDF-adapted version of the Modified Sholl Analysis (Fernandez et al., 2008
) was developed. A traditional Sholl array using a series of 10 μm-spaced concentric rings was centered upon the dorsal horn bifurcation of the sLNv terminal arbor and extended forward for 100 μm with assays conducted at zeitgeber time 2–4, i.e. 2–4 h after lights on. To provide for compatibility with the GS system, this assay examined PDF reactive puncta (≥1 μm in diameter) within each 10 μm concentric ring. Puncta were counted as a reflective index of spatial synaptic arbor complexity. Values were determined for each concentric ring throughout the arbor in both left and right hemispheres and averaged for n = 1 for each individual animal.
Statistics
Statistical analyses were performed using GraphPad InStat 3 (GraphPad Software, Inc., San Diego, CA, USA). Unpaired, nonparametric, two-tailed Mann–Whitney tests were used to compare means of control and dfmr1, as well as means of eGS treated with vehicle or RU486 in the PDF-adapted Modified Sholl Analysis. While unpaired, parametric student’s t-tests could have been applied in certain instances, the cumulative consideration of data sets examining each concentric ring in a given treatment paradigm did vary with regard to normality. Thus, it was deemed more appropriate to globally apply the more conservative, nonparametric measures. Significance levels in figures are represented as p < 0.05 (*); p < 0.01 (**); p < 0.001 (***). All error bars represent standard error of the mean (SEM).
Over-Elaboration of sLNv Synaptic Arbors in dfmr1-null Mutants
In the Drosophila FraX model, dFMRP loss results in overgrowth of sLNv synaptic arbors in the dorsal brain (Dockendorff et al., 2002
; Morales et al., 2002
; Reeve et al., 2005
, 2008
; Sekine et al., 2008
; Gatto and Broadie, 2009
). The increased architectural complexity can be readily visualized with expression of the UAS-mCD8::GFP membrane marker transgene driven by the LNv-specific PDF-GAL4, comparing control and dfmr1-null mutant (Figure 1
A). Co-immunostaining for the LNv-specific neuropeptide PDF reveals a punctate pattern of varicosities distributed throughout the sLNv synaptic arbor interspersed along the axonal branches. Likewise, expression of the UAS-synaptotagmin-GFP transgene, an integral synaptic vesicle marker, specifically labels synaptic boutons within the sLNv arbor revealing the same characteristic synaptic overgrowth in dfmr1-null mutants (Figure 1
B). Double-labeling shows an array of synaptotagmin-GFP boutons within the synaptic arbor, overlapping and co-localized with the anti-PDF array. These analyses show that PDF labeling serves as an excellent indicator of synaptic boutons in both control and dfmr1 nulls (Figure 1
B). Co-localization of PDF with synaptotagmin-GFP was seen in 77 ± 3% of puncta in control and 71 ± 2% in dfmr1 mutants (p = 0.13, n = 7 for both). PDF was independently present in 19 ± 3% of control puncta vs. 23 ± 2% in dfmr1 (p = 0.21), and synaptotagmin-GFP was observed alone in 4 ± 1% of control and 6 ± 0.3% of dfmr1 puncta (p = 0.13). We conclude that examining the spatial distribution of PDF puncta serves as a reflective index of both the axonal arbor morphological complexity and synaptic bouton distribution in the sLNvs.
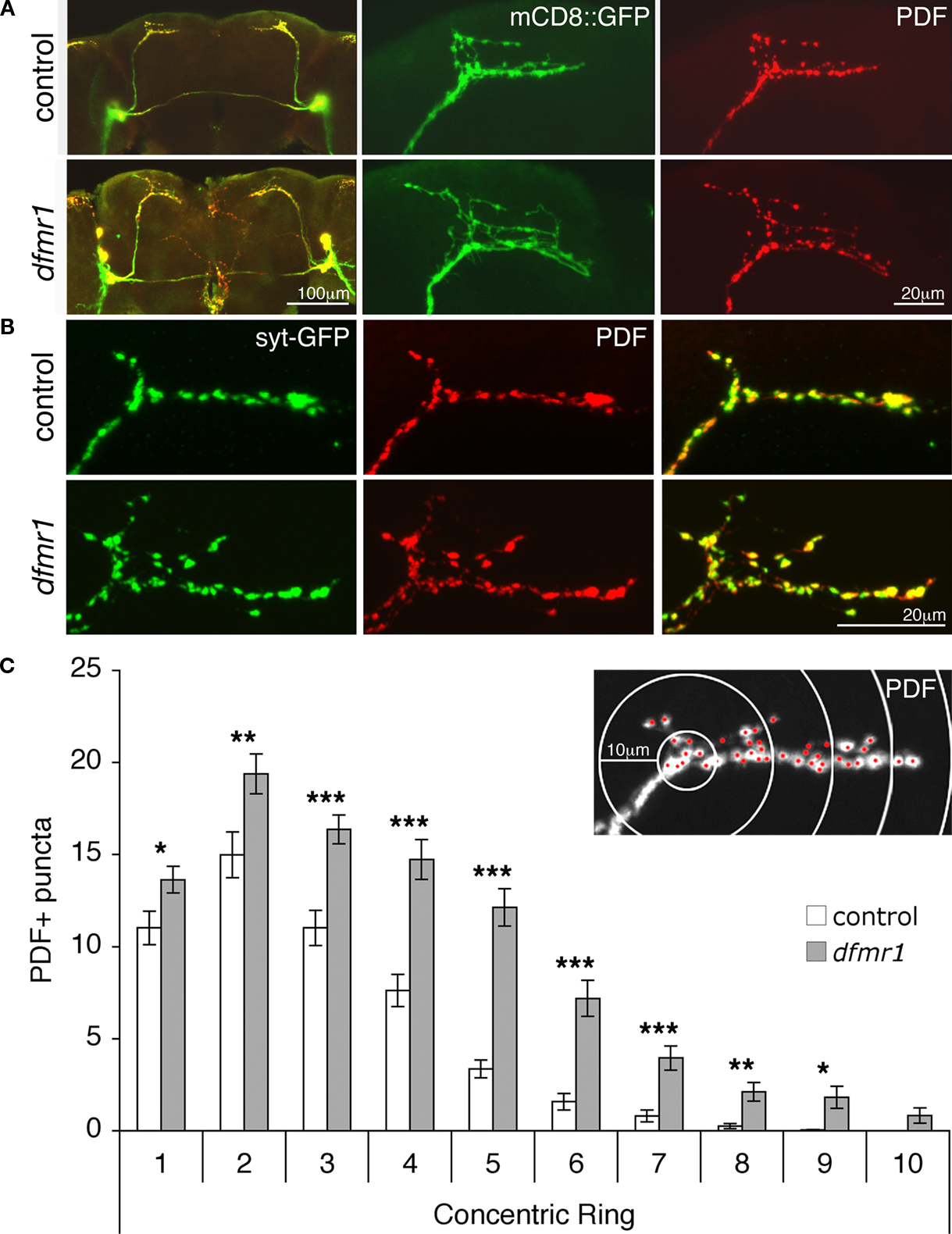
Figure 1. sLNv synaptic architecture defects in the absence of dFMRP. (A) Representative images of adult Drosophila brains isolated from animals expressing a UAS-mCD8::GFP (green) transgene under the control of the ventrolateral neuron (LNv) specific PDF-GAL4 driver in both genetic background control and homozygous dfmr150M null (dfmr1) mutants. Co-immunohistochemistry was done for the LNv neuropeptide PDF (red). At left, low magnification images show both the large LNv axonal projections with the established posterior optic tract connecting both hemispheres and the small LNv (sLNv) axonal tracts that defasiculate and arborize in the protocerebrum. At right, high magnification views focus solely on the terminal sLNv synaptic arbors. (B) Images of sLNv synaptic arbors from animals expressing UAS-synaptotagmin-GFP (syt-GFP, green) under the control of PDF-GAL4 in both control and dfmr1-null backgrounds. Brains show co-immunohistochemistry for PDF (red). (C) Sholl Analysis quantification of the spatial distribution of synaptic boutons throughout the sLNv arbor in both genetic background controls and dfmr1 nulls based on counting PDF-reactive (PDF+) puncta (inset; ≥1 μm in diameter, demarcated in red) starting from the point of dorsal horn bifurcation. Bars indicate mean ± SEM Significance levels: p < 0.05 (*); p < 0.01 (**); p < 0.001 (***).
To quantify the synaptic defects in dfmr1 mutants, we employed a PDF-adapted modified Sholl Analysis to record sLNv synapse architecture (Figure 1
C, inset) (Fernandez et al., 2008
). The approach involves aligning a traditional Sholl array of 10 μm-spaced concentric rings centered upon the dorsal horn bifurcation of the sLNv terminal arbor, extending the array for 100 μm, and reporting the number of PDF synaptic boutons within each ring. This method reliably characterizes the robust differential distribution of synaptic boutons in dfmr1 mutants compared to control (Figure 1
C). These quantified analyses reveal that dfmr1-null animals harbor increased numbers of synaptic boutons throughout the sLNv terminal (total puncta: 51 ± 4 control vs. 92 ± 5 dfmr1; n = 26 for both; p < 0.0001) projecting, at minimum, 20 μm further toward the midline (average maximal ring occupancy: 6 ± 0.2 control vs. 8 ± 0.3 dfmr1; p < 0.0001) and with a greatly increased average number of boutons beyond 50 μm (3 ± 1 control vs. 16 ± 3 dfmr1; p < 0.0001). Moreover, the number of synaptic boutons in every concentric ring is significantly elevated in dfmr1 nulls throughout the arbor (Figure 1
C): 10 μm ring (11 ± 1 control, 14 ± 1 dfmr1; p = 0.039), 20 μm ring (15 ± 1 control, 19 ± 1 dfmr1; p = 0.004); 30 μm ring (11 ± 1 control, 16 ± 1 dfmr1; p = 0.0001), 40 μm ring (8 ± 1 control, 15 ± 1 dfmr1; p < 0.0001), 50 μm ring (3 ± 0.5 control, 12 ± 1 dfmr1; p < 0.0001), 60 μm ring (2 ± 0.5 control, 7 ± 1 dfmr1, p < 0.0001), 70 μm ring (1 ± 0.3 control, 4 ± 0.7 dfmr1; p < 0.0001), 80 μm ring (0.3 ± 0.1 control, 2 ± 0.5 dfmr1; p = 0.002), 90 μm ring (0.04 ± 0.04 control, 2 ± 0.6 dfmr1; p = 0.023), and the most distal 100 μm ring (0 control, 1 ± 0.4 dfmr1). These results show that both the number and spatial distribution of boutons in the sLNv synaptic arbor is altered in the absence of dFMRP, with more boutons occupying a larger territory beyond the normal extent of the sLNv arborization.
Gene-Switch Conditional Control of dFMRP During Development and at Maturity
To examine the temporal requirement(s) of dFMRP in regulating synapse architecture and to determine whether dFMRP reintroduction could rescue synaptic defects, we exploited the transgenic conditional GS system (Osterwalder et al., 2001
; Roman et al., 2001
). The GS method employs a hormone-responsive modified UAS-GAL4 system to drive inducible, targeted, tissue-specific transgene expression. We controlled a UAS-dfmr1 transgene with the pan-neuronal elav-GS GAL4 by treating with the activating progesterone analog, RU486 (mifepristone), in the dfmr1-null mutant background to examine the influence of dFMRP reintroduction on sLNv morphology and synaptic elaboration (Figure 1
) (Gatto and Broadie, 2008
). In defining treatment paradigms, analyses were performed on wild-type control (w1118), dfmr1-null and dfmr1-null animals harboring both the elav-GS GAL4 driver and UAS-dfmr1 transgene (eGS animals, henceforth) treated with RU486 (eGS + RU) or only ethanol vehicle (eGS + ET) as indicated (Figure 2
A). We first confirmed that RU486 induced elav-GS GAL4 does drive dFMRP transgene expression pan-neuronally in the brain, albeit not uniformly (Figure 2
A). Close examination likewise confirmed elav expression in the sLNvs nuclei (Figure 2
B), establishing that sLNvs are elav-positive. These findings indicate that the GS method allows control of dFMRP expression in neurons widely throughout the brain, including within the sLNv neurons.
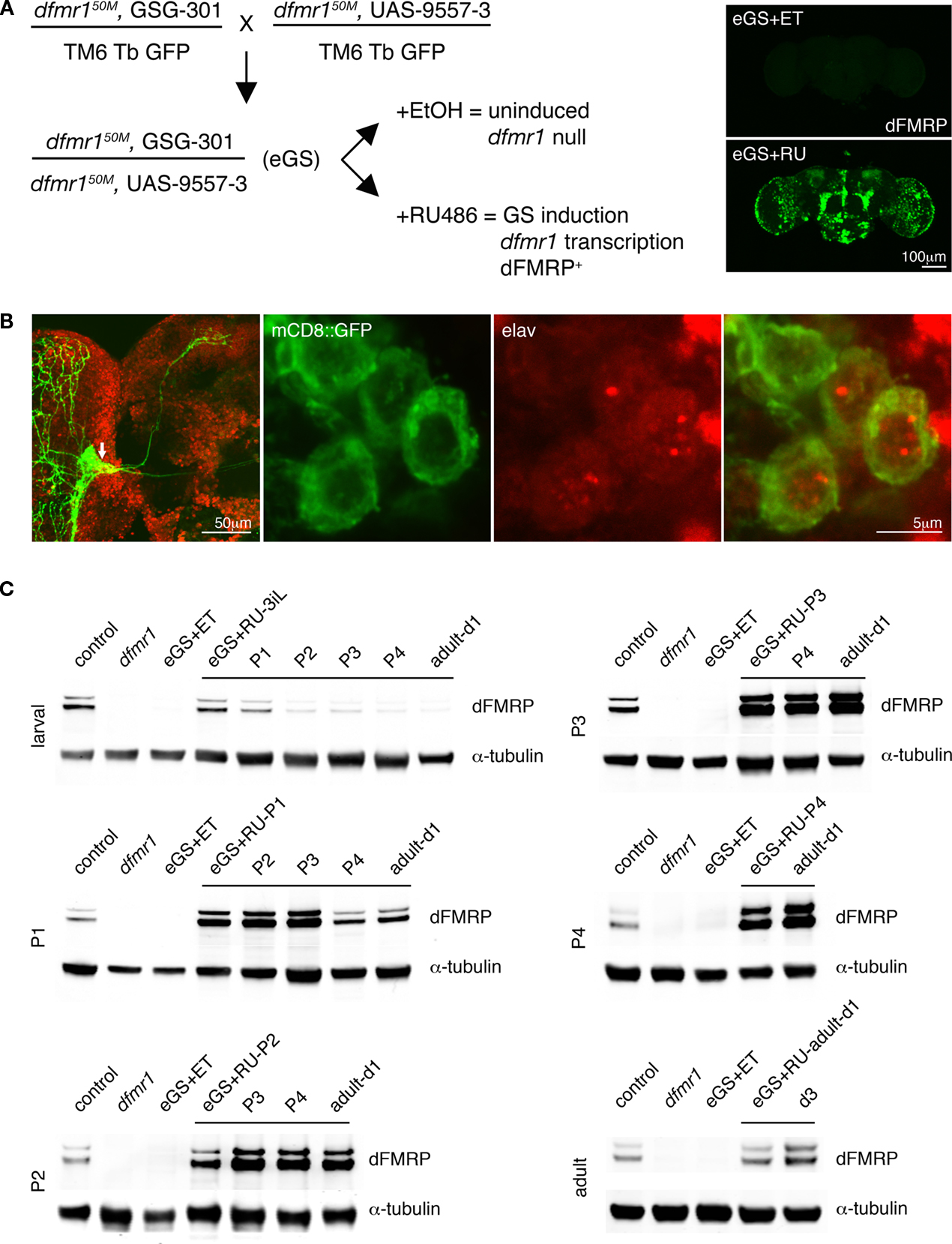
Figure 2. Gene-Switch conditional induction of dFMRP expression during staged periods throughout development and at maturity. (A) The transgenic crossing and treatment scheme used to mediate temporal control of dFMRP expression in the dfmr150M-null background using the Gene-Switch (GS) system. Homozygous dfmr1-null animals harboring the neuron-specific elav-GS driver (GSG-301) and UAS-dfmr1 transgene responder (UAS-9557-3) (eGS) were treated with vehicle (EtOH) alone or RU486 to generate the two matched populations for comparisons. Whole brain imaging (inset, right) illustrates typical patterns of dFMRP immunostaining after adult-onset induction with RU486. (B) Representative images of adult Drosophila brains expressing a UAS-mCD8::GFP (green) transgene under the control of the PDF-GAL4 driver, co-immunostained for elav (red). Low magnification image on the left; higher magnification images on the right. Elav is expressed within the sLNv cell bodies (arrow, left), in a punctate pattern within the neuronal nuclei (right). (C) Representative Western blots illustrating the dFMRP induction achieved using stage-specific RU486 treatment paradigms, indicated to the left of each series, assessed at ∼24 h intervals from initiation. Genotypes as indicated: w1118 (control), homozygous dfmr150M null allele (dfmr1) and dfmr150M, elav-GSG-301/dfmr150M, UAS-9557-3 (eGS). Treatments as indicated: eGS with ethanol vehicle (eGS + ET) and RU486 (eGS + RU). Blots were probed for dFMRP, with α-tubulin employed as a loading control.
The expression of dFMRP is strongly modulated as a function of age both during development and adult aging, with peak expression at the late stages of brain maturation (Gatto and Broadie, 2008
; Tessier and Broadie, 2008
; Bushey et al., 2009
). Therefore, RU486 treatment windows were assessed at all time points during development and at maturity for both endogenous and induced dFMRP expression levels (Figure 2
C). Progressive interventions were performed to drive dFMRP transgene expression from the earliest stages of development, through intermediate and later stages, culminating with adult-onset expression. The earliest induction was to constitutively feed RU486 throughout larval development (instars 1–3) and then remove animals from treatment. Controlled RU486 dosage was used to match endogenous dFMRP levels, measured in wandering third instar larvae at 94 ± 11% (n = 3) of the corresponding control levels (Figure 2
C). Following removal of the drug, dFMRP level rapidly declined during pupal stages (P1–P4) and was almost entirely absent by eclosion into the adult allowing transient early dFMRP expression.
Sequential RU486 treatments were used during each day of pupal metamorphosis to drive dFMRP expression starting at pupal day 1 (P1), P2, P3 and P4 (Figure 2
C). Induction at P1 was achieved by bath application of RU486 on the late wandering third instar larvae, just prior to puparium formation. This treatment drives dFMRP expression starting in P1 and extending into the early stages of pupal development (Figure 2
C). We previously reported a quantified half-life of dFMRP driven by larval RU486 induction of ∼25 h (Gatto and Broadie, 2008
). In the pupae, induced dFMRP persists longer, presumably owing to restrictions on drug clearance and metabolism in the pupae, so that pupal RU486 treatments result in broader windows of expression. In each of the later pupal interventions, the pupal case cuticle was cut to introduce RU486 via brief (2 min) bath application for absorption (Figure 2
C). Treatments were performed just prior to the onset of day P2, P3 and P4 to drive dFMRP expression, which was then monitored over subsequent 24 h intervals progressing to adult eclosion.
The final intervention was to drive dFMRP expression only at maturity in an otherwise dfmr1-null mutant condition. Larvae and pupae were reared until eclosion in the absence of RU486, and no dFMRP expression was detectable (Figure 2
C). Following eclosion, adult animals were then fed RU486 to drive dFMRP levels matching wild type after 24 h of treatment. Adult onset expression was maintained for an additional 72 h course to assess the consequences of continued adult dFMRP expression (Figure 2
C).
Early Developmental and Adult-Onset dFMRP Expression do not Alleviate dfmr1-null Defects
Using the conditional GS system, we systematically investigated when reintroduction of dFMRP could restore the gross sLNv synaptic architecture defects of the dfmr1 null (Figure 1
). We examined how each inductive paradigm might modulate synaptic structure, as quantified with our modified Sholl Analysis of synaptic bouton spatial distribution (Figure 1
C). We first examined animals in which normal dFMRP levels were driven throughout larval life, the period of neurogenesis and neural fate specification. This developmentally early intervention provided no detectable restorative effect on the organization of the sLNvs arborization or synaptic bouton disposition as compared to the eGS + ET animals, which phenocopy the dfmr1 null alone (Figure 3
A). When dFMRP is introduced during larval stages only, the resultant adults display PDF puncta within the sLNv arbors that remain unchanged with respect to total number (eGS + ET: 88 ± 6, n = 10 vs. eGS + RU: 85 ± 6, n = 10; p = 0.97) and spatial distribution throughout the Sholl array (Figure 3
A, right). We conclude that providing dFMRP transiently during early stages of neural development provides no benefit for correcting the hallmark synaptic defects caused by the loss of dFMRP as assayed at maturity.
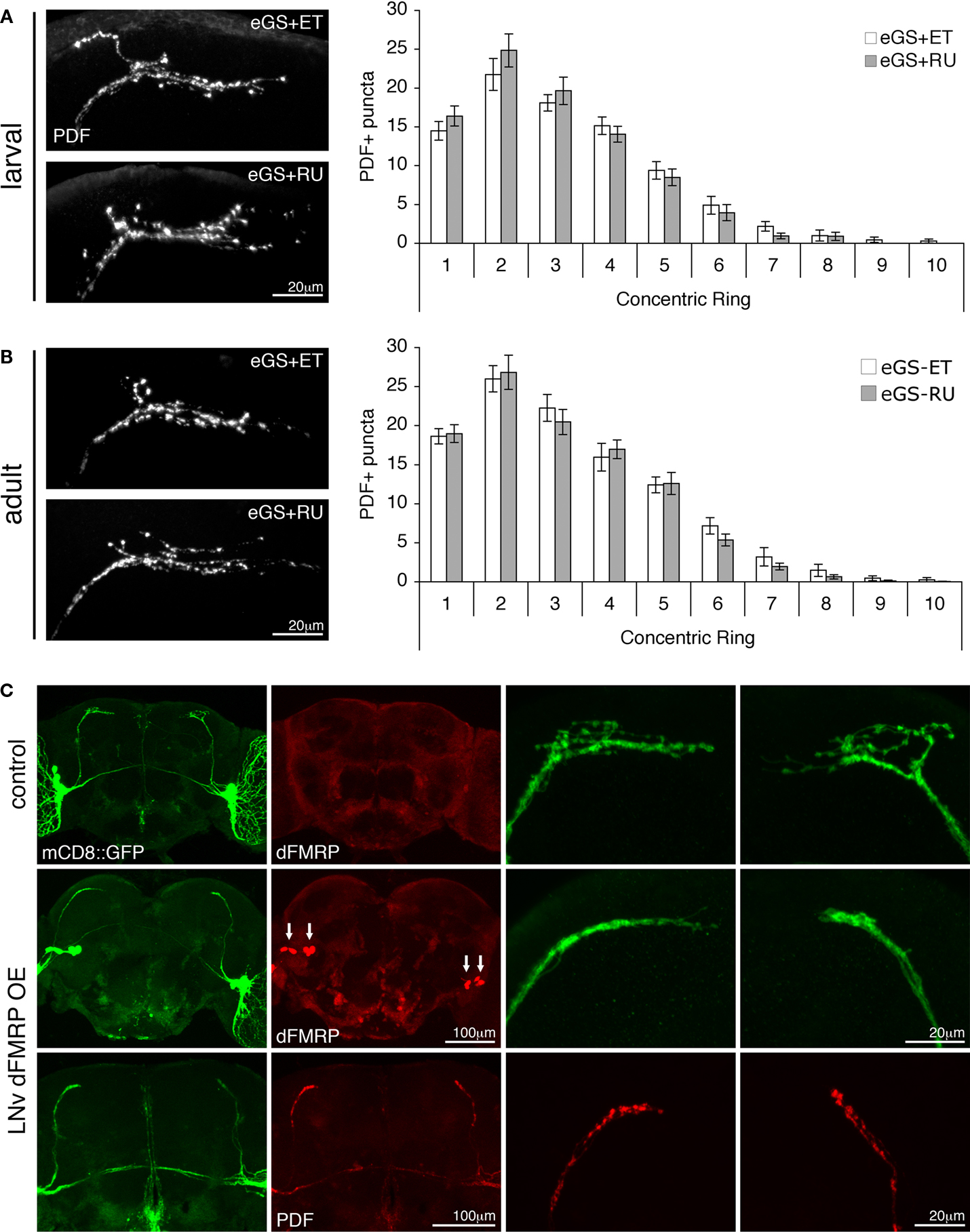
Figure 3. dFMRP induction during early development and at maturity provides no rescue of dfmr1-null mutant sLNv defects. (A) Comparison of the dfmr1-null eGS ethanol vehicle (eGS + ET) and RU486 (eGS + RU) treatment throughout larval development (instars L1–L3). At left, representative images shown of adult sLNv terminal arbors immunostained for PDF. At right, quantification from the Sholl Analysis indicating no change with RU486 induction. (B) Analyses of adult-onset dFMRP induction. At left, representative images shown from control and RU486-induced animals (treated d1–3) with sLNv synaptic arbors immunostained for PDF. At right, quantification from the Sholl Analysis indicating no change with RU486 induction. (C) Representative images of ventrolateral clock neurons expressing either UAS-mCD8::GFP (green) alone under the control of PDF-GAL4 (control: PDF-GAL4/UAS-mCD8::GFP) or in conjunction with, and thus overexpressing, UAS-dfmr1 (LNv dFMRP OE: PDF-GAL4/UAS-mCD8::GFP; UAS-9557-3, dfmr150M/+). Co-immunolabeling for dFMRP (red) illustrates endogenous (top row) and transgenic (middle row) levels in the brain, and specifically in the sLNv cell bodies (arrows). High magnification views illustrate the sLNv synaptic arbor collapse caused by cell-autonomous over-expression of dFMRP. Examination of collapsed arbors via co-immunolabelling for PDF (red, bottom row) revealed the persistence of PDF-positive presynaptic boutons.
We next assayed whether adult-onset dFMRP induction could rescue synaptic defects in dfmr1-null animals (Figure 3
B). This determination is particularly important for the design of FraX intervention strategies, as it addresses the question of whether the synaptic overgrowth characteristic of the disease is reversible at a state of maturity. eGS dfmr1-null animals were raised in the absence of RU486 induction preventing dFMRP expression during development and then switched to an RU486-containing diet at eclosion. eGS animals with sustained induction for the following 3 days showed no detectable reversal of the overgrown sLNv synaptic terminal area, the over-elaborated synaptic branch arborization, or the over-proliferation of synaptic boutons as compared to the eGS + ET animals, which phenocopy the dfmr1 null alone (Figure 3
B). When reintroduced only in the adult, the aged adult animals expressing dFMRP display PDF synaptic puncta within the sLNvs arbors that remain unchanged with respect to total number (eGS + ET: 77 ± 4, n = 15 vs. eGS + RU: 73 ± 4, n = 15; p = 0.63) and spatial distribution throughout the Sholl array (Figure 3
B, right). We conclude that providing dFMRP at maturity, following completion of neural development, provides no benefit for correcting the hallmark synaptic defects caused by the loss of dFMRP.
Despite reintroduction of dFMRP throughout either the larval or adult phase, no palliative effect was observed in the dfmr1-null sLNv synaptic connections. Therefore, we next tested whether phenotypic reversal of these defects would be identifiable upon dFMRP elevation in these cells. We compared two conditions: (1) UAS-mCD8::GFP alone, and (2) UAS-mCD8::GFP co-expressed with UAS-dfmr1, specifically driven in sLNvs (Figure 3
C). dFMRP in the control is expressed at a uniform low level in neurons, including the sLNv clock neurons (Figure 3
C, top row, red), whereas targeted dFMRP, exploiting the PDF-GAL4 driver, elevates expression only in these cells (Figure 3
C, middle row, red, arrows). This targeted, constitutive expression of dFMRP within the sLNvs results in collapse and retraction of the terminal synaptic arbor, evident in the reduction of synaptic area encompassed, loss of synaptic varicosities and complete lack of outward projecting axonal branches (Figure 3
C, green). Examination of the PDF distribution within these collapsed terminal arbors revealed that, although the gross anatomy of the synapse was radically altered, discrete PDF-positive synaptic puncta remained apparent (Figure 3
C, bottom row, red). These results suggest that the presynaptic potential of the sLNvs remains intact. In contrast to the changes resulting from constitutive lateral neuron-specific dFMRP overexpression, timed pan-neuronal dFMRP introduction with the GS system, either early or late, causes no such change in the synaptic arborization of the dfmr1 null sLNvs, which remain over-extended and over-elaborated. Even over-expression of dFMRP in the adult (Figures 2
A,C, after full 72 h RU486 treatment: 246 ± 14% control level, n = 4) failed to cause a reduction in the synaptic arbor of the null mutant, showing that the over-expression effect is likewise restricted to a specific developmental window. These results together strongly suggest that GS-induced dFMRP expression has not yet been provided at the appropriate stage.
A Critical Window of dFMRP Requirement During Late Brain Development
The above results suggest that the requirement for dFMRP function in synaptic patterning likely occurs in the period of late brain development during pupal metamorphosis. This timing is consistent with elevated endogenous dFMRP expression during the late pupal period, and the role of dFMRP during this period in activity-dependent synaptic refinement (Tessier and Broadie, 2008
). As the pupal period lasts 4 days at 25°C (P1–P4), we subdivided this period by inducing dFMRP at the start of each consecutive day in turn in otherwise dfmr1-null mutant animals (Figure 2
B). The consequences on sLNv clock neuron synaptic arborizations were then evaluated in mature adults (d1–2) in each case. The results of these studies are shown in Figure 4
.
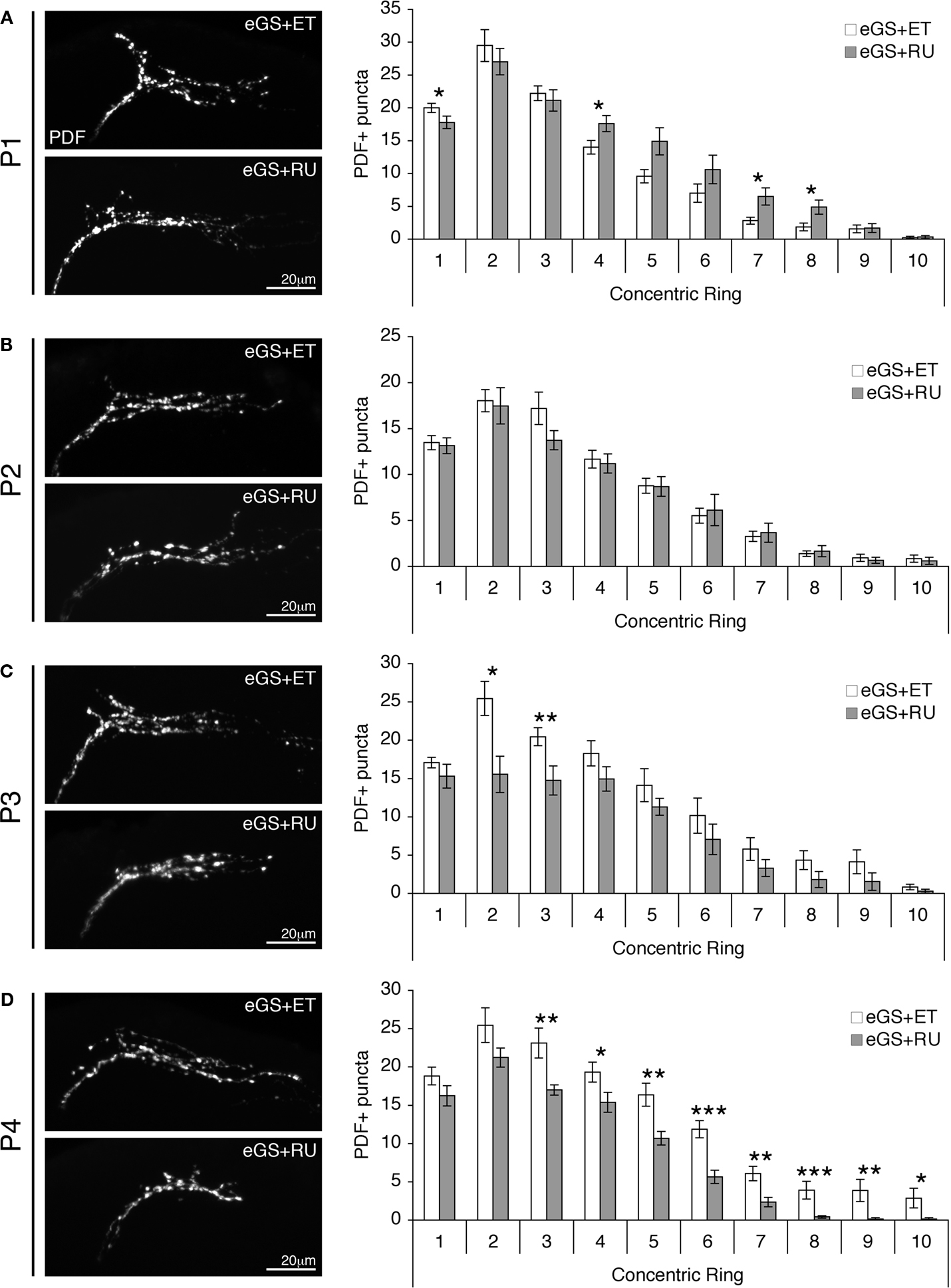
Figure 4. dFMRP induction only during late brain maturation restores normal sLNv synaptic architecture. (A) Representative images of adult sLNv synaptic arbors immunostained for PDF after pupal day 1 (P1) inductive treatment with ethanol vehicle (eGS + ET) or RU486 (eGS + RU). At right, quantification from the PDF-adapted modified Sholl Analysis, indicating a moderately exacerbated mutant phenotype. Significance levels: p < 0.05 (*). (B) Representative images of adult sLNv synapses immunostained for PDF after P2 inductive treatment with ethanol vehicle (eGS + ET) or RU486 (eGS + RU). At right, quantification showing no change with dFMRP induction. (C,D) Representative images are displayed of adult sLNv terminal synapses immunostained for PDF after P3 (C) or P4 (D) treatment with ethanol vehicle (eGS + ET) or RU486 (eGS + RU). Quantifications show significant rescue of the synaptic defects. Significance levels: p < 0.05 (*); p < 0.01 (**); p < 0.001 (***).
We began with RU486 drug delivery to drive dFMRP induction at the earliest stage of pupal development, with induction at pupal day 1 (P1; Figure 4
A). This early phase dFMRP expression provided no correction of the dfmr1-null sLNv synapses compared to the eGS + ET animals, which phenocopy the dfmr1 null alone. Indeed, this inappropriate early induction exacerbated the mutant phenotype, resulting in an elevated number of PDF synaptic boutons redistributed within 30% of the sLNv axonal arbor based on the Sholl Analysis (Figure 4
A, right). While the total number of boutons was unchanged (p = 0.32; n = 11 eGS + ET, n = 14 eGS + RU), the spatial distribution of boutons was significantly altered, with an increased number of boutons in the 40 μm concentric ring (eGS + ET: 14 ± 1 vs. eGS + RU: 18 ± 1; p = 0.025), 70 μm ring (eGS + ET: 3 ± 0.5 vs. eGS + RU: 7 ± 1; p = 0.037), and 80 μm ring (eGS + ET: 2 ± 0.6 vs. eGS + RU: 5 ± 1; p = 0.035). The overall frequency of boutons in the more distal domains was shifted from 82% to 93% at 70 μm and from 63% to 93% at 80 μm in eGS + ET compared to eGS + RU (Figure 4
A, right). Similarly, induction at P2 provided no improvement of the dfmr1-null sLNv architecture compared to the eGS + ET animals (Figure 4
B). P2 induction of dFMRP provided no significant influence upon the synaptic arborization, with no change in the total number of PDF boutons (eGS + ET: 81 ± 4, n = 15 vs. eGS + RU: 77 ± 7, n = 15; p = 0.27) or their spatial distribution (Figure 4
B, right). We conclude that induction of dFMRP during the first half of pupal development does not restore normal synaptic connectivity in the null mutant animals.
In contrast to all of the preceding interventions, reintroduction of dFMRP specifically within the latter half of pupal development provided the key to structural reparation of the dfmr1-null sLNv synaptic defects. At P3, dFMRP induction resulted in partial rescue of the synaptic arbor, including the number of synaptic boutons and the extent of spatial overgrowth (Figure 4
C, left). Based on the Sholl Analysis, there was a highly significant reduction in the total number of PDF boutons present in the synaptic terminal arbor (p = 0.009; n = 12 eGS + ET, n = 10 eGS + RU), with the restoration effect most pronounced in the region immediately adjacent to the dorsal horn bifurcation (Figure 4
C, right). PDF-reactive boutons were reduced in both the 20 μm concentric ring (eGS + ET: 25 ± 2, vs. eGS + RU: 16 ± 2; p = 0.013) and the 30 μm domain (eGS + ET: 20 ± 1 vs. eGS + RU: 15 ± 2; p = 0.007). The most dramatic rescue was effected upon dFMRP induction at the very end of development, during pupal day 4 (P4; Figure 4
D). Throughout the sLNv synaptic arbors the total number of PDF boutons was very significantly reduced (p < 0.0001; n = 12 eGS + ET, n = 15 eGS + RU), with spatially specific resolution achieved over more than 80% of the arbor area (Figure 4
D, right). The distal concentric rings from 30 to 100 μm displayed the following decrements in PDF boutons: 30 μm (eGS + ET: 23 ± 2 vs. eGS + RU: 17 ± 1, p = 0.003), 40 μm (eGS + ET: 19 ± 1 vs. eGS + RU: 15 ± 1, p = 0.026), 50 μm (eGS + ET: 16 ± 2 vs. eGS + RU: 11 ± 1, p = 0.004), 60 μm (eGS + ET: 12 ± 1 vs. eGS + RU: 6 ± 1, p = 0.0006), 70 μm (eGS + ET: 6 ± 1 vs. eGS + RU: 2 ± 1, p = 0.005), 80 μm (eGS + ET: 4 ± 1 vs. eGS + RU: 0.4 ± 0.2, p = 0.0004), 90 μm (eGS + ET: 4 ± 1 vs. eGS + RU: 0.2 ± 0.2, p = 0.001) and 100 μm (eGS + ET: 3 ± 1 vs. eGS + RU: 0.2 ± 0.2, p = 0.011). We conclude that dFMRP plays a highly specific role in the very latest stages of synaptic development and refinement that is required to properly restrict the number and specificity of synaptic connections, and that dFMRP is capable of operating in this mechanism only during this strictly defined developmental window of opportunity.
Loss of FMRP compromises the synaptic connectivity of the circadian clock circuit and impairs the behavioral output of rhythmic circadian activity patterns, making this system ideal for investigating the temporal requirements for FMRP function (Elia et al., 2000
; Gould et al., 2000
; Dockendorff et al., 2002
; Inoue et al., 2002
; Morales et al., 2002
; Reeve et al., 2005
, 2008
; Miano et al., 2008
; Sekine et al., 2008
; Zhang et al., 2008
; Bushey et al., 2009
). Using our well-established Drosophila FraX model (Zhang et al., 2001
; Bolduc et al., 2008
; Gatto and Broadie, 2008
; Pan et al., 2008
; Tessier and Broadie, 2008
; Repicky and Broadie, 2009
) and the well-characterized Drosophila clock circuitry (Helfrich-Forster, 2005
; Chang, 2006
; Nitabach and Taghert, 2008
), this study was aimed at identifying the critical period(s) of dFMRP function in the synaptic organization of clock neurons. The GS method provides tight temporal control of transgene expression specifically within neurons (Osterwalder et al., 2001
; Roman et al., 2001
), allowing us to dissect the exact timing of the dFMRP requirement. We have identified a single responsive time window, pupal days 3–4, during which reintroduction of dFMRP provides rescue of the arbor overgrowth and excessive synapse establishment characterizing the dfmr1-null mutant. Introduction of dFMRP either before or after this period provides no benefit. We conclude, therefore, that dFMRP function in sculpting synaptic connections is locked into this transient developmental time window. As these studies were conducted with pan-neuronal expression, we cannot yet distinguish between a cell autonomous function of dFMRP in sLNv clock neurons compared to potential roles of dFMRP in neighboring neurons.
We have previously investigated the temporal requirements of dFMRP at the peripheral glutamatergic neuromuscular junction (NMJ) (Gatto and Broadie, 2008
). In dfmr1-null mutants, this synaptic arbor is over-elaborated with expanded synaptic area, excess synaptic branching and supernumerary synaptic boutons (Zhang et al., 2001
), thus closely resembling the phenotype in the central brain clock neurons shown here. Employing GS inductive paradigms to drive dFMRP in the dfmr1-null mutant background, these studies similarly demonstrate a temporally restricted role for dFMRP in sculpting synaptic architecture (Gatto and Broadie, 2008
). Re-introduction of dFMRP only during the period of normal NMJ synaptogenesis enabled the correct development and maintenance of standard, sustainable terminal structure. In contrast, dFMRP introduction at maturity provided very little rescue of the mutant overgrown synaptic defects, with only weak, partial reduction in the number of synaptic boutons (Gatto and Broadie, 2008
). The partial rescue achieved likely reflects the fact that synaptogenesis continues throughout the larval period in Drosophila, and therefore development and maturity are not as well separated as in the brain clock circuit. Overall, these previous findings at the NMJ strongly support our current results of a transient development requirement for dFMRP within the sLNv clock neurons.
Interestingly, a host of FraX-associated phenotypes are known to be developmentally transient, also suggesting temporally-restricted FMRP function. In the Drosophila FraX model, dfmr1-null mushroom body neurons display heightened axonal and dendritic complexity, with excessive synaptic connectivity (Pan et al., 2004
). These defects become evident only very late in pupal brain development, with the appearance of abnormally long axonal branches and associated excessive synaptic processes (Tessier and Broadie, 2008
). Upon initial use following eclosion, activity-dependent synaptic pruning occurs in wild-type but completely fails in dfmr1-null mutant neurons. Following this normal dFMRP-dependent developmental window, delayed and inappropriate pruning occurs in the absence of dFMRP (Tessier and Broadie, 2008
). This refinement process takes days in Drosophila, but occurs over the course of weeks in mice. Similar to the Drosophila findings, in Fmr1-null mouse layer V somatosensory barrel cortex, the increase in synaptic dendrite spine length and density manifests during postnatal week 1, attenuates by week 2 and is undetectable by week 4 (Nimchinsky et al., 2001
; Galvez and Greenough, 2005
). Likewise, Fmr1-null ascending projections connecting layers III and IV display aberrant transmission strength, experience-dependent plasticity and axonal arborization at postnatal week 2, which also resolves by postnatal week 3–4 (Bureau et al., 2008
). These studies suggest a transient FMRP role in synaptic development and refinement. It should be noted that differences could yet arise between the Drosophila and murine FraX models with regard to the temporal requirements for FMRP. Mice harbor the two Fmr1 paralogs, Fxr1 and Fxr2 (Siomi et al., 1995
; Zhang et al., 1995
), which could influence the effects of Fmr1 deletion during development. However, the protracted postnatal period required for circuit refinement in mammals needs to be carefully considered to define developmental vs. adult FMRP requirement. We eagerly await the establishment of similar mouse conditional Fmr1 expression models to provide further insight into the etiology of FraX.
In the Drosophila FraX model, the P3–P4 critical period definition for sLNv synaptic structuring is entirely consistent with our recent finding that dFMRP levels are transiently elevated during this same period (Tessier and Broadie, 2008
), with subsequent rapid decrement following maturation (Tessier and Broadie, 2008
; Bushey et al., 2009
). A similar transient period of FMRP elevation is reported in rodents, likewise corresponding to the restricted period of synaptogenesis and synaptic refinement (Khandjian et al., 1995
; Singh et al., 2007
; Singh and Prasad, 2008
). During the corresponding period, dFMRP has been shown to regulate a similar stage-dependent mechanism of activity-dependent axonal pruning and synapse elimination in the Drosophila mushroom body (Tessier and Broadie, 2008
). Assuming a conserved mechanism within the clock circuit, this role would explain the temporally-restricted dFMRP function in establishing appropriate sLNv synaptic connections. Consistent with this transient critical window for dFMRP function, assays to ameliorate dfmr1-null clock defects by decreased metabotropic glutamate receptor (mGluR) signaling, via treatment with mGluR antagonists such as 2-methyl-6-phenylethynyl-pyridine (MPEP), fail to resolve circadian impairments with treatment either during larval development or in the adult (McBride et al., 2005
). We suggest that such previous pharmacological treatment attempts have inadvertently missed the therapeutic target window of late brain development.
FMRP is an RNA-binding, polysome-associated protein that influences mRNA trafficking/stability (Zalfa et al., 2005
; Zhang et al., 2007
; Dictenberg et al., 2008
; Estes et al., 2008
) and regulates protein translation (Laggerbauer et al., 2001
; Li et al., 2001
; Sung et al., 2003
; Zalfa et al., 2003
; Lu et al., 2004
; Qin et al., 2005
; Tessier and Broadie, 2008
; Bechara et al., 2009
). In the Drosophila brain, dFMRP negatively regulates cytoskeleton organizing proteins such as Futsch, the mictrobule-binding MAP1B homolog, and Chickadee, the actin-binding Profilin homolog (Zhang et al., 2001
; Reeve et al., 2005
; Tessier and Broadie, 2008
). In sLNv clock neurons, overexpression of Chickadee mimics dfmr1-null architectural defects, while concurrently decreasing Chickadee levels in the dfmr1 null suppresses the phenotypes (Reeve et al., 2005
). Importantly, dfmr1 mutants display only a developmentally transient elevation of Chickadee expression at P4 into post-eclosion day 1 (Tessier and Broadie, 2008
). Thus, targeted reintroduction of dFMRP at this time point would prevent the elevated protein levels, providing for normal synaptic architecture and connectivity. In parallel, dFMRP also modulates the actin cytoskeleton by regulating the small GTPase Rac1 (Lee et al., 2003
), CYFIP/Sra-1 (cytoplasmic FMRP interacting protein) (Schenck et al., 2003
) and the actin depolymerizing factor/Cofilin (Castets et al., 2005
; Le Clainche and Carlier, 2008
), which all may contribute to the normalization of sLNv architecture. In fact, the dFMRP N-terminal region specifically interacts with CYFIP, cooperatively regulating sLNv axonal projections and synaptic development (Reeve et al., 2008
). Thus, we posit that the temporally-specific regulation of these cytoskeleton-modifying dFMRP targets is fundamentally important in establishing and/or maintaining proper synaptic connections within the clock circuit.
The PDF cells have been described as “GABA-responsive wake-promoting” neurons in the clock circuit (Parisky et al., 2008
). Suppression of neuronal activity in these cells increases sleep, while coordinately increasing LNv excitability decreases sleep. sLNvs express the Rdl GABAA receptor, and sLNv-specific Rdl knockdown impairs sleep while Rdl over-expression yields more rapid sleep onset and enhanced sleep (Parisky et al., 2008
). These findings are particularly relevant to the Drosophila FraX model, as it has been recently reported that dfmr1 nulls display increased sleep, while dFMRP over-expression decreases sleep (Bushey et al., 2009
). However, dfmr1 nulls have been shown to under express all known Drosophila GABAA receptor subunits (D’Hulst et al., 2006
). Consistently, the Fmr1 mutant mouse has reduced GABAA receptors (D’Hulst et al., 2006
; Gantois et al., 2006
), associated with both structural and functional alterations of the GABAergic circuits in the neocortex and hippocampus, respectively (D’Antuono et al., 2003
; Selby et al., 2007
). These changes may correlate with recent accounts of neuronal hyperexcitability in both Drosophila and mouse FraX models (Gibson et al., 2008
; Repicky and Broadie, 2009
). A small molecule screen for rescue of dfmr1-mutant phenotypes identified GABA, nipecotic acid (GABA reuptake inhibitor) and creatinine (suspected GABAA receptor activator), again implicating GABAergic signaling (Chang et al., 2008
). In dfmr1 nulls, GABA treatment specifically relieved misregulation of the MAP1B homolog, Futsch, mushroom body β-lobe midline crossing and courtship defects. Together, these data suggest that manipulation of GABA signaling may alleviate PDF neuron and circadian behavioral phenotypes. With the emergence of information to direct appropriate temporal targeting of such interventions, implementation of this and/or any therapeutic option should be more effective and ultimately more successful.
The authors declare that the research presented was completed in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are especially grateful to Dr. Haig Keshishian (Yale University) for the elav-Gene-Switch line that made this work possible. We thankfully acknowledge the Drosophila Bloomington Stock Center and the Iowa Developmental Studies Hybridoma Bank for essential genetic lines and antibodies, respectively. We would also like to acknowledge members of the Broadie Lab, especially Dr. Charles Tessier, Dr. Sarah Repicky and Mr. R. Lane Coffee, for insightful discussion and critical feedback during manuscript preparation. This work was supported in part by a Postdoctoral Fellowship from the FRAXA Research Foundation and the National Institutes of Health through the NIH Roadmap for Medical Research Training Grant T32 MH075883 to C.L.G. and R01 grant MH084989 to K.B.
McBride, S. M., Choi, C. H., Wang, Y., Liebelt, D., Braunstein, E., Ferreiro, D., Sehgal, A., Siwicki, K. K., Dockendorff, T. C., Nguyen, H. T., McDonald, T. V., and Jongens, T. A. (2005). Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45, 753–764.
Nitabach, M. N., Wu, Y., Sheeba, V., Lemon, W. C., Strumbos, J., Zelensky, P. K., White, B. H., and Holmes, T. C. (2006). Electrical hyperexcitation of lateral ventral pacemaker neurons desynchronizes downstream circadian oscillators in the fly circadian circuit and induces multiple behavioral periods. J. Neurosci. 26, 479–489.