1
Interdisziplinäres Zentrum für Neurowissenschaften, SFB488 Junior Group, Institut für Neuroanatomie, Universitätsklinikum Heidelberg, Heidelberg, Germany
2
Wilhelm Johannsen Centre for Functional Genome Research, Department of Cellular and Molecular Medicine, The Panum Institute, Copenhagen University, Copenhagen, Denmark
3
Santaris Pharma, Hørsholm, Denmark
4
Copenhagen Institute of Technology, Aalborg University, Ballerup, Denmark
Recent studies using primary neuronal cultures have revealed important roles of the microRNA pathway in the regulation of neuronal development and morphology. For example, miR-134 is involved in dendritogenesis and spine development in hippocampal neurons by regulating local mRNA translation in dendrites. The in vivo roles of microRNAs in these processes are still uninvestigated, partly due to the lack of tools enabling stable in vivo delivery of microRNAs or microRNA inhibitors into neurons of the mammalian brain. Here we describe the construction and validation of a vector-based tool for stable delivery of microRNAs in vivo by use of recombinant adeno-associated virus (rAAV). rAAV-mediated overexpression of miR-134 in neurons of the postnatal mouse brain provided evidence for a negative role of miR-134 in dendritic arborization of cortical layer V pyramidal neurons in vivo, thereby confirming previous findings obtained with cultured neurons. Our system provides researchers with a unique tool to study the role of any candidate microRNA in vivo and can easily be adapted to microRNA loss-of-function studies. This platform should therefore greatly facilitate investigations on the role of microRNAs in synapse development, plasticity and behavior in vivo.
microRNAs (miRNAs) have emerged as important post-transcriptional regulators of gene expression. Mature miRNAs are processed from primary transcripts (pri-miRNAs) by two RNase III enzymes, first by Drosha yielding a ∼70 nt (nucleotide) precursor miRNA (pre-miRNA) followed by Dicer processing of the pre-miRNA into a ∼22 nt miRNA duplex. Usually, only one strand of the miRNA duplex defines the functional miRNA, which is guided to the 3′-untranslated region (3′-UTR) of target mRNAs by the RNA-induced silencing complex (RISC). Here, miRNAs bind to partially complementary regions, thereby exerting their function as regulators of gene expression (Bartel, 2004
). Dicer cleaves double-stranded RNA (dsRNA) in 21–23 nt intervals independent of the specific sequence (Zamore et al., 2000
; Elbashir et al., 2001
; Zhang et al., 2002
), thus the site of Drosha cleavage of pri miRNAs pre-determines cleavage specificity within the pre-miRNAs. However, relatively little is known about the elements of individual pri miRNAs that are required for Drosha processing. Drosha forms a complex, the Microprocessor, with the dsRNA binding protein DGCR8 which facilitate binding of Drosha to the pri-miRNAs (Denli et al., 2004
; Gregory et al., 2004
; Han et al., 2004
). DGCR8 recognition motifs within pri-miRNAs appear to be largely determined by structural features (Han et al., 2006
). In particular, Drosha processing requires both dsRNA elements at the base of the miRNA stem-loop structure and flanking single-stranded RNA (ssRNA) tails (Lee et al., 2003
; Zeng and Cullen, 2005
; Han et al., 2006
).
miRNAs have been implicated in many distinct processes in the nervous system, ranging from neuronal differentiation (Krichevsky et al., 2006
; Kim et al., 2007
; Shibata et al., 2008
), neuronal stem cell commitment (Rybak et al., 2008
), brain development (Giraldez et al., 2005
; Leucht et al., 2008
; Maller Schulman et al., 2008
), neurite outgrowth (Barik, 2008
; Yu et al., 2008
) to synaptic plasticity (Schratt et al., 2006
; Siegel et al., 2009
). In addition, evidence for the involvement of miRNAs in a range of neurological diseases has been obtained (Abelson et al., 2005
; Kim et al., 2007
; Hébert et al., 2008
; Packer et al., 2008
; Stark et al., 2008
). We have recently shown that the brain specific miRNA, miR-134, regulates spine morphogenesis of primary hippocampal neurons in culture by regulating the local translation of LimK1 mRNA, a regulator of actin filament dynamics (Schratt et al., 2006
). More recently, we found that myocyte enhancing factor 2 (Mef2) dependent transcription of miR-134 is required for activity-dependent dendritic outgrowth of primary hippocampal neurons, through miR-134 mediated regulation of the translational repressor Pumilio2 (Pum2) (Fiore et al., 2009
). The potentially important roles of miRNAs in synapse development and function indicated by these in vitro studies highlight the need for the construction of tools that enable efficient in vivo miRNA manipulation. In vivo delivery of synthetic miRNA duplexes is possible, but this approach is limited owing to the low stability of RNA oligonucleotides in vivo, the lack of regulated expression and the inefficient uptake of the oligonucleotides by the neurons.
Here, we describe the construction of a vector based tool, which allows stable and efficient delivery of miRNAs in vivo by use of rAAV. We expressed miRNAs from chimeric hairpins located in the 3′-UTR of enhanced GFP (eGFP) on an adeno-associated virus (AAV) vector. The resulting co-expression of miRNAs and eGFP allowed tracing of neurons in vivo in which miRNAs had been delivered. Using this tool, we found that miR-134 delivery into cortical layer V neurons impaired dendritogenesis in the mouse brain in vivo. In principle, rAAV-directed expression and inhibition of miRNAs should allow us to investigate the in vivo effect of any miRNA during mammalian neuron development in a rapid, specific and cost-effective manner.
DNA Constructs
The chimeric hairpins were engineered by polynucleotide cloning into the 3′-UTR of eGFP on pAAV-6P-SEWB using the BsrGI/HindIII sites (Figure 1 in Supplementary Material). Cloning of pGL3-LimK1-3′UTR is described in Schratt et al. (2006)
. The miR-134 binding site in the pGL3-LimK1-3′UTR construct was mutated to a miR-134 perfect binding site using the QuikChange II Site-Directed Mutagenesis kit (Stratagene), resulting in pGL3 LimK1 3′UTR 134pbds. The rat Hs3st2-3′UTR was amplified by use of PCR from cDNA made from rat brain total RNA (Ambion), comprising a region from 225 nts to 736 nts downstream of the coding region (Ensemble transcript ID: ENSRNOT00000023773). The PCR product was cloned into psiCHECK-2 (Promega) using the NotI/XhoI sites, resulting in the psiCHECK-Hs3st2-3′UTR construct. The miR-99a binding site in the psiCHECK-Hs3st2 3′UTR construct was mutated to a perfect miR-99a binding site by overlapping extension PCR (An et al., 2005
), resulting in psiCHECK-Hs3st2 3′UTR-99apbds. All primer sequences are listed in Supplementary Materials.
Cell Culture, Transfection and Virus Infection of Primary Neurons
Dissociated primary cortical and hippocampal neurons from embryonic day 18 (E18) Sprague Dawley rats (Charles River Laboratories, Sulzfeld, Germany) were prepared and cultured as described in Schratt et al. (2004)
. The neurons were transfected by mixing 1 μg total DNA/RNA per well of a 24 well plate with 100 μl of a 1:50 dilution of Lipofectamine 2000 (Invitrogen) in Neurobasal Medium (Invitrogen). After 20 min incubation at RT the transfection mixtures were diluted 1:5 in Neurobasal Medium and applied onto the neurons for 2 h.
HEK293T cells were cultured in MEM media (Invitrogen) supplied with 10% fetal bovine serum, 1 mM glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin. HEK293 cells were transfected using the calcium phosphate method. A final CaCl2 concentration of 0.1 M was used and an incubation time of 5 h.
Primary neurons (75.000 hippocampal neurons, 250.000 cortical neurons per 24-well) were transduced with rAAV by applying the viral particles into the culture media using a volume of the viral stock resulting in a visual GFP signal after 4–5 days and ∼100% transduction efficiency. rAAV titers were estimated to 0.6–2.7 × 108 IFU/ml based on HEK293 cell titration experiments.
Preparation of Infectious rAAV
Infectious rAAV was generated by a 1:1:1 co-transfection of pAAV 6P-SEWB (Shevtsova et al., 2005
) or pAAV-6P-SEWB derivatives with helper plasmids (pDP1 and pDP2) (Grimm et al., 2003
) into HEK293 cells using 13 μg of each plasmid per 15 cm cell culture dish. Following incubation for 2.5–3 days, the HEK293 cells were harvested for virus purification.
rAAV crude lysates were prepared by resuspending harvested HEK293 cells in PO buffer (20 mM Tris, 150 mM NaCl, pH 8.0) followed by lysis by three freeze-thaw cycles. After filtering the virus containing supernatant using a 0.45 μm syringe filter unit, the crude lysates were centrifuged to remove cell debris originating from the HEK293 cells. rAAV was purified by use of the Iodixanol density step gradient method (Zolotukhin et al., 1999
). The detailed protocol can be found in the “Methods” section in Supplementary Material.
In Vivo Injection
The experimental protocol for in vivo injection was approved by the Regierungspräsidium Karlsruhe (AZ 35-9185.81/G-170/07) and designed to minimize suffering and reduce the number of animals used. Postnatal day 0 (P0) C57BL/6 mice (Charles River Laboratories, Sulzfeld, Germany) were cryoanesthetized and injected with 2 μl purified rAAV stock into each lateral ventricle (2 mm ventral of lambda, ±0.7 mm from midline, depth: 1.8 mm) using a 10-μl Hamilton microliter syringe. Individual experiments were performed on pups from the same litter, which were previously tattooed on the footpads to identify the groups injected with rAAV carrying different pAAV-6P-SEWB derivatives. Following injection, the pups were placed on a 37°C warming pad and returned to the mother after regaining normal activity and color.
Immunohistochemistry
Injected mice were killed at P21, the brains were dissected and fixed ON in 4% paraformaldehyde, 4% sucrose in PBS at 4°C. Coronal brain sections (100 μm) were washed 3× 5 min in PBS, pre incubated for 1 h at RT in blocking buffer (10% normal goat serum, 0.25% TritonX-100 in PBS) followed by ON incubation at 4°C with rabbit anti-GFP antibody (1:2000; Invitrogen, A6455). After wash in blocking buffer at RT (2× 2 min and 2× 20 min) the brain sections were incubated for 2 h at RT with goat Alexa-488 coupled anti-rabbit antibody (1:250; Invitrogen, A11034). After another round of washing in blocking buffer the sections were counterstained with Hoechst and mounted for microscopy.
Image Analysis
Image capture and image analysis was performed with the experimenter blinded to the experimental conditions. In vivo dendritogenesis assays were performed on cortical layer V pyramidal neurons imaged from 100 μm immunostained coronal brain sections obtained from mice injected with rAAV. Projection images were made from seven 20× z-stack images with an interval of 5.5 μm and a resolution of 1024 × 1024 pixels using a confocal laser scanning microscope (LSM 5 Pascal, Zeiss, Germany). To analyze dendritic branching, a grid of 10 concentric circles spaced by 25 μm was placed centered on the soma of the neurons and the number of dendritic crossings (intersections) with each circle was counted. The data was obtained from three independent litters of mice, each litter providing two to three brains per experimental condition. On average, seven different neurons were imaged per experimental condition, resulting in the following data set: (a) AAV vector: 9 brains, 69 neurons, (b) control1.1: 8 brains, 61 neurons, (c) miRNA134.1: 8 brains, 57 neurons.
Western Blotting
For western blotting primary hippocampal neurons were transduced with purified rAAV at 10–11 DIV and the cells were lysed and prepared for blotting 8 days later. Western blot was performed as described in Siegel et al. (2009)
using the following primary antibodies: mouse anti-LimK1 antibody (1:2000; Transduction Laboratories, L13020), mouse anti-β-actin antibody (1:10000; Sigma, A5441) and rabbit anti-Pum2 antibody (1:2000; NOVUS Biologicals, NB100-387). For recognition of the primary antibodies HRP-conjugated goat anti-rabbit antibody (1:20000; Calbiochem, 401315) or HRP-conjugated rabbit anti-mouse antibody (1:20000; Calbiochem, 402335) was used. Sizes of protein bands were determined using the Precision Plus Protein Dual Color Standard (BIO-RAD).
Luciferase Assay
Primary cortical neurons were transfected at 4 DIV and luciferase assays were performed 3 days later using the Dual-Luciferase Reporter Assay System (Promega). For assays using luciferase reporter constructs containing a miRNA perfect binding site 500 ng of the AAV-6P-SEWB derivatives were co-transfected along with 125 ng of the luciferase reporter construct per well of a 24 well plate. When using luciferase reporter constructs containing miRNA wild type target sites only 12.5 ng of the luciferase construct was used. pre-miR control (Ambion, Negative control #1) and 99a pre-miR (Ambion) were used in a final concentration of 20 nM.
Quantitative Real-Time PCR (qPCR)
RNA was purified using QIAZOL (Qiagen) and treated with TURBO DNase (Ambion) to remove DNA contamination. Quantitative real-time PCR was performed with a 7300 Real Time PCR System (Applied Biosystems) using TaqMan MicroRNA Assays (Applied Biosystems) and iTaq SybrGreen Supermix with ROX (BIO-RAD) for the detection of miRNAs and mRNAs, respectively. Primers for detection of U6 snRNA and β-3-tubulin can be found in the “Methods” section in Supplementary Material.
Northern blot
RNA was purified as for Quantitative real-time PCR. Northern blots for detection of small RNAs were performed as described in Schratt et al. (2006)
. As molecular marker we used the Decade Marker system (Ambion). Sequences of northern probes used can be found in the “Methods” section in Supplementary Material.
Engineering of AAv Plasmids Expressing microRNAs from Chimeric Hairpins
In the interest of overexpressing miRNAs in vivo and guiding this expression to neurons, we used pAAV-6P-SEWB as a cloning vector, an AAV vector expressing eGFP under control of the synapsin promoter (Shevtsova et al., 2005
). For in vivo delivery, we packed rAAV particles using two helper plasmids expressing AAV serotype-1 and -2 capsid proteins (Grimm et al., 2003
), respectively, which are serotypes leading to widespread transduction of neurons throughout the neonatal mouse brain after intraventricular injection (Passini and Wolfe, 2001
; Passini et al., 2003
; Broekman et al., 2006
). Close to 100% transduction efficiency was observed when transducing primary cortical and hippocampal neurons using rAAV serotype 1/2, compared to only 20–25% efficiency when using Lipofectamine as transfection agent (unpublished observations). Therefore, rAAV could be used to perform biochemical assays in dissociated neurons for validation of the potential of rAAV-expressed miRNAs to regulate target gene expression. Due to the small promoter fragment used (ca. 600 bp), we also observed eGFP expression driven by the synapsin promoter upon transfection of the pAAV-6P-SEWB vector into non-neuronal HEK293 cells. This allowed us to use HEK293 cells for many of our mechanistic studies.
The exact flanking sequences of miRNA hairpin stem-loop structures required for Drosha processing of individual pri miRNAs are unknown, though 125 nts on each side of the hairpin appears to be sufficient in many cases (Chen et al., 2004
). Interestingly, the well studied miR-30a hairpin requires only ∼20 nts on each side at its base in the pri-miRNA for efficient cleavage by Drosha (Figure 1
A). Processing is abolished by disruption of dsRNA structures of this flanking region (Lee et al., 2003
; Zeng and Cullen, 2003
), suggesting that these structures are recognized by the Microprocessor complex. To bypass the uncertainty of elements required for Drosha cleavage of individual pri-miRNAs, we used the flanking elements of the miR-30a hairpin required for Drosha processing to design chimeric hairpins expressing candidate miRNAs. Importantly, a similar miR-30 hairpin design was recently described for the expression of artificial siRNAs in RNA interference studies (Dickins et al., 2005
; Stegmeier et al., 2005
). Two different designs of chimeric hairpins were engineered. The first design (Figure 1
A, miRNA134.1, miRNA99a.1, control1.1 and control2.1) was obtained by substituting the miR-30a sequence within the miR-30a precursor with a miRNA or control sequence of interest. Subsequently, the 3′end of the stem of the chimeric hairpins, the so-called star sequence, was modulated in a way that the overall secondary structure of the engineered hairpins mimicked the secondary structure of the miR-30a precursor. For construct miRNA134.1 and miRNA99a.1, the miR-134 and miR 99a mature sequences were inserted into the miR-30a backbone, respectively. Control1.1 and control2.1 contained sequences (AACCTTGTGGTCCTTAGGTGCG and cel-miR-67, respectively) which are not natural components of the small RNA pool of rodents, and therefore served as important negative controls for potential non-specific effects of small RNA overexpression in general (Grimm et al., 2006
; Narvaiza et al., 2006
). Based on the GC content, miR-134, miR-99a and control1 all display weaker 5′-end base pairing to their complement strands when inserted into the chimeric hairpins compared to their 3′-ends. Therefore, the sense strand of these duplexes should be preferentially loaded into the RISC complex (Khvorova et al., 2003
; Schwarz et al., 2003
). The second design (Figure 1
B, miRNA134.2 and control1.2) is a modification of the first hairpin design (Figure 1
A), where the single stranded terminal loop originating from the miR-30a precursor was substituted by the single stranded terminal loop from the miR 134 hairpin. The secondary structure of the stem near the single stranded terminal loop was designed to mimic the secondary structure of the miR-134 hairpin in this region.
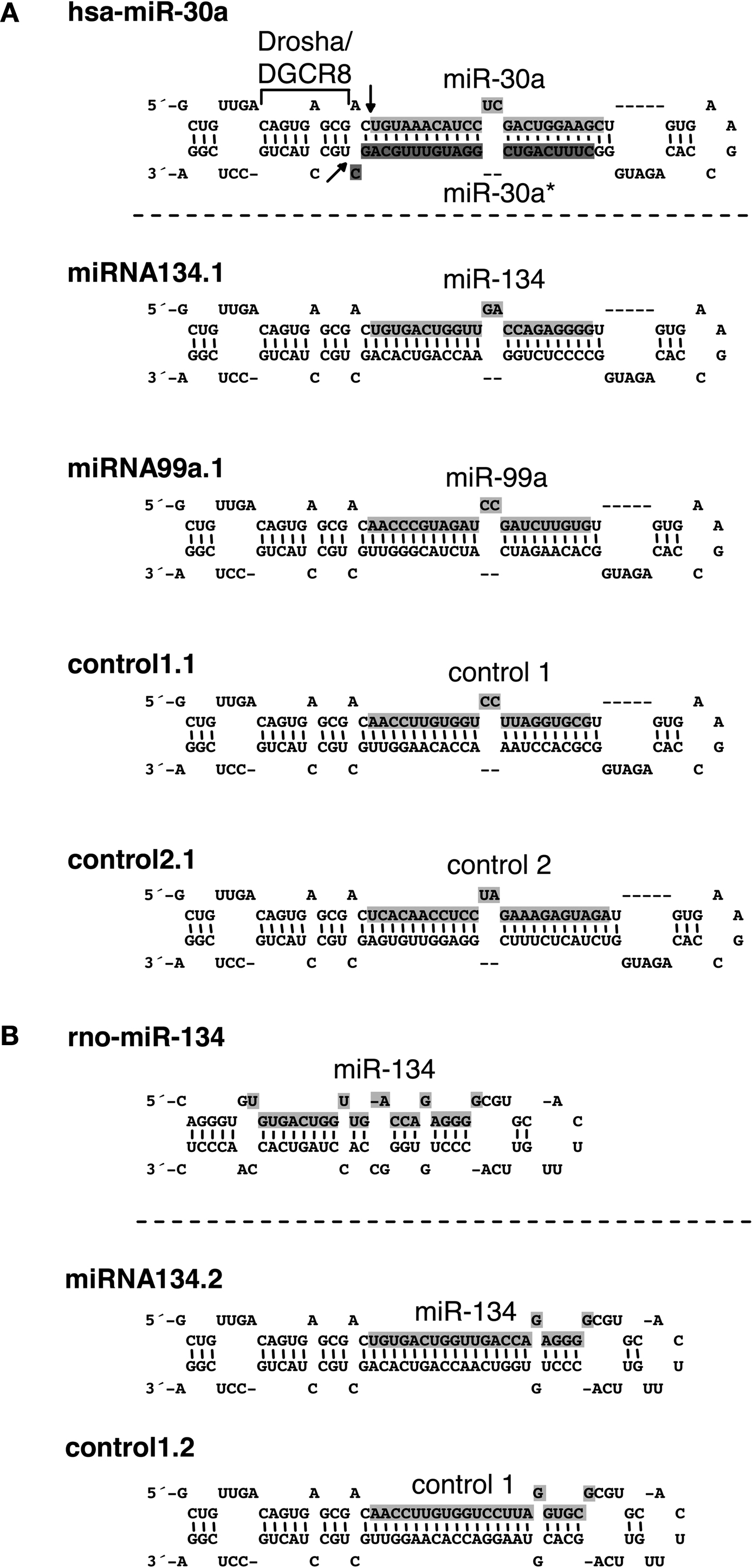
Figure 1. Secondary structures and sequences of chimeric hairpins. (A) Top: structure and sequence of the hsa-miR-30a hairpin including flanking sequences required for Drosha processing. Light grey shaded sequence: miR 30a mature sequence. Dark grey shaded sequence: miR-30a* mature sequence. Drosha/DGCR8: flanking region of the miR-30a hairpin required for Drosha processing. Arrows: site of Drosha cleavage. Bottom: structure and sequence of chimeric hairpins, design 1 (see text for details). Shaded sequence: miRNAs or control sequences expressed from the chimeric hairpins. (B) Top: structure and sequence of the rno-miR-134 hairpin. Shaded sequence: miR-134 mature sequence. Bottom: Structure and sequence of chimeric hairpins, design 2 (see text for details). Shaded sequence: miRNAs or controls expressed from the chimeric hairpins.
The chimeric hairpins were cloned into the 3′-UTR of eGFP on pAAV-6P-SEWB immediately at the end of the eGFP coding region (Figure 1 in Supplementary Material). This allowed co-expression of eGFP and miRNAs or control sequences expressed from the chimeric hairpins. The pAAV-6P-SEWB derivatives all efficiently expressed eGFP, though slightly less efficiently than the parental pAAV-6P-SEWB vector, presumably due to the insertion of a hairpin. Nevertheless, we inferred that the stability of the eGFP transcript was not significantly reduced either due to insertion of extra sequence in the 3′-UTR or due to a possible cleavage of the 3′-UTR by Drosha.
Validation of microRNA Expression from Chimeric Hairpins
To examine the capacity of the chimeric hairpins to express miRNAs and control sequences we first transfected the miRNA134.1, control1.1 and control2.1 constructs into primary cortical neurons and analyzed the expression by northern blot (Figure 2
A, left panel). An approximately fourfold higher level of miR-134 was detected in the cortical neurons transfected with the miRNA134.1 construct, which originated from a transfection efficiency of only 20–25%. Interestingly, a large fraction of the product expressed from the miRNA134.1 clone displayed a slightly larger size than endogenously expressed miR-134 detected in the control conditions. Control1.1 and control2.1 expressed small RNA products of the expected sizes. Importantly, expression from the constructs was also observed by northern blot when transducing primary cortical neurons with the constructs packed into virus particles (Figure 2
A, right panel). We estimated transduction efficiency to be close to 100%, suggesting that miR-134 levels were fourfold lower in individual virus-transduced cells compared to transfected cells. As further validation of miR-134 expression from the chimeric hairpin, we transfected the miRNA134.1 clone into primary cortical neurons and analyzed the expression by qPCR (Figure 2
B) using specific stem-loop primers. Thereby, we observed an approximately 10-fold induction of detectable miR-134 compared to the condition transfected with AAV vector. Expression of miR-134 after transduction of primary cortical neurons with rAAV carrying the miRNA134.1 construct was in addition followed over time and analyzed by qPCR (Figure 2 in Supplementary Material). Here, a continuous increase of detectable miR-134 was observed over a period of 11 days, displaying an approximately fivefold up-regulation at the peak of expression.
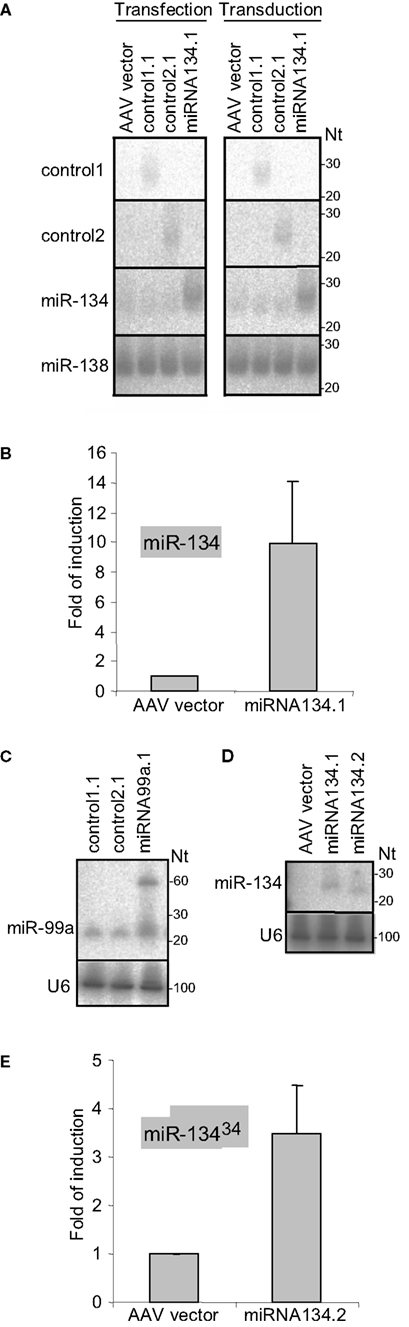
Figure 2. Processing of the chimeric hairpins. (A) Expression of miR-134 and control sequences from chimeric hairpins, design 1. Left panel: 10 μg of the indicated plasmids were transfected into 4 DIV primary cortical neurons per 10 cm tissue culture dish. RNA was prepared for northern blotting at 7 DIV and assayed for the indicated miRNAs. Right panel: 1 DIV primary cortical neurons were induced with rAAV (crude lysate) carrying the indicated plasmids. RNA was extracted at 7 DIV for northern blot assaying for the indicated miRNAs. ImageJ software was used to extract expression data and the levels of miR-134 were normalized against the levels of endogenously expressed miR-138. (B,E) Expression of miR-134 from the miRNA134.1 (B) and the miRNA134.2 (E) construct. 4 DIV primary cortical neurons were transfected with 250 ng the indicated plasmids per well of a 24 well plate. At 7 DIV, RNA was harvested and qPCR analysis was performed. miR-134 levels were normalized against the levels of endogenously expressed U6 snRNA. Fold of induction is expressed relative to the miR-134 levels in cells transfected with AAV vector, which are arbitrarily set to 1. Results are shown as means + s.d. [(B): n = 2; (E): n = 2]. (C,D) Expression of miR-99a and miR-134 from the miRNA99a.1 (C) and the miRNA134.2 (D) construct, respectively. HEK293 cells were transfected with 8 μg of the indicated plasmids per 10 cm cell culture dish. RNA was harvested 2 days later and subjected to northern blot analysis. Expression data were extracted using ImageJ software. miR-99a levels (C) were normalized against the levels of endogenously expressed U6 snRNA.
To test the general applicability of our approach, we designed an expression construct for an additional neuronal miRNA, miR-99a. miR-99a displays high endogenous levels in our primary cortical neuron cultures which preclude efficient detection of miR-99a overexpression by northern blot (data not shown). Therefore, we validated expression obtained with the miR-99a construct by transfecting HEK293 cells, which display lower levels of miR-99a. Using northern blot, we observed a ∼twofold upregulation of detectable miR-99a from miRNA99a.1 compared to the endogenous level of miR-99a observed in the control conditions (Figure 2
C). The product expressed from the miRNA99a.1 construct displayed a similar size as the endogenously expressed miR-99a. In addition, a clear ∼60 nt long precursor band of the expected size was observed, showing efficient processing of the chimeric hairpin by Drosha.
We observed that miR-134 expressed from the miRNA134.1 construct migrated slightly slower during gel electrophoresis compared to endogenously expressed miR-134 (Figure 2
A), suggesting that the miR-30a loop might have some influence on Dicer processing. Therefore, we examined the constructs of single stranded terminal loops originating from either the miR-30a hairpin (miRNA134.1) or the miR-134 hairpin (miRNA134.2) for processing. Using northern blot, we found that miRNA134.2 expressed mature miR-134 after transfection into HEK293 cells (Figure 2
D). However, the product displayed a slightly smaller size than that expressed from the miR134.1 construct run in parallel resembling more the size of the endogenous miR-134 (Figure 2
A). The dsRNA stem in the miRNA134.2 construct is 2 nts shorter than in miRNA134.1, and thus both the nature of the single stranded loop and the length of the dsRNA stem could influence processing. After transfection into primary cortical neurons we were able to detect miR-134 expression from the miRNA134.2 construct using qPCR (Figure 2
E), though we observed a lower degree of overexpression compared to miRNA134.1 (Figure 2
B). In summary, our results show that the Microprocessor recognition element originating from the miR-30a hairpin is sufficient to ensure processing of the chimeric hairpins independent of the nature of the single stranded terminal loop.
Efficient production of neural miRNAs from the engineered chimeric hairpins can be achieved irrespective of the individual miRNA sequence inserted into the miR-30a backbone. This indicates that most, if not all, miRNAs can be efficiently expressed by our chimeric hairpin approach.
Functionality of microRNAs Expressed from Chimeric Hairpins
Next we asked whether the miRNA products expressed from the chimeric hairpins could recognize target sites in the 3′-UTR of target mRNAs and thereby regulate their translation and/or abundance. We performed luciferase assays using luciferase reporter constructs in which the 3′-UTR of selected target mRNAs (LimK1, Hs3st2) had been cloned downstream of the luciferase coding region. These constructs were co-transfected along with the chimeric hairpins into primary cortical neurons, and luciferase activity was measured 3 days later. To validate the functionality of the product expressed from the miRNA134.1 clone we used a luciferase construct, pGL3-LimK1-3′UTR-134pbds, carrying the LimK1 3′-UTR in which the miR-134 target site had been mutated from an imperfect to a perfect match binding site. This perfect match binding site makes the construct more susceptible to binding of miR-134, and therefore should be more efficiently regulated than the construct carrying the wild type miR-134 target site. Using pGL3-LimK1-3′UTR-134pbds, we observed a reduction of luciferase activity upon miRNA134.1 co-transfection compared to control1.1 and control2.1 transfected cells (50% and 42%, respectively) (Figure 3
A). Therefore, the product expressed from the miRNA134.1 construct efficiently targeted the miR-134 perfect match binding site in the luciferase construct. On the other hand, we did not detect reduced luciferase activity of the pGL3 LimK1 3′UTR reporter, which contains the wild type miR 134 target site, when co-transfecting it along with the miRNA134.1 construct (Figure 3 in Supplementary Material). To investigate the effects on endogenous target protein levels, we transduced 10–11 DIV primary hippocampal neurons with rAAV carrying the miRNA134.1 construct and prepared cell extracts 8 days later. We used western blot analysis to determine protein levels of the two validated miR-134 target genes, LimK1 and Pum2 (Schratt et al., 2006
; Fiore et al., 2009
). We observed reduced Pum2 protein levels compared to the control conditions (Figure 3
B). This strongly suggests that the product of the miRNA134.1 construct is a functional miR-134, which most likely binds directly to the cognate target site in the 3′-UTR of the Pum2 transcript, thereby inhibiting Pum2 protein production. We further detected a less consistent but moderate reduction in the LimK1 protein levels by western blot (data not shown).
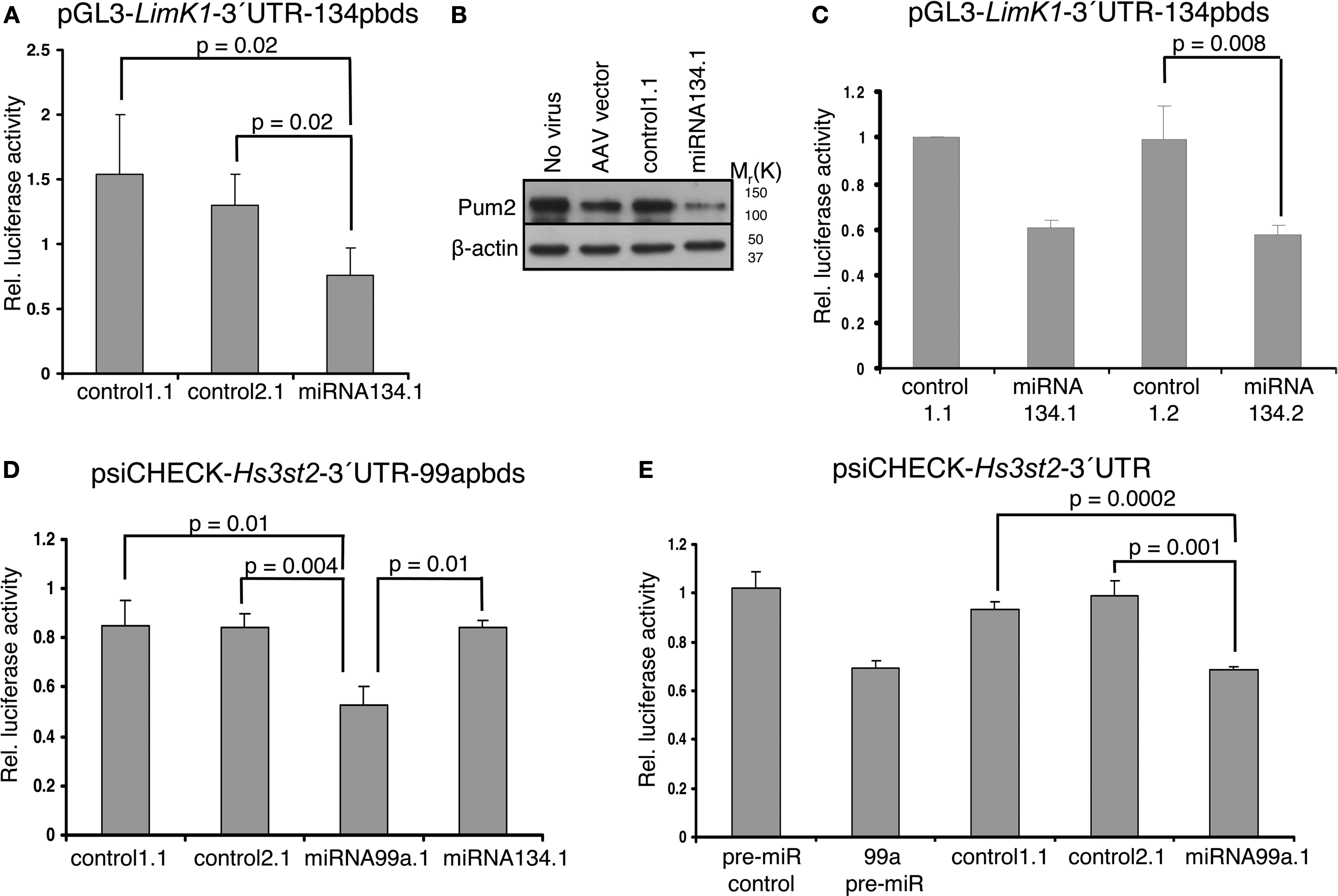
Figure 3. Functionality of products expressed from miRNA134. 1, 2 and miRNA99a.1. (A,C) Products of the miRNA134.1 (A) and miRNA134.2 (C) constructs regulate expression of a luciferase reporter construct carrying a miR 134 perfect binding site. The indicated plasmids (or AAV vector) were co transfected along with the pGL3-LimK1-3′UTR-134pbds construct into primary cortical neurons for luciferase assay analysis (see Materials and Methods for details). Values are expressed relative to the internal Renilla luciferase activity and normalized to the activity of the luciferase reporter when co-transfected with (A) AAV vector and (C) control1.1, which both are arbitrarily set to 1. Results are shown as means + s.d. [(A): n = 4; (C): n = 3]. Indicated p values were calculated using Student′s t-test. (B) miR-134 expressed from the miRNA134.1 construct affects the Pum2 protein levels. 10–11 DIV hippocampal neurons were transduced with purified rAAV carrying the indicated plasmids. Eight days later whole cell extracts were prepared and subjected to western blot analysis probing with Pum2 antibody and a β-actin antibody as loading control. Here we show one out of three representative blots, on average displaying a significant 64% reduction in Pum2 protein levels. (D,E) miR-99a expressed from miRNA99a.1 regulates expression of luciferase reporter constructs carrying a miR-99a perfect binding site (psiCHECK-Hs3st2-3′UTR-99apbds) (D) as well as a construct carrying the Hs3t2-3′UTR containing a wild type miR-99a target site (psiCHECK-Hs3st2-3′UTR) (E). The indicated plasmids, AAV vector or synthetic pre-miRs were co-transfected along with luciferase reporter constructs into primary cortical neurons for luciferase assay (see Materials and Methods for details). Values are expressed relative to the internal Firefly luciferase activity and normalized to the activity of the luciferase reporter when co-transfected with (D) AAV vector and (E) synthetic pre-miR control, which arbitrarily are set to 1. Results are shown as means + s.d. [(D): n = 3; (E): n = 3]. Indicated p-values were calculated using Student’s t-test.
We next wanted to test the functionality of miR-134 expressed from miRNA134.2, which yielded slightly less mature miR-134 as detected by qPCR (Figure 2
E vs. Figure 2
B), but of similar size as endogenous miR-134. Therefore, we co-transfected the miRNA134.2 construct and its control, control1.2, along with the luciferase reporter construct pGL3-LimK1-3′UTR-134pbds into primary cortical neurons. Here, we detected a decrease of luciferase activity (∼41%) in the miRNA134.2 condition compared to the control1.2 condition (Figure 3
C). Interestingly, despite the lower expression levels, the product expressed from the miRNA134.2 clone was equally efficient in repressing luciferase activity compared to the miRNA134.1 construct assayed in parallel. This suggests that at least some of the product expressed from miRNA134.1 might be less functional, possibly due to aberrant Dicer processing leading to a slightly longer miR-134 form.
A putative target gene of miR-99a is heparan sulfate (glucosamine) 3-O-sulfotransferase 2 (Hs3st2) predicted by the microRNA.org resource (Betel et al., 2008
). We cloned the Hs3st2 3′UTR downstream of the luciferase coding region on psiCHECK-2, resulting in the psiCHECK-Hs3st2-3′UTR construct. We then mutated the miR 99a putative binding site to a miR-99a perfect binding site and initially used this new construct, psiCHECK Hs3st2 3′UTR 99apbds, to validate the functionality of the product expressed from the miRNA99a.1 clone. The product expressed from the miRNA99a.1 construct was capable of reducing luciferase activity (∼38%) compared to products expressed from control1.1, control2.1 and miRNA134.1 assayed in parallel (Figure 3
D). In addition, the product of the miRNA99a.1 construct targeted the miR-99a wild type binding site in the Hs3st2-3′UTR, since co-transfecting of the psiCHECK-Hs2st2-3′UTR construct along with miRNA99a.1 into primary cortical neurons similarly reduced luciferase activity compared to co-transfection of control1.1 and control2.1 (Figure 3
E). The observed reduction was comparable to the reduction of luciferase activity obtained when co-transfecting synthetic pre-miR 99a along with the luciferase construct. This implies that Hs3st2 is indeed a target of miR-99a, and more importantly, that the miRNA99a.1 construct expresses functional miR-99a. Taken together, we could demonstrate efficient expression of two neuronal miRNAs, miR-134 and miR-99a, with our approach.
miR-134 Expression Impairs Dendritogenesis In Vivo
Infection with the virus particles described here should in principle allow for stable miRNA overexpression in postmitotic neurons in vivo, thereby providing a tool to study the physiological roles of neuronal miRNAs in vivo. We initially focused on miR-134, since this miRNA has been shown to play important roles in the regulation of neuronal morphology in primary neuronal cultures, yet its in vivo role is unknown (Schratt et al., 2006
; Fiore et al., 2009
). For these studies we used purified rAAV carrying the miRNA134.1 construct as well as rAAV carrying either the AAV vector or the control1.1 construct as negative controls. We injected the virus particles into the lateral ventricles of P0 mice and harvested their brains after 3 weeks for analysis. To study the effect of miR-134 on dendritogenesis, we focused on cortical layer V pyramidal neurons since the density of viral infection in the hippocampal granular layer was too high to permit the effective analysis of individual neurons. Projection images of the cortical layer V pyramidal neurons (Figure 4
A) were analyzed using Sholl analysis (see Materials and Methods for details). Only the basal dendritic arbor was considered, since the large apical dendrites were often mechanically severed during the cutting of the brains. We detected an 11% and 13% reduction in dendritic complexity (as expressed by total dendritic length) of basal dendrites of the cortical layer V pyramidal neurons infected with miRNA134.1 compared to neurons infected with the AAV vector or control1.1, respectively (Figure 4
B). The reduction was seen along the length of the dendrites as illustrated by the Sholl profile in Figure 4
C. Thus, miR-134 overexpression interferes with dendritogenesis of cortical layer V pyramidal neurons, suggesting an important function of miR-134 in neuronal morphology in vivo. Notably, ectopic expression of miR-134 in dissociated hippocampal neurons similarly interfered with activity-dependent growth of dendrites (Fiore et al., 2009
).
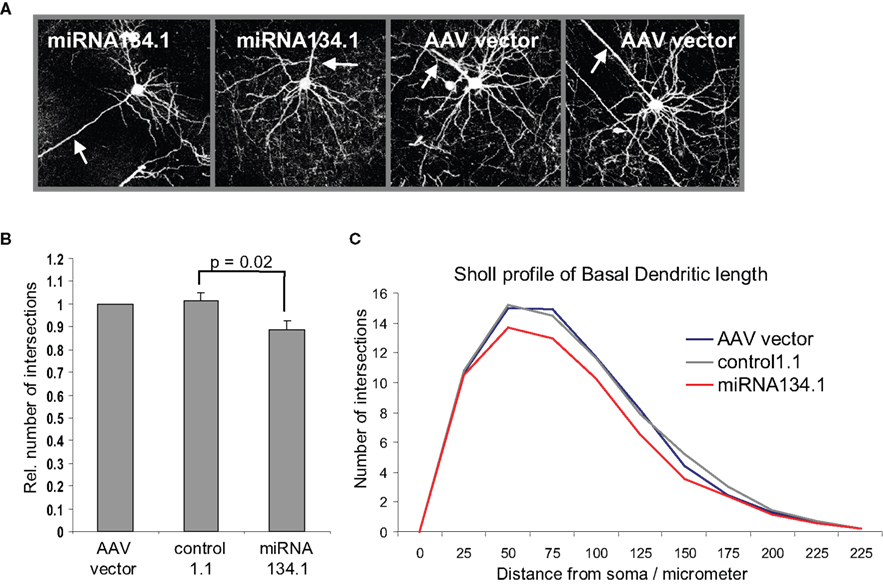
Figure 4. miR-134 affects dendritogenesis in vivo. P0 mice were injected with purified rAAV carrying miRNA134.1, AAV vector or control1.1. Brains were harvested at P21, fixed ON and cut into 100 μm coronal brain sections. The sections were immunostained using an eGFP antibody and mounted for confocal microscopy. (A) Representative projection images of cortical layer V pyramidal neurons infected with AAV vector or miRNA134.1. Arrows point out the location of the primary apical dendrite for orientation. (B) miR-134 negatively affects dendritogenesis in vivo. Dendritic branching of cortical layer V pyramidal neurons was analyzed by Sholl analysis (see Materials and Methods for details), only considering the basal dendrites. The number of dendritic intersections is normalized to the number of intersections of neurons infected with AAV vector, which is arbitrarily set to 1. Results are shown as means + s.d. (n = 3 independent experiments; at least six neurons each from two to three different brains per experiment; (a) AAV vector: 9 brains, 69 neurons, (b) control1.1: 8 brains, 61 neurons, (c) miRNA134.1: 8 brains, 57 neurons.). Indicated p-value was calculated using Student’s t-test. (C) Sholl profile of the basal dendritic length of cortical layer V pyramidal neurons. The number of dendritic intersections with each concentric circles is plotted against the distance of the individual circles from soma, 25 μm interval (Between-Subjects ANOVA: p = 0.002 and 0.013 comparing the miRNA134.1 condition to the control1.1 and AAV condition, respectively).
In this study, we describe a rAAV-based tool for stable expression of miRNAs, which enables studies of miRNA function in post-mitotic neurons in vitro and in vivo. We designed miRNA expressing chimeric hairpins using elements from the well characterized miR 30a hairpin known to ensure Drosha processing (Figure 1
) (Lee et al., 2003
; Zeng and Cullen, 2003
), thereby bypassing our limited knowledge about sequence requirements for Drosha processing of individual miRNAs. We tested our designs on two neuronal miRNAs, miR-134 and miR-99a, which were both efficiently processed (Figures 2
A,C). Therefore, the processing of the designed chimeric hairpins seems completely independent of the inserted miRNA sequences, thereby likely allowing efficient processing and expression of any miRNA of interest.
In the interest of guiding miRNA expression to all neuronal cell types in vivo, we positioned the chimeric hairpins under the control of the synapsin promoter within the AAV vector (Shevtsova et al., 2005
). However, our system is easily adaptable for diverse studies in the postnatal mammalian brain. For example, the delivery of miRNAs can be directed to specific brain regions based on promoter choice. Furthermore, the introduction of inducible promoters (Guo et al., 2008
; Stieger et al., 2009
) would allow a precise temporal control over miRNA expression, thereby facilitating studies on synaptic plasticity which are often confounded by developmental effects. Compared to classical genetics, the AAV technology offers an acute and inexpensive alternative for the study of neuronal miRNA function. In principle, the same platform can be used to introduce vector based competitive miRNA inhibitors, such as miRNA sponges containing several miRNA target sites in tandem (Ebert et al., 2007
). Our tool, in addition, improves miRNA studies in vitro using primary neurons as a model system. In contrast to conventional transfection-based methods, rAAV infection makes primary neurons amenable to biochemistry, as illustrated here by the detection of reduced Pum2 protein levels by western blot after the introduction of miR-134 (Figure 3
B).
miRNA target sites can be divided into two categories. Category one targets mainly rely on strong base pairing to the 5′-end of miRNAs (7mer seed match) whereas category two targets display weak seed pairing compensated by strong base pairing to the 3′ end (Brennecke et al., 2005
; Bartel, 2009
). miR-134 and miR 99a expressed from the chimeric hairpins showed affinity towards their target sites in the 3′-UTR of Pum2 (Fiore et al., 2009
) and Hs3st2 (Figures 3
B,E), respectively, both of which belong to category one targets. This observed functionality reflects correct 5′-end Drosha processing of the miRNAs. The miR-134 target site in the LimK1 3′ UTR (Schratt et al., 2006
) is a category two target site. We detected minimal regulatory effect on this target gene by miR-134 expressed from its chimeric hairpin (data not shown). One could speculate that category two targets are lower affinity targets than category one targets, and if so, the obtained expression of miRNAs from our hairpins might not be strong enough to ensure regulation of category two targets. To increase the miRNA expression levels, improvements in hairpin design, changing the location of the chimeric haripins in the 3′-UTR of eGFP and/or insertion of more than one copy of the chimeric hairpins could be alternative options.
Processing of the chimeric hairpins only relied on the Drosha site from miR-30a hairpin independently of the nature of the terminal loop (Figure 2
D). This allows replacement of the terminal loop, if its identity is of biological relevance. We observed different sizes of miR-134 expressed from constructs containing either the terminal loop from the miR-30a hairpin, miRNA134.1, or from the miR-134 hairpin, miRNA134.2 (Figure 2
D). This probably reflects differences in Dicer processing, either due to differences in length of the dsRNA stem of the two constructs or the nature of their terminal loops. miR-134 expressed from the miRNA134.2 construct resembled more closely the size of endogenous miR 134, whereas a significant fraction of miR-134 expressed from the miRNA134.1 was slightly larger, ∼2 nts longer (Figures 2
A,D). Since the two constructs regulated luciferase expression to a similar extent (Figure 3
C), equal levels of functional miR-134 appears to be expressed from the two clones. Two possibilities could explain our findings. First, miRNA134.1 expresses two isoforms of miR-134 of different lengths, and only the shorter form is biologically active. Second, miRNA134.1 expresses higher levels of the longer miR-134 isoform that is only partially active. Further studies are required to distinguish between these possibilities.
Here, we provide evidence that miR-134 expression impairs dendritogenesis of mouse cortical layer V pyramidal neurons in vivo. To our knowledge, this is the first demonstration of an in vivo role of a specific miRNA in the control of neuronal morphology in post mitotic neurons. In analogy, Mef2 induction of miR-134 is required for activity dependent dendritogenesis of primary hippocampal neurons in culture, an effect mediated by miR-134 dependent downregulation of Pum2 protein levels (Fiore et al., 2009
). Interestingly, both overexpression and inhibition of miR 134 impair activity-dependent dendritogenesis, implying that the role of miR-134 is to fine-tune Pum2 levels within a physiologically beneficial level. Given that in vivo overexpression of miR-134 impaired dendritogenesis under normal developmental conditions, neurons within cortical circuits in vivo probably receive sufficient synaptic input to reveal the activity-dependent function of miR-134. Whether the effect of miR-134 on dendritogenesis in vivo is also due to deregulated Pum2 expression remains to be investigated. Surprisingly, in preliminary experiments we found no significant effect on spine morphology of hippocampal and cortical layer V pyramidal neurons in vivo after delivery of miR-134 expressed from miRNA134.1 (data not shown). This could be explained by our findings that LimK1, whose regulation by miR-134 is critical for spine growth, is not efficiently regulated by rAAV-expressed miR 134 (data not shown). The clarification of this and other issues will have to await the development of miR-134 loss-of-function tools.
In summary, we present a tool for the stable delivery of any miRNA in vivo. This tool should be extremely useful for future studies addressing the role of miR-134 and other miRNAs in nervous system function.
The authors declare that the research was conducted in the absence of any commercial or financial relationships potentially constructing a conflict of interest.
We kindly thank M. Veith for providing the pGL3-LimK1-3′UTR 134pbds construct. pAAV-6P-SEWB and helper plasmids, pDP1 and pDP2, were gifted by M. Schwarz, MPIMF Heidelberg, and we gratefully thank him for the initial help with rAAV production and in vivo injection. R. Fiore performed rAAV titer measurements. We thank T. Wüst and D. Maeda for excellent technical assistance. This work was supported by grants from the Danish Medical Research Council (S. Kauppinen), the Deutsche Forschungsgemeinschaft (SFB488; G. Schratt), the National Institute on Drug Abuse (1R21DA025102-01; G. Schratt), and the Human Frontier Science Program (Career Developmental Award; G. Schratt).
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuralcircuits/paper/10.3389/neuro.04/016.2009/
Abelson, J. F., Kwan, K. Y., O’Roak, B. J., Baek, D. Y., Stillman, A. A., Morgan, T. M., Mathews, C. A., Pauls, D. L., Rasin, M. R., Gunel, M., Davis, N. R., Ercan-Sencicek, A. G., Guez, D. H., Spertus, J. A., Leckman, J. F., Dure, L. S.,4th, Kurlan, R., Singer, H. S., Gilbert, D. L., Farhi, A., Louvi, A., Lifton, R. P., Sestan, N., and State, M. W. (2005). Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science 310, 317–320.
Hébert, S. S., Horre, K., Nicolai, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N., Kauppinen, S., Delacourte, A., and De Strooper, B. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–6420.
Passini, M. A., Watson, D. J., Vite, C. H., Landsburg, D. J., Feigenbaum, A. L., and Wolfe, J. H. (2003). Intraventricular brain injection of adeno-associated virus type 1 (AAV1) in neonatal mice results in complementary patterns of neuronal transduction to AAV2 and total long-term correction of storage lesions in the brains of beta-glucuronidase-deficient mice. J. Virol. 77, 7034–7040.
Siegel, G., Obernosterer, G., Fiore, R., Oehmen, M., Bicker, S., Christensen, M., Khudayberdiev, S., Leuschner, P. F., Busch, C. J., Kane, C., Hubel, K., Dekker, F., Hedberg, C., Rengarajan, B., Drepper, C., Waldmann, H., Kauppinen, S., Greenberg, M. E., Draguhn, A., Rehmsmeier, M., Martinez, J., and Schratt, G. M. (2009). A functional screen implicates microRNA 138 dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat. Cell Biol. 11, 705–716.