1
Department of Biology and Volen Center for Complex Systems, Brandeis University, Waltham, MA, USA
2
Department of Physiology and Neuroscience, New York University School of Medicine, New York, NY, USA
Injection of NMDAR antagonist into the thalamus can produce delta frequency EEG oscillations in the thalamocortical system. It is surprising that an antagonist of an excitatory neurotransmitter should trigger such activity, and the mechanism is unknown. One hypothesis is that the antagonist blocks excitation of GABAergic cells, thus producing disinhibition. To test this hypothesis, we investigated the effect of NMDAR antagonist (APV) on cells of the nucleus reticularis (nRT) in rat brain slices, a thalamic nucleus that can serve as a pacemaker for thalamocortical delta oscillations and that is composed entirely of GABAergic neurons. We found, unexpectedly, that nRT cells are hyperpolarized by APV. This occurs because these cells have an unusual form of NMDAR (probably NR2C) that contributes inward current at resting potential in response to ambient glutamate. The hyperpolarization produced by APV is sufficient to deinactivate T-type calcium channels, and these trigger rhythmic bursting at delta frequency. The APV-induced delta frequency bursting is abolished by dopamine D2 receptor antagonist, indicating that dopamine and NMDAR antagonist work synergistically to stimulate delta frequency bursting. Our results have significant implications concerning the electrophysiological basis of schizophrenia and bring together the NMDAR hypofunction, dopamine, and GABA theories of the disease. Our results suggest that NMDAR hypofunction and dopamine work synergistically on the GABAergic cells of the nRT to generate the delta frequency EEG oscillations, a thalamocortical dysrhythmia (TCD) in the awake state that is an established abnormality in schizophrenia.
In rats, injection of NMDAR antagonist into the thalamus can make regions of the cortex become overactive, as indicated by immediate early gene activation (Vaisanen et al., 2004
) and delta frequency oscillations (Buzsaki, 1991
). Consistent with this, NMDAR antagonist leads to activation of the human thalamus (Langsjo et al., 2003
). It is unclear how an antagonist of an excitatory neurotransmitter can produce such action. NMDARs can excite inhibitory cells (Grunze et al., 1996
), and it has therefore been suggested that antagonist might reduce firing in the GABAergic cells in the nucleus reticularis of the thalamus (nRT), and, by disinhibition, increase firing in principal cells (Sharp et al., 2001
). However, the fact that NMDAR antagonist also induces c-fos expression in the nRT, is not consistent with these cells becoming inactive (Vaisanen et al., 2004
). To resolve this issue, we have recorded directly from the nRT and observed the effects of NMDAR antagonist on firing patterns.
An additional motivation for our work is the relevance of NMDAR antagonist action to schizophrenia. The NMDAR hypofunction theory of schizophrenia has strong experimental support, notably the ability of NMDAR antagonist to produce many symptoms of the disease when given to normal subjects (Krystal et al., 1994
; Umbricht et al., 2000
). A well-established symptom of schizophrenia is enhanced delta oscillations in the EEG and MEG (Boutros et al., 2008
). Thus the fact that such oscillations can be induced in cortex by injection of NMDAR antagonist into the thalamus is of considerable interest. Such slow oscillations, which normally occur during slow-wave sleep (McCormick and Bal, 1997
), may generate symptoms of neuropsychiatric disease if they occur during wakefulness (Llinas et al., 1999
).
Brain Slices Preparation
Long-Evans rats (3 weeks old, Charles River, Wilmington, MA, USA) were housed under a 12-h light/dark cycle in a temperature- and humidity-controlled environment with free access to food and water. The animals were sacrificed under fluothane anesthesia. The brains were rapidly removed and were cut into 300–350 μm thick horizontal slices with a vibratome (Leica VT 1000S, Nussloch, Germany) in an oxygenated ice-cold solution containing (in mM): NaCl 124, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, Dextrose 10, CaCl2 2.5, and MgSO4 4. Slices containing the nRT were collected and incubated for at least 1 h before transferring into the chamber for recording. Experimental protocols were approved by the institutional animal care and use committees at the Brandeis University.
Electrophysiology
Patch electrodes had resistances of 3–5 MΩ. For current-clamp recording, electrodes were filled with an internal solution containing (in mM): KMeSO4 120, KCl 12, MgCl2 1, EGTA 2, CaCl2 0.2, HEPES 10, Mg-ATP 2, and Na-GTP 0.4. pH was adjusted to 7.2–7.4 with KOH. For voltage-clamp recording, the internal solution contained (in mM): CsCl 43, CsMeSO4 92, TEA 5, EGTA 2, MgCl2 1, HEPES 10, and ATP 4. pH was adjusted to 7.2–7.4 with CsOH, and the final osmolarity was ∼290 mOsm. Brain slices were immersed in oxygenated artificial cerebrospinal fluid with a flow rate of 2–3 ml/min. The nRT was visually identified using dark field illumination and a CCD camera. Both voltage-clamp and current-clamp recordings were performed with an Axopatch 200B amplifier (Molecular Devices, Foster City, CA, USA). Signals were digitized at a sampling rate of 5 kHz and were filtered at 2 kHz using a data acquisition program (Igor Pro 5.0, Wavemetrics, Oregon, USA). The suitability of voltage-clamp in nRT cells was verified previously by studying the kinetics of transient-inward T-type Ca2+ currents (Sun et al., 2001
), indicating that nRT cells are electronically compact enough for voltage-clamp analysis. Liquid junction potential was corrected offline in current-clamp recording. The recordings were performed at ∼33°C. For extracellular single-unit recording, a glass electrode filled with ACSF (<1 MΩ resistance) was placed in the nRT. Single-unit activity was identified by the stable amplitude of spikes.
The decay of NMDAR-EPSC was fit by the sum of two exponential functions: A(t) = Aslowexp(−t/τslow) + Afastexp(−t/τfast), in which τslow and τfast are the decay time constants of the slow and fast component and Aslow and Afast are their respective amplitudes. The decay time constants were averaged using τslow[Aslow/(Aslow + Afast)] + τfast[Afast/(Aslow + Afast)]. The I-V curves of the NMDAR-EPSCs were fit by the equation: I = [agmax(V−Vr)]/[a + [Mg2+]oexp(−VδZF/RT)], in which V is holding potential, Vr is reversal potential, gmax is the conductance at +40 mV, a is the dissociation constant in the absence of transmembrane voltage, and δ represents the fraction of membrane voltage at the blocking site (Anchisi et al., 2001
).
Dopamine Depletion
The depletion of catecholamine was achieved as previously described (O’Donnell and Grace, 1993
; Otmakhova and Lisman, 1996
). In brief, rats were injected with reserpine (s.c., 5 mg/kg) 24 h before sacrificing to deplete catecholamine stores. Then, at least 1 h before recordings, 100 μM of tyrosine hydroxylase inhibitor, DL-α-methyl-π-tyrosine methyl ester hydrochloride, was added to ACSF to block new synthesis of dopamine and noradrenalin. The inhibitor was present throughout the incubation and experiments. The depletion of dopamine by >90% was verified previously by high-performance liquid chromatography (O’Donnell and Grace, 1993
).
Statistics
The data are presented as mean ± standard error. The two-tailed paired t test was used for two-group comparisons. ANOVA followed by Tukey’s test was used for multigroup comparisons. The difference was considered significant when P < 0.05.
Whole-cell recordings were made from the nRT GABAergic neurons of 2- to 3-week-old rats. The average resting membrane potential was −70.3 ± 0.8 mV. Application of NMDAR antagonist, DL-2-Amino-5-phosphonopentanoic acid (APV, 50 μM), hyperpolarized these cells by −6.4 ± 1.3 mV (P < 0.05, n = 13, Figures 1
A,B). Hyperpolarization is known to alter the firing mode of nRT neurons. At normal resting potential, cells fire in tonic mode. If cells are hyperpolarized by current injection, this deinactivates T-type Ca2+ channels (Llinas and Jahnsen, 1982
; Huguenard and Prince, 1992
), which allows rhythmic bursting (Llinas and Geijo-Barrientos, 1988
; Contreras et al., 1993
) (Figure 1
C). We thus examined whether APV-induced hyperpolarization was able to induce bursting. Figure 1
D shows that the firing mode is indeed changed from tonic spiking to bursting when the cell is hyperpolarized by APV (8 out of 13 cells).
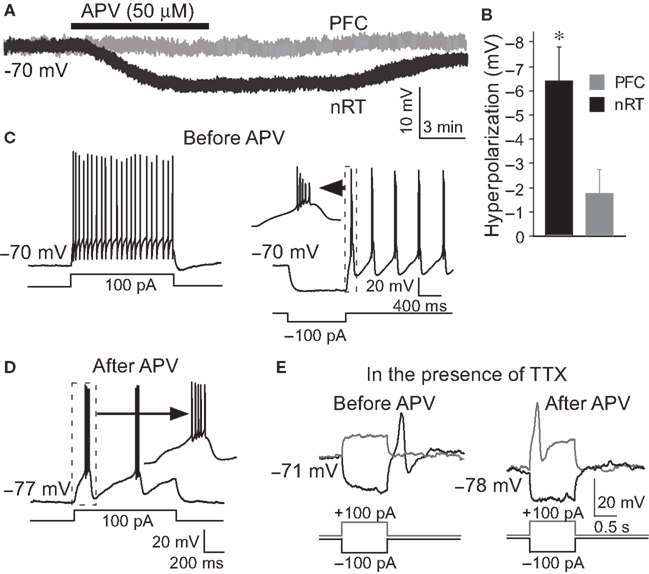
Figure 1. APV hyperpolarized nRT GABAergic neurons and changed their firing mode. (A) 50 μM APV hyperpolarized nRT GABAergic neurons, but not PFC pyramidal neurons. (B) Group data show quantification of APV-induced hyperpolarization in nRT and PFC. (C) Before APV application, a depolarizing pulse induced tonic spiking (left panel), whereas hyperpolarization induced a rebound burst (right panel). (D) In the presence of APV, the same depolarizing current induced bursting activity. (E) In 0.5 μM tetrodotoxin (TTX), before APV application, hyperpolarizing pulse induced a rebound calcium spike; a depolarizing pulse did not induce active response in nRT neurons (left panel). After APV was applied, a depolarizing pulse induced a calcium spike (right panel).
We further examined the subset (5 of 13 cells) of cells in which NMDAR antagonist did not induce bursting. In those cells, a very small additional hyperpolarization (−3.2 ± 1.2 mV) by negative current injection could induce bursting. This is in contrast to the situation before APV application when a larger hyperpolarization was required to induce bursting (−8.1 ± 0.5 mV, P < 0.01).
Blocking NMDARs has several consequences that could produce bursting, for example reducing calcium influx (Murthy et al., 2000
) or inhibiting protein synthesis (Sutton et al., 2006
). On the other hand, the bursting might arise simply because of the hyperpolarization and the resulting deinactivation of T channels (Jahnsen and Llinas, 1984b
). To clarify this issue, we injected positive currents to reverse the APV-induced hyperpolarization without removing the NMDAR blockade. As shown in Figure 2
, this switched the cell back to tonic firing mode. Taken together, these results argue that APV facilitates bursting activity in nRT neurons by hyperpolarizing resting potential.
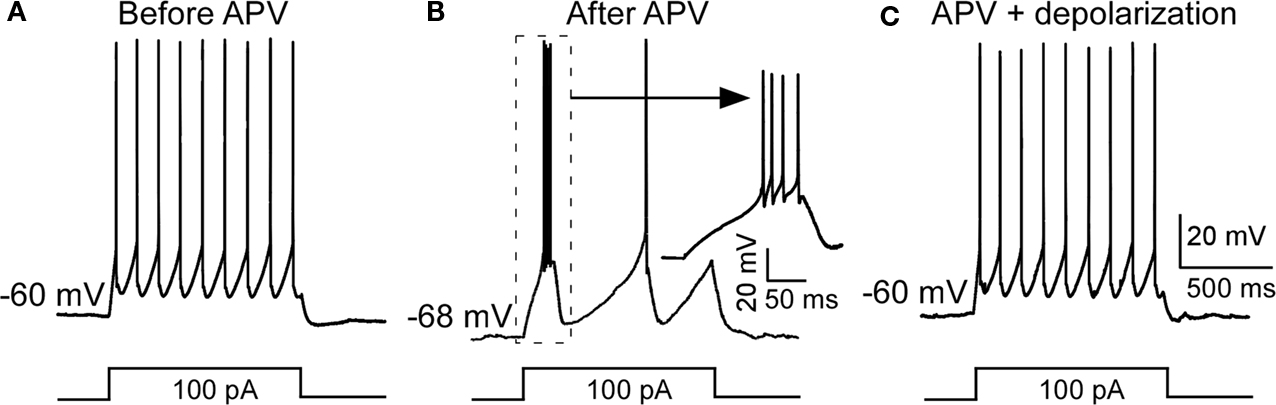
Figure 2. Depolarization restored APV-induced change in firing mode. (A) A depolarizing pulse induced tonic spike before APV application. (B) APV hyperpolarized membrane potential and induced burst firing. (C) Positive DC current injection reversed the APV-induced hyperpolarization and restored firing to the tonic mode.
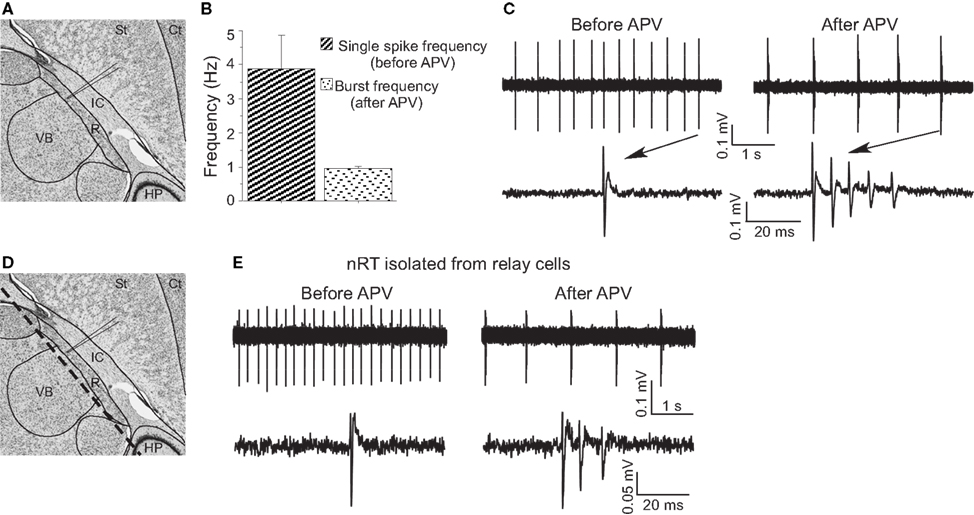
Figure 3. APV-induced delta frequency bursting was studied using extracellular recording. (A) Recording site in horizontal brain slices (edited from http://brainmaps.org/
). St, striatum; Ct, cortex; IC, internal capsule; VB, ventrobasal complex; HP, hippocampus; R, nucleus reticularis of the thalamus (nRT). (B) Group data show single-spike frequency before APV application and burst frequency after APV application. (C) Representative traces show APV-induced delta frequency oscillation (upper panels). Spikes are shown with higher temporal resolution (lower panels). (D) The same schematic drawing as in (A) shows the dashed line where knife cuts were made to isolate nRT from relay cells. (E) APV also changed firing mode in surgically isolated nRT.
To further study the effect of APV on firing properties, we used extracellular recording (Figure 3
A) because this gives a better indicator of normal firing patterns than does whole-cell recording (which can lead to changes in intracellular concentrations). Under control conditions, cells fired tonic spikes at 3.88 ± 0.99 Hz (n = 5) (Figure 3
C), consistent with previous work (Bal and McCormick, 1993
). In APV, this single-spike pattern was replaced by rhythmic bursting (five out of five experiments) (Figure 3
C). Each burst contained three to five spikes, with an interspike interval less than 10 ms, comparable to burst properties in vitro ((Llinas and Geijo-Barrientos, 1988
) and Figure 1
C) and in vivo (Contreras et al., 1993
). The burst frequency (1.01 ± 0.11 Hz) in APV was in the delta frequency range (Figure 3
B). Such spontaneous bursting was not seen in whole-cell recording, presumably because of washout of some essential factor.
To determine whether network properties are involved in the oscillation, we applied APV after blocking action potentials with TTX (0.5 μM). Under these conditions, APV still produced a hyperpolarization (−5.8 ± 1.8 mV, n = 6, P < 0.05) and allowed generation of the Ca2+ spike that underlies bursting (Figure 1
E). As an additional test (Figure 3
D), we made a knife cut in slices to isolate nRT cells from relay cells. In the isolated nRT, the firing rate before APV application (4.1 ± 0.5 Hz) and the bursting after APV (1.0 ± 0.2 Hz, n = 3) were comparable to those in slices made without the knife cut (Figure 3
E). Recordings from relay cells showed a comparable hyperpolarization, but usually did not show bursting properties in APV (Figure S1 in Supplementary Material). This difference may stem from different types of T channels in the two cell types (Talley et al., 1999
). Taken together, these results indicate that NMDAR antagonist produces bursting in nRT cells by a mechanism that does not require network properties.
We sought to understand why NMDAR antagonist produces a large hyperpolarization in nRT cells. Experiments in the hippocampus show that ambient extracellular glutamate is sufficient to partially activate NMDARs (Sah et al., 1989
; Herman and Jahr, 2007
; Le Meur et al., 2007
). However, near resting potential, the NR1/NR2A and NR1/NR2B receptors present in the hippocampus are blocked by Mg2+ (Sah et al., 1989
). nRT neurons contain mRNA and protein for NR2C at unusually high levels (Monyer et al., 1994
; Wenzel et al., 1997
; Karavanova et al., 2007
). This isoform has much less Mg2+ block near resting potential than do NR2A or NR2B (Monyer et al., 1992
; Kuner and Schoepfer, 1996
; Wenzel et al., 1997
). Thus, the large hyperpolarization of nRT by APV might be due to a block of a basal inward current carried by NR2C. However, nRT also contains NR2A and NR2B subunits. If NR2C dominates function, the current-voltage curve of the channels should show low rectification because the voltage-dependent Mg2+ block is low. To measure the rectification of NMDARs in the nRT, we evoked NMDAR-mediated EPSCs (Figure 4
A) in the presence of CNQX (25 μM) and bicuculline (25 μM) by stimulating the internal capsule, which primarily activates glutamatergic corticofugal fibers projecting to the nRT (Jones, 2002
). As shown in Figure 4
B, the current-voltage curve in the range of −30 to −70 mV has much less rectification in nRT neurons than in cortical neurons. For instance, at membrane potential of −60 mV, the NMDAR current (normalized to the value at +40 mV) was 0.54 ± 0.06 in nRT (n = 6), much larger than the 0.13 ± 0.03 value in cortical neurons (n = 8, P < 0.05). The value of 0.54 (which inversely relates to rectification) is in reasonable agreement with the value of 0.4 for NR1/NR2C measured in an expression system [estimated from the experiment of (Monyer et al., 1994
)]. The decay time of the EPSC in nRT neurons (243 ± 31 ms; n = 6) (Figure 4
A) is characteristic of NR2C (382 ± 45 ms), but much shorter than that of NR2D (4.8 ± 0.9 s), another subunit with low Mg2+ block (Monyer et al., 1994
). In summary, nRT cells contain NMDARs with low rectification, a property consistent with the presence of NR2C.
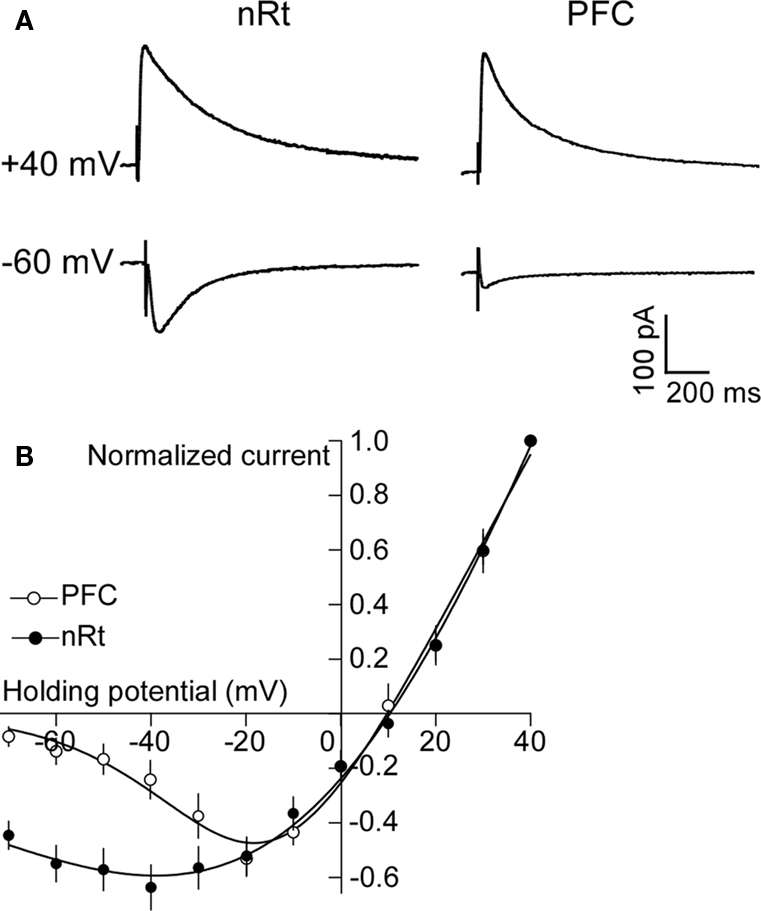
Figure 4. NMDARs in nRT neurons have low rectification. (A) Synaptically evoked NMDAR currents at +40 and −60 mV in PFC and nRT. (B) Plot of current-voltage relationship (I-V curve) of NMDAR in PFC and nRT (normalized to current at +40 mV).
In the cortex and hippocampus, NR2C is absent in pyramidal cells (though present in a subpopulation of interneurons) (Monyer et al., 1994
; Karavanova et al., 2007
). Thus, the APV-induced hyperpolarization should be smaller in cells that do not contain NR2C. Consistent with this prediction, PFC pyramidal cells show only a small hyperpolarization after APV application (−1.8 ± 0.9 mV, n = 5; P > 0.05, Figures 1
A,B), as do hippocampal pyramidal neurons (Sah et al., 1989
).
NMDAR antagonists can produce many of the symptoms of schizophrenia (Krystal et al., 1994
; Umbricht et al., 2000
). The ability of NMDAR antagonist injected into the thalamus (Buzsaki, 1991
) to evoke delta oscillations may relate to the enhancement of delta frequency EEG power in schizophrenia (Knott et al., 2001
; Jeanmonod et al., 2003
, for a meta-analysis, see Boutros et al., 2008
). Given the importance of dopamine in schizophrenia, we wondered whether dopamine has a role in APV-induced bursting. Thalamic cells contain D2-type receptors (Mrzljak et al., 1996
; Khan et al., 1998
), and the nRT in the rat is one of the few thalamic regions that are densely innervated by dopaminergic axons originating from substantia nigra (Anaya-Martinez et al., 2006
; Garcia-Cabezas et al., 2009
). Application of the D2R agonist, quinpirole (10 μM), had no effect on resting potential in nRT (−0.5 ± 1.2 mV, n = 8, P > 0.05; Figure 5
A). Surprisingly, the D2R antagonist, eticlopride (10 μM), produced a large depolarization (7.6 ± 2.5 mV, n = 7, P < 0.05; Figure 5
B). These results could be explained on the basis of D2R hyperpolarizing action in the thalamus (Lavin and Grace, 1998
) and the assumption that basal dopamine in the nRT is sufficient to saturate or nearly saturate the D2-type receptors. A lower level of basal dopamine would be expected in the ventrobasal (VB) thalamic nucleus because the density of dopaminergic axon is very much lower (Garcia-Cabezas et al., 2009
). Consistent with this prediction and a previous study (Lavin and Grace, 1998
), eticlopride (10 μM) had no effect in VB neurons (0.6 ± 1.1 mV, n = 5, P > 0.05), whereas quinpirole (10 μM) produced a hyperpolarization (−5.0 ± 1.6 mV, n = 7, P < 0.05; Figure S2 in Supplementary Material). To further test the existence of basal dopamine in the nRT, we depleted dopamine using the reserpine method (see Materials and Methods). After applying this procedure, we found that the dopamine agonist, quinpirole (10 μM), now produced a substantial hyperpolarization in nRT neurons (−6.5 ± 1.7 mV, n = 11, P < 0.05; Figure S3 in Supplementary Material). Taken together, these results indicate that, in the normal slice, D2Rs in the nRT are saturated or nearly saturated by basal dopamine.
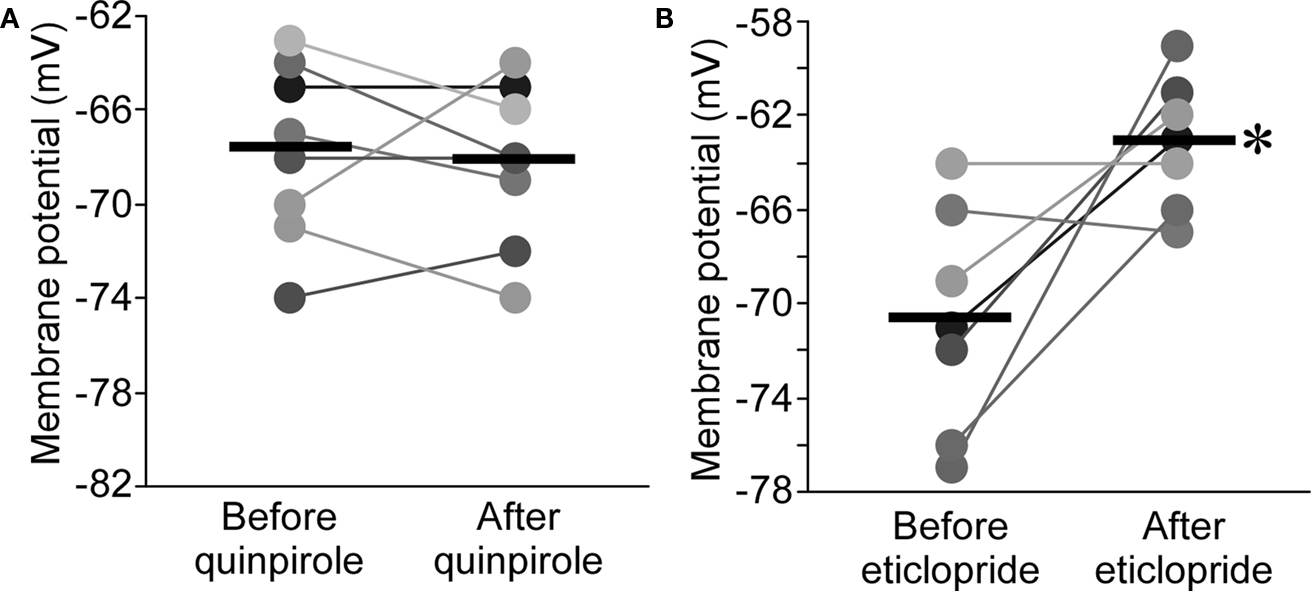
Figure 5. D2R agonist had no effects on resting potential of nRT neurons (A), whereas D2R antagonist significantly depolarized nRT neurons (B).
To explore the functional role of this basal dopamine, we tested whether dopamine antagonist could block the bursting produced by NMDAR antagonist. After the induction of bursting with APV, the application of D2 antagonist (eticlopride; 10 μM) depolarized the cell (5.2 ± 1.1 mV, n = 8, P < 0.05) and blocked bursting observed intracellularly in six out of eight experiments (Figure 6
A) and extracellularly (Figure 6
B) in four out of five experiments. These findings indicate that the synergistic action of dopamine and NMDAR antagonist is required to produce delta frequency bursting (Figure 7
). The ability of D2R antagonist to block this bursting may contribute to the therapeutic action of D2R antagonists (Neborsky et al., 1981
).
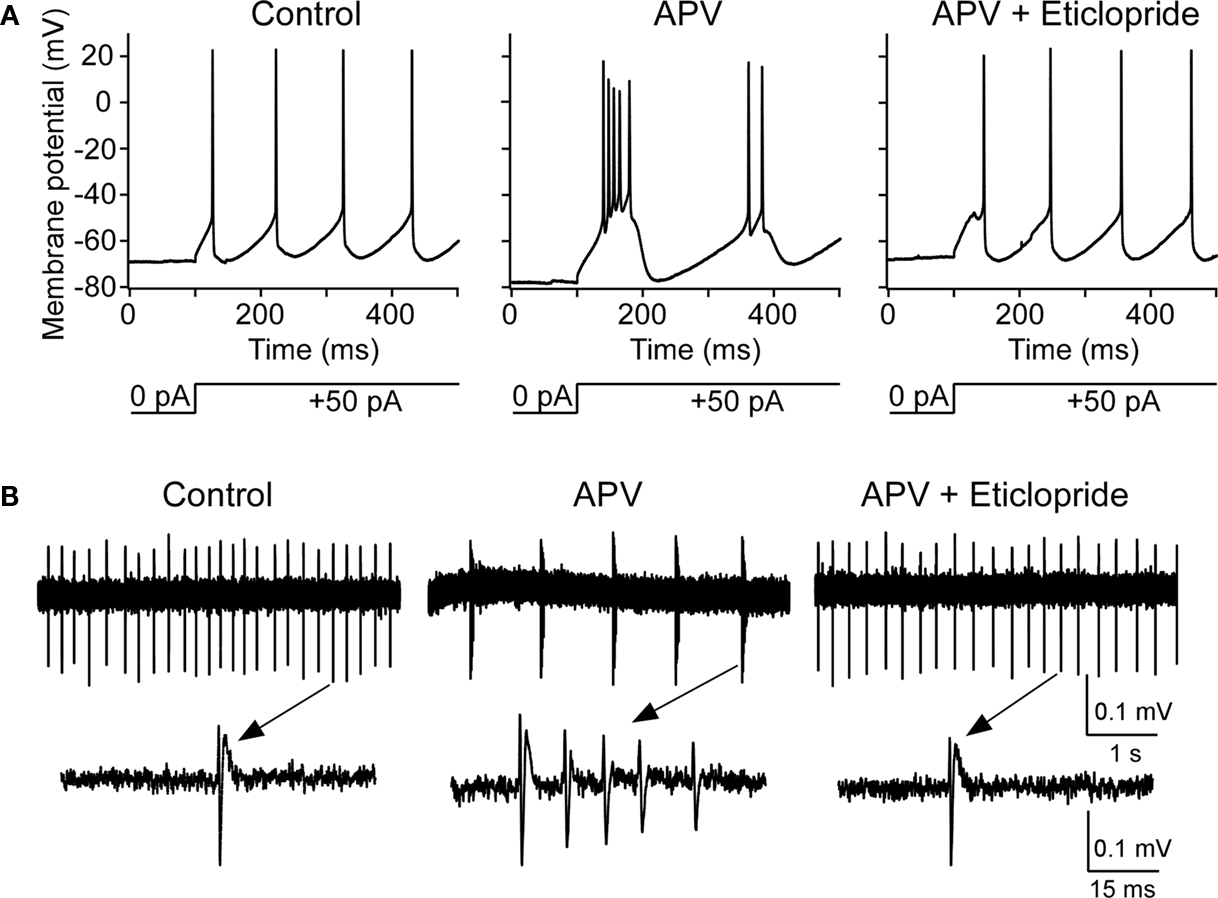
Figure 6. D2 receptor antagonist abolishes APV-induced bursting in nRT cells. (A) Intracellular studies show that, before APV application, depolarizing pulse induced tonic spikes (left panel); after APV application, the same pulse induced bursting (middle panel). After subsequent application of D2 antagonist eticlopride (10 μM), burst mode was switched back to tonic firing mode (right panel). (B) Similar to above but using extracellular recording (upper panels) as demonstrated in Figure 2
. Spikes are shown with higher temporal resolution (lower panels).
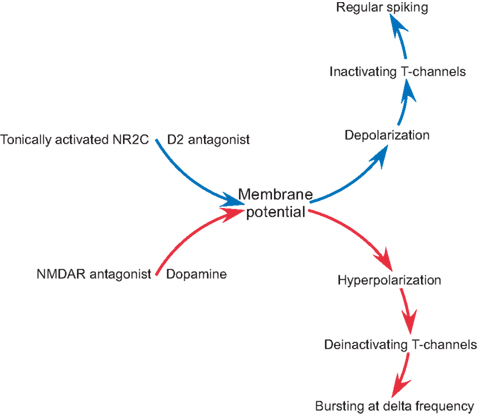
Figure 7. A diagram summarizing the role of NMDAR and dopamine in intrinsic conductance and firing pattern of nRT neurons.
Our results provide evidence for a novel mechanism by which NMDAR antagonist produces delta oscillations in the nRT. This mechanism is intrinsic to the nRT and results from a hyperpolarization produced by the antagonist. Critical for this hyperpolarization is the presence of NMDARs that have low rectification in the voltage range near resting potential. These channels, like other NMDARs, are partially activated by ambient glutamate (Sah et al., 1989
; Herman and Jahr, 2007
; Le Meur et al., 2007
). Other NMDARs (NR2A and NR2B) have strong rectification because of voltage-dependent Mg2+ block. Thus, ambient glutamate, which is mainly maintained by non-vesicular release from either glia or neuron (Featherstone and Shippy, 2008
), has little effect on resting potential because the channels are blocked by Mg2+ in this voltage range. In contrast, the NMDARs in the nRT have weak rectification and therefore contribute to resting potential. Thus, block of these channels produces hyperpolarization. This hyperpolarization has special significance in the nRT because these cells have T-type Ca2+ channels. Hyperpolarization deinactivates T-type channels and thereby allows them to produce Ca2+ spikes (which trigger bursts of action potentials, Figure 7
). The overall effect is to convert the firing mode from tonic spiking to delta frequency bursting (Jahnsen and Llinas, 1984a
; Llinas and Geijo-Barrientos, 1988
).
Several lines of evidence suggest that the unusual properties of NMDARs in the nRT are attributable to NR2C. First, the thalamus is one of the few brain regions that contain high amounts of NR2C. This has been established both by in situ hybridization (Monyer et al., 1994
; Wenzel et al., 1997
) and by knockin of an NMDAR with a beta-galactosidase marker (Karavanova et al., 2007
). The expression of this marker shows that NR2C protein is more highly expressed in the thalamus (including the nRT) than in most other brain regions. Our results show that the NMDARs in the nRT have the low-rectification characteristic of NR2C (Monyer et al., 1992
). This low rectification stems from their low Mg2+ block (Monyer et al., 1992
). NMDARs in the nRT have a deactivation time of several hundred milliseconds, similar to that of NR2C in expression systems (Monyer et al., 1992
). Such kinetics make it unlikely that the receptors channels contain NR2D (another low rectifying form) because NR2D deactivates much more slowly (in seconds). These results make it likely that NR2C is the main contributor to NMDAR function in the nRT, though some unknown modification of other NMDAR subunits cannot be excluded.
The delta frequency bursting induced by NMDAR antagonist in the nRT requires dopamine release from the dopaminergic axons that innervate the thalamus (Anaya-Martinez et al., 2006
; Garcia-Cabezas et al., 2009
). This is demonstrated by the fact that APV-induced bursting is blocked by D2R antagonist. Thus, delta bursting arises through a synergistic action of dopamine and NMDAR hypofunction.
Implications for Schizophrenia
The NMDAR hypofunction theory (Javitt and Zukin, 1991
; Coyle, 1996
; Olney et al., 1999
) of schizophrenia is supported by the fact that NMDAR antagonists produce many of the symptoms of schizophrenia when given to normal subjects (Krystal et al., 1994
; Umbricht et al., 2000
). A well-established finding in schizophrenia is that delta (and theta) power in the EEG is elevated in the awake state [for a meta-analysis, see (Boutros et al., 2008
)]. This abnormality has been observed with both EEG and MEG recording and is localized in frontal, central, and temporal cortical lobes (Llinas et al., 1999
; Fehr et al., 2001
; Rockstroh et al., 2007
; Boutros et al., 2008
). Thus, it is of considerable interest that delta oscillation in the cortex can be evoked by APV injection into the rat thalamus (Buzsaki, 1991
). It has long been thought that the nRT is a pacemaker for the thalamocortical system (Steriade et al., 1985
, 1987
; Buzsaki, 1991
; Bal and McCormick, 1993
). Recent experiments confirm the pacemaker role by making genetic deletions of SK2 or Kv3 channels in the nRT; this prevents rhythmic bursting in nRT and produces a dramatic reduction of delta power in the cortical EEG (Cueni et al., 2008
; Espinosa et al., 2008
).
We have found that APV-induced delta oscillations are produced in the nRT through processes intrinsic to the nRT. Given the pacemaker role of the nRT described above, our results thus point to the thalamus as the origin of low-frequency EEG abnormalites in schizophrenia. This is consistent with several other lines of evidence for thalamic involvement in schizophrenia (Andreasen, 1997
; Popken et al., 2000
), and the thalamic origin of EEG abnormalities in schizophrenia has been previously suggested (Sharp et al., 2001
; Behrendt, 2006
). It has further been proposed that schizophrenia is a thalamocortical dysrhythmia (Llinas et al., 1999
, 2005
), a class of disease caused by hyperpolarization of thalamic cells and the resulting low-frequency oscillation. Our results advance this hypothesis by suggesting a specific mechanism for producing the hyperpolarization and generating the dysrhythmia.
Delta oscillations normally occur throughout the cortex during slow-wave sleep (McCormick and Bal, 1997
). Their occurrence in the awake state in frontal and temporal regions is important because these oscillations could have a causal role in producing cognitive abnormalities associated with schizophrenia. Given the gating role of nRT in sensory processing (Andreasen, 1997
; Krause et al., 2003
), abnormal delta oscillations in the nRT may contribute to sensory dysfunction, e.g., prepulse inhibition (Behrendt, 2006
). During the delta oscillations of sleep, the high-frequency gamma oscillation necessary for brain function occurs during only a fraction of the slow oscillation cycle (Steriade et al., 1996
; Destexhe et al., 2007
). This is thought to contribute to impaired cognitive function during sleep and may similarly impair function either through direct action or by disinhibition of neighboring cortical regions (Llinas et al., 2005
).
Our results bring together several major theories of schizophrenia. The NMDAR hypofunction theory has substantial support (Javitt and Zukin, 1991
; Coyle, 1996
; Pilowsky et al., 2006
; Lisman et al., 2008
). NMDAR hypofunction is likely to have multiple origins (Coyle, 2006
; Allen et al., 2008
). It is noteworthy that postmortem studies show that thalamic NR1 and NR2C transcripts are markedly decreased in schizophrenia (Ibrahim et al., 2000
). The dopamine hyperfunction theory is based on the therapeutic action of D2R antagonists (Snyder, 1976
; Seeman, 1987
) and has received support from PET studies of dopamine release (Laruelle et al., 1996
). Consistent with the dopamine theory, we find that D2R antagonist blocks the delta oscillations induced by NMDAR antagonist. The GABAergic neuron theory is based on the observation that, in schizophrenia, fast-spiking GABAergic neurons have a lowered level of parvalbumin and GAD67 (Akbarian et al., 1995
; Benes and Berretta, 2001
; Lewis et al., 2001
). Our results tie together these hypotheses by showing how dopamine and NMDAR hypofunction work synergistically on the GABAergic neurons of the nRT to produce abnormal oscillatory activity (Figure 7
).
An important question that remains to be studied is how the induction of cortical oscillations by NMDAR antagonist depends on age. We have found that oscillations can be induced in the nRT in young rats, but downstream cortical damage occurs only in adult rats (Nakki et al., 1996
), consistent with the finding that psychosis is produced by NMDAR antagonists only in adults (White et al., 1982
). These findings may be related to the delayed onset of schizophrenia. In agreement with the existence of a developmental process in the thalamocortical system (Sharp et al., 1992
), bursting in nRT does not spread to relay cells in slices from young animals (Figure S1 in Supplementary Material), but does in older animals (Buzsaki, 1991
). Thus, the biophysics of the interaction between the nRT and relay cells is likely to provide insight into the mechanism of developmental processes in schizophrenia. A recent study shows that HCN channels in relay cells are developmentally regulated; thus relay cells in young animal are not able to fire rhythmic bursting because they do not have enough Ih conductance (Kanyshkova et al., 2009
). This supports the idea that the propagation of delta oscillations from the nRT to cortex may depend on the properties of relay cells, and explain the absence of delta oscillation in relay cells in young animals.
The great deal of information known about the physiology and neuromodulation of the thalamus (McCormick, 1989
; Cox et al., 1995
; Turner and Salt, 2003
) is likely to be useful in developing strategies for blocking abnormal oscillations and in elucidating the possible effects of schizophrenia candidate genes (Winterer and Weinberger, 2004
) in the generation of abnormal thalamic oscillations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict or interest.
We thank Drs. Joe Coyle and Robbie Greene for comments on the manuscript and Dr. Edwin Richard for proofreading the manuscript. This work was supported by National Institute of Health Conte Center Grant 5P50 MH060450-08 to John E. Lisman and by NIH grants NS13724 to R.R.L.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuralcircuits/paper/10.3389/neuro.04/020.2009/
Kanyshkova, T., Pawlowski, M., Meuth, P., Dube, C., Bender, R. A., Brewster, A. L., Baumann, A., Baram, T. Z., Pape, H. C., and Budde, T. (2009). Postnatal expression pattern of HCN channel isoforms in thalamic neurons: relationship to maturation of thalamocortical oscillations. J. Neurosci. 29, 8847–8857.
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., Heninger, G. R., Bowers, M. B. Jr., and Charney, D. S. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 51, 199–214.
Laruelle, M., Abi-Dargham, A., van Dyck, C. H., Gil, R., D’Souza, C. D., Erdos, J., McCance, E., Rosenblatt, W., Fingado, C., Zoghbi, S. S., Baldwin, R. M., Seibyl, J. P., Krystal, J. H., Charney, D. S., and Innis, R. B. (1996). Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl. Acad. Sci. U.S.A. 93, 9235–9240.