Watson School of Biological Sciences, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, NY, USA
The last decade has witnessed the identification of single-gene defects associated with an impressive number of mental retardation syndromes. Fragile X syndrome, the most common cause of mental retardation for instance, results from disruption of the FMR1 gene. Similarly, Periventricular Nodular Heterotopia, which includes cerebral malformation, epilepsy and cognitive disabilities, derives from disruption of the Filamin A gene. While it remains unclear whether defects in common molecular pathways may underlie the cognitive dysfunction of these various syndromes, defects in cytoskeletal structure nonetheless appear to be common to several mental retardation syndromes. FMR1 is known to interact with Rac, profilin, PAK and Ras, which are associated with dendritic spine defects. In Drosophila, disruptions of the dFmr1 gene impair long-term memory (LTM), and the Filamin A homolog (cheerio) was identified in a behavioral screen for LTM mutants. Thus, we investigated the possible interaction between cheerio and dFmr1 during LTM formation in Drosophila. We show that LTM specifically is defective in dFmr1/cheerio double heterozygotes, while it is normal in single heterozygotes for either dFmr1 or cheerio. In dFmr1 mutants, Filamin (Cheerio) levels are lower than normal after spaced training. These observations support the notion that decreased actin cross-linking may underlie the persistence of long and thin dendritic spines in Fragile X patients and animal models. More generally, our results represent the first demonstration of a genetic interaction between mental retardation genes in an in vivo model system of memory formation.
Mental retardation is a common condition affecting 3% of the population (Hagberg et al., 1981
; Shea, 2006
) and is caused by several etiologies (Aicardi, 1998
; Raymond and Tarpey, 2006
). Multiple single-gene syndromes have been identified in the last decade. Among them, Fragile X syndrome is the most frequent (Hinton et al., 1995
; Skinner et al., 2005
) and is caused by the absence of FMRP (Wohrle et al., 1992
; De Boulle et al., 1993
; Hornstra et al., 1993
; Trottier et al., 1994
), which is encoded by the FMR1 gene. As observed in human (Cianchetti et al., 1991
; Cornish et al., 1999
) and in several animal models, mutations in FMR1 homologs have yielded memory defects (Fmr1, 1994
; Maes et al., 1994
; Kooy et al., 1996
; McBride et al., 2005
; Bolduc et al., 2008
) and long thin dendritic spines (Rudelli et al., 1985
; Hinton et al., 1991
; Wisniewski et al., 1991
; Comery et al., 1997
; Irwin et al., 2002
). Consistent with the latter observation, Drosophila FMRP homologue has been linked to known actin modifying molecules such as Rac in flies (Billuart and Chelly, 2003
; Schenck et al., 2003
), profilin in flies(Reeve et al., 2005
), PAK in mice (Hayashi et al., 2007
) and Ras in mice(Hu et al., 2008
), but the molecular dysfunction underlying this defect in synaptic structure remains unknown.
Regulation of actin cytoskeleton is impaired in many human mental retardation syndromes (Inlow and Restifo, 2004
) and appears crucially involved in synaptic plasticity in various cellular models of memory (Dillon and Goda, 2005
). Similarly, induction of long-term potentiation in hippocampal neurons, a cellular model for synaptic plasticity, leads to modifications of dendritic spine shape (Engert and Bonhoeffer, 1999
; Matus, 2000
). The Filamin A gene is involved in actin cytoskeleton remodeling (Flanagan et al., 2001
; Stossel et al., 2001
). Filamin A is expressed in neurites of embryonic rat hippocampal neurons (Fox et al., 1998
). Disruption of Filamin A impairs neuronal migration probably because ligand binding no longer induces actin reorganization (Fox et al., 1998
; Bellenchi et al., 2007
). Interestingly, Filamin A mutations have been found in patients with Periventricular Nodular Heterotopia (PNH), and these patients suffer various degrees of cognitive dysfunction and epilepsy (Battaglia et al., 1997
; Fox et al., 1998
). Finally, two patients with PNH were recently reported to have mutations in FMR1 rather than in Filamin A (Moro et al., 2006
).
Working on genetic mechanisms of memory in Drosophila (Bolduc and Tully, 2009
), we too have noted a potential link between FMR1 and Filamin A. We recently have shown that disruptions of dFmr1, the fly homolog of FMR1, show (i) neuroanatomical, learning and memory deficits when disrupted early in development, and (ii) deficits specific to LTM formation when disrupted only in adults (Bolduc et al., 2008
). Independently, a behavioral screen for LTM mutants identified the joy strain, which carries a genetic lesion in cheerio, the fly ortholog of Filamin A (Dubnau et al., 2003
). Given these observations, we hypothesized that Fmr1 and Filamin A may interact in activity-dependent remodeling of actin cytoskeleton. We have tested this hypothesis by evaluating genetic interaction (38) between dFmr1 and cheerio during olfactory memory formation in Drosophila (Quinn et al., 1974
; Tully and Quinn, 1985
; Tully et al., 1994
; Restifo, 2005
). Here, we show that (i) cheerio is expressed in the adult fly brain, (ii) cheerio expression is upregulated in the joy mutant, (iii) LTM specifically is impaired in cheerio mutants, (iv) LTM specifically is impaired in dFmr1; cheerio double heterozygotes and (v) Filamin A is abnormally downregulated in the dFmr1 mutant during LTM formation. These data show for the first time that regulators of protein translation (FMRP) and cytoskeletal structure (Filamin A) function together during LTM formation, thereby presenting a plausible molecular mechanism for a link between dendritic spine morphology and cognitive dysfunction in mental retardation syndromes.
Drosophila Strains
Flies were raised and disposed of as per Cold Spring Harbor Laboratory regulations under the supervision of Dr. Tim Tully. The cherjoy mutant was previously generated in our laboratory as part of behavioral screen for LTM mutants (Cold Spring Harbor Laboratory) using P-element mutagenesis (Dubnau et al., 2003
). The cherjoy mutant carries a P-element insertion within the cheerio gene. These mutants were “genotyped” using PCR primers (in bold):. 12944000→ATTTTCATTAATTGCGAAATGCCGCGACAAAACGCAGTGTGAACTGCAGCACTTACAAAAACTACAGTGAAACGGTGCTTAGATAAATCATATATCTAATTTCTGTACATAGTAGTTGGTTGAAAAAGTTAACTGCTCTATTATCACTAAATTTGAATTGTCCAGTTCTGCATTTATAGCTTCTAATGTGTAGCATTCAATCCTTTAAAAAAAAGGTGTTTCTCAGAATGATAAACTCCTAGGAATTCCTAAAAATCTACGGTTATCTTTCATTCTTTTCAAACGTAAAATAAGGGAATATTTTAAAGACTTCGATTAAGGGAATTTGATTTTTGAATTTGATTCGCGACAATCAATTTTCGAATGACCGGGAGTTTCCTGAAACTTCATCATAGTGAAGTTCGCCTGTGGCTCACGCACACTGCCGCACTCTATTCGCACACACACACACACACACACCTGTTGATGGCAAACGTCGTAGTGTGCGGCTGGCTGTTGGGCGTAGAAATGTAAGTTGCTAAAGTTGTGCTCCAAATTGTTATTGTTGCTATTGCTGGTCGTATAAACACACGCGTTTTGCTGATTTTGCTGCGAATTTGGCGCGTTCTTTCTGCCGTGACTGTTGCTGTTGTTGTAGTAGTGGTTGCCCGTATTACTGCTGTTGTTATTGTTGCTGAGGAATATGGTCGCCTGCGTCGTTTGACCGTTTGTATAATTCGCGTTTTGCACCAATTCACCGCCGTGCGTTAAATTTGCCGATCCGGGACCGTCAGCGTTGTTACTCGAGCACGTACCACCGTTAAATAAATTCTTATTTTTAAACGTTGAAGΔTTTGTCGCCGATCACCGGCATTCGGATCCGCCATATCAGCTGATTTGCTGAATAAAGAGAGGGGCGAGAGAGCGGCAAACAAAAAATAAAACAAGAGAGCATTTTCTTTCTCTTTAACATTTTGAGAATTGTGTTCTCTTAAAAATGTCTTTTTCTCAGCCAGCACACAAAACCAGTTTTCGAAAGAAAATTATTAACAGATTCAAGTTCAGTTTCAGTGTGAGAGCGTAAAGTTAAATATGTATATTTTATAACTTATAAGAACTTCGATATGCCCCAAAGTATGCACCAATAATAATCGGCTTTTCAAAGAGTGGTTACCCAAAATAAAGTTTTTTGTTAAAATATAACAGGTTTATACGACATTATTCCATTCCATTCAATTTTTCTATTTCAATTCCATTATGTTTTTTTTTACTTTATATTTATTTAAAATTTTAAAAAACACCCAATATTAATCTGCCATCAAAACAACATTAGTAGCATAAGCGTAAGCTCGGAGCGATAATAATATTACCCAGTAAATCAGTGACTAATAAACCAGTCATAGAATAAAACGATTCATTTTATTCGTTTTAGTGCACAAAAGCCATTGGTAATGTTTAAAAATATTTGCTATTTTAGTCGGCAGCATTACCATTCGTATATATGTATGTAGTTATATTCGTATATATAGGGGGGCCAAGCCCTGGAATTCTCAAATCCCGACCCTTTTTCAGAACGTGAACCCAAATAAAGTGACTCATGATCCCCTTGCACACCCACACCAAC←12945600.
The Drosophila cherEPSΔ5 mutant was obtained from Dr. Lynn Cooley (Yale University) (Sokol and Cooley, 2003
). Drosophila w1118(isoCJ1) were used for wild-type control. The Drosophila Fmr1B55 was obtained from Dr. Kendall Broadie (Vanderbilt University). Flies were outcrossed for six generations to w1118(isoCJ1) flies to equilibrate genetic backgrounds.
Genetic crosses
Double heterozygote +,cherΔ5/Fmr1B55, + flies were generated by mating cherΔ5/Tm6b,Sb males to Fmr1B55/Fmr1B55 females. All progeny were trained and tested together, and then the Sb+ + ,cherΔ5/Fmr1B55, + progeny were used to calculate the performance index (PI). Double heterozygote+, cherJoy/Fmr1B55, +flies were generated by mating+, cherJoy/+, cherJoy males and Fmr1B55, +/Fmr1B55, +females.
Pavlovian Learning Assay
In general, Drosophila were raised at 22oC and placed at 25oC overnight prior to behavioral experiments. Adult Drosophila less than 3-days old were subjected to Pavlovian olfactory conditioning for (i) one training session (learning), (ii) 10 training sessions without a rest interval (massed training) or (iii) 10 training sessions with 15 min rest between each (spaced training). After training, flies were stored at 18oC and then conditioned responses were tested at a 1 or 4-day retention intervals (Tully and Quinn, 1985
). About 150 flies were trapped inside a training chamber, 95% of the inside of which was covered with an electrifiable copper grid. Flies were allowed 90 s to acclimate and then were exposed sequentially to two odors, 3-octanol (OCT) and 4-methylcyclohexanol (MCH), carried through the chamber in a current of air (750 mL/min). Flies first were exposed for 60 s to the conditioned stimulus (CS + ; either OCT or MCH), during which time they received the unconditioned stimulus (US; twelve 1.25-s pulses of 60 V DC electric shock at 5-s interpulse intervals). After the CS+ presentation, the chamber was ventilated with fresh air for 45 s. Then, flies were exposed for 60 s to a second, control stimulus (CS-; either MCH or OCT), which was not paired with electric shock. After the CS- presentation, the chamber was again flushed with fresh air for 45 s. Relative concentration of OCT and MCH were adjusted so that naïve flies distributed themselves 50:50 in the T-maze.
To test for conditioned odor avoidance immediately after Pavlovian conditioning, flies were tapped gently from the training chamber into an elevator-like compartment that transports them to the choice point of the T-maze. Ninety seconds later, flies were exposed to two converging currents of air from opposite arms of the T-maze, one carrying OCT and the other MCH. Flies were allowed to choose between the CS+ and CS− for 120 s, at which time they were trapped inside their respective arms of the T-maze (by sliding the elevator out of register), anesthetized and counted. For massed training, groups of flies received ten, instead of one, training sessions, with no rest interval in between. For spaced training, groups of flies received ten training sessions, with a 15-min rest interval between each. Conditioned avoidance responses were tested one or 4 days after training as for immediate memory (above). PI and statistical test are performed using JMP software (SAS). All graphs depict mean ± SEM (PRISM).
Sensorimotor Controls
To rule out sensori-motor explanations for poor performance in the Pavlovian learning task, olfactory acuity and shock reactivity were assessed as in Boynton and Tully (Boynton and Tully, 1992
) and Dura (Dura et al., 1993
), respectively. For olfactory acuity, flies were placed in a T-maze and given the choice between an odor versus air. The odors are naturally aversive, and flies usually avoid the T-maze’s arm containing odor. For shock reactivity, flies were placed in a T maze and given a choice between an electrified grid in one T maze arm and an unconnected grid in the other. After the flies distributed themselves for 2 min, they were anesthetized and counted, and the PI was calculated for post-training experiments.
Western Blot Analysis
Approximately 2 mL of flies were collected in a 15 mL Falcon tube and flash-frozen in liquid nitrogen. Flies then were shaken, and fly heads were separated from bodies using 25 and 40 sieves. Fly heads were dispensed to a mortar and pylon on dry ice and pulverized, transferring the to a microcentrifuge tube to be homogenized using Invitrogen extraction buffer. Following homogenization, protein content was measured on Ependorf BioPhotometer. Protein solution was diluted 1:10 in water, and 10 mL of that solution was added to 1 mL of Bradford solution. 50 ug of total protein was loaded per lane with extraction buffer. Electrophoresis was conducted as suggested in the Invitrogen manual for 55 min using a 3–8% gradient gel. Blotting was conducted for 1 h at room temperature. For FMRP staining, the 5A11 antibody 1:500 (Developmental Hybridoma) was used in combination with WesternBreeze kit. For cheerio (Filamin A) staining, Rat Anti-Filamin C-terminal (1:5000) was obtained as a generous gift from Dr. Lynn Cooley (Sokol and Cooley, 2003
). Actin (Sigma) was as a loading control (1:5000). Quantification was done with six measurements per lane and obtained using ImageJ software. Flies were collected immediately after training.
Immunohistochemistry
Two- to five-day-old flies were dissected and processed as described previously in Xia (Xia et al., 2005
). For consistency, only females were selected. All genotypes were dissected similarly and processed in parallel. On day 1, flies were dissected in PBS and then brain were transferred to 4% PA for fixation at room temperature for 10 min and placed in a vacuum for 15 min in 0.2% Triton in 4% PA. Blocking and penetration were done in penetration/blocking buffer for 2 h at 4°C. Brains then were transferred to dilution buffer containing the primary antibody and placed overnight at 4°C. On day 2, brains were washed with wash buffer 4 times (10 min/time). Brains were transferred to secondary antibody and incubated overnight at 4°C in the dark. On day 3, brains were washed again 4 times (10 mins/time) and then mounted in a well made of 2 stacked reinforcer O-rings. The well was filled with approximately 7 uL of Focus-Clear solution and covered by a cover slip. Images were acquired using LSM software from Zeiss at 20×. The average thickness of a brain was around 100 um. For GFP imaging, brains were dissected, vacuumed and then left in penetration/blocking buffer overnight. On day 2, they were washed and mounted.
To assess mushroom body morphology, anti-FasII antibody 1D4 (Developmental Studies Hybridoma Bank, University of Iowa) was used at a concentration of 1:20 (Michel et al., 2004
). Anti-FMRP antibody 5A11 (Developmental Studies Hybridoma Bank, University of Iowa) was used at a concentration of 1:100 (Inoue et al., 2002
). Rat anti-Filamin A (c-terminal) (a generous gift from Dr. Lynn Cooley) was used at a concentration of 1:3000 (Sokol and Cooley, 2003
). An equivalent cheerio imaging result was observed using the P-element enhancer-trap line, Kyoto 105280 crossed to UAS-GFP. The secondary anti-mouse Cy3 antibody (Jackson Lab) was used at a concentration of 1:200.
Cheerio Expression is Aberrant in Mutants
We took advantage of a cheerio mutant previously generated in a study of ring canal formation (Sokol and Cooley, 2003
). The cherΔ5 mutant was derived from an imprecise excision of the EP(3) 3175 P-element insertion; homozygous females display defective germline cell packaging and border cell migration (Sokol and Cooley, 2003
). We first asked if Cheerio was expressed in the adult fly brain (Figures 1
A,B). With western blot analysis, we detected two isoforms of cheerio in adult fly brain, as was observed previously in egg chambers (Sokol and Cooley, 2003
). Immunohistochemical analysis of wild-type adult brain revealed Cheerio expression mostly in the cytoplasm of cells lying at the base of the brain and midline (Figure 1
B), highest in areas corresponding to the ventro-caudal region of the subesophageal ganglia and the median bundle. With both methods, Cheerio expression in brain was (i) decreased in the of the loss-of-function homozygous mutant, cherΔ5 and (ii) increased in the joy memory mutant (Figures 1
A–C). As was reported originally by Sokol et al., who generated this antibody, we could detect some anti-cheerio signal with western blot but not with immunohistochemistry in cherΔ5 (Sokol and Cooley, 2003
).
Considering the cerebral malformation in PNH patients, we examined, in wild-type and mutant flies, the structural integrity of the mushroom body (alpha and beta lobes), a neuroanatomical site important for olfactory memory (Pascual and Preat, 2001
; Didelot et al., 2006
; Yu et al., 2006
; Krashes et al., 2007
; Lu et al., 2007
; Qian et al., 2007
). We did not observe any gross morphological defects in cherΔ5/cherΔ5 or cherjoy/cherjoy homozygous mutants. (Figure 1
D).
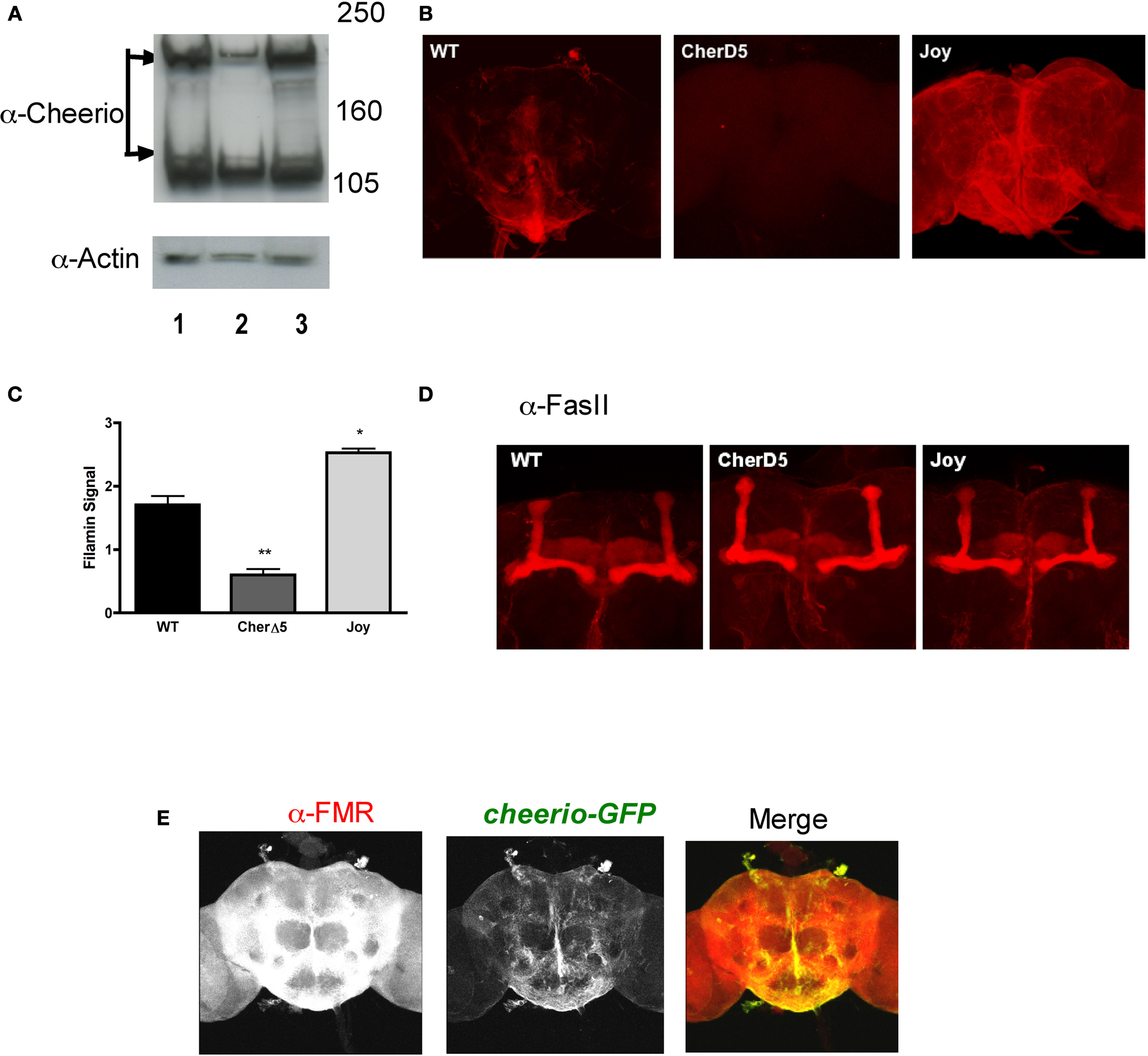
Figure 1. Expression of Filamin A is aberrant in cheerio mutants but does not affect the gross morphology of mushroom body. (A) Western blot analysis for Cheerio reveals two different size products as noted before (Sokol and Cooley, 2003
). Compared to wild-type brains (lane 1), Cheerio expression is lower in cherΔ5 homozygotes (lane 2) and higher in cherJoy homozygotes (lane 3). All lines were loaded with 50 ug of total protein. Anti-Actin was used as loading control. All heads were collected similarly and processed in parallel. Protein mass markers are in kD. (B) Immunohistochemistry for Filamin A in dissected adult brain for wild-type (WT), homozygous cherΔ5 (CherD5) and homozygous cherJoy (Joy) flies (N = 10 brains per genotype were evaluated; representative examples are depicted). All brains are shown at 20× magnification. Brains were dissected similarly, processed in parallel and imaged under identical LSCM conditions. (C) Quantification of Filamin A expression from western blot analysis. Scores shown are averages of both α-cheerio bands for each genotype. (D) Mushroom body architecture, visualized by Fas II immunostaining, is normal among wild-type (WT), cherΔ5 (CherD5) and cher joy (Joy) flies (N = 10 brains per genotype were evaluated; representative examples are depicted). All brains are shown at 20× magnification. Brains were dissected similarly and processed in parallel and imaged under identical LSCM conditions. (E) Immunohistochemical co-localization of Fmr1 (mouse α-FMR in red channel) and Cheerio (cheerio-GFP in green channel) in dissected wild-type fly brains. All brains were dissected similarly, processed in parallel and imaged under identical LSCM conditions). N = 10 brains per genotype; all brains are 20× magnification.
LTM is Disrupted in Cheerio Mutants
Previous studies in Drosophila have established that memory formation after Pavlovian olfactory learning proceeds through several genetically distinct temporal phases (STM: short-term memory, MTM: middle-term memory, ARM: anesthesia-resistant memory and LTM; Tully et al., 1994
). One-day memory after spaced training (ten training sessions with a 15-min rest interval between each) is composed of a cycloheximide-sensitive LTM component and a cycloheximide-insensitive ARM component. In contrast, 1-day memory after massed training (ten training sessions with no rest intervals) is composed of only ARM (Tully et al. 1994
; Yin et al., 1994
).
In both the cherΔ5/cherΔ5 and cherjoy/cherjoy homozygous mutants, 1-day memory after spaced training was defective (P = 0.0208 and P < 0.0001, respectively; cf. Dubnau et al., 2003
), while that after massed training was not (P = 0.701 and P = 0.074, respectively; Figures 2
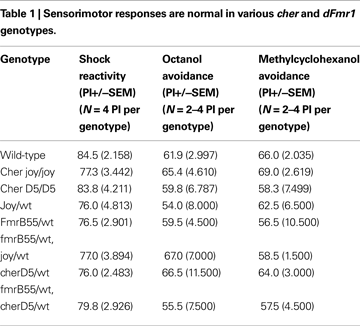
A,B). To rule out sensorimotor defects as a possible explanation for mutants’ abnormal behavioral performance, we assessed “task-relevant” olfactory acuity and shock reactivity and found no significant differences between mutants and control (Table 1
). Even though LTM and ARM both can be detected 1 day after spaced training, ARM is decremental while LTM persists; 4 days after spaced training only LTM is present in wild-type flies (Tully et al. 1994
; Figures 2
C,D). 4-day memory after spaced training in both cherΔ5/cherΔ5 and cherjoy/cherjoy homozygous was significantly lower than normal (P = 0.0034 and P = 0.0165) and near zero (Figures 2
C,D), suggesting the defect in 1-day memory likely was produced by the absence of LTM.
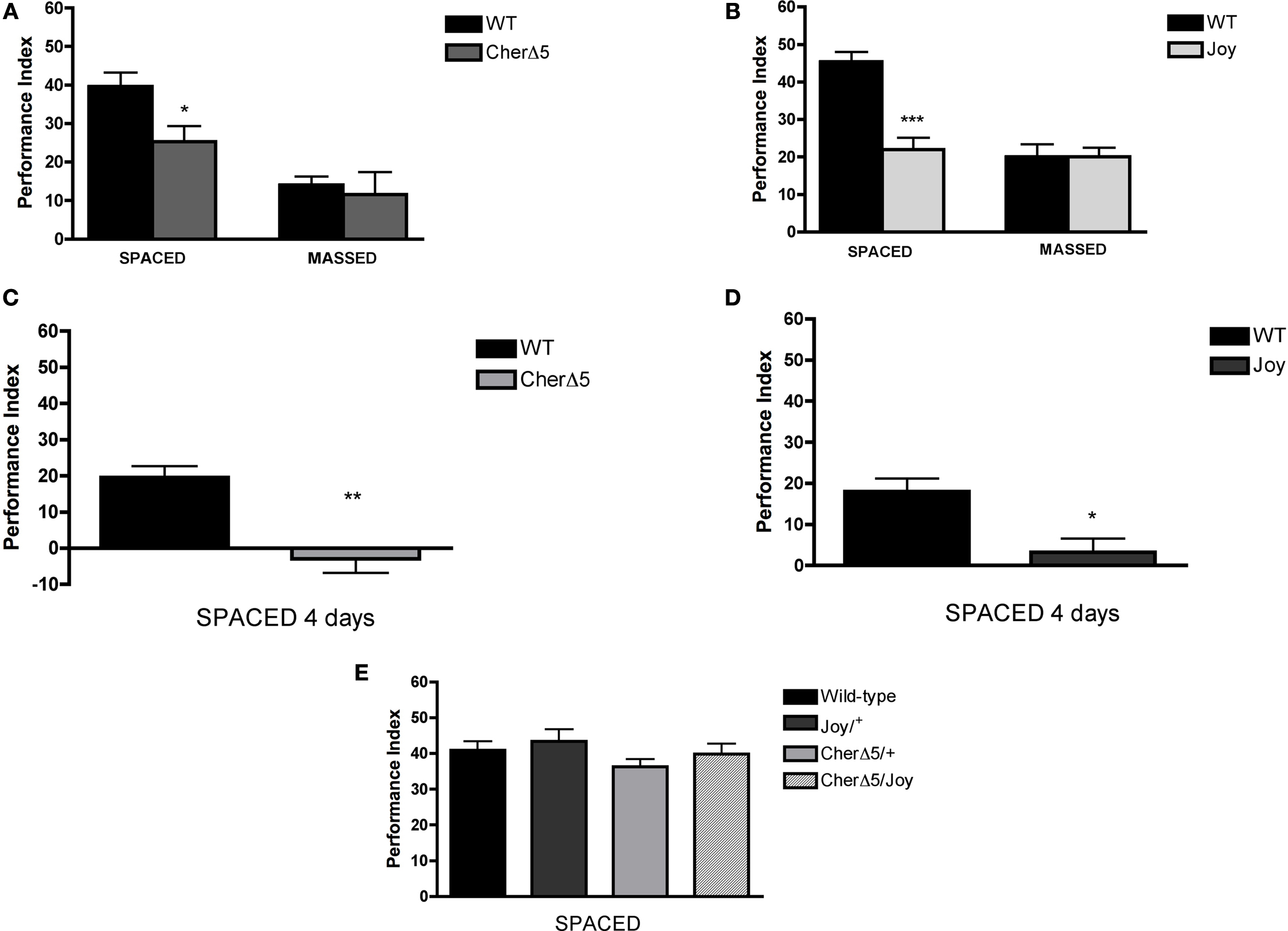
Figure 2. LTM is defective in cheerio mutants. (A) One-day memory after spaced training is lower than normal in the loss-of-function homozygous mutant, cherΔ5 (CherD5; P = 0.0208; N = 8 PIs per genotype). In contrast, 1-day memory after massed training is normal in the cherΔ5 mutant (P = 0.701; N = 8 PIs per genotype). (B) One-day memory after spaced training is lower than normal in the gain-of-function mutant, cherJoy (Joy; P < 0.0001; N = 8 PIs per genotype). In contrast, 1-day memory after massed training is normal in the cherJoy mutant (P = 0.4149; N = 8 PIs per genotype). (C) 4-day memory after spaced training is lower than normal in the cherΔ5 (CherΔ5) mutant (P = 0.0034; N = 4 PIs per genotype). (D) 4-day memory after spaced training is lower than normal in the cher joy (joy) mutant (P = 0.0165; N = 4 PIs per genotype). (E) One-day memory after spaced training is normal in cherΔ5/+ (cherΔ5/+) or cherjoy/+ (joy/+) heterozygotes and in the cherΔ5/cherjoy (cherΔ5/joy) heteroallelic (P = 0.3447; N = 8 PIs per genotype). See text for discussion.
The cherΔ5 and cherjoy mutations had opposite effects on cheerio gene expression (Figures 1
A–C) though both disrupted LTM. This suggested the interesting possibilities either that cherjoy might act as a dominant negative mutation or alternatively that the two mutations might act additively in heteroallelic mutants. We found that 1-day memory after spaced training was normal in cherjoy/+ and cherΔ5/+ heterozygotes and in the cherjoy/cherΔ5 heteroallelic mutant (Figure 2
E). Thus, it appears that the cherjoy mutation does not act as a dominant negative and rather that the two mutations act additively in the heteroallelic mutant, thereby restoring LTM presumably by normalizing the expression level of Cheerio.
Fmr1 and Cheerio Interact Genetically During LTM Formation
Because both dFmr1 and cheerio mutants are defective in LTM, we sought to investigate a possible genetic interaction using double heterozygotes. Consistent with this hypothesis, the expression patterns of FMRP and Cheerio overlapped largely in the ventro-caudal suboesophageal ganglia and in the midline bundle (Figure 1
E). We assessed LTM formation in two different double heterozygotes, cherΔ5/+;+/Fmr1B55 and cherJoy/+;+/Fmr1B55. In both cases, 1-day memory after spaced training was significantly reduced (P < 0.0001 and P < 0.0001, respectively) but that after massed training was not (P = 0.7478 and P = 0.5314, respectively). In contrast, 1-day memory after both spaced and massed training were normal for each of the single heterozygotes (cherΔ5/+, cherjoy/+ and Fmr1B55/+) (Figures 3
A,B). In the double heterozygotes, shock reactivity and olfactory acuity were normal (Table 1
), and 4-day memory after spaced training was significantly reduced and near zero (Figures 3
C,D).
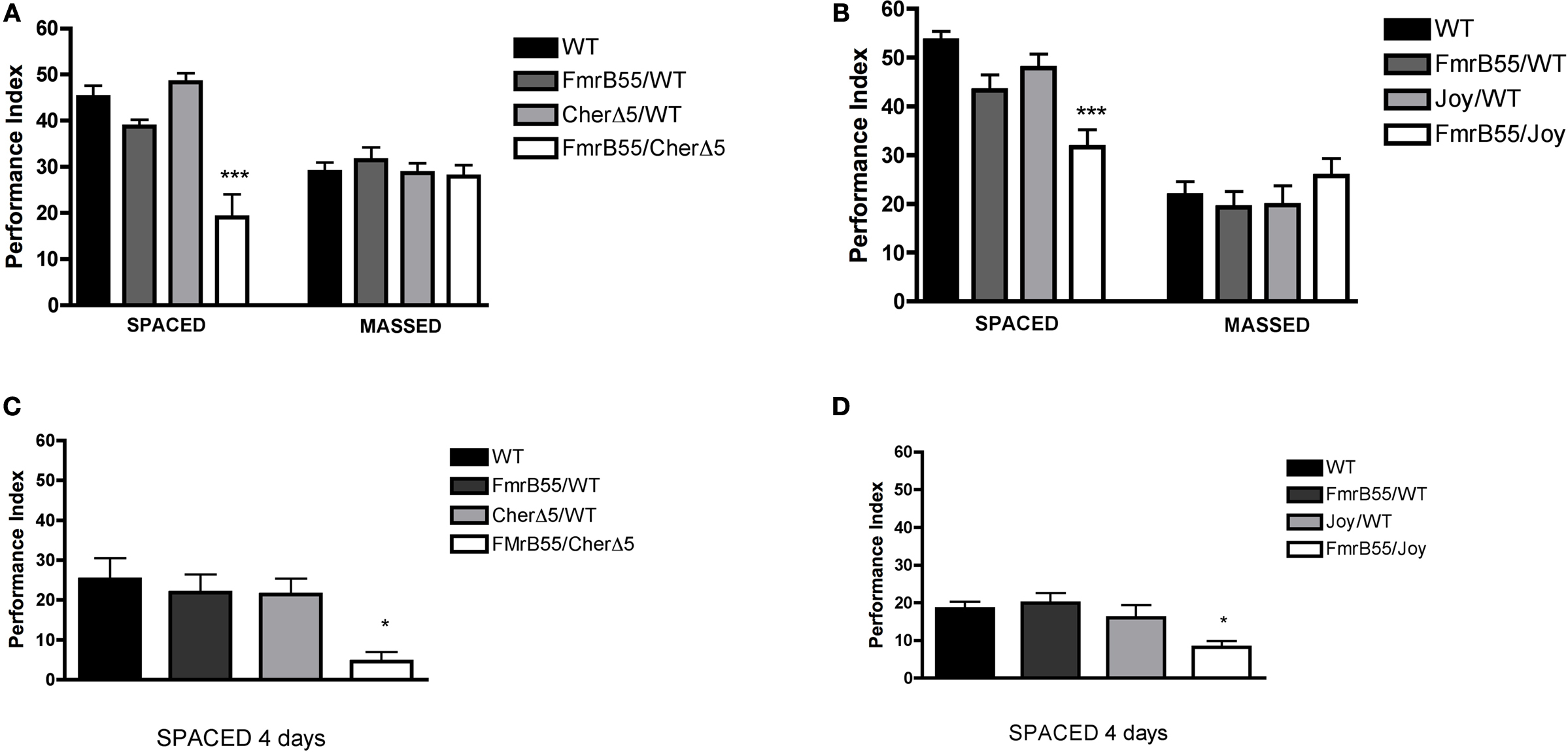
Figure 3. dFmr1 and cheerio interact during long-term memory formation. (A) One-day memory after spaced training is defective in the cherΔ5,+/+, dFmr1B55 double heterozygote (FMrB55/CherD5; P < 0.0001) but not in either single heterozygote (FmrB55/WT or CherD5/WT). In contrast, 1-day memory after massed training is normal among the cherΔ5,+/+, dFmr1B55 double heterozygote (FMrB55/CherD5) and both single heterozygotes (FmrB55/WT or CherD5/WT). N = 8 PIs per genotype. (B) One-day memory after spaced training is defective in the cherjoy,+/+, dFmr1B55 double heterozygote (FMrB55/Joy; P < 0.0001) but not in either single heterozygote (FmrB55/WT or Joy/WT). In contrast, 1-day memory after massed training is normal among the cherjoy,+/+, dFmr1B55 double heterozygote (FMrB55/Joy) and both single heterozygotes (FmrB55/WT or Joy/WT). N = 8 PIs per genotype. (C) 4-day memory after spaced training is defective in the cherΔ5,+/+, dFmr1B55 double heterozygote (FMrB55/CherD5; ANOVA P = 0.0057, Tukey P < 0.01) but not in either single heterozygote (FmrB55/WT or CherD5/WT). N = 10 PIs per genotype (D) 4-day memory after spaced training is defective in the cherjoy,+/+, dFmr1B55 double heterozygote (FMrB55/Joy; ANOVA P = 0.0072 Tukey P < 0.05) but not in either single heterozygote (FmrB55/WT or Joy/WT). N = 12 PIs per genotype.
Filamin a Expression is Misregulated in the dFmr1 Mutant During LTM Formation
Given FMRP’s role in regulating activity-dependent synaptic processes(Vanderklish and Edelman, 2002
; Weiler et al., 2004
; Muddashetty et al., 2007
), we thought to check Cheerio expression levels in the dFmr1 mutant after Pavlovian olfactory learning. After spaced training, we observed no obvious change in Cheerio expression in wild-type flies but a striking and significant decrease in Cheerio expression in the dFMR1 mutant (Figures 4
A,B). Relative deficiency of synaptic proteins have been observed (Muddashetty et al., 2007
) before and has led to the occlusion hypothesis stating that mass protein synthesis in absence of FMRP leads to relative decrease of synaptic protein (Bolduc et al., 2008
; Kelleher and Bear, 2008
). After massed training, Cheerio levels were reduced significantly but similarly in both wild-type flies and the dFMR1 mutant (Figures 4
C,D). These data provide a plausible molecular correlate to the differential performance of dFMR1/+;cheerio/+ double heterozygotes after spaced versus massed training.
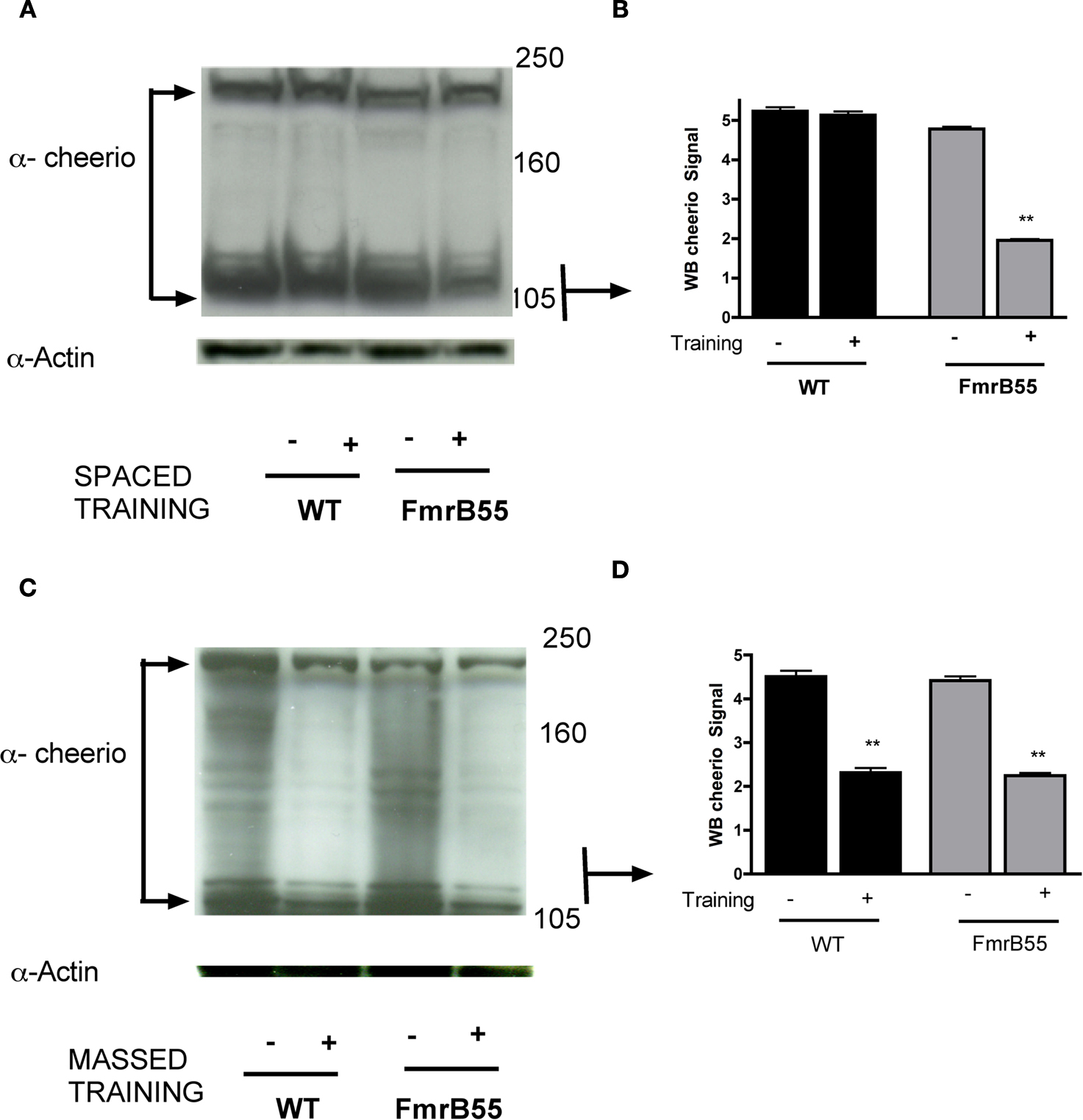
Figure 4. Activity-dependent expression of Filamin A is aberrant after spaced training in the dFMR1 mutant. (A) Western blot analysis of Cheerio level before (−) or after (+) spaced training in wild-type flies (WT) or the dFMR1 (FmrB55) mutant. All lanes were loaded with 50-ug total protein. Anti-actin (lowest band for each lane) was used as loading control. All heads were collected similarly and processed in parallel. Protein mass markers are in kD. (B) Quantification of western blot analysis for the smaller isoform of Cheerio (105 kD). In wild-type (WT) flies, Cheerio short-form levels are similar before (−) and after (+) spaced training. In the dFMR1 mutant (FmrB55), however, the Cheerio short-form is significantly reduced. (C) Western blot analysis of Cheerio before (−) or after (+) massed training in wild-type flies (WT) or the dFMR1 (FmrB55) mutant. All lanes were loaded with 50-ug total protein. Anti-actin (lowest band for each lane) was used as loading control. All heads were collected similarly and processed in parallel. Protein mass markers are in kD. (D) Quantification of western blot analysis for the smaller isoform of Cheerio (105 kD). The Cheerio short-form levels are similar before (−) massed training in both wild-type (WT) and dFMR1 mutant (FmrB55) flies. After massed training, Cheerio short-term is significantly reduced in both wild-type (WT) and dFMR1 mutant (FmrB55) flies.
Guided by the clinical suggestion that FMRP and Filamin A might have an intersecting function in regulating changes in neuronal cytoskeletal structure(Moro et al., 2006
), we have shown that aberrant expressions levels (increased in cherjoy or decreased in cherJoy) of cheerio, the fly homolog of Filamin A, are associated with specific defects in LTM memory formation. Such an observation has been reported before in the MeCP2 mouse model of Rett syndrome. Whereas Rett syndrome is associated with a reduction in MeCP2 levels (Amir et al., 1999
) and mouse MeCP2 null mutation recapitulates several Rett-like symptoms (Guy et al., 2001
), overexpression also leads to neurological defects (Collins et al., 2004
). We also have shown that both decreased or increased levels of FMRP are associated with memory defects in Drosophila (Bolduc et al., 2008
).
The cheerio and dFmr1 genes functionally interact during LTM formation. This interaction was observed specifically after spaced training and not after massed training. Given FMRP’s role in the regulation of protein translation (Li et al., 2001
; Khandjian et al., 2004
; Stefani et al., 2004
), we took this clue to the molecular level to quantify Cheerio expression levels in dFmr1 mutants. After spaced training, Cheerio expression did not change in wild-type flies, but the Cheerio short isoform specifically was reduced in the dFmr1 mutant. After massed training, this Cheerio short isoform is reduced in both wild-type and dFmr1 mutants. Thus, spaced training appears normally to disinhibit regulation of Cheerio short-form, and this disinhibition appears aberrant in the dFmr1 mutant. Further work will be required to understand this molecular mechanism. Nonetheless, the genetic interaction observed here for LTM formation may explain the similarities between patients with periventricular nodules and Fragile X syndrome.
A role for cheerio and dFmr1 during LTM formation suggests a shared molecular mechanism among the clinically and etiologically different mental retardation syndromes (Berry-Kravis and Huttenlocher, 1992
; Berry-Kravis and Sklena, 1993
), which show defects in dendritic spines morphology. Other examples could include dysregulation of PAK, observed in Neurofibromatosis 1 NF1 and Fragile X syndrome (Tang et al., 1998
; Hayashi et al., 2007
), and an abnormality in mTOR signaling observed in Tuberous sclerosis complex TSC1, TSC2 and NF1 (Johannessen et al., 2005
).
In Fragile X patients, the usual mushroom shaped dendritic spines are decreased in number and density and, instead, an excess of elongated thin spines is observed (Rudelli et al., 1985
; Hinton et al., 1991
; Irwin et al., 2002
). Although no study has yet linked Filamin A directly to the cytoskeletal structure of dendritic spines, cell membrane shape, in general, has been shown to depend on Filamin A. Serum starved melanoma cells lacking Filamin A fail to form a three-dimensional orthogonal network of cytoskeletal elements, for instance, after serum application. Instead, they form a dense mat of long actin filaments (Flanagan et al., 2001
) – reminiscent of the long thin dendritic spines. Our results suggest that aberrant levels of Cheerio expression during LTM formation could lead to decreased actin cross-linking, thereby generating abnormally shaped dendritic spines in Fragile X patients. Interestingly, a case of PNH and severe mental retardation has been reported to result from a duplication of Filamin A (Fink et al., 1997
). At abnormally high concentrations, Filamin A causes actin arrangements into parallel, instead of orthogonal, arrays (Hartwig and Stossel, 1975
). Further studies with high resolution imaging, such as the use two photon-mediated release of caged glutamate, will be needed to look at dendritic spines in animal models of Fragile X and PNH. In addition, pharmacological rescue of Fragile X mutants using other cross-linkers could be tested. Indeed, alpha-actinin has been shown to interact with Filamin in a recent report by Esue et al. (2009)
.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This project was supported by NIH and Dart Neuroscience LLC (to T.T.) and a CCHCSP Career Development Award (to F.B.).
Fox, J. W., Lamperti, E. D., Eksioglu, Y. Z., Hong, S. E., Feng, Y., Graham, D. A., Scheffer, I. E., Dobyns, W. B., Hirsch, B. A., Radtke, R. A., Berkovic, S. F., Huttenlocher, P. R., and Walsh, C. A. (1998). Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325.
Irwin, S. A., Idupulapati, M., Gilbert, M. E., Harris, J. B., Chakravarti, A. B., Rogers, E. J., Crisostomo, R. A., Larsen, B. P., Mehta, A., Alcantara, C. J., Patel, B., Swain, R. A., Weiler, I. J., Oostra, B. A., and Greenough, W. T. (2002). Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile X knockout mice. Am. J. Med. Genet. 111, 140–146.
McBride, S. M., Choi, C. H., Wang, Y., Liebelt, D., Braunstein, E., Ferreiro, D., Sehgal, A., Siwicki, K. K., Dockendorff, T. C., Nguyen, H. T., McDonald, T. V., and Jongens, T. A. (2005). Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45, 753–764.
Weiler, I. J., Spangler, C. C., Klintsova, A. Y., Grossman, A. W., Kim, S. H., Bertaina-Anglade, V., Khaliq, H., de Vries, F. E., Lambers, F. A., Hatia, F., Base, C. K., and Greenough, W. T. (2004). Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc. Natl. Acad. Sci. U.S.A. 101, 17504–17509.
Wohrle, D., Kotzot, D., Hirst, M. C., Manca, A., Korn, B., Schmidt, A., Barbi, G., Rott, H. D., Poustka, A., Davies, K. E. and Steinbach, P. (1992). A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am. J. Hum. Genet. 51, 299–306.