1
Center for Nanomedicine and Tissue Engineering, A.O. Ospedale Niguarda Ca’ Granda, Milan, Italy
2
Materials Science Department, University of Milan-Bicocca, Milan, Italy
3
Biotechnology and Biosciences Department, University of Milan-Bicocca, Milan, Italy
4
Consorzio Nazionale Interuniversitario per le Scienze Fisiche della Materia UdR Milan-Bicocca, Milan, Italy
The understanding of phenomena involved in the self-assembling of bio-inspired biomaterials acting as three-dimensional scaffolds for regenerative medicine applications is a necessary step to develop effective therapies in neural tissue engineering. We investigated the self-assembled nanostructures of functionalized peptides featuring four, two or no glycine-spacers between the self-assembly sequence RADA16-I and the functional biological motif PFSSTKT. The effectiveness of their biological functionalization was assessed via in vitro experiments with neural stem cells (NSCs) and their molecular assembly was elucidated via atomic force microscopy, Raman and Fourier Transform Infrared spectroscopy. We demonstrated that glycine-spacers play a crucial role in the scaffold stability and in the exposure of the functional motifs. In particular, a glycine-spacer of four residues leads to a more stable nanostructure and to an improved exposure of the functional motif. Accordingly, the longer spacer of glycines, the more effective is the functional motif in both eliciting NSCs adhesion, improving their viability and increasing their differentiation. Therefore, optimized designing strategies of functionalized biomaterials may open, in the near future, new therapies in tissue engineering and regenerative medicine.
The “bottom up” approach for creating new biomaterials in nanomedicine and tissue engineering is achieving a consistent approval among the scientific community (Gazit, 2007
). A considerable number of interdisciplinary publications, from physics to biology, describe guidelines to fabricate new nano-biomaterials for specific applications (Reddi, 2000
; Drotleff et al., 2004
; Hirst and Smith, 2004
; Tsai et al., 2007
; Ulijn and Smith, 2008
). Molecular self-assembly became an alternative paradigm to create functional nanostructures using spontaneous diffusion and specific association of molecules directed by weak interactions (Weigel et al., 2006
). Taking advantage of the extensive research regarding protein folding and stability, self-assembling peptides are likely to become innovative nano-structured biomaterials designed at the molecular level. In peptide based biomaterials the main effort is driven towards the definition of the rules linking peptide sequences to their molecular structure organization and thus their functional properties (Zhang et al., 2005
). Self-assembling peptides can be prepared via solid-phase synthesis with precise control over sequence, molecular dimension and functionalization. Zhang and his co-workers developed short amphiphilic peptides (e.g. RADA16-I) able to mimic the extra cellular matrix, so to be intriguing scaffolds for tissue engineering applications (Yokoi et al., 2005
). Depending on the tissue hosting the self-assembled implant (for example bone, cartilage, myocardium and nervous tissue), biomechanical and biochemical properties of the required implant may be tuned. Moreover the bio-mimetic scaffold has also to adopt its nanostructure without harming the cells to be carried and implanted (Drury and Mooney, 2003
). The 12- to 16-mer peptide class discovered by Zhang folds into double layered β-sheets to form nanofibers featuring folded molecular structures similar to amyloid proteins (Yokoi et al., 2005
). In this peptide class the process of assembling is triggered by physiological ranges of pH and temperature. This characteristic make them appealing for application in nanomedicine because they can be injected directly into the injury site warranting a minimum invasive surgery (Zhang et al., 2005
). Numerous publications provided insights on this self-assembling peptide class and a wide variety of new biomaterials was then developed for in vitro and in vivo applications (Davis et al., 2006
; Gelain et al., 2007b
; Gambaretto et al., 2008
; Semino, 2008
). Recently, a “second generation” of self-assembling peptides obtained via functionalization of the self-assembling backbone sequences with specific bioactive motifs was introduced: the functionalized peptides (FPs). FPs can be tailored for the specific tissue to be regenerated (Gelain et al., 2007a
). Rational modifications of the peptides sequence aim at directly controlling the peptide molecular aggregation within the fibril matrix and its bioactivity. Recent studies indicate that nanoscale features within a matrix influence cell adhesion, morphology and proliferation (Berry et al., 2004
). Therefore, a wide range of FPs have been developed, adding different biological motifs at the C-terminal of RADA16-I (Gelain et al., 2007a
). The standard functionalization strategy comprises a two glycine spacer (Gs) placed between the RADA16-I sequence and the biological motif in order to confer a satisfactory exposure to cell membrane receptors. However, the “side effects” of this spacer over scaffold biological functionality and peptide self-assembling propensity has never been investigated. Therefore we performed a comprehensive set of analyses over self-assembling RADA16-I core peptides linked to functional motifs via Gs of different lengths.
In more details, in order to assess the spacer’s relevance over scaffold gelation and its biological influence over neural stem cells (NSCs) adhesion, we synthesized and purified RADA16-I-GGPFSSKTK (2G-BMHP1), a functionalized self-assembly peptide already proved to stimulate NSC adhesion and differentiation in vitro (Gelain et al., 2007a
), RADA16-I-PFSSTKT (0G-BMHP1), lacking any Gs, and RADA16-I-GGGGPFSSTKT (4G-BMHP1), comprising a 4-glycines long spacer,. We investigated the nanostructure of each peptide via atomic force microscope (AFM). To evaluate their process of self-assembly, Fourier Transform Infrared (FTIR) and Raman spectra of peptide dehydrated solutions at different pHs were measured. We investigated about the stability of nano-structure in a “not assembling condition” (pH 2) and in physiological condition (pH 8). Additionally FTIR spectra were collected during thermal treatments in order to assess structural changes, and consequently thermal stability, in the assembled nanostructures. To confirm part of the hypotheses brought up by the characterization experiments, we tested the effectiveness of the different functionalization strategies assessing the adhesion, proliferation and survival of NSCs in vitro.
Our structural analysis suggests that the three peptides show a similar nanostructure, but the lack of Gs affects both the stability of the cross-β structure and a proper functional motif exposure. On the other hand longer Gs preserved and enhanced the peptide functionalization. Our study shows how deepening the understanding in nanostructure assembly and functionalization is crucial for this class of biomaterials deemed to become powerful scaffolds of neural regenerative medicine applications.
Synthesis and Purification
The self-assembling peptides RADA16-I (Ac-RADARADARADARADA-CONH2), 2G-BMHP1 (Ac-RADARADARADARADAGGPFSSTKT-CONH2), 0G-BMHP1 (Ac-RADARADARADARADAPFSSTKT-CONH2) and 4G-BMHP1 (Ac-RADARADARADARADAGGGGPFSSTKT-CONH2), were F-moc synthesized via CEM liberty microwave automated synthesizer. Sample masses were checked via MALDI-TOF mass-spectrometer (Applied biosystems), HPLC purified (Waters), lyophilized (Labconco) and dissolved at 1% (w/v) concentration in distilled water (solution pH = 3).
AFM Measurements
Functionalized peptides at a concentration of 1% w/v solution were diluted (in a ratio of 1:100) in sterile water, 5 μl of these solutions were placed on mica muscovite substrates and kept at room temperature for 1 min. The mica surfaces were then rinsed with Millipore-filtered water to remove loosely bound peptides and dried under a gentle stream of gaseous nitrogen.
Atomic force microscope images were collected in tapping™ mode by a MultiMode Nanoscope IIIa (Digital Instruments) under dry nitrogen atmosphere using single-beam silicon cantilever probes (Veeco RTESP: resonance frequency 300 KHz, nominal tip radius of curvature 10 nm, forces constant 40 N/m). For high spatial resolution a cantilever probe with a resonance frequency 320 KHz, nominal tip radius of curvature 2–5 nm, forces constant 42 N/m (Veeco TESP-SS) was also used. If necessary, data sets were subjected to a first-order flattening. Measured fiber dimensions with RSTEP were corrected because of the convolution effect arising from the finite size of the AFM tip. Figure S4 in Supplementary Material shown the method used for the measurements. Being the observed tabular nanofiber heights (between 1 and 1.5 nm) far lower than the tip radius (10 nm) the observed widths were corrected with the formula (Jun et al., 2004
; Taraballi et al., 2009
):

where Δx is the width broadening effect, h is the nanofiber height, and rt is for tip radius.
FTIR Spectroscopy
Each peptide was dissolved at a final concentration of 0.1% (w/v) in different buffers with a specific pH to test the assembling process. Two buffers were employed to analyze the peptide structure: buffer 1 at pH 8 (NaOH 5 mM); buffer 2 at pH 2 (HCl 1.2 mM). The sample pH was measured by a digital pH-meter. Moreover, in order to displace the samples pH, other two buffers were used: buffer 3 (NaOH 15 mM) to shift pH from 2 to 8; buffer 4 (HCl 25 mM) to shift the pH from 8 to 2. We adopted this notation for sake of simplicity during all through the text. Buffer 3 was used to resuspend the sample in buffer 1 after the evaporation of buffer 1.The same procedure was used for buffer 4, after the evaporation of buffer 2.
Fourier Transform Infrared spectroscopy was employed for the peptide characterization, since it enable to detect their secondary structure and the formation of β-sheet intermolecular interactions in peptide assembly (Seshadri et al., 1999
). In addition, it can work at the same peptide concentration used in their vitro and in vivo experiments, while Circular Dichroism (CD) spectroscopy requires lower concentrations different from those of their standard use.
Fourier Transform Infrared spectroscopy measurements in attenuated total reflection (ATR) were performed using a single reflection diamond element (Golden Gate, Specac, USA). The FTIR spectrometer FTS-40A (Bio-Rad, Digilab Division, Cambridge, MA, USA), equipped with a nitrogen cooled mercury cadmium telluride detector and an air purging system, was employed under the following conditions: 2 cm−1 spectral resolution, 20 kHz scan speed, 1000 scan co-addition, and triangular apodization. 5 μl of each peptides sample at pH 2 and pH 8 were deposited on the ATR plate and spectra were recorded after solvent evaporation to allow the formation of a hydrated peptide film. After these measurements, the same samples were re-suspended using buffer 3 (for the pH 2 samples) or buffer 4 (for pH 8 samples), and the spectra were recorded again after the solvent evaporation. The spectra of 0.1 mg of the same peptide in lyophilized state was also recorded.
The ATR/FTIR spectra were reported after normalization on the Amide I band area to compensate for possible differences in the peptide content. To better resolve the Amide I band into its secondary structures, the second derivative analysis of the absorption spectra was performed following the Savitsky–Golay procedure (5 points) after a binomial smoothing (11 points) of the spectra.
Raman Micro-Spectroscopy
Raman spectra were collected on the same samples used for FTIR measurements in order to compare directly the information from the two different spectroscopic techniques. Samples were deposited on a silicon substrate following the same procedure described above, giving the best signal-to-noise ratio with no interference from water phonon spectrum. Measurements were performed using the 632.8 nm line of a He-Ne laser as light source and collecting the scattered light signal in backscattering geometry by a Raman spectrometer (Labram Dilor). Signal was detected by means of a CCD (Jobin-Yvon Spectrum One 3000), with a spectral resolution of 1 cm−1, by averaging 10 spectra obtained with an acquisition time of 180 s each. Background from the silicon Micro-Raman spectrum, overgrow the region that start from 900 to 1000 cm−1 for this reason the investigated spectral range goes from 1000 to 1800 cm−1 and contains all the main features of the peptide spectra (De Gelder et al., 2007
). Spectra were then normalized to the peak at ∼1003 cm−1 from ring mode of phenylalanine. The high-energy spectral region around 2900 cm−1 was also analyzed, looking at the relative intensities, to observe the solvent effects on the CH2, CH3, and aromatic CH2 stretching modes.
Thermal Treatment Monitored by FTIR Spectroscopy
For thermal unfolding studies, peptide samples were dissolved in deutered water (D2O) at 4 mg/ml concentration and 15 μl of the above solutions were placed in a temperature-controlled cell (Wilmad, Buena, NJ, USA), made of two BaF2 windows separated by a 100 μm Teflon spacer. The samples were then heated at a constant rate of 0.2°C/min over the range from 30°C to 100°C and FTIR measurements of peptide solutions were recorded in the transmission mode under the same condition employed for ATR/FTIR spectra. The absorption spectra of the peptides were obtained after subtraction of the D2O solvent absorption from that of the peptide solutions. The Amide I components, reflecting the different secondary structures and β-sheet intermolecular interaction in peptide assemblies, were identified via second derivative analysis of the measured the absorption spectrum (Susi and Byler, 1986
) following the Savitsky–Golay procedure (5 points) after a binomial smoothing (11 points) of the spectrum (GRAMS/32 software). During thermal treatment, the dissolution of the intermolecular β-sheet structures was reported as percentage variation of the 1618 cm−1 band intensity, evaluated from the second derivatives of the absorption spectra (Javor et al., 2008
).
Cell Cultures
Neural stem cell cultures were established and expanded as previously described. Briefly, neural precursors isolated from the sub ventricular zone (SVZ) of 8-week-old CD-1 albino mice striata, at passage 10, were used. Cell proliferation was performed in Neurocult basal medium serum-free medium (StemCellTechnologies, Canada), in the presence of basic fibroblast growth factor (βFGF from PeproTech, Rocky Hill, NJ, USA) and epidermal growth factor (EGF from PeproTech) at final concentrations of 10 ng/ml and 20 ng/ml. The medium without growth factors was used as a basal medium. Cell seeding (at a concentration of 2–8 × 104 cells/cm2) was performed 2 days after the last mechanical dissociation in order to seed the maximum percentage of stem cells. Cells were seeded on the top-surface of each assembled nanofibrous scaffold (scaffold thickness of approximately 100–150 μm) prepared as previously described (Gelain et al., 2006
) in 96 multiwells (Falcon). Scaffolds were obtained from 1% v/w peptide solutions.
Positive and negative controls consisted of Cultrex-BME® substrate (R&D systems) (0.15 mg/ml solution) and untreated bottom well surfaces respectively. NSCs were cultured with basal medium supplemented with βFGF (10 ng/ml), added to enhance neuronal progeny differentiation. After 3 days, the medium was shifted to a medium containing Leukemia Inhibitory Factor (LIF, Chemicon) (20 ng/ml) and Brain Derived Neurotrophic Factor (BDNF, Peprotech) (20 ng/ml) to pursue the neuronal and glial population maturation in NSC progeny. Serum-free cell culture media were chosen to avoid random serum proteins adsorption within the scaffolds. Cells were fed every 3 days with the same fresh culture medium. Phase contrast images of the adhering cells were acquired via inverted microscope (Axiovert, Zeiss) at 1 week after seeding. To quantify cell viability and proliferation scaffolds and NSCs were processed with CellTiter 96® Aquesous Proliferation Assay (Promega, Madison, WI, USA) at 7 days in vitro. Briefly, 20 μl of CellTiter solution were added into each well and incubated for 2 h. Absorbance values (n = 5) were measured by using a Vmax microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 490 nm wavelength. Values, reported as means ± standard error of the mean, were blanked to their respective controls consisting of same substrates and cell culture media without cells. The linear response between the cell number and absorbance at 490 nm of the incubated kit solution was previously confirmed at cell concentrations ranging from 1 × 103 to 5 × 105 cells per well (data not shown). On all possible coupled combinations of two different experimental groups we performed paired t-tests to assess the statistical significance of their differences.
In differentiation tests cells were stained with lineage-specific antibodies for Nestin (Chemicon, Hofheim, Germany) positive progenitor cells, β-Tubulin (Covance, Richmond, CA, USA) positive neurons, Glial Fibrillary Acidic Protein (Chemicon) positive astrocytes, GalC (Chemicon) and O4 (Chemicon) positive oligondendrocytes. Primary antibodies were then stained with secondary ALEXA 488 goat anti-mouse (Molecular Probes) and CY3 Anti-Rabbit IgG antibodies (Jackson Immuno Research). Cell nuclei were counterstained with DAPI (Molecular Probes).
Quantitative analyses were performed by counting 100–300 cells for each of six randomly chosen fields over a total of two independent experiments. Phase contrast and fluorescence images of the adhering cells were acquired via inverted microscope (Axiovert, Zeiss) at 1 week after seeding. Paired t-tests were adopted for different substrate results of the same marker.
Peptides Design
We tested three self-assembling peptides (Figure 1
) with an identical functional motif (PFSSTKT) at the C-terminal of the RADA16-I peptide, a self-assembling sequence capable of driving the assembly process. The only difference among these peptides is the presence of two (2G-BMHP1) or four glycines (4G-BMHP1) or none (0G-BMHP1) between the RADA16-I sequence and the functional motif as depicted in Figure 1
(glycines are colored in green). The addiction of further glycines between self-assembling sequence and biological motifs was not taken into account because longer peptide sequences could influence their synthesis yields thus limiting the significance and applicability of the findings. The presence of side chains with minimal steric hindrances in glycines is likely to provide higher degrees of freedom to the overall molecules in comparison to those normally imposed by the side chain of other aminoacids. Thus Gs were used as hinges between the two peptide segments (functional motif and self-assembling core) not to compromise the self-assembly of the RADA16-I part and the exposure of the biological motif. Nonetheless we tested a similarly FP deprived of the Gs spacer (0G-BMHP1) to investigate the contribution of the Gs. Each peptide was dissolved in MilliQ solution, and then PBS (pH 7.4) was added to test their propensity of forming hydrogel scaffolds at the macroscale. Each peptide formed a hydrogel when PBS was added.
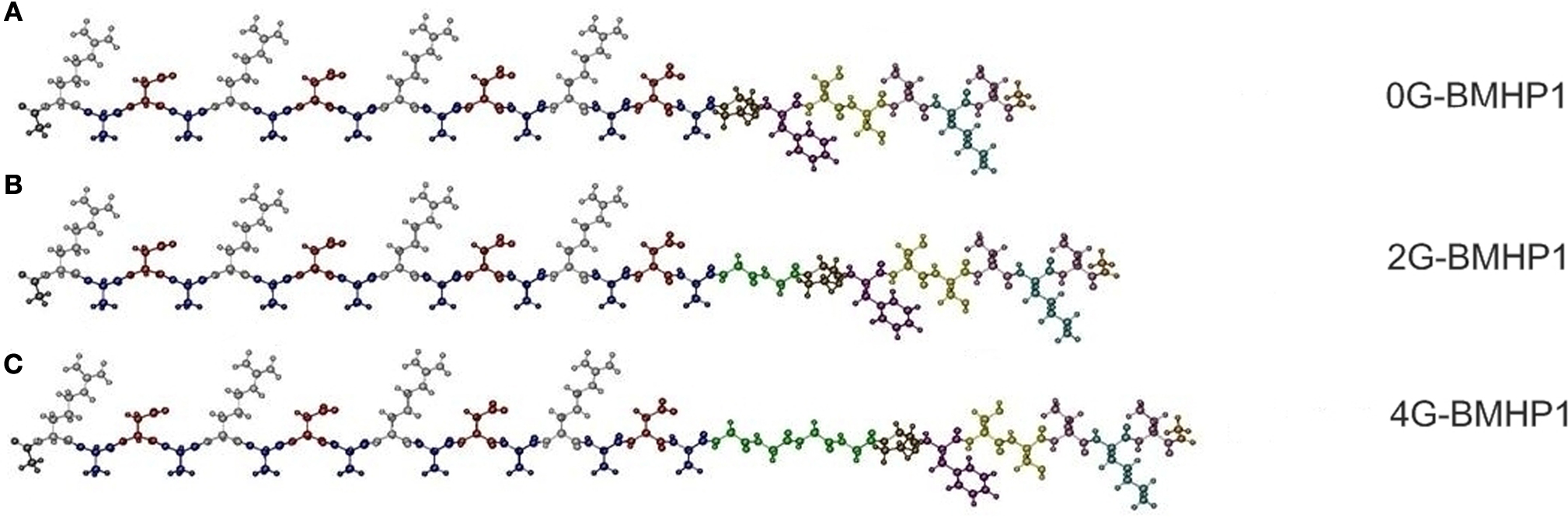
Figure 1. The functionalized self-assembling peptides used in this study. (A) 0G-BMHP1, (B) 2G-BMHP1, (C) 4G-BMHP1. A spacer of two (B) or four glycines (C) (colored in green) was inserted between the self-assembling cores and the functional motif PFSSTKT.
AFM Measurements
Atomic force microscope measurements (in tapping mode) were performed to image the nanostructure of the self-assembling peptides. 2G-BMHP1, 0G-BMHP1 and 4G-BMHP1 formed nanofibers in MilliQ water with comparable dimensions as showed in Figure 2
. In particular the nanostructure of the peptides looks similar to the RADA16-I structure, indeed the average nanofiber widths are 14.0 ± 1.4 nm for 2G-BMHP1, 14.2 ± 1.4 nm for 4G-BMHP1 and 13.0 ± 1.6 nm for 0G-BMHP1. The instrument error (∼10%) has the same order of magnitude of the theoretical width difference among three peptides, then a width variation introduced by the Gs spacer cannot be demonstrated. Nevertheless peptide nanofibers are significantly wider than those of RADA16-I (9.4 ± 1.2 nm) (Taraballi et al., 2009
), an expected finding given by the added functionalized tails flanking the self-assembled cores. Higher resolution measurements of the same peptides were taken via AFM tips with a smaller radius of curvature (2 nm) and confirmed the previous observations (see Insets).

Figure 2. AFM images of FPs solutions at a concentration of 0.05% (w/v). 2G-BMHP1 (B) and 4G-BMHP1 (C) self-assemble into nanofibers (average width: ∼15 nm). 0G-BMHP1 (A) self-organizes into nanofibers shorter than (B) and (C) but featuring similar width. Inserts show high-resolution images of single or few fibers of each peptide at a concentration of 0.01% (w/v).
ATR/FTIR Spectroscopy
Fourier Transform Infrared spectra of the peptide assemblies displayed several absorption bands in the mid-infrared region. Among them we focused our attention on the Amide I band (from 1700 to 1600 cm−1), due to the C=O stretching vibration of the peptide bond, since this particular mode is sensitive to the secondary structure and the β-sheet intermolecular interactions of the peptides (Seshadri et al., 1999
; Natalello et al., 2008
). Second derivatives of the absorption spectra were performed to better resolve the different spectral components overlapped in the Amide I region (Susi and Byler, 1986
). ATR/FTIR measured spectra of each peptide in the different buffer conditions are reported in Figure 3
A, and their second derivative spectra in Figure 3
B.
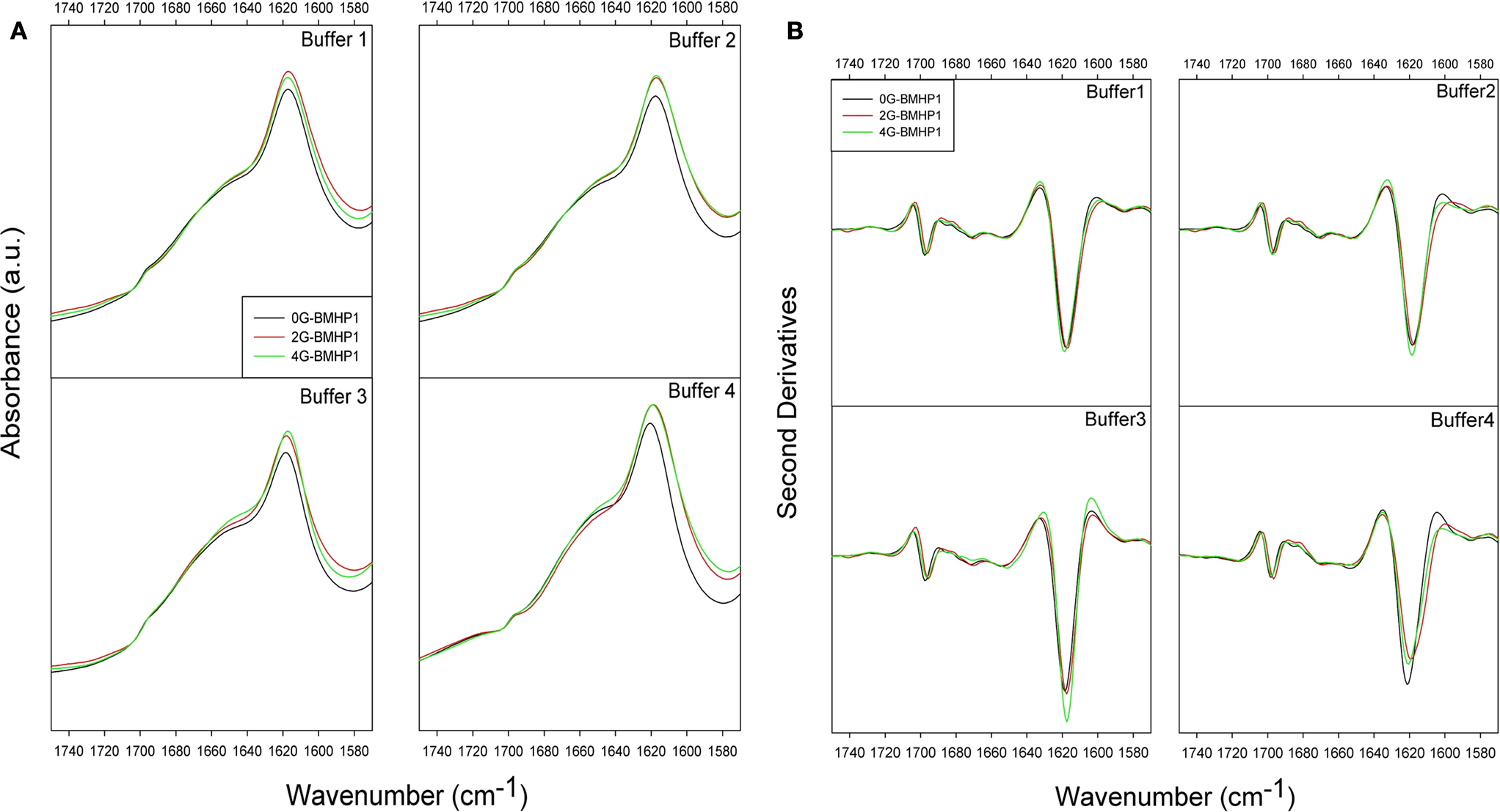
Figure 3. ATR/FTIR absorption spectra of self-assembling peptides. (A) ATR/FTIR absorption spectra of self-assembling peptides. (B) Second derivatives of ATR/FTIR spectra in different buffer conditions. Spectra show the two intermolecular β-sheet bands around 1618 and 1696 cm−1. The highest intensity of the β-sheet components is observed for 4G-BMHP1 in assembling conditions of buffer 3 ((B), buffer 3). 0G-BMHP1 displays an up shift of the 1618 cm−1 component in buffer 4 ((B), buffer 4), indicating a loosely packed assembly.
All peptides display two components centered at ∼1618 cm−1 and at ∼1696 cm−1, typically observed in aggregates as due to intermolecular anti-parallel β-sheets. A broad component around 1653–1649 cm−1 was also observed and assigned to random coil structures, in agreement with the CD analysis (data not shown) (Ye et al., 2008
) and hydrogen/deuterium exchange experiment monitored by FTIR (see Figure S1 in Supplementary Material).
The most significant differences among the tested peptides were observed in the second derivative spectra after pH displacements (buffer 3 and buffer 4). Indeed, among peptides exposed to buffer 3 (assembling condition from pH 2 to pH 8), 4G-BMHP1 displayed the highest 1618 cm−1 peak, indicating that this peptide has the highest amount of intermolecular β-sheet structures. Furthermore, after dilution in buffer 4, the intermolecular β-sheet low frequency band of 0G-BMHP1 was found to up-shift of 2 cm−1, indicating a loosely packed assembly of the peptide deprived of the Gs spacer under this condition.
Raman Spectroscopy
Raman spectra of the hydrated films of the investigated peptides in the 1600–1800 cm−1 range (Amide I region) and in the 2500–3100 cm−1 range are shown in Figures 4
A,B, respectively. The region at lower energy, from 580 cm−1 to 1100 cm−1 (not shown in figure), contains the complex modes of proline (νC–N and νC–C) at around 850–950 cm−1. This region is almost completely dominated by the second order phonon spectrum of the substrate and it is not suitable for reliable analyses. Hints about the secondary structure of peptides can be obtained from the analysis of the amide I region from 1600 to 1700 cm−1. Indeed, the peak conformation and a noticeable asymmetry of the amide I band suggest a distribution of secondary structures. An important peak component above 1700 cm−1 arises from C=O stretching from aspartic acids and is observed in the amide I band especially when the peptides (in particular 4G-BMHP1 and 2G-BMHP1) are dissolved in buffer 1 and buffer 4.
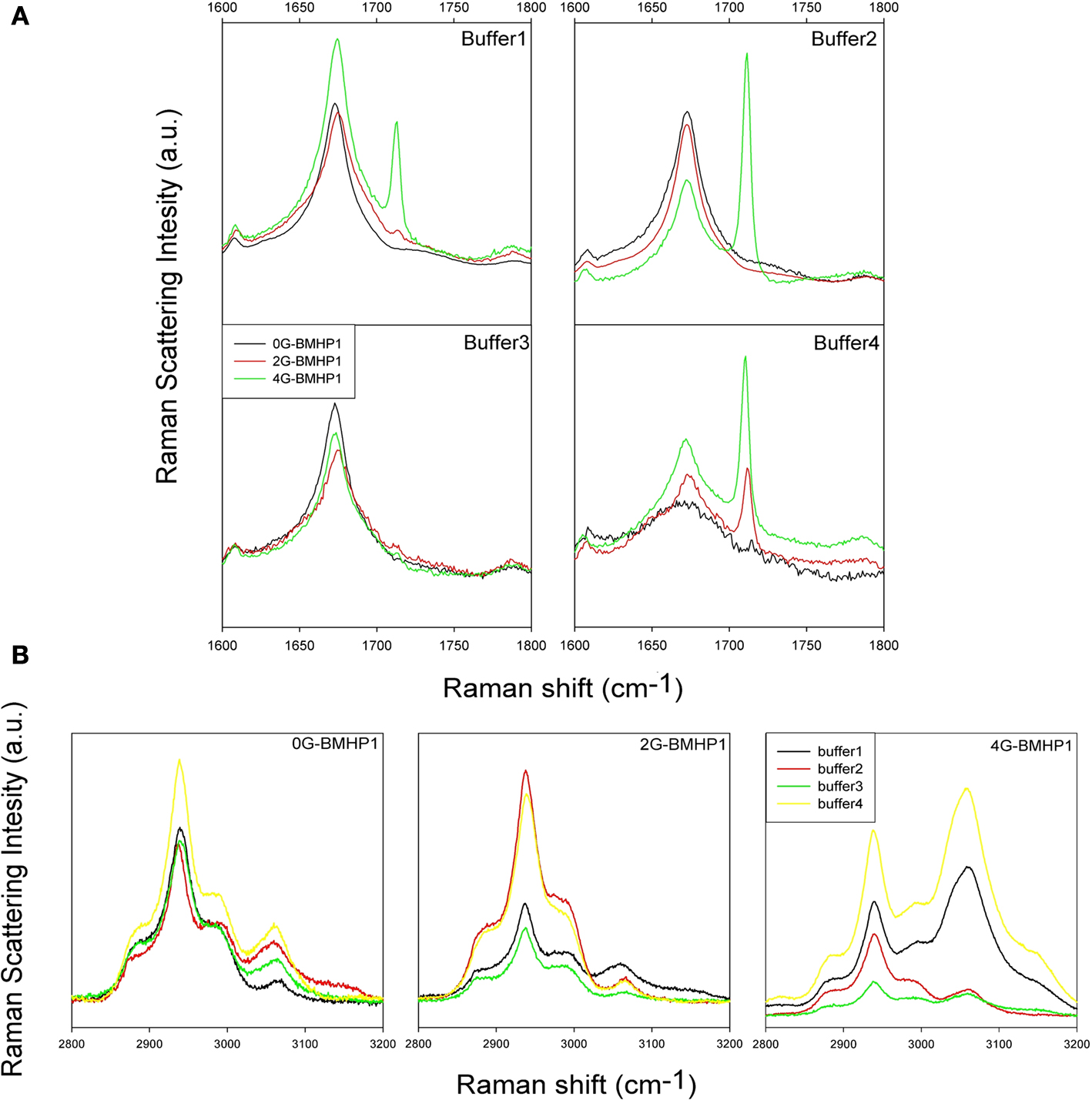
Figure 4. Raman Spectra of tested self-assembling peptides. (A) Raman Spectra (1600–1800 cm−1 region) of the tested self-assembling peptides. Each spectrum shows the peptide Amide I region in all buffered solutions. In buffer 1, buffer 2 and buffer 3, each peptide shows a similar conformation of Amide I peak, suggesting a similar β-sheet conformation of the peptide centered in 1675 cm−1. In buffer 4 the pH shift alters the self-assembled structure of 0G-BMHP1 thus affecting the shape of its Amide I peak and suggesting a wider unordered component. (B) Raman Spectra of 2500–3100 cm−1 region of the tested self-assembling peptides (region of νCH2) exposed to different buffers. The change in the relative intensity between the peak at ∼2940 cm−1 (νCH2) and the peak at ∼3061 cm−1 νCH2 of Phe can be appreciated in case of peptide 4G BMHP1.
Buffer 4 induced significant changes in the shape of amide I region in 0G-BMHP1. In Figure 4
A the amide I peak of 0G-BMHP1 shows a broader shape than those obtained with the other peptides. Moreover, the amide I peak of 0G-BMHP1 shifted from 1675 (other buffers) to 1672 cm−1 (buffer 4).
In Figure 4
B the peak at 2940 cm−1 is due to the CH2 stretching, while the peak at 3060 cm−1 is ascribable to CH2 stretching of Phe (De Gelder et al., 2007
). Therefore, the latter mode is a sensible probe of the status of phenylalanine, containing the sole aromatic ring in the peptide molecules located within the added functional motifs. The analysis of this region can provide useful indications for understanding the folding of the added functional tails. Noteworthy, another phenylalanine phonon mode previously assigned to ring breathing is observed at 1003 cm−1 (Taraballi et al., 2009
) but its partial overlapping with the substrate spectrum compelled us to consider it as insufficiently reliable.
In Figure 5
only 4G-BMHP1 shows a relevant spectral modification related to the buffer conditions. In fact the intensity ratio between peaks at 2940 and 3060 cm−1 is completely interchanged passing from buffer 2 or 3 (i.e. favoring self-assembling) to respectively buffer 1 or 4. The intensity ratio (3061 cm−1/2940 cm−1) in the “not assembling” conditions is ∼1.3 while in the assembling conditions is ∼0.5. The drastic effect on the functional motif in 4G-BMHP1 evinces a larger mobility of the functional motif that makes it more sensitive to the solvent conditions.
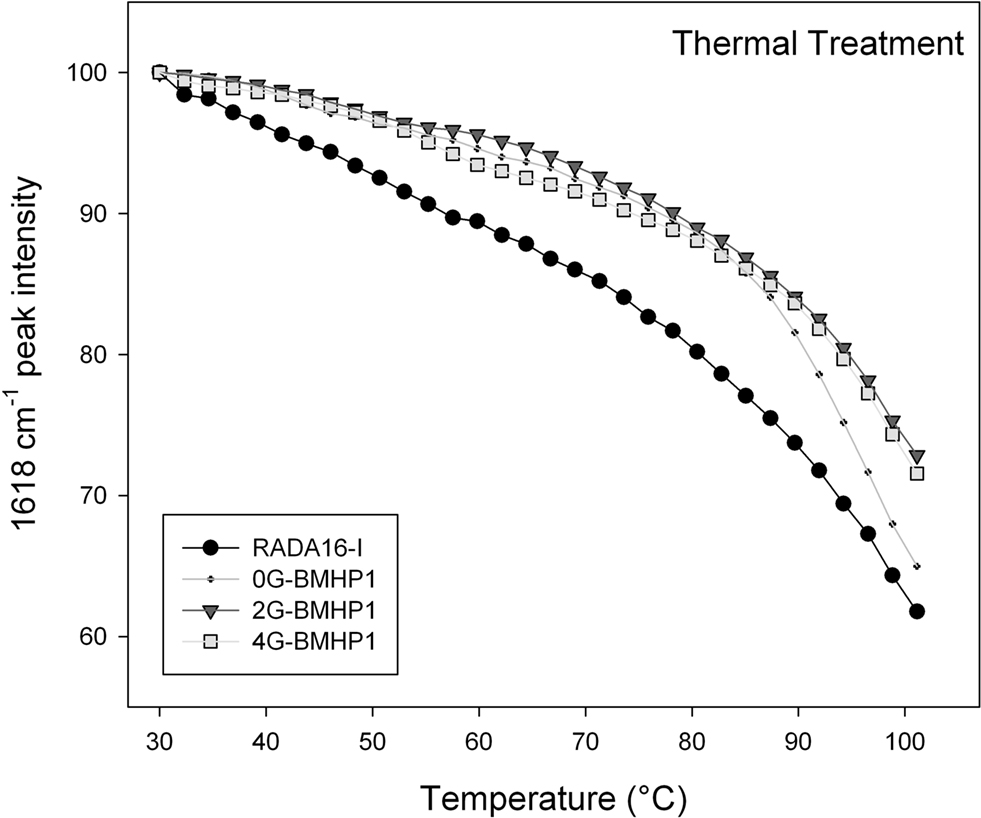
Figure 5. Thermal stability of the self-assembled peptides. Temperature dependence of the intermolecular β-sheet band intensity at 1618 cm−1 of the self-assembly peptides in D2O solution, from 30°C to 100°C. The FPs show a higher stability in comparison with RADA16-I. Noteworthy, 0G-BMHP1 at 100°C reduces its 1618 cm−1 peak intensity at ∼65% similarly to RADA16-I, while 2G-BMHP1 and 4G-BMHP1 at the same temperature decrease to ∼74%. Standard deviation of the data from independent experiments is smaller than the symbol size.
Thermal Stability
To assess the stability of the self-assembled molecular structures we performed a thermal treatment on each tested peptide (see Materials and Methods for details) and on the RADA16-I, included as a positive control. Each peptide was dissolved in D2O and incubated overnight at 4°C. The disassembly of the intermolecular β-sheet structure, induced by thermal treatment, was monitored by FTIR spectroscopy. As the temperature increased, the intermolecular β-sheet band at 1618 cm−1 decreased in intensity and shifted toward higher wavenumbers (see Figure S2 in Supplementary Material), a result that indicated, respectively, a disassembly of the structure and a reduction of its compactness. In addition, after cooling back to 30°C, the peptide assemblies displayed a significant down shift of the 1618 cm−1 band. This effect can be ascribed to the hydrogen/deuterium (H/D) exchange that occurred during the thermal cycle. Therefore, the 1618 cm−1 band variation gives information on the overall stability of the assembled structures. In Figure 5
the 1618 cm−1 peak intensity is reported as a function of temperature. The tested peptides kept their intermolecular β-sheet structure up to 80°C and showed similar thermal unfolding profiles. In particular, RADA16-I self-assembled structure was found to be less stable than those of the FP, as shown by the faster decrease of the 1618 cm−1 band. At approximately 100°C, 2G-BMHP1 and 4G-BMHP1 lost 28% of their initial β-sheet structure, while 0G-BMHP1 and RADA16-I lost, respectively, 34% and 39% of this component.
In Vitro NSC Adhesion and Differentiation
We adopted our well-established in vitro protocol (Gelain et al., 2007a
) to evaluate differences, if any, occurred in NSC adhesion and proliferation depending on the FP substrate used. Negative control (untreated wells) (Figure 6
E) and positive controls (Cultrex) (Figure 6
D) were used. At day 0 the absorbance value was 0.21 ± 0.05 (n = 5). One week after seeding NSC proliferated and adhered as expected with 2G-BMHP1 (Figure 6
B), less intensively in case of 0G-BMHP1 (Figure 6
A), i.e. with isolated clusters of poorly branched cells, and remarkably more for 4G-BMHP1 (Figure 6
C) where branched and differentiating cells formed an almost confluent monolayer. Quantification of NSC proliferation and survival confirmed these observations by showing significant differences between FPs with longer spacers and negative controls (Figure 6
F). On the other hand the absence of any glycine-spacer between the self-assembling sequence and the functional motifs seems to impair the bioactivity of the latter (no significant difference of absorbance values between 0G-BMHP1 and negative control).
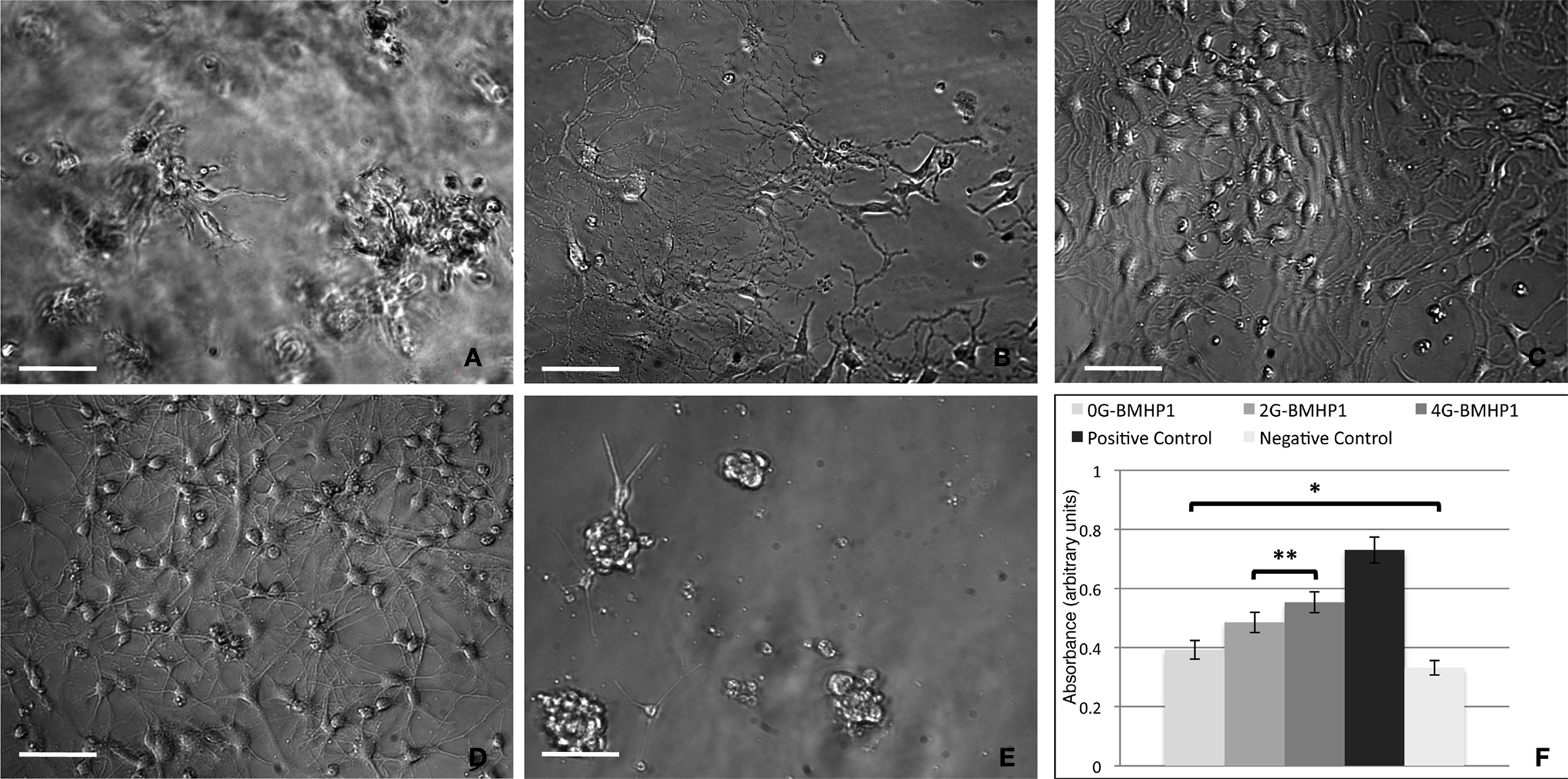
Figure 6. Phase contrast images of differentiating neural stem cells (7 days in vitro) over the tested scaffolds. (A) 0G-BMHP1, (B) 2G-BMHP1, (C) 4G-BMHP1 SAPeptides. (D) Positive and (E) negative controls. 4G-BMHP1 coaxes NSCs to differentiate and survive most effectively while, in case of 0G-BMHP1, small clusters of poorly adhered NSCs testify a possible poor availability of the BMHP1 functional motif for cell membrane receptor binding and consequently cell differentiation pathways activation. Additionally, the “sinking” of NSCs within the assembled scaffold of 0G-BMHP1 (A) testifies a possible lower mechanical stiffness insufficient to bear the weight of NSCs seeded over the top surface of the substrate. Scale bars are 100 μm. CellTiter results (F) show significant differences for all possible coupled experimental groups (P < 0.05) except for (*) 0G-BMHP1 vs negative control, and (**) 2G-BMHP1 vs 4G-BMHP1. Values are reported as means ± standard error of the mean.
The same in vitro protocol was adopted to test the degree of differentiation of NSC vs Gly-spacer length at 7 days in vitro (Figure 7
). Fractions of GFAP+ cells increased with spacer length and, inversely, Nestin+ cells decreased. In case of both markers, NSCs differentiated over all SAPs show values significantly different from positive control. Nonetheless percentages of β-Tubulin+ and GalC/O4+ cells did not correlate with the length of the spacers.
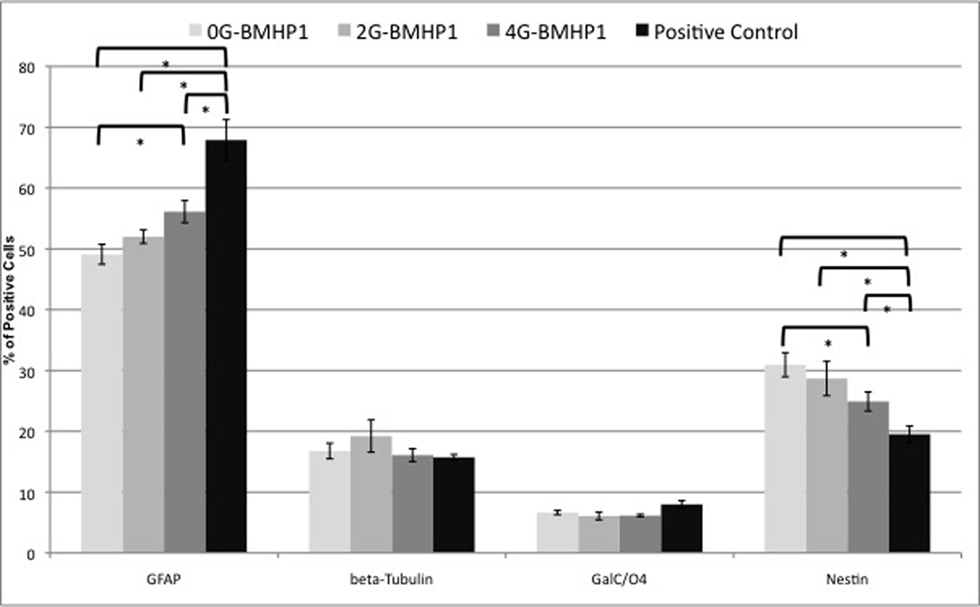
Figure 7. Quantitative NSCs differentiation assay. Data are expressed as percentages of cells positive for GFAP, β-Tubulin, GALC/O4 and Nestin markers over the total number of cells counted in the same fields (see Materials and Methods for details). NSCs have been differentiated for 7 days in vitro. By increasing the length of the Gly-spacer, the percentage of Nestin+ significantly decreased. Despite that GFAP+ cell fraction increased inversely. β-Tubulin+ and GalC/O4+ cell percentages did not significantly differ among the tested substrates and assembled SAPs. NSCs cultured over plastic well surfaces (Negative Control), being composed of clusters of mainly Nestin+ cells, were not included in this graph. Values are expressed as means ± standard error of the mean. * = significantly different values (P < 0.05).
Our accurate structural investigation led up to a better understanding of new designing rules for new self-assembling peptides suited for neuroregenerative applications.
The different peptides tested in our work differ for the presence of a couple of glycine-spacers between the self-assembly sequence and the biological motif. In previous publications(Gelain et al., 2007a
) the Gs spacer was introduced as an hinge between the two part of SAPpeptide, ground on the hypothesis that the presence of lateral spacer of glycines should confer higher mobility to the bioactive motifs (Gelain et al., 2007a
). But, to the best of our knowledge no structural study validated this hypothesis with functionalized self-assembling peptides.
Our AFM studies revealed that the nanostructure of each tested peptide is similar to that one of RADA16-I (Figure 2
). The only difference found was the length of the nanofibers: in fact Figure 2
shows that the nanofibers formed by 0G-BMHP1 are shorter than those formed by 2G-BMHP1 and 4G-BMHP1. Our hypothesis is that the self-assembled nanofibers of 0G-BMHP1, lacking of Gs, show either higher stiffness and allows closer electrostatic interactions between the added functional motif and the self-assembling core, thus respectively making the nanofibers less resistant to solvent stresses and interfering with the stability of the self-assembling cores. We performed Raman and FTIR spectroscopy measurements to validate our hypothesis and to understand the direct contribution conferred by the Gs to the stability of self-assembled nanostructures.
The ATR/FTIR measurements carried out on hydrated films from the peptide solutions show that all the tested peptides assembled similarly forming anti-parallel β-sheets, as indicated by the Amide I components at 1618 cm−1 and at 1696 cm−1 (see Figure 3
). Therefore, the presence or the absence of the chosen Gs spacers does not seem to prevent the formation of intermolecular β-sheet structures in the tested solvent conditions. Additionally, our data point out that 0G-BMHP1 nanostructure is less stable than those of the other two FPs for two main reasons: (i) when exposed to buffer 4, the shift of the 1618 cm−1 component toward higher wavenumbers suggests a relaxation of its intermolecular β-sheet structure and (ii) the thermal treatment indicate a lower stability of this peptide, which is the most sensitive to temperature. FTIR spectra of peptide 4G-BMHP1 in buffer 3 show a more intense component of β-sheets, a result in agreement with the thermal treatment analysis, in which 4G-BMHP1 appeared to be more stable than 0G-BMHP1 and RADA16-I. The FTIR data are in accordance with those of the Raman spectroscopy.
Longer glycine-spacers increase the peptide degrees of freedom, leading to an extended intermolecular β-sheet structure driven by the self-assembling sequence, thus contributing to the stability of the overall molecular structure. This improvement in the structural stability, even when compared with that one of RADA16-I, is likely to be shared by other FPs with longer glycine-spacers. In addition, the four-Gs improved the exposure of the functional motif, as indicated by Raman spectroscopy and by the incremented viability of cultured NSCs.
Raman results provide three main indications confirming our model: (i) the analysis of the amide I mode shows that the total lack of the glycine-spacer (data of 0G-BMHP1) affects the stability of the β-structure, in agreement with ATR-FTIR results; on the other hand, (ii) no relevant effect is observed in this region as a function of the spacer length, up to 4 glycines, suggesting that a long spacer is not necessarily detrimental to the assembling features of the peptide; (iii) the analysis of the of the CH2 bound to the Phe aromatic ring modes evinces that longer glycine-spacers make the bioactive motifs sensibly more exposed to the solvent, in agreement with the NSC results in vitro. In fact this is clearly visible in Figures 6
C,F, proving the potential of these peptides as scaffolds for cell cultures and in vivo transplantation therapies. 0G-BMHP1 fails to favour NSC proliferation and adhesion and, at the same time, the cells fall in the hydrogel scaffold suggesting that the biomechanical nanostructured substrate stiffness is insufficient for supporting seeded cells. This finding is in accordance with AFM results showing nanofibers with different lengths in the case of 0G-BMHP1. Nonetheless, while 4G-BMHP1 shows similar nanostructures to those of 2G-BMHP1, it is also more effective in stimulating NSC adhesion and, consequently, enhancing their survival in a differentiating medium. Indeed quantitative data of the differentiation of NSCs progenies show an important influence of the number of glycines over the fraction of differentiating astrocytes and nestin positive immature cells. Other phenotypes did not seem to be dramatically influenced by the functional motif exposure, however this may be due to the specific two-dimensional cell assay we developed. It is indeed widely known that neurons and oligodendrocytes may preferentially differentiate over the astrocyte layer that spontaneously develops in standard two-dimensional cultures, thus de facto decreasing the influence of the functional motifs incorporated in the self-assembled scaffold underneath. Thanks to the enhanced functional motif exposure to the solvent and to its higher degrees of freedom we can reasonably postulate that the statistical probability for the PFSSTKT functional motif of being available to cell membrane receptor binding in the desired spatial orientation for peptide-protein docking is improved.
This study describes an ensemble of complementary approaches useful to investigate the self-assembled structures of peptides derived from the functionalization of RADA16-I. It elucidates not only how the FPs are more stable than RADA16-I in respect to temperature and pH variations, but that the insertion of longer glycines-spacers between the self-assembling cores and the functional motifs improves the effectiveness of SAPeptides functionalization, thus providing better tools for in vitro studies in cell biology and for applications in neuroregenerative therapies in vivo.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank to Carolina Volontè and Diego Silva for their precious help in the cell culture and scaffold characterization experiments. First and corresponding authors thank CARIPLO foundation and Regione Lombardia for their financial support. Silvia Maria Doglia acknowledges the financial support of the F.A.R. (Fondo d’Ateneo per la Ricerca) and A.N. the post-doctoral fellowship of the University of Milano-Bicocca.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/neuroengineering/paper/10.3389/neuro.16/001.2010/
Davis, M. E., Hsieh, P. C., Takahashi, T., Song, Q., Zhang, S., Kamm, R. D., Grodzinsky, A. J., Anversa, P., and Lee, R. T. (2006). Local myocardial insulin-like growth factor 1 (IGF-1) delivery with biotinylated peptide nanofibers improves cell therapy for myocardial infarction. Proc. Natl. Acad. Sci. U.S.A. 103, 8155–8160.
Natalello, A., Prokorov, V. V., Tagliavini, F., Morbin, M., Forloni, G., Beeg, M., Manzoni, C., Colombo, L., Gobbi, M., Salmona, M., and Doglia, S. M. (2008). Conformational plasticity of the Gerstmann–Straussler–Scheinker disease peptide as indicated by its multiple aggregation pathways. J. Mol. Biol. 381, 1349–1361.