 Polyxeni Kalisperati1†
Polyxeni Kalisperati1† Evangelia Spanou1†
Evangelia Spanou1† Ioannis S. Pateras2
Ioannis S. Pateras2 Penelope Korkolopoulou3
Penelope Korkolopoulou3 Anastasia Varvarigou4
Anastasia Varvarigou4 Ioannis Karavokyros5
Ioannis Karavokyros5 Vassilis G. Gorgoulis2,6,7Panayiotis G. Vlachoyiannopoulos8Stavros Sougioultzis1*
Vassilis G. Gorgoulis2,6,7Panayiotis G. Vlachoyiannopoulos8Stavros Sougioultzis1*- 1Gastroenterology Unit, Department of Pathophysiology, School of Medicine, National and Kapodistrian University, Athens, Greece
- 2Department of Histology and Embryology, School of Medicine, National and Kapodistrian University, Athens, Greece
- 31st Department of Pathology, Laiko Hospital, School of Medicine, National and Kapodistrian University of Athens, Athens, Greece
- 4Department of Pediatrics, University of Patras Medical School, Patras, Greece
- 51st Department of Surgery, Laiko Hospital, University of Athens, School of Medicine, Athens, Greece
- 6Biomedical Research Foundation of the Academy of Athens, Athens, Greece
- 7Faculty of Biology, Medicine and Health Manchester Cancer Research Centre, Manchester Academic Health Sciences Centre, The University of Manchester, Manchester, UK
- 8Department of Pathophysiology, School of Medicine, National and Kapodistrian University, Athens, Greece
Helicobacter pylori (H. pylori) is a Gram negative bacterium that colonizes the stomach of almost half human population. It has evolved to escape immune surveillance, establishes lifelong inflammation, predisposing to genomic instability and DNA damage, notably double strand breaks. The epithelial host cell responds by activation of DNA damage repair (DDR) machinery that seems to be compromised by the infection. It is therefore now accepted that genetic damage is a major mechanism operating in cases of H. pylori induced carcinogenesis. Here, we review the data on the molecular pathways involved in DNA damage and DDR activation during H. pylori infection.
Introduction
Rudolf Virchow was the first to propose a potential association between chronic inflammation and cancer, based on his observations of inflammatory cells presence in human malignant tissues. Eventually, inflammation was established as an important hallmark of cancer (Hanahan and Weinberg, 2011). Approximately, 20–25% of cancer cases are attributed to chronic inflammation (Ames et al., 1995; Mantovani et al., 2008).
Accumulating evidence suggest that chronic inflammation either non-infectious such as in autoimmune disorders, or as a result of pathogen infection is connected to cancer development. Characteristic examples of autoimmune disorders that may promote tumorigenesis are celiac disease and Sjögren syndrome (Austad et al., 1967; Voulgarelis et al., 2012). Several infectious agents have been associated with carcinogenesis including hepatitis B and C viruses, strongly associated with hepatocellular carcinoma, human papilloma virus (HPV) with cervical cancer and H. pylori with gastric cancer (Plummer et al., 2016). Infection-related cancer is caused either by direct tumorigenic effect of the pathogen, or indirectly through the accompanying inflammation and the subsequent genomic instability (Cortes-Bratti et al., 2001; Coussens and Werb, 2002; Scanu et al., 2015).
Inflammation as well as several endogenous or exogenous agents, may result in DNA lesions which in turn promote DNA repair pathways and interception of DNA replication, a mechanism known as replication stress (Berti and Vindigni, 2016). In particular, inflammation induces a vicious cycle among continuous injury and repair, resulting in aberrant cell proliferation that favors replication stress. Inflammation-induced replication stress enhances DNA damage, which subsequently induces DNA damage response (DDR), genomic instability and finally tumorigenesis (Coussens and Werb, 2002).
Herein, we review the interplay between pathogens and especially H. pylori, inflammation, genomic instability, and tumorigenesis.
Double Strand Breaks (DSBs) and DNA Damage Response (DDR) Pathway-Overview
Human cells and their genome are under constant attack by DNA-damaging agents, resulting in 10s of 1000s of DNA lesions daily (Lindahl and Barnes, 2000). DSBs are lethal and difficult to repair (Khanna and Jackson, 2001; Rouse and Jackson, 2002; Harper and Elledge, 2007; Jackson and Bartek, 2009).
The cell responds to DNA lesions by activating a complex mechanism, named DDR, which detects and then initiates signaling in order to repair DNA (Harrison and Haber, 2006; Harper and Elledge, 2007). There are several DDR pathways, according to the type of DNA lesion. As far as DSBs are concerned, non-homologous end-joining (NHEJ) and homologous recombination (HR), are the major DNA repair mechanisms (Lieber, 2008; San Filippo et al., 2008).
Double strand breaks are detected by the DNA damage sensors namely MRN complex (Mre11-Rad50-Nbs1) which in turn orderly recruits other agents of the DDR pathway (Lee and Paull, 2005; Jackson and Bartek, 2009; Polo and Jackson, 2011; Roos and Kaina, 2013).
In brief, DSBs promote activation of ATM and ATR, resulting in H2AX histone phosphorylation at Ser 139 (γH2AX) an initial step toward DNA repair (Shiloh, 2003; Bartek and Lukas, 2007; Cimprich and Cortez, 2008; Cook et al., 2009; Xiao et al., 2009). γH2AXformation activates the transducers Chk1 and Chk2 leading to p53 activation (Kastan and Bartek, 2004; Bartek and Lukas, 2007; Riley et al., 2008). As soon as p53 is activated, cell cycle arrest is triggered, in order for the DNA lesion to be repaired. Alternatively, if the lesion cannot be repaired apoptosis or premature senescence are promoted. All the above p53-induced responses are tumor suppressing.
A novel model of tumorigenesis was proposed by Bartkova et al. (2005) and Gorgoulis et al. (2005), suggesting that DNA replication stress enhance DSBs formation, leading to genomic instability and selective pressure for p53 mutations, abrogating the tumor suppressing actions of p53 (Halazonetis et al., 2008).
If the DDR pathway is ineffective either by overload or p53 mutations, faithful DNA repair is compromised resulting in genomic instability (Negrini et al., 2010). Notably, the DDR pathway, through its upstream kinase ATM, also keeps in check another major tumor suppressor factor, namely p14ARF (Velimezi et al., 2013; Wallace et al., 2014) that functions as a second anti-tumor barrier to DDR activation (Evangelou et al., 2013). Thus p53 inactivation can have detrimental effect(s) since both these anti-tumor routes are compromised.
Inflammation, Genomic Instability, and Tumorigenesis
Chronic inflammation results in inflammatory cell infiltration and production of cytokines in tissues. Inflammatory cells of innate immunity such as macrophages, MDSCs, neutrophils, and dendritic cells are known to present tumor enhancing activity (Gabrilovich and Nagaraj, 2009; Grivennikov et al., 2010; Terzic et al., 2010).
Recently, Pereira-Lopes et al. (2015) reported that macrophage functions, such as proliferation, ROS and cytokine production, are mediated through DDR and in particular, by the NBS1 protein, part of the MRN sensor complex. They studied mice carrying NBS1 hypomorphic protein and found impairment of DDR, accumulation of DSBs, defects in macrophage proliferation, increased production of cytokines and increased rate of macrophage senescence. Taken together their findings suggest the role of DDR in controlling immunopathology and facilitating tissue repair during inflammation and infections (Colonna, 2015).
Inflammatory tissues contain several growth factors, such as epidermal growth factor (EGF), platelet derived growth factor (PDGF), fibroblast growth factor (FGF), TGF-α, TGF-β, insulin-like growth factors 1 and 2 that directly promote cell proliferation, replication stress and eventually DSBs formation (Cianfarani et al., 1998; Jakowlew, 2006; Aivaliotis et al., 2012). In addition, cytokines such as IL-1β, IL-6, TNF-α and IFN-γ induce the formation of RONS that trigger DNA mutations and epigenetic alterations affecting the proteins responsible for cell cycle control or survival (Colotta et al., 2009; Jurk et al., 2014; Kiraly et al., 2015).
A characteristic example of the interaction between inflammation, cytokines and DDR is the SASP. SASP refers to the production of various substances by the senescent cell, including inflammatory cytokines (e.g., IL-6, IL-8), proteases and growth factors (Bavik et al., 2006; Coppe et al., 2008; Malaquin et al., 2013) that act as inflammatory stimulators and affect neighboring non-senescent cells, in a paracrine fashion. Depending on the microenvironment, SASP may act either as anti-tumorigenic factor favoring senescence, or as oncogenic stimulant (Gorgoulis and Halazonetis, 2010). Specifically, SASP factors enhance senescence in normal and low grade preneoplastic cells but promote tumor development in high grade preneoplastic or cancerous cells. SASP is considered a delayed response, taking days to operate and is driven by DDR which is constantly active in senescent cells. It is a means of long-term modulation of the microenvironment by senescent cells, and despite the lack of mechanistic details thus far, it adds to the notion that, inflammation, DDR, senescence and cytokines contribute to cancer development (Coppe et al., 2008; Rodier et al., 2009).
Pathogens and DDR
Many virulence factors induce host DNA damage and DDR activation. Viruses such as EBV, Herpes-Simplex virus 1,2 (HSV 1,2), cytomegalovirus (CMV), hepatitis C virus (HCV), HPV, are all associated with cancerous pathologies, triggering DDR activation (Georgakilas et al., 2010; Pateras et al., 2015).
Bacteria such as Escherichia coli, Campylobacter jejuni, Streptococcus bovis, and H. pylori are known to induce DNA damage in host cells (Nougayrede et al., 2006; Liyanage et al., 2010). Especially, S. bovis is strongly associated with colorectal cancer. It is still unknown whether bacteria or their toxins induce tumorigenesis directly by cell transformation (Cortes-Bratti et al., 2001; Scanu et al., 2015) or indirectly due to the accompanying chronic inflammation (Coussens and Werb, 2002) or whether both mechanisms operate.
H. pylori and its Implication in DDR and Carcinogenesis
In 2005, the Nobel Prize in Physiology or Medicine was awarded to Barry Marshall and Robin Warren, for their discovery of the bacterium H. pylori and its role in gastritis and peptic ulcer disease (Marshall and Warren, 1984). The bacterium is the major cause of peptic ulcer, gastric cancer, and lymphoma of Mucosa Associated Lymphoid Tissue (MALT) (Marshall and Warren, 1984; Parsonnet et al., 1997; Parsonnet and Isaacson, 2004). H. pylori colonizes the gastric mucosa of almost half the world population and although provokes a robust inflammatory response of the gastric mucosa, evades eradication leading to lifelong infection.
Despite the fact that H. pylori is associated with persistent gastric mucosa inflammation, the great majority of the infected population (∼80%) remains asymptomatic. Three important factors are associated with the development of symptomatic H. pylori disease: (1) H. pylori’s virulence factors (2) host susceptibility and response and (3) environmental cofactors, including smoking and diet (Atherton, 2006).
H. pylori’s Virulence Factors
Helicobacter pylori’s CagA, encoded by the cagA gene within the CagPAI of H. pylori genome, is the most important of its virulent factors. CagA(+) strains induce enhanced inflammation of the gastric mucosa and have stronger association with gastric cancer. CagA protein is injected by the bacterium through a type IV secretion system (TFSS) and hijacks the host cell molecular machinery by interfering with multiple host signaling pathways including NFκB and MAPKs (Keates et al., 2001), affecting apoptosis, cell growth and motility (Odenbreit et al., 2000; Lin et al., 2010; Murata-Kamiya, 2011). CagA(+) strains also induce higher levels of expression of the proinflammatory cytokines TNFα, IL-1β, and IL-8 that trigger oxidative stress and oxidative DNA damage in the infected mucosa, thus promoting genomic instability and tumorigenesis (Blaser et al., 1995; Peek et al., 1995; Eftang et al., 2012). Other H. pylori virulence factors, such as VacA and NapA, also augment the inflammatory response and oxidative stress (Kim et al., 2007).
Host Response to H. pylori Infection
Helicobacter pylori-induced host inflammatory response rather enhances than attenuates its pathogenicity. Both innate and adaptive host responses are activated during the infection (Hardbower et al., 2014). Neutrophils and macrophages trigger ROS and NO production, create oxidative stress and tissue damage. The bacterium survives oxidative stress by the production of the enzymes Nap A, catalase and superoxide catalase; thus, leading to persistent immunocyte infiltration in the gastric mucosa and further enhancement of oxidative burst (Wilson et al., 1996; Ramarao et al., 2000).
Adaptive immune response in H. pylori infection is mainly mediated by Th1 and Th17 T cells that secrete INFγ and IL-17 and are thought to further induce inflammatory cells influx and perpetuate mucosal damage (D’Elios et al., 1997; Szabo et al., 1999). VacA, Cag A, gamma-glutamyltranspeptidase, and arginase produced by H. pylori alter T-cells responses by interfering with T cell proliferation, activation and apoptosis (Gebert et al., 2003; Gerhard et al., 2005; Wang et al., 2010; Larussa et al., 2015). Interestingly, the bacterium, through dendritic cells, can induce T-regulatory(Treg)/TGF-β activation that blunts Th1/Th17 responses, early in the course of the infection. It has been suggested that the attenuation of Treg responses during progression of the infection shifts the balance leading to more inflammation and more oxidative DNA damage (Harris et al., 2008; Kao et al., 2010; Wang et al., 2010).
Furthermore, host polymorphisms affecting cytokines and especially IL-1β, TNF-α, and IL-10 are considered as important factors of host susceptibility to peptic ulceration and gastric adenocarcinoma, linking local inflammatory responses with tumorigenesis (El-Omar et al., 2000; Machado et al., 2001; Furuta et al., 2002; Hwang et al., 2002; Rad et al., 2004). Individuals infected with more virulent strains of H. pylori and carriers of multiple cytokine polymorphisms are considered to be more susceptible to gastric cancer development.
H. pylori Promotes Gastric Carcinogenesis
Since 1990s when epidemiologic studies provided evidence for the association between H. pylori and gastric cancer and the bacterium was classified as human carcinogen (Forman et al., 1991; Nomura et al., 1991; Parsonnet et al., 1991), our knowledge on the mechanisms of H. pylori induced carcinogenesis has been greatly improved and still evolving, due to the rigorous research in this field.
Several reports also suggest a putative role of EBV in gastric carcinogenesis, more evident in gastric stump and non antral tumors (Murphy et al., 2009). Recent data from the Cancer Genome Atlas (TCGA) project led to the classification of gastric cancer into four subtypes: EBV related, microsatellite unstable, genomically stable, and CIN tumors; each type is characterized by specific molecular defects and elevated expression of DDR pathways (Cancer Genome Atlas Research Network, 2014). Although the significance of EBV relationship to gastric carcinogenesis is largely unexplored, studies clearly show that eradication of H. pylori reduces the risk of gastric cancer development, especially in patients without precancerous lesions (Wong et al., 2004; Ford et al., 2014).
According to the Correa model, H pylori infection induced gastritis progresses to gastric cancer through the premalignant stages of gastric atrophy, IM, and dysplasia (Correa, 1992). More recent evidence suggests that H. pylori related IM may result from SPEM, a metaplastic epithelium developing during the infection as a result of parietal cells loss and trans-differentiation of chief cells. Persistent inflammation may advance SPEM into a more proliferative metaplasia and to adenocarcinoma development (Weis and Goldenring, 2009). Nevertheless, the majority of H. pylori carriers are asymptomatic and although almost 20% acquire preneoplastic changes, approximately only 2% will develop gastric cancer and lymphoma (Peek and Blaser, 2002).
Gastric carcinogenesis in H. pylori infection is affected by several host and bacterial virulence factors. Dysregulation of cellular homeostasis in gastric mucosa is the main result of host response to the pathogen, favoring chronic inflammation, DNA lesions, and finally tissue damage (Preston-Martin et al., 1990; Algood et al., 2007). BMDCs that migrate to gastric mucosa in the context of H. pylori infection may aid to the neoplastic process, although evidence is mainly from animal model studies (Houghton et al., 2004; Bessede et al., 2014).
A dysregulation of apoptosis, including its induction or inhibition may play a key-role in H. pylori tumorigenesis. Induction of apoptosis results to gastric atrophy, hypochlorhydria and possibly to BMDCs recruitment, promoting tumor growth. Inhibition of apoptosis attenuates defense mechanisms of host against DNA damage, accumulates genetic errors and promotes cell malignant transformation (Sougioultzis et al., 2003; Ricci et al., 2011).
It should be mentioned that both experimental and human evidence suggest that H. pylori also induces mitochondrial DNA (mtDNA) mutagenesis which seems to enhance oxidative stress and contribute to gastric cancer development (Machado et al., 2009).
H. pylori Triggers DDR
Bartkova et al. (2005) as well as Gorgoulis et al. (2005) have demonstrated that phosphorylated 53BP1, ATM, H2AX, Chk2 and p53, are indicative of DDR activation in premalignant lesions, following DSBs formation. Xie et al. (2014) studied γH2AX expression in human gastric tissue samples, irrespective of H. pylori infection. The levels of γH2AX gradually increased from chronic gastritis, IM, to dysplasia and were higher in the presence of H. pylori. In gastric cancer γH2AX was also expressed, but in lower levels than in the aforementioned premalignant lesions (Xie et al., 2014).
Toller et al. (2011), examined the activation of DDR pathway, in transformed cells (AGS) infected with H. pylori. They concluded that H. pylori infection induces the formation of DSBs through a direct host–pathogen contact and triggers DDR pathway, as assessed by the phosphorylation of H2AX. DSBs due to H. pylori are continuously repaired, although prolonged infection may compromise DDR and result to unrepaired breaks (Toller et al., 2011).
Hanada et al. (2014), reported that ATM is activated in formalin fixed H. pylori infected human mucosa tissue samples, as well as in H. pylori infected AGS cultured cells. DDR activation was also confirmed by γH2AX expression in H. pylori infected AGS cells that co-expressed ATM. They also observed that both CagA+ and CagA- strains induce DNA damage, albeit CagA+ strains are related with a greater DNA damage and more potent DDR activity (Hanada et al., 2014).
Koeppel et al. (2015) recently reported that H. pylori induces a specific pattern of DNA damage in infected cells, different from other mutagenic agents, mostly affecting chromosomal ends, resulting in telomeres loss and CIN, genetic alterations that are implicated in gastric carcinogenesis. They also observed that H. pylori compromises DDR by inhibiting several factors involved, such as ATR, MRE11, and NBS1. In another series of experiments Hartung et al. (2015)showed that DSBs in H. pylori infected cells are introduced by XPF/XPG endonucleases in a type IV secretion system (T4SS)-dependent manner that requires NF-κB/RelA activation. Interestingly, DSBs induced by the bacterium promote NF-κB target gene transactivation and host cell survival (Hartung et al., 2015; Koeppel et al., 2015).
Although limited, both in vitro and in vivo evidence suggest, that H. pylori infection promotes DSBs formation either directly through host–pathogen contact or indirectly due to the accompanying chronic inflammation, eventually resulting to DDR activation (Table 1). Persistent infection and associated inflammation may compromise the DDR pathway leading to mutations of p53, unrepaired DNA damage and even tumorigenesis. Indeed, p53 mutation is a frequent (32%) and early event in sporadic gastric cancer (Ochiai et al., 1996; Petitjean et al., 2007) (Figure 1).

TABLE 1. Reviewed bibliography on DDR activation and H. pylori.
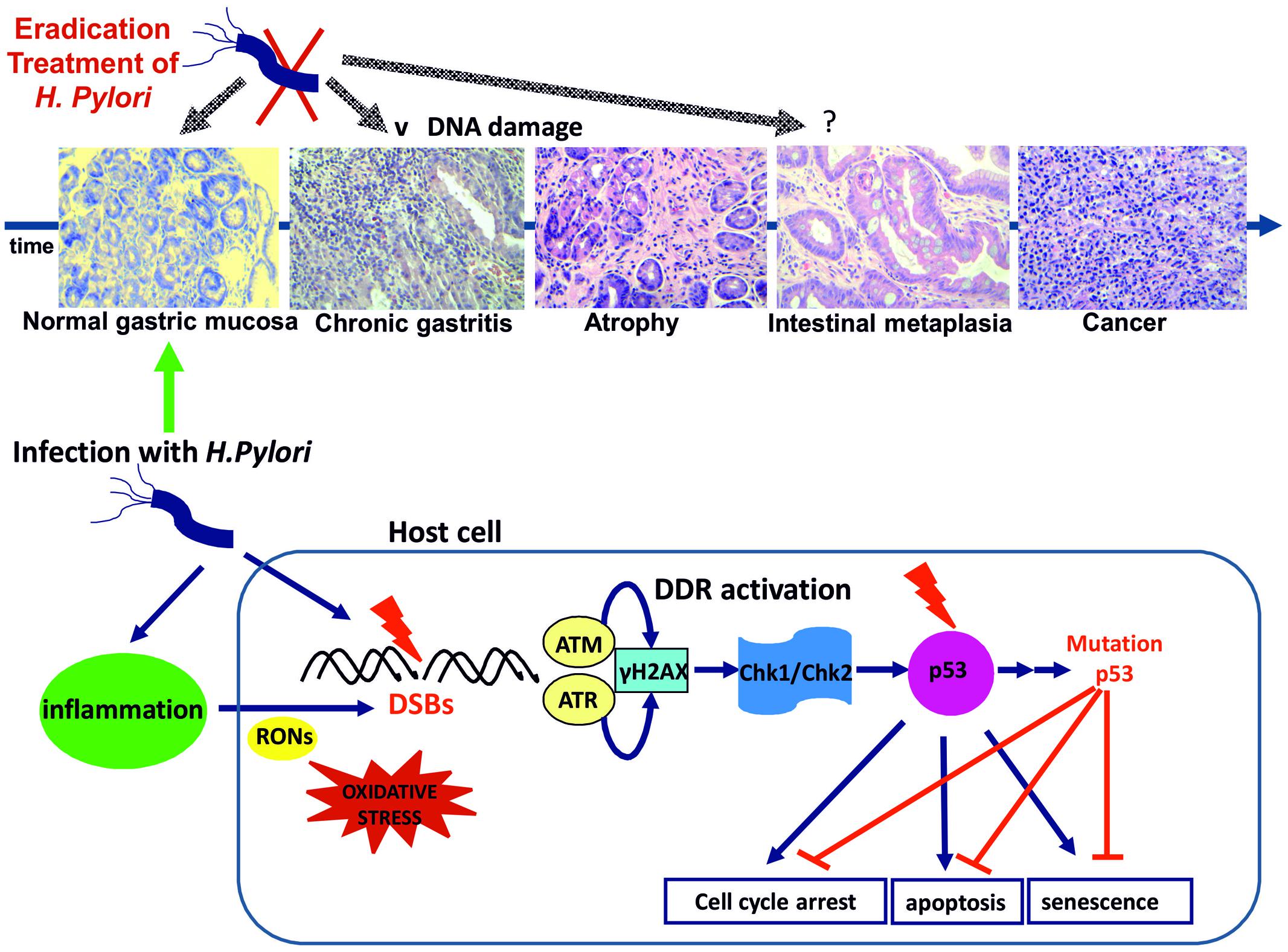
FIGURE 1. Helicobacter pylori infection results to DSBs possibly through a direct pathogen-host interaction. Inflammation through oxidative/nitrosative stress seems to play a significant role. DSBs formation triggers DDR activation through ATM and ATR activation and subsequent phosphorylation of H2AX at Ser 139 (γH2AX). γH2AX formation activates the transducers Chk1 and Chk2 leading to p53 activation and promoting cell cycle arrest for DNA lesion repair or alternatively induction of apoptosis or premature senescence. DNA replication stress results to genomic instability and selective pressure for p53 mutations abrogating the tumor suppressing actions of p53 (Bartek and Lukas, 2007). H pylori infection induced gastritis progresses to gastric cancer through the premalignant stages of gastric atrophy, IM, and dysplasia according to the Correa model (representative histology slides, eosin-hematoxylin photos x200). H. pylori eradication at the early stages of gastritis and atrophy may result to attenuation of DNA damage and DDR activation. IM is considered as “a point of no return” and p53 mutations are common in the metaplastic epithelium (Ochiai et al., 1996; Xie et al., 2014).
Future Perspectives
There is little doubt nowadays that H. pylori is a risk factor for gastric cancer development. Gastric mucosa inflammation, oxidative burst and the resultant altered epithelial cell turnover are implicated. Accumulating evidence, as briefly outlined above, suggest that the bacterium causes DNA damage to the host cells and triggers DDR. Interestingly, H. pylori seems to compromise the integrity of DDR by affecting various proteins of the pathway and, in parallel, maintains its niche by promoting cell survival through DSBs induced NFκB gene-transactivation (Hartung et al., 2015; Koeppel et al., 2015). Hence, H. pylori triggers the DDR and immune response crosstalk promoting a vicious cycle of DNA damage and persistent inflammation that fuels tumorigenesis.
DNA damage repair activity seems to decrease after eradication of H. pylori but probably persists in areas of IM which is considered “a point of no return,” meaning that it is not reversible after H. pylori clearance and frequently contains p53 mutations. Studying the DDR pathway in H. pylori related IM with mutated vs. wild p53 will likely provide useful molecular data on the development of the metaplastic-precancerous epithelium and lead to targeted therapeutic interventions for gastric cancer prevention.
Author Contributions
PKa, ES, IP designed the report. PKa, ES, IP, AV, and IK reviewed the bibliography. PKa and ES collected the data and wrote the paper. PKa, ES, IP, and VG performed the immunohistochemical analysis. PKo performed the pathological analysis. SS, PV, and VG supervised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
BMDCs, bone marrow derived cells; CIN, chromosomal instability; DDR, DNA damage repair; DSBs, double strand breaks; EBV, Epstein–Barr virus; H. pylori, Helicobacter pylori; IM, intestinal metaplasia; MDSCs, myeloid derived suppressor cells; NBS, Nijmegen breakage syndrome; RONS, reactive oxygen and nitrogen species; SASP, senescence-associated secretory phenotype; SSBs, single strand breaks; SPEM, spasmolytic polypeptide expressing metaplasia; Treg, T-regulatory cells; γH2AX, phosphorylated H2AX.
References
Aivaliotis, I. L., Pateras, I. S., Papaioannou, M., Glytsou, C., Kontzoglou, K., Johnson, E. O., et al. (2012). How do cytokines trigger genomic instability? J. Biomed. Biotechnol. 2012:536761. doi: 10.1155/2012/536761
Algood, H. M., Gallo-Romero, J., Wilson, K. T., Peek, R. M. Jr., and Cover, T. L. (2007). Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol. Med. Microbiol. 51, 577–586. doi: 10.1111/j.1574-695X.2007.00338.x
Ames, B. N., Gold, L. S., and Willett, W. C. (1995). The causes and prevention of cancer. Proc. Natl. Acad. Sci. U.S.A. 92, 5258–5265. doi: 10.1073/pnas.92.12.5258
Atherton, J. C. (2006). The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu. Rev. Pathol. 1, 63–96. doi: 10.1146/annurev.pathol.1.110304.100125
Austad, W. I., Cornes, J. S., Gough, K. R., McCarthy, C. F., and Read, A. E. (1967). Steatorrhea and malignant lymphoma. The relationship of malignant tumors of lymphoid tissue and celiac disease. Am. J. Dig. Dis. 12, 475–490. doi: 10.1007/BF02233180
Bartek, J., and Lukas, J. (2007). DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 19, 238–245. doi: 10.1016/j.ceb.2007.02.009
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434, 864–870. doi: 10.1038/nature03482
Bavik, C., Coleman, I., Dean, J. P., Knudsen, B., Plymate, S., and Nelson, P. S. (2006). The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 66, 794–802. doi: 10.1158/0008-5472.CAN-05-1716
Berti, M., and Vindigni, A. (2016). Replication stress: getting back on track. Nat. Struct. Mol. Biol. 23, 103–109. doi: 10.1038/nsmb.3163
Bessede, E., Staedel, C., Acuna Amador, L. A., Nguyen, P. H., Chambonnier, L., Hatakeyama, M., et al. (2014). Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 33, 4123–4131. doi: 10.1038/onc.2013.380
Blaser, M. J., Perez-Perez, G. I., Kleanthous, H., Cover, T. L., Peek, R. M., Chyou, P. H., et al. (1995). Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 55, 2111–2115.
Cancer Genome Atlas Research Network (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. doi: 10.1038/nature13480
Cianfarani, S., Tedeschi, B., Germani, D., Prete, S. P., Rossi, P., Vernole, P., et al. (1998). In vitro effects of growth hormone (GH) and insulin-like growth factor I and II (IGF-I and -II) on chromosome fragility and p53 protein expression in human lymphocytes. Eur. J. Clin. Invest. 28, 41–47. doi: 10.1046/j.1365-2362.1998.00247.x
Cimprich, K. A., and Cortez, D. (2008). ATR: an essential regulator of genome integrity. Nat. Rev. 9, 616–627. doi: 10.1038/nrm2450
Colonna, M. (2015). DNA damage response impacts macrophage functions. Blood 126, 2440–2442. doi: 10.1182/blood-2015-09-668970
Colotta, F., Allavena, P., Sica, A., Garlanda, C., and Mantovani, A. (2009). Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081. doi: 10.1093/carcin/bgp127
Cook, P. J., Ju, B. G., Telese, F., Wang, X., Glass, C. K., and Rosenfeld, M. G. (2009). Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 458, 591–596. doi: 10.1038/nature07849
Coppe, J. P., Patil, C. K., Rodier, F., Sun, Y., Munoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6:2853–2868. doi: 10.1371/journal.pbio.0060301
Correa, P. (1992). Human gastric carcinogenesis: a multistep and multifactorial process–First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 52, 6735–6740.
Cortes-Bratti, X., Frisan, T., and Thelestam, M. (2001). The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 39, 1729–1736. doi: 10.1016/S0041-0101(01)00159-3
Coussens, L. M., and Werb, Z. (2002). Inflammation and cancer. Nature 420, 860–867. doi: 10.1038/nature01322
D’Elios, M. M., Manghetti, M., De Carli, M., Costa, F., Baldari, C. T., Burroni, D., et al. (1997). T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 158, 962–967.
Eftang, L. L., Esbensen, Y., Tannaes, T. M., Bukholm, I. R., and Bukholm, G. (2012). Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiol. 12:9. doi: 10.1186/1471-2180-12-9
El-Omar, E. M., Carrington, M., Chow, W. H., McColl, K. E., Bream, J. H., Young, H. A., et al. (2000). Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404, 398–402. doi: 10.1038/35006081
Evangelou, K., Bartkova, J., Kotsinas, A., Pateras, I. S., Liontos, M., Velimezi, G., et al. (2013). The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 20, 1485–1497. doi: 10.1038/cdd.2013.76
Ford, A. C., Forman, D., Hunt, R. H., Yuan, Y., and Moayyedi, P. (2014). Helicobacter pylori eradication therapy to prevent gastric cancer in healthy asymptomatic infected individuals: systematic review and meta-analysis of randomised controlled trials. BMJ 348:g3174. doi: 10.1136/bmj.g3174
Forman, D., Newell, D. G., Fullerton, F., Yarnell, J. W., Stacey, A. R., Wald, N., et al. (1991). Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ 302, 1302–1305. doi: 10.1136/bmj.302.6788.1302
Furuta, T., El-Omar, E. M., Xiao, F., Shirai, N., Takashima, M., and Sugimura, H. (2002). Interleukin 1beta polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterology 123, 92–105. doi: 10.1053/gast.2002.34156
Gabrilovich, D. I., and Nagaraj, S. (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174. doi: 10.1038/nri2506
Gebert, B., Fischer, W., Weiss, E., Hoffmann, R., and Haas, R. (2003). Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301, 1099–1102. doi: 10.1126/science.1086871
Georgakilas, A. G., Mosley, W. G., Georgakila, S., Ziech, D., and Panayiotidis, M. I. (2010). Viral-induced human carcinogenesis: an oxidative stress perspective. Mol. Biosyst. 6, 1162–1172. doi: 10.1039/b923958h
Gerhard, M., Schmees, C., Voland, P., Endres, N., Sander, M., Reindl, W., et al. (2005). A secreted low-molecular-weight protein from Helicobacter pylori induces cell-cycle arrest of T cells. Gastroenterology 128, 1327–1339. doi: 10.1053/j.gastro.2005.03.018
Gorgoulis, V. G., and Halazonetis, T. D. (2010). Oncogene-induced senescence: the bright and dark side of the response. Curr. Opin. Cell Biol. 22, 816–827. doi: 10.1016/j.ceb.2010.07.013
Gorgoulis, V. G., Vassiliou, L. V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434, 907–913. doi: 10.1038/nature03485
Grivennikov, S. I., Greten, F. R., and Karin, M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. doi: 10.1016/j.cell.2010.01.025
Halazonetis, T. D., Gorgoulis, V. G., and Bartek, J. (2008). An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. doi: 10.1126/science.1140735
Hanada, K., Uchida, T., Tsukamoto, Y., Watada, M., Yamaguchi, N., Yamamoto, K., et al. (2014). Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect. Immun. 82, 4182–4189. doi: 10.1128/IAI.02368-14
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hardbower, D. M., Peek, R. M. Jr., and Wilson, K. T. (2014). At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J. Leukoc. Biol. 96, 201–212. doi: 10.1189/jlb.4BT0214-099R
Harper, J. W., and Elledge, S. J. (2007). The DNA damage response: ten years after. Mol. Cell 28, 739–745. doi: 10.1016/j.molcel.2007.11.015
Harris, P. R., Wright, S. W., Serrano, C., Riera, F., Duarte, I., Torres, J., et al. (2008). Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 134, 491–499. doi: 10.1053/j.gastro.2007.11.006
Harrison, J. C., and Haber, J. E. (2006). Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 40, 209–235. doi: 10.1146/annurev.genet.40.051206.105231
Hartung, M. L., Gruber, D. C., Koch, K. N., Gruter, L., Rehrauer, H., Tegtmeyer, N., et al. (2015). H. pylori-induced dna strand breaks are introduced by nucleotide excision repair endonucleases and promote nf-kappab target gene expression. Cell Rep. 13, 70–79. doi: 10.1016/j.celrep.2015.08.074
Houghton, J., Stoicov, C., Nomura, S., Rogers, A. B., Carlson, J., Li, H., et al. (2004). Gastric cancer originating from bone marrow-derived cells. Science 306, 1568–1571. doi: 10.1126/science.1099513
Hwang, I. R., Kodama, T., Kikuchi, S., Sakai, K., Peterson, L. E., Graham, D. Y., et al. (2002). Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology 123, 1793–1803. doi: 10.1053/gast.2002.37043
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jakowlew, S. B. (2006). Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 25, 435–457. doi: 10.1007/s10555-006-9006-2
Jurk, D., Wilson, C., Passos, J. F., Oakley, F., Correia-Melo, C., Greaves, L., et al. (2014). Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nature Commun. 2:4172. doi: 10.1038/ncomms5172
Kao, J. Y., Zhang, M., Miller, M. J., Mills, J. C., Wang, B., Liu, M., et al. (2010). Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138, 1046–1054. doi: 10.1053/j.gastro.2009.11.043
Kastan, M. B., and Bartek, J. (2004). Cell-cycle checkpoints and cancer. Nature 432, 316–323. doi: 10.1038/nature03097
Keates, S., Sougioultzis, S., Keates, A. C., Zhao, D., Peek, R. M. Jr., Shaw, L. M., et al. (2001). cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 276, 48127–48134.
Khanna, K. K., and Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247–254. doi: 10.1038/85798
Kim, J. M., Kim, J. S., Lee, J. Y., Kim, Y. J., Youn, H. J., Kim, I. Y., et al. (2007). Vacuolating cytotoxin in Helicobacter pylori water-soluble proteins upregulates chemokine expression in human eosinophils via Ca2+ influx, mitochondrial reactive oxygen intermediates, and NF-kappaB activation. Infect. Immun. 75, 3373–3381. doi: 10.1128/IAI.01940-06
Kiraly, O., Gong, G., Olipitz, W., Muthupalani, S., and Engelward, B. P. (2015). Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLoS Genet. 11:e1004901. doi: 10.1371/journal.pgen.1004901
Koeppel, M., Garcia-Alcalde, F., Glowinski, F., Schlaermann, P., and Meyer, T. F. (2015). Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 11, 1703–1713. doi: 10.1016/j.celrep.2015.05.030
Larussa, T., Leone, I., Suraci, E., Imeneo, M., and Luzza, F. (2015). Helicobacter pylori and T helper cells: mechanisms of immune escape and tolerance. J. Immunol. Res. 2015:981328. doi: 10.1155/2015/981328
Lee, J. H., and Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554. doi: 10.1126/science.1108297
Lieber, M. R. (2008). The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 283, 1–5. doi: 10.1074/jbc.R700039200
Lin, W. C., Tsai, H. F., Kuo, S. H., Wu, M. S., Lin, C. W., Hsu, P. I., et al. (2010). Translocation of Helicobacter pylori CagA into Human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer Res. 70, 5740–5748. doi: 10.1158/0008-5472.CAN-09-4690
Lindahl, T., and Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 65, 127–133. doi: 10.1101/sqb.2000.65.127
Liyanage, N. P., Manthey, K. C., Dassanayake, R. P., Kuszynski, C. A., Oakley, G. G., and Duhamel, G. E. (2010). Helicobacter hepaticus cytolethal distending toxin causes cell death in intestinal epithelial cells via mitochondrial apoptotic pathway. Helicobacter 15, 98–107. doi: 10.1111/j.1523-5378.2010.00749.x
Machado, A. M., Figueiredo, C., Touati, E., Máximo, V., Sousa, S., Michel, V., et al. (2009). Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin. Cancer Res. 15, 2995–3002. doi: 10.1158/1078-0432.CCR-08-2686
Machado, J. C., Pharoah, P., Sousa, S., Carvalho, R., Oliveira, C., Figueiredo, C., et al. (2001). Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 121, 823–829. doi: 10.1053/gast.2001.28000
Malaquin, N., Vercamer, C., Bouali, F., Martien, S., Deruy, E., Wernert, N., et al. (2013). Senescent fibroblasts enhance early skin carcinogenic events via a paracrine MMP-PAR-1 axis. PLoS ONE 8:e63607. doi: 10.1371/journal.pone.0063607
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008). Cancer-related inflammation. Nature 454, 436–444. doi: 10.1038/nature07205
Marshall, B. J., and Warren, J. R. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315. doi: 10.1016/S0140-6736(84)91816-6
Murata-Kamiya, N. (2011). Pathophysiological functions of the CagA oncoprotein during infection by Helicobacter pylori. Microbes Infect. 13, 799–807. doi: 10.1016/j.micinf.2011.03.011
Murphy, G., Pfeiffer, R., Camargo, M. C., and Rabkin, C. S. (2009). Meta-analysis shows that prevalence of Epstein–Barr Virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 137, 824–833. doi: 10.1053/j.gastro.2009.05.001
Negrini, S., Gorgoulis, V. G., and Halazonetis, T. D. (2010). Genomic instability–an evolving hallmark of cancer. Nat. Rev. 11, 220–228. doi: 10.1038/nrm2858
Nomura, A., Stemmermann, G. N., Chyou, P. H., Kato, I., Perez-Perez, G. I., and Blaser, M. J. (1991). Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 325, 1132–1136. doi: 10.1056/NEJM199110173251604
Nougayrede, J. P., Homburg, S., Taieb, F., Boury, M., Brzuszkiewicz, E., Gottschalk, G., et al. (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851. doi: 10.1126/science.1127059
Ochiai, A., Yamauchi, Y., and Hirohashi, S. (1996). p53 mutations in the non-neoplastic mucosa of the human stomach showing intestinal metaplasia. Int. J. Cancer 69, 28–33. doi: 10.1002/(SICI)1097-0215(19960220)69:1<28::AID-IJC6>3.0.CO;2-Y
Odenbreit, S., Puls, J., Sedlmaier, B., Gerland, E., Fischer, W., and Haas, R. (2000). Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500. doi: 10.1126/science.287.5457.1497
Parsonnet, J., Friedman, G. D., Orentreich, N., and Vogelman, H. (1997). Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40, 297–301. doi: 10.1136/gut.40.3.297
Parsonnet, J., Friedman, G. D., Vandersteen, D. P., Chang, Y., Vogelman, J. H., Orentreich, N., et al. (1991). Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325, 1127–1131. doi: 10.1056/NEJM199110173251603
Parsonnet, J., and Isaacson, P. G. (2004). Bacterial infection and MALT lymphoma. N. Engl. J. Med. 350, 213–215. doi: 10.1056/NEJMp038200
Pateras, I. S., Havaki, S., Nikitopoulou, X., Vougas, K., Townsend, P. A., Panayiotidis, M. I., et al. (2015). The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 154, 36–56. doi: 10.1016/j.pharmthera.2015.06.011
Peek, R. M. Jr., and Blaser, M. J. (2002). Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2, 28–37. doi: 10.1038/nrc703
Peek, R. M. Jr., Miller, G. G., Tham, K. T., Perez-Perez, G. I., Zhao, X., Atherton, J. C., et al. (1995). Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab. Invest. 73, 760–770.
Pereira-Lopes, S., Tur, J., Calatayud-Subias, J. A., Lloberas, J., Stracker, T. H., and Celada, A. (2015). NBS1 is required for macrophage homeostasis and functional activity in mice. Blood 126, 2502–2510. doi: 10.1182/blood-2015-04-637371
Petitjean, A., Mathe, E., Kato, S., Ishioka, C., Tavtigian, S. V., Hainaut, P., et al. (2007). Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629. doi: 10.1002/humu.20495
Plummer, M., de Martel, C., Vignat, J., Ferlay, J., Bray, F., and Franceschi, S. (2016). Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob. Health 4, e609–e616. doi: 10.1016/S2214-109X(16)30143-7
Polo, S. E., and Jackson, S. P. (2011). Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433. doi: 10.1101/gad.2021311
Preston-Martin, S., Pike, M. C., Ross, R. K., Jones, P. A., and Henderson, B. E. (1990). Increased cell division as a cause of human cancer. Cancer Res. 50, 7415–7421.
Rad, R., Dossumbekova, A., Neu, B., Lang, R., Bauer, S., Saur, D., et al. (2004). Cytokine gene polymorphisms influence mucosal cytokine expression, gastric inflammation, and host specific colonisation during Helicobacter pylori infection. Gut 53, 1082–1089. doi: 10.1136/gut.2003.029736
Ramarao, N., Gray-Owen, S. D., and Meyer, T. F. (2000). Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol. Microbiol. 38, 103–113. doi: 10.1046/j.1365-2958.2000.02114.x
Ricci, V., Romano, M., and Boquet, P. (2011). Molecular cross-talk between Helicobacter pylori and human gastric mucosa. World J. Gastroenterol. 17, 1383–1399. doi: 10.3748/wjg.v17.i11.1383
Riley, T., Sontag, E., Chen, P., and Levine, A. (2008). Transcriptional control of human p53-regulated genes. Nat. Rev. 9, 402–412. doi: 10.1038/nrm2395
Rodier, F., Coppe, J. P., Patil, C. K., Hoeijmakers, W. A., Munoz, D. P., Raza, S. R., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979. doi: 10.1038/ncb1909
Roos, W. P., and Kaina, B. (2013). DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 332, 237–248. doi: 10.1016/j.canlet.2012.01.007
Rouse, J., and Jackson, S. P. (2002). Interfaces between the detection, signaling, and repair of DNA damage. Science 297, 547–551. doi: 10.1126/science.1074740
San Filippo, J., Sung, P., and Klein, H. (2008). Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257. doi: 10.1146/annurev.biochem.77.061306.125255
Scanu, T., Spaapen, R. M., Bakker, J. M., Pratap, C. B., Wu, L. E., Hofland, I., et al. (2015). Salmonella manipulation of host signaling pathways provokes cellular transformation associated with gallbladder carcinoma. Cell Host Microbe 17, 763–774. doi: 10.1016/j.chom.2015.05.002
Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155–168. doi: 10.1038/nrc1011
Sougioultzis, S., Foukas, P. G., Tzivras, M., Kourtessas, D., Gorgoulis, V. G., Davaris, P., et al. (2003). Alterations in the proliferating compartment of gastric mucosa during Helicobacter pylori infection: the putative role of epithelial cells expressing p27(kip1). Mod. Pathol 16, 1076–1085. doi: 10.1097/01.MP.0000093626.15701.76
Szabo, I., Brutsche, S., Tombola, F., Moschioni, M., Satin, B., Telford, J. L., et al. (1999). Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 18, 5517–5527. doi: 10.1093/emboj/18.20.5517
Terzic, J., Grivennikov, S., Karin, E., and Karin, M. (2010). Inflammation and colon cancer. Gastroenterology 138, 2101.e5–2114.e5. doi: 10.1053/j.gastro.2010.01.058
Toller, I. M., Neelsen, K. J., Steger, M., Hartung, M. L., Hottiger, M. O., Stucki, M., et al. (2011). Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. U.S.A. 108, 14944–14949. doi: 10.1073/pnas.1100959108
Velimezi, G., Liontos, M., Vougas, K., Roumeliotis, T., Bartkova, J., Sideridou, M., et al. (2013). Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 15, 967–977. doi: 10.1038/ncb2795
Voulgarelis, M., Ziakas, P. D., Papageorgiou, A., Baimpa, E., Tzioufas, A. G., and Moutsopoulos, H. M. (2012). Prognosis and outcome of non-Hodgkin lymphoma in primary Sjogren syndrome. Medicine 91, 1–9. doi: 10.1097/MD.0b013e31824125e4
Wallace, M. D., Southard, T. L., Schimenti, K. J., and Schimenti, J. C. (2014). Role of DNA damage response pathways in preventing carcinogenesis caused by intrinsic replication stress. Oncogene 33, 3688–3695. doi: 10.1038/onc.2013.339
Wang, Y. H., Gorvel, J. P., Chu, Y. T., Wu, J. J., and Lei, H. Y. (2010). Helicobacter pylori impairs murine dendritic cell responses to infection. PLoS ONE 5:e10844. doi: 10.1371/journal.pone.0010844
Weis, V. G., and Goldenring, J. R. (2009). Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer 12, 189–197. doi: 10.1007/s10120-009-0527-6
Wilson, K. T., Ramanujam, K. S., Mobley, H. L., Musselman, R. F., James, S. P., and Meltzer, S. J. (1996). Helicobacter pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology 111, 1524–1533. doi: 10.1016/S0016-5085(96)70014-8
Wong, B. C., Lam, S. K., Wong, W. M., Chen, J. S., Zheng, T. T., Feng, R. E., et al. (2004). Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA 291, 187–194. doi: 10.1001/jama.291.2.187
Xiao, A., Li, H., Shechter, D., Ahn, S. H., Fabrizio, L. A., Erdjument-Bromage, H., et al. (2009). WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 457, 57–62. doi: 10.1038/nature07668
Keywords: Helicobacter pylori (H. pylori), double strand breaks (DSBs), DNA damage repair (DDR), γH2AX, eradication, genomic instability, tumorigenesis, chronic inflammation
Citation: Kalisperati P, Spanou E, Pateras IS, Korkolopoulou P, Varvarigou A, Karavokyros I, Gorgoulis VG, Vlachoyiannopoulos PG and Sougioultzis S (2017) Inflammation, DNA Damage, Helicobacter pylori and Gastric Tumorigenesis. Front. Genet. 8:20. doi: 10.3389/fgene.2017.00020
Received: 17 October 2016; Accepted: 08 February 2017;
Published: 27 February 2017.
Edited by:
Linda Pattini, Politecnico di Milano, ItalyReviewed by:
Dominic C. Voon, Kanazawa University, JapanAristides G. Eliopoulos, University of Crete, Greece
Fabrizio D’Adda Di Fagagna, FIRC Institute of Molecular Oncology, Belize
Copyright © 2017 Kalisperati, Spanou, Pateras, Korkolopoulou, Varvarigou, Karavokyros, Gorgoulis, Vlachoyiannopoulos and Sougioultzis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stavros Sougioultzis, ssougiou@med.uoa.gr
†These authors have contributed equally to this work.