

 Dan Song
Dan Song

- Department of Clinical Pharmacology, China Medical University, Shenyang, People's Republic of China
The involvement of glycogenolysis, occurring in astrocytes but not in neurons, in learning is undisputed (Duran et al., 2013). According to one school of thought the role of astrocytes for learning is restricted to supply of substrate for neuronal oxidative metabolism. The present “perspective” suggests a more comprehensive and complex role, made possible by lack of glycogen degradation, unless specifically induced by either (1) activation of astrocytic receptors, perhaps especially β-adrenergic or (2) even small increases in extracellular K+ concentration above its normal resting level. It discusses (1) the known importance of glycogenolysis for glutamate formation, requiring pyruvate carboxylation; (2) the established role of K+-stimulated glycogenolysis for K+ uptake in cultured astrocytes, which probably indicates that astrocytes are an integral part of cellular K+ homeostasis in the brain in vivo; and (3) the plausible role of transmitter-induced glycogenolysis, stimulating Na+,K+-ATPase/NKCC1 activity and thereby contributing both to the post-excitatory undershoot in extracellular K+ concentration and the memory-enhancing effect of transmitter-mediated reduction of slow neuronal afterhyperpolarization (sAHP).
Gold and Korol (2012) discussed a key role for glycogenolysis in memory establishment and reviewed evidence that moderate levels of systemically released adrenaline enhance and higher levels impair learning. They concluded that memory-enhancing effects mainly reflected increases in blood glucose levels, supporting brain processes engaged during memory establishment. It is well known that glucose and other substrates like short-medium chain fatty acids may improve memory, even under pathological conditions (Watson and Craft, 2004; Henderson and Poirier, 2011).
The intentions of this perspective article are (1) to more strongly emphasize that the glycogenolytic effect in brain is caused by noradrenaline released within the central nervous system and to explain why the stimulation is not restricted to neurons, (2) to emphasize that a need for glycogenolysis during the establishment of memory was first shown by Marie E. Gibbs' pioneering studies, which were overlooked in the Gold/Korol paper, (3) to elaborate on additional recent observations regarding K+ uptake in astrocytes, which support the importance of astrocytic glycogenolysis during learning and are consistent with early concepts by Marie Gibbs and her colleagues, and (4) to refer to an additional Gibbs paper, showing that brain injection of not only lactate but also octanoate or β-hydroxybutyrate, but not glucose, can rescue learning disability induced by previous administration of oligomeric β-amyloid (Aβ) 1–42.
With few exceptions, systemically released adrenaline does not enter the brain (Weil-Malherbe et al., 1959). Small amounts of adrenaline, together with larger amounts of the slightly different molecule, noradrenaline, are produced in locus coeruleus and a few additional neuronal nuclei in the brain stem, from which noradrenergic nerve fibers reach the entire central nervous system (reviewed by Hertz et al., 2004). Transmitter release both in genuine synapses and from non-synaptic axonal release sites (Beaudet and Descarries, 1978), secures that noradrenaline effects can be exerted on all brain cell types (O'Donnell et al., 2012). This includes astrocytes (Bekar et al., 2008), where glycogenolysis is a key target (Hertz et al., 2010). Recently, glycogen's importance for learning has been substantiated by Duran et al. (2013), demonstrating severe disturbances in long-term memory formation and learning-dependent synaptic plasticity in mice lacking brain glycogen synthase.
One group of authors regards transfer to neurons of glycogen-derived lactate as the main reason for the memory-enhancing effect of glycogenolysis. Suzuki et al. (2011) showed that extracellular lactate levels in rat hippocampus increase during learning, and that this increase, memory and long-term potentiation (LTP) were abolished by glycogenolytic inhibitors. Disruption of the expression of astrocytic lactate transporters also caused amnesia and LTP impairment, which could be rescued by L-lactate, but not by glucose. Specific knock-down of the neuronal monocarboxylate transporter MCT2 led to amnesia, against which neither L-lactate nor glucose could protect. These findings are consistent with a proposed concept, the astrocyte-to-neuron lactate shuttle (ANLS), that lactate release from astrocytes and its uptake in neurons might be important for brain function (Pellerin and Magistretti, 2012). Newman et al. (2011) confirmed memory impairment by inhibition of glycogenolysis. Again, the impairment could be counteracted by either lactate or glucose, and blockade of the neuronal monocarboxylate transporter impaired memory, with no reversal by either lactate or glucose. Since the ability of neurons to metabolize glucose should be intact, the authors further advanced the concept of astrocyte-to-neuron transfer of lactate by concluding that lactate might be a specially important substrate for neurons during working memory by rapidly providing additional energy. Consistent with this concept Dringen et al. (1993) had shown in cultured astrocytes that glycogenolysis causes release of lactate, not of glucose, and that glycogen is continuously re-synthesized in the presence of glucose. However, the rate of glycogen turnover in the living brain is very modest compared to that of glucose breakdown (Öz et al., 2012). Furthermore, although the observed learning deficits reported by both Suzuki et al. (2011) and Newman et al. (2011) are solid, it can not be excluded that inactivation of transporters may have additional effects. Effects by alpha-cyano-4-hydroxycinnamate (4-CIN) may include inhibition of mitochondrial entry of pyruvate (McKenna et al., 2001; Rae et al., 2012), and MCT2 knock-down might have a similar effect (Hashimoto et al., 2008). Also, although an Alzheimer study by Gibbs et al. (2009) showed that lactate, octanoate, and β-hydroxybutyrate could rescue Aβ-impaired memory, the authors concluded that this was due to an effect on astrocytic metabolism.
Many studies by Marie E. Gibbs describe the importance of noradrenaline-stimulated glycogenolysis for one-trial aversive learning in the day-old chick, a precocious animal. The first of these resembles the papers discussed above in emphasizing metabolic aspects (O'Dowd et al., 1994). It demonstrates dependence on iodoacetate-inhibitable glycolysis, regardless whether glucose or glycogen is metabolized, but it also shows time periods with decreased glycogen content in the brain. Later, attention was drawn toward the importance of glycogenolysis for glutamate synthesis (Figure 1), in studies culminating in the demonstrations (Gibbs et al., 2006, 2007) that (1) administration of the glycogenolytic inhibitor DAB prevented a normally observed rise in cellular glutamate level soon after training, (2) no compensatory decrease occurred in contents of other amino acid interacting with the tricarboxylic acid (TCA) cycle, indicating de novo synthesis of glutamate (or inhibited breakdown), and (3) learning was inhibited by intracranial injection of DAB only during highly specific time periods. These included periods when glycogen content was decreased, but also a time period around 30 min post-training, when no such decrease could be shown. The importance of glycogen to support glutamatergic transmission has also been shown in a cell culture study by different authors (Sickmann et al., 2009).
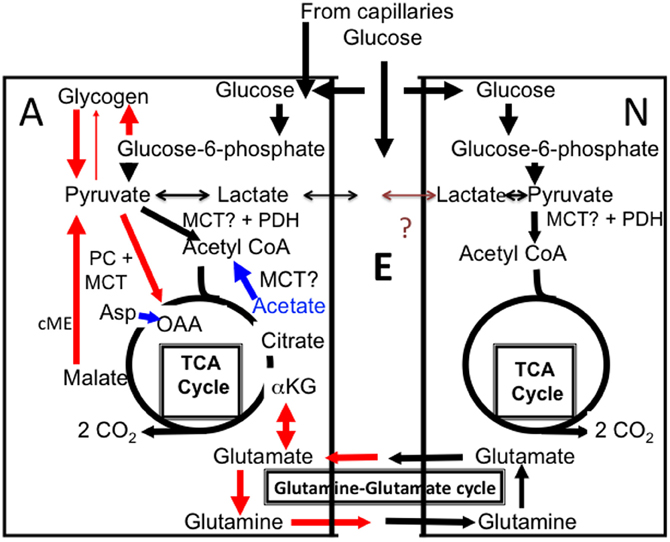
Figure 1. Cartoon of glucose metabolism in astrocytes (A, left) and neurons (N, right), with extracellular space shown as E. Glucose is accumulated both in neurons and astrocytes and metabolized via glucose-6-phosphate to pyruvate and in astrocytes also used for glycogen synthesis. Pyruvate is mainly metabolized via pyruvate dehydrogenase (PDH) to acetyl coenzyme A (acetyl CoA) that, in the mitochondrial tricarboxylic acid (TCA) cycle, condenses with oxaloacetate (OAA) to form citrate (pathway shown in black). Lactate can also be formed from pyruvate. Some of this lactate may be released from the cell, although glycogen-derived lactate may be formed and released slowly in vivo. According to some of the authors discussed in the present “perspective” release of glycogen-derived lactate may be followed by its uptake into neurons and use as an important metabolic substrate for neurons during learning. The postulated net flux from astrocytes to neurons has never been proven and a brown question mark is shown in the Figure. Citrate is further metabolized via α-ketoglutarate (α-KG) to eventually regenerate oxaloacetate, which condenses with another molecule of acetyl CoA, and so on. During the process two molecules of CO2 are released. The purpose of this cycle is solely generation of ATP. In astrocytes additional pathways are also operating (red), which is a major reason for the importance of astrocytes, including astrocytic glycogenolysis. Glucose is used for synthesis of glycogen, which is metabolized to glucose-6-phosphate (by a different route than glucose and therefore selectively inhibitable by DAB), and from there by the same pathway as glucose, to pyruvate. Pyruvate can be carboxylated to form a “new” molecule of oxaloacetate, which condenses with acetyl CoA to form a “new” molecule of citrate, from which α-ketoglutarate and glutamate can be formed and transferred to neurons via glutamine release (red). Glutamate released as a transmitter to the extracellular space is virtually quantitatively taken up by astrocytes. Eighty-five percent of the accumulated amount is converted to glutamine by the astrocyte-specific glutamine synthetase and carried back to neurons in the “glutamine–glutamate (Gln–Glu) cycle” where it can be re-utilized as transmitter glutamate or converted to γ-aminobutyric acid (GABA). The remaining 15% is degraded in astrocytes after conversion by cytosolic malic enzyme (cME) to α-ketoglutarate, then to malate, which exits the cycle and is decarboxylated to pyruvate. This pyruvate can then be oxidized in the TCA cycle via acetyl CoA, and astrocytic formation of glutamate combined with subsequent glutamate degradation supplies almost as much energy (ATP) as degradation via PDH. Noradrenergic agonists subtype-specifically stimulate glycogen synthesis (via α2-adrenergic receptors) and glycogenolysis (via β-adrenergic receptors), as well as pyruvate carboxylase activity (via α2-adrenergic receptors). Elevated extracellular concentrations of K+ also stimulate both glycogenolysis and pyruvate carboxylation. Lactate can function as a pyruvate precursor or product in both astrocytes and neurons, whereas acetate is an astrocyte-specific precursor of acetyl CoA, but not a precursor for pyruvate carboxylation. However, if both acetate and aspartate (Asp) are supplied (indicated in blue), oxaloacetate, formed from aspartate, and acetyl CoA, formed from acetate, can sustain net production of citrate, α-ketoglutarate and glutamate. Entry of pyruvate or acetate into the mitochondrial TCA cycle requires a transporter, here simply indicated as MCT.
Both human and rodent brain astrocytes in vivo oxidatively degrade glucose at a rate, which per cell volume is at least as high as in neurons (reviewed by Hertz, 2011). More than one half of glucose oxidation in astrocytes occurs via formation and degradation of glutamate in a complex process, which includes neuronal glutamate uptake and subsequent release, followed by a mainly astrocytic uptake (Danbolt, 2001) and intense glutamate metabolism in astrocytes (Bauer et al., 2012; McKenna, 2012). Figure 1 shows that glutamate synthesis requires pyruvate entry into the astrocytic TCA cycle via two pathways, (1) the conventional pyruvate dehydrogenase-mediated pathway (black in the Figure) and (2) pyruvate carboxylation, catalyzed by pyruvate carboxylase (red in the Figure). This enzyme is not expressed in neurons, but is abundant in astrocytes (Shank et al., 1985; Hutson et al., 2008), where glutamate production is rapid (Yu et al., 1983). Most generated glutamate is transferred via glutamine to neurons and used as precursor for transmitter glutamate and GABA (Figure 1). In the brain in vivo the rate of this transfer equals the rate of glucose utilization, i.e., it is extremely fast (reviewed by Hyder and Rothman, 2012). Although about 85% of the flow of glutamine from astrocytes to neurons is compensated for by a flow in the opposite direction of glutamate and some GABA, 15% is not but depends upon continuous glutamate production in astrocytes. Over time the transfer of newly synthesized glutamate equals the subsequent metabolic degradation of GABA and glutamate, to a large extent in astrocytes (Hertz, 2011; McKenna, 2012), However, fluctuations occurring in tissue glutamate levels during brain function (Mangia et al., 2012) must indicate altered glutamate production and/or degradation rates. Established rates of glycogenolysis (see above) are not high enough to enable glycogen to supply the pyruvate carboxylase-dependent one half of the glutamate molecule. Rather, it is required for the function of the pyruvate carboxylase, a question that will be re-discussed toward the end of this communication.
As in the papers by Suzuki et al. (2011) and Newman et al. (2011) memory could in the Gibbs studies be rescued after DAB administration by lactate and by glucose, but only during precisely limited time intervals (tabulated in Gibbs et al., 2008). An amnestic effect of DAB administration 30 sec after training was prevented by glucose administration immediately after training or by lactate injection either immediately after training or anytime during the period 10–20 min post-training. The DAB challenges with glucose/lactate provide per se no information about the cell type(s) in which the supplied substrates were metabolized. Acetate, which is generally considered an astrocyte-specific metabolic substrate (Muir et al., 1986; Waniewski and Martin, 1998; but see also Rae et al., 2012), could rescue at the same times as lactate. However, this was only the case, when aspartate (which alone had no effect) was administered together with acetate. As can be seen from Figure 1 (blue), such co-administration enables glutamate formation even when pyruvate carboxylation is inhibited, since aspartate is a precursor for oxaloacetate, and oxaloacetate is the initial glutamate precursor generated by pyruvate carboxylation. Consistent with this explanation, administration of glutamine itself at similar time also rescued memory (Gibbs et al., 2008).
Glycogenolysis occurs in response to stimulation of the β-adrenergic receptor, as a β2-adrenergic effect in the day-old chick (Gibbs et al., 2008) and a β1-adrenergic effect in human brain (Quach et al., 1988). The subtype difference reflects species differences in β-adrenergic subtype expression in fowl and mammals (Fernández-López et al., 1997). The glycogen molecule is unique in being stable under control conditions, but rapidly degraded in response to certain transmitters (Magistretti, 1988), and to even very small increases in extracellular K+ concentrations from their resting level (Hof et al., 1988).
Besides responding to β-adrenergic receptor activation (Quach et al., 1988; Subbarao and Hertz, 1990; Gibbs et al., 2008), glycogenolysis is also evoked by some other transmitters, including serotonin (Quach et al., 1982; Magistretti, 1988; Kong et al., 2002; Darvesh and Gudelsky, 2003). In mature cultured astrocytes stimulation of β1-adrenoceptors activates protein kinase A (via Gs) and causes an in increase in free cytosolic calcium concentration ([Ca2+]i) after a Gs-Gi switch (Du et al., 2010) with the latter probably responsible for most of the glycogenolytic effect. Addition of 5 mM extracellular K+ concentrations also increases [Ca2+]i in astrocytes, secondary to an Na+,K+-ATPase effect (Xu et al., 2013). A different possibility for glycogenolytic stimulation (Choi et al., 2012) is activation of astrocytic soluble adenylate cyclase (sAC) by depolarization-induced entry of bicarbonate [carried by a Na+-bicarbonate cotransporter (NBC)], which increases cAMP (and thus activates protein kinase A), but the cAMP effect is minuscule. Glycogen synthesis is essential for continued glycogenolysis. In astrocytes this process is also stimulated by noradrenaline, acting on a different subtype receptor, the α2-adrenoceptor (Hertz et al., 2007; Hutchinson et al., 2008). The α2-adrenergic receptor is at least as densely expressed in astrocytes (per mg mRNA) as in freshly isolated neurons (Hertz et al., 2010). The importance for learning of a continuous glycogen synthesis/glycogenolysis process by temporally selective, interspersed periods of β-adrenergic and α2-adrenergic activity has recently been clearly demonstrated by Gibbs and Hutchinson (2012). A distinct decrease of glycogen content in brain immediately after learning, but not around 30 min (Gibbs et al., 2007), in spite of the ability of DAB to inhibit at both times, may also reflect that serotonin acts as the major glycogenolysis-inducing transmitter right after training, whereas noradrenaline does so at 30 min (Gibbs et al., 2008). Serotonin is not known to stimulate glycogen synthesis.
A seminal paper by DiNuzzo et al. (2012) suggested that glycogenolysis may be required for astrocytic contributions to clearance of excess extracellular K+ resulting from neuronal K+ release during excitation. This suggestion greatly expanded the potential role of glycogenolysis in memory formation. Unless excessively high extracellular K+ concentrations are reached, clearance of elevated extracellular K+ occurs by activation of Na+,K+-ATPases. In both astrocytes and neurons Na+,K+-ATPase activity is regulated in conventional manners by ion sensitivities, i.e., at its intracellular site by Na+ stimulation and at its extracellular site by K+ stimulation. Due to enzyme affinities, determined by subtype expression levels of its α and β subunits and of the auxiliary protein FXYD (Delprat et al., 2006), only the astrocytic enzyme is stimulated by increases in extracellular K+ concentration beyond its normal resting level (Grisar et al., 1980; Hajek et al., 1996). The astrocytic contribution to clearance of elevated extracellular K+ seems to be essential for K+ homeostasis in brain (Walz, 2000; Somjen et al., 2008; MacAulay and Zeuthen, 2012; Wang et al., 2012). However, in contrast to the excitation-induced increase in intracellular Na+ in neurons, no Na+ enters astrocytes spontaneously, but Xu et al. (2013) showed in cultured astrocytes that slightly elevated extracellular K+ activates a glycogenolysis-dependent Na+,K+-ATPase/endogenous ouabain signaling pathway leading to astrocytic Na+ entry. Without this activation elevated K+ concentrations are unable to stimulate the astrocytic Na+,K+-ATPase. Activation of glycogenolysis by elevated extracellular K+ concentration allows initial K+ reaccumulation in astrocytes, followed by K+ release through Kir1.4 channels and neuronal K+ uptake when the stimulation of the astrocytic uptake ceases after K+ concentration normalization. A role of this process in learning is likely but not yet established.
The astrocytic Na+,K+-ATPase is also specifically stimulated by isoproterenol, a non-subtype-specific β-adrenergic agonist (Hajek et al., 1996). The neuronal enzyme is stimulated by noradrenaline but not by isoproterenol, and in both cell types the stimulation is inhibited by increased extracellular K+ concentrations (Hajek et al., 1996). Although neuronal α2-adrenergic stimulation may also be important for memory-enhancing effects by α2-adrenergic stimulation (Caetano et al., 2012), β-adrenergic effects are essential (Oh et al., 2009). At non-elevated extracellular K+ concentration β1-adrenergic stimulation enhances regulatory volume increase in mouse astrocytes during hypertonic stimulation, again via a glycogenolytic effect (Xu, Song, Du, Yan, Hertz and Peng, unpublished results). The β1-adrenergic stimulation of volume increase relies on Na+,K+-ATPase-dependent stimulation of a combined uptake of H2O, Na+, K+ and 2Cl−, mediated by the co-transporter NKCC1 (Hamann et al., 2010). In brain slices extracellular hypertonicity reduces excitatory activity (Huang and Somjen, 1997), an effect similar to that of slow neuronal afterhyperpolarization (sAHP). This is important, because Na+,K+-ATPase operation during clearance of excess K+ from the extracellular space may create extracellular hypertonicity, as demonstrated after neuronal activity by Dietzel et al. (1982, 1989). The neuronal effect of hyperosmolarity must be mediated via astrocytes, since acute hyperosmotic stimulus-induced Fos expression in neurons depends on initial activation of astrocytes (Yuan et al., 2010). Reduction of sAHP by β-adrenergic activity [and in some situations by activation of other transmitter receptors (Araneda and Andrade, 1991; Fisahn et al., 2005)] is thought to be immensely important for memory-enhancing effects, especially of noradrenaline and isoproterenol (Oh et al., 2009; Zaitsev and Anwyl, 2012). During the isoproterenol-induced regulatory volume increase glycogenolysis again fulfills Na+,K+-ATPase's signaling requirements and thus enables the NKCC1 cotransporter to create an undershoot in extracellular K+ concentration and to enhance regulatory volume increase. Since isoproterenol-mediated stimulation of Na+,K+-ATPase activity is prevented at elevated K+, sAHP can only be activated after termination of extracellular K+ clearance.
More than 35 years ago Gibbs and Ng (1977) demonstrated inhibition of memory when Na+,K+-ATPase activity is prevented by conventional inhibitory concentrations of ouabain (Figure 2). The study also showed that adrenaline could counteract the inhibition. Additional papers by the Gibbs group have further demonstrated temporal and hemispheric details of the amnestic effects of ouabain administration (Watts and Mark, 1971; Robertson et al., 1978; Bell and Gibbs, 1979; Tucker and Gibbs, 1979; Crowe et al., 1989; Gibbs et al., 2003). The β2-adrenergic agonist zinterol was not available at that time, but recent experiments by Marie E. Gibbs have shown that zinterol also counteracts the inhibitory effect of ouabain (personal communication). This observation provides in vivo support for the importance of β-adrenergic interaction with Na+,K+-ATPase function during learning, with stimulation of regulatory volume increase and concomitant reduction of sAHP being the most likely explanation for the rescue of memory. In this context, it should be noted that Gibbs and Ng (1976) suggested 37 years ago that “the possible hyperpolarization of membrane potential associated with sodium pump activity may serve to mark the labile memory trace” until changes into more permanent memory take place. More recent studies (Disterhoft et al., 1986; Oh et al., 2009) have shown that in many types of learning the opposite occurs: after learning sAHP becomes reduced even in the absence of transmitter activity, so the neuron becomes more easily recruited, and transmitter application has no further effect. Nevertheless, Gibbs and Ng (1976) remain the first to have contemplated an association between neuronal afterhyperpolarization and learning.
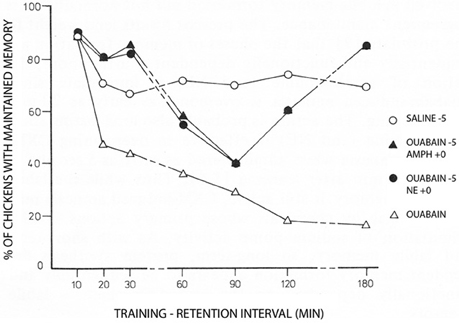
Figure 2. Day-old chicks were during training exposed to a red bead tainted with the aversively tasting drug anthranilate and to an untainted blue bead. The Figure shows the percentage of trained chickens, which have learned to avoid the red color, which at the memory test was untainted. Under control conditions ~70% remember. This number was rapidly reduced by administration of ouabain 5 min before training (with saline added to the controls at the same time) to reach <20% after 2–3 h. However, the amnesic effect of ouabain was initially, and after a transient dip in memory, also at later times prevented by noradrenaline (NE) given at the time of learning (summarized on the Figure as ‘ouabain −5, NE +0). Amphetamine administered using the same time schedule, had a similar effect, probably due to noradrenaline release. Each point represents the average of measurements in 18–20 birds. For further details, see Gibbs and Ng (1977), from where the Figure is reproduced, with permission. For later procedural improvements see, e.g., Gibbs et al. (2008).
An ability of either elevated K+ concentrations (Kaufman and Driscoll, 1993) or either noradrenaline or clonidine, an α2-adrenergic receptor agonist (Chen and Hertz, 1999), to stimulate rate of pyruvate carboxylation might suggest that signaling events akin to those involved in regulation of K+ uptake also might be involved during activation of pyruvate carboxylase. As discussed above, α2-adrenergic receptor stimulates glycogen synthesis. The studies on pyruvate carboxylation by Chen and Hertz (1999) were carried out in glucose-free medium, so glycogen may have become absent, rendering it impossible to know if β-adrenergic stimulation would otherwise have been effective.
In conclusion, glycogen's unaltered degradation under control conditions, and the immediate response to a slight increase in extracellular K+ concentrations or transmitter release may explain its importance for glutamate formation, cellular K+ homeostasis and probably also for reduction of the excitation-inhibitory sAHP. Need of glycogenolysis for glutamate synthesis by astrocytes, contributions by astrocytes to clearance of extracellular K+ increase following neuronal activity, and astrocytic participation in sAHP reduction all seem to contribute to glycogen's unique, pluralistic role in integrative establishment of memory. Both in vivo studies and culture studies have concurred in the importance of glycogenolysis, and thus of astrocytic metabolism, for glutamate production. Its role in K+ clearance from the extracellular space following neuronal activity has not yet been directly established in vivo. However, there is abundant supportive in vivo evidence (see Xu et al., 2013). Possible importance of glycogenolysis for reduction of sAHP could be investigated in vivo, as could roles of glycogenolysis for interactions between learning and sAHP reduction. The robust evidence for glycogen's importance in learning shown by Duran et al. (2013), together with information reviewed in this perspective suggest that such studies could make essential contributions toward understanding “how multiple diverse functions are integrated to produce complex behaviors,” such as learning.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Araneda, R., and Andrade, R. (1991). 5-Hydroxytryptamine 2 and 5-hydroxytryptamine 1A receptors mediate opposing responses on membrane excitability in rat association cortex. Neuroscience 40, 399–412.
Bauer, D. E., Jackson, J. G., Genda, E. N., Montoya, M. M., Yudkoff, M., and Robinson, M. B. (2012). The glutamate transporter, GLAST, participates in a macromolecular complex that supports glutamate metabolism. Neurochem. Int. 61, 566–574.
Bekar, L. K., He, W., and Nedergaard, M. (2008). Locus coeruleus alpha-adrenergic-mediated activation of cortical astrocytes in vivo. Cereb. Cortex 18, 2789–2795.
Bell, G. A., and Gibbs, M. E. (1979). Interhemispheric engram transfer in chick. Neurosci. Lett. 113, 163–168.
Beaudet, A., and Descarries, L. (1978). The monoamine innervation of rat cerebral cortex: synaptic and nonsynaptic axon terminals. Neuroscience 3, 851–860.
Caetano, M. S., Jin, L. E., Harenberg, L., Stachenfeld, K. L., Arnsten, A. F., and Laubach, M. (2012). Noradrenergic control of error perseveration in medial prefrontal cortex. Front. Integr. Neurosci. 6:125. doi: 10.3389/fnint.2012.00125
Chen, Y., and Hertz, L. (1999). Noradrenaline effects on pyruvate decarboxylation: correlation with calcium signaling. J. Neurosci. Res. 58, 599–606.
Choi, H. B., Gordon, G. R., Zhou, N., Tai, C., Rungta, R. L., Martinez, J., et al. (2012). Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron 75, 1094–1104.
Crowe, S. F., Ng, K. T., and Gibbs, M. E. (1989). Memory formation processes in weakly reinforced learning. Pharmacol. Biochem. Behav. 33, 881–887.
Darvesh, A. S., and Gudelsky, G. A. (2003). Activation of 5-HT2 receptors induces glycogenolysis in the rat brain. Eur. J. Pharmacol. 464, 135–140.
Delprat, B., Bibert, S., and Geering, K. (2006). FXYD proteins: novel regulators of Na, K-ATPase. Med. Sci. (Paris) 22, 633–638.
Dietzel, I., Heinemann, U., Hofmeier, G., and Lux, H. D. (1982). Stimulus-induced changes in extracellular Na+ and Cl− concentration in relation to changes in the size of the extracellular space. Exp. Brain Res. 46, 73–84.
Dietzel, I., Heinemann, U., and Lux, H. D. (1989). Relations between slow extracellular potential changes, glial potassium buffering, and electrolyte and cellular volume changes during neuronal hyperactivity in cat brain. Glia 2, 25–44.
DiNuzzo, M., Mangia, S., Maraviglia, B., and Giove, F. (2012). The role of astrocytic glycogen in supporting the energetics of neuronal activity. Neurochem. Res. 37, 2432–2438.
Disterhoft, J. F., Coulter, D. A., and Alkon, D. L. (1986). Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc. Natl. Acad. Sci. U.S.A. 83, 2733–2737.
Dringen, R., Gebhardt, R., and Hamprecht, B. (1993). Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res. 623, 208–214.
Du, T., Li, B., Li, H., Li, M., Hertz, L., and Peng, L. (2010). Signaling pathways of isoproterenol-induced ERK1/2 phosphorylation in primary cultures of astrocytes are concentration-dependent. J. Neurochem. 115, 1007–1023.
Duran, J., Saez, I., Gruart, A., Guinovart, J. J., and Delgado-García, J. M. (2013). Impairment in long-term memory formation and learning-dependent synaptic plasticity in mice lacking glycogen synthase in the brain. J. Cereb. Blood Flow Metab. doi: 10.1038/jcbfm.2012.200. [Epub ahead of print].
Fernández-López, A., Revilla, V., Candelas, M. A., González-Gil, J., Díaz, A., and Pazos, A. (1997). A comparative study of α2- and beta-adrenoceptor distribution in pigeon and chick brain. Eur. J. Neurosci. 9, 871–883.
Fisahn, A., Heinemann, S. F., and McBain, C. J. (2005). The kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurones. J. Physiol. 562, 199–203.
Gibbs, M. E., Anderson, D. G., and Hertz, L. (2006). Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 54, 214–222.
Gibbs, M. E., Andrew, R. J., and Ng, K. T. (2003). Hemispheric lateralization of memory stages for discriminated avoidance learning in the chick. Behav. Brain Res. 139, 157–165.
Gibbs, M. E., Gibbs, Z., and Hertz, L. (2009). Rescue of Aβ (1-42)-induced memory impairment in day-old chick by facilitation of astrocytes oxidative metabolism: implications for Alzhermer's disease. J. Neurochem. 109, 230–236.
Gibbs, M. E., Hutchinson, D., and Hertz, L. (2008). Astrocytic involvement in learning and memory consolidation. Neurosci. Biobehav. Rev. 32, 927–944.
Gibbs, M. E., and Hutchinson, D. S. (2012). Rapid turnover of glycogen in memory formation. Neurochem. Res. 37, 2456–2463.
Gibbs, M. E., Lloyd, H. G., Santa, T., and Hertz, L. (2007). Glycogen is a preferred glutamate precursor during learning in 1-day-old chick: biochemical and behavioral evidence. J. Neurosci. Res. 85, 3326–3333.
Gibbs, M. E., and Ng, K. T. (1976). Diphenylhydantoin facilitation of labile, protein-independent memory. Brain Res. Bull. 1, 203–208.
Gibbs, M. E., and Ng, K. T. (1977). Counteractive effects of norepinephrine and amphetamine on quabain-induced amnesia. Pharmacol. Biochem. Behav. 6, 533–537.
Gold, P. E., and Korol, D. L. (2012). Making memories matter. Front. Integr. Neurosci. 6:116. doi: 10.3389/fnint.2012.00116
Grisar, T., Franck, G., and Schoffeniels, E. (1980). Glial control of neuronal excitability in mammals: II. Enzymatic evidence: two molecular forms of the Na+, K+-ATPase in brain. Neurochem. Int. 2C, 311–320.
Hajek, I., Subbarao, K. V., and Hertz, L. (1996). Acute and chronic effects of potassium and noradrenaline on Na+, K+-ATPase activity in cultured mouse neurons and astrocytes. Neurochem. Int. 28, 335–342.
Hamann, S., Herrera-Perez, J. J., Zeuthen, T., and Alvarez-Leefmans, F. J. (2010). Cotransport of water by the Na+-K+-2Cl− cotransporter NKCC1 in mammalian epithelial cells. J. Physiol. 568, 123–135.
Hashimoto, T., Hussien, R., Cho, H. S., Kaufer, D., and Brooks, G. A. (2008). Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS ONE 3:e2915. doi: 10.1371/journal.pone.0002915
Henderson, S. T., and Poirier, J. (2011). Pharmacogenetic analysis of the effects of polymorphisms in APOE, IDE and IL1B on a ketone body based therapeutic on cognition in mild to moderate Alzheimer's disease; a randomized, double-blind, placebo-controlled study. BMC Med. Genet. 12:137. doi: 10.1186/1471-2350-12-137
Hertz, L. (2011). Astrocytic energy metabolism and glutamate formation–relevance for 13C-NMR spectroscopy and importance of cytosolic/mitochondrial trafficking. Magn. Reson. Imaging 29, 1319–1329.
Hertz, L., Chen, Y., Gibbs, M. E., Zang, P., and Peng, L. (2004). Astrocytic adrenoceptors: a major drug target in neurological and psychiatric disorders? Curr. Drug Targets CNS Neurol. Disord. 3, 239–267.
Hertz, L., Lovatt, D., Goldman, S. A., and Nedergaard, M. (2010). Adrenoceptors in brain: cellular gene expression and effects on astrocytic metabolism and [Ca2+]i. Neurochem. Int. 57, 411–420.
Hertz, L., Peng, L., and Dienel, G. A. (2007). Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 27, 219–249.
Hof, P. R., Pascale, E., and Magistretti, P. J. (1988). K+ at concentrations reached in the extracellular space during neuronal activity promotes a Ca2+-dependent glycogen hydrolysis in mouse cerebral cortex. J. Neurosci. 8, 1922–1928.
Huang, R., and Somjen, G. G. (1997). Effect of hypertonia on voltage-gated ion currents in freshly isolated hippocampal neurons, and on synaptic currents in neurons in hippocampal tissue slices. Brain Res. 748, 157–167.
Hutchinson, D. S., Summers, R. J., and Gibbs, M. E. (2008). Energy metabolism and memory processing: role of glucose transport and glycogen in responses to adrenoceptor activation in the chicken. Brain Res. Bull. 76, 224–234.
Hutson, S. M., Cole, J. T., Sweatt, A. J., and LaNoue, K. F. (2008). Is the anaplerotic enzyme pyruvate carboxylase (PC) only expressed in astrocytes? J. Neurochem. 104, S58.
Hyder, F., and Rothman, D. L. (2012). Quantitative fMRI and oxidative neuroenergetics. Neuroimage 62, 985–994.
Kaufman, E. E., and Driscoll, B. F. (1993). Evidence for cooperativity between neurons and astroglia in the regulation of CO2 fixation in vitro. Dev. Neurosci. 15, 299–305.
Kong, E. K., Peng, L., Chen, Y., Yu, A. C., and Hertz, L. (2002). Up-regulation of 5-HT2B receptor density and receptor-mediated glycogenolysis in mouse astrocytes by long-term fluoxetine administration. Neurochem. Res. 27, 113–120.
MacAulay, N., and Zeuthen, T. (2012). Glial K+ clearance and cell swelling: key roles for cotransporters and pumps. Neurochem. Res. 37, 2299–2309.
Magistretti, P. J. (1988). Regulation of glycogenolysis by neurotransmitters in the central nervous system. Diabete Metab. 14, 237–246.
Mangia, S., Giove, F., and Dinuzzo, M. (2012). Metabolic pathways and activity-dependent modulation of glutamate concentration in the human brain. Neurochem. Res. 37, 2554–2561.
McKenna, M. C. (2012). Substrate competition studies demonstrate oxidative metabolism of glucose, glutamate, glutamine, lactate and 3-hydroxybutyrate in cortical astrocytes from rat brain. Neurochem. Res. 37, 2613–2626.
McKenna, M. C., Hopkins, I. B., and Carey, A. (2001). Alpha-cyano-4-hydroxycinnamate decreases both glucose and lactate metabolism in neurons and astrocytes: implications for lactate as an energy substrate for neurons. J. Neurosci. Res. 66, 747–754.
Muir, D., Berl, S., and Clarke, D. D. (1986). Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain Res. 380, 336–340.
Newman, L. A., Korol, D. L., and Gold, P. E. (2011). Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 6:28427. doi: 10.1371/journal.pone.0028427
O'Donnell, J., Zeppenfeld, D., McConnell, E., Pena, S., and Nedergaard, M. (2012). Norepinephrine: a neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem. Res. 37, 2496–2512.
O'Dowd, B. S., Gibbs, M. E., Ng, K. T., Hertz, E., and Hertz, L. (1994). Astrocytic glycogenolysis energizes memory processes in neonate chicks. Brain Res. Dev. Brain Res. 78, 137–141.
Oh, M. M., McKay, B. M., Power, J. M., and Disterhoft, J. F. (2009). Learning-related postburst afterhyperpolarization reduction in CA1 pyramidal neurons is mediated by protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 106, 1620–1625.
Öz, G., Tesfaye, N., Kumar, A., Deelchand, D., Eberly, L., and Seaquist, E. (2012). Brain glycogen content and metabolism in subjects with type 1 diabetes and hypoglycemia unawareness. J. Cereb. Blood Flow Metab. 32, 256–263.
Pellerin, L., and Magistretti, P. J. (2012). Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 32, 1152–1166.
Quach, T. T., Duchemin, A. M., Rose, C., and Schwartz, J. C. (1988). [3H]glycogenolysis in brain slices mediated by beta-adrenoceptors: comparison of physiological response and [3H]dihydroalprenolol binding parameters. Neuropharmacology 27, 629–635.
Quach, T. T., Rose, C., Duchemin, A. M., and Schwartz, J. C. (1982). Glycogenolysis induced by serotonin in brain: identification of a new class of receptor. Nature 298, 373–375.
Rae, C., Fekete, A. D., Kashem, M. A., Nasrallah, F. A., and Bröer, S. (2012). Metabolism, compartmentation, transport and production of acetate in the cortical brain tissue slice. Neurochem. Res. 37, 2541–2553.
Robertson, S., Gibbs, M. E., and Ng, K. T. (1978). Sodium pump activity, amino acid transport and long-term memory. Brain Res. Bull. 3, 53–58.
Shank, R. P., Bennett, G. S., Freytag, S. O., and Campbell, G. L. (1985). Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 329, 364–367.
Sickmann, H. M., Walls, A. B., Schousboe, A., Bouman, S. D., and Waagepetersen, H. S. (2009). Functional significance of brain glycogen in sustaining glutamatergic neurotransmission. J. Neurochem. 109, 80–86.
Somjen, G. G., Kager, H., and Wadman, W. J. (2008). Computer simulations of neuron-glia interactions mediated by ion flux. J. Comput. Neurosci. 25, 349–365.
Subbarao, K. V., and Hertz, L. (1990). Effect of adrenergic agonists on glycogenolysis in primary cultures of astrocytes. Brain Res. 536, 220–226.
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., et al. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823.
Tucker, A. R., and Gibbs, M. E. (1979). Saccharin aversion memory in rats: inhibition of cycloheximide-resistant memory by ouabain. Physiol. Behav. 23, 341–346.
Walz, W. (2000). Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 36, 291–300.
Wang, F., Smith, N. A., Xu, Q., Fujita, T., Baba, A., Matsuda, T., et al. (2012). Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+. Sci. Signal. 5, ra26.
Waniewski, R. A., and Martin, D. L. (1998). Preferential utilization of acetate by astrocytes is attributable to transport. J. Neurosci. 18, 5225–5233.
Watson, G. G., and Craft, S. (2004). Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer's disease. Eur. J. Pharmacol. 490, 97–113.
Watts, M. E., and Mark, R. F. (1971). Drug inhibition of memory formation in chickens. II. Short-term memory. Proc. R. Soc. Lond. B Biol. Sci. 178, 455–464.
Weil-Malherbe, H., Axelrod, J., and Tomchick, R. (1959). Blood-brain barrier for adrenaline. Science 129, 1226–1227.
Xu, J. N., Song, D., Xue, Z. X., Gu, L., Hertz, L., and Peng, L. (2013). Requirement of glycogenolysis for uptake of increased extracellular K+ in astrocytes – potential implications for K+ homeostasis and glycogen usage in brain. Neurochem. Res. 38, 472–485.
Yu, A. C., Drejer, J., Schousboe, A., and Hertz, L. (1983). Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 41, 1484–1487.
Yuan, H., Gao, B., Duan, L., Jiang, S., Cao, R., Xiong, Y. F., et al. (2010). Acute hyperosmotic stimulus-induced Fos expression in neurons depends on activation of astrocytes in the supraoptic nucleus of rats. J. Neurosci. Res. 88, 1364–1373.
Keywords: astrocyte, glutamate, glycogen, learning, slow neuronal afterhyperpolarization
Citation: Hertz L, Xu J, Song D, Du T, Yan E and Peng L (2013) Brain glycogenolysis, adrenoceptors, pyruvate carboxylase, Na+,K+-ATPase and Marie E. Gibbs' pioneering learning studies. Front. Integr. Neurosci. 7:20. doi: 10.3389/fnint.2013.00020
Received: 25 January 2013; Accepted: 15 March 2013;
Published online: 03 April 2013.
Edited by:
Sharif A. Taha, University of Utah Medical School, USAReviewed by:
Eliana Scemes, Albert Einstein College of Medicine, USAMaiken Nedergaard, New York Medical College, USA
Copyright © 2013 Hertz, Xu, Song, Du, Yan and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Liang Peng, Department of Clinical Pharmacology, China Medical University, No. 92, Beier Road, Heping District, Shenyang 110001, People's Republic of China. e-mail:aGtraWQwOEB5YWhvby5jb20=