 Manigandan Krishnan1
Manigandan Krishnan1 Ayishal B. Mydeen2
Ayishal B. Mydeen2 Mohammed M. Nakhal2
Mohammed M. Nakhal2 Marwa F. Ibrahim2
Marwa F. Ibrahim2 Richard L. Jayaraj1†
Richard L. Jayaraj1† Milos R. Ljubisavljevic3
Milos R. Ljubisavljevic3 Mohammad I. K. Hamad2*
Mohammad I. K. Hamad2* Fatima Y. Ismail1,4*
Fatima Y. Ismail1,4*- 1Department of Pediatrics, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates
- 2Department of Anatomy, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates
- 3Department of Physiology, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates
- 4Department of Neurology (Adjunct), Johns Hopkins University School of Medicine, Baltimore, MD, United States
Introduction: Mutations in the methyl-CpG-binding protein-2 gene (MECP2), which cause Rett syndrome (RTT), disrupt neuronal activity; however, the impact of the MECP2 loss-of-function on the cytoarchitecture of medial entorhinal cortex layer II (MECII) neurons—crucial for spatial memory and learning—remains poorly understood.
Methods: In this study, we utilized Golgi staining and neuron tracing in the Mecp2+/− mouse model of RTT to investigate the pyramidal and stellate cell alterations in MECII.
Results and discussion: Our findings revealed that pyramidal cells displayed a significant reduction in apical dendritic length, soma size, and spine density, while basal dendrites showed increased dendritic complexity and branching. On the other hand, stellate cells exhibited dendritic hypertrophy along with increased soma size, primary dendrites, and localized increase in dendritic intersections, despite an overall reduction in total dendritic length and spine density. These findings underscore the notion that MECP2 loss-of-function can disrupt MECII pyramidal and stellate cell cytoarchitecture in a cell-type-specific manner, emphasizing its critical role in maintaining proper dendritic morphology in circuits, which is crucial for learning and memory.
1 Introduction
Dendrites are specialized neural structures that receive and integrate synaptic inputs from presynaptic neurons, playing a crucial role in shaping the neuronal networks that underpin cognitive and behavioral processes (Wu et al., 1999; Prigge and Kay, 2018; Hamad et al., 2023). Dysregulation in dendritic architectures such as length, branching patterns, and spine density can impair synaptic networks and lead to region- and cell-type-specific neuronal dysfunction, which in turn influences cognitive and behavioral traits (Armstrong et al., 1995; Moretti et al., 2005; Valnegri et al., 2015). Many neurodevelopmental diseases, such as autism spectrum disorder (ASD) (Geschwind and Levitt, 2007), fragile X syndrome (Contractor et al., 2015), trisomy 21 Down syndrome (Klein and Haydar, 2022), and epilepsy (Leifeld et al., 2022), are associated with unusual changes in the structure of dendrites. Among these ailments is RTT, a severe and progressive condition caused by X chromosome MECP2 gene mutations (Amir et al., 1999; Gulmez Karaca et al., 2019; Good et al., 2021), with an estimated frequency of 1 in 10,000. RTT almost entirely affects women (Hagberg, 1985; Neul et al., 2010) and is characterized by cognitive, learning, memory, and movement disabilities (Armstrong, 2005; Guy et al., 2007).
Maintaining dendritic structure and function across growth and development depends on the Mecp2 protein, a transcriptional regulator in neurons (Shahbazian et al., 2002; Gonzales and LaSalle, 2010; Neul et al., 2010). RTT syndrome patients have varied and region-specific dendritic structural alterations, especially in layers 2, 3, and 5 of the motor cortex, which and prefrontal cortex exhibit the most marked decrease in dendritic complexity and spine density (Leontovich et al., 1999), however, less dramatic alterations are seen in the hippocampal and visual cortex (Belichenko et al., 1997; Armstrong, 2005). Mouse models of MECP2 mutations recapitulate these region-specific alterations. Mecp2-null mice showed a significant decrease in the branching of dendrites and the number of spines on pyramidal neurons in the somatosensory (Layers 2, 3, and 5) and motor (Layer 5) areas of the brain (Fukuda et al., 2005). In the Mecp2T158A/y partial mutation models, older animals showed changes in the dendrites of the somatosensory area and smaller cell bodies in the sensorimotor cortex, which were connected to later behavioral issues (Wang et al., 2013).
The brain region crucial for memory is the entorhinal cortex (EC), which acts as an interface between the neocortex and the hippocampus. The MECII, which is a part of the EC, has six layers (L1 to L6), and these layers play a major role in processing spatial and sensory inputs for navigation and memory formation (Howard et al., 2014). The MECII has two different cell types—pyramidal and stellate cells—which differ not only in their gene and protein expression markers but also in their structural characteristics and projection targets (Alonso and Klink, 1993). MECII pyramidal cells selectively express calbindin (a protein that binds calcium) and Wolfram syndrome 1 (Wfs1)—a gene involved in endoplasmic reticulum homeostasis. They send signals mainly to the hippocampal CA1 area and mostly to other parts of the cortex and subcortex (Brun et al., 2008; Kitamura et al., 2014, 2017). Whereas MECII stellate cells typically express the transcription factor reelin, primarily innervating dentate gyrus-granular cells and hippocampal CA3 pyramidal cells, thereby playing a vital role in spatial memory and contextual learning (Kitamura et al., 2014; Ray et al., 2014; Hamad et al., 2021). Research reported that MECII are among the earliest neurons affected by Alzheimer's disease (AD) by disrupting the link between the neocortex and hippocampus, contributing to early memory loss (Khan et al., 2014).
Given RTT-associated cognitive deficits and the MECII's role in spatial and memory circuits, we hypothesize that MECP2 loss-of-function might lead to cell-type-specific dendritic alterations in MECII, potentially contributing to the pathophysiology of RTT phenotypes. Therefore, we aimed to investigate how loss of MECP2 affects the changes in the dendrites of MECII pyramidal and stellate cells by using a Mecp2+/− mouse model. Dendritic architecture was assessed using Golgi staining, high-resolution imaging, and morphometric analysis to better understand how MECP2 loss leads to the cell-type-specific disruptions in MECII.
2 Material and methods
2.1 Animals and maintenance
Heterozygous female Mecp2+/− mice (8 weeks of age, strain: 003890, B6.129P2(C)- Mecp2tm1.1Bird/J) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). The 8-week-old wild-type (WT) C57BL/6 of both sexes were procured from the Laboratory Animal Research Facility at the United Arab Emirates University. All experimental mice were housed in a climate-controlled environment with a 12-h light/dark cycle and had unrestricted access to food and water. The temperature was maintained at 25°C.
2.2 Experimental design
We selected 12-month-old female Mecp2+/− mice (n = 6) that consistently exhibited hind-limb clasping (Supplementary Figures 1D, E) for subsequent morphological analysis of the MECII region. Respective age-matched WT female mice (n = 6) were used as normal controls. The animal experimental protocols were approved by the Animal Use Ethics Committee of the United Arab Emirates University (ethical approval number: ERA_2021_8444).
2.3 Neuronal dendrite golgi-cox staining
The morphology of neuronal dendrites was analyzed using the FD Rapid GolgiStain Kit (FD NeuroTechnologies, Inc.; PK401 Cell Systems Biology). Tissue preparation and staining were performed according to the manufacturer's instructions. At the end of the experiment, the body weights of WT and Mecp2+/− controls were recorded. Mice were decapitated, their brains were gently washed with water, weighed, and then incubated with equal volumes of Golgi-Cox impregnation solution (Solutions A and B) for 2 weeks in the dark. Subsequently, the brains were immersed in Solution C for 1 week. A vibratome was utilized to obtain 120 μm parasagittal sections that were cut from both hemispheres, extending to the medial border of the MEC region. The absence of a broad white band surrounding the external capsule was a distinguishing characteristic of these sections. A maximum of 16 slices were obtained per mouse, including the MEC region from both hemispheres. The brain slices were then rehydrated in milli Q water, stained with Solutions D and E for 10 min, washed again in milli Q water, dehydrated, and cover slipped with a water-based mounting medium.
The MEC area for morphological assessment was defined using the mouse brain atlas (Paxions, 2008). For WT mice, off-sagittal sections were aligned to the following Bregma coordinates: anteroposterior (AP) = −5.0 mm, mediolateral (ML) = +3.5 mm, and dorsoventral (DV) = +3.0 mm (Figures 1A, B) and for Mecp2 mice, the sections were aimed at the Bregma coordinates: AP = −5.0 mm, ML = +2.9 mm, and DV = +3.0 mm (Figures 1A, B). These locations were chosen to uniformly target the MEC in all animals. To identify the layers (L1 to L6) within the MEC, we followed cytoarchitectural criteria as described previously (Canto et al., 2008; Witter et al., 2017), which separate layers according to cell density, soma size, and general cytoarchitecture. L1 consists of multipolar neurons (MPNs). Dense clusters of tiny stellate neurons are defined in L2; a narrow band of pyramidal neurons are defined in L2 and L3; moderately spiny pyramidal-shaped neurons in L4; more widely dispersed bigger pyramidal cells are defined in L5 and MPNs are located throughout L6.
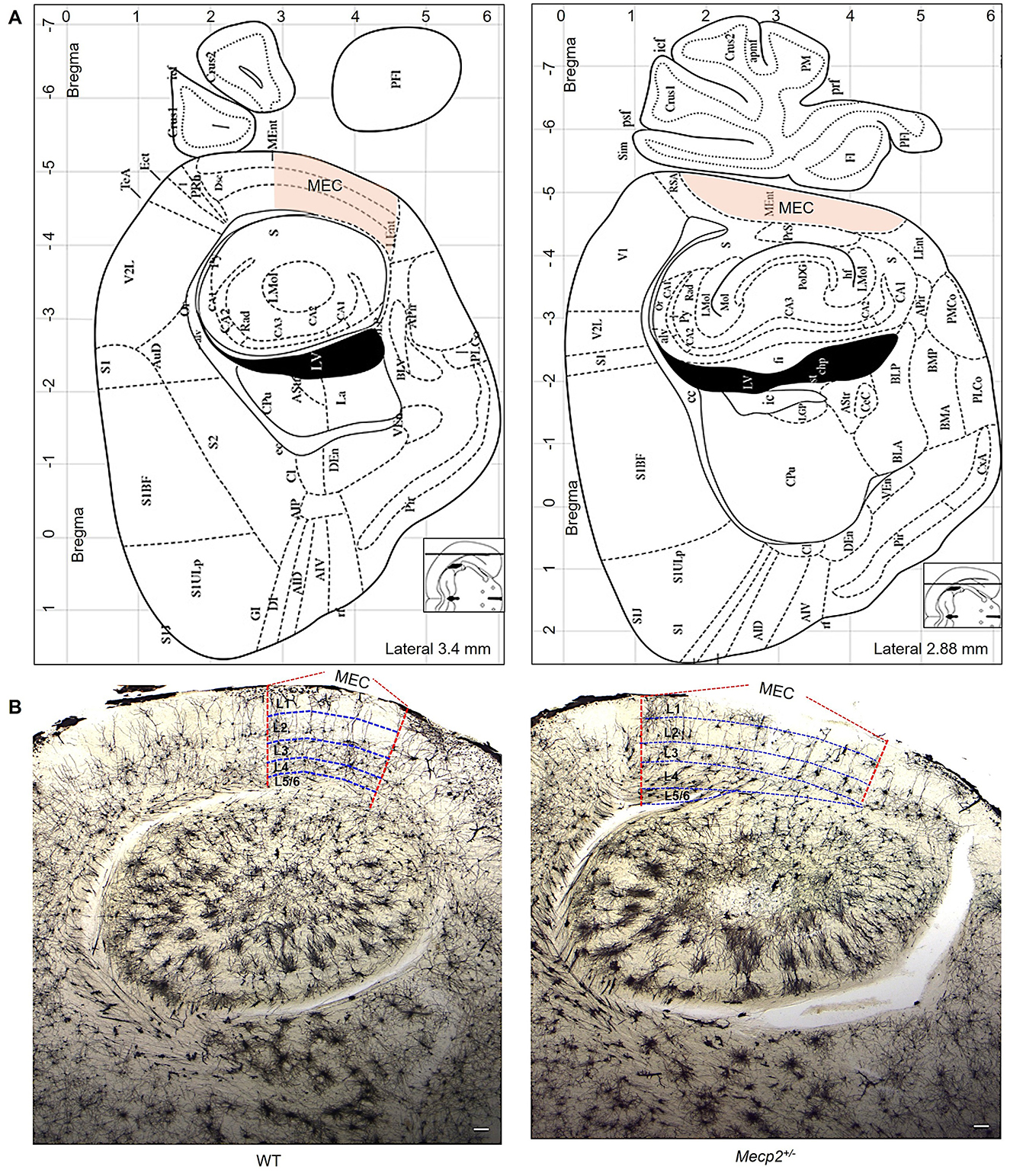
Figure 1. The MEC area is utilized to measure dendritic morphology and trace neurons. (A) The MEC (orange hue) area utilized to select neurons for dendritic morphology evaluations was established using the mouse brain atlas (Paxions, 2008). (B) Golgi staining shows dendritic arbor structures and branching patterns in sagittal slices of WT and Mecp2+/− mice (2.5× magnification, scale bar: 1000 μm). Vertical lines (red color) represent MEC areas, and horizontal lines (blue color) indicate MEC layers (L1 to L6), as established by previous findings (Witter et al., 2017).
2.4 Identification and 3D reconstruction of pyramidal and stellate cells
In this study, pyramidal and stellate cells from MECII regions were identified and reconstructed using the Neurolucida™ system (MicroBrightField, MBF Bioscience Inc., Williston, VA, USA) at 1000 × magnification, following previously described methods (Hamad et al., 2024). A detailed analysis of the morphology of each neuron was conducted to characterize the architecture of the dendrites and the projections of the axons. Pyramidal cells were identified by their unique apical dendrite and several small basal dendrites. These dendrites are highly spiny, resembling those of pyramidal cells in the somatosensory and motor cortices. To quantify their morphological parameters, soma size, apical dendritic length, mean basal dendritic length (total basal dendritic length divided by the number of primary dendrites), total apical dendritic segments, mean basal dendritic segments, and dendritic spine density (number of spines/10 μm of dendrites) were analyzed.
Stellate cells were recognized by a larger number of primary dendrites, high dendritic spine density, and axonal projections extending to the white matter rather than forming local branches. Although these neurons exhibit a multipolar arrangement, they differ markedly from inhibitory interneurons, which typically branch locally (Karube et al., 2004) and possess four primary dendrites with relatively few spines (Kawaguchi et al., 2006). In contrast, stellate cells project their axons distally within MECII and toward the white matter and hippocampus (Kitamura et al., 2014; Fuchs et al., 2016), while possessing eight spiny primary dendrites. To quantify their morphological parameters, we calculated soma size, dendritic spine density (number of spines/10 μm of dendrites), mean dendritic length (total dendritic length divided by the number of primary dendrites), mean number of dendritic segments (total dendritic segments divided by the number of primary dendrites), and the number of primary dendrites.
Sholl analysis was used to assess dendritic complexity in both stellate and pyramidal cells in MECII of Mecp2+/− mice, as compared to wild-type (WT) controls. This approach involves drawing concentric circles (or spheres) at 10 μm intervals around the soma and counting the number of dendritic intersections. This methodology facilitates the identification of specific regions where dendritic complexity changes (Sholl, 1953; Zagrebelsky et al., 2020).
2.5 Statistical analysis
All statistical analyses were conducted using SigmaPlot Version 12. Comparisons between the two groups were performed using Student's unpaired t-test when the normality test (Shapiro-Wilk) was passed; otherwise, the Mann–Whitney test was used. Data are presented as the mean ± standard error of the mean (SEM). Results were considered statistically significant at p < 0.05.
3 Results
3.1 Mecp2+/− mice show altered dendritic morphology of MECII pyramidal cells
We chose to study the morphology of MECII neurons in the Mecp2+/− mouse model because MECII plays a crucial role in connecting the hippocampus with other cortical regions that are essential for spatial navigation and memory (Howard et al., 2014). As shown in Figures 2A, G, Mecp2+/− mice exhibited a significant reduction in mean apical dendritic length (381.41 ± 24.26 μm) when compared to WT (563.1 ± 38.66 μm), but the number of mean apical dendritic segments (Figure 2H) remained unchanged in Mecp2+/− (17.35 ± 1.18), and WT mice (16.9 ± 1.41). To determine in which dendritic compartment of the apical dendrites the reduction of complexity has occurred, we performed Sholl analyses, which revealed an increase in mean apical dendritic intersections specifically at 40 μm (3.18 ± 0.26) from the soma in Mecp2+/− mice (Figures 2F, I, J) when compared to WT (2.42 ± 0.27 at 40 μm). The total number of apical intersections remained unchanged.
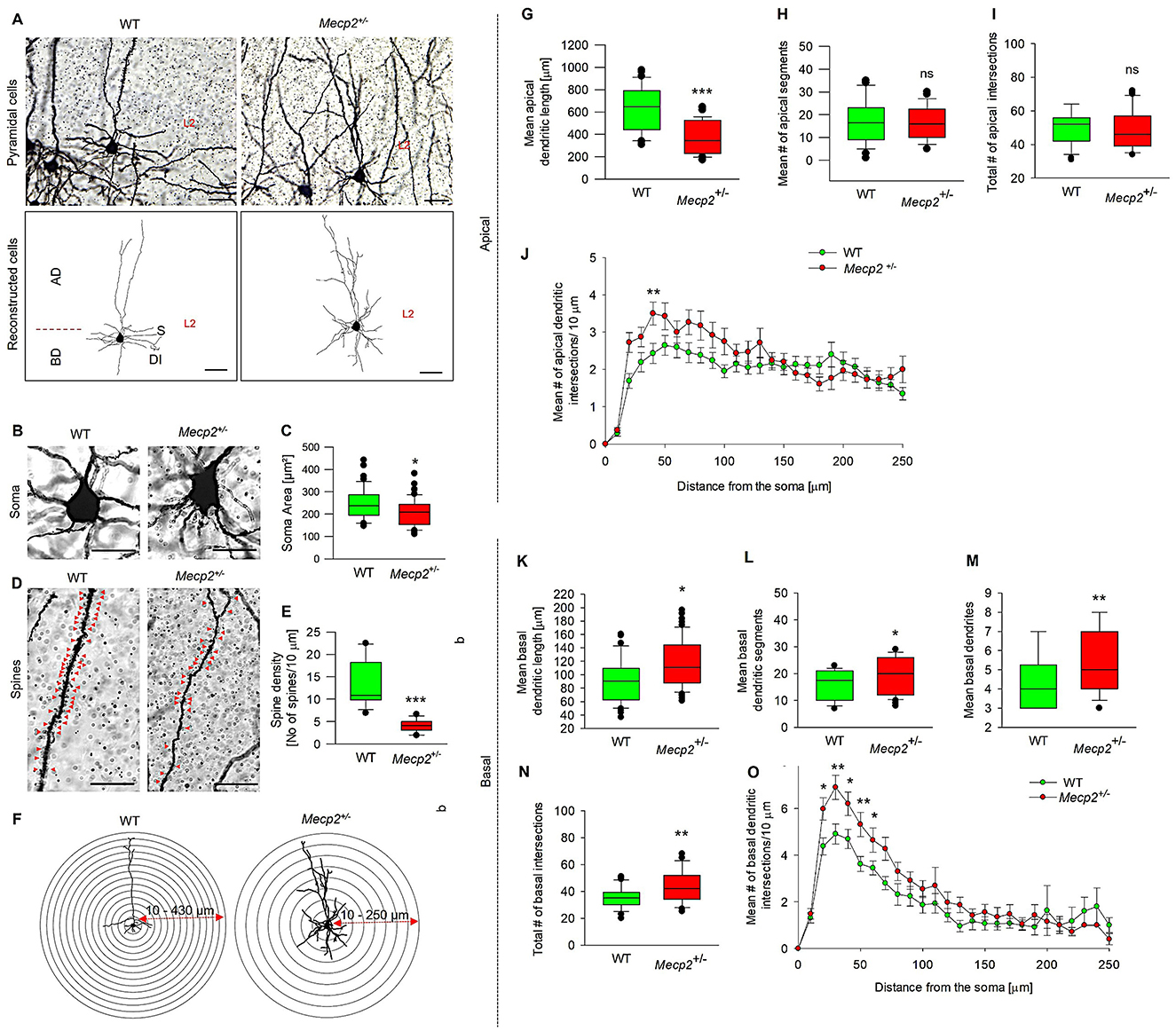
Figure 2. Quantitative morphometric analysis of dendrites in MECII pyramidal cells of Mecp2+/− mice. (A) Golgi staining reveals dendritic arbor structures and branching patterns of pyramidal cells in sagittal sections of WT and Mecp2+/− mice (40× magnification, scale bars: 100 μm), together with their corresponding 2D reconstructed pyramidal neuron images. AD-apical dendrites, BD-basal dendrites, S-soma, and DI-dendritic intersections, and L2-Layer2. Golgi-stained image showing (B) Soma size and (C) mean soma area, (D) spine distributions and (E) dendritic spine densities of WT and Mecp2+/− mice. Representative stereotype images were obtained with a 63× oil immersion objective (20 μm, mean ± SEM (n = 6), number of cells analyzed = 12 cells/group, *p < 0.05, ***p < 0.001 by Mann-Whitney t-test). (G) Mean apical dendritic length, (H) mean apical dendritic segments, and (I, J, F) Sholl analysis of dendritic intersections in Mecp2+/− mice compared to WT. Concentric circles are centered on the soma and spaced at 30 μm intervals, ranging from 10 to 430 μm for WT and 10 to 250 μm for Mecp2+/−, and (K) Quantitative analysis of mean basal dendritic length, (K) Mean basal dendritic segments, (L) Mean basal dendrites, (M) and (N, O, F) Basal dendritic intersections in Mecp2+/− mice compared to WT. Data are presented as mean ± SEM (n = 6 independent preparations; number of cells analyzed/group = 42 cells (WT) and 53 cells (Mecp2+/−), *p < 0.05, **p < 0.01, ***p < 0.001, ns non-significant by Mann-Whitney t-test).
Considering the apical dendritic changes observed in pyramidal cells of Mecp2+/− mice, we further investigated the dendritic patterns in the basal region of these neurons. Figure 2A depicts our qualitative morphometric study of basal dendrites in pyramidal cells, which indicated substantial alterations in the dendritic structure of Mecp2+/− mice. Mecp2 heterozygosity resulted in a longer mean basal dendritic length (115.72 ± 5.56 μm, Figure 2K) compared to WT (97.73 ± 6.34 μm). In addition, we noticed a significant increase in the mean basal dendritic segments (20.62 ± 1.36, Figure 2L), mean basal dendrites (5.50 ± 0.23, Figure 2M), and total number of basal dendritic intersections (49.25 ± 3.68, Figure 2N) compared to WT (mean basal dendritic segments, 5.76 ± 0.96; mean basal dendrites, 4.61 ± 0.29; and total number of basal dendritic intersections, 33.73 ± 2.11). Sholl analysis of dendritic complexity revealed that Mecp2+/− mice exhibited increased basal dendritic intersections starting at 20 μm from the soma and extending to 60 μm (Figures 2F, O). Given the deficiencies in pyramidal cell development and differentiation in Mecp2+/− mice, we next examined soma size and spine density patterns. Figures 2B, C indicate that Mecp2+/− animals have a considerably reduced soma area (208.9 ± 8.47 μm2) compared to WT mice (245.3 ± 10.59 μm2). Spine distribution and density quantification results (Figures 2D, E) revealed a substantial drop in spine numbers (4.11 ± 0.39 spine density/10 μm) in Mecp2+/− animals, compared to WT mice with 13.16 ± 1.50 spine density/10 μm. These morphological alterations in MECII pyramidal cells in Mecp2+/− mice might contribute to altered synaptic connectivity.
3.2 Mecp2+/– mice show increased dendritic morphology of MECII stellate cells
We then explored the effect of Mecp2+/− on the dendritic patterns of stellate cells (Figure 3), which are part of the grid cell network within the MECII and are also implicated in spatial navigation and memory. Figures 3A, B show that Mecp2 heterozygosity was associated with a reduction in the mean dendritic length of stellate cells to 130.55 ± 5.95 μm from that of WT (138.61 ± 7.67 μm), while mean dendritic segments (Figure 3C) remained unchanged in both groups (Mecp2+/−, 25.24 ± 0.89; WT, 26.56 ± 1.63). However, dendritic hypertrophy was observed in Mecp2+/− (Figure 3H), as indicated by an increased number of primary dendrites (9.84 ± 0.92) when compared to WT (5.21 ± 0.38). Furthermore, Sholl analysis of dendritic complexity revealed a distance-specific increase in mean dendritic intersections in Mecp2+/− mice at 20 μm from the soma, extending to 50 μm (Figures 3J, K). However, the total number of dendritic intersections in MECII stellate cells (Figure 3I) did not differ between Mecp2+/− (77.61 ± 4.24) and WT mice (65.62 ± 5.77). Notably, we observed severe dendritic alterations in MECII stellate cells. Further, we investigated the soma size and spine density changes in the Mecp2+/− mice and observed that soma size significantly increased to 311.3 ± 13.31 μm (Figures 3D, F) compared to WT (236.8 ± 9.95 μm). Spine distribution and density analyses (Figures 3E, G) showed a decrease in the spine numbers in Mecp2+/− mice (20.54 ± 2.38 spine density/10 μm) compared to the WT (32.75 ± 2.46 spine density/10 μm). Collectively, Mecp2 loss-of-function can result in structural abnormalities in MEC stellate cells on L2, as evidenced by dropped spine density and altered dendritic structures.
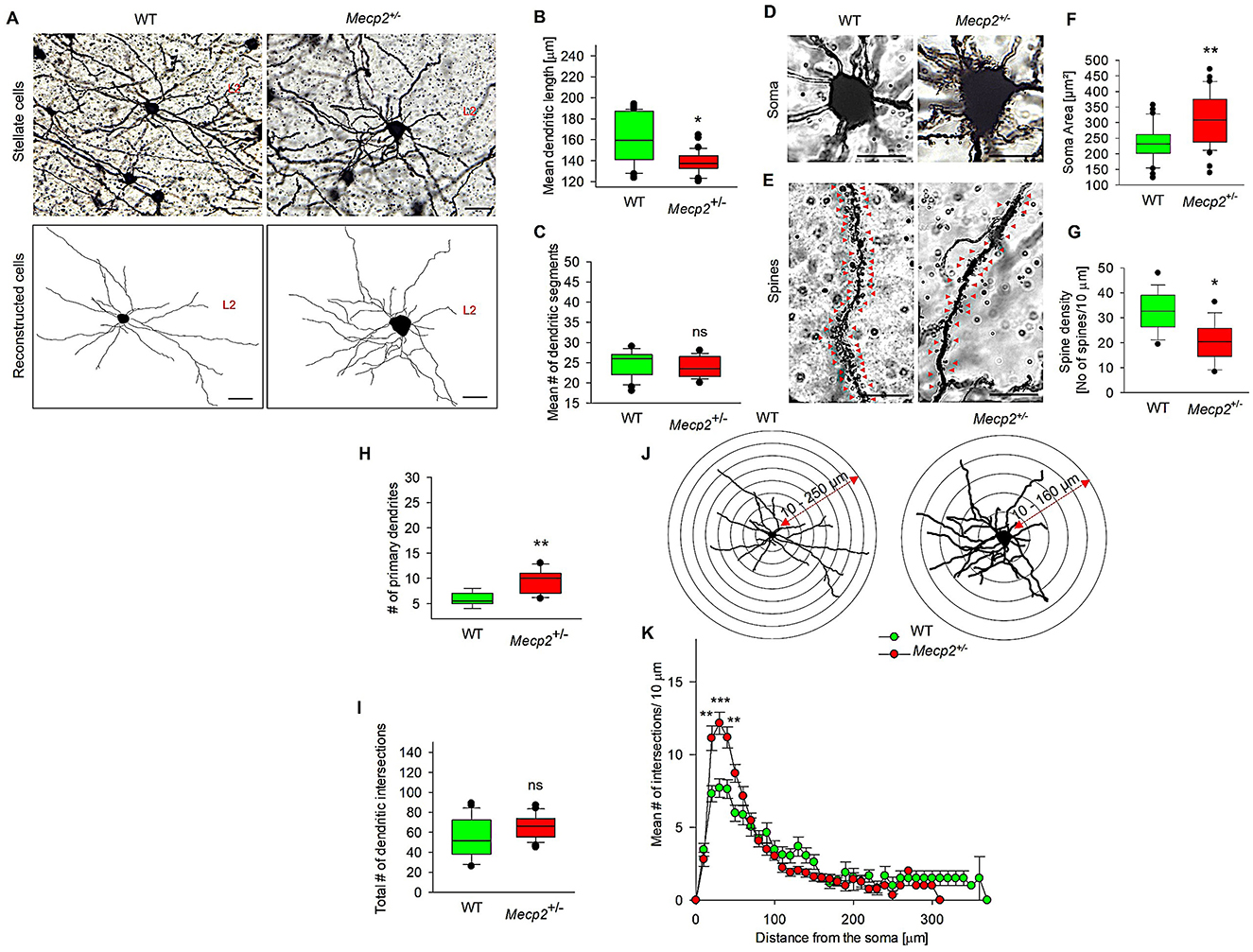
Figure 3. Quantitative morphometric analysis of dendrites in MECII stellate cells of Mecp2+/− mice. (A) Representative Golgi staining of stellate cells in both WT and Mecp2+/− mice, along with their corresponding 2D reconstructed images, showed variations in dendritic phenotypes (40× magnification, scale bar: 100 μm, L2: Layer 2). (B) Mean dendritic length, (C) Mean dendritic segments. Golgi-stained image showing (D) Soma size and (E) Spine distributions, (F) Mean soma area, and (G) dendritic spine densities of WT and Mecp2+/− mice. Representative stereotype images of soma and spine densities were obtained with a 63× oil immersion objective (20 μm, mean ± SEM (n = 6), number of cells analyzed = 12 cells/group, *p < 0.05, **p < 0.01 by Mann-Whitney t-test). (H) Mean primary dendrites, (I) Total dendritic intersections, (J) Sholl analysis of dendritic intersections in the stellate cells of WT and Mecp2+/− mice. Concentric circles are centered on the soma and spaced at 30 μm intervals, ranging from 10 to 250 μm for WT and 10 to 160 μm for Mecp2+/− and (K) Sholl analysis showing increased dendritic intersections in Mecp2+/− mice at 20–50 μm from the soma. Data are presented as mean ± SEM (n = 6 independent preparations); number of cells analyzed/group = 38 cells (WT) and 40 cells (Mecp2+/−); Comparisons: *p < 0.05, **p < 0.01, ***p < 0.001, and ns non-significant by the Mann-Whitney t-test with Mecp2+/− mice).
4 Discussion
Loss-of-function in the MECP2 gene produces RTT, a neurodevelopmental condition characterized by extensive anatomical and functional deficits in the brain. Aberrant neuronal structures are one of the suggested underlying mechanisms of RTT pathology (Chapleau et al., 2009; Albizzati et al., 2024). However, these abnormalities may vary substantially and are often cell-type and region-specific (Asaka et al., 2006; Smrt et al., 2007; Katz et al., 2012; Wang et al., 2013).
Mecp2 mutant animal models and brain slices from RTT patients (Bauman et al., 1995; Guy et al., 2001, 2007) have consistently shown dendritic abnormalities such as decreased size, dendritic complexity, and spine density in the motor, frontal, temporal, visual, hippocampus, and somatosensory areas (Dunn and MacLeod, 2001; Moretti et al., 2005; Belichenko et al., 2009; Chapleau et al., 2009; Zhang et al., 2016). Though the dendritic pathology in these brain regions is well-documented, much less is known about MECII, a region vital for memory and sensory integration, and it is anatomically linked to both the hippocampus and other cortical regions (Kitamura et al., 2015, 2017; Witter et al., 2017; Osanai et al., 2023). Thus, an investigation into how MECP2 affects the normal architectures of MECII pyramidal and stellate cells could provide deeper information about the structural deficits linked to RTT pathophysiology could pave the way for targeted therapeutic strategies. RTT animal models with inducible MECP2 gene loss exhibit global brain shrinkage and a reduction in neuronal cell body size at late juvenile or adult stages (Nguyen et al., 2012). We observed a significant decrease in body and brain weight in Mecp2+/− mice at 12 months of age, which aligns with postmortem reports of reduced brain weight in RTT patients. These reductions are thought to result from halted halted development or neuronal shrinkage, rather than neuronal degeneration (Armstrong et al., 1999).
In this study, we systematically examined the morphology of selected neuronal populations (such as pyramidal and stellate cells) in the MECII region of Mecp2+/− mice exhibiting significant hind limb clasping (a RTT pathology), which is consistent with the reported motor phenotype in MECP2 loss-of-function animal models (Chao et al., 2010). Our findings show substantial cytoarchitectural disruptions in the MEC pyramidal (L2) and stellate cells (L2) of the RTT mice, emphasizing the function of Mecp2 in regulating the morphological integrity within the MEC—a region crucial for memory and spatial navigation (Howard et al., 2014). We found that apical dendrites of the pyramidal cells (L2) showed significant shortening in Mecp2+/− animals, aligning with earlier research that loss of Mecp2 impairs dendritic growth and development (Kishi and Macklis, 2004; Chapleau et al., 2009). On the other hand, apical dendritic number remained unaffected, indicating that Mecp2 functional loss may selectively affect dendritic elongation rather than branching. Even though the overall cellular connectivity may still be impaired, we noticed a localized increase in the apical dendritic intersections close to the soma at 40 μm, suggesting the compensatory dendritic remodeling (Belichenko et al., 2009).
Unlike apical dendrites, the morphology of basal dendrites in pyramidal cells at L2 exhibited increased complexity—as evidenced by the greater dendritic complexity observed between 20 and 60 μm from the soma (domain that integrates cortical inputs) in the Mecp2+/− mice. It's well-documented that neurons in the L2 are the major projections toward EC > dentate gyrus > CA2/CA3 areas of the hippocampus, and any morphological changes in L2 neurons may impact EC-hippocampal neural connections (Witter and Amaral, 1991; Witter et al., 2017). In line with this agreement, as the changes mainly happen in the basal region, we suggest that they affect the signals terminating in the L2 rather than those arriving far away in L1, which could impact the overall function within MEC-hippocampus signaling or compensatory remodeling because of RTT pathology (Fukuda et al., 2005). Shrinkage of soma size is associated with neurotrophic or metabolic dysfunctions, particularly in BDNF signaling—a well-recognized downstream target of MeCP2 (Chang et al., 2006). In this study, we observed a reduction in the soma size of pyramidal cells, consistent with the above findings of RTT models.
Another key component of the grid cell network in the MECII region is stellate cells, which regulate the synaptic information to other neurons. In this study, Mecp2+/− mice showed modestly reduced mean dendritic length but exhibited dendritic hypertrophy, as evinced by the greater number of primary dendrites in stellate cells at L2. This dendritic reorganization may indicate a malformed developmental trajectory or compensatory sprouting (Belichenko et al., 2009). Increased dendritic intersections from Sholl analysis near 20–50 μm from the soma, again revealing distance-specific remodeling without an increase in total dendritic complexity. Since spines are crucial for synaptic excitatory response, their loss is likely to alter the neuronal connectivity and integrity, leading to several behavioral deficits (Chapleau et al., 2009). In this study, we found that MECII pyramidal and stellate cells exhibited fewer dendritic spines—where synaptic connections form. This reduction in spine density could indicate synaptic dysfunction; such features are commonly reported in the RTT pathology involving MeCP2 dysfunction (Zoghbi, 2003; Chahrour et al., 2008). In contrast to the pyramidal cells, the soma size was significantly enlarged in stellate cells, suggesting cell-type-specific effects of Mecp2 functional loss. This diverse effect may result from different gene regulation or metabolic demands across several neuronal types and needs future investigations (Chahrour and Zoghbi, 2007).
In summary, pyramidal and stellate cells in the MECII region showed marked structural alterations, including dendritic morphology, soma size and spine density, suggesting that Mecp2 loss-of-function can disrupt synaptic connectivity between the MEC-hippocampal axis (Table 1). Overall, our study highlights how Mecp2 dysfunction can differentially alter dendritic cytoarchitectures in these MECII cell types, offering insights into the neural basis of spatial and memory dysfunction in RTT pathology.
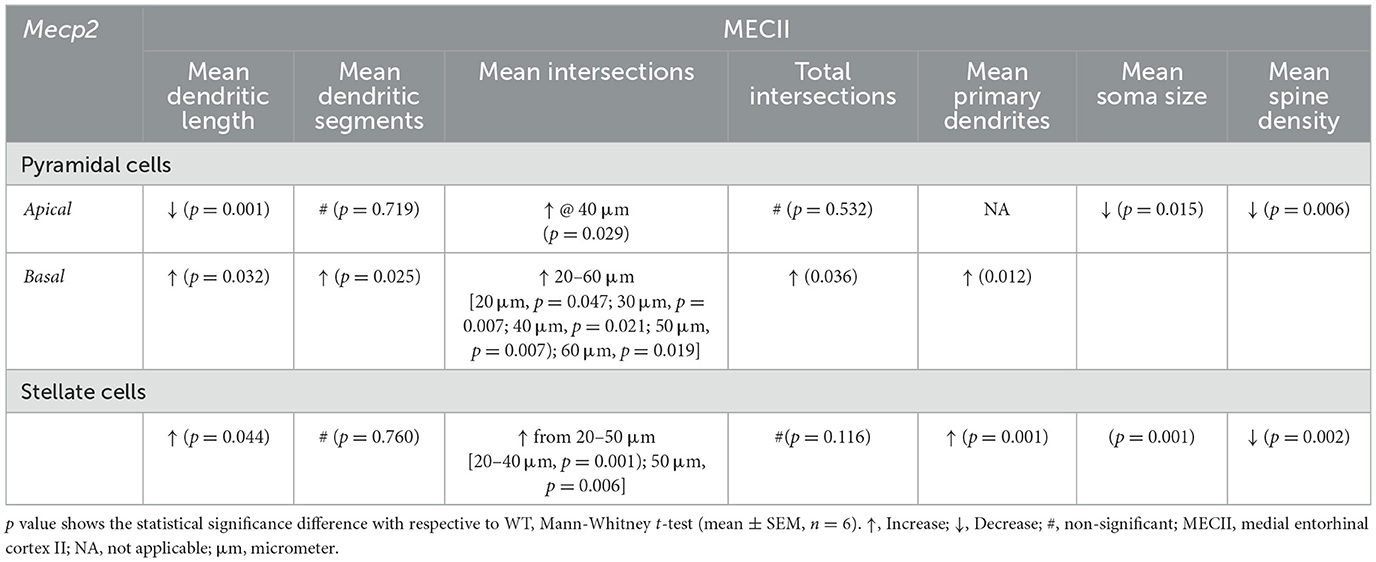
Table 1. Mecp2+/− mice exhibits structural changes in their MECII pyramidal and stellate cells.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by United Arab Emirates Animal Comittee (ethical approval number: ERA_2021_8444). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MK: Data curation, Formal analysis, Methodology, Resources, Software, Validation, Writing – original draft, Writing – review & editing. ABM: Data curation, Methodology, Validation, Writing – original draft. MMN: Data curation, Formal analysis, Methodology, Software, Writing – original draft. MFI: Formal analysis, Methodology, Validation, Writing – original draft. RLJ: Data curation, Methodology, Writing – original draft. MRL: Conceptualization, Data curation, Funding acquisition, Investigation, Project administration, Resources, Validation, Visualization, Writing – review & editing, Writing – original draft. MIKH: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Writing – original draft, Writing – review & editing. FYI: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the United Arab Emirates University startup grant numbers 12M142 and 31M525 to MIKH, the UAEU UPAR grant number 12M159 to MIKH; and UAEU CMHS Grant number 12M117 to FYI and MRL.
Acknowledgments
The authors extend their gratitude to Prof. Sandeep Subramanya and Dr. Balaji Venkataraman from the Department of Physiology, College of Medicine and Health Sciences, UAEU, for their assistance with thermal cycler and southern blotting.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnana.2025.1580435/full#supplementary-material
References
Albizzati, E., Breccia, M., Florio, E., Cabasino, C., Postogna, F. M., Grassi, R., et al. (2024). Mecp2 knock-out astrocytes affect synaptogenesis by interleukin 6 dependent mechanisms. iScience 27:109296. doi: 10.1016/j.isci.2024.109296
Alonso, A., and Klink, R. (1993). Differential electroresponsiveness of stellate and pyramidal-like cells of medial entorhinal cortex layer II. J. Neurophysiol. 70, 128–143. doi: 10.1152/jn.1993.70.1.128
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., Zoghbi, H. Y., et al. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Armstrong, D., Dunn, J. K., Antalffy, B., and Trivedi, R. (1995). Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 54, 195–201. doi: 10.1097/00005072-199503000-00006
Armstrong, D. D. (2005). Neuropathology of Rett syndrome. J. Child. Neurol. 20, 747–753. doi: 10.1177/08830738050200082401
Armstrong, D. D., Dunn, J. K., Schultz, R. J., Herbert, D. A., Glaze, D. G., Motil, K. J., et al. (1999). Organ growth in Rett syndrome: a postmortem examination analysis. Pediatr. Neurol. 20, 125–129. doi: 10.1016/S0887-8994(98)00124-6
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227. doi: 10.1016/j.nbd.2005.07.005
Bauman, M. L., Kemper, T. L., and Arin, D. M. (1995). Microscopic observations of the brain in Rett syndrome. Neuropediatrics 26, 105–108. doi: 10.1055/s-2007-979737
Belichenko, P. V., Hagberg, B., and Dahlstrom, A. (1997). Morphological study of neocortical areas in Rett syndrome. Acta. Neuropathol. 93, 50–61. doi: 10.1007/s004010050582
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C., et al. (2009). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258. doi: 10.1002/cne.22009
Brun, V. H., Leutgeb, S., Wu, H. Q., Schwarcz, R., Witter, M. P., Moser, E. I., et al. (2008). Impaired spatial representation in CA1 after lesion of direct input from entorhinal cortex. Neuron 57, 290–302. doi: 10.1016/j.neuron.2007.11.034
Canto, C. B., Wouterlood, F. G., and Witter, M. P. (2008). What does the anatomical organization of the entorhinal cortex tell us? Neural. Plast. 2008:381243. doi: 10.1155/2008/381243
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341-348. doi: 10.1016/j.neuron.2005.12.027
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269. doi: 10.1038/nature09582
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S., et al. (2009). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 35, 219–233. doi: 10.1016/j.nbd.2009.05.001
Contractor, A., Klyachko, V. A., and Portera-Cailliau, C. (2015). Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. doi: 10.1016/j.neuron.2015.06.017
Dunn, H. G., and MacLeod, P. M. (2001). Rett syndrome: review of biological abnormalities. Can. J. Neurol. Sci. 28, 16–29. doi: 10.1017/S0317167100052513
Fuchs, E. C., Neitz, A., Pinna, R., Melzer, S., Caputi, A., Monyer, H., et al. (2016). Local and distant input controlling excitation in layer II of the medial entorhinal cortex. Neuron 89, 194–208. doi: 10.1016/j.neuron.2015.11.029
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., and Goto, Y. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 64, 537–544. doi: 10.1093/jnen/64.6.537
Geschwind, D. H., and Levitt, P. (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 17, 103–111. doi: 10.1016/j.conb.2007.01.009
Gonzales, M. L., and LaSalle, J. M. (2010). The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 12, 127–134. doi: 10.1007/s11920-010-0097-7
Good, K. V., Vincent, J. B., and Ausio, J. (2021). MeCP2: the genetic driver of Rett syndrome epigenetics. Front. Genet. 12:620859. doi: 10.3389/fgene.2021.620859
Gulmez Karaca, K., Brito, D. V. C., and Oliveira, A. M. M. (2019). MeCP2: a critical regulator of chromatin in neurodevelopment and adult brain function. Int. J. Mol. Sci. 20:4577. doi: 10.3390/ijms20184577
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hagberg, B. (1985). Rett's syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta. Paediatr. Scand. 74, 405–408. doi: 10.1111/j.1651-2227.1985.tb10993.x
Hamad, M. I. K., Daoud, S., Petrova, P., Rabaya, O., Jbara, A., Al Houqani, S., et al. (2024). Reelin differentially shapes dendrite morphology of medial entorhinal cortical ocean and island cells. Development 151:dev202449. doi: 10.1242/dev.202449
Hamad, M. I. K., Emerald, B. S., Kumar, K. K., Ibrahim, M. F., Ali, B. R., Bataineh, M. F., et al. (2023). Extracellular molecular signals shaping dendrite architecture during brain development. Front. Cell. Dev. Biol. 11:1254589. doi: 10.3389/fcell.2023.1254589
Hamad, M. I. K., Petrova, P., Daoud, S., Rabaya, O., Jbara, A., Melliti, N., et al. (2021). Reelin restricts dendritic growth of interneurons in the neocortex. Development 148:dev199718. doi: 10.1242/dev.199718
Howard, L. R., Javadi, A. H., Yu, Y., Mill, R. D., Morrison, L. C., Knight, R., et al. (2014). The hippocampus and entorhinal cortex encode the path and Euclidean distances to goals during navigation. Curr. Biol. 24, 1331–1340. doi: 10.1016/j.cub.2014.05.001
Karube, F., Kubota, Y., and Kawaguchi, Y. (2004). Axon branching and synaptic bouton phenotypes in GABAergic nonpyramidal cell subtypes. J. Neurosci. 24, 2853–2865. doi: 10.1523/JNEUROSCI.4814-03.2004
Katz, D. M., Berger-Sweeney, J. E., Eubanks, J. H., Justice, M. J., Neul, J. L., Pozzo-Miller, L., et al. (2012). Preclinical research in Rett syndrome: setting the foundation for translational success. Dis. Model. Mech. 5, 733–745. doi: 10.1242/dmm.011007
Kawaguchi, Y., Karube, F., and Kubota, Y. (2006). Dendritic branch typing and spine expression patterns in cortical nonpyramidal cells. Cereb. Cortex 16, 696–711. doi: 10.1093/cercor/bhj015
Khan, U. A., Liu, L., Provenzano, F. A., Berman, D. E., Profaci, C. P., Sloan, R., et al. (2014). Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat. Neurosci. 17, 304–311. doi: 10.1038/nn.3606
Kishi, N., and Macklis, J. D. (2004). MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 27, 306–321. doi: 10.1016/j.mcn.2004.07.006
Kitamura, T., Ogawa, S. K., Roy, D. S., Okuyama, T., Morrissey, M. D., Smith, L. M., et al. (2017). Engrams and circuits crucial for systems consolidation of a memory. Science 356, 73–78. doi: 10.1126/science.aam6808
Kitamura, T., Pignatelli, M., Suh, J., Kohara, K., Yoshiki, A., Abe, K., et al. (2014). Island cells control temporal association memory. Science 343, 896–901. doi: 10.1126/science.1244634
Kitamura, T., Sun, C., Martin, J., Kitch, L. J., Schnitzer, M. J., Tonegawa, S., et al. (2015). Entorhinal cortical ocean cells encode specific contexts and drive context-specific fear memory. Neuron 87, 1317–1331. doi: 10.1016/j.neuron.2015.08.036
Klein, J. A., and Haydar, T. F. (2022). Neurodevelopment in Down syndrome: concordance in humans and models. Front. Cell. Neurosci. 16:941855. doi: 10.3389/fncel.2022.941855
Leifeld, J., Forster, E., Reiss, G., and Hamad, M. I. K. (2022). Considering the role of extracellular matrix molecules, in particular reelin, in granule cell dispersion related to temporal lobe epilepsy. Front. Cell. Dev. Biol. 10:917575. doi: 10.3389/fcell.2022.917575
Leontovich, T. A., Mukhina, J. K., Fedorov, A. A., and Belichenko, P. V. (1999). Morphological study of the entorhinal cortex, hippocampal formation, and basal ganglia in Rett syndrome patients. Neurobiol. Dis. 6, 77–91. doi: 10.1006/nbdi.1998.0234
Moretti, P., Bouwknecht, J. A., Teague, R., Paylor, R., and Zoghbi, H. Y. (2005). Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 14, 205–220. doi: 10.1093/hmg/ddi016
Neul, J. L., Kaufmann, W. E., Glaze, D. G., Christodoulou, J., Clarke, A. J., Bahi-Buisson, N., et al. (2010). Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol. 68, 944–950. doi: 10.1002/ana.22124
Nguyen, M. V., Du, F., Felice, C. A., Shan, X., Nigam, A., Mandel, G., et al. (2012). MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J. Neurosci. 32, 10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012
Osanai, H., Nair, I. R., and Kitamura, T. (2023). Dissecting cell-type-specific pathways in medial entorhinal cortical-hippocampal network for episodic memory. J. Neurochem. 166, 172–188. doi: 10.1111/jnc.15850
Prigge, C. L., and Kay, J. N. (2018). Dendrite morphogenesis from birth to adulthood. Curr. Opin. Neurobiol. 53, 139–145. doi: 10.1016/j.conb.2018.07.007
Ray, S., Naumann, R., Burgalossi, A., Tang, Q., Schmidt, H., Brecht, M., et al. (2014). Grid-layout and theta-modulation of layer 2 pyramidal neurons in medial entorhinal cortex. Science 343, 891–896. doi: 10.1126/science.1243028
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/S0896-6273(02)00768-7
Sholl, D. A. (1953). Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 87, 387–406. doi: 10.1038/171387a0
Smrt, R. D., Eaves-Egenes, J., Barkho, B. Z., Santistevan, N. J., Zhao, C., Aimone, J. B., et al. (2007). Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol. Dis. 27, 77–89. doi: 10.1016/j.nbd.2007.04.005
Valnegri, P., Puram, S. V., and Bonni, A. (2015). Regulation of dendrite morphogenesis by extrinsic cues. Trends Neurosci 38, 439-447. doi: 10.1016/j.tins.2015.05.003
Wang, I. T., Reyes, A. R., and Zhou, Z. (2013). Neuronal morphology in MeCP2 mouse models is intrinsically variable and depends on age, cell type, and Mecp2 mutation. Neurobiol. Dis. 58, 3–12. doi: 10.1016/j.nbd.2013.04.020
Witter, M. P., and Amaral, D. G. (1991). Entorhinal cortex of the monkey: V. Projections to the dentate gyrus, hippocampus, and subicular complex. J. Comp. Neurol. 307, 437–459. doi: 10.1002/cne.903070308
Witter, M. P., Doan, T. P., Jacobsen, B., Nilssen, E. S., and Ohara, S. (2017). Architecture of the entorhinal cortex a review of entorhinal anatomy in rodents with some comparative notes. Front. Syst. Neurosci. 11:46. doi: 10.3389/fnsys.2017.00046
Wu, G. Y., Zou, D. J., Rajan, I., and Cline, H. (1999). Dendritic dynamics in vivo change during neuronal maturation. J. Neurosci. 19, 4472–4483. doi: 10.1523/JNEUROSCI.19-11-04472.1999
Zagrebelsky, M., Tacke, C., and Korte, M. (2020). BDNF signaling during the lifetime of dendritic spines. Cell Tissue Res. 382, 185–199. doi: 10.1007/s00441-020-03226-5
Zhang, Y., Cao, S. X., Sun, P., He, H. Y., Yang, C. H., Chen, X. J., et al. (2016). Loss of MeCP2 in cholinergic neurons causes part of RTT-like phenotypes via alpha7 receptor in hippocampus. Cell Res. 26, 728–742. doi: 10.1038/cr.2016.48
Keywords: Rett syndrome, Mecp2 mouse model, dendritic branching, pyramidal cells, stellate cells, medial entorhinal cortex
Citation: Krishnan M, Mydeen AB, Nakhal MM, Ibrahim MF, Jayaraj RL, Ljubisavljevic MR, Hamad MIK and Ismail FY (2025) Altered dendritic morphology of MEC II pyramidal and stellate cells in Rett syndrome mice. Front. Neuroanat. 19:1580435. doi: 10.3389/fnana.2025.1580435
Received: 20 February 2025; Accepted: 23 May 2025;
Published: 24 June 2025.
Edited by:
Alberto Munoz, Complutense University of Madrid, SpainReviewed by:
Giulia Quattrocolo, Norwegian University of Science and Technology, NorwayGarret Anderson, University of California, Riverside, United States
Copyright © 2025 Krishnan, Mydeen, Nakhal, Ibrahim, Jayaraj, Ljubisavljevic, Hamad and Ismail. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad I. K. Hamad, bS5oYW1hZEB1YWV1LmFjLmFl; Fatima Y. Ismail, ZmF0aW1hLmlzbWFpbEB1YWV1LmFjLmFl
†Present address: Richard L. Jayaraj, Institute of Sciences in Emergency Medicine, Department of Emergency Medicine, Guangdong Provincial People's Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, Guangzhou, China