 Selin Neseliler1,2
Selin Neseliler1,2 Darshana Narayanan1
Darshana Narayanan1 Yaihara Fortis-Santiago1
Yaihara Fortis-Santiago1 Donald B. Katz1*† and Susan J. Birren2,3*†
Donald B. Katz1*† and Susan J. Birren2,3*†- 1 Department of Psychology, Brandeis University, Waltham, MA, USA
- 2 Department of Biology, Brandeis University, Waltham, MA, USA
- 3 National Center for Behavioral Genomics, Brandeis University, Waltham, MA, USA
Acute inhibition of acetylcholine (ACh) has been shown to impair many forms of simple learning, and notably conditioned taste aversion (CTA). The most adhered-to theory that has emerged as a result of this work – that ACh increases a taste’s perceived novelty, and thereby its associability – would be further strengthened by evidence showing that enhanced cholinergic function improves learning above normal levels. Experimental testing of this corollary hypothesis has been limited, however, by side-effects of pharmacological ACh agonism and by the absence of a model that achieves long-term increases in cholinergic signaling. Here, we present this further test of the ACh hypothesis, making use of mice lacking the p75 pan-neurotrophin receptor gene, which show a resultant over-abundance of cholinergic neurons in sub-regions of the basal forebrain (BF). We first demonstrate that the p75−/− abnormality directly affects portions of the CTA circuit, locating mouse gustatory cortex (GC) using a functional assay and then using immunohistochemisty to demonstrate cholinergic hyper-innervation of GC in the mutant mice – hyper-innervation that is unaccompanied by changes in cell numbers or compensatory changes in muscarinic receptor densities. We then demonstrate that both p75−/− and wild-type (WT) mice learn robust CTAs, which extinguish more slowly in the mutants. Further testing to distinguish effects on learning from alterations in memory retention demonstrate that p75−/− mice do in fact learn stronger CTAs than WT mice. These data provide novel evidence for the hypothesis linking ACh and taste learning.
Introduction
The cholinergic system is implicated in the performance of many survival behaviors, notably including feeding (Bermudez-Rattoni, 2004). Perturbations of the basal forebrain (BF), the primary source of cortical and limbic system acetylcholine (ACh, Hecker and Mesulam, 1994; Semba, 2004), impair both expression of naïve preferences for one taste over another (Pratt et al., 2007), and the learning of new preferences in paradigms such as conditioned taste aversion (CTA, whereby animals learn to dislike/avoid tastes associated with gastric distress, Gutierrez et al., 1999b; Gonzalez et al., 2000; Semba, 2000; Bermudez-Rattoni, 2004). Similarly, pharmacological antagonism of muscarinic cholinergic synapses within parts of the taste system that receive input from sub-regions of BF, including gustatory (insular) cortex (GC) and basolateral amygdala (BLA), also hinder CTA learning (Naor and Dudai, 1996; GC, Berman et al., 2000; Gutierrez et al., 2003).
This work, and microdialysis studies suggesting that presentation of a new taste causes release of ACh in GC (Miranda et al., 2000), form the basis of a powerful theory implicating ACh as the signal for taste novelty (Miranda et al., 2000; Ranganath and Rainer, 2003; Jeewajee et al., 2008; Nunez-Jaramillo et al., 2008), and thus as a vital part of strong CTA (Bermudez-Rattoni, 2004). These studies would be much strengthened, however, by the complimentary data, showing that enhancement of cholinergic activity in the taste system improves taste learning. At least one study has in fact suggested that cholinergic agonism allows learning to otherwise ineffective stimuli (Clark and Bernstein, 2009), but related data concerning normal learning are difficult to collect and interpret, both because the reduction of consumption typically used to measure CTA suffers from a floor effect (making enhanced learning difficult to detect) and also because cholinergic agonism can result in seizures and profoundly disrupt pathways that may be unrelated to CTA (Olney et al., 1983; Naor and Dudai, 1996). What would be highly useful in this regard is a model organism with chronic, non-traumatic elevations of cholinergic function; while developmental cholinergic manipulation would, like all other methods of cholinergic manipulation, have the potential to cause secondary effects in other systems, it would be simple in this model organism to test the corollary hypothesis of the ACh theory, namely that increases in cholinergic function improve taste learning.
Perturbed function via non-traumatic developmental processes (i.e., in which neurons are not suddenly removed from a system accustomed to their presence) can be achieved in mice by genetic manipulation of gene expression. One such mouse model provides a particularly useful phenotype for studies of cholinergic function: the p75 knockout mouse contains a targeted deletion of the p75 low affinity, pan-neurotrophin receptor gene, which, in the normal adult brain, is selectively expressed in BF cholinergic neurons (Hartikka and Hefti, 1988). Although the loss of this receptor has a range of effects, the most notable of these is increased numbers of cholinergic neurons (and decreased numbers of GABAergic neurons) in BF (Van der Zee et al., 1996; Naumann et al., 2002; Lin et al., 2007). If this increase in cholinergic neuron number in fact results in a taste system that is “hyper-cholinergic,” then the cholinergic theory of taste novelty would predict that p75−/− mice should condition more strongly than normal mice.
Here, we performed this test, first by functionally defining the GC in mice and demonstrating that the p75−/− phenotype results in reliable cholinergic hyper-innervation of this region, and then by showing significantly supra-normal learning in the mutants. Our examinations of these mice therefore provide novel evidence for the hypothesis that ACh controls taste associability.
Materials and Methods
Subjects
Adult male and female wild-type (WT; 57BL/6J) and strain-matched p75−/− mice (Lee et al., 1992) from the Jackson Laboratory (Bar Harbor, ME, USA) served as subjects in this study. All animals were maintained on a 12 h light/12 h dark cycle. Animals were housed individually with free access to standard food pellet and maintained on a 23.5 h water deprivation schedule for the duration of training and experimentation. All the behavioral experiments were carried out between 12:00 and 4:00 pm.
Cortical Cannulation
Adult mice were initially anesthetized via intraperitoneal (ip) injections of a ketamine/xylazine cocktail (ketamine, 100 mg/kg; xylazine, 10 mg/kg) and placed in a stereotaxic frame. General anesthesia throughout the surgery was maintained using isoflurane inhalant (0.5% isoflurane at 0.5 l/min oxygen). A scalp midline incision was made, after which the scalp was retracted and the skull leveled. Two small holes were drilled in the skull so that guide cannulae (23-gage, 10 mm in length) could be lowered into putative GC under stereotaxic guidance (AP: +1.48 mm and ML ± 3.10 mm relative to bregma; DV −1.8 mm from the surface of the brain). Cannulae were stabilized using Vetbone and dental acrylic. Stainless steel stylets (30-gage, 10 mm in length) were inserted into the guide cannulae to ensure patency.
Muscimol Dose Response
Mice were allowed to recover for a minimum of 7 days following surgery. After recovery, mice were maintained on a 30 min/day water restriction protocol for 6 days in the home cage, to ensure stable intake of water. The last three of these days, water intake (g) from the cage lick-spout was measured, and the average water intake for each mouse was calculated.
On test day, mice received infusions of one of five different doses of muscimol (MP Biomedicals, LLC, Ohio 1, 0.75, 0.5, 0.25, and 0 μg/μl, diluted in 0.3 μl saline). Fifteen minutes after the infusion, mice were given 30 min access to water through a lick-spout in their home cages. The effect of muscimol on the water intake of each mouse was then quantified [infusion day (g)/average water intake (g) × 100]. Motor behavior and water consumption were compared with mice that received saline infusion.
Our results show 0.5 μg/μl muscimol to be the smallest concentration that resulted in a significant change in the water intake of the mice [t(7) = 2.644, p = 0.033, two-tailed]. In accordance with these results, we used 0.5 μg/μl muscimol in 0.3 μl saline for identifying GC in the CTA protocol.
Drug Delivery
Mice were secured in experimenter’s hand and infusion cannulae, connected via polyethylene tubes to 10 μl Hamilton syringes in an infusion pump (Harvard Apparatus, Massachusetts, MA, USA), were inserted to ~0.2 mm beyond the bottom of the previously implanted guide cannulae. Muscimol (dosing determined as described above) was bilaterally infused into the GC at a rate of 0.15 μl/min for 2 min (for a total of 0.3 μl). Infusion cannulae were left in place for an additional minute to allow for diffusion from the tip of the injecture (Stone et al., 2005), after which they were removed slowly, to ensure that negative pressure did not suck infusate up into the guide cannula.
Identifying Gustatory Cortex
Adult mice were adapted to water restriction as described above. On the training day, mice received intra-cranial infusions of either 0.5 μg/μl muscimol in 0.3 μl saline or saline alone, and were returned to their home cages. Fifteen minutes later, mice were given 30 min of ad lib access to a novel palatable solution (100 mM NaCl). Immediately afterward, they were given intraperitoneal injections of LiCl (0.15 M, 2% body weight), which induced gastric malaise. All animals received 15 min access to water 2 h after the termination of the training session unless indicated otherwise; by this time, mice were observed to be drinking normally, which led us to conclude that there was no need to interpolate a rest day between training and test: thus, 24 h after training, mice were once again given 30 min of ad lib access to 100 mM NaCl in a testing session. Because basal consumption was highly variable, and because p75−/− mice as a group drank slightly but significantly more than WT mice (see Table 1), the acquisition of CTA was quantified in terms of a normalized comparison between NaCl solution intake in the training and testing sessions [(NaCl intake on the testing day/NaCl intake on the infusion day) × 100]. Subsequent testing (see below) demonstrated that the observed differences in basal consumption had by themselves little impact on learning.
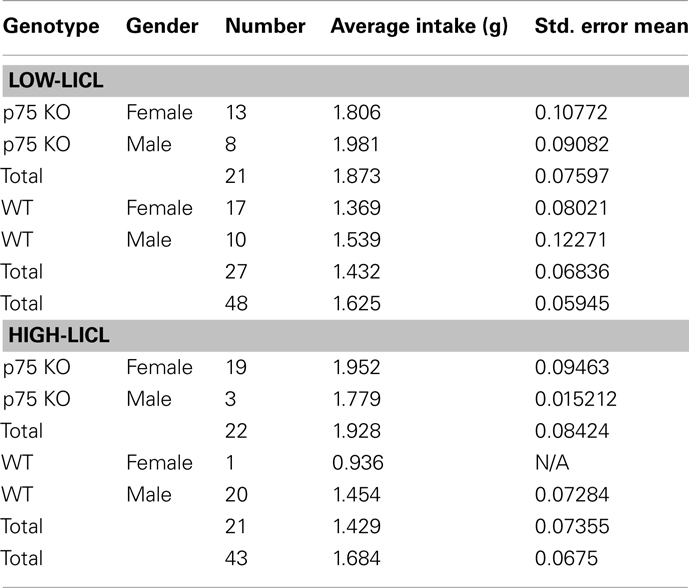
Table 1. Breakdown of groups used in behavioral tests.
Between-Strain Comparison of Conditioned Taste Aversion
Mature adult WT and p75−/− mice (43 mice for high-LiCl and 48 mice for low-LiCl experiments) were adapted to the water deprivation protocol for 6 days and then given the CTA protocol as described above; Table 1 provides more details on the groups of adult mice (genders and strains) used in these experiments. No muscimol was administered in these experiments, and 10 mM saccharin was used instead of NaCl. For the “low-LiCl” experiments, the concentration of intraperitoneally administered injection of LiCl was reduced to 1% of body weight. To more completely characterize the induced aversions, testing sessions were repeated for 5 more days (six testing sessions in all); across this period, the induced aversion gradually faded, allowing evaluation of extinction of learning.
Histology
Mice were anesthetized with isoflurane followed by an injection of a ketamine/xylazine/acepromazine cocktail. After deep anesthesia was achieved, the mice were perfused with ice-cold saline followed by ice-cold 80–100 ml of 4% paraformaldehyde (PFA), 0.1 M phosphate buffer, pH 7.4. Brains were rapidly removed, post-fixed overnight at 4°C in PFA alone, and maintained in 30% sucrose at 4°C until sectioning for cannula placement, ChAT staining, or NeuN staining.
Identification of gustatory cortex
A subset of mice implanted with cannulae received fluorescent muscimol (0.5 μg/μl, BODIPY, TMR-X conjugate, Invitrogen, CA, USA delivered through the method described above) prior to perfusion, to visualize the diffusion of muscimol. From these animals, 100 μm coronal slices were cut starting either at the corpus callosum intersection or at the first appearance of cannulae tracks (whichever was more anterior). PBS-soaked sections were imaged immediately after slicing, through the 4× objective on an Olympus IX-81 inverted fluorescence microscope (Allen et al., 2008). The image of the whole coronal brain section was captured using an Orca-ER digital CCD camera (Hamamatsu, Japan) and Volocity software (PerkinElmer, Waltham, MA, USA). Images were then overlaid on figures from a mouse brain atlas (Paxinos and Franklin, 2001) using Adobe Illustrator CS3. This allowed for identification of mouse GC, based on the location of effective muscimol infusion sites and of effective spread of the infused muscimol.
The diffusion of effective infusions of fluorescent muscimol (i.e., those that blocked CTA) was estimated (and plotted on a lateral view of the mouse brain) to be around the diameter of the cannulae (635 μm). Diffusion was roughly elliptical, due to the presence of the cannulae themselves, averaging 635 μm in the horizontal axis and 228 μm dorsal–ventral axis.
ChAT and NeuN staining
Twelve consecutive coronal slices (40 μm thick) through GC, beginning where the corpus callosum first intersects anteriorly (corresponding to +1.10 relative to bregma) were cut on a vibratome and collected in PBS. Every third slice was used for the following immunostaining procedure. Fifteen consecutive coronal slices (40 μm thick) through gustatory thalamus, beginning where the corpus callosum first intersects caudally (corresponding to −2.46 relative to bregma) were cut on a vibratome and collected in PBS. Starting from the fifth slice (corresponding to −2.30 relative to bregma), every third slice was used for the following immunostaining procedure.
Sections were gently agitated for 30 min at room temperature in a preblock solution containing 0.1% NP-40 with 10% donkey serum in PBS, and incubated overnight in primary antibody solution diluted in the preblock solution. The primary antibodies used were mouse anti-neuron-specific protein NeuN (1:1000; Chemicon, Temecula) for the identification and quantification of the neuronal somas and goat anti-choline acetyltransferase (ChAT, 1:1000; Chemicon) for detecting cholinergic cell bodies and fibers. The sections were also stained with a rabbit anti-human p75 neurotrophin receptor (p75, 1:1000; Promega, Madison, WI, USA) to confirm that the p75−/− mice did not express this receptor.
After being washed three times in PBS (20 min each), the sections were incubated in secondary antibodies diluted in preblock solution (Rhodamine-, Cy5-, and FITC-conjugated secondary antibodies respectively; Jackson ImmunoResearch, West Grove, PA, USA) for 3 h. After another three washes, the sections were mounted on Superfrost Plus (Fisher Scientific) slides in n-propyl gallate. The sections were kept at 4°C in the dark until imaged.
Whole brain slices were imaged using the 4× objective on an OlympusIX-81 inverted fluorescence microscope fitted with fluorescein, rhodamine, and Cy5 filters. Images for each whole coronal brain section were captured using an Orca-ER CCD digital camera (Hamamatsu, Japan) and Volocity software (Improvision, Lexington, KY, USA). Slice locations were matched to corresponding images from the mouse atlas (Paxinos and Franklin, 2001).
Uncompressed 16-bit gray-scale images were made of NeuN- and ChAT-stained slices at 2 μm intervals using an automated focus drive. Imaging was done at 20× magnification resulting in images of 436.20 μm × 332.35 μm regions of GC, and 872.4 μm × 332.35 μm regions of gustatory thalamus. Images were exported to ImageJ (NIH, USA) for analysis. From each series of 20 images in a stack, the six images were used to z-project the stack into a single image. Using the Z-projection through the maximum intensity values for each image stack ensured that all the fibers and cell somas were present in one image, allowing analysis of the total number of cells, and of fiber density and length, averaged across slices and across mice.
ChAT density analysis
Images were first converted to 8-bit in ImageJ and the Feature J software plug-in was used to detect axons using Hessian-based matrices (Grider et al., 2006). The resulting eigen-image was converted into a binary image by thresholding using the Isodata algorithm implemented in Image J. The calculated threshold was adjusted as needed (in increments of not more than 10 pixels) to optimally detect all axons with high background signals. The fraction of the thresholded image covered with the fibers was calculated using the Analyze Particles tool. The thresholded images were then skeletonized (i.e., the width of each fiber was reduced to a single pixel) and axonal length was re-quantified in isolation from potential artifacts of staining intensity.
NeuN staining
For automated counting of neuron number, the watershed algorithm was implemented on thresholded images and particle analysis were carried out using the analyze particles tool. Only particles bigger than 150 square pixels were included in the automated count. Manual counting analysis of cells was performed on a subset of the data, using the cell counter plug-in on the original image. The cell counts obtained manually and with the automated method from 12 different images were found to be closely correlated. Therefore, the automated counting was deemed satisfactory, and carried out on the other images.
Quantitative Real-Time PCR
RNA was prepared from GC of WT and p75−/− mice using the RNeasy mini kit (Qiagen, Valencia, CA, USA). RNA was reverse transcribed into cDNA using MMLV-reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and random hexamer primers [46]. Real-time PCR was performed in triplicate for each sample using a Rotor-Gene 3000 (Qiagen, Valencia, CA, USA). Primer sets were designed using BLAST (NIH, USA). Primer sets used in this study were:
GAPDH f 5′-AACT TTT GGC ATT GTG GAA GG-3′, GAPDH-r 5′-GCA TGC AGG GAT GAT CT-3′, M1f 5′-CAT GGA GTC CCT CAC ATC CT-3′, M1-r 5′-TGT ATT TGGT GGA GCT TTT GG-3′, M2-f 5′-TAC CCA GTT AAG CGG ACC AC-3′, M2-r 5′-CCC GTC TTC CAC AGT CCT TA-3′, M4-f 5′-ATC GAG ATC GTA CCT GCC AC-3′, M4-r 5′-AAT GGC AAA GAT TGT CCG AG-3′.
The two ΔΔCT method was used for real-time PCR analysis (Livak and Schmittgen, 2001). For each primer set, PCR reactions were run in triplicate and normalized to the average of triplicate reactions run with glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Statistics
Data were analyzed using SPSS 15.0 (SPSS Inc., Chicago, IL, USA). Mean values, standard errors, independent t-test, ANOVAs, and Pearson Correlations were calculated to compare the WT mice with the p75−/− mice.
Results
Identification of Primary Gustatory Cortex in the Mouse
We first investigated whether the p75−/− mouse was an appropriate model in which to explore the impact of long-term, developmentally based cholinergic hyper-innervation on taste learning by examining whether there was increased innervation of CTA-relevant regions. Researchers have reported an increased number of cholinergic neurons in the BFs of p75−/− mice (Van der Zee et al., 1996; Naumann et al., 2002; Lin et al., 2007). This expansion of the number of cholinergic neurons appears to lead to increased cholinergic innervation in some, but not all sub-regions of forebrain structures such as the hippocampus (Yeo et al., 1997), but the regions projecting to insular cortex (where GC resides) have not been examined, nor has it been conclusively shown that increases in BF neurons result in cortical hyper-innervation. We therefore first set out to examine GC in the p75−/− mouse.
While gustatory sub-regions within insular cortex have been described using both structural and functional techniques in the rat (Yamamoto, 2006), the equivalent areas in the mouse have not been conclusively identified (Tokita et al., 2009). To isolate the appropriate region, we made use of the fact that GC is known to be vital for CTA learning (Yamamoto et al., 1995; Naor and Dudai, 1996; Yasoshima and Yamamoto, 1997; Berman and Dudai, 2001; Grossman et al., 2008; Bertrand et al., 2009), while the regions surrounding GC are not known to have any involvement in CTA. We were able to use a functional assay to pinpoint the taste-relevant part of insular cortex, identifying it as that part in which CTA was inhibited by localized infusions of the GABA-a agonist muscimol.
Intra-cranial cannulae were inserted bilaterally into presumptive GC (identified on the basis of anatomical landmarks) of WT mice in stereotaxic survival surgeries (see Materials and Methods; Figure 1). After at least 7 days of recovery, the mice were introduced to the behavioral protocol, wherein we tested their ability to develop conditioned aversions to taste stimuli when spiking of neurons in putative GC was silenced by infusions of muscimol (Krupa et al., 1998). First, a pilot group was used to ascertain a safe dose of muscimol – one that only minimally interfered with normal feeding behavior. Mice received bilateral infusions of 0, 0.25, 0.5, 0.75, or 1.0 μg/μl muscimol in 0.3 μl saline vehicle 15 min before being given access to a water-filled lick-spout for 30 min. The highest of these concentrations strongly inhibited water consumption, consistent with non-specific effects of the blocker on gustatory behaviors (data not shown). Lower concentrations had minimal effects on water consumption, however, and so we carried out CTA experiments using 0.50 μg/μl muscimol infusions. This, the lowest concentration that had any effect on water consumption in our pilot experiments [t(7) = 2.644, p = 0.033], has been shown to effectively block action potential generation in vivo in the region surrounding the infusion cannula (Krupa et al., 1998).
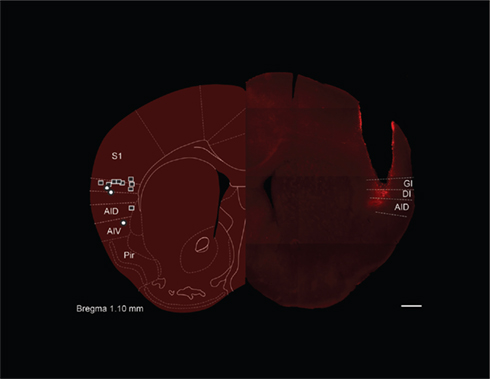
Figure 1. Gustatory cortex in the mouse. The left hemisphere of this figure is a schematic diagram of the likely coronal plane of mouse (reprinted with permission from Paxinos and Franklin, 2001) gustatory cortex (GC), selected for its homology to the known location of rat GC (Katz et al., 2001). The super-imposed squares and circles show the location of cannula tips for muscimol and control mice, respectively, in the GC localization experiments – most tips are found in granular (GI) or dysgranular (DI) insular cortex; also noted are dorsal and ventral agranular insular (AID and AIV) as well as piriform cortex (Pir). The right hemisphere shows a photomicrograph from one mouse subject (with the approximate demarcations of granular, dysgranular, and agranular cortex noted). The cannula track is readily visible, as is the localized spread of fluorescent muscimol infused through that cannula just before perfusion. Scale bar = 500 μm.
We tested the acquisition of CTA in a naïve group of cannulated mice. These mice received 0.3 μl of 0.5 μg/μl muscimol while control mice received infusions of vehicle. Mice then consumed 100 mM NaCl out of a lick-spout ad lib for 30 min, and their consumption was measured in grams. The drinking session was immediately followed by an ip injection of the emetic LiCl, after which mice were returned to their home cages for the night. The following day the mice were offered 30 min of access to the same taste. Figure 2A shows the result of this experiment: control mice developed substantial aversions to the emesis-paired taste, measured as a decrease in consumption between the training and testing sessions. Muscimol infusions completely blocked this decrement in consumption [t(12) = 2.24, p = 0.045, two-tailed t-test]; in fact, consumption of NaCl increased between training and testing for this group – a result that is consistent with what is known about cessation of neophobia with experience (Domjan and Gillan, 1976). Given the anatomical (Fortis-Santiago et al., 2010) and functional evidence (Wang et al., 2006), as well as our direct imaging evidence (using fluorescent muscimol, see Figure 1), all of which suggests the spread of such muscimol infusions is highly circumscribed, we conclude that our implantation coordinates correctly localize mouse GC. This conclusion received further support from control experiments demonstrating that infusion of muscimol in mice in which the cannulae were placed outside of IC had no effect on acquisition of CTA (p > 0.1). Figure 2B shows a parasaggital view of the most effective muscimol infusion sites, along with estimations of muscimol spread based on fluorescence measurements (see Materials and Methods); our localization of mouse GC is essentially identical to that region identified on the basis of tracer injections into taste thalamus (Chen et al., 2011).
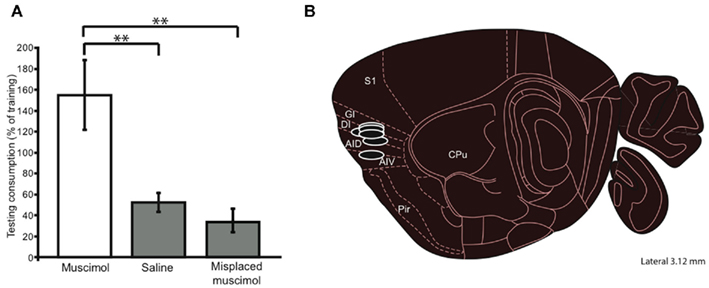
Figure 2. Functional test of mouse GC localization. (A) A muscimol concentration that had relatively little impact on ad lib consumption (i.e., 0.50 μg/μl), infused into putative GC just before a CTA training session (a single pairing of orally administered NaCl and ip injected LiCl), inhibited taste learning – these mice did not reduce their NaCl consumption in the testing session (y-axis). Mice receiving control (saline) infusions into putative GC learned normally, consuming much less NaCl after training, as did controls receiving muscimol infusions into non-GC sites. Thus we can conclude that the infusion cannulae have correctly targeted mouse GC. (B) In a parasagittal view, the locations, and approximate spreads, of muscimol infusions that were most effective at blocking CTA. Error bars, here and in every figure, represent the standard error of the mean, ** = p < 0.01; see text for further details. S1, somatosensory cortex; GI, DI, AID, AIV, gustatory insular cortex (granular, dysgranular, agranular dorsal and ventral); Pir, piriform cortex; CPU, caudate putamen.
Cholinergic Hyper-Innervation of IC in the p75−/− Mouse
We next examined cholinergic innervation of identified GC in p75−/− and WT mice. Cholinergic fiber analysis was carried out in 40 μm coronal slices made through GC. Choline acetyltransferase (ChAT) density was calculated as the fraction of the image covered with fibers. Data were averaged across slices and across mice. This analysis of ChAT density revealed a significantly heavier cholinergic innervation of GC in p75−/− mice compared to WT controls [t(9) = 3.988, p = 0.003; two-tailed; Figure 3A].
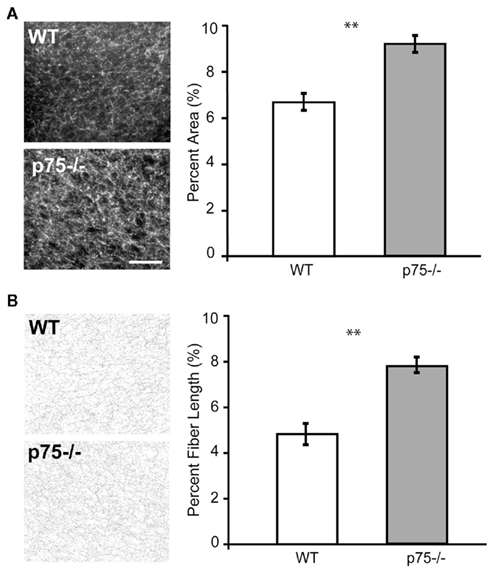
Figure 3. p75−/− mice showed cholinergic hyper-innervation in GC compared to wild-type mice. (A) The two photomicrographs show ChAT-stained sections through GC. More fibers are visible in p75−/− mice than in wild-type. The group data are summarized in the panel to the right, which shows that in the p75−/− mouse, a higher percentage of the space in the images (y-axis) was taken up by ChAT-stained fibers than in the wild-type (WT) mouse. (B) The same analysis done on skeletonized images (see Materials and Methods for details), which allows us to rule out confounding explanations having to do with the possibility of brighter staining. The group data demonstrates that, when stained fibers are reduced to equi-luminant, equi-thick lines, these cholinergic fibers are longer in p75−/− mice than in wild-type mice. ** = p < 0.01, scale bar = 100 μm.
A concern with such analyzes is that they may fail to distinguish between true increases in number/length of fibers and increased intensity of staining of individual fibers. We therefore re-analyzed the slices to evaluate the length of ChAT-positive fibers on skeletonized images using ImageJ software (NIH, USA). Such pre-processing eliminates information relating to intensity. These fiber-length measurements confirmed the results of the previous analysis, showing a significant increase in cholinergic innervation in the GC of p75−/− mice compared to WT controls [t(9) = 3.648, p = 0.005; two-tailed; Figure 3B].
Loss of p75 expression did not have gross secondary effects on GC in these mice beyond cholinergic hyper-innervation itself. Despite the increase in both BF cholinergic neuron number and cholinergic innervation of the IC, we found no change in the total number of cortical neurons in the GC of adult p75−/− mice compared to WT control animals. Coronal slices used for the fiber-length analysis were co-labeled with the NeuN antibody to identify all neuronal cell bodies in the slice. The number of neurons per section was counted using the watershed algorithm and the analyze particles tools in ImageJ (Figure 4A). There was no difference in total cortical neuron number between regionally matched slices taken from p75−/− and WT animals [t(8) < 1].
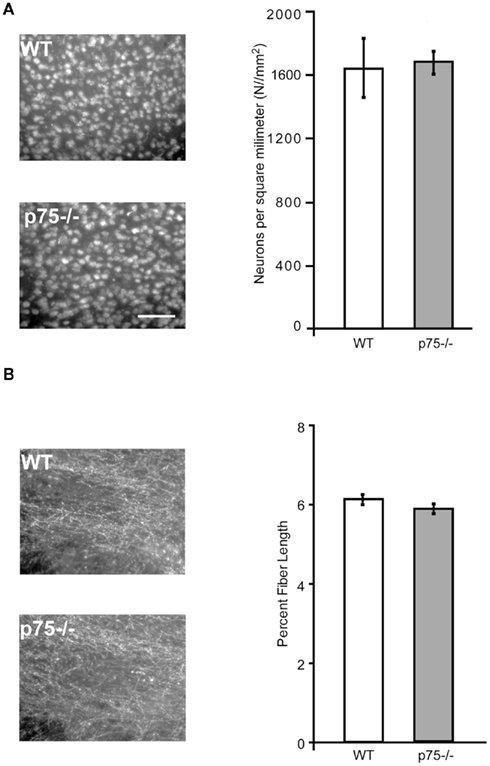
Figure 4. p75−/− and wild-type mice are comparable in cortical neuron number and cholinergic innervation of gustatory thalamus. (A) The photomicrographs show NeuN staining, which labels all neurons in wild-type and p75−/− GC slices. The group data at right demonstrates that the two strains did not differ in the number of neurons in GC, indicating that cholinergic hyper-innervation did not change the number of cortical neurons. (B) Like Figure 3B, this figure shows ChAT staining of slices harvested from p75−/− and wild-type mice – this time from gustatory thalamus, which receives cholinergic innervation from brainstem rather than basal forebrain. The photomicrographs (left) and group data (right) reveal no major p75/wild-type differences in cholinergic innervation of this important part of the mouse gustatory system. Scale bar = 100 μm.
We examined the specificity of p75-dependent cholinergic increases in GC by comparing these data to measurements of cholinergic fiber length in the gustatory thalamus. The parvicellular division of the ventroposterior medial thalamic nucleus conveys gustatory information and contains extensive cholinergic innervation. However, this innervation is predominantly derived from the brain stem (notably the pedunculopontine tegmental nuclei, Parent and Descarries, 2008) rather than from the BF. We therefore predicted that thalamic innervation would be normal in p75−/− mice. In line with this expectation, and in contrast to the cortical innervation pattern, we found no difference in ChAT staining in the gustatory thalamic nuclei of p75−/− mice [t(2) = 1.39, p = 0.30, Figure 4B].
Furthermore, we found only limited evidence that the increased cholinergic innervation of GC causes compensatory changes in muscarinic receptors. We examined mRNA expression of the major CNS muscarinic receptors – M1, M2, and M4 – in RNA isolated from the GC of WT and p75−/− mice using RT-PCR. We found no change in mRNA levels of the M1 and M4 receptors (Figure 5), but we did observe a significant decrease in signal for M2 receptor mRNA: M1, t(11) = 0.11, p = 0.915; M2 t(11) = 2.598, p = 0.025, M4 t(11) = 0.074, p = 0.404.
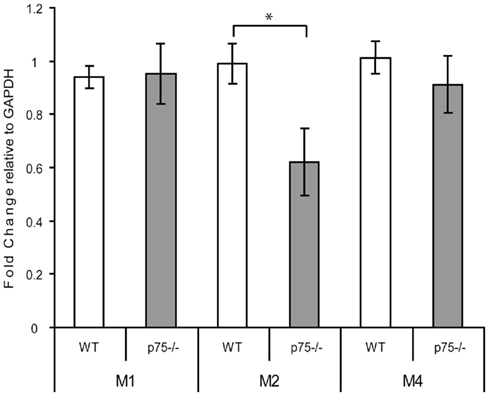
Figure 5. p75−/− and wild-type GC differed in expression of the M2, but not M1 or M4, cholinergic receptor mRNAs. RNA was isolated from GC of wild-type and p75−/− mice and the levels of M1, M2, and M4 receptor mRNA were measured relative to expression of GAPDH using real-time PCR. Levels of mRNA are expressed relative to GAPDH mRNA expression levels, showing no change in M1 and M4 levels and a decrease in M2 mRNA. * = p < 0.05.
The Impact of Cholinergic Hyper-Innervation on Taste Learning
Based on the above data, and the literature linking reductions of cholinergic function to learning impairments, we hypothesized that p75−/− mice should form stronger memories than WT mice on a standard taste learning task. Our evidence reveals that this is in fact the case.
To test this prediction, we returned to the CTA paradigm. Groups of p75−/− and WT mice were adapted to the testing chamber, given a single training session involving a pairing of 10 mM saccharin with ip injections of 0.15 M LiCl (2% body weight), and tested for their post-training consumption of the conditioned taste. Consumption was followed across several post-training days, so that the speed with which behavior returned to baseline (i.e., extinction of learning) could also be assessed. Figure 6A shows the result of this experiment, with consumption normalized to pre-learning saccharine consumption. Strong initial learning (significant reduction of consumption in testing session compared to baseline), appeared to extinguish more quickly in WT mice than p75 mutants – by extinction session 5, WT mice drank 119% of their naïve (training day) consumption, reflecting the fact that consumption in that initial session was affected by mild neophobia for the novel taste (Domjan and Gillan, 1976), whereas p75−/− mice had re-attained 93% of naïve consumption. A two-way mixed ANOVA of these data revealed, as expected, a significant effect of day [F (5,195) = 44.16, p < 0.001] as well as a day × mouse interaction [F (5,195) = 2.83, p < 0.02] consistent with the appearance of Figure 6A, in which p75−/− and WT mice drank similarly little saccharin in the testing session, and WT mice drank more than p75−/− mice in each of the extinction sessions. Pairwise post hoc tests were somewhat equivocal and difficult to interpret, with significant strain differences appearing on extinction days 2 and 5.
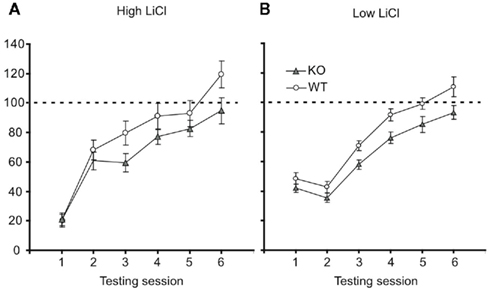
Figure 6. p75−/− mice learn stronger CTAs than wild-type mice. (A) With the standard dose of LiCl (0.15 M, 2% of body weight), both wild-type (open ovals), and p75−/− mice (closed triangles) learn strong CTAs, reducing their consumption of saccharin in the first testing session to ~20% of training-session consumption (y-axis). Across further testing sessions (x-axis) both groups re-learned to consume saccharin (i.e., the CTA underwent extinction), but this occurred faster for wild-type mice (see text for statistics). (B) When the LiCl dose was reduced to 1% of body weight, the learning was accordingly milder. By lifting consumption away from the floor, it became possible to observe a difference in initial learning: p75 mice learned stronger CTAs (i.e., consumed less saccharin) than wild-type mice. This difference was maintained through several extinction trials. See text for statistical details.
It is tempting to conclude that p75 mice learned stronger aversions than WT mice, on the basis of the fact that their learning extinguished more slowly. The failure to observe differences in initial learning (i.e., in testing session 1) could well be explained as a function of a floor effect: CTA is powerful and evolutionarily important, and “normal-strength” learning consists of a near elimination of consumption; increases in that strength should therefore be difficult to detect. In fact, when the ANOVA was repeated without the initial testing session, the session × strain interaction vanished (F < 1), demonstrating that the interaction was wholly dependent on the initial testing session, and suggesting that the rate of extinction did not actually differ as a function of cholinergic innervation. The alternative possibility must also be considered, however – the possibility that the p75-deficient and WT mice developed equivalent CTAs, but that p75−/− mice show an “extinction deficit.” Acquisition and extinction of learning are known to involve distinct network and sub-cellular mechanisms (Berman and Dudai, 2001), and while extinction has not been specifically linked to cholinergic function, it is possible that cholinergic hyper-innervation of GC targets precisely this process.
To distinguish between these possibilities, we performed a second learning experiment on an additional sample of p75−/− and WT mice. This experiment was identical to the first, but the volume of injected LiCl was cut in half – a change that reduces the intensity of the induced emesis, and thus reduces the strength of the learned aversion (Sakai and Yamamoto, 1997). It was assumed that this reduction would “lift” consumption off of the “floor,” allowing any inter-strain differences in initial learning to be more easily observed.
Figure 6B shows the result of this experiment: at the smaller LiCl dose, consumption is now far from the “floor” in all sessions for both strains, and p75 mice can now be seen to learn stronger aversions in the first testing session and all sessions thereafter. While modest, this difference confirms that the effect observed in Figure 6A does not reflect a selective difference in extinction learning. A two-way mixed effect ANOVA was performed on the testing session data: a significant main effect of session [F (5,175) = 68.78, p < 0.001] revealed that extinction occurred for both types of mice; a significant main effect of strain [F (1,35) = 5.32, p < 0.03], meanwhile, reveals that p75−/− mice did indeed learn stronger aversions than WT mice, consuming less fluid across testing sessions. While we observed substantial variability in basal consumption in the p75−/− group, subsequent analysis revealed that these large differences were positively (r = 0.21) but insignificantly (tz < 1) related to consumption in the testing session. This suggests that mice that consumed more saccharin during training did not learn stronger CTAs (and thus removes basal consumption differences as a possible confound to the results).
The utter lack of a strain × session interaction, both in this ANOVA (F < 1) and that comparing extinction days 1–5 in the high-LiCl experiment, reveals that the difference between p75−/− and WT mice did not vary significantly between session. While the difference between the groups was small, it was consistent from the first test until the last test in any session that was not confounded with a floor effect, which is to say that the extinction differences followed directly from differences in initial learning (statistical texts make it clear that it is inappropriate to probe for specific session-specific strain differences when a significant interaction is not present, see, e.g., Howell, 2007). Thus, we conclude that p75−/− mice learned stronger taste aversions that extinguished at normal rates.
Discussion
Acute and long-term disruptions of the cholinergic system, including lesions of the BF and infusion of inhibitors into targets of BF including GC, are known to impair a rodent’s ability to learn a CTA. Here, we used a genetic approach to confirm a key but largely untested implication of these findings: we show that cholinergic hyper-innervation of the taste system leads to stronger than normal taste aversion learning. ChAT immunostaining confirmed that GC of p75−/− mice, identified using a functional assay, contains a greater number of cholinergic fibers than that of WT mice. Subsequent analyses ruled out the possibility that the increase was due to increased staining intensity rather than fiber length, and revealed that p75 mutants were similar to WT mice with regard to numbers of neurons in GC, post-synaptic M1 and M4 ACh receptor expression in GC (but not M2, see below), and cholinergic innervation in gustatory regions that receive their cholinergic input via the brainstem rather than BF (i.e., the gustatory thalamus). These mice thus show specific cholinergic hyper-innervation of the taste system, and thus would be predicted to show stronger than WT learning, a prediction that was borne out in our behavioral testing.
The assays that we used to determine cholinergic hyper-innervation of the taste system centered on GC, providing confirmation that increases in BF cholinergic neuron numbers (previously described for this mutant, Lin et al., 2007) resulted in an increase in the number of cholinergic fibers in the taste system. This does not imply that the actions of ACh, or the cholinergic changes observed in p75−/− mice, are restricted to GC. Cholinergic activity at the brain stem, amygdala, and cortical levels of the taste system has been implicated in CTA (Bermudez-Rattoni, 2004), and it is possible that all of these regions are hyper-innervated in the p75 behavioral phenotype. Most notably, the BLA – and amygdala–cortical connectivity – are known to be deeply involved in CTA (Bermudez-Rattoni, 2004; Grossman et al., 2008), and cholinergic hyper-innervation may well affect this circuitry although given the extremely high density of cholinergic innervation in the wild-type BLA it is hard to know if increases can be detected (see Muller et al., 2011). Cholinergic hyper-innervation has also been shown in the hippocampus of the p75-deficient mice (Yeo et al., 1997), but it is unlikely that this previously described effect underlies the results shown here, as hippocampal activity appears to exert an inhibitory influence on taste learning (Stone et al., 2005).
It may reasonably be asked whether altered cholinergic innervation – that is, the increased number and length of cholinergic fibers – is in fact the only reasonable explanation for our behavioral findings. For instance, we did observe a down-regulation of the M2 receptor, inhibition of which increases ACh release in the cortex through a presynaptic mechanism and enhances passive avoidance learning (Carey et al., 2001). It is therefore possible that down-regulation of the M2 receptor could function to further augment the effect of hyper-innervation, contributing partially or wholly to our observed enhancements in taste learning. This hypothesis fails to accord with experiments involving cortical application of M2 inhibitors, however, which report either no impact (Ramirez-Lugo et al., 2003) or impairments of CTA (Naor and Dudai, 1996). The interpretation of these latter studies is complicated by the possibility that the manipulations used to inhibit M2 also inhibited post-synaptic cortical neurons (Amar et al., 2010; Brown, 2010); regardless, while decreases in M2 receptor expression level in the p75−/− mice could potentially contribute to increased GC ACh levels and the enhancement of CTA seen in this study, either directly or indirectly, the current state of the field suggests that it is unlikely that loss of M2 receptors is a key mechanism for compensation of cholinergic hyper-innervation.
Finally, while the most prominent abnormality in p75−/− mice is in the BF, changes in this region involve both an increase in the number of cholinergic neurons and a decrease in GABAergic neurons (Lin et al., 2007). Furthermore, the cholinergic abnormality itself may well result in additional, secondary changes in neuronal structure and circuit function, implicating other neurotransmitter systems. Any (and all) of these effects could potentially have played a role in driving the observed behavioral phenomenon; most notably, cholinergic modulation may ultimately impact dopaminergic and glutamatergic function, known to be involved in proper conditioning (Fenu et al., 2001; Jiménez and Tapia, 2004). All methods of perturbing ACh have such secondary impacts, however: the effect of lesions, and even of temporary inactivations, are inevitably non-local (Honey and Sporns, 2008; Alstott et al., 2009), and even the behavioral effects of selective BF cholinergic immuno-lesions may reflect the involvement of other BF cell groups (Gutierrez et al., 1999a,b); p75-Saporin, which cleanly targets cholinergic neurons, still causes non-specific damage to the distributed neuronal networks into which these neurons are connected.
Perhaps even more dramatic, cholinergic agonism can result in seizures, via profound disruption of extra-cholinergic pathways (Olney et al., 1983; Naor and Dudai, 1996), making it a particularly difficult preparation to work with. To our knowledge, one study has successfully examined taste learning using cholinergic agonism (specifically, carbachol); this study reported, consistent with our results, that boosting cortical cholinergic function allowed rats to acquire CTAs to normally ineffective familiar stimuli (Clark and Bernstein, 2009). Thus, while no single method can provide direct proof of cholinergic function in this system in the absence of potentially confounding indirect effects, the use of the p75−/− mice provides important independent evidence that increased cholinergic function results in enhanced taste learning.
In fact, the genetic approach may offer certain advantages over other methods. For one thing, it largely eliminates issues of spatial variability: the hyper-innervation observed here, at least as far as GC was concerned, appeared to be uniformly and broadly distributed; thus, our results are less subject to variability borne of regional differences in targeting dependent on the precise placement of infusion cannulae. In addition, developmental compensation of critical functions in knockout mice may stabilize the ancillary circuits that are acutely perturbed under more acute manipulations of ACh. p75−/− mice become cholinergically hyper-innervated through slow, non-traumatic developmental processes and, thus, do not experience acute perturbations of cholinergic and other systems; they retain fundamental abilities to feed, are not seizure-prone, and show normal behaviors including odorant recognition responses (Barrett et al., 2010). In many regards they appear largely normal – while there are reports of behavioral impairments in one type of spatial learning task in the p75-deficient mice (Wright et al., 2004), other researchers report enhancement of spatial learning (Greferath et al., 2000) and, at the cellular level, of hippocampal long-term potentiation (Barrett et al., 2010).
One question that remains to be answered has to do with the psychological effect of the knockout phenotype. Our behavioral results clearly show that p75-deficient mice condition more strongly than WT mice – while the results using a high dose of LiCl could be interpreted to suggest a p75-WT difference in extinction rather than learning, our data make it clear that saccharin was observably more aversive to trained p75−/− mice in every session that was not contaminated by a floor effect for consumption, including the first testing session in the low-LiCl experiment. Thus, the inter-strain extinction differences largely follow from differences in initial conditioning. Our conditioning data do not, however, enable us to say whether the observed behavioral effects represent a simple associative learning abnormality as opposed to a more permanent change. Since our knockout mice failed to achieve the same level of extinction as WT mice even across six testing sessions, we cannot say for sure that CTA in mice with cholinergic hyper-innervation did not permanently elevate a “fear-related component” of the response to saccharin. Future work will address this question.
More centrally with regard to the theory of cholinergic involvement in learning, our data do not speak to the issue of whether the p75−/− mice truly found the tastes to be more novel (i.e., less familiar) as opposed to more salient (i.e., more potent and intense). Either of these effects would result in stronger learning (Schmajuk et al., 1996; Berridge and Robinson, 1998) without reflecting a direct change to general learning circuitry. Several lines of evidence link cholinergic function to novelty, however. Familiarizing a rodent with a taste causes a down-regulation of taste-related ACh release (Miranda et al., 2000), whereas acute inhibition of cholinergic function inhibits the normal behavioral effects of such familiarization (Naor and Dudai, 1996; Berman et al., 2000; Miranda et al., 2000). Conversely, increasing cholinergic activity enhances both taste novelty and salience (Clark and Bernstein, 2009). It is therefore reasonable to speculate that p75−/− mice treat the taste as somehow “even less familiar than a novel taste” and thus are more likely to associate this taste with malaise than WT mice. Future work assessing the effect of familiarization on the inter-strain differences should shed light on this issue.
With few exceptions (one being mentioned above), the current understanding of the role of the cholinergic system in an animal’s response to novel tastes is based upon experimental perturbations that destroy or inhibit key cells or signaling in the neural circuit, resulting in attenuation of gustatory behaviors such as CTA. Less common, but of great value in assessing the role of this system, are findings of enhanced functions following manipulations that increase cholinergic signaling. Our use of a knockout mouse strain that links cholinergic hyper-innervation to stronger CTA demonstrates that a developmental increase in the BF cholinergic projection to GC, brought about through genetic manipulation, is effective in regulating the strength of conditioning. In addition to providing independent evidence for cholinergic regulation of CTA, this study confirms the power of genetic approaches for studying taste behaviors (Masugi et al., 1999; Jacobson et al., 2006), and suggests new areas for investigation that include the developmental role of cholinergic signaling during the maturation of gustatory circuitry.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by NIH NS057305 to Susan J. Birren, CD006666 to Donald B. Katz, and by 2P30NS045713 for Core Facilities for Neurobiology. The authors wish to thank Jeanine Hinterneder and Suzanne Paradis for critical readings of the manuscript, and our editor and reviewers for a truly enjoyable submission and revision process.
References
Allen, T. A., Narayanan, N. S., Kholodar-Smith, D. B., Zhao, Y., Laubach, M., and Brown, T. H. (2008). Imaging the spread of reversible brain inactivations using fluorescent muscimol. J. Neurosci. Methods 171, 30–38.
Alstott, J., Breakspear, M., Hagmann, P., Cammoun, L., and Sporns, O. (2009). Modeling the impact of lesions in the human brain. PLoS Comput. Biol. 5, e1000408. doi: 10.1371/journal.pcbi.1000408
Amar, M., Lucas-Meunier, E., Baux, G., and Fossier, P. (2010). Blockade of different muscarinic receptor subtypes changes the equilibrium between excitation and inhibition in rat visual cortex. Neuroscience 169, 1610–1620.
Barrett, G. L., Reid, C. A., Tsafoulis, C., Zhu, W., Williams, D. A., Paolini, A. G., Trieu, J., and Murphy, M. (2010). Enhanced spatial memory and hippocampal long-term potentiation in p75 neurotrophin receptor knockout mice. Hippocampus 20, 145–152.
Berman, D. E., and Dudai, Y. (2001). Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science 291, 2417–2419.
Berman, D. E., Hazvi, S., Neduva, V., and Dudai, Y. (2000). The role of identified neurotransmitter systems in the response of insular cortex to unfamiliar taste: activation of ERK1-2 and formation of a memory trace. J. Neurosci. 20, 7017–7023.
Bermudez-Rattoni, F. (2004). Molecular mechanisms of taste-recognition memory. Nat. Rev. Neurosci. 5, 209–217.
Berridge, K. C., and Robinson, T. E. (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 28, 309–369.
Bertrand, D., Yannick, S., Mathilde, B., Frederic, L., Nadine, R., and Guillaume, F. (2009). Critical role of insular cortex in taste but not odour aversion memory. Eur. J. Neurosci. 29, 1654–1662.
Brown, D. A. (2010). Muscarinic acetylcholine receptors (mAChRs) in the nervous system: some functions and mechanisms. J. Mol. Neurosci. 41, 340–346.
Carey, G. J., Billard, W., Binch, H. III., Cohen-Williams, M., Crosby, G., Grzelak, M., Guzik, H., Kozlowski, J. A., Lowe, D. B., Pond, A. J., Tedesco, R. P., Watkins, R. W., and Coffin, V. L. (2001). SCH 57790, a selective muscarinic M(2) receptor antagonist, releases acetylcholine and produces cognitive enhancement in laboratory animals. Eur. J. Pharmacol. 431, 189–200.
Chen, X., Gabitto, M., Peng, Y., Ryba, N. J., and Zuker, C. S. (2011). A gustotopic map of taste qualities in the mammalian brain. Science 333, 1262–1266.
Clark, E. W., and Bernstein, I. L. (2009). Boosting cholinergic activity in gustatory cortex enhances the salience of a familiar conditioned stimulus in taste aversion learning. Behav. Neurosci. 123, 764–771.
Domjan, M., and Gillan, D. (1976). Role of novelty in the aversion for increasingly concentrated saccharin solutions. Physiol. Behav. 16, 537–542.
Fenu, S., Bassareo, V., and Di Chiara, G. (2001). A role for dopamine D1 receptors of the nucleus accumbens shell in conditioned taste aversion learning. J. Neurosci. 21, 6897–6904.
Fortis-Santiago, Y., Rodwin, B. A., Neseliler, S., Piette, C. E., and Katz, D. B. (2010). State dependence of olfactory perception as a function of taste cortical inactivation. Nat. Neurosci. 13, 158–159.
Gonzalez, C. L., Miranda, M. I., Gutierrez, H., Ormsby, C., and Bermudez-Rattoni, F. (2000). Differential participation of the NBM in the acquisition and retrieval of conditioned taste aversion and morris water maze. Behav. Brain Res. 116, 89–98.
Greferath, U., Bennie, A., Kourakis, A., Bartlett, P. F., Murphy, M., and Barrett, G. L. (2000). Enlarged cholinergic forebrain neurons and improved spatial learning in p75 knockout mice. Eur. J. Neurosci. 12, 885–893.
Grider, M. H., Chen, Q., and Shine, H. D. (2006). Semi-automated quantification of axonal densities in labeled CNS tissue. J. Neurosci. Methods 155, 172–179.
Grossman, S. E., Fontanini, A., Wieskopf, J. S., and Katz, D. B. (2008). Learning-related plasticity of temporal coding in simultaneously recorded amygdala-cortical ensembles. J. Neurosci. 28, 2864–2873.
Gutierrez, H., Gutierrez, R., Silva-Gandarias, R., Estrada, J., Miranda, M. I., and Bermudez-Rattoni, F. (1999a). Differential effects of 192IgG-saporin and NMDA-induced lesions into the basal forebrain on cholinergic activity and taste aversion memory formation. Brain Res. 834, 136–141.
Gutierrez, H., Gutierrez, R., Ramirez-Trejo, L., Silva-Gandarias, R., Ormsby, C. E., Miranda, M. I., and Bermudez-Rattoni, F. (1999b). Redundant basal forebrain modulation in taste aversion memory formation. J. Neurosci. 19, 7661–7669.
Gutierrez, R., Rodriguez-Ortiz, C. J., De La Cruz, V., Nunez-Jaramillo, L., and Bermudez-Rattoni, F. (2003). Cholinergic dependence of taste memory formation: evidence of two distinct processes. Neurobiol. Learn. Mem. 80, 323–331.
Hartikka, J., and Hefti, F. (1988). Development of septal cholinergic neurons in culture: plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J. Neurosci. 8, 2967–2985.
Hecker, S., and Mesulam, M. M. (1994). Two types of cholinergic projections to the rat amygdala. Neuroscience 60, 383–397.
Honey, C. J., and Sporns, O. (2008). Dynamical consequences of lesions in cortical networks. Hum. Brain Mapp. 29, 802–809.
Jacobson, L. H., Kelly, P. H., Bettler, B., Kaupmann, K., and Cryan, J. F. (2006). GABA(B(1)) receptor isoforms differentially mediate the acquisition and extinction of aversive taste memories. J. Neurosci. 26, 8800–8803.
Jeewajee, A., Lever, C., Burton, S., O’Keefe, J., and Burgess, N. (2008). Environmental novelty is signaled by reduction of the hippocampal theta frequency. Hippocampus 18, 340–348.
Jiménez, B., and Tapia, R. (2004). Biochemical modulation of NMDA receptors: role in conditioned taste aversion. Neurochem. Res. 29, 161–168.
Katz, D. B., Simon, S. A., and Nicolelis, M. A. (2001). Dynamic and multimodal responses of gustatory cortical neurons in awake rats. J. Neurosci. 21, 4478–4489.
Krupa, D. J., Brisben, A. J., Katz, D. B., and Nicolelis, M. A. L. (1998). Role of SI cortex in thalamic processing of complex somatosensory stimuli. Abstr. Soc. Neurosci. 24, 132.
Lee, K. F., Li, E., Huber, L. J., Landis, S. C., Sharpe, A. H., Chao, M. V., and Jaenisch, R. (1992). Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69, 737–749.
Lin, P. Y., Hinterneder, J. M., Rollor, S. R., and Birren, S. J. (2007). Non-cell-autonomous regulation of GABAergic neuron development by neurotrophins and the p75 receptor. J. Neurosci. 27, 12787–12796.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408.
Masugi, M., Yokoi, M., Shigemoto, R., Muguruma, K., Watanabe, Y., Sansig, G., van der Putten, H., and Nakanishi, S. (1999). Metabotropic glutamate receptor subtype 7 ablation causes deficit in fear response and conditioned taste aversion. J. Neurosci. 19, 955–963.
Miranda, M. I., Ramirez-Lugo, L., and Bermudez-Rattoni, F. (2000). Cortical cholinergic activity is related to the novelty of the stimulus. Brain Res. 882, 230–235.
Muller, J. F., Mascagni, F., and McDonald, A. J. (2011). Cholinergic innervation of pyramidal cells and parvalbumin-immunoreactive interneurons in the rat basolateral amygdala. J. Comp. Neurol. 519, 790–805.
Naor, C., and Dudai, Y. (1996). Transient impairment of cholinergic function in the rat insular cortex disrupts the encoding of taste in conditioned taste aversion. Behav. Brain Res. 79, 61–67.
Naumann, T., Casademunt, E., Hollerbach, E., Hofmann, J., Dechant, G., Frotscher, M., and Barde, Y. A. (2002). Complete deletion of the neurotrophin receptor p75NTR leads to long-lasting increases in the number of basal forebrain cholinergic neurons. J. Neurosci. 22, 2409–2418.
Nunez-Jaramillo, L., Jimenez, B., Ramirez-Munguia, N., Delint-Ramirez, I., Luna-Illades, C., Tapia, R., and Bermudez-Rattoni, F. (2008). Taste novelty induces intracellular redistribution of NR2A and NR2B subunits of NMDA receptor in the insular cortex. Brain Res. 1215, 116–122.
Olney, J. W., de Gubareff, T., and Labruyere, J. (1983). Seizure-related brain damage induced by cholinergic agents. Nature 301, 520–522.
Parent, M., and Descarries, L. (2008). Acetylcholine innervation of the adult rat thalamus: distribution and ultrastructural features in dorsolateral geniculate, parafascicular, and reticular thalamic nuclei. J. Comp. Neurol. 511, 678–691.
Paxinos, G., and Franklin, K. B. J. (2001). The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press.
Pratt, W. E., Spencer, R. C., and Kelley, A. E. (2007). Muscarinic receptor antagonism of the nucleus accumbens core causes avoidance to flavor and spatial cues. Behav. Neurosci. 121, 1215–1223.
Ramirez-Lugo, L., Miranda, M. I., Escobar, M. L., Espinosa, E., and Bermudez-Rattoni, F. (2003). The role of cortical cholinergic pre- and post-synaptic receptors in taste memory formation. Neurobiol. Learn. Mem. 79, 184–193.
Ranganath, C., and Rainer, G. (2003). Neural mechanisms for detecting and remembering novel events. Nat. Rev. Neurosci. 4, 193–202.
Sakai, N., and Yamamoto, T. (1997). Conditioned taste aversion and c-fos expression in the rat brainstem after administration of various USs. Neuroreport 8, 2215–2220.
Schmajuk, N. A., Gray, J. A., and Lam, Y. W. (1996). Latent inhibition: a neural network approach. J. Exp. Psychol. Anim. Behav. Process. 22, 321–349.
Semba, K. (2000). Multiple output pathways of the basal forebrain: organization, chemical heterogeneity, and roles in vigilance. Behav. Brain Res. 115, 117–141.
Semba, K. (2004). Phylogenetic and ontogenetic aspects of the basal forebrain cholinergic neurons and their innervation of the cerebral cortex. Prog. Brain Res. 145, 3–43.
Stone, M. E., Grimes, B. S., and Katz, D. B. (2005). Hippocampal inactivation enhances taste learning. Learn. Mem. 12, 579–586.
Tokita, K., Inoue, T., and Boughter, J. D. Jr. (2009). Afferent connections of the parabrachial nucleus in C57BL/6J mice. Neuroscience 161, 475–488.
Van der Zee, C. E., Ross, G. M., Riopelle, R. J., and Hagg, T. (1996). Survival of cholinergic forebrain neurons in developing p75NGFR-deficient mice. Science 274, 1729–1732.
Wang, Y.-Y., Fontanini, A., and Katz, D. B. (2006). Temporary basolateral amygdala lesions disrupt acquisition of socially transmitted food preferences in rats. Learn. Mem. 13, 794–800.
Wright, J. W., Alt, J. A., Turner, G. D., and Krueger, J. M. (2004). Differences in spatial learning comparing transgenic p75 knockout, New Zealand Black, C57BL/6, and Swiss Webster mice. Behav. Brain Res. 153, 453–458.
Yamamoto, T. (2006). Neural substrates for the processing of cognitive and affective aspects of taste in the brain. Arch. Histol. Cytol. 69, 243–255.
Yamamoto, T., Fujimoto, Y., Shimura, T., and Sakai, N. (1995). Conditioned taste aversion in rats with excitotoxic brain lesions. Neurosci. Res. 22, 31–49.
Yasoshima, Y., and Yamamoto, T. (1997). Rat gustatory memory requires protein kinase C activity in the amygdala and cortical gustatory area. Neuroreport 8, 1363–1367.
Keywords: p75 knockout mouse, cholinergic system, conditioned taste aversion, taste learning
Citation: Neseliler S, Narayanan D, Fortis-Santiago Y, Katz DB and Birren SJ (2011) Genetically induced cholinergic hyper-innervation enhances taste learning. Front. Syst. Neurosci. 5:97. doi: 10.3389/fnsys.2011.00097
Received: 11 August 2011; Accepted: 09 November 2011;
Published online: 01 December 2011.
Edited by:
Milagros Gallo, University of Granada, SpainCopyright: © 2011 Neseliler, Narayanan, Fortis-Santiago, Katz and Birren. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Donald B. Katz and Susan J. Birren, Department of Psychology, Brandeis University, Volen 206/MS 013, Waltham, MA 02454, USA. e-mail:ZGJrYXR6QGJyYW5kZWlzLmVkdQ==;YmlycmVuQGJyYW5kZWlzLmVkdQ==
†Donald B. Katz and Susan J. Birren have contributed equally to this work.