 Yuwei Wang
Yuwei Wang David W. Grainger
David W. Grainger- 1College of Pharmacy, California Health Science University, Clovis, CA, United States
- 2Department of Pharmaceutics and Pharmaceutical Chemistry, Salt Lake City, UT, United States
- 3Department of Biomedical Engineering, University of Utah, Salt Lake City, UT, United States
Nearly a half-century after original liposome discovery as a prospective lipid pharmaceutical carrier, the global liposomal drug delivery market has increased dramatically, with an annual market growth rate of 13.2%, valued at ∼$6,993 million by 2027. As an intrinsically complex delivery system, liposomal formulations face much greater characterization and regulatory review challenges than traditional small molecule drugs and biologics. Due to rapid liposomal drug development, both European Medicines Agency (EMA) and US Food and Drug Administration (FDA) now provide regulatory guidance for new liposomal drug application reviews. The expanding global liposome drug market and associated driving forces for increased research and development (R&D) in novel liposomal products are key factors propelling liposomal drug interests. We review and compare EU and US regulations on liposomal drug submissions, and provide insights into regulatory strategies throughout the entire liposomal drug development process. This addresses current gaps noted between liposome-based drug development in research labs and current regulatory guidance for liposomal drug approvals in order to facilitate more efficient, less costly, and less risky complex drug development.
Global Liposome Drug Markets
According to the most recent 2021 reports, the liposomal drug delivery market is predicted to reach nearly $7 billion by 2027, nearly double that from 2019 ($3.595 billion), with a compound annual growth rate of 8.8% over 2020–2027. (The Insight Partners, 2021) Other market data indicate that liposomal application extensions from pharmaceutical products into cosmetics, food, and agriculture should stimulate global market growth rate increases to 13.2% over the period 2021 to 2028. (Data Bridge, 2021) Common clinically used, marketed liposomal drug products (Figure 1) are administered intravenously and intramuscularly, now covering diverse medical needs in cancer therapy, fungal disease, viral vaccines, photodynamic ocular ablative therapy, and local analgesia and anesthesia. (Bulbake et al., 2017; Wang and Grainger, 2019) Liposomal forms of doxorubicin, paclitaxel, amphotericin B, and other active pharmaceutical agents are approved for clinical use. (Bulbake et al., 2017) Over 30 other formulations are in current clinical trials for expanded medical applications. Liposomal cosmetic and nutritional products are also of increasing interest. Following the 1995 precedent inaugural liposome drug approval (Doxil™), (Barenholz, 2012) and patent expiration in 2009, follow-on generic versions of this product and other approved liposome first-in-human products were anticipated. However, this has been much slower than anticipated; reasons for delayed generic liposomal drugs are clarified below.

FIGURE 1. Current commercial liposomal drugs used clinically.
Forces Driving R&D in Liposomal Drug Development
The North American region dominates liposome drug delivery markets, due primarily to favorable reimbursement policies to payors for these expensive therapeutics. Key pharmaceutical companies exploit prior liposomal product approval and marketing success, and have invested both know-how and billions of dollars supporting innovative research to drive rapid development of liposome-based drug delivery technology. Critically, these industries build robust approval processes and increasingly automated manufacturing pipelines to efficiently develop and commercialize liposomal drug products through established technology platforms. Resulting vibrant R&D, experience, production facilities, and well-established commercialization strategies enhance new liposomal drug capabilities and products, de-risking pathways to markets.
Addressing unmet, significant clinical needs with new products creates new markets, consistently the driving force for pharmaceutical innovations. Liposomal drugs are no exception. Global increases in patients with diseases addressed by targeted liposomal drug therapies propel global liposomal drug market growth. (Data Bridge, 2021) Worldwide increases in liposomal drug development in recent decades are shown to result from the rising prevalence of chronic diseases that liposomal drug delivery feasibly treat. In the US, the Centers for Disease Control and Prevention reported in 2019 that nearly 60% of US citizens have at least one chronic disease, and 40% of people suffer at least two chronic diseases. (CDC, 2022) The Asia Pacific region has considerable potential for liposomal drug development due to the increasing incidence of chronic disease.
North America’s well-established pipeline shows considerable investment in liposomal drug R&D. Increasing numbers of FDA-approved liposomal drugs and increasing clinical trials for new formulations and indications propel clinical interest and use. Liposome-based delivery technology, including stealth liposomes, gas-filled liposomes, non-PEGylated liposomes, and DepoFoam™ technology, and newer liposomes for dedicated immunotherapy, are exploited. (Filipczak et al., 2020; Salehi et al., 2020) Liposome delivery systems now have decades of clinical use, providing both drug physical protection and enhanced circulatory and biodistribution properties to encapsulated drugs, improving pharmacokinetics and pharmacodynamics. Liposomes also have the potential to target delivery, avoid certain tissue exposures, and facilitate drug penetration through biological barriers. Longer circulating half-life, improved organ distribution, higher intracellular drug amounts, improved therapeutic effects, increased maximum tolerated doses, and reduced toxicities are frequently reported. (Deodhar and Dash, 2018; Salehi et al., 2020; Ren et al., 2021; Andra et al., 2022).
Standard nucleic acid drug technologies based on plasmid DNA, messenger RNA, small interfering RNA, microRNA, antisense oligonucleotides, and emerging innovative CRISPR/Cas9 therapies (Filipczak et al., 2020) increasingly employ liposomal delivery strategies. These nucleic acids have high negative charge density, significantly inhibiting cell uptake, requiring diverse methods for cellular entry. For in vivo delivery, viral vectors have been a traditional nucleic acid delivery approach. Given their in vivo properties, liposomal drug delivery systems are well-suited for nucleic acid drug delivery as a well-developed non-viral vector alternative. One recent clinical example is Patisiran (ONPATTRO®), approved and marketed in 2018 by Alnylam Pharmaceuticals (United States). Patisiran is the first-in-human RNA interference (RNAi) therapy approved globally as a liposome formulation, encapsulating a slightly modified siRNA against transthyretin (TTR), facilitating targeted therapy primarily to the liver to reduce TTR accumulation in tissues. (Urits et al., 2020) Administered traditionally via parenteral routes, liposome delivery systems are now being developed for several additional administration methods (e.g., oral, transdermal, and blood-brain barrier delivery). (Guimarães et al., 2021).
EMA and FDA Regulations That Affect Complex Drug Product Development
Changes in regulatory policies by numerous national agencies significantly impact current and future R&D investment, product and market trends for any drug; liposomes as complex drugs are particularly impacted. For example, the US FDA and Health Canada policies encourage liposomal formulations for anti-fungal and anti-cancer treatments. India’s government reduces drug prices for treating infectious and chronic diseases, providing incentives for investment into corresponding drug formulation development that affect complex drug market growth.
Facing consistent increases in liposomal drug formulation technology and therapeutic interests, regulatory agencies are compelled to produce transparent policies guiding approvals for these complex drug products. Liposomes have been problematic within the FDA’s regulatory framework that distinguishes drugs, devices, biologics, or their combinations. Liposomes are classified as complex generic drug products. Substantial R&D challenges are widely recognized for asserting therapeutic equivalence of complex generic drug products, prompting governments to revise regulatory processes. (United States Government Accountability Office, 2017) In response, the FDA has endeavored to harmonize policies and processes for regulating complex generic drug products, including liposomes. New product-specific guidances (PSGs) now regularly appear for generic complex products. (U.S. Food and Drug Administration, 2022), stimulating several similar international regulatory initiatives. (European Commission’s Joint Research Centre (JRC) and the Global Coalition for Regulatory Science Research (GCRSR), 2019; International Pharmaceutical Regulators Programme (IPRP), 2022) This focus in the US is largely responsive to US congressional pressure to increase competition and reduce prices for complex products (U.S. Food and Drug Administration, 2017).
Non-biological complex drugs (NBCDs) are defined as medicinal products comprising synthetic components and frequently nanoparticulate structures representing in totality the active pharmaceutical ingredient and whose physicochemical properties cannot be completely characterized or readily analytically quantified. (Lionberger, 2019) Because these properties may also be substantially changed by each manufacturing process, resulting NBCD product performance is possibly variable. (Crommelin and de Vlieger, 2015; Crommelin et al., 2015; Colombo et al., 2018) As NBCDs, liposomes are now covered by the recently issued 2018 draft guidance (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2018) affirming and updating science-based regulatory procedures for this NBCD. (Zhang, 2020) This alters the first liposome guidance published in 2002 (U.S. Food and Drug Administration, 2002) describing ‘liposome drug products: chemistry, manufacturing, and controls; human pharmacokinetics and bioavailability; and labeling documentation, but explicitly providing no information on clinical efficacy and safety studies, and nonclinical pharmacology and/or toxicology studies. A later guidance specifically focused on product development of follow-on (generic) versions of doxorubicin liposomes through the FDA 505(j) pathway. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2014) The latest guidance document issues the FDA Center for Drug Evaluation and Research working group instructions for new drug applications (NDAs) and abbreviated new drug applications (ANDAs) regulatory submissions for liposome drug products. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2018) Further guiding recommendations can be applied to liposome drugs intended to be marketed under Biologics License Applications (BLAs). Figure 2 describes the complex drug development pathway.
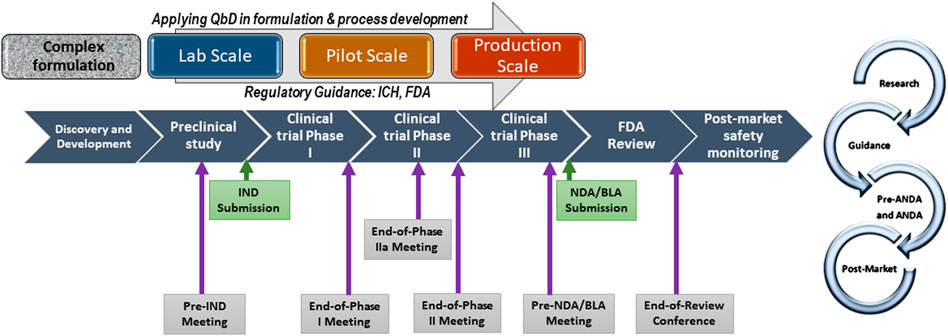
FIGURE 2. Complex drug development pathway with scale-up process development, QbD, and specific meetings with FDA. IND: Investigational New Drug, NDA: New Drug Application; BLA: Biologic Licensing Application (https://www.fda.gov/drugs/generic-drugs/fy-2018-gdufa- science-and-research-outcomes).
EMA does not yet have specific liposomes guidelines. In the EU, both innovator (i.e., a patented new chemical entity in a novel formulation or manufacturing process, with first-in-human approval from a regulatory authority and sold under a brand name), and follow-on liposome products follow a centralized procedure, with no specific guidance documents available for innovator liposome products. A “reflection paper” issued in 2009/2013 describes follow-on versions of doxorubicin liposomes to assist in generating relevant quality, nonclinical and clinical data to support a marketing authorization of intravenous liposomal products developed with reference to an innovator liposomal product. (European Medicines Agency, 2013a) EMA’s requirement document focuses on parenteral liposome products.
One US follow-up product is now approved (Lipodox® as a generic version of Doxil®), but EMA rejected Lipodox because no bioequivalence for free doxorubicin in both Lipodox and Caelyx® (i.e., European brand name for Doxil: produced by the same United States manufacturer) could be asserted. (Committee for Medicinal Products for Human Use, 2011) EMA and FDA issued divergent regulatory decisions for the identical generic (follow-on) liposome product. Elsewhere globally (e.g., India, Taiwan, Argentina, and China), follow-on doxorubicin and amphotericin liposome products are marketed, yet specific registration criteria for these liposome products are not apparent. FDA efforts increasingly focus on complex products, typically more challenging to develop due to technological and regulatory uncertainty (Office of Generic Drugs, 2020).
The current FDA liposomal guidance does not cover clinical efficacy and safety, and nonclinical pharmacology/toxicology studies, so liposome regulatory comparisons best focus on Chemistry Manufacturing and Controls (CMC) between EU and US regulatory requirements. FDA’s guidance provides much more detail regarding CMC. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2018) Both EMA and FDA guidance mention that any process change, no matter how minor, requires comparative investigations after liposomal product marketing authorization. So, applicants should best avoid making any changes to liposomal manufacturing after approval. Both agencies agree that liposomal pharmacokinetics studies are influenced by many factors, such as target patient population and proposed drug indication. Hence, study design should be analyzed on a case-by-case basis. They recommend liposome drug product applicants seek product-specific advice on questions regarding study design, data requirements, or any post-approval changes. Consistent communication with the regulatory agency, commensurate with product development and manufacturing timelines, is critical as with any complex drug product.
Insights Into Best Practices Throughout the Entire Liposomal Drug Development Process
Formulation Design. When designing liposome formulations, and in fact, for all innovator drug products, it is always easier to employ excipients/lipid components already listed in the FDA’s Inactive Ingredient Database (IID). The IID provides names, dosage forms, and the maximum excipient amounts used in approved drugs with a dosage form listed for each excipient. Formulators should try to use excipients listed within the IID-listed amount range for the intended route of administration. Otherwise, information submitted for a novel lipid ingredient should be comparable to a drug substance. Full GMP compliance is required for all clinical studies in most countries. All excipients must be compendia grade from the beginning to avoid questions or impacts from possible excipient impurities. Changes in grade and source of excipients during development must be addressed for potential consequences to product safety and efficacy. When formulation composition is presented in ranges, several items must be specified (The Insight Partners, 2021): product development studies, (Data Bridge, 2021) rationale for selected ranges, (Bulbake et al., 2017) how critical excipient source affects finished product quality. These ranges should evaluate and be supported by drug development data.
It is prudent to follow FDA-recommended descriptions of liposomal formulations from initiation of drug development. The FDA requests amounts of each lipid component used in the formulation based on the product’s final form. In addition, the FDA recommends expressing each lipid’s molar ratio to drug substance for each lipid in the finished formulation. Drug substance amounts are included in any liposomal formulation.
In small-scale formulation development phases, sterile filtration (filter pore size: 0.22 µm) must be tested early. Each component in the formulation must be shown compatible with filter materials. Some liposome components could interact with filter materials during filtration, resulting in clogs and high pressure, compromising liposomal integrity and structure, and causing possible failure of follow-on filter integrity tests. Passing filter integrity tests is required for drug product release.
1. Analytical Methods Development for Liposome Product Characterization. Liposome Drug Formulation Characterization. Analytical methods for liposome drug formulations require the following properties by regulatory agencies: morphology, particle size, surface characteristics (e.g., pegylation), zeta potential, drug product viscosity, parameters of the contained drug, encapsulation efficiency, drug-to-lipid ratio, liposome phase transition temperature, in vitro drug release in physiologically/clinically relevant media, and stability studies, such as drug leakage rates from the liposomes throughout shelf-life, or liposome integrity changes in response to salt concentration, pH, temperature, etc. A complementary method is suggested in FDA draft guidelines (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2017) indicating that inherent differences among various analytical techniques may produce different measurement endpoints, and should be considered, especially in measuring critical quality attributes (CQAs). Taking liposome particle size as an example, both transmission electron microscopy and dynamic light scattering are used for size measurement, but the basis of both analytical methods must be described to account for possible differences.
2. Drug Product Specification. In each submission, both agencies require a detailed description and complementary diagram of the liposome’s structure, including ligands, core-shell structures, or surface coatings. When using ligands (e.g., peptides, proteins) for targeted delivery, control of ligand orientation and conformational state is emphasized in EMA’s guideline. (Committee for Medicinal Products for Human Use (CHMP), 2013) Impact of coating heterogeneity and random ligand orientation on biodistribution and PK may affect liposome safety and efficacy. FDA provides example attributes to include in liposome drug product release specifications: physicochemical properties (particle size, zeta-potential, physical stability, etc.), encapsulated and free drug substance in the product, degradation products related to lipids or drug substance, lipid content, in vitro drug release, and residual amount of organic solvents used in fabrication. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2018) EMA’s document states that in vitro drug substance release should be tested in physiologically/clinically relevant media, and suggests performing tests at variable temperatures and pH. (European Medicines Agency, 2013a), (Committee for Medicinal Products for Human Use (CHMP), 2013) FDA guidance also requires in vitro release studies in physiological media (e.g., human plasma). A complete in vitro drug release profile must reach a plateau where no significant increase over at least three consecutive time points and minimally 85% of the labeled amount of encapsulated drug is released. However, if a highly stable liposome formulation shows no release profile, FDA allows an accelerated release study in non-physiological conditions using a quality control release test. Lastly, an In Vitro/In Vivo Correlation (IVIVC) is highly recommended by FDA. (Shen and Burgess, 2015) In the event that a complete IVIVC cannot be obtained, correlations between in vitro release and in vivo pharmacokinetic data should be summarized in the submission to justify the in vitro quality control release test. FDA sees a fully validated in vitro release method as quality testing reflecting product clinical performance. (Solomon et al., 2017) By measuring free drug versus encapsulated drug, the in vitro release study characterizes liposome integrity which may significantly impact the drug product’s clinical performance. As stated in FDA’s draft guidance: “Ideally, the dissolution/in vitro release method should be able to discriminate batches that are not bioequivalent to the pivotal clinical batch, which will have demonstrated efficacy and safety” (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2017).
Process Development. FDA recommends that applicants use a detailed process flow diagram detailing each process unit operation. Ranges of process parameters or process controls must be identified and supported by development studies. Identification of product CQAs and critical materials attributes (CMAs) used to design and implement the in-process manufacturing controls is critical, and earlier is better. Process controls include in-process controls (IPC) and in-process samples (IPS); the difference is that the process cannot proceed until the IPC is validated to be within the specified range. The FDA will request methods that remove uncaptured (free) drug molecules from formulations during review submissions. FDA draft guidance defines empty liposomes or those missing or having incomplete surface modifications (e.g., coating of targeting moiety) as impurities that must be quantified. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2017) Production processes must be validated at manufacturing scale for consistency and reproducibility.
Liposomal formulations cannot be terminally sterilized, so liposomal injectable products must be aseptically filtered. At least two filtrations are required at process completion; one for sterile filtration, a second for bioburden filtration. The FDA emphasizes sterilizing filtration process in their guidance, and seeks verification in regulatory submissions. Membrane pressure must be recorded during filtration and ensured not to exceed maximum membrane pressure; the filter member integrity test is required for each process filtration step. Liposomes must be fully characterized post-filtration to ensure that integrity and structure remain unchanged. High liposomal suspension concentrations should be avoided for filtration.
A manufacturing process must meet three criteria before entering production: 1) robustness (repeatability), 2) scalability, and 3) validatability. FDA guidance emphasizes validation as a lifecycle process, utilizing statistics to continuously refine and monitor the manufacturing process. (U.S. Food and Drug Administration, 2011) This lifecycle process is executed in three stages: 1) process design, 2) process qualification, 3) continued process verification. The FDA’s 2011 guidance (U.S. Food and Drug Administration, 2011) emphasizes a more scientific, data-driven approach, contrasting their 1987 topical guideline describing a more binary, yes-or-no, one-and-done approach applied to validate a process (U.S. Food and Drug Administration, 1987).
Both EMA and FDA authorities agree that small changes to liposome manufacturing processes can significantly influence end-product performance. Applicants should avoid making changes to approved manufacturing processes. Liposome production scale-up is very challenging, but standard scale-up methods are used in industry (e.g., alcohol injection and crossflow techniques). (Filipczak et al., 2020) Employing mature manufacturing methods is less risky under regulatory review; FDA emphasizes maturity of nanotechnology manufacturing methods in its guidance. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2017) Manufacturing scale-up timing, from lab-scale proof-of-concept to pilot scale, is essential to properly define both process and controls for scaling up production for manufacturing clinical trial batches or commercial products. With accumulated manufacturing experience and increased risk assessment during development phases, researchers should continually optimize manufacturing processes and associated control strategies over time. Should manufacturing changes in formulation, container closure system, manufacturing facility, or equipment be made after approval, each change must be carefully documented with supporting data and reported to agencies for further review and approval.
Stability Studies. Other current regulatory guidance documents related to stability studies and testing conditions to support new drug product applications all apply to liposomal drugs. Specifically to liposomal products, each lipid used in liposomal formulation requires validation in stability studies and stress testing. Stress testing usually includes stability after exposure to high and low temperatures, light, pH, and oxygen, which may induce lipid degradation. Both the chemical stability of encapsulated drug and of each lipid component in the liposome formulation must be assessed to determine storage conditions and retest periods. Due to increasing surface coatings use in nanomedicines, EMA published a guidance explicitly focusing on surface coatings. (Committee for Medicinal Products for Human Use (CHMP), 2013) This guidance requests stability studies of coating moieties to detach or degrade relevant to efficacy and safety during storage and in use. (Committee for Medicinal Products for Human Use (CHMP), 2013) In vivo impact on PK and biodistribution attributed to coating materials should be studied. The guidance asks researchers to consider potential surface functional groups formed by coating detachment or degradation, biodistribution of the released coatings, and their metabolic fate.
For liposome product stability validation, FDA requires three studies: microbiological, physical, and chemical product stability. Both EU and US guidance indicate that products requiring dilution or reconstitution must complete in-use stability verification for product storage and use under pharmacy preparation conditions. Liposome suspensions as infusion products must be diluted to large volumes using infusion diluent before administration. Liposome morphology and structure should remain stable if the concentration is reduced below the self-assembled concentration after dilution or upon entering the bloodstream. A liposomal infusion product has considerable contact time with infusion tubing under infusion pump pressure; in-use stability studies may be performed under these actual administration conditions. Specific guidance for lyophilized parenteral liposome drug products, either small molecule or biologics drugs, is unavailable. Therefore, regulatory requirements should be identical to all other lyophilized parenteral products.4 For liposomal lyo-products, reconstitution conditions, the in-use period after reconstitution, and storage condition after reconstitution and dilution must be stated in both labeling and preparation instructions. For non-ready-to-use products, EMA emphasizes describing the process robustness/repeatability for reconstitution or pharmacy preparation.
Excess reserve samples beyond those needed for initial stability studies must be planned in case of testing failures or additional parameters requiring further testing. Ensuring sufficient stability samples available in advance avoids the costly need to change batches during these studies, which may trigger reviewer questions. Commonly, twice the number of samples needed for initial stability studies are held in reserve. Retaining reasonable amounts of IPC samples and products from each batch is recommended to address potential needs and establish connections between early development lots to later or commercial-scale lots, including any manufacturing process changes, scale-up, and site transfers.
Quality by Design (QbD) Requirements. QbD is a powerful tool to ensure the quality of medicines in their design, development, and manufacturing using statistical, analytical and risk management methods. In 2011, EMA and FDA instituted a program for joint agency assessment of quality sections relevant to QbD. (European Medicines Agency and U.S. Food and Drug Administration, 2013) Their goal is to ensure consistent implementation and harmony between the EU and US of ICH Q8 Pharmaceutical Development, Q9 Quality Risk Management, Q10 Pharmaceutical Quality Systems, and Q11 Development and Manufacture of Drug Substances during assessment processes. (European Medicines Agency, 2013b) Both agencies integrate ICH guidelines for technical requirements for developing pharmaceutical quality systems for pharmaceutical product registration.
Through the launched pilot program, EMA and FDA reached agreements on wide ranging QbD issues, such as Design Space verification, Design Space and risk assessment level of detail in applications, continuous process verification, and continuous manufacturing. In ICH Q8(R2), QbD is defined as “A systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.” (ICH, 2009) QbD methods identify Quality Target Product Profiles (QTPP), translate them into CQAs and CMAs desired, and then identify critical process parameters (CPPs) to produce the drug product in good quality consistently. (Bhattacharyya et al., 2019) In ICH Q8, Design Space is defined as “The multidimensional combination and interaction of input variables and process parameters that have been demonstrated to provide assurance of quality. Working within the design space is not considered a change. Movement out of the design space is considered to be a change and would normally initiate a regulatory post-approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval.” (ICH, 2009).
Using Design-of-Experiments (DoE) with QbD and an appropriate model with a thorough statistical analysis, one could effectively identify design space, the combination of conditions yielding optimal results. (Politis et al., 2017), (Jain et al., 2019) ICH’s Integrated Implementation Training Workshop about Implementation of Q8/Q9/Q10 emphasized that presentation of Design Space in regulatory filings should include critical and non-critical parameters. Process parameters and CQAs should be kept within critical parameter ranges, and non-critical parameter ranges also support the filing review. (ICH, 2010) In FDA’s internal policy, MAPP 5016.1, (Office of Pharmaceutical Science and Office of New Drug Quality Assessment, 2011) CMC reviewers are instructed to review submissions in terms of QTPP, CQA, product design and product understanding, process design and understanding, product and process control strategies. (Torres, 2015) The proposed QbD design space is subject to regulatory review. Regulatory agencies will not consider any formulation or manufacturing change occurring while working within the design space as a change. Significant variations occur when related to a new design space or extension of an approved one. The established design space is confirmed by performing qualification runs, implementing them in the manufacturing process, and including them in the continued process verification (CPV) program until new changes are introduced. Like CQAs, the design space must be re-evaluated when further information becomes available using the lifecycle approach. Application of QbD frameworks and concepts specifically for liposome formulation and process development is recently reviewed (Wang and Grainger, 2019).
Preclinical Studies. Both FDA and EMA state that a traditional way of measuring total drug concentration in plasma may not reflect liposomal drug therapeutic performance at targeted organs for pharmacokinetics (PK) studies. The PK studies should quantify encapsulated and unencapsulated drug release in tissue, which affects toxicity and efficacy. Before starting nonclinical PK studies, reliable analytical methods for quantifying total, encapsulated and free drug amounts in plasma and tissue samples should be carefully established and validated for regulatory review. EU guidance indicates that liposomes can fracture during tissue processing, resulting in analytical error. (European Medicines Agency, 2013a) Attention to validating analytical methods development for preclinical PK studies is therefore critical.
In the Biopharmaceutics section regarding drug release characteristics, the FDA recommends conducting comparative studies between the liposome drug formulation and non-liposomal approved product with the same active ingredient. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2018) The FDA draft guidance emphasizes understanding of in vivo release mechanisms based on material physicochemical properties as one risk factor for assessment during product development. (Center for Drug Evaluation and Research and U.S. Food and Drug Administration, 2017) Both agencies suggest that the applicant consult their review division for product-specific advice on study design.
Conclusions and Expert Opinions
Given rapid increases in liposomal drug products, applications and markets, product competition has escalated. Establishing clear guidance by regulatory agencies is critical for expediently guiding technological advances and informing drug product complexity. More extensive regulatory agency efforts are required to consistently guide rapid liposomal drug product development. Policies that better predict technologies, products, therapies, and risks, and match regulatory requirements and compliance to producers’ needs will improve efficiency prospect for success, de-risk translation, and foster innovation. Knowing regulatory agencies’ requirements for liposomal drug products is critical for efficient liposomal R&D. Drug development companies should communicate with regulatory agencies early in development phases to direct regulatory review to specific product innovations, and gain feedback and input on proper development strategies. Liposome companies should better invest in educating their research, operations, and marketing personnel in regulatory requirements. Bridging research scientists, manufacturing engineers, regulatory personnel, and new product marketing with a common regulatory strategy will improve the efficiency of liposome-based drug product development to address expanding markets.
Author Contributions
DWG and YW contributed to manuscript writing, revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andra, V. V. S. N. L., Pammi, S. V. N., Bhatraju, L. V. K. P., and Ruddaraju, L. K. (2022). A Comprehensive Review on Novel Liposomal Methodologies, Commercial Formulations, Clinical Trials and Patents. BioNanoSci. 12, 274–291. doi:10.1007/s12668-022-00941-x
Barenholz, Y. (2012). Doxil - the First FDA-Approved Nano-Drug: Lessons Learned. J. Controlled Release 160 (2), 117–134. doi:10.1016/j.jconrel.2012.03.020
Bhattacharyya, S., Adhikari, H., and Regmi, D. (2019). A Brief Review on QbD Approach on Liposome and the Requirements for Regulatory Approval. Rese. Jour. Pharm. Technol. 12 (8), 4057. doi:10.5958/0974-360X.2019.00699.1
Bulbake, U., Doppalapudi, S., Kommineni, N., and Khan, W. (2017). Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 9, 12. doi:10.3390/pharmaceutics9020012
CDC (2022). National Center for Chronic Disease Prevention and Health Promotion (NCCDPHP). Chronic Diseases in America. Available at: https://www.cdc.gov/chronicdisease/resources/infographic/chronic-diseases.htm (Accessed February 25, 2022).
Center for Drug Evaluation and ResearchU.S. Food and Drug Administration (2014). Draft Guidance on Doxorubicin Hydrochloride. Beltsville, MD: U.S. Food and Drug Administration.
Center for Drug Evaluation and ResearchU.S. Food and Drug Administration (2017). Drug Products, Including Biological Products, that Contain Nanomaterials.
Center for Drug Evaluation and ResearchU.S. Food and Drug Administration (2018). Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability, and Labeling Documentation.
Colombo, S., Beck-Broichsitter, M., Bøtker, J. P., Malmsten, M., Rantanen, J., and Bohr, A. (2018). Transforming Nanomedicine Manufacturing toward Quality by Design and Microfluidics. Adv. Drug Deliv. Rev. 128, 115–131. doi:10.1016/j.addr.2018.04.004
Committee for Medicinal Products for Human Use (2011). CHMP Assessment Report Doxorubicin SUN. Eur. Medicines Agency.
Committee for Medicinal Products for Human Use (Chmp) (2013). Surface Coatings: General Issues for Consideration Regarding Parenteral Administration of Coated Nanomedicine Products.
Crommelin, D. J. A., and de Vlieger, J. S. B. (2015). Non-Biological Complex Drugs. doi:10.1007/978-3-319-16241-6
Crommelin, D. J. A., Shah, V. P., Klebovich, I., McNeil, S. E., Weinstein, V., Flühmann, B., et al. (2015). The Similarity Question for Biologicals and Non-biological Complex Drugs. Eur. J. Pharm. Sci. 76 (76), 10–17. doi:10.1016/j.ejps.2015.04.010
Data Bridge (2021). Market Research. Liposomal Drugs Market – Global Industry Trends and Forecast to 2028 | Data Bridge Market Research. Available at: https://www.databridgemarketresearch.com/reports/global-liposomal-drugs-market (Accessed February 25, 2022).
Deodhar, S., and Dash, A. K. (2018). Long Circulating Liposomes: Challenges and Opportunities. Ther. Deliv. 9 (12), 857–872. doi:10.4155/tde-2018-0035
European Commission’s Joint Research Centre (Jrc) and the Global Coalition for Regulatory Science Research (Gcrsr) (2019). Global Summit on Regulatory Science 2019 Nanotechnology and Nanoplastics.
European Medicines Agency (2013). Data Requirements for Intravenous Liposomal Products Developed with Reference to an Innovator Liposomal Product. Availale at: https://www.ema.europa.eu/en/data-requirements-intravenous-liposomal-products-developed-reference-innovator-liposomal-product-0 (Accessed February 15, 2022).
European Medicines Agency (2013). European Medicines Agency-Food and Drug Administration Pilot Programme for Parallel Assessment of Quality-By-Design Applications: Lessons Learnt and Questions and Answers Resulting from the First Parallel Assessment. Available at: https://www.ema.europa.eu/documents/other/european-medicines-agency-food-drug-administration-pilot-programme-parallel-assessment-quality_en.pdf (Accessed February 25, 2022).
European Medicines AgencyU.S. Food and Drug Administration (2013). EMA-FDA Pilot Program for Parallel Assessment of Quality-By-Design Applications: Lessons Learnt and Q&A Resulting from the First Parallel Assessment.
Filipczak, N., Pan, J., Yalamarty, S. S. K., and Torchilin, V. P. (2020). Recent Advancements in Liposome Technology. Adv. Drug Deliv. Rev. 156, 4–22. doi:10.1016/j.addr.2020.06.022
Guimarães, D., Cavaco-Paulo, A., and Nogueira, E. (2021). Design of Liposomes as Drug Delivery System for Therapeutic Applications. Int. J. Pharmaceutics 601, 120571. doi:10.1016/j.ijpharm.2021.120571
ICH (2010). ICH Quality Implementation Working Group - Integrated Implementation Training Workshop. Workshop A: Des. Space. Switzerland: ICH.
ICH (2009). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Pharmaceutical Development Q8(R2).
International Pharmaceutical Regulators Programme (IPRP) (2022). International Pharmaceutical Regulators Programme (IPRP). Available at: http://www.iprp.global/working-group/nanomedicines (Accessed March 6, 2022).
Jain, A., Hurkat, P., and Jain, S. K. (2019). Development of Liposomes Using Formulation by Design: Basics to Recent Advances. Chem. Phys. Lipids 224, 104764. doi:10.1016/j.chemphyslip.2019.03.017
Lionberger, R. A. (2019). Innovation for Generic Drugs: Science and Research under the Generic Drug User Fee Amendments of 2012. Clin. Pharmacol. Ther. 105 (4), 878–885. doi:10.1002/cpt.1364
Politis, S., Colombo, P., Colombo, G., and M. Rekkas, D. (2017). Design of Experiments (DoE) in Pharmaceutical Development. Drug Dev. Ind. Pharm. 43 (6), 889–901. doi:10.1080/03639045.2017.1291672
Office of Generic Drugs (2020). Office of Generic Drugs, 2019 Annual Report, Ensuring Access to Safe, Affordable, and Effective Generic Drugs. U.S. Food Drug Adm.
Office of Pharmaceutical Science and Office of New Drug Quality Assessment (2011). Manual of Policies and Procedures (MAPP 5016.1): Applying ICH Q8 (R2), Q9, and Q10 Principles to CMC Review FDA.
Ren, D., Cao, W., Liu, X., Han, Q., Fan, W., Li, G., et al. (2021). Case Report: Use of Liposomal Amphotericin B in Low Doses in Patients with Visceral Leishmaniasis. Front. Med. 8, 766400. doi:10.3389/fmed.2021.766400
Salehi, B., Mishra, A. P., Nigam, M., Kobarfard, F., Javed, Z., Rajabi, S., et al. (2020). Multivesicular Liposome (Depofoam) in Human Diseases. Iran J. Pharm. Res. 19 (2), 9–21. doi:10.22037/ijpr.2020.112291.13663
Shen, J., and Burgess, D. J. (2015). In Vitro-In Vivo Correlation for Complex Non-oral Drug Products: Where Do We Stand? J. Controlled Release 219, 644–651. doi:10.1016/j.jconrel.2015.09.052
Solomon, D., Gupta, N., Mulla, N. S., Shukla, S., Guerrero, Y. A., and Gupta, V. (2017). Role of In Vitro Release Methods in Liposomal Formulation Development: Challenges and Regulatory Perspective. AAPS J. 19 (6), 1669–1681. doi:10.1208/s12248-017-0142-0
The Insight Partners (2021). Liposome Drug Delivery Market Size Worth $6,992.95 Million, Globally, by 2027 at 8.8% CAGR - Exclusive Report by the Insight Partners. Available at: https://finance.yahoo.com/news/liposome-drug-delivery-market-size-134300312.html (Accessed February 25, 2022).
Torres, M. (2015). Challenges in Implementing Quality by Design - an Industry PerspectiveBioProcess International. Available at: https://bioprocessintl.com/analytical/downstream-development/challenges-in-implementing-quality-by-design-an-industry-perspective/(Accessed February 25, 2022).
United States Government Accountability Office (2017). U.S. Government Accountability Office (GAO). Report to Congressional Requesters on Generic Drugs — FDA Should Make Public its Plans to Issue and Revise Guidance on Nonbiological Complex Drugs.
Urits, I., Swanson, D., Swett, M. C., Patel, A., Berardino, K., Amgalan, A., et al. (2020). A Review of Patisiran (ONPATTRO) for the Treatment of Polyneuropathy in People with Hereditary Transthyretin Amyloidosis. Neurol. Ther. 9 (2), 301–315. doi:10.1007/s40120-020-00208-1
U.S. Food and Drug Administration (2017). Generic Drug User Fee Amendments of 2017, Title Iii, Fda Reauthorization Act of 2017, Pub. L. 115-52, §§ 301-307, 131 Stat, 1020–1028.
U.S. Food and Drug Administration (2011). HHS Guidance for Industry Process Validation: General Principles and Practices (2011).
U.S. Food and Drug Administration (2002). Liposome Drug Products Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation. FDA Draft Guidance.
U.S. Food and Drug Administration (2022). Upcoming Product-specific Guidances for Complex Generic Drug Product Development | FDA. Available at: https://www.fda.gov/drugs/guidances-drugs/upcoming-product-specific-guidances-complex-generic-drug-product-development (Accessed March 4, 2022).
Wang, Y., and Grainger, D. W. (2019). Lyophilized Liposome-Based Parenteral Drug Development: Reviewing Complex Product Design Strategies and Current Regulatory Environments. Adv. Drug Deliv. Rev. 151-152, 56–71. doi:10.1016/j.addr.2019.03.003
Keywords: FDA, EMA, regulatory policy, NBCD, drug development, manufacture process, quality, QbD
Citation: Wang Y and Grainger DW (2022) Regulatory Considerations Specific to Liposome Drug Development as Complex Drug Products. Front. Drug. Deliv. 2:901281. doi: 10.3389/fddev.2022.901281
Received: 21 March 2022; Accepted: 04 April 2022;
Published: 28 April 2022.
Edited by:
Heather Sheardown, McMaster University, CanadaReviewed by:
Zhongjian Chen, Shanghai Skin Disease Hospital, ChinaCopyright © 2022 Wang and Grainger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David W. Grainger, ZGF2aWQuZ3JhaW5nZXJAdXRhaC5lZHU=