
- Division of Pharmacotherapeutics, Keio University Faculty of Pharmacy, Tokyo, Japan
MicroRNAs (miRNAs) are small non-coding RNAs which function as endogenous silencers of various target genes. miRNAs are expressed in a tissue-specific manner and playing important roles in cell proliferation, apoptosis, and differentiation during mammalian development. Links between miRNAs and the initiation and progression of human diseases including cancer are becoming increasingly apparent. Recent studies have revealed that some miRNAs such as miR-9, miR-29 family, and miR-34 family are differentially expressed in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. These miRNAs are also reported to act as tumor suppressors during human carcinogenesis. In this review, we discuss about miRNAs which are important in the molecular pathogenesis of both cancer and neurodegeneration. Cancer and neurodegenerative disorder may be influenced by common miRNA pathways that regulate differentiation, proliferation, and death of cells.
The Biogenesis of miRNA
MicroRNAs (miRNAs) are ~22 nucleotide (nt) non-coding RNAs that can post-transcriptionally downregulate the expression of various target genes. Currently, ~1500 human miRNAs have been identified in the human genome, each of which potentially controls hundreds of target genes. As shown in Figure 1, miRNA genes are generally transcribed from transcription start sites (TSSs) by RNA polymerase II (pol II) to form primary transcripts (pri-miRNAs). Pol II-transcribed pri-miRNAs are capped with 7-methylguanosine and are polyadenylated. The nuclear RNase III enzyme Drosha and its co-factor DGCR8 process pri-miRNAs into ~60-nt precursor miRNAs (pre-miRNAs), which form an imperfect stem-loop structure. Pre-miRNAs are transported into the cytoplasm by exportin 5 and are subsequently cleaved by Dicer into mature miRNAs, which are then loaded into the RNA-induced silencing complex (RISC). The miRNA/RISC complex downregulates specific gene products by translational repression via binding to partially complementary sequences in the 3′-untranslated regions (UTRs) of the target mRNAs or by directing mRNA degradation via binding to perfectly complementary sequences (He and Hannon, 2004). One strand of the miRNA duplex, which is derived from pre-miRNA, remains stably associated with RISC and guides it mainly, but not exclusively, to the 3′-UTR of the target mRNAs. Targeting can also occur in the 5′-UTR of the target mRNAs (Lewis et al., 2005; Lytle et al., 2007; Qin et al., 2010). In addition, a recent study has revealed that miRNAs are not only able to downregulate specific gene products but also can activate the expression of a target gene (Vasudevan et al., 2007). miRNAs are expressed in a tissue-specific manner and play important roles in cell proliferation, apoptosis, and differentiation during mammalian development. Links between miRNAs and the development of human malignancies are becoming increasingly apparent (Calin and Croce, 2006,2007; Saito et al., 2009a).
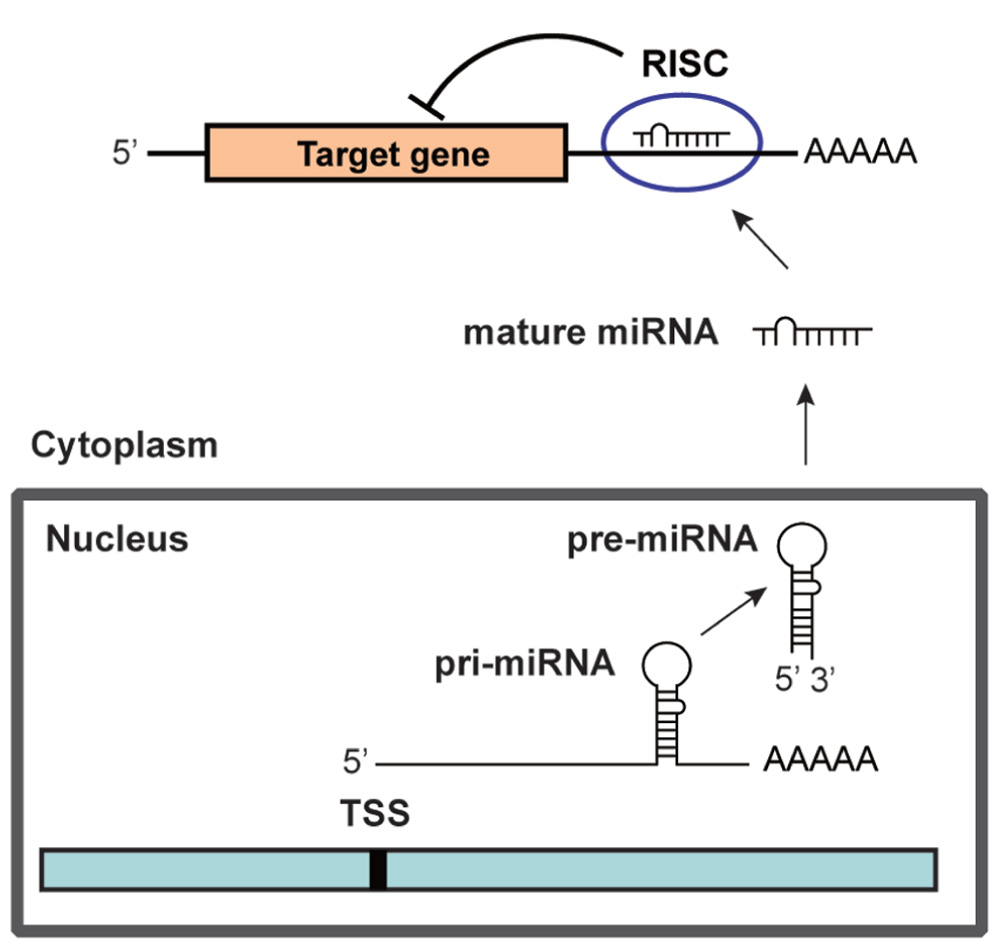
FIGURE 1. The biogenesis of miRNA.
miRNA as Tumor Suppressors and Oncogenes
The finding that predicted targets of miRNAs are enriched for genes involved in transcriptional regulation, cell proliferation, and apoptosis implies that aberrant expression of miRNAs might contribute to the initiation and progression of human cancer. Onco-miRNAs and suppressor-miRNAs can represent two different looks of the same gene, behaving as oncogenes or tumor suppressors depending on tissue type and specific targets (Fabbri et al., 2007b). Calin et al. (2004) examined the mapping of 186 human miRNAs and compared their locations to genomic regions involved in cancers. They found that miRNA genes are frequently located at fragile sites which include loss of heterozygosity (LOH), amplification, and breakpoints, indicating that these miRNAs may be dysregulated in human cancers. Some miRNAs are downregulated in various human cancers, which suggests that they may function as tumor suppressors. let-7 is downregulated in lung cancer and targets a critical oncogene RAS (Johnson et al., 2005). miR-15 and miR-16 are downregulated in chronic lymphocytic leukemia and target an antiapoptotic gene BCL2 (Cimmino et al., 2005). miR-143 and miR-145 are downregulated in colorectal and breast cancer (Michael et al., 2003; Iorio et al., 2005).
On the other hand, some miRNA genes are overexpressed in cancers, indicating that they may have roles as oncogenes and accelerate the development of cancer. miR-155 and its host gene BIC are highly expressed in several types of B cell lymphoma (Eis et al., 2005). High expression of precursor miR-155 is reported in children with Burkitt lymphoma (Metzler et al., 2004). Moreover, recent studies have shown that miR-155 is overexpressed in several types of human solid tumors including breast, colon, and lung cancer (Iorio et al., 2005; Volinia et al., 2006; Yanaihara et al., 2006). These studies strongly suggest that miR-155 is an oncogenic miRNA. The miR-17-92 cluster, which is located on chromosome 13q31, is activated by the oncogene c-Myc which is an important regulator of cell growth and is often mutated or amplified in human cancers. The miR-17-92 cluster is highly expressed in breast, colon, lung, pancreatic, and prostate cancer, as well as in B cell lymphoma (Hayashita et al., 2005; He et al., 2005; Volinia et al., 2006). c-Myc binding upregulates the transcription factor E2F1 and two miRNAs in the miR-17-92 cluster, miR-17-5p and miR-20a, target E2F1 (O’Donnell et al., 2005). This suggests that there is a negative feedback loop involving c-Myc, E2F1, miR-17-5p, and miR-20a whereby c-Myc induces expression of E2F1 and the post-transcriptional repressors of E2F1, miR-17-5p, and miR-20a. Volinia et al. (2006) showed that miR-20a also targets the tumor suppressor transforming growth factor beta receptor 2 (TGFBR2). Chan et al. (2005) showed that miR-21 is strongly overexpressed in the highly malignant brain tumor glioblastoma. They also revealed that knockdown of miR-21 in cultured glioblastoma cells induces activation of caspases and leads to increased apoptotic cell death, suggesting that miR-21 is an antiapoptotic factor in human glioblastoma cells. miR-21 is upregulated in breast, colon, lung, pancreatic, prostate, and stomach cancer (Iorio et al., 2005; Volinia et al., 2006). Moreover, miR-21 is highly overexpressed in human cholangiocarcinoma and modulates gemcitabine-induced apoptosis by directly altering PTEN expression (Meng et al., 2007). Taken together, these findings indicate that miRNAs have critical roles in the mechanism of human carcinogenesis and that aberrant expression of miRNAs may contribute to the development of human cancer.
miRNA in Cancers and Neurodegenerative Disorders
Cancer and neurodegenerative disease may be influenced by common signaling pathways regulating the balance of cell survival and death. Recent studies suggest that the molecular machinery involved in maintaining neural function in neurodegenerative disease may be shared with oncogenic pathways. p53 is one of the most extensively studied proteins because of its role in cancer prevention, and has been recently shown to be involved in aging and Alzheimer’s disease (AD). The observation that aging and AD interfere with proteins controlling duplication and cell cycle may lead to the speculation that, in senescent neurons, aberrations in proteins generally dealing with cell cycle control and apoptosis could affect neuronal plasticity and functioning rather than cell duplication (Lanni et al., 2012).
Recent studies have revealed that some miRNAs are aberrantly expressed in the brains of patients with neurodegenerative diseases such as AD, Parkinson’s disease (PD) and Huntington’s disease (HD; Junn and Mouradian, 2012). This suggests that miRNAs play critical roles in neurodegeneration as well as cancer. Specific miRNAs may regulate the expression of their target proteins that are involved in the development of neurodegenerative diseases. Interestingly, several miRNAs differentially expressed in neurodegenerative diseases such as miR-9, miR-29 family, and miR-34 family are considered to be potential tumor suppressor miRNAs (Table 1). Cancer and neurodegenerative disorder may be influenced by common miRNA pathways that regulate differentiation, proliferation and death of cells.
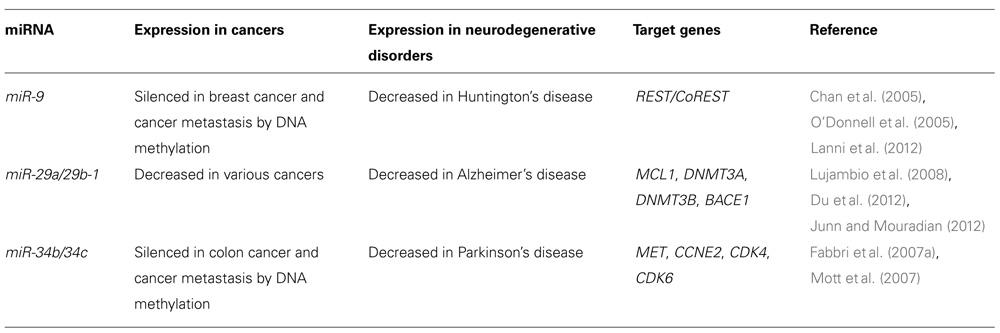
TABLE 1. miRNAs differentially expressed in cancers and neurodegenerative disorders.
miR-9
Aberrant DNA methylation at CpG island promoters of tumor suppressor genes is one of the most important mechanisms of human carcinogenesis. Recent studies have shown that miR-9 is silenced by aberrant CpG island methylation in various human cancers including breast cancer, cancer metastasis, gastric cancers, and lung cancers, suggesting that miR-9 is a potential tumor suppressor miRNA (Lehmann et al., 2008; Lujambio et al., 2008; Du et al., 2012; Heller et al., 2012).
Huntington’s disease is an autosomal dominant neurodegenerative disease caused by CAG trinucleotide repeat expansion in huntingtin, which encodes Huntingtin (Htt). Patients with HD experience abnormal motor movements, cognitive decline, and psychiatric disturbances that frequently result in premature death. Although Htt is ubiquitously expressed, patients with HD show predominantly CNS manifestations. One putative mechanism underlying the transcriptional changes is aberrant cellular distribution of the transcriptional repressor RE1-silencing transcription factor (REST). REST expression is highest in pluripotent stem cells and decreases upon restriction to neural progenitor cells and subsequently to neurons. REST silences neuronal gene expression in non-neuronal cells. In mature, healthy neurons, REST is expressed at low levels and primarily sequestered in the cytoplasm in part through interaction with Htt. However, in patients with HD, mutant Htt fails to bind REST, enabling its nuclear translocation. In the nucleus, REST can bind RE1 consensus sequences and recruit corepressors including mSin3, REST corepressor 1 (CoREST), and methyl CpG binding protein 2 (MeCP2) to inactivate neuron-specific genes (Zuccato et al., 2003).
Packer et al. (2008) have reported that levels of several miRNAs with upstream RE1 sites are decreased in HD patient cortices relative to healthy controls. One of these, miR-9 and miR-9*, which decreased early in HD, are processed from the same primary transcript from three genomic loci (miR-9-1, miR-9-2, and miR-9-3). miR-9-1 and miR-9-3 both have upstream RE1 sequences that can be occupied by REST. Interestingly, miR-9/miR-9*, targets two components of the REST complex: miR-9 targets REST and miR-9* targets CoREST. These data provide evidence for a double negative feedback loop between the REST silencing complex and miR-9/miR-9* (Packer et al., 2008).
miR-29 Family
A recent study has revealed that miR-29 family (miR-29a, miR-29b, and miR-29c) target the de novo DNA methyltransferases DNMT3A and DNMT3B and expression levels of miR-29 family were suppressed in lung cancer. The reduced expression of the miR-29 family induced overexpression of DNMT3A and DNMT3B, resulting in aberrant DNA methylation in lung cancer (Fabbri et al., 2007a). In addition, Mott et al. (2007) have demonstrated that MCL1, encoding an antiapoptotic BCL2 family protein, is one of the targets of miR-29 family, and that miR-29 miRNAs regulate apoptosis by targeting MCL1. These findings suggest that miR-29 family act as tumor suppressors by targeting DNMT3A, DNMT3B, and MCL1.
Mutations in the APP and PSEN genes cause amyloid β (Aβ) accumulation and familial AD. However, little is known about the mechanisms that contribute to Aβ accumulation in the vast majority of sporadic AD cases. BACE1/β-secretase cleavage of APP is the rate-limiting step for Aβ peptide production. Increased BACE1 expression is observed in patients with sporadic AD, and several mechanisms for this upregulation have been proposed. A link between BACE1 levels, Aβ load, and AD pathology has been reported, suggesting that increased BACE1 expression is indeed an important risk factor for sporadic AD (Li et al., 2004).
Hebert et al. (2008) investigated changes in miRNA expression profiles of sporadic AD patients and found that several miRNAs potentially involved in the regulation of APP and BACE1 expression appeared to be decreased in their brain. They have shown that miR-29a, miR-29b-1, and miR-9 can regulate BACE1 expression as their targets. The miR-29a/b-1 cluster was significantly decreased in AD patients displaying overexpression of BACE1 protein. Similar correlations between expression of this cluster and BACE1 were found during brain development and in primary neuronal cultures. They provided evidence for a potential causal relationship between miR-29a/b-1 expression and Aβ generation in a cell culture model and proposed that loss of specific miRNAs can contribute to increased BACE1 and Aβ levels in sporadic AD (Hebert et al., 2008). A recent study has shown that miR-29c also regulates BACE1 protein expression (Zong et al., 2011). These findings suggest that miR-29 family regulate BACE1 expression and play important roles in the pathogenesis of AD.
miR-34 b/34c
miR-34a was identified as a target of p53 and induces a G (He and Hannon, 2004) cell cycle arrest, senescence, and apoptosis (He et al., 2007; Tazawa et al., 2007). miR-34a expression is silenced in several types of cancer including pancreatic cancer due to aberrant CpG methylation of its promoter. Re-expression of miR-34a in a pancreatic cancer cell line induced senescence and cell cycle arrest by targeting CDK6, indicating that miR-34a represents a tumor suppressor gene which is inactivated by CpG methylation in pancreatic cancer (Lodygin et al., 2008). miR-34b and miR-34c are also reported to be direct targets of p53 and silenced by aberrant CpG island methylation in colorectal cancer (Toyota et al., 2008).
Parkinson’s disease is the second most common neurodegenerative disorder, characterized by the presence of protein inclusions or Lewy bodies and a progressive loss of dopaminergic neurons in the midbrain. Minones-Moyano et al. (2011) have evaluated miRNA expression deregulation in PD brain samples. miRNA expression profiles revealed decreased expression of miR-34b and miR-34c in brain areas with variable neuropathological affectation at clinical stages of PD. Downregulation of miR-34b/c was detected in pre-motor stages of PD. Depletion of miR-34b or miR-34c in differentiated SH-SY5Y dopaminergic neuronal cells resulted in a moderate reduction in cell viability that was accompanied by altered mitochondrial function and dynamics, oxidative stress and reduction in total cellular adenosine triphosphate content. Moreover, they have shown that DJ1 and Parkin are indirect targets of miR-34b/c. miR-34b/c downregulation induced a decrease in the expression of DJ1 and Parkin, two proteins associated to familial forms of PD. DJ1 and Parkin expression was reduced in PD brain samples displaying strong miR-34b/c downregulation. These data suggest that early deregulation of miR-34b/c, which are direct targets of p53, in PD triggers downstream transcriptome alterations underlying mitochondrial dysfunction and oxidative stress, which ultimately compromise cell viability (Minones-Moyano et al., 2011). As mentioned above, p53 is also involved in the pathogenesis of AD. These findings indicate that p53 may play important roles in the initiation and progression of both AD and PD via different miRNA-mediated mechanisms.
miRNA-Mediated Therapy for Cancer and Neurodegenerative Disease
The distinct connection between aberrant expression of miRNAs and human diseases suggests that miRNAs could be therapeutic targets. Epigenetic changes such as DNA methylation and histone modification modulate chromatin structure and gene expression in mammalian development and in human diseases (Egger et al., 2004). Many miRNAs are expressed in a tissue- and tumor-specific manner, implying that some miRNAs are subject to epigenetic control. We have recently shown that ~5% of human miRNAs are upregulated more than threefold by treatment of T24 bladder cancer cells with the DNA demethylating agent 5-aza-2′-deoxycytidine (5-Aza-CdR) and the histone deacetylase (HDAC) inhibitor 4-phenylbutyric acid (PBA). In particular, miR-127, which is embedded in a CpG island, is remarkably induced by a decrease in DNA methylation levels and an increase in active histone marks around the promoter region of the miR-127 gene. In addition, activation of miR-127 by epigenetic treatment induced downregulation of its target oncogene BCL6 (Saito et al., 2006). We have also demonstrated that treatment of gastric cancer cells with 5-Aza-CdR and PBA induces activation of miR-512-5p which is located at Alu repeats on chromosome 19. Activation of miR-512-5p by epigenetic treatment induces suppression of MCL1, resulting in apoptosis of gastric cancer cells (Saito et al., 2009b). These results indicate that chromatin remodeling by epigenetic treatment can directly modulate expression of miRNAs that are involved in the pathogenesis of cancer and neurodegenerative disease. In addition, recent study has shown that locked-nucleic-acid-modified oligonucleotide (LNA-antimiR) effectively antagonizes specific miRNAs and could be used to silence miRNAs that are overexpressed in human diseases (Elmen et al., 2008). Because the link between miRNAs and cancer and neurodegeneration has only just begun to be understood, there could be a large number of critical miRNAs and their target genes. Further studies are necessary to identify the specific miRNAs that could be therapeutic targets and/or molecular markers for human cancers and neurodegenerative disorders.
Conflict of Interest Statement:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Grant-in-Aid for Young Scientists A (23680090 to Yoshimasa Saito) and Grant-in-Aid for Scientific Research C (24590993 to Hidetsugu Saito) from Japan Society for Promotion of Science (JSPS), Takeda Science Foundation (to Yoshimasa Saito), Sagawa Foundation for Promotion of Cancer Research (to Yoshimasa Saito), and Inaida Foundation (to Hidetsugu Saito).
References
Calin, G. A., and Croce, C. M. (2006). MicroRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866.
Calin, G. A., and Croce, C. M. (2007). Chromosomal rearrangements and microRNAs: a new cancer link with clinical implications. J. Clin. Invest. 117, 2059–2066.
Calin, G. A., Sevignani, C., Dumitru, C. D., Hyslop, T., Noch, E., Yendamuri, S., Shimizu, M., Rattan, S., Bullrich, F., Negrini, M., and Croce, C. M. (2004). Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. U.S.A. 101, 2999–3004.
Chan, J. A., Krichevsky, A. M., and Kosik, K. S. (2005). MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 65, 6029–6033.
Cimmino, A., Calin, G. A., Fabbri, M., Iorio, M. V., Ferracin, M., Shimizu, M., Wojcik, S. E., Aqeilan, R. I., Zupo, S., Dono, M., Rassenti, L., Alder, H., Volinia, S., Liu, C. G., Kipps, T. J., Negrini, M., and Croce, C. M. (2005). miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. U.S.A. 102, 13944–13949.
Du, Y., Liu, Z., Gu, L., Zhou, J., Zhu, B. D., Ji, J., and Deng, D. (2012). Characterization of human gastric carcinomarelated methylation of 9 miR CpG islands and repression of their expressions in vitro and in vivo. BMC Cancer 12, 249. doi: 10.1186/1471-2407-12-249
Egger, G., Liang, G., Aparicio, A., and Jones, P. A. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463.
Eis, P. S., Tam, W., Sun, L., Chadburn, A., Li, Z., Gomez, M. F., Lund, E., and Dahlberg, J. E. (2005). Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. U.S.A. 102, 3627–3632.
Elmen, J., Lindow, M., Schutz, S., Lawrence, M., Petri, A., Obad, S., Lindholm, M., Hedtjarn, M., Hansen, H. F., Berger, U., Gullans, S., Kearney, P., Sarnow, P., Straarup, E. M., and Kauppinen, S. (2008). LNA-mediated microRNA silencing in non-human primates. Nature 452, 896–899.
Fabbri, M., Garzon, R., Cimmino, A., Liu, Z., Zanesi, N., Callegari, E., Liu, S., Alder, H., Costinean, S., Fernandez-Cymering, C., Volinia, S., Guler, G., Morrison, C. D., Chan, K. K., Marcucci, G., Calin, G. A., Huebner, K., and Croce, C. M. (2007a). MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. U.S.A. 104, 15805–15810.
Fabbri, M., Ivan, M., Cimmino, A., Negrini, M., and Calin, G. A. (2007b). Regulatory mechanisms of microRNAs involvement in cancer. Expert Opin. Biol. Ther. 7, 1009–1019.
Hayashita, Y., Osada, H., Tatematsu, Y., Yamada, H., Yanagisawa, K., Tomida, S., Yatabe, Y., Kawahara, K., Sekido, Y., and Takahashi, T. (2005). A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 65, 9628–9632.
He, L., and Hannon, G. J. (2004). MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531.
He, L., He, X., Lim, L. P., de Stanchina, E., Xuan, Z., Liang, Y., Xue, W., Zender, L., Magnus, J., Ridzon, D., Jackson, A. L., Linsley, P. S., Chen, C., Lowe, S. W., Cleary, M. A., and Hannon, G. J. (2007). A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134.
He, L., Thomson, J. M., Hemann, M. T., Hernando-Monge, E., Mu, D., Goodson, S., Powers, S., Cordon-Cardo, C., Lowe, S. W., Hannon, G. J., and Hammond, S. M. (2005). A microRNA polycistron as a potential human oncogene. Nature 435, 828–833.
Hebert, S. S., Horre, K., Nicolai, L., Papadopoulou, A. S., Mandemakers, W., Silahtaroglu, A. N., Kauppinen, S., Delacourte, A., and De Strooper, B. (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. U.S.A. 105, 6415–6420.
Heller, G., Weinzierl, M., Noll, C., Babinsky, V., Ziegler, B., Altenberger, C., Minichsdorfer, C., Lang, G., Dome, B., End-Pfutzenreuter, A., Arns, B. M., Grin, Y., Klepetko, W., Zielinski, C. C., and Zochbauer-Muller, S. (2012). Genome-wide miRNA expression profiling identifies miR-9-3 and miR-193a as targets for DNA methylation in non-small cell lung cancers. Clin. Cancer Res. 18, 1619–1629.
Iorio, M. V., Ferracin, M., Liu, C. G., Veronese, A., Spizzo, R., Sabbioni, S., Magri, E., Pedriali, M., Fabbri, M., Campiglio, M., Menard, S., Palazzo, J. P., Rosenberg, A., Musiani, P., Volinia, S., Nenci, I., Calin, G. A., Querzoli, P., Negrini, M., and Croce, C. M. (2005). MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 65, 7065–7070.
Johnson, S. M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K. L., Brown, D., and Slack, F. J. (2005). RAS is regulated by the let-7 microRNA family. Cell 120, 635–647.
Junn, E., and Mouradian, M. M. (2012). MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 133, 142–150.
Lanni, C., Racchi, M., Memo, M., Govoni, S., and Uberti, D. (2012). p53 at the crossroads between cancer and neurodegeneration. Free Radic. Biol. Med. 52, 1727–1733.
Lehmann, U., Hasemeier, B., Christgen, M., Muller, M., Romermann, D., Langer, F., and Kreipe, H. (2008). Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J. Pathol. 214, 17–24.
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20.
Li, R., Lindholm, K., Yang, L. B., Yue, X., Citron, M., Yan, R., Beach, T., Sue, L., Sabbagh, M., Cai, H., Wong, P., Price, D., and Shen, Y. (2004). Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. U.S.A. 101, 3632–3637.
Lodygin, D., Tarasov, V., Epanchintsev, A., Berking, C., Knyazeva, T., Korner, H., Knyazev, P., Diebold, J., and Hermeking, H. (2008). Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 7, 2591–2600.
Lujambio, A., Calin, G. A., Villanueva, A., Ropero, S., Sanchez-Cespedes, M., Blanco, D., Montuenga, L. M., Rossi, S., Nicoloso, M. S., Faller, W. J., Gallagher, W. M., Eccles, S. A., Croce, C. M., and Esteller, M. (2008). A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. U.S.A. 105, 13556–13561.
Lytle, J. R., Yario, T. A., and Steitz, J. A. (2007). Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. U.S.A. 104, 9667–9672.
Meng, F., Henson, R., Wehbe-Janek, H., Ghoshal, K., Jacob, S. T., and Patel, T. (2007). MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647–658.
Metzler, M., Wilda, M., Busch, K., Viehmann, S., and Borkhardt, A. (2004). High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer 39, 167–169.
Michael, M. Z., O’Connor, S. M., van Holst Pellekaan, N. G., Young, G. P., and James, R. J. (2003). Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol. Cancer Res. 1, 882–891.
Minones-Moyano, E., Porta, S., Escaramis, G., Rabionet, R., Iraola, S., Kagerbauer, B., Espinosa-Parrilla, Y., Ferrer, I., Estivill, X., and Marti, E. (2011). MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 20, 3067–3078.
Mott, J. L., Kobayashi, S., Bronk, S. F., and Gores, G. J. (2007). mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 26, 6133–6140.
O’Donnell, K. A., Wentzel, E. A., Zeller, K. I., Dang, C. V., and Mendell, J. T. (2005). c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843.
Packer, A. N., Xing, Y., Harper, S. Q., Jones, L., and Davidson, B. L. (2008). The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 28, 14341–14346.
Qin, W., Shi, Y., Zhao, B., Yao, C., Jin, L., Ma J., and Jin, Y. (2010). miR-24 regulates apoptosis by targeting the open reading frame (ORF) region of FAF1 in cancer cells. PLoS ONE 5, e9429. doi: 10.1371/journal.pone.0009429
Saito, Y., Liang, G., Egger, G., Friedman, J. M., Chuang, J. C., Coetzee, G. A., and Jones, P. A. (2006). Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9, 435–443.
Saito, Y., Suzuki, H., and Hibi, T. (2009a). The role of microRNAs in gastrointestinal cancers. J. Gastroenterol. 44(Suppl. 19), 18–22.
Saito, Y., Suzuki, H., Tsugawa, H., Nakagawa, I., Matsuzaki, J., Kanai, Y., and Hibi, T. (2009b). Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene 28, 2738–2744.
Tazawa, H., Tsuchiya, N., Izumiya, M., and Nakagama, H. (2007). Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 104, 15472–15477.
Toyota, M., Suzuki, H., Sasaki, Y., Maruyama, R., Imai, K., Shinomura, Y., and Tokino, T. (2008). Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 68, 4123–4132.
Vasudevan, S., Tong, Y., and Steitz, J. A. (2007). Switching from repression to activation: microRNAs can up-regulate translation. Science 318, 1931–1934.
Volinia, S., Calin, G. A., Liu, C. G., Ambs, S., Cimmino, A., Petrocca, F., Visone, R., Iorio, M., Roldo, C., Ferracin, M., Prueitt, R. L., Yanaihara, N., Lanza, G., Scarpa, A., Vecchione, A., Negrini, M., Harris, C. C., and Croce, C. M. (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 103, 2257–2261.
Yanaihara, N., Caplen, N., Bowman, E., Seike, M., Kumamoto, K., Yi, M., Stephens, R. M., Okamoto, A., Yokota, J., Tanaka, T., Calin, G. A., Liu, C. G., Croce, C. M., and Harris, C. C. (2006). Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9, 189–198.
Zong, Y., Wang, H., Dong, W., Quan, X., Zhu, H., Xu, Y., Huang, L., Ma, C., and Qin, C. (2011). miR-29c regulates BACE1 protein expression. Brain Res. 1395, 108–115.
Keywords: microRNA, cancer, neurodegenerative disorder, miR-9, miR-29a/29b-1, miR-34b/34c
Citation: Saito Y and Saito H (2012) MicroRNAs in cancers and neurodegenerative disorders. Front. Gene. 3:194. doi:10.3389/fgene.2012.00194
Received: 26 July 2012; Paper pending published: 14 August 2012;
Accepted: 11 September 2012; Published online: 26 September 2012.
Edited by:
Peng Jin, Emory University School of Medicine, USAReviewed by:
Francesca Fanini, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, ItalyJun Yasuda, The Japanese Foundation for Cancer Research – Cancer Institute, Japan
Copyright: © 2012 Saito and Saito. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Yoshimasa Saito, Division of Pharmacotherapeutics, Keio University Faculty of Pharmacy, 1-5-30 Shibakoen, Minato-ku, Tokyo 105-8582, Japan. e-mail: yoshimasa.saito@gmail.com