 Pawel Paszek
Pawel PaszekA single cell is inherently noisy. This noise is observed as a variability or heterogeneity between individual cells' responses in an isogenic population, and emerges from fundamental physical process governing state of the cell over time. In practice, states of two seemingly identical cells may be different in the same environment; and in fact the behavior of the population average my not correspond to any of the individual cells. Recent decades brought a technological breakthrough in many areas in our ability to measure and interpret cellular heterogeneity, including live-cell imaging (Spiller et al., 2010) and genome-wide epigenetic and expression analyses (in particular next generation sequencing) (Chattopadhyay et al., 2014). The emerging picture is that the cellular noise is not a nuisance, but a ubiquitous functional trait that could perhaps be therapeutically exploited. Here we discuss relevant technological advances as well as postulate the need for more quantitative and integrated temporal single cell biology approaches to study cellular heterogeneity.
Quantifying Cellular Noise
Recent technological advances in single-cell bioassays transformed our ability to measure cellular noise and heterogeneity. They vary in the number of parameters that can be simultaneously monitored, the spatio-temporal resolution, and the ability to provide quantitative understanding (Table 1).
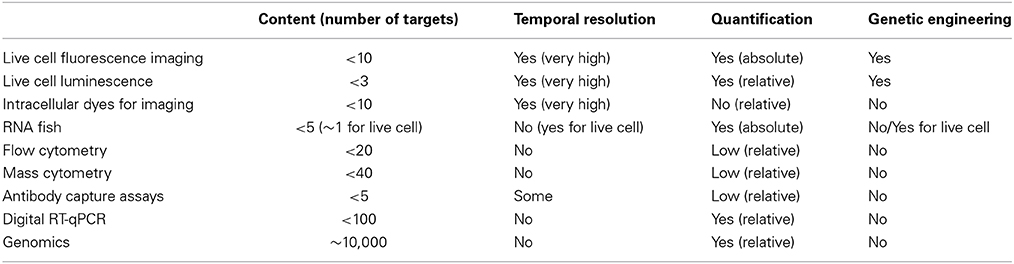
Table 1. Review of single-cell biology approaches.
“Seeing is believing” and thus the fundamental breakthrough to measure cellular noise has been the use of genetically encoded fluorescent or luminescent probes for live cell microscopy (Spiller et al., 2010). This is by far the most temporarily and spatially resolved, and quantitative approach (Locke and Elowitz, 2009), which over last few decades provided a step change in our ability to visualize heterogeneity, for example noisy gene expression (Molina et al., 2013) or all-or-none cellular sensing mechanisms (Tay et al., 2010). The key applications involve genetically engineered systems for expression of protein fusions for monitoring of cellular dynamics, or promoter-driven reporters for analysis of transcriptional responses. In fact, the combination of both, for example simultaneous monitoring of RNA (by labeling individual transcripts, see below) and protein level in real-time provides more statistical power for dynamical correlation studies of gene expression noise (Larson et al., 2013). More advanced imaging methods such as Förster Resonance Energy Transfer (FRET) and Fluorescence Recovery After Photobleaching (FRAP) also exist. They allow detailed measurements of molecular interactions and intra-cellular movement, and ultimately absolute quantifications of number of reacting molecules, for example via Fluorescence Correlation Spectroscopy (FCS) (Spiller et al., 2010). The advance of live-cell imaging have been aided with the development of tracking algorithms for analysis of time-laps movies, an essential part of the data quantification pipeline (Shen et al., 2006; Zambrano et al., 2014) as well as microfluidic systems for manipulations of individual cells on the microscope (Yin and Marshall, 2012). This together enabled automatic high-throughput analyses of hundreds of cells at a time, removing the analysis bottleneck. The imaging approach however is quite limited by the number of probes that can be simultaneously resolved and applications monitoring more that three processes are still rare (Gerlich et al., 2001). Better more spectrally resolved fluorophores are requited, although combination of fluorescent and luminescent probes allows further multi-parametrization. At the same time, engineering of model systems is not trivial. Viral (Payne, 2007) and Bacterial Artificial Chromosome (Gong et al., 2010) systems are commonly used to stably introduce transgenes into cells, with the latter providing more contextual expression, especially for reporter (promoter-dependent) systems. However, more physiological approaches such as CRISPR also emerged allowing labeling endogenous genes in their full epigenetic context (Jinek et al., 2012). Moving from cell lines into animal models proves to be even a more challenging task, simply because of the Mendellian genetics affecting transgene transmissions and thus limiting ability to multiplex different probes.
A notable part of the imaging approach has been the development of RNA fluorescence in situ hybridization (FISH) methods. They rely on targeting transcripts with fluorescently labeled oligonucleotide probes in fixed cells (Raj et al., 2008). RNA FISH allows spatial visualization and absolute quantification of individual transcripts in cells without the need for genetic engineering, and thus contextual expression can be measured. As with live-cell microscopy, physical properties of fluorescent probes only allow simultaneous measurements of limited number of probes (Singer et al., 2014). The static nature of RNA FISH measurement is it largest limitation; however a substantial effort has been made toward visualization of transcripts in living cells. A key development arrived with a MS2 system based on a RNA-binding protein (derived from bacteriophage MS2) that is capable of targeting transcripts, when tagged with a specific recognition motif (Bertrand et al., 1998). Subsequently MS2 system has been successfully transferred into an animal model with a development of transgenic mouse expressing MS2 probe for β-actin RNA (Lionnet et al., 2011). While gaining the temporal resolution, live-cell RNA FISH requires genetic engineering. Methods for labeling of endogenous transcripts in living cell also have been introduced, for example using human RNA-binding protein PUMILIO1, which recognizes the target RNA sequence rather than the secondary structure as in case of MS2 protein (Ozawa et al., 2007). However, multiplexing for live cell RNA imaging is still limited by the lack of well-characterized RNA-binding motifs, and sensitivity required for detection of relative small number of target molecules.
Toward High-Content Measurements: Targeting Proteins
The natural drive in the field has been toward multi-parameter measurements that are beyond capability of live-cell imaging. These initially stemmed from the antibody-based recognition systems, which were historically used to phenotype cellular populations. After years of development, current antibody based fluorescent-activated flow cytometry (FACS) systems now enable measurements of up to 20 parameters simultaneously (Chattopadhyay et al., 2006). This has been possible due to developments of vast array of chemically or biologically derived dyes as well as advances in laser technology and equipment (Bendall et al., 2012). An interesting approach is the combination of flow cytometry with microscopy by ImageStream cytometers, which in addition of standard antibody-based phenotyping provides additional parameters by enabling visualization of protein localization or cell morphology (Rao et al., 2012). The most recent innovation includes mass cytometry, theoretically capable of measuring up to 100 parameters (practically up to 40), which uses purified isotopes rather than fluorescent dyes to label antibodies, and therefore can be resolved by mass spectroscopy (Bjornson et al., 2013). Unfortunately, single-cell methods for traditional label-free mass spectrometry are not fully developed yet, only allowing analysis of selected, usually very highly abundant proteins or metabolites (Rubakhin et al., 2011). Most recent developments in the antibody-based approaches involve miniaturization of assays into microfluidic capture devices (Lu et al., 2013). These essentially enable measuring proteins secreted by individual cells in sophisticated micro-plate systems, and thus facilitate temporal analyses. In general, the antibody-based methods are capable of providing a quite rich single-cell data, with some degree of quantification, however there still relatively limited to much fewer than 100 simultaneous measurements.
The Single-Cell Genomic Revolution
A major step change in the field occurred when the well-established “omics” technology, in particular next generation sequencing was applied to single cells. This, over few recent years transformed our ability to measure cellular heterogeneity from monitoring few sometimes arbitrarily selected markers into unbiased genome-wide analyses (Eberwine et al., 2014). In addition to providing ability to find and phenotype “rare” cells, single-cell genomic data provided more statistical power for association studies. Of the “omics” methods, single-cell transcriptomics (scRNA-seq) is the most advanced and widely used in the field. In just few years, scRNA-seq enabled characterizing transcriptional landscapes of immune (Shalek et al., 2014), cancer (Ramskold et al., 2012) and embryonic (Islam et al., 2011) cells, among others. The method enabled high content decomposition of cellular heterogeneity within a healthy (Jaitin et al., 2014) or cancer (Patel et al., 2014) tissues, as well as demonstrated single-cell splicing events and random mono-allelic gene expression patterns (Deng et al., 2014). What follows now is the development of methods for studying the epigenome regulation led by establishment of single-cell bisulfite sequencing for measurements of DNA methylation (Smallwood et al., 2014) or nucleosome mapping (Small et al., 2014). While whole genome single-cell DNA amplifications are possible, there is still a sensitivity gap for studying DNA-protein interactions or histone modifications due to additional purification steps required (Gilfillan et al., 2012).
Single-cell genomics is still very much an emerging field and thus many questions remain about reproducibility of data and normalization standards required (Wu et al., 2014). A simple fact that a minimal amount of material can be collected from a cell (~10 pg of DNA and ~20 pg of RNA) implies the need for ample amplification before the measurement can be taken. The amplification process naturally introduces bias toward more highly abundant molecules, affecting the dynamical range of the measurement and specifically detection of low-level expressing targets. Many normalization and quality-control protocols are used to improve quantitative aspects of this analysis. The most rigorous controls involve combination of cell and molecular barcoding (so called unique molecular identifiers, UMI); with allows labeling of individual transcripts prior to amplification and thus detecting individual transcript molecules (Jaitin et al., 2014). Other methods include external RNA spike-in controls for amplification bias, as well as RNA FISH or digital RT-qPCR for transcript distribution normalizations. The latter is capable of multiplexing up to 96 transcripts in 96 single cells when combined with microfluidics, for example C1 system (Wu et al., 2014), and thus it is a single-cell method with its own merit. The depth of sequencing required for single cell analyses is also debatable. Initial studies used depths of the order of standard population level experiments (~10 mln reads), however recent studies utilizing UMI barcoding suggest that as low as 50 k reads per cell is sufficient to measure up to 10 k unique transcript molecules per cell (Jaitin et al., 2014). This allows massive parallelization with thousands of cell analyzed simultaneously and thus reducing the cost. Despite this, single-cell sequencing methods require further optimization as current estimations suggest that only a small fraction of transcripts in a cell can be harvested and measured.
Toward More Integrated Temporal Analyses
The picture emerging from the single-cell methods is that of unprecedented, previously unobservable levels of noise and heterogeneity in single cells. This noise emerges via all-on-nothing (Tay et al., 2010) or graded (Warmflash et al., 2012) transcription factor activation in fluctuating environments, the downstream complex dynamics involving pulsatile and oscillatory patterns (Paszek et al., 2010; Levine et al., 2013), the cross-talk with other intracellular signaling systems and cell-to-cell communication, which ultimately drives heterogeneous gene and protein expression patterns at the genome wide-scale (Shalek et al., 2014). However, despite the recent revolution in single-cell biology very few methods exist that is capable of integrating different measurements. These are needed for the association studies between different levels of cellular organization, and ultimately the understanding of how the noise propagates in individual cells and tissues.
However, is the static measurement going to be sufficient for studying associations? One could expect that integrated genome-wide studies would enable correlating for example epigenetic, transcriptomic, and proteomic heterogeneity. However, existing analyses already suggested that this correlation might be in fact smaller than expected (Taniguchi et al., 2010). More importantly, the ultimate task of resolving how noise and heterogeneity contributes to disease, which is often associated with heterogeneous and complex genetic traits may be even more challenging.
In the inference of dynamical systems the statistical power and causality comes from the quantitative and temporal rather than static measurements under informative perturbation. This is best exemplified by a number of theoretical analyses and inference of cellular noise (Bowsher and Swain, 2014). In the near future no one expects a temporal resolution in genomic data, therefore the breakthrough has to come from elsewhere [although in some biological systems, pseudo-temporal ordering of cells undergoing differentiation programmes can be obtained; Trapnell et al., 2014]. One approach is the integration of existing temporal approaches, e.g., live-cell imaging with genomic and proteomic end-point assays, for example using microfluidic systems or micro dissection techniques for cell isolation. However, it is the integration of different temporal approaches that perhaps can provide a step-change in the field. While simultaneous RNA and protein visualization has already been achieved (Larson et al., 2013), much richer dataset are required. One avenue to provide those temporally resolved data is integrating imaging and antibody-based (e.g., capture) assays. In addition, more precise ways to perturb cell are also required, for example such as optogenetic (Deisseroth, 2011), and nanowire approaches (Shalek et al., 2010). These will allow dissecting contributions of complex intracellular networks that eventually control the genome-wide heterogeneity.
Biological systems are inherently complex, and a noisy cell is their common denominator. The single cell biology approach is ideally suited to resolve this noise and heterogeneity with further developments of more integrated and quantitative approaches.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Pawel Paszek holds a BBSRC David Phillips Research Fellowship (BB/I017976/1). This work was also supported by the Wellcome Trust (097820/Z/11/B).
References
Bendall, S. C., Nolan, G. P., Roederer, M., and Chattopadhyay, P. K. (2012). A deep profiler's guide to cytometry. Trends Immunol. 33, 323–332. doi: 10.1016/j.it.2012.02.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertrand, E., Chartrand, P., Schaefer, M., Shenoy, S. M., Singer, R. H., and Long, R. M. (1998). Localization of ASH1 mRNA particles in living yeast. Mol. Cell 2, 437–445.
Bjornson, Z. B., Nolan, G. P., and Fantl, W. J. (2013). Single-cell mass cytometry for analysis of immune system functional states. Curr. Opin. Immunol. 25, 484–494. doi: 10.1016/j.coi.2013.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bowsher, C. G., and Swain, P. S. (2014). Environmental sensing, information transfer, and cellular decision-making. Curr. Opin. Biotechnol. 28, 149–155. doi: 10.1016/j.copbio.2014.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chattopadhyay, P. K., Gierahn, T. M., Roederer, M., and Love, J. C. (2014). Single-cell technologies for monitoring immune systems. Nat. Immunol. 15, 128–135. doi: 10.1038/ni.2796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chattopadhyay, P. K., Price, D. A., Harper, T. F., Betts, M. R., Yu, J., Gostick, E., et al. (2006). Quantum dot semiconductor nanocrystals for immunophenotyping by polychromatic flow cytometry. Nat. Med. 12, 972–977. doi: 10.1038/nm1371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deisseroth, K. (2011). Optogenetics. Nat. Methods 8, 26–29. doi: 10.1038/nmeth.f.324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, Q., Ramskold, D., Reinius, B., and Sandberg, R. (2014). Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196. doi: 10.1126/science.1245316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eberwine, J., Sul, J. Y., Bartfai, T., and Kim, J. (2014). The promise of single-cell sequencing. Nat. Methods 11, 25–27. doi: 10.1038/nmeth.2769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gerlich, D., Beaudouin, J., Gebhard, M., Ellenberg, J., and Eils, R. (2001). Four-dimensional imaging and quantitative reconstruction to analyse complex spatiotemporal processes in live cells. Nat. Cell Biol. 3, 852–855. doi: 10.1038/ncb0901-852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilfillan, G. D., Hughes, T., Sheng, Y., Hjorthaug, H. S., Straub, T., Gervin, K., et al. (2012). Limitations and possibilities of low cell number ChIP-seq. BMC Genomics 13:645. doi: 10.1186/1471-2164-13-645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gong, S., Kus, L., and Heintz, N. (2010). Rapid bacterial artificial chromosome modification for large-scale mouse transgenesis. Nat. Protoc. 5, 1678–1696. doi: 10.1038/nprot.2010.131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Islam, S., Kjallquist, U., Moliner, A., Zajac, P., Fan, J. B., Lonnerberg, P., et al. (2011). Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 21, 1160–1167. doi: 10.1101/gr.110882.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jaitin, D. A., Kenigsberg, E., Keren-Shaul, H., Elefant, N., Paul, F., Zaretsky, I., et al. (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779. doi: 10.1126/science.1247651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larson, D. R., Fritzsch, C., Sun, L., Meng, X., Lawrence, D. S., and Singer, R. H. (2013). Direct observation of frequency modulated transcription in single cells using light activation. Elife 2:e00750. doi: 10.7554/eLife.00750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levine, J. H., Lin, Y., and Elowitz, M. B. (2013). Functional roles of pulsing in genetic circuits. Science 342, 1193–1200. doi: 10.1126/science.1239999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lionnet, T., Czaplinski, K., Darzacq, X., Shav-Tal, Y., Wells, A. L., Chao, J. A., et al. (2011). A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat. Methods 8, 165–170. doi: 10.1038/nmeth.1551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Locke, J. C., and Elowitz, M. B. (2009). Using movies to analyse gene circuit dynamics in single cells. Nat. Rev. Microbiol. 7, 383–392. doi: 10.1038/nrmicro2056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, Y., Chen, J. J., Mu, L., Xue, Q., Wu, Y., Wu, P. H., et al. (2013). High-throughput secretomic analysis of single cells to assess functional cellular heterogeneity. Anal. Chem. 85, 2548–2556. doi: 10.1021/ac400082e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Molina, N., Suter, D. M., Cannavo, R., Zoller, B., Gotic, I., and Naef, F. (2013). Stimulus-induced modulation of transcriptional bursting in a single mammalian gene. Proc. Natl. Acad. Sci. U.S.A. 110, 20563–20568. doi: 10.1073/pnas.1312310110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ozawa, T., Natori, Y., Sato, M., and Umezawa, Y. (2007). Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat. Methods 4, 413–419. doi: 10.1038/nmeth1030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paszek, P., Jackson, D. A., and White, M. R. (2010). Oscillatory control of signalling molecules. Curr. Opin. Genet. Dev. 20, 670–676. doi: 10.1016/j.gde.2010.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patel, A. P., Tirosh, I., Trombetta, J. J., Shalek, A. K., Gillespie, S. M., Wakimoto, H., et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. doi: 10.1126/science.1254257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Payne, C. K. (2007). Imaging gene delivery with fluorescence microscopy. Nanomedicine (Lond.) 2, 847–860. doi: 10.2217/17435889.2.6.847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raj, A., Van Den Bogaard, P., Rifkin, S. A., Van Oudenaarden, A., and Tyagi, S. (2008). Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 5, 877–879. doi: 10.1038/nmeth.1253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramskold, D., Luo, S., Wang, Y. C., Li, R., Deng, Q., Faridani, O. R., et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782. doi: 10.1038/nbt.2282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rao, R. R., Li, Q., Gubbels Bupp, M. R., and Shrikant, P. A. (2012). Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity 36, 374–387. doi: 10.1016/j.immuni.2012.01.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rubakhin, S. S., Romanova, E. V., Nemes, P., and Sweedler, J. V. (2011). Profiling metabolites and peptides in single cells. Nat. Methods 8, S20–S29. doi: 10.1038/nmeth.1549
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shalek, A. K., Robinson, J. T., Karp, E. S., Lee, J. S., Ahn, D. R., Yoon, M. H., et al. (2010). Vertical silicon nanowires as a universal platform for delivering biomolecules into living cells. Proc. Natl. Acad. Sci. U.S.A. 107, 1870–1875. doi: 10.1073/pnas.0909350107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shalek, A. K., Satija, R., Shuga, J., Trombetta, J. J., Gennert, D., Lu, D., et al. (2014). Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369. doi: 10.1038/nature13437
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shen, H., Nelson, G., Nelson, D. E., Kennedy, S., Spiller, D. G., Griffiths, T., et al. (2006). Automated tracking of gene expression in individual cells and cell compartments. J. R. Soc. Interface 3, 787–794. doi: 10.1098/rsif.2006.0137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singer, Z. S., Yong, J., Tischler, J., Hackett, J. A., Altinok, A., Surani, M. A., et al. (2014). Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol. Cell 55, 319–331. doi: 10.1016/j.molcel.2014.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Small, E. C., Xi, L., Wang, J. P., Widom, J., and Licht, J. D. (2014). Single-cell nucleosome mapping reveals the molecular basis of gene expression heterogeneity. Proc. Natl. Acad. Sci. U.S.A. 111, E2462–E2471. doi: 10.1073/pnas.1400517111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smallwood, S. A., Lee, H. J., Angermueller, C., Krueger, F., Saadeh, H., Peat, J., et al. (2014). Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820. doi: 10.1038/nmeth.3035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spiller, D. G., Wood, C. D., Rand, D. A., and White, M. R. (2010). Measurement of single-cell dynamics. Nature 465, 736–745. doi: 10.1038/nature09232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taniguchi, Y., Choi, P. J., Li, G. W., Chen, H., Babu, M., Hearn, J., et al. (2010). Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538. doi: 10.1126/science.1188308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tay, S., Hughey, J. J., Lee, T. K., Lipniacki, T., Quake, S. R., and Covert, M. W. (2010). Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 466, 267–271. doi: 10.1038/nature09145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., et al. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. doi: 10.1038/nbt.2859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Warmflash, A., Zhang, Q., Sorre, B., Vonica, A., Siggia, E. D., and Brivanlou, A. H. (2012). Dynamics of TGF-beta signaling reveal adaptive and pulsatile behaviors reflected in the nuclear localization of transcription factor Smad4. Proc. Natl. Acad. Sci. U.S.A. 109, E1947–E1956. doi: 10.1073/pnas.1207607109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, A. R., Neff, N. F., Kalisky, T., Dalerba, P., Treutlein, B., Rothenberg, M. E., et al. (2014). Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 11, 41–46. doi: 10.1038/nmeth.2694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yin, H., and Marshall, D. (2012). Microfluidics for single cell analysis. Curr. Opin. Biotechnol. 23, 110–119. doi: 10.1016/j.copbio.2011.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zambrano, S., Bianchi, M. E., and Agresti, A. (2014). High-throughput analysis of NF-kappaB dynamics in single cells reveals basal nuclear localization of NF-kappaB and spontaneous activation of oscillations. PLoS ONE 9:e90104. doi: 10.1371/journal.pone.0090104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: single-cell analysis, single-cell genomics, cellular heterogeneity, biological noise, measuring noise
Citation: Paszek P (2014) From measuring noise toward integrated single-cell biology. Front. Genet. 5:408. doi: 10.3389/fgene.2014.00408
Received: 29 September 2014; Paper pending published: 16 October 2014;
Accepted: 03 November 2014; Published online: 19 November 2014.
Edited by:
Daniel Hebenstreit, University of Warwick, UKReviewed by:
Jongrae Kim, University of Leeds, UKCopyright © 2014 Paszek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:cGF3ZWwucGFzemVrQG1hbmNoZXN0ZXIuYWMudWs=