 Tímea Pócza1†
Tímea Pócza1† Vince Kornél Grolmusz1,2†János Papp1,2
Vince Kornél Grolmusz1,2†János Papp1,2 Henriett Butz1,2,3Attila Patócs1,2,3
Henriett Butz1,2,3Attila Patócs1,2,3 Anikó Bozsik1,2*
Anikó Bozsik1,2*- 1Department of Molecular Genetics, National Institute of Oncology, Budapest, Hungary
- 2Hereditary Cancers Research Group, Hungarian Academy of Sciences, Semmelweis University, Budapest, Hungary
- 3Department of Laboratory Medicine, Semmelweis University, Budapest, Hungary
In addition to single nucleotide variations and small-scale indels, structural variations (SVs) also contribute to the genetic diversity of the genome. SVs, such as deletions, duplications, amplifications, or inversions may also affect coding regions of cancer-predisposing genes. These rearrangements may abrogate the open reading frame of these genes or adversely affect their expression and may thus act as germline mutations in hereditary cancer syndromes. With the capacity of disrupting the function of tumor suppressors, structural variations confer an increased risk of cancer and account for a remarkable fraction of heritability. The development of sequencing techniques enables the discovery of a constantly growing number of SVs of various types in cancer predisposition genes (CPGs). Here, we provide a comprehensive review of the landscape of germline SV types, detection methods, pathomechanisms, and frequency in CPGs, focusing on the two most common cancer syndromes: hereditary breast- and ovarian cancer and gastrointestinal cancers. Current knowledge about the possible molecular mechanisms driving to SVs is also summarized.
Introduction
The genetic diversity of the human genome is based on several types of variations from single nucleotide polymorphisms to large genomic rearrangements (LGRs). Chromosomal rearrangements comprise various types of structural variations (SVs). Some of them are copy-neutral (balanced) rearrangements, such as inversions and translocations, while others modify the dosage of chromosomal regions. These latter groups consist of copy number variations (CNVs), which are gains or losses of DNA fragments (deletions, duplications, or amplifications) constituting approximately 5% of the genome and providing the major source of genetic diversity (Zhang et al., 2009). Although CNVs may involve larger chromatin structures, the majority of them are subtle alterations of submicroscopic size generally ranging from 100 bp to 3 Mb (Zhang et al., 2009).
As opposed to recurrent genomic rearrangements, which are mainly gross recombinational events between chromosomal arms encompassing the same genomic interval in unrelated individuals, SVs in cancer predisposition genes (CPGs) are mainly non-recurrent events. This means that there is no particular genomic region for the breakpoints; these can arise at any chromosomal position. Only slight hot spots or grouping of breakpoints can be discerned with especially complex chromatin architectural structures or within pseudogene regions (Puget et al., 2002).
Structural variations appear either as somatic variations, especially in tumors, or can be generated in the germline. In this case, they are heritable. When SVs fall in functionally relevant regions of the genome, especially when they alter the open reading frame of coding genes, they seriously compromise gene function. This is especially remarkable in CPGs, where these changes contribute to the hereditary mutation profile and confer an increased risk for cancer. Noteworthy, SVs can also affect non-coding genes involved in cancer susceptibility (lncRNAs, microRNAs, and other types of small RNAs), some of which also have exon/intron structures.
Cancer predisposition genes are mainly tumor suppressors, which generally act recessively: both alleles should be lost for developing a phenotype. The Knudson two-hit model sets out that the first hit is an inherited germline mutation, which is followed by a subsequent somatically acquired second hit for tumor generation (Knudson, 1971). The second hit for tumorigenesis frequently appears in one individual’s lifetime, causing dominantly appearing cancer disease phenotypes.
The pathogenicity of the SVs is not directly obvious in all cases. While the majority are clear-cut mutations, there are cases, especially in certain duplications and inversions, where additional functional tests are required to assess their effect on clinical outcomes.
Continuously evolving sequencing technologies enable the detection of various types of SVs, which were formerly missed by conventional detection techniques. This allows the identification of an emerging number of rearrangements, which further broadens the spectrum of these variations.
Here, we provide a comprehensive review of germline SV types, detection methods, pathogenic mechanisms, and frequency in CPGs, focusing on three common cancer syndromes. These are hereditary breast- and ovarian cancer (HBOC) and two types of hereditary gastrointestinal cancers, i.e., familial adenomatous polyposis (FAP) and hereditary non-polyposis colorectal cancer (HNPCC or Lynch syndrome). Our work also covers rare hereditary cancer syndromes, each possessing a strong heritability factor and associated with acknowledged tumor suppressor genes.
SVs – Underlying Causative Molecular Mechanisms
The molecular mechanisms generating rearrangements may be replicational or recombinational events and are mostly related to repair processes.
Non-allelic homologous recombination (NAHR) has long been acknowledged as the principal molecular process for SV generation (Sen et al., 2006). A decade ago, novel mechanisms were also discovered as possible casual events. Below, we summarize the most relevant molecular mechanisms calling forth SVs and present them in Figure 1 through selected examples of already proved rearrangements.
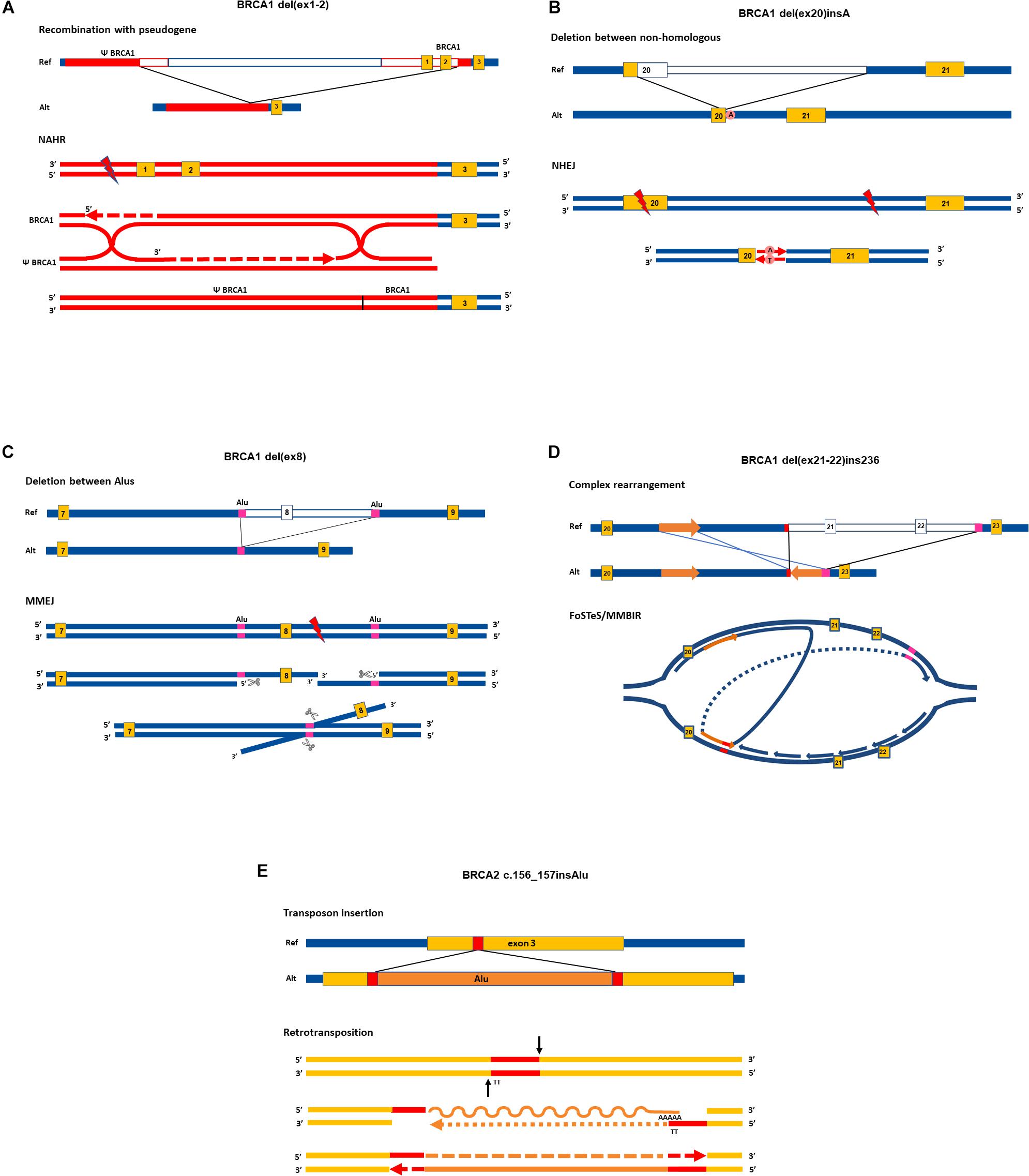
Figure 1. Proposed rearrangement types in cancer susceptibility genes. Upper parts of the panels show the allelic structure in sense orientation of elected rearrangements. Lower parts of the panels depict the most probable molecular mechanisms giving rise to the respective rearrangements, focusing on the main successive steps. Enzymes and auxiliary proteins mediating the mechanisms are not indicated. Yellow boxes indicate exons, red and magenta lines denote homologies, empty lines in reference allele mark deleted regions in alternative allele. Red lightning signs stand for double-strand break. Graphical representation of exons and introns is not to scale. The running name of the rearrangements are indicated above each graph, and exact names are given in the caption with HGVS nomenclature. All SVs taken as examples are registered variations in the LOVD database. NAHR, non-allelic homologous recombination, NHEJ, non-homologous end joining; MMEJ, microhomology-mediated end-joining; MMBIR, microhomology-mediated brake-induced repair; FoSTeS, fork stalling and template switching. Ref, reference allele; Alt, alternative allele. (A) NG_005905.2:g.61201_98134del. Running name: BRCA1 del(ex1-2) (Puget et al., 2002). At the position of the DNA double-strand break, the 5′ ends are resected and one of the overhanging 3′ ends invades into the D-loop of the homolog Psi-BRCA1 region annealing with its complementary strand. Synthesis proceeds further for hundreds of base pairs, harnessing the ectopic homology as a template. Extensive homology between BRCA1 and its pseudogene enables the formation of double Holliday junction. The resolution of the double cross with recombination event results in a hybrid region of Psi-BRCA1 and BRCA1. Single-strand nicks are sealed by polymerase (dashed arrows) and ends are rejoined by ligase. (B) LRG_292t1:c.5213_5278-2753delinsA. Running name: BRCA1 del(ex20)insA (Belogianni et al., 2004; Bozsik et al., 2020). DNA stretch between the two double-strand breaks is deleted and the two exposed ends are rejoined by NHEJ without homology requirement. Error-prone polymerase seals the nick by editing the sequence with an additional adenine residue at the synapsis. (C) LRG_292t1:c.442-1102_547+252del. Running name: BRCA1 del(ex8) (Bozsik et al., 2020). DNA ends are processed at the site of the double-strand break: 5′ strands are resected by exonucleases (marked with scissors). The overhanging 3′ strands find 26 base pairs with exact microhomology between nearby 300-bp-long and almost completely homolog AluSx and AluSp sequences (marked with purple boxes). The two strands anneal by the microhomology, and the protruding 3′ strand is eliminated by flap trimming (incision is marked with scissors). The DNA ends are rejoined by ligase. (D) LRG_292t1:c.5278-492_5407-128delins236. Running name: BRCA1 del(ex21-22)ins236 (Zikan et al., 2008; Bozsik et al., 2020). Replication forks stalls at structural hindrance caused by palindrome sequence. The 3′ end of the newly synthesized strand disassembles and reanneals to the complementer strand of the same replication bubble with the help of small homology of few nucleotides (indicated with red line). The synthesis proceeds in the reverse direction for 236 base pairs (marked with an orange arrow) and reanneals to the original template using another stretch of microhomology (marked with magenta line) skipping the intervening region. (E) NG_012772.1:g.8686_8687insAlu. Running name: BRCA2 c.156-157insAlu (Peixoto et al., 2011). RNA sequence from the AluYa5 inserts into the exon three through target primed reverse transcription method. Endonuclease incises (black arrows) at the ends of the target site (marked with red) liberating 3′ end with TT nucleotides. This serves as a complementer template for the polyA tail of the AluYa5 RNA to anneal. Reverse transcription priming is provided by the free 3′ end of the gene. After reverse transcription (indicated by dotted arrow) gaps are filled by polymerase. The RNA strand is lysed and exchanged with DNA also by 3′→5′ synthesis action of polymerase (dashed arrow). Lygase seals the ends. At the end of the retrotransposition process, the Alu sequence is inserted into the exon with the flanking duplication of the target site.
Non-allelic homologous recombination happens between false homologous alleles. This phenomenon, which is called illegitimate recombination, is the main source of recurrent rearrangements. The mechanism usually takes place during meiosis and mitosis and requires an extensive chromosomal homology region of several kilobases similar to conventional recombination (Weckselblatt and Rudd, 2015). Segmental duplications or low-copy repeats throughout the genome serve as a target for ectopic (non-allelic) alignment of the chromosomal regions (Bailey et al., 2002). Additionally, the human genome contains several thousands of transposon-related remnants and may even be located on different chromosomes, which might cause such illegitimate recombinations (Arkhipova and Yushenova, 2019). NAHR can take place between homologous chromosomes, but it can also be intrachromosomal (between sister chromatids) or take place during an intrachromatidal event. NAHR between chromosomal arms calls forth duplications and deletions. Intrachromatidal recombinations between direct repeats result in deletion, whereas inverted repeats serve as a target for inversions. Recurrent rearrangement in CPGs through NAHR is feasible in the case of extensive homology served by long pseudogene regions. Smaller-scale rearrangements are more likely to happen as a result of repair events. Homologous recombinational repair, occurring mainly during the S phase of the cell cycle, can also mispair with ectopic regions with similar sequences and can generate deletion and duplication of chromosomal fragments through a recombination-like resolution of the Holliday junction (Hastings et al., 2009b; Figure 1A). Non-homologous end joining (NHEJ) is a more error-prone end-joining repair, generally requiring no homology (Zhao et al., 2020). This mechanism is proposed for deletions, where no, or only a few base pairs of homology are detectable at the junction points. The insertion of additional nucleotides at the junction point (so-called scars) is characteristic of this repair (Figure 1B).
The breakpoints of the majority of CNVs encompassing exons of CPGs fall in Alu repetitive sequences. This indicates that Alu elements have a substantial role in the generation of exon-scale chromosomal rearrangements. Novel findings argue that NAHR events require more extensive homology than the typical 300 bp of Alu sequences (Kowalczykowski, 2015). Instead, for rearrangements between small stretches of homologies microhomology-mediated end-joining (MMEJ) mechanism was suggested (McVey and Lee, 2008; Sfeir and Symington, 2015; Sinha et al., 2016). This is a special repair mechanism at double-strand breaks, which involves 5′ strand resection and annealing of the 3′ overhangs mediated by nearby/proximal small homologies of 5–50 bases (Tournier et al., 2004; Figure 1C).
The combination of chromosomal segments, which are sometimes even distantly positioned, can occur as a result of replication-based molecular mechanisms: Fork Stalling and Template Switching (FoSTeS) and Microhomology-mediated Break-Induced Repair (MMBIR) (Hastings et al., 2009a). Despite the difference in the molecular background, these processes are not distinguishable by the resulted product: both give rise to complex rearrangements. Both mechanisms are preceded by stalled replication forks: the DNA polymerase is stopped either by palindrome loops and structural hindrance (FoSTeS) or breaks in the template strand (MMBIR) (Hastings et al., 2009a). The 3′ end disengages and anneals to another replication fork through microhomology of only a few (<6 bp) bases. The new fork, though positioned adjacently, may be distant in the chromosome, or even can be located on another chromosome. The polymerase uses this new strand as a template and replicates a stretch of this region before the strand reanneals to the original fork. Moreover, the 3′ end invasion to new replication forks can be repeated several times between different chromosomal regions before reannealing, thus entailing multiple, distantly located fragments coming together in juxtapositions (Colnaghi et al., 2011). This mechanism can also give rise to deletions, duplications with misaligned homology, and even reversions when the leading strand anneals to the lagging strand. The typical FoSTeS/MMBIR-resulted rearrangement in CPGs is a characteristic pattern of some kilobase deletion combined with a short stretch of reverse oriented duplication of a neighboring intronic segment (Figure 1D).
Retrotransposition is also a way for copy number gain. The main mobile elements in the human genome are Long INterspersed Element-1 (L1), SINE-VNTR-Alu (SVA), and Alu, the copy number of which continuously expands with replicative copy-and-paste retrotransposition. The mechanism involves the reverse transcription of the RNA of these elements and insertion of the cDNA copy into a new genomic position with the help of a special endonuclease (Goodier, 2016; Figure 1E). L1 elements are the only autonomous transposons in the current human genome. Alu and SVA elements have no capacity for reverse transcription themselves but harness the enzymatic activity of L1 elements for moving (Hancks and Kazazian, 2016).
It is important to note, that the results of the different mechanisms are overlapping. Therefore, the underlying molecular event is not unequivocally identifiable by the inspection of the rearrangement pattern.
Pathomechanisms of SVs in Cancer-Predisposing Genes
There is a wide array of mechanisms by which a CNV can abrogate cancer gene function. The most typical is the deletion of one or more exons coding for indispensable domains or structural elements of the protein. Moreover, out-of-frame deletions, generated at any position of the open reading frame, result in premature termination codon (PTC) on the transcript, which either codes for a truncated protein or is eliminated by nonsense-mediated decay. When the deletion affects the promoter region, the regulation of the gene expression may be compromised. For example, a 10 kb promoter deletion in the APC gene affecting promoter 1B reduces the expression of APC-1B (Yamaguchi et al., 2016). If the rearrangements on the chromosome are more extensive, the deletion may cover the whole coding gene.
Contrary to the effect of deletions, the pathogenic effect of duplications is not straightforward. Breakpoint characterizations are needed for the detection of their exact positions and orientations for interpreting their genetic consequences. Out-of-frame tandem duplications generate PTCs and interfere with protein function similarly to deletions. The duplication of in-frame exons, resulting in two tandem copies of certain protein regions, theoretically, does not necessarily cause a severe adverse effect on the protein, if domain positions and functions are not affected. The same applies to promoter duplications, which sometimes also involve the first coding exons of the gene: in this case, there is at least one correct copy of the whole gene and optimal choice between the two promoters can help to evade the generation of an altered transcript. For example, the tandem duplication of 357 kb upstream of the BRCA1 gene, reaching up to BRCA1 exons 1–19, was evaluated as a benign variation (Du et al., 2018).
A special form of gene silencing is transcriptional interference (read-through). As an example, MSH2 silencing can occur due to the deletion of 3′ exons of the upstream EPCAM gene (Ligtenberg et al., 2009). The EPCAM deletion eliminates the transcription termination signal, thus the RNA polymerase goes further towards the neighboring MSH2 gene, preventing the binding of transcription factors to the MSH2 promoter, consequently hindering its transcriptional initiation. Furthermore, the long transcript usually ends up in a PTC, directing this fused RNA towards nonsense-mediated decay, resulting in a deletion effect on both EPCAM and MSH2 (Kovacs et al., 2009).
Retroelement (RE) insertions are rare events of great consequence regarding the functional abrogation on CPGs. Insertion of a RE (L1, SVA, or Alu elements) into exons or intron regions of genes may cause exon skipping, exonization (Schmitz and Brosius, 2011), PTC generation, or transcriptional interference (Kaer et al., 2011). Aberrant splicing as a result of RE insertion into splice regulation regions was also described. A prominent example for this latter effect is the insertion of an Alu-like element in MLH1 intron 7, which interferes via a canonical splice donor site, leading to complete disruption of mRNA splicing (Li et al., 2020).
SV Detection Methods
Precise determination of SVs is not an easy task. Due to their heterogeneity, there is no one standard procedure that allows the correct identification of both deletions, insertion, and copy number alterations involving multiallelic loci. Generally, molecular biological methods providing quantitative differences can be used for the detection of SVs (Cantsilieris et al., 2013; Butz and Patocs, 2019). Based on the size of SVs, different assays are available. Two widely employed approaches in routine clinical practice are hybridization-based and PCR-based techniques.
Fluorescent in-situ hybridization (FISH) is typically used for the identification of large genomic alterations, such as gross chromosomal abnormalities, but current advances in the technique enable the detection of CNVs with sizes as small as 50 kb. Fluorescently labeled DNA probes complementary to the sequence of specific regions are hybridized to metaphase chromosomes or interphase nuclei (Bayani and Squire, 2004). A state-of-the-art version of FISH providing even better resolution (5–500 kb) is fiber-FISH, where probes are visualized on mechanically stretched chromosomes (Ceulemans et al., 2012). This technique is especially appropriate for determining complex CNVs. Applying different fluorescence dyes, multiple DNA targets can be tested simultaneously, allowing for whole-genome analysis (multi-color FISH) (Ceulemans et al., 2012).
Another hybridization-based method is Southern blotting. Recently, due to its highly labor-intensive workflow, radioisotope labeling, and the requirement of high quantity and quality DNA, it has been mostly replaced by other techniques.
Of these approaches, microarrays, which belong to high-throughput techniques, are used to analyze the expression, genotype, or copy number of multiple genes simultaneously. In germline testing, array-based genotyping platforms (i.e., single nucleotide polymorphism-SNP arrays) are applied (Zhang et al., 2009). SNP arrays covering the entire genome or selected genetic regions using disease-specific SNP panels are also employed. The principle is based on the hybridization of fluorescently labeled probes detecting each genotype. By virtue of the intensity of the fluorescence signal, hetero-, hemi-, and homozygous variants may be distinguished, so the presence of either deletions or insertions of SVs can be determined. Loss of heterozygosity can be demonstrated by a parallel evaluation of normal and somatic DNAs of the same patient.
Array comparative genome hybridization (array CGH) uses a small glass slide (chip) that contains thousands of probes specific for certain regions of the genome. Fragmented sample and reference DNA are labeled with different fluorescent dyes, combined, and hybridized to the DNA probes on the array slide. After detection of the two fluorescent signals, the results are given as the ratio of test DNA to reference DNA at each probe. Depending on the chosen platform’s design and probe density, the resolution of CGH can vary from whole chromosomes to a few kilobases in size (Davies et al., 2005). In clinical practice, this is the most frequently employed cytogenetic assay. It is applied for the analysis of LGRs as well as submicroscopic structural alterations with unclear clinical importance. There are several databases (Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources; DECIPHER (Firth et al., 2009); International Standards for Cytogenomic Arrays Consortium; ISCA Consortium (Riggs et al., 2013), which aid in the interpretation of results. In tumor genetics, CGH is useful for the detection of somatic changes, including tumor heterogeneity and somatic mosaicism.
Optical genome mapping, an accurate high-throughput assay originally designed for aiding contiguous genome assemblies, is also applicable for the identification of all classes of SVs in the human genome (involving balanced events). Ultra-long, linearized DNA molecules are fluorescently labeled and digested with a combination of restriction enzymes. The nicks at the cleavages are optically detected as fluorescent signal discontinuities, which give a characteristic high-resolution restriction pattern for the DNA sequence. Dedicated software can assemble DNA stretches according to pattern similarities and comparative analysis of the strands enables the detection of divergent regions >500 bp caused by SVs. Optical mapping offers a significantly higher resolution than karyotyping on a similar scale to fiber-FISH.
Copy number changes can also be detected based on relative quantitations by qPCR or QMPSF (Quantitative Multiplex PCR of Short Fluorescent fragments). In both cases, the quantity of the examined region is compared to that of control regions with surely two copies. With the qPCR analysis, deletions are readily detectable by the difference of Ct values, each unit of Ct corresponding to two copy differences (Schmittgen and Livak, 2008). Limitations for duplications, however, do exist since the detection of a 2:3 ratio is not feasible. In contrast, QMPSF, where the area under the curve of the sample and control peaks yielded by multiplex PCR are compared, is amenable also for the detection of duplications (Ceulemans et al., 2012).
Inverse PCR is a suitable method for the verification of single inversions with known breakpoints. The principle of the detection is, that a PCR product is generated only when the primers hybridizing to the same strand in a reference template get into opposing orientations as a result of inversion (Wagner et al., 2002). New inversions may be discovered by allelic dropout test following long-range PCR. It is based on the phenomenon, that amplicons covering an inversion breakpoint appear as spurious deletions of one allele, presenting as stretches of homozygosity spanning the position of the inversion variant (Rhees et al., 2014). The detection of insertions is similarly demanding since their insertion point reside mainly in introns, which are not genotyped routinely. cDNA-level analysis of the genes may shed light on a part of these rearrangements since some of them generate new exons (exonization) (Schmitz and Brosius, 2011).
Multiplex ligation-dependent probe amplification (MLPA) is a semi-high-throughput technique developed to detect copy number alteration of up to 50 genomic DNA sequences in a single multiplex PCR-based mode (Kozlowski et al., 2008). Both internal control probes and positive-negative control samples have to be used during the analysis. First normalizing to internal controls (positions that are typically not affected by copy number alterations) in each sample, and then normalizing to control samples yield the relatively quantitative determination of the dosage in each probed locus. MLPA is an efficient way for detecting large deletions even in the hemizygote state. In a molecular genetic analysis of hereditary cancer syndromes, assays and complex reagents are available, and some of them have been already approved for in vitro diagnostic applications1. It has to be noted, that sequence polymorphisms within the ligation site can disturb the ligation sufficiently to cause a false positive deletion call (Serizawa et al., 2010).
Next-generation sequencing (NGS) is a high-throughput technology allowing simultaneous sequencing of multiple DNA samples. Currently, NGS-based procedures are the most widely used techniques in the routine molecular genetic diagnosis of hereditary cancer syndromes (Sarkadi et al., 2018). These approaches allow simultaneous determination of germline mutations and somatic alterations in sporadic tumors but have several requirements both from the sample and investigator sides (Robson et al., 2015). Complex laboratory workflow followed by bioinformatic analysis is needed for data mining (Krumm et al., 2012). Genotyping based on read depth analysis allows absolute copy number determination. Basically, the number of sequencing reads that map to a specific region is proportional to the number of copies of this region in the genome. The original hypothesis applies the Poisson distribution of sequencing reads, which means that a region assumed to be deleted or duplicated has fewer or more mapped reads than expected, respectively. Regarding instrumentation and sequencing chemistry, a wide selection of NGS analyses can be performed. In everyday practice, targeted gene panel sequencing (i.e., cancer panels, metabolic panels, pharmacogenetic panels, etc.) and whole exome sequencing are the most widely employed. Copy number determination from exome sequencing data is challenging because the coverage of coding exomes by sequencing reads is not uniform and can be biased by sequence capture design (Robson et al., 2015; Deans et al., 2017). Recent advances in computational approaches allow increasingly accurate determination of SVs and by unraveling the whole sequence, the correct breakpoints can also be determined (Oliver et al., 2015).
In summary, there are numerous methods available for the determination of SVs, but there remains no gold standard approach. Based on clinical practice, a combination of these techniques (i.e., array CGH and MLPA, NGS and MLPA, or qPCR) would allow the best diagnostic accuracy. The introduction of NGS technology and the development of computational data analysis will significantly increase the throughput and improve the accuracy of determining SVs.
Examples of SVs in Cancer-Predisposing Genes
The size of germline deletions affecting cancer-predisposing genes ranges from few hundreds of base pairs to several kilobases (Bozsik et al., 2020). Chromosome regions characterized by abundant directly oriented repeats, especially Alu sequences, are markedly prone to deletions primarily through the MMEJ mechanism (Smith et al., 2016). Frequently occurring deletion types are single exon deletions and multi-exon deletions. A typical example for the former is BRCA1 del(ex8) (Sluiter and van Rensburg, 2011; Bozsik et al., 2020; Figure 1B) and for the latter CHEK2 del(ex9-10) (Cybulski et al., 2007), and both variants cause frameshifts at the transcript level. The deletion of the full BRCA1 gene, del(ex1-24) has been previously detected in various populations (de la Hoya et al., 2006; Engert et al., 2008; Fachal et al., 2014). Pseudogene regions of a gene can serve as long sequence stretches with considerable homology for NAHR events, thus providing hot-spots for illegitimate recombinations that often result in deletions. There are several rearrangements with different breakpoints between BRCA1 and its pseudogene (Psi-BRCA1), generating a ∼37 kb deletion involving the BRCA1 promoter and exons 1–2 (Puget et al., 2002). Similarly, the PMS2 locus also has multiple pseudogenes, especially PMS2CL, which has an almost 100% sequence identity with PMS2 exons 12–15. This exact sequence homology enables dynamic gene conversions and recombinations between the two regions (Kohlmann and Gruber, 1993). Concerning genotype-phenotype correlations, there is no evidence for HBOC genes, BRCA1 and BRCA2; whole exon deletions manifest in a more severe phenotype of the disease than smaller-scale indels (Gad et al., 2003; Walsh et al., 2006; Smith et al., 2016; Bozsik et al., 2020). Whole exon deletions in Lynch syndrome genes, MLH1 and MSH2, however, are associated with a slightly earlier age of onset for colorectal cancer than small truncating variants, but this difference does not reach the nominal significance of 0.05 (Smith et al., 2016). In contrast, whole gene deletions may have altered phenotypic consequences in some syndromes: whole NF1 deletions tend to cause a more severe phenotype, whereas whole NF2 deletions generally result in a milder phenotype than truncating point mutations (Smith et al., 2016). Nevertheless, genetic alterations affecting additional causative genes may correlate with disease phenotype: in the case of a 7.4 Mb deletion encompassing NF2 and neighboring genes corresponds to a more severe phenotype (Smith et al., 2016). In another example of contiguous gene deletion, germline 10q chromosomal deletion resulted in the loss of both PTEN and BMPR1A, and this corresponds to distinct pathological features of polyposis syndromes, underlining the complex interactions of these genes in tumorigenesis (Delnatte et al., 2006). Similarly, contiguous gene deletion within the 2p16-p21 chromosomal region, encompassing MSH2, MSH6, EPCAM, and 24 additional genes, causes Lynch syndrome with distinct phenotypic features (Salo-Mullen et al., 2018).
The majority of genomic duplications are directly oriented tandem repeats in CPGs (Sluiter and van Rensburg, 2011) and genome-wide (Newman et al., 2015) as well. The most prevalent tandem duplication in the BRCA1 locus is BRCA1 dup(ex13), which was detected in high frequency in nearly all European populations (The BEDSG, 2000). The bulk of single-exon or multi-exon duplications in CPGs reported so far are unambiguously pathogenic, although duplications encompassing the whole promoter together with a various number of downstream exons are evaluated as variants with unknown significance. For example, the examination of the BRCA1 dup(ex1-2) variant by Fachal et al. (2014) failed to identify any aberrant transcripts (Fachal et al., 2014). Pathogenicity of other exon duplications detected by dosage-sensitive genotyping tests must also be confirmed by precise breakpoint assessment, as it was done for BRCA2 dup(ex22-24) by van Luttikhuizen et al. (2020). They revealed, that the duplicated region was arranged in tandem and direct orientation, generating a PTC (van Luttikhuizen et al., 2020).
Particular types of copy gains include the insertion of mobile REs. Qian et al. (2017), conducted a large pan-cancer study on a panel of 26 genes and found that RE insertions were identifiable in 10 of the 26 genes tested (Qian et al., 2017). Indeed, RE insertions were detected in several genes (BRCA1/2, APC, ATM, PMS2, MLH1, and MSH2) by other groups studying hereditary breast and gastrointestinal cancers (Kaer et al., 2011; Li et al., 2020). Insertions of Alu repetitive motifs into exonic or intronic regions are the most prevalent transposition events in cancer-predisposing genes (Kaer et al., 2011). An Alu insertion in exon 3 of the BRCA2 gene caused exon 3 skipping, and this is a founder mutation in the Portuguese population (Machado et al., 2007; Peixoto et al., 2011). In the Lynch syndrome-associated gene PMS2, insertion of an SVA nonautonomous retrotransposon element in intron 7 causes partial exonization of SVA using cryptic splice sites (van der Klift et al., 2012). APC, the germline susceptibility gene for FAP is disrupted by the insertion of an L1 sequence into exon 16 (Miki et al., 1992).
The detection of inversions can be challenging, therefore, their contribution to the SV pool is underestimated. In HNPCC, a 10 Mb paracentric inversion involving exons 1–7 of the MSH2 gene was described first (Chen, 2008). This inversion was found to be a frequent cause of Lynch syndrome in a US population, accounting for an appreciable percentage of the mutational burden of this gene (Rhees et al., 2014). Later, another cryptic paracentric inversion of exons 2–6 of the same gene was detected (Liu et al., 2016). Germline inversion has also been shown for the MLH1 locus, another major susceptibility gene in HNPCC. In this latter case, the inversion breakpoints are in intron 15 of MLH1 and intron 3 of the neighboring LRRFIP2 genes, generating two fusion transcripts between MLH1 and LRRFIP2 (Morak et al., 2011).
Translocations of whole chromosome arms are not typical events for disrupting tumor suppressor genes. However, two isolated cases with different chromosomal arm interchanges were described so far, each affecting the APC gene—a constitutional reciprocal translocation t(5;10) (van der Luijt et al., 1995) and a t(5;7) translocation (Sahnane et al., 2016).
The combination of rearrangement types manifests in complex genomic rearrangements. Despite these rearrangements possessing more than one junction point, they often arise from one molecular event, typically FoSTeS/MMBIR in cancer susceptibility genes. The characteristic pattern of deletion together with reverse duplication of some hundred base pairs occurs in various independent CNVs. BRCA1 del(ex21-22) with reverse-oriented insertion of 236 bp of an intronic repeat is a founder complex CNV in the Czech population (Zikan et al., 2008; Ticha et al., 2010; Figure 1D and Table 1) and has also been reported as a recurrent variant in other European countries (Sluiter and van Rensburg, 2011; Bozsik et al., 2020). A complex recombination event characterized by the deletion of exons 5–10 and the insertion of a 35-bp nucleotide stretch in inverted orientation derived from the intron 3 sequence of the BRCA1 gene is also a Czech founder mutation (Ticha et al., 2010). A deletion of exons 6–8 of MLH1, with the retention of 349-bp of intron 6 is also a complex rearrangement reported in one patient with colorectal cancer (McVety et al., 2005).
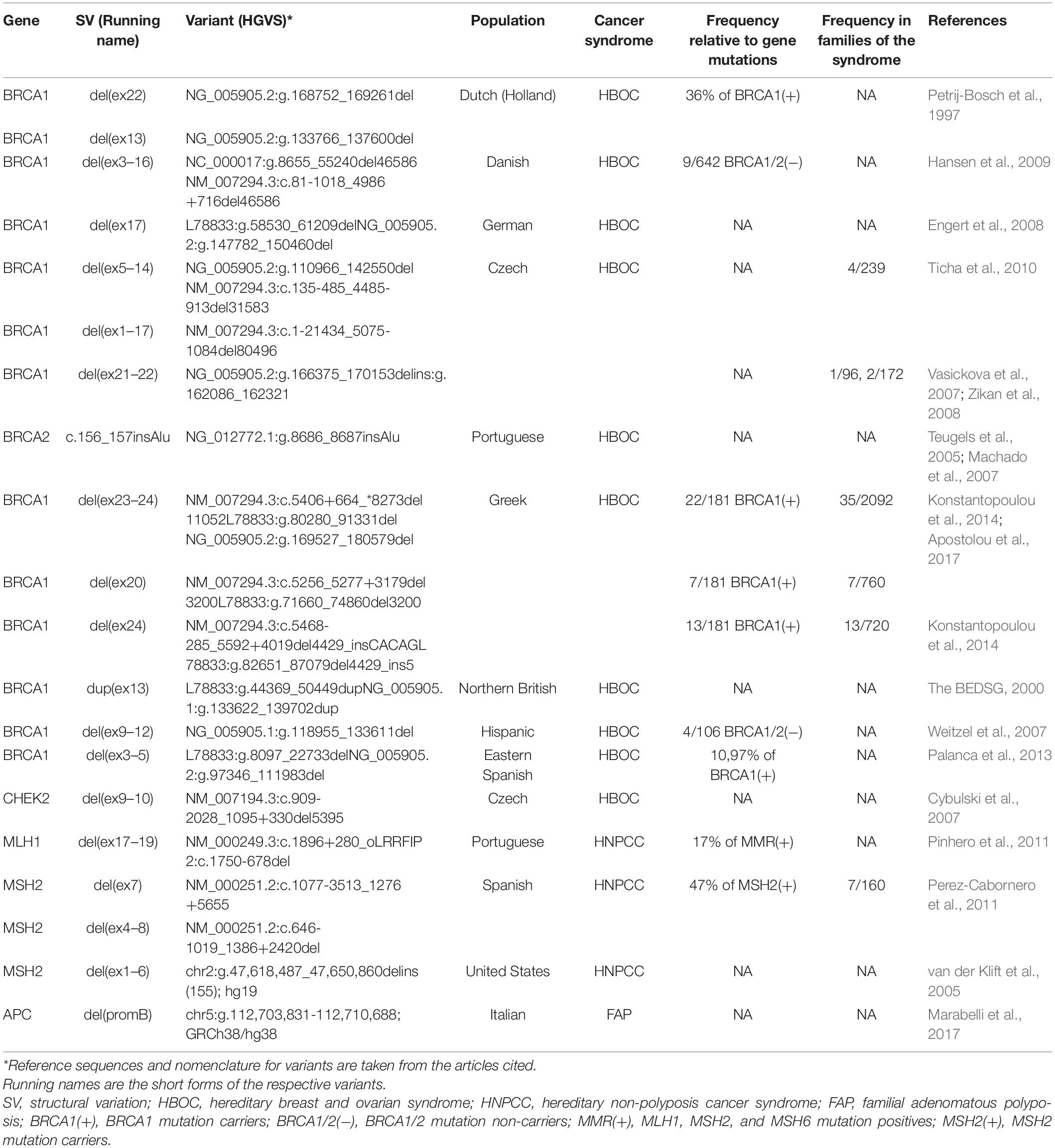
Table 1. Examples of founder SVs and frequencies in various populations.
Frequency of Germline SVs in Cancer-Predisposing Genes
The type and frequency of germline SVs in cancer susceptibility genes show wide differences across various populations. This is conceivably due in part to the different detection methods applied, the various and sometimes limited number of patients tested, as well as the founder alterations specific to certain populations. Founder variants are genetic alterations with a common origin, which are generated in an ancestor and spread through generations in an isolated ethnic group, thus these are recurrent and characteristic of a population. For example, haplotype analysis revealed, that the recurrent BRCA1 deletion of exons 23 and 24 is a Greek founder mutation (Apostolou et al., 2017). Similarly, a recurrent exon 22 deletion in the BRCA1 gene was found in the Netherlands (Sluiter and van Rensburg, 2011). Duplication of BRCA1 exon 13 of Northern British origin (The BEDSG, 2000), as well as the above-mentioned deletion of exon 21-22 in BRCA1 of Czech origin, are also frequently occurring CNVs in all populations in Europe (Ticha et al., 2010; Sluiter and van Rensburg, 2011; Bozsik et al., 2020). A large screening study in the US revealed, that 70.8% of all BRCA1 rearrangements of Western and Northern European origin are made up of five founder CNVs (Judkins et al., 2012). Table 1 summarizes some examples of founder pathogenic SVs and their frequencies in the source populations.
Deletions are the most prevalent CNV types in cancer susceptibility genes (Sluiter and van Rensburg, 2011; Mancini-DiNardo et al., 2019; Bozsik et al., 2020), contributing to approximately 80–85% of all rearrangements (Mancini-DiNardo et al., 2019). In contrast, duplications account for only 10–15% of rearrangements (Mancini-DiNardo et al., 2019; Bozsik et al., 2020). The predominance of molecular processes resulting in genomic deletions compared to duplications might explain the observed difference between the frequencies of these alterations (Hastings et al., 2009b). RE insertions also represent a significant SV type as they accounted for one in every 325 unique pathogenic variants detected in a large pan-cancer study (Qian et al., 2017). In this cohort, 92% of all RE events were retrotransposition of Alu elements, while the most frequently affected genes with unique RE insertions were BRCA2 (45.9%) and ATM (16.2%) (Qian et al., 2017). Mechanistically, there is no reason for the observed predominance of BRCA2 in RE events.
A hereditary pan-cancer gene panel survey of 376,159 individuals in the US revealed 3,461 LGRs in 27 genes (Mancini-DiNardo et al., 2019). In general, SVs accounted for 7.2% of all pathogenic variants detected. The largest proportion of pathogenic LGRs were identified in BRCA1 (27.4%), followed by PMS2 (11.7%), CHEK2 (11.1%), and MSH2 (8.9%) (Mancini-DiNardo et al., 2019). In a separate study focusing on point-mutation-negative HNPCC patients, 11% of cases harbored large rearrangements in four predisposition genes (MLH1, MSH2, MSH6, and PMS2) among which 29.6% affected the MSH2 gene (van der Klift et al., 2005). Similarly, 15% of the point mutation-negative patients with classical FAP had a genomic deletion in APC (Michils et al., 2005). On the contrary, various studies in different populations focusing on BRCA1/2 mutation-negative HBOC patients revealed that large rearrangements of BRCA1 and BRCA2 genes contributed to only 2–3% of the cases (Agata et al., 2006; Preisler-Adams et al., 2006; Thomassen et al., 2006).
The proportion of pathogenic SVs in a given locus as a fraction of the clear-cut mutations of the gene shows a different ratio pattern. Table 2 summarizes the reported SV ratios in the most relevant susceptibility genes of various cancer predisposition syndromes. Typically, SVs represent 10% (ranging from 0.1 to 60.7%) of the acknowledged mutations of these genes. The high extreme was detected in the STK11 gene, where 30–60% of all mutations are CNVs (Aretz et al., 2005; Mancini-DiNardo et al., 2019). The ratios are also high in the case of MSH2 and PMS2, where large deletions account for ∼20 and ∼25% of mutations, respectively (Kohlmann and Gruber, 1993; Mancini-DiNardo et al., 2019). MUTYH is the less abundant in SVs with its ratio of 0.1% relative to all mutations of the genes (Mancini-DiNardo et al., 2019). The differences with regard to SV frequencies in different genes are conceivably a function of their genetic surroundings and chromosomal complexity. For example, the higher proportion of Alu repeats may contribute to the higher rate of genomic rearrangements in MSH2 compared to that of MLH1 (van der Klift et al., 2005). The prevalence of BRCA1 rearrangements over BRCA2 is explained also by the differences in the ratio of intronic Alu repeats between the two genes (Judkins et al., 2012; Sluiter and van Rensburg, 2011). Similarly, PMS2, due to its extensive pseudogene regions is an especially good subject for rearrangements through recombinations (Smith et al., 2016).
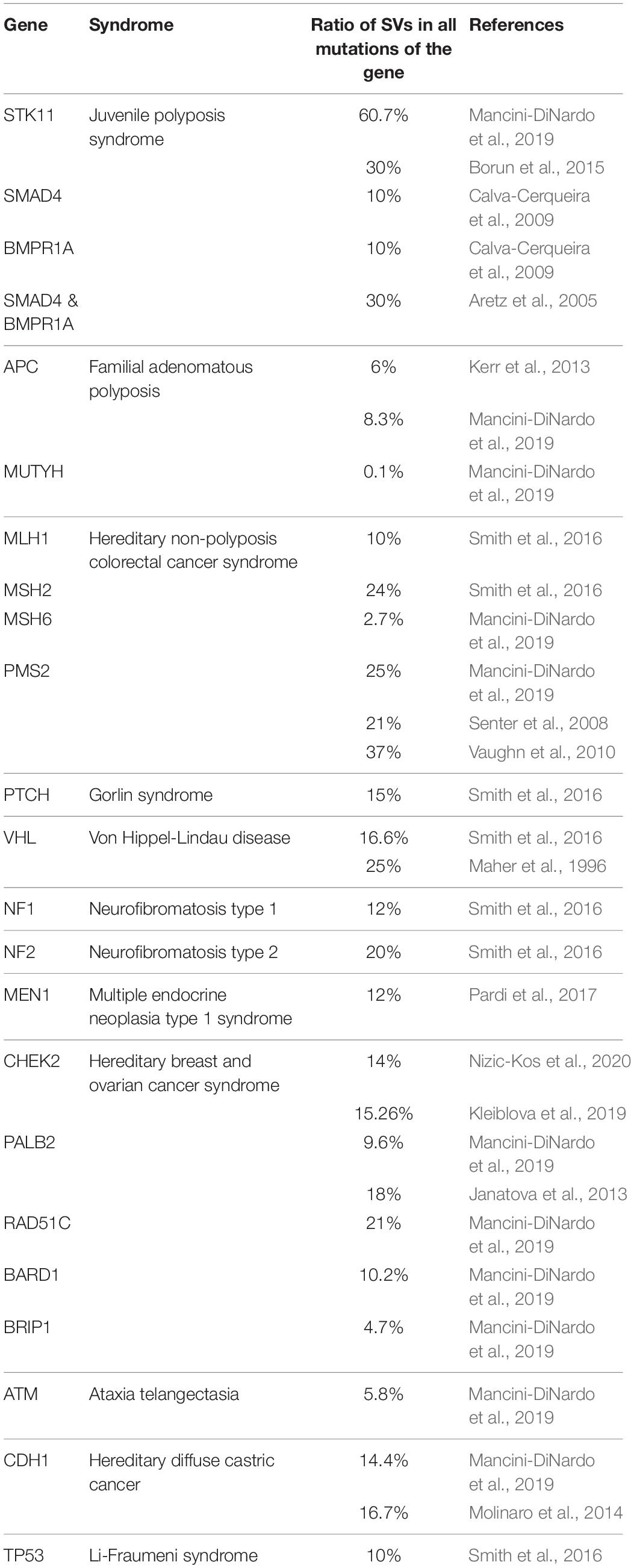
Table 2. Relative ratios of germline SVs compared to all mutations of the susceptibility gene in various cancer syndromes.
CNVs in BRCA1 and BRCA2 genes, being the most penetrant HBOC predisposition loci, are extensively studied in various populations worldwide. To date, more than 100 LGRs have been characterized in BRCA1, whereas much fewer have been characterized in BRCA2 (Sluiter and van Rensburg, 2011). The CNV ratios of BRCA1 and BRCA2 genes relative to all pathogenic mutations detected in HBOC probands of various ethnicities are visualized on the histogram in Figure 2. On average, CNVs account for 10% of all pathogenic mutations of the BRCA1 gene; the differences in ratios in various ethnicities are mainly attributed to founder mutations. BRCA2 locus has only low contribution (<0.5%) to CNVs (Hogervorst et al., 2003; de la Hoya et al., 2006; Thomassen et al., 2006; Engert et al., 2008; Ticha et al., 2010; Fachal et al., 2014). A remarkable percentage of BRCA2 large rearrangements have only been detected in Portugal, where the c.156_157insAlu founder mutation constitutes the bulk of the cases (Machado et al., 2007). An elevated ratio of BRCA2 CNVs is also observed in male breast cancer patients (Tournier et al., 2004).
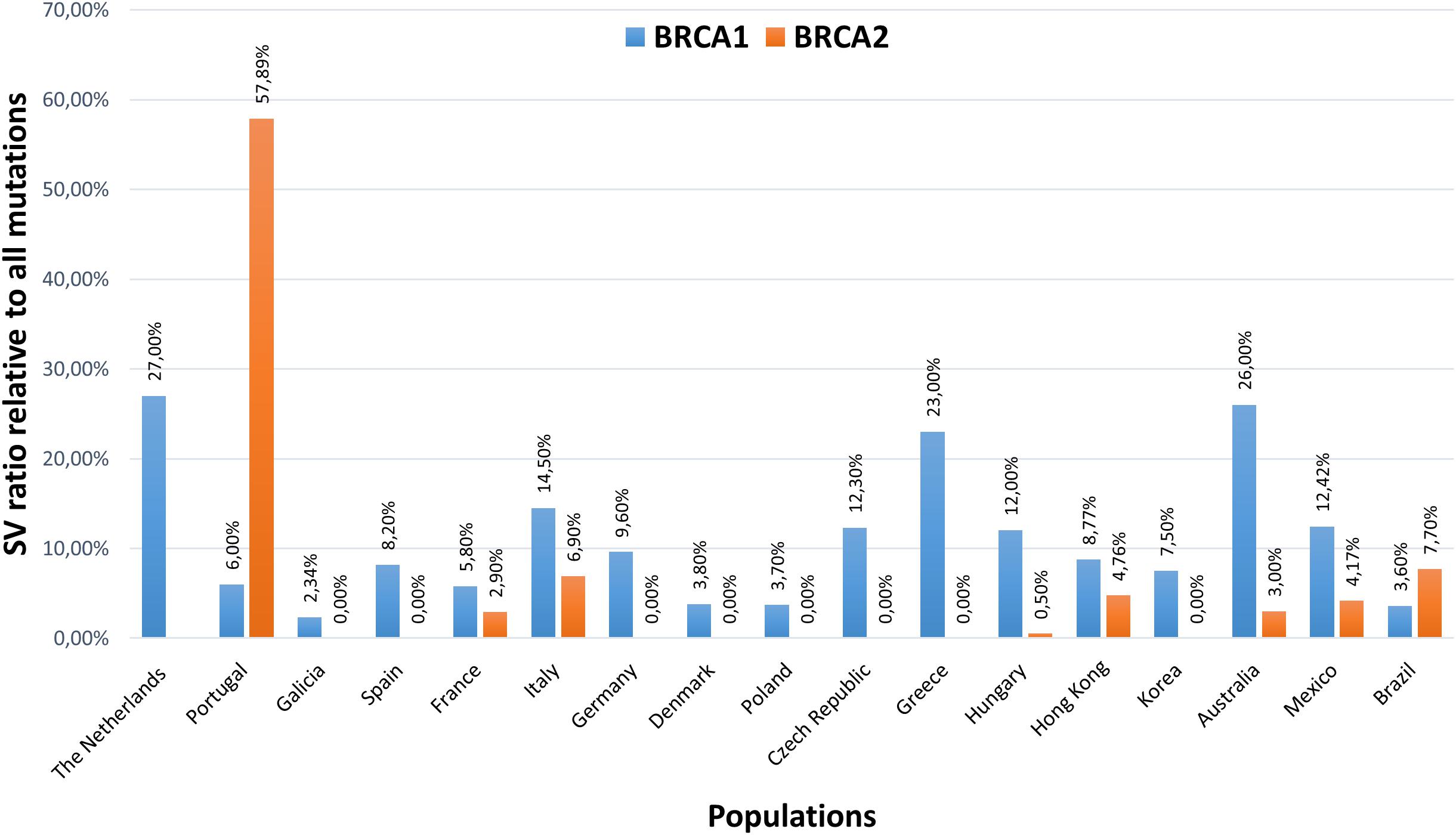
Figure 2. Structural variation ratios of BRCA1 and BRCA2 genes relative to all pathogenic mutations detected in HBOC probands of various ethnicities. The Netherlands (Hogervorst et al., 2003), Portugal (Peixoto et al., 2011), Galicia (Fachal et al., 2014), Spain (de la Hoya et al., 2006), France (Caux-Moncoutier et al., 2011), Italy (Concolino et al., 2018), Germany (Engert et al., 2008), Denmark (Thomassen et al., 2006), Poland (Rudnicka et al., 2013), Czech Republic (Ticha et al., 2010), Greece (Armaou et al., 2009), Hungary (Bozsik et al., 2020), Hong Kong (Kwong et al., 2015), Korea (Seong et al., 2014), Australia (James et al., 2015), Mexico (Lopez-Urrutia et al., 2019), Brazil (Palmero et al., 2018).
Additional association studies are seeking to identify further pathogenic CNVs in breast cancer contributing to the disease phenotype. Kumaran et al. (2017), identified 200 common germline CNVs associated with breast cancer in a whole-genome sequencing study of 422 breast cancer cases and 348 controls (Kumaran et al., 2017). Moreover, they also confirmed, that germline CNVs conferred dosage effects on gene expression in breast tissue (Kumaran et al., 2017). Similarly, another study of genome-wide germline CNVs identified 275 unique rearrangements that potentially contribute to breast cancer initiation and/or progression (Masson et al., 2014).
Summary
Within germline SVs in CPGs for hereditary cancer syndromes, copy number changes are the prevailing alterations. The predominant CNVs are deletions, affecting various portions of the genes. The main structural source of these deletions is intronic Alu sequences. Double-strand break repairs and additional molecular mechanisms harness these sequence homologies and may result in copy changes through ectopic alignments. Studies conducted in different populations confirmed that SVs generally account for 7–10% of all mutations in CPGs, thus their contribution of mutational burden is significant. Precise detection of these types of alterations is essential to provide an optimal genetic diagnosis. Differences in neighboring genetic architecture, as well as various applied detection techniques, may contribute to the wide range of variations in the exact ratios of pathogenic CNVs compared to point mutations (Mancini-DiNardo et al., 2019) [etc.].
The association with the clinico-pathological phenotype is straightforward in the majority of germline SVs, however, in a few cases, and especially within some duplications, pathogenic effects cannot be addressed unambiguously. Moreover, several studies proposed that copy number changes, although larger in size, do not necessarily associate with a more severe pathological phenotype than smaller-scale indels (Walsh et al., 2006; Rhees et al., 2014; Smith et al., 2016; Bozsik et al., 2020). On the other hand, extensive rearrangements affecting the whole gene together with several neighboring genes may elicit a complex phenotype due to the putative interfering effects of the respective proto-oncogenes and tumor suppressors (Delnatte et al., 2006; Smith et al., 2016). However, since the number of cases harboring such rearrangements is limited, further studies are needed to ascertain these correlations.
Due to the continuous development of dosage-sensitive detection modes and validations, an increasing number of germline structural rearrangements are being discovered in several CPGs. Note that synthesis of the data highlighted differences regarding structural variation types and frequencies between the studied CPGs. This has raised the possibility, that some rearrangement types, mainly inversions and insertions, may be underrepresented as a consequence of genotyping insufficiency, and a significant portion of heritability may remain unexplained with current genotyping assays. For example, in NGS sequencing results spurious deletions, not validated as real copy number losses may be a consequence of allelic dropout or failed alignment of the reads due to possible breakpoints of other types of rearrangements. Genotyping techniques that also enable sequencing of introns are preferred since the majority of rearrangement breakpoints reside in these regions. Equally important, several deletions affecting more genes may manifest in a multilocus phenotype, modulating the typical symptoms of diseases. Therefore, careful evaluation of the syndromic spectrum is warranted for determining the genotyping eligibility criteria.
Databases
The following curated databases register the detected SVs: InSight LOVD for gastrointestinal hereditary tumors (http://insight-database.org). Breast-and ovarian cancer: ENIGMA, BRCA Exchange (https://brcaexchange.org/variants) and LOVD Fanconi anemia mutation database (https://databases.lovd.nl/shared/genes/BRCA1). General: Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home). Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
Author Contributions
AB, TP, VG, and AP wrote and carried out the original draft preparation. TP, JP, HB, and AP reviewed and edited the manuscript. AP carried out the funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Hungarian Research Grants TUDFO/51757/2019-ITM and TKP2020-NKA-26.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
SV, structural variation; CNV, copy number variation; LGR, large genomic rearrangement; CPG, cancer predisposition gene; HBOC, hereditary breast- and ovarian cancer; FAP, familial adenomatous polyposis; HNPCC, hereditary non-polyposis colorectal cancer
Footnotes
References
Agata, S., Viel, A., Della Puppa, L., Cortesi, L., Fersini, G., Callegaro, M., et al. (2006). Prevalence of BRCA1 genomic rearrangements in a large cohort of Italian breast and breast/ovarian cancer families without detectable BRCA1 and BRCA2 point mutations. Genes Chromosomes Cancer 45, 791–797. doi: 10.1002/gcc.20342
Apostolou, P., Pertesi, M., Aleporou-Marinou, V., Dimitrakakis, C., Papadimitriou, C., Razis, E., et al. (2017). Haplotype analysis reveals that the recurrent BRCA1 deletion of exons 23 and 24 is a Greek founder mutation. Clin. Genet. 91, 482–487. doi: 10.1111/cge.12824
Aretz, S., Stienen, D., Uhlhaas, S., Loff, S., Back, W., Pagenstecher, C., et al. (2005). High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum. Mutat. 26, 513–519. doi: 10.1002/humu.20253
Arkhipova, I. R., and Yushenova, I. A. (2019). Giant transposons in eukaryotes: is bigger better? Genome Biol. Evol. 11, 906–918. doi: 10.1093/gbe/evz041
Armaou, S., Pertesi, M., Fostira, F., Thodi, G., Athanasopoulos, P. S., Kamakari, S., et al. (2009). Contribution of BRCA1 germ-line mutations to breast cancer in Greece: a hospital-based study of 987 unselected breast cancer cases. Br. J. Cancer 101, 32–37. doi: 10.1038/sj.bjc.6605115
Bailey, J. A., Gu, Z., Clark, R. A., Reinert, K., Samonte, R. V., Schwartz, S., et al. (2002). Recent segmental duplications in the human genome. Science 297, 1003–1007. doi: 10.1126/science.1072047
Bayani, J., and Squire, J. A. (2004). Fluorescence in situ Hybridization (FISH). Curr. Protoc. Cell Biol. Chapter 22:Unit 22.4. doi: 10.1002/0471143030.cb2204s23
Belogianni, I., Apessos, A., Mihalatos, M., Razi, E., Labropoulos, S., Petounis, A., et al. (2004). Characterization of a novel large deletion and single point mutations in the BRCA1 gene in a Greek cohort of families with suspected hereditary breast cancer. BMC Cancer 4:61. doi: 10.1186/1471-2407-4-61
Borun, P., De Rosa, M., Nedoszytko, B., Walkowiak, J., and Plawski, A. (2015). Specific Alu elements involved in a significant percentage of copy number variations of the STK11 gene in patients with Peutz-Jeghers syndrome. Fam. Cancer 14, 455–461. doi: 10.1007/s10689-015-9800-5
Bozsik, A., Pocza, T., Papp, J., Vaszko, T., Butz, H., Patocs, A., et al. (2020). Complex characterization of germline large genomic rearrangements of the BRCA1 and BRCA2 genes in high-risk breast cancer patients-novel variants from a Large National Center. Int. J. Mol. Sci. 21:4650. doi: 10.3390/ijms21134650
Butz, H., and Patocs, A. (2019). “Brief summary of the most important molecular genetic methods (PCR, qPCR, microarray, next-generation sequencing, etc.),” in Genetics of Endocrine Diseases and Syndromes, eds P. Igaz and A. Patócs (Cham: Springer), 33–52.
Calva-Cerqueira, D., Chinnathambi, S., Pechman, B., Bair, J., Larsen-Haidle, J., and Howe, J. R. (2009). The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin. Genet. 75, 79–85. doi: 10.1111/j.1399-0004.2008.01091.x
Cantsilieris, S., Baird, P. N., and White, S. J. (2013). Molecular methods for genotyping complex copy number polymorphisms. Genomics 101, 86–93. doi: 10.1016/j.ygeno.2012.10.004
Caux-Moncoutier, V., Castera, L., Tirapo, C., Michaux, D., Remon, M. A., Lauge, A., et al. (2011). EMMA, a cost- and time-effective diagnostic method for simultaneous detection of point mutations and large-scale genomic rearrangements: application to BRCA1 and BRCA2 in 1,525 patients. Hum. Mutat. 32, 325–334. doi: 10.1002/humu.21414
Ceulemans, S., van der Ven, K., and Del-Favero, J. (2012). Targeted screening and validation of copy number variations. Methods Mol. Biol. 838, 311–328. doi: 10.1007/978-1-61779-507-7_15
Chen, J. M. (2008). The 10-Mb paracentric inversion of chromosome arm 2p in activating MSH2 and causing hereditary nonpolyposis colorectal cancer: re-annotation and mutational mechanisms. Genes Chromosomes Cancer 47, 543–545. doi: 10.1002/gcc.20556
Colnaghi, R., Carpenter, G., Volker, M., and O’Driscoll, M. (2011). The consequences of structural genomic alterations in humans: genomic disorders, genomic instability and cancer. Semin. Cell Dev. Biol. 22, 875–885. doi: 10.1016/j.semcdb.2011.07.010
Concolino, P., Rizza, R., Mignone, F., Costella, A., Guarino, D., Carboni, I., et al. (2018). A comprehensive BRCA1/2 NGS pipeline for an immediate Copy Number Variation (CNV) detection in breast and ovarian cancer molecular diagnosis. Clin. Chim. Acta 480, 173–179. doi: 10.1016/j.cca.2018.02.012
Cybulski, C., Wokolorczyk, D., Huzarski, T., Byrski, T., Gronwald, J., Gorski, B., et al. (2007). A deletion in CHEK2 of 5,395 bp predisposes to breast cancer in Poland. Breast Cancer Res. Treat. 102, 119–122. doi: 10.1007/s10549-006-9320-y
Davies, J. J., Wilson, I. M., and Lam, W. L. (2005). Array CGH technologies and their applications to cancer genomes. Chromosome Res. 13, 237–248. doi: 10.1007/s10577-005-2168-x
de la Hoya, M., Gutierrez-Enriquez, S., Velasco, E., Osorio, A., Sanchez de Abajo, A., Vega, A., et al. (2006). Genomic rearrangements at the BRCA1 locus in Spanish families with breast/ovarian cancer. Clin. Chem. 52, 1480–1485. doi: 10.1373/clinchem.2006.070110
Deans, Z. C., Costa, J. L., Cree, I., Dequeker, E., Edsjo, A., Henderson, S., et al. (2017). Integration of next-generation sequencing in clinical diagnostic molecular pathology laboratories for analysis of solid tumours; an expert opinion on behalf of IQN Path ASBL. Virchows Arch. 470, 5–20. doi: 10.1007/s00428-016-2025-7
Delnatte, C., Sanlaville, D., Mougenot, J. F., Vermeesch, J. R., Houdayer, C., Blois, M. C., et al. (2006). Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am. J. Hum. Genet. 78, 1066–1074. doi: 10.1086/504301
Du, C., Mark, D., Wappenschmidt, B., Bockmann, B., Pabst, B., Chan, S., et al. (2018). A tandem duplication of BRCA1 exons 1-19 through DHX8 exon 2 in four families with hereditary breast and ovarian cancer syndrome. Breast Cancer Res. Treat. 172, 561–569. doi: 10.1007/s10549-018-4957-x
Engert, S., Wappenschmidt, B., Betz, B., Kast, K., Kutsche, M., Hellebrand, H., et al. (2008). MLPA screening in the BRCA1 gene from 1,506 German hereditary breast cancer cases: novel deletions, frequent involvement of exon 17, and occurrence in single early-onset cases. Hum. Mutat. 29, 948–958. doi: 10.1002/humu.20723
Fachal, L., Blanco, A., Santamarina, M., Carracedo, A., and Vega, A. (2014). Large genomic rearrangements of BRCA1 and BRCA2 among patients referred for genetic analysis in Galicia (NW Spain): delimitation and mechanism of three novel BRCA1 rearrangements. PLoS One 9:e93306. doi: 10.1371/journal.pone.0093306
Firth, H. V., Richards, S. M., Bevan, A. P., Clayton, S., Corpas, M., Rajan, D., et al. (2009). DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 84, 524–533. doi: 10.1016/j.ajhg.2009.03.010
Gad, S., Bieche, I., Barrois, M., Casilli, F., Pages-Berhouet, S., Dehainault, C., et al. (2003). Characterisation of a 161 kb deletion extending from the NBR1 to the BRCA1 genes in a French breast-ovarian cancer family. Hum. Mutat. 21:654. doi: 10.1002/humu.9148
Goodier, J. L. (2016). Restricting retrotransposons: a review. Mob. DNA 7:16. doi: 10.1186/s13100-016-0070-z
Hancks, D. C. Jr., and Kazazian, H. H. Jr. (2016). Roles for retrotransposon insertions in human disease. Mob. DNA 7:9. doi: 10.1186/s13100-016-0065-9
Hansen, T., Jonson, L., Albrechtsen, A., Andersen, M. K., Ejlertsen, B., and Nielsen, F. C. (2009). Large BRCA1 and BRCA2 genomic rearrangements in Danish high risk breast-ovarian cancer families. Breast Cancer Res. Treat. 115, 315–323. doi: 10.1007/s10549-008-0088-0
Hastings, P. J., Ira, G., and Lupski, J. R. (2009a). A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 5:e1000327. doi: 10.1371/journal.pgen.1000327
Hastings, P. J., Lupski, J. R., Rosenberg, S. M., and Ira, G. (2009b). Mechanisms of change in gene copy number. Nat. Rev. Genet. 10, 551–564. doi: 10.1038/nrg2593
Hogervorst, F. B., Nederlof, P. M., Gille, J. J., McElgunn, C. J., Grippeling, M., Pruntel, R., et al. (2003). Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 63, 1449–1453.
James, P. A., Sawyer, S., Boyle, S., Young, M. A., Kovalenko, S., Doherty, R., et al. (2015). Large genomic rearrangements in the familial breast and ovarian cancer gene BRCA1 are associated with an increased frequency of high risk features. Fam. Cancer 14, 287–295. doi: 10.1007/s10689-015-9785-0
Janatova, M., Kleibl, Z., Stribrna, J., Panczak, A., Vesela, K., Zimovjanova, M., et al. (2013). The PALB2 gene is a strong candidate for clinical testing in BRCA1- and BRCA2-negative hereditary breast cancer. Cancer Epidemiol. Biomark. Prev. 22, 2323–2332. doi: 10.1158/1055-9965.EPI-13-0745-T
Judkins, T., Rosenthal, E., Arnell, C., Burbidge, L. A., Geary, W., Barrus, T., et al. (2012). Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer 118, 5210–5216. doi: 10.1002/cncr.27556
Kaer, K., Branovets, J., Hallikma, A., Nigumann, P., and Speek, M. (2011). Intronic L1 retrotransposons and nested genes cause transcriptional interference by inducing intron retention, exonization and cryptic polyadenylation. PLoS One 6:e26099. doi: 10.1371/journal.pone.0026099
Kerr, S. E., Thomas, C. B., Thibodeau, S. N., Ferber, M. J., and Halling, K. C. (2013). APC germline mutations in individuals being evaluated for familial adenomatous polyposis: a review of the Mayo Clinic experience with 1591 consecutive tests. J. Mol. Diagn. 15, 31–43. doi: 10.1016/j.jmoldx.2012.07.005
Kleiblova, P., Stolarova, L., Krizova, K., Lhota, F., Hojny, J., Zemankova, P., et al. (2019). Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int. J. Cancer 145, 1782–1797. doi: 10.1002/ijc.32385
Knudson, A. G. Jr. (1971). Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U.S.A. 68, 820–823. doi: 10.1073/pnas.68.4.820
Kohlmann, W., and Gruber, S. B. (1993). “Lynch syndrome,” in GeneReviews((R)), eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Konstantopoulou, I., Tsitlaidou, M., Fostira, F., Pertesi, M., Stavropoulou, A. V., Triantafyllidou, O., et al. (2014). High prevalence of BRCA1 founder mutations in Greek breast/ovarian families. Clin. Genet. 85, 36–42. doi: 10.1111/cge.12274
Kovacs, M. E., Papp, J., Szentirmay, Z., Otto, S., and Olah, E. (2009). Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum. Mutat. 30, 197–203. doi: 10.1002/humu.20942
Kowalczykowski, S. C. (2015). An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 7:a016410. doi: 10.1101/cshperspect.a016410
Kozlowski, P., Jasinska, A. J., and Kwiatkowski, D. J. (2008). New applications and developments in the use of multiplex ligation-dependent probe amplification. Electrophoresis 29, 4627–4636. doi: 10.1002/elps.200800126
Krumm, N., Sudmant, P. H., Ko, A., O’Roak, B. J., Malig, M., Coe, B. P., et al. (2012). Copy number variation detection and genotyping from exome sequence data. Genome Res. 22, 1525–1532. doi: 10.1101/gr.138115.112
Kumaran, M., Cass, C. E., Graham, K., Mackey, J. R., Hubaux, R., Lam, W., et al. (2017). Germline copy number variations are associated with breast cancer risk and prognosis. Sci. Rep. 7:14621. doi: 10.1038/s41598-017-14799-7
Kwong, A., Chen, J., Shin, V. Y., Ho, J. C., Law, F. B., Au, C. H., et al. (2015). The importance of analysis of long-range rearrangement of BRCA1 and BRCA2 in genetic diagnosis of familial breast cancer. Cancer Genet. 208, 448–454. doi: 10.1016/j.cancergen.2015.05.031
Li, Y., Salo-Mullen, E., Varghese, A., Trottier, M., Stadler, Z. K., and Zhang, L. (2020). Insertion of an Alu-like element in MLH1 intron 7 as a novel cause of Lynch syndrome. Mol. Genet. Genomic Med. 8:e1523. doi: 10.1002/mgg3.1523
Ligtenberg, M. J., Kuiper, R. P., Chan, T. L., Goossens, M., Hebeda, K. M., Voorendt, M., et al. (2009). Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat. Genet. 41, 112–117. doi: 10.1038/ng.283
Liu, Q., Hesson, L. B., Nunez, A. C., Packham, D., Williams, R., Ward, R. L., et al. (2016). A cryptic paracentric inversion of MSH2 exons 2-6 causes Lynch syndrome. Carcinogenesis 37, 10–17. doi: 10.1093/carcin/bgv154
Lopez-Urrutia, E., Salazar-Rojas, V., Brito-Elias, L., Coca-Gonzalez, M., Silva-Garcia, J., Sanchez-Marin, D., et al. (2019). BRCA mutations: is everything said? Breast Cancer Res. Treat. 173, 49–54. doi: 10.1007/s10549-018-4986-5
Machado, P. M., Brandao, R. D., Cavaco, B. M., Eugenio, J., Bento, S., Nave, M., et al. (2007). Screening for a BRCA2 rearrangement in high-risk breast/ovarian cancer families: evidence for a founder effect and analysis of the associated phenotypes. J. Clin. Oncol. 25, 2027–2034. doi: 10.1200/JCO.2006.06.9443
Maher, E. R., Webster, A. R., Richards, F. M., Green, J. S., Crossey, P. A., Payne, S. J., et al. (1996). Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J. Med. Genet. 33, 328–332. doi: 10.1136/jmg.33.4.328
Mancini-DiNardo, D., Judkins, T., Kidd, J., Bernhisel, R., Daniels, C., Brown, K., et al. (2019). Detection of large rearrangements in a hereditary pan-cancer panel using next-generation sequencing. BMC Med. Genomics 12:138. doi: 10.1186/s12920-019-0587-3
Marabelli, M., Gismondi, V., Ricci, M. T., Vetro, A., Abou Khouzam, R., Rea, V., et al. (2017). A novel APC promoter 1B deletion shows a founder effect in Italian patients with classical familial adenomatous polyposis phenotype. Genes Chromosomes Cancer 56, 846–854. doi: 10.1002/gcc.22488
Masson, A. L., Talseth-Palmer, B. A., Evans, T. J., Grice, D. M., Hannan, G. N., and Scott, R. J. (2014). Expanding the genetic basis of copy number variation in familial breast cancer. Hered. Cancer Clin. Pract. 12:15. doi: 10.1186/1897-4287-12-15
McVety, S., Younan, R., Li, L., Gordon, P. H., Wong, N., Foulkes, W. D., et al. (2005). Novel genomic insertion– deletion in MLH1: possible mechanistic role for non-homologous end-joining DNA repair. Clin. Genet. 68, 234–238. doi: 10.1111/j.1399-0004.2005.00486.x
McVey, M., and Lee, S. E. (2008). MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 24, 529–538. doi: 10.1016/j.tig.2008.08.007
Michils, G., Tejpar, S., Thoelen, R., van Cutsem, E., Vermeesch, J. R., Fryns, J. P., et al. (2005). Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): a Belgian study. Hum. Mutat. 25, 125–134. doi: 10.1002/humu.20122
Miki, Y., Nishisho, I., Horii, A., Miyoshi, Y., Utsunomiya, J., Kinzler, K. W., et al. (1992). Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 52, 643–645.
Molinaro, V., Pensotti, V., Marabelli, M., Feroce, I., Barile, M., Pozzi, S., et al. (2014). Complementary molecular approaches reveal heterogeneous CDH1 germline defects in Italian patients with hereditary diffuse gastric cancer (HDGC) syndrome. Genes Chromosomes Cancer 53, 432–445. doi: 10.1002/gcc.22155
Morak, M., Koehler, U., Schackert, H. K., Steinke, V., Royer-Pokora, B., Schulmann, K., et al. (2011). Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 48, 513–519. doi: 10.1136/jmedgenet-2011-100050
Newman, S., Hermetz, K. E., Weckselblatt, B., and Rudd, M. K. (2015). Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am. J. Hum. Genet. 96, 208–220. doi: 10.1016/j.ajhg.2014.12.017
Nizic-Kos, T., Krajc, M., Blatnik, A., Stegel, V., Skerl, P., Novakovic, S., et al. (2020). Bilateral disease common among slovenian CHEK2-positive breast cancer patients. Ann. Surg. Oncol. [Epub ahead of print]. doi: 10.1245/s10434-020-09178-y
Oliver, G. R., Hart, S. N., and Klee, E. W. (2015). Bioinformatics for clinical next generation sequencing. Clin. Chem. 61, 124–135. doi: 10.1373/clinchem.2014.224360
Palanca, S., de Juan, I., Perez-Simo, G., Barragan, E., Chirivella, I., Martinez, E., et al. (2013). The deletion of exons 3-5 of BRCA1 is the first founder rearrangement identified in breast and/or ovarian cancer Spanish families. Fam. Cancer 12, 119–123. doi: 10.1007/s10689-012-9579-6
Palmero, E. I., Carraro, D. M., Alemar, B., Moreira, M. A. M., Ribeiro-Dos-Santos, A., Abe-Sandes, K., et al. (2018). The germline mutational landscape of BRCA1 and BRCA2 in Brazil. Sci. Rep. 8:9188. doi: 10.1038/s41598-018-27315-2
Pardi, E., Borsari, S., Saponaro, F., Bogazzi, F., Urbani, C., Mariotti, S., et al. (2017). Mutational and large deletion study of genes implicated in hereditary forms of primary hyperparathyroidism and correlation with clinical features. PLoS One 12:e0186485. doi: 10.1371/journal.pone.0186485
Peixoto, A., Santos, C., Pinheiro, M., Pinto, P., Soares, M. J., Rocha, P., et al. (2011). International distribution and age estimation of the Portuguese BRCA2 c.156_157insAlu founder mutation. Breast Cancer Res. Treat. 127, 671–679. doi: 10.1007/s10549-010-1036-3
Perez-Cabornero, L., Borras Flores, E., Infante Sanz, M., Velasco Sampedro, E., Acedo Becares, A., Lastra Aras, E., et al. (2011). Characterization of new founder Alu-mediated rearrangements in MSH2 gene associated with a Lynch syndrome phenotype. Cancer Prev. Res. 4, 1546–1555. doi: 10.1158/1940-6207.CAPR-11-0227
Petrij-Bosch, A., Peelen, T., van Vliet, M., van Eijk, R., Olmer, R., Drusedau, M., et al. (1997). BRCA1 genomic deletions are major founder mutations in Dutch breast cancer patients. Nat. Genet. 17, 341–345. doi: 10.1038/ng1197-341
Pinhero, R., Pazhekattu, R., Marangoni, A. G., Liu, Q., and Yada, R. Y. (2011). Alleviation of low temperature sweetening in potato by expressing Arabidopsis pyruvate decarboxylase gene and stress-inducible rd29A : a preliminary study. Physiol. Mol. Biol. Plants 17, 105–114. doi: 10.1007/s12298-011-0056-8
Preisler-Adams, S., Schonbuchner, I., Fiebig, B., Welling, B., Dworniczak, B., and Weber, B. H. (2006). Gross rearrangements in BRCA1 but not BRCA2 play a notable role in predisposition to breast and ovarian cancer in high-risk families of German origin. Cancer Genet. Cytogenet. 168, 44–49. doi: 10.1016/j.cancergencyto.2005.07.005
Puget, N., Gad, S., Perrin-Vidoz, L., Sinilnikova, O. M., Stoppa-Lyonnet, D., Lenoir, G. M., et al. (2002). Distinct BRCA1 rearrangements involving the BRCA1 pseudogene suggest the existence of a recombination hot spot. Am. J. Hum. Genet. 70, 858–865. doi: 10.1086/339434
Qian, Y., Mancini-DiNardo, D., Judkins, T., Cox, H. C., Brown, K., Elias, M., et al. (2017). Identification of pathogenic retrotransposon insertions in cancer predisposition genes. Cancer Genet. 21, 159–169. doi: 10.1016/j.cancergen.2017.08.002
Rhees, J., Arnold, M., and Boland, C. R. (2014). Inversion of exons 1-7 of the MSH2 gene is a frequent cause of unexplained Lynch syndrome in one local population. Fam. Cancer 13, 219–225. doi: 10.1007/s10689-013-9688-x
Riggs, E. R., Wain, K. E., Riethmaier, D., Savage, M., Smith-Packard, B., Kaminsky, E. B., et al. (2013). Towards a universal clinical genomics database: the 2012 international standards for cytogenomic arrays consortium meeting. Hum. Mutat. 34, 915–919. doi: 10.1002/humu.22306
Robson, M. E., Bradbury, A. R., Arun, B., Domchek, S. M., Ford, J. M., Hampel, H. L., et al. (2015). American society of clinical oncology policy statement update: genetic and genomic testing for cancer susceptibility. J. Clin. Oncol. 33, 3660–3667. doi: 10.1200/JCO.2015.63.0996
Rudnicka, H., Debniak, T., Cybulski, C., Huzarski, T., Gronwald, J., Lubinski, J., et al. (2013). Large BRCA1 and BRCA2 genomic rearrangements in Polish high-risk breast and ovarian cancer families. Mol. Biol. Rep. 40, 6619–6623. doi: 10.1007/s11033-013-2775-0
Sahnane, N., Bernasconi, B., Carnevali, I., Furlan, D., Viel, A., Sessa, F., et al. (2016). Disruption of the APC gene by t(5;7) translocation in a Turcot family. Cancer Genet. 209, 107–111. doi: 10.1016/j.cancergen.2015.12.003
Salo-Mullen, E. E., Lynn, P. B., Wang, L., Walsh, M., Gopalan, A., Shia, J., et al. (2018). Contiguous gene deletion of chromosome 2p16.3-p21 as a cause of Lynch syndrome. Fam. Cancer 17, 71–77. doi: 10.1007/s10689-017-0006-x
Sarkadi, B., Grolmusz, V. K., Butz, H., Kovesdi, A., Liko, I., Nyiro, G., et al. (2018). [Evolution of molecular genetic methods in the clinical diagnosis of hereditary endocrine tumour syndromes]. Orv. Hetil. 159, 285–292. doi: 10.1556/650.2018.31036
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Schmitz, J., and Brosius, J. (2011). Exonization of transposed elements: a challenge and opportunity for evolution. Biochimie 93, 1928–1934. doi: 10.1016/j.biochi.2011.07.014
Sen, S. K., Han, K., Wang, J., Lee, J., Wang, H., Callinan, P. A., et al. (2006). Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 79, 41–53. doi: 10.1086/504600
Senter, L., Clendenning, M., Sotamaa, K., Hampel, H., Green, J., Potter, J. D., et al. (2008). The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135, 419–428. doi: 10.1053/j.gastro.2008.04.026
Seong, M. W., Cho, S. I., Kim, K. H., Chung, I. Y., Kang, E., Lee, J. W., et al. (2014). A multi-institutional study of the prevalence of BRCA1 and BRCA2 large genomic rearrangements in familial breast cancer patients. BMC Cancer 14:645. doi: 10.1186/1471-2407-14-645
Serizawa, R. R., Ralfkiaer, U., Dahl, C., Lam, G. W., Hansen, A. B., Steven, K., et al. (2010). Custom-designed MLPA using multiple short synthetic probes: application to methylation analysis of five promoter CpG islands in tumor and urine specimens from patients with bladder cancer. J. Mol. Diagn. 12, 402–408. doi: 10.2353/jmoldx.2010.090152
Sfeir, A., and Symington, L. S. (2015). Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 40, 701–714. doi: 10.1016/j.tibs.2015.08.006
Sinha, S., Villarreal, D., Shim, E. Y., and Lee, S. E. (2016). Risky business: microhomology-mediated end joining. Mutat. Res. 788, 17–24. doi: 10.1016/j.mrfmmm.2015.12.005
Sluiter, M. D., and van Rensburg, E. J. (2011). Large genomic rearrangements of the BRCA1 and BRCA2 genes: review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res. Treat. 125, 325–349. doi: 10.1007/s10549-010-0817-z
Smith, M. J., Urquhart, J. E., Harkness, E. F., Miles, E. K., Bowers, N. L., Byers, H. J., et al. (2016). The contribution of whole gene deletions and large rearrangements to the mutation spectrum in inherited tumor predisposing syndromes. Hum. Mutat. 37, 250–256. doi: 10.1002/humu.22938
Teugels, E., De Brakeleer, S., Goelen, G., Lissens, W., Sermijn, E., and De Greve, J. (2005). De novo Alu element insertions targeted to a sequence common to the BRCA1 and BRCA2 genes. Hum. Mutat. 26:284. doi: 10.1002/humu.9366
The BEDSG (2000). The exon 13 duplication in the BRCA1 gene is a founder mutation present in geographically diverse populations. The BRCA1 Exon 13 duplication screening group. Am. J. Hum. Genet. 67, 207–212.
Thomassen, M., Gerdes, A. M., Cruger, D., Jensen, P. K., and Kruse, T. A. (2006). Low frequency of large genomic rearrangements of BRCA1 and BRCA2 in western Denmark. Cancer Genet. Cytogenet. 168, 168–171. doi: 10.1016/j.cancergencyto.2005.12.016
Ticha, I., Kleibl, Z., Stribrna, J., Kotlas, J., Zimovjanova, M., Mateju, M., et al. (2010). Screening for genomic rearrangements in BRCA1 and BRCA2 genes in Czech high-risk breast/ovarian cancer patients: high proportion of population specific alterations in BRCA1 gene. Breast Cancer Res. Treat. 124, 337–347. doi: 10.1007/s10549-010-0745-y
Tournier, I., Paillerets, B. B., Sobol, H., Stoppa-Lyonnet, D., Lidereau, R., Barrois, M., et al. (2004). Significant contribution of germline BRCA2 rearrangements in male breast cancer families. Cancer Res. 64, 8143–8147. doi: 10.1158/0008-5472.CAN-04-2467
van der Klift, H., Wijnen, J., Wagner, A., Verkuilen, P., Tops, C., Otway, R., et al. (2005). Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 44, 123–138. doi: 10.1002/gcc.20219
van der Klift, H. M., Tops, C. M., Hes, F. J., Devilee, P., and Wijnen, J. T. (2012). Insertion of an SVA element, a nonautonomous retrotransposon, in PMS2 intron 7 as a novel cause of Lynch syndrome. Hum. Mutat. 33, 1051–1055. doi: 10.1002/humu.22092
van der Luijt, R. B., Tops, C. M., Khan, P. M., van der Klift, H. M., Breukel, C., van Leeuwen-Cornelisse, I. S., et al. (1995). Molecular, cytogenetic, and phenotypic studies of a constitutional reciprocal translocation t(5;10)(q22;q25) responsible for familial adenomatous polyposis in a Dutch pedigree. Genes Chromosomes Cancer 13, 192–202. doi: 10.1002/gcc.2870130309
van Luttikhuizen, J. L., Bublitz, J., Schubert, S., Schmidt, G., Hofmann, W., Morlot, S., et al. (2020). From a variant of unknown significance to pathogenic: reclassification of a large novel duplication in BRCA2 by high-throughput sequencing. Mol. Genet. Genomic Med. 8:e1045. doi: 10.1002/mgg3.1045
Vasickova, P., Machackova, E., Lukesova, M., Damborsky, J., Horky, O., Pavlu, H., et al. (2007). High occurrence of BRCA1 intragenic rearrangements in hereditary breast and ovarian cancer syndrome in the Czech Republic. BMC Med. Genet. 8:32. doi: 10.1186/1471-2350-8-32
Vaughn, C. P., Robles, J., Swensen, J. J., Miller, C. E., Lyon, E., Mao, R., et al. (2010). Clinical analysis of PMS2: mutation detection and avoidance of pseudogenes. Hum. Mutat. 31, 588–593. doi: 10.1002/humu.21230
Wagner, A., van der Klift, H., Franken, P., Wijnen, J., Breukel, C., Bezrookove, V., et al. (2002). A 10-Mb paracentric inversion of chromosome arm 2p inactivates MSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosomes Cancer 35, 49–57. doi: 10.1002/gcc.10094
Walsh, T., Casadei, S., Coats, K. H., Swisher, E., Stray, S. M., Higgins, J., et al. (2006). Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295, 1379–1388. doi: 10.1001/jama.295.12.1379
Weckselblatt, B., and Rudd, M. K. (2015). Human structural variation: mechanisms of chromosome rearrangements. Trends Genet. 31, 587–599. doi: 10.1016/j.tig.2015.05.010
Weitzel, J. N., Lagos, V. I., Herzog, J. S., Judkins, T., Hendrickson, B., Ho, J. S., et al. (2007). Evidence for common ancestral origin of a recurring BRCA1 genomic rearrangement identified in high-risk Hispanic families. Cancer Epidemiol. Biomark. Prev. 16, 1615–1620. doi: 10.1158/1055-9965.EPI-07-0198
Yamaguchi, K., Nagayama, S., Shimizu, E., Komura, M., Yamaguchi, R., Shibuya, T., et al. (2016). Reduced expression of APC-1B but not APC-1A by the deletion of promoter 1B is responsible for familial adenomatous polyposis. Sci. Rep. 6:26011. doi: 10.1038/srep26011
Zhang, F., Gu, W., Hurles, M. E., and Lupski, J. R. (2009). Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 10, 451–481. doi: 10.1146/annurev.genom.9.081307.164217
Zhao, B., Rothenberg, E., Ramsden, D. A., and Lieber, M. R. (2020). The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 21, 765–781. doi: 10.1038/s41580-020-00297-8
Keywords: germline mutation, structural variations, cancer–predisposing genes, copy number variation, large genomic rearrangement, structural variation
Citation: Pócza T, Grolmusz VK, Papp J, Butz H, Patócs A and Bozsik A (2021) Germline Structural Variations in Cancer Predisposition Genes. Front. Genet. 12:634217. doi: 10.3389/fgene.2021.634217
Received: 27 November 2020; Accepted: 08 March 2021;
Published: 14 April 2021.
Edited by:
Judit Bene, University of Pécs, HungaryReviewed by:
Silvia R. Rogatto, University of Southern Denmark, DenmarkPilar Garre, San Carlos University Clinical Hospital, Spain
Tamás I. Orbán, Hungarian Academy of Sciences (MTA), Hungary
Copyright © 2021 Pócza, Grolmusz, Papp, Butz, Patócs and Bozsik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anikó Bozsik, bozsik.aniko@oncol.hu; genetikus@gmail.com
†These authors have contributed equally to this work