 Dominick Matteau
Dominick Matteau Anthony Duval1
Anthony Duval1 Vincent Baby
Vincent Baby Sébastien Rodrigue
Sébastien Rodrigue- 1Département de biologie, Université de Sherbrooke, Sherbrooke, QC, Canada
- 2Centre de diagnostic vétérinaire de l’Université de Montréal, Université de Montréal, Saint-Hyacinthe, QC, Canada
Mesoplasma florum is an emerging model organism for systems and synthetic biology due to its small genome (∼800 kb) and fast growth rate. While M. florum was isolated and first described almost 40 years ago, many important aspects of its biology have long remained uncharacterized due to technological limitations, the absence of dedicated molecular tools, and since this bacterial species has not been associated with any disease. However, the publication of the first M. florum genome in 2004 paved the way for a new era of research fueled by the rise of systems and synthetic biology. Some of the most important studies included the characterization and heterologous use of M. florum regulatory elements, the development of the first replicable plasmids, comparative genomics and transposon mutagenesis, whole-genome cloning in yeast, genome transplantation, in-depth characterization of the M. florum cell, as well as the development of a high-quality genome-scale metabolic model. The acquired data, knowledge, and tools will greatly facilitate future genome engineering efforts in M. florum, which could next be exploited to rationally design and create synthetic cells to advance fundamental knowledge or for specific applications.
Introduction
Mollicutes form a group of bacteria characterized by the absence of a cell wall and exceptionally small genomes. During the past decades, the field of molecular and cellular biology experienced significant advances, leading to a heightened interest for this class of bacteria. As new molecular data was generated, more particularly about the mycoplasmas, the idea that these microorganisms could actually be the simplest self-replicating life forms existing on Earth was becoming increasingly plausible (Morowitz and Tourtellotte, 1962; Morowitz, 1984). The minimal genome concept started to emerge: what is the smallest set of genes required for autonomous life, and what functions do they encode? Are there many or only one possible combination of genes composing a minimal genome? If we could understand the function of every single gene in a cell, we would have a better comprehension of the most fundamental principles of life (Peterson et al., 2001; Glass et al., 2017; Lachance et al., 2019). Just as the study of the hydrogen atom was fundamental in developing the laws of quantum physics, examining the simplest autonomous cells presented itself as the most logical starting point for this endeavor (Morowitz and Tourtellotte, 1962; Morowitz, 1984). An impressive number of Mollicutes species were isolated during the 1980s and 1990s, including many species associated with plants and insects (Whitcomb and Tully, 1995; Pettersson and Johansson, 2002). Unlike most mycoplasmas, which are typically parasitic, many of these species appeared to be commensals, coexisting in a mutually beneficial relationship with a variety of animal hosts. Many of these isolates also showed no strict requirement of sterols or cholesterol for growth in vitro, and were initially regrouped under the genus name Acholeplasma (Tully, 1979; Tully, 1983; Tully et al., 1990; Tully et al., 1993). This was the case for Mesoplasma florum, a bacterium that has become an interesting model organism for the fields of systems and synthetic biology.
What is Mesoplasma florum?
M. florum is a small (0.5–0.6 µm), ovoid, near-minimal and non-pathogenic bacterium of the Mollicutes class (Figure 1A) initially described for the first time as Acholeplasma florum in 1984 by McCoy and colleagues (McCoy et al., 1984). The species was named after its recovery site-the flowers of healthy plants found in Florida, United States. M. florum L1, the type strain of the species, was isolated from flowers of a lemon tree (Citrus limon) (McCoy et al., 1980; McCoy et al., 1984). Since M. florum grew in culture media without sterols it was originally classified in the genus Acholeplasma (Tully, 1979; Tully, 1983; Clark et al., 1986; Tully et al., 1990). However, this species was reassigned to the Mesoplasma genus in 1993 according to new physiological and molecular evidence, including phylogenetic clustering based on 16S rRNA sequence analysis (Tully et al., 1993). M. florum is in fact closely related to a phylogenetically distinct group of mycoplasmas called the mycoides cluster (Figure 1B). This cluster notably includes Mycoplasma mycoides and Mycoplasma capricolum, two well-known model organisms for the fields of systems and synthetic biology (Sirand-Pugnet et al., 2007; Glass et al., 2017; Lachance et al., 2019). Yet, in contrast to M. mycoides and M. capricolum, M. florum has never been associated with any disease, and no virulence factor has been identified in its genome. As for other members of the class Mollicutes, M. florum does not have a cell wall and its genome is particularly small, varying from 738,512 (BARC 787) to 830,640 bp (W20) depending on the exact strain, with an average GC content of about 27% (Baby et al., 2018b). M. florum genes are predominantly oriented according to the direction of DNA replication, frequently expressed as polygenic transcriptional units, and occupy most of the genome space, typical of bacterial genomes (Baby et al., 2018b; Matteau et al., 2020). This bacterium also uses an alternative genetic code (the Mycoplasma and Spiroplasma code) in which the canonical UGA stop codon rather codes for the incorporation of a tryptophan (Navas-Castillo et al., 1992). This distinctive feature is also present in mycoplasmas of the mycoides cluster as well as in the phylogenetically related Mollicute Spiroplasma citri, the causative agent of the Citrus stubborn disease (Saglio et al., 1973).
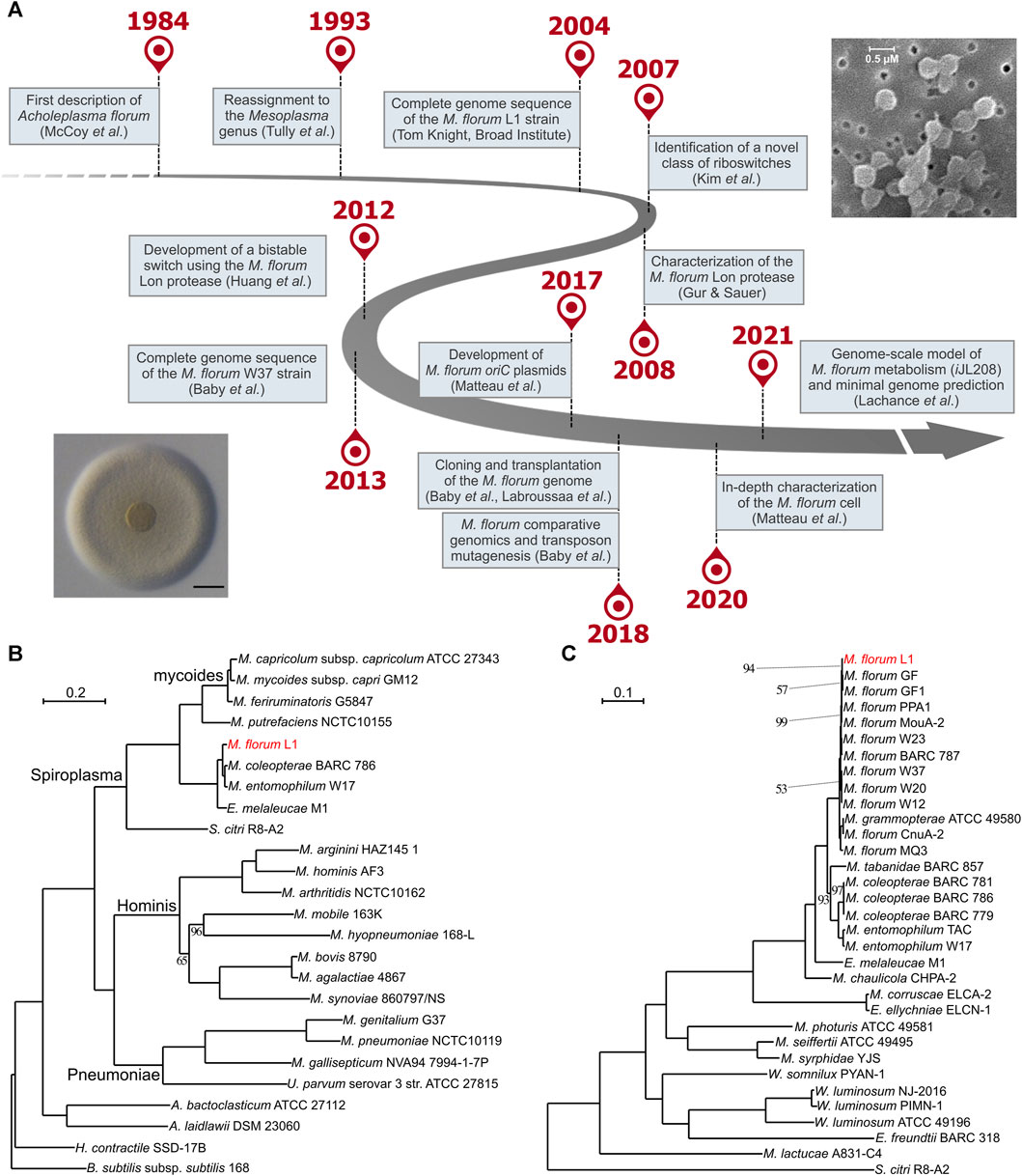
FIGURE 1. Forty years of research on Mesoplasma florum. (A) Important milestones in M. florum research timeline. Representative picture of an M. florum L1 colony displaying the typical “fried-egg” morphology (adapted from Labroussaa et al., 2016, Vol. 44, No. 17, pp. 8501–8511, by permission of Oxford University Press; scale bar: 100 µm) as well as M. florum L1 cells observed by scanning electron microscopy (Baby et al., 2013) are also depicted. (B) and (C) Maximum-likelihood phylogenetic trees of the Mollicutes (B) and the Mesoplasma/Entomoplasma genera (C) inferred using concatenated alignments of 109 and 229 conserved proteins, respectively. Trees were constructed using RAxML (Stamatakis, 2014) with 150 bootstrap replicates as determined using the autoFC bootstopping criterion. Bootstrap replicate values are of 100 unless specified otherwise. Bacillus subtilis and S. citri were used as outgroups. See Table 1 for additional information on strains and genomes included in the Mesoplasma/Entomoplasma phylogenetic tree.
Among all previously isolated M. florum strains, the L1 strain is the most extensively studied. Compared to most Mollicutes, M. florum L1 shows a remarkably fast growth rate, corresponding to a doubling time of ∼32 min at the optimal growth temperature (34°C) (Matteau et al., 2020). In comparison, M. mycoides subspecies capri has a doubling time of ∼60 min in similar conditions (Gibson et al., 2010; Hutchison et al., 2016), whereas for M. capricolum subspecies capricolum and Mycoplasma pneumoniae this value is estimated to be around 90 min and 8–20 h, respectively (Seto and Miyata, 1998; Yus et al., 2009; Wodke et al., 2013). Since Mollicutes have experienced massive gene loss events through evolution, they have lost the capacity to synthesize many metabolites, resulting in an important simplification of their metabolism (Sirand-Pugnet et al., 2007). In M. florum, for example, most of the biosynthesis occurs through salvage pathways, and the energy production relies exclusively on glycolysis and fermentation since no respiratory system is present (Lachance et al., 2021). Consequently, this bacterium, as for most Mollicutes, requires a very rich medium to palliate its metabolic deficiencies in vitro. The most common growth medium for M. florum is the ATCC 1161, a complex and undefined medium containing horse serum, yeast extract, and heart infusion broth. Other similar media such as SP5 have also been used (Whitcomb et al., 1982; McCoy et al., 1984; Pollack and Williams, 1996; Matteau et al., 2015; Matteau et al., 2020; Baby et al., 2018a). M. florum L1 colonies display the typical Mollicutes “fried-egg” appearance on solid medium (Figure 1A) (McCoy et al., 1984; Tully et al., 1994; Labroussaa et al., 2016), and batch cultures growing in ATCC 1161 display the four typical bacterial growth phases (lag, exponential, stationary, and decline), reaching up to ∼1010 cells/mL at the end of the exponential growth phase (Matteau et al., 2015; Matteau et al., 2020). M. florum growth rate is however highly limited by the concentration of horse serum and yeast extract present in the medium, clearly demonstrating the dependance of this bacterium on pre-assembled building blocks for its metabolism (Lachance et al., 2021). The end of the exponential phase also coincides with an important drop in the pH of the medium, most likely due to the accumulation of lactate and acetate fermentation products (Pollack and Williams, 1996; Matteau et al., 2020; Lachance et al., 2021). This decrease in the medium’s pH is likely to be responsible for the progressive death of the M. florum cell population after the stationary phase. Indeed, no significant mortality is observed when the exponential phase is maintained using a continuous culture device (Matteau et al., 2015).
Where does M. florum primarily live?
M. florum is hypothesized to live primarily inside the gastrointestinal tract of insects, which would provide continuous access to complex nutrients such as sugars, lipids, peptides, and other metabolites required for growth. The continuous flow of the digestive tract would also prevent the accumulation of fermentation products and the possible acidification of the milieu, acting similar to a continuous culture device (Matteau et al., 2015). This natural habitat would also explain the presence of this bacterium on plant surfaces as insects would carry them from site to site and excrete the microbe through their feces (Whitcomb et al., 1982; McCoy et al., 1984; Tully et al., 1990). The extracellular polysaccharide layer surrounding M. florum cells, which was shown to occupy for up to 5% of the total M. florum biomass, probably contributes to the survivability of this microorganism on plant surfaces (Matteau et al., 2020; Lachance et al., 2021). Mainly composed of galactose and glucose, this capsule-like structure might provide a physical protection against desiccation, and therefore participate in the dissemination of M. florum across insect populations. The possibility that M. florum uses plants as secondary hosts like some pathogenic spiroplasmas seems rather unlikely since no such observation has ever been reported and M. florum has never been isolated in the context of a plant disease.
The full range of hosts susceptible to M. florum colonization and the possibility of a predominant association with specific insect types are still not well-defined. Although a few strains were directly recovered from the gut content of insects such as soldier beetles (Cantharidae) as well as Vespid wasps (Monobia quadridens) (Clark et al., 1986; Tully et al., 1987), most of the previously described M. florum strains were originally isolated from plant flowers (Table 1). This prevents their direct association with an insect host. Still, the isolation source of closely related species, especially species of the Mesoplasma and Entomoplasma genera, suggests that M. florum could potentially be found in a wide variety of insects, including firefly beetles (Ellychnia corrusca), goldenrod soldier beetles (Chauliognathus pennsylvanicus), as well as tabanid (Tabanus catenatus) and syrphid (Syrphidae) flies (Clark et al., 1986; Tully et al., 1987; Tully et al., 1989; Tully et al., 1994). Furthermore, Mesoplasma and Entomoplasma have intermixed relationships and recent phylogenetic data suggest that they should no longer be taxonomically separated (Gasparich and Chih-Horng, 2019) (Figure 1C).
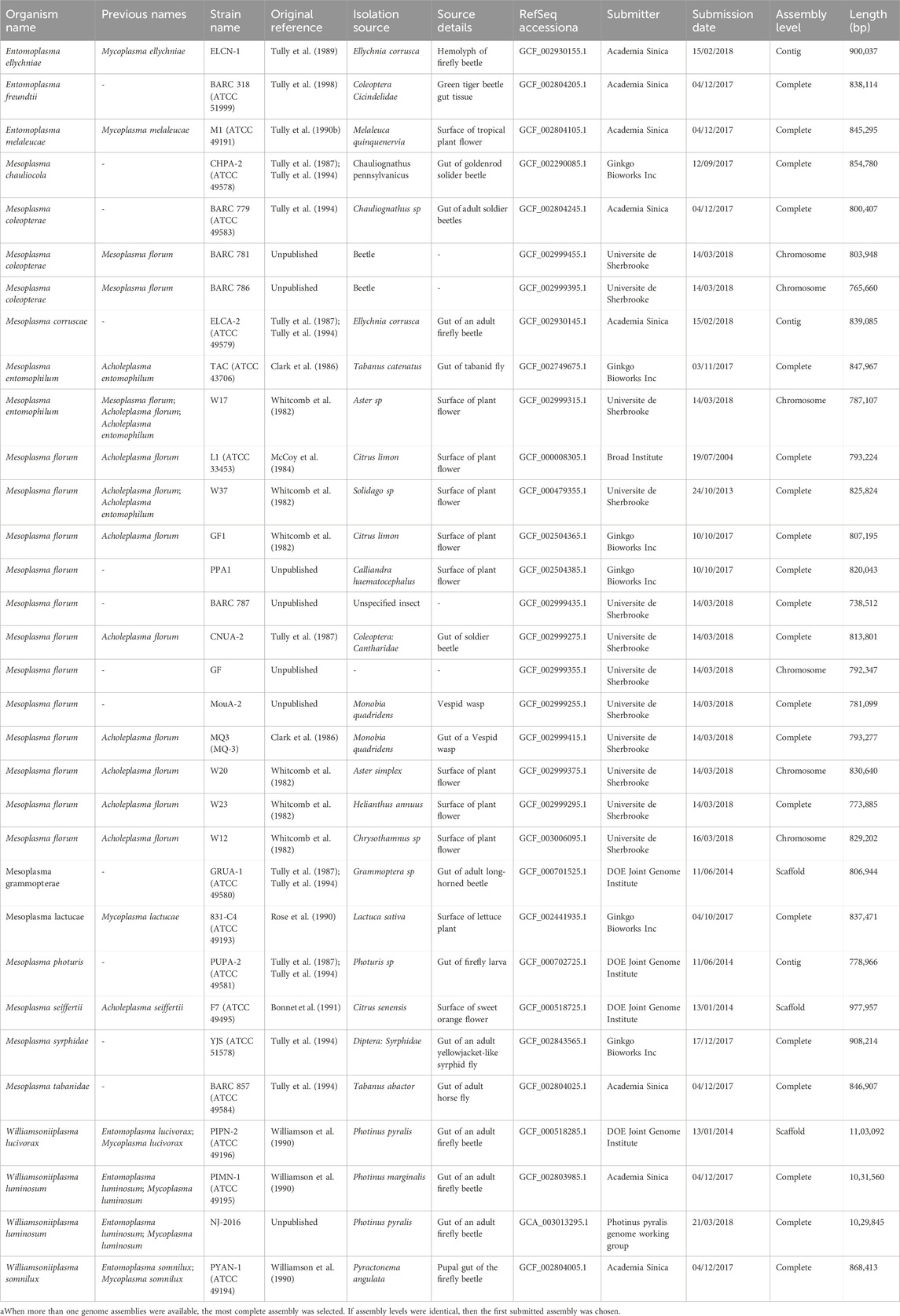
TABLE 1. List of Mesoplasma and Entomoplasma strains with genome assemblies deposited on the RefSeq database.
While we cannot completely rule out the possibility that M. florum could be pathogenic in certain hosts or under yet unidentified circumstances, its ecological niche seems quite different from related pathogenic mycoplasmas of the mycoides cluster. Since the growth of M. florum is dramatically impaired at 37°C (McCoy et al., 1984; Matteau et al., 2020), the probability that it infects warm-blooded animals similar to M. mycoides or M. capricolum is indeed very low. Recent data suggest that mycoplasmas of the mycoides cluster rather gained the ability to infect animals like other mycoplasmas through convergent evolution, in which a common ancestor experienced important gene losses and acquisitions, notably by exchanging genes with the Hominis and Pneumoniae lineages (Lo et al., 2018). Whether M. florum simply benefits from its hosts or rather perform advantageous metabolic activities, for example, by degrading or secreting particular metabolites in the gut, remains also to be determined. It has been shown that some bacteria of the Entomoplasmatales clade play important roles in the digestive system of attine fungus-farming leaf-cutting ants (Sapountzis et al., 2015; Sapountzis et al., 2018). In any cases, M. florum or its predecessor had to adapt and develop strategies to compete for the available resources. Its small size might in fact be advantageous in that context. With an average cell diameter of 0.5–0.6 µm (Figure 1A), M. florum is estimated to have a total cell volume of only 0.08–0.10 µm3, which is nearly 50 times smaller than Escherichia coli (Volkmer and Heinemann, 2011; Dai and Zhu, 2018; Matteau et al., 2020). This causes M. florum cells to have a surface area to volume ratio approximately 2.5 times higher than E. coli, as well as a relatively higher biomass fraction allocated to lipids (∼18%). These characteristics probably facilitate the importation of complex nutrients from the environment that are required for biosynthesis reactions and ATP production. Given its scavenger lifestyle, nutrient acquisition certainly occupies a critical role in M. florum metabolism. Transport reactions actually represent about a third (84/277) of the total number of reactions included in the recently published genome-scale model (GEM) of M. florum (Lachance et al., 2021). This is also reflected by the capacity of M. florum L1 to import and process various sugars, including glucose, fructose, sucrose, trehalose, and maltose (Lachance et al., 2021). Since the glycolysis is the only way of producing ATP in M. florum, being able to degrade various sugars might be important to survive in the insect gut, especially if the hosts diet is variable across individuals or between feeding periods. Interestingly, genes responsible for carbohydrate transport and metabolism are among the most variable between M. florum strains (Baby et al., 2018b), suggesting that some strains might be more fit to certain diets. Since the phylogeny of those strains could not be linked to their geographical origin or isolation source (Baby et al., 2018b), nutritional preferences of M. florum primary hosts could be one of many important actors driving the evolution of this species.
Another important consideration about very small cells is the limited amount of material that their volume can accommodate. This is well exemplified by the very small genomes of Mollicutes, which can be as small as 580 kbp in the case of Mycoplasma genitalium (Su and Baseman, 1990; Fraser et al., 1995). At 0.5–0.6 µm of diameter, M. florum cells are in fact only 5,000 to 6,000 times larger than a hydrogen atom, and weight just about 100 fg (Morowitz and Tourtellotte, 1962; Morowitz, 1984; Sundararaj et al., 2004; Matteau et al., 2020). With only ∼800 kbp, the M. florum chromosome obviously requires fewer nucleotides and most probably less energy than for E. coli to replicate, especially since both organisms have approximately the same number of genome copies per cell (Bionumbers, 2015; Matteau et al., 2020). The number of RNA and protein molecules is also much lower in M. florum compared to E. coli, corresponding to roughly 10 times fewer molecules per cell for both constituents. Yet, if we normalize these values per unit of volume, M. florum and E. coli show similar proteins and RNA concentrations (Sundararaj et al., 2004; Milo, 2013; Bionumbers, 2015; Matteau et al., 2020). Combined with the low metabolic cost predicted for M. florum biomass synthesis reactions, which are mainly fueled by the import, assembly, and rearrangement of premade molecular building blocks, these physical limitations might decrease the amount of energy needed to complete a round of cellular division. This probably contributes to the fast growth rate of M. florum, and could explain why little amounts of sugars are sufficient to sustain its growth in vitro (Lachance et al., 2021). The main protease responsible for the degradation of incomplete proteins that are expressed from mRNA lacking stop codons is also 8 to 16 times more processive in M. florum compared to E. coli (Gur and Sauer, 2008). This could allow a more efficient recycling of the amino acids incorporated into incomplete proteins. This protease (Lon) was notably used in metabolic engineering applications (Zhou et al., 2023) as well as to develop artificial gene circuits in other bacteria (Huang et al., 2012; Cameron and Collins, 2014; Sakkos et al., 2021; Szydlo et al., 2022). Of course, other factors most likely come into play to explain the fast-growing phenotype of M. florum compared to other Mollicutes. Not spending resources and energy on the expression of virulence factors is probably one of them. Allocating most of its resources on protein expression might also help, as nearly half of all protein molecules present in the M. florum cell are associated with translation and other related processes (Matteau et al., 2020; Lachance et al., 2021). More precisely, the estimated ribosome concentration in M. florum is roughly ten times higher than the values reported for M. pneumoniae, but comparable to concentrations estimated in M. mycoides and E. coli (Sundararaj et al., 2004; Kühner et al., 2009; Yus et al., 2009; Bakshi et al., 2012; Wodke et al., 2013; Breuer et al., 2019; Matteau et al., 2020). Rather than adopting complex survival strategies like M. pneumoniae and other slow-growing pathogenic mycoplasmas, M. florum appears to focus on rapid biomass production to thrive in its natural environment. The reconstruction of a GEM that accounts for protein expression constraints (ME-model) (Lloyd et al., 2018) and its comparison with protein abundances previously estimated for M. florum might provide additional clues on the relationship between protein allocation and growth rate in Mollicutes.
Is the genome of M. florum minimal?
Although the M. florum genome has been streamlined by evolution (Sirand-Pugnet et al., 2007), previous studies showed that it is not minimal, at least not under laboratory conditions (Baby et al., 2018b; Lachance et al., 2021). Even if Mollicutes have some of the smallest genomes found in nature, a considerable fraction of their genome is dispensable in rich media. Most non-essential elements consist of genes or regulatory elements important for fitness and robustness of the cells in their natural habitat, which generally provide much more challenging and variable physicochemical conditions compared to laboratory settings. In M. genitalium, for example, approximately 100 of its 485 predicted protein-coding genes were found to be non-essential using random transposition mutagenesis experiments (Hutchison et al., 1999; Glass et al., 2006). Another good example is JCVI-syn3.0, the currently closest approximation of a minimal organism (Hutchison et al., 2016). This artificial bacterium harbors a synthetic chromosome of only 531 kbp and 438 protein-coding genes based on the M. mycoides subspecies capri genome, which represents an impressive reduction of roughly 50% compared to the original sequence. Still, around 25% of the remaining genes in JCVI-syn3.0 and derivative strains are of unknown function (Hutchison et al., 2016; Glass et al., 2017; Breuer et al., 2019), highlighting our current gap of knowledge in the biology of even the simplest forms of life.
What could be the M. florum minimal genome, and would it be any different from JCVI-syn3.0? In M. florum, essential genes have been studied using two different but complementary methods, i.e., comparative genomics and random transposon mutagenesis (Baby et al., 2018b). By comparing the genomic sequence of 13 M. florum strains, two main groups were revealed, one comprising most of the M. florum representatives (10/13), and a second one containing only three strains, namely, W17, BARC 781, and BARC 786. Interestingly, these three strains were recently renamed based on their average nucleotide identity with other Mesoplasma species (Table 1). Nonetheless, the genomes of W17, BARC 781, and BARC 786 were found to be highly syntenic with the other representatives, and a core set of 546 homologous gene cluster families was observed in all compared genomes (Baby et al., 2018b). This corresponds to approximately 80% of all protein coding genes present in each strain, which was found to vary between 651 and 740 among strains. Unsurprisingly, more than 25% of the conserved M. florum genes are related to translation, a functional category that was observed to be significantly enriched in the core genome compared to the entire gene sets. Still, transposon mutagenesis performed in the M. florum L1 strain showed that a total of 430 genes out of 720 can be interrupted by transposon, including 320 core genes (Baby et al., 2018b). No transposon was observed in the remaining 290 genes, which are most likely essential in M. florum L1 or could have been missed given the transposon insertion density of the study. The number of putatively essential genes was however increased to 332 upon re-analysis of the transposition insertion data by considering the insertion position of the transposons within M. florum open-reading frames (Lachance et al., 2021). All analyzed genomes were predicted to encode 29 tRNA genes, as well as two virtually identical copies of the rRNA gene loci, although one copy is probably sufficient for growth (Asai et al., 1999; Hutchison et al., 2016).
Gene conservation and essentiality data have been used to propose minimal genome scenarios for M. florum L1. One scenario would be to remove all non-core genes from its genome, which should yield a ∼645 kbp genome coding for 585 genes if all intergenic and non-coding elements are retained (Baby et al., 2018b). However, 25 non-core protein coding genes were identified to be essential for M. florum L1 in ATCC 1161 medium. Including these genes in the minimal genome design would thus increase the chances of producing a viable cell. The 110 genes interrupted by transposons and absent from the core genome thus represent interesting first-step candidates for genome streamlining. Yet, this genome would probably be far from minimal since a majority (∼55%) of core genes can be interrupted by transposons without severely impacting M. florum growth. On the other hand, keeping only the genes in which no transposon was detected is a dubious strategy since synthetic lethality interactions are likely to occur, resulting in a non-viable cell when certain combinations of genes are simultaneously deleted. Given the phylogenetic proximity between M. florum and M. mycoides (Figure 1B), another possible scenario would be to include the 409 M. florum L1 protein-coding genes in which an ortholog was found in JCVI-syn3.0. Intriguingly, this Syn3.0 inspired minimal genome would contain 401 of the 585 M. florum L1 core genes, but would lack 57 genes identified as essential in M. florum (Baby et al., 2018b). Conversely, 69 gene families unique to M. mycoides JCVI-syn3.0 would not be present in that design.
Even if we combine the 57 essential genes found only in M. florum L1 with the 409 protein-coding genes shared between M. florum and JCVI-syn3.0, it remains difficult to predict if this synthetic design will be viable. Genome design rules remain poorly understood, and most synthetic genome projects rely on trial-and-error approaches, involving long and fastidious rounds of optimization. For instance, to create JCVI-syn3.0, it took not only many rounds of genome design, transposon mutagenesis, and debugging, but also an extensive knowledge of the biochemical data available in the literature as well as an impressive amount of time and resources (Sleator, 2010; Sleator, 2016; Hutchison et al., 2016). Systems biology approaches that can integrate multiple layers of information and systematically evaluate genome designs represent promising tools in that context (Chalkley et al., 2019; Rees-Garbutt et al., 2020a; Rees-Garbutt et al., 2020b). Such approaches were recently used to further explore the minimal gene set of M. florum and compare it with JCVI-syn3.0 (Lachance et al., 2021). This required the reconstruction of a high-quality metabolic GEM for M. florum, consisting of 370 reactions, 208 genes, and 351 metabolites (iJL208). This model was experimentally validated using growth data on various sugars as well as gene expression and essentiality data, which were all in good agreement with the model predictions (Lachance et al., 2021). Gene essentiality data and metabolic constraints defined by the model allowed the prediction of a 562 kbp minimal genome containing 535 protein-coding genes. Since this prediction also considered the 387 previously identified M. florum transcription units (Matteau et al., 2020), its viability is more likely than previously mentioned hypothetical scenarios. Interestingly, this minimal genome contains 97 more protein-coding genes than JCVI-syn3.0, which could be due to real biological differences between the two organisms or simply be caused by prediction inaccuracies given the current gaps of knowledge in M. florum and Mollicutes biology. While this prediction shares 343 protein-coding genes with JCVI-syn3.0, it contains 129 genes unique to M. florum as well as 63 genes exclusively shared with JCVI-syn1.0, the parent strain of JCVI-syn3.0. This suggests that different minimal genome compositions probably exist, even for closely related species. However, most genes unique to M. florum are currently of unknown function, which complicates further investigation. Still, many protein-coding genes unique to M. florum or shared with JCVI-syn1.0 are associated with metabolic functions, notably transport and carbohydrate metabolism (Lachance et al., 2021). We can therefore imagine that different pathways could be used by minimal genomes to produce energy and fulfill cellular needs. Some minimal genome configurations could thus be more optimal than others. Indeed, 19 genes initially discarded in JCVI-syn3.0 were later reintroduced to resolve important morphological and growth defects, creating a more robust cell named JCVI-syn3A (Breuer et al., 2019; Pelletier et al., 2021). Among these genes, two are present in the minimal M. florum genome prediction. However, the construction of synthetic M. florum genomes will ultimately be needed to test and validate these computational predictions.
Can we engineer the genome of M. florum?
The M. florum genome engineering toolbox is not as sophisticated as those available for E. coli or Saccharomyces cerevisiae. However, there is a growing number of methods that can be used for modifying the M. florum genome. Given its relative simplicity, Tn5 transposon mutagenesis was the first approach used in M. florum (Baby et al., 2018b). This system had previously been used in many bacterial species, including M. mycoides (Goryshin et al., 2000; Karas et al., 2014; Hutchison et al., 2016). Given the natural M. florum antibiotic susceptibility profile (Matteau et al., 2017), the widely used tetM gene conferring resistance to tetracycline was chosen as the selection marker in the transposon. The transformation of this transposon by electroporation resulted in tetracycline resistant M. florum colonies on ATCC 1161 plates (Baby et al., 2018b). Despite of a relatively high variability in the method efficiency, this allowed the creation of a collection comprising 2,806 individually picked transposon insertion mutants in which 430 of the 720 M. florum genes were found to be interrupted (Baby et al., 2018b; Lachance et al., 2021). Similar to the E. coli Keio collection (Baba et al., 2006), this library of gene-inactivated M. florum mutants represents an invaluable resource to study the biology of this near-minimal bacterium, especially for finding function to currently unassigned genes. This approach could also be repeated using different growth conditions to obtain additional information on the function of specific genes.
Another way to deliver genetic material into the genome is through the transformation of plasmids. Unfortunately, no natural plasmid has yet been reported to replicate in M. florum, and artificial plasmids developed in M. mycoides, M. capricolum, and S. citri have been shown to be incompatible with this species (Matteau et al., 2017). These plasmids harbor a partial or complete copy of the host chromosomal origin of replication (oriC) to replicate in their host. The oriC contains short DNA sequences known as DnaA boxes essential for the recognition by the DnaA protein, which is responsible for initiating DNA replication in bacteria (Messer, 2002). In Mollicutes, DnaA boxes are generally located within the two intergenic regions flanking the dnaA gene (Cordova et al., 2002; Lartigue et al., 2003; Ishag et al., 2017; Matteau et al., 2017). Artificial plasmids have recently been constructed using the M. florum predicted oriC (Matteau et al., 2017). The tetM gene was included in all tested M. florum oriC plasmids. While both intergenic regions surrounding the dnaA gene were shown to be essential for replication, contrasting with observations in S. citri (Lartigue et al., 2002), the presence of a copy of the dnaA gene was not. Plasmids containing both dnaA intergenic regions (pMflT-o3 and pMflT-o4) were stably maintained for more than 85 generations with or without antibiotics selection. Interestingly, M. florum oriC plasmids could successfully be transformed by electroporation or polyethylene glycol (PEG) transformation, as well as by conjugation from an E. coli strain using the RP4 system (Matteau et al., 2017). These plasmids allowed the validation of two additional selection markers, pac and aadA1, conferring resistance to puromycin and streptomycin/spectinomycin, respectively. While the pac marker had previously been used in other Mollicutes (Algire et al., 2009; Krishnakumar et al., 2010; Maglennon et al., 2013), this was the first reported use of the aadA1 marker in a Mollicutes species. The functionality of this cassette also confirmed the recognition of PN25 promoter by the M. florum σ70 factor (Brunner and Bujard, 1987), which had not been used in the context of the tetM marker. This result is consistent with the sequence of the M. florum consensus promoter, which is, similar to E. coli, characterized by a strongly conserved−10 box of sequence TAWAAT (Matteau et al., 2020). However, in M. florum, the -35 box is highly degenerated. The M. florum oriC plasmids represent basic molecular tools that will help the validation of additional DNA parts in this bacterium, as well as facilitate the development of more sophisticated approaches to engineer its genome.
Since oriC plasmids are replicated using the same mechanism as the chromosome, they are maintained at very low copy numbers in the cells. In M. florum, these plasmids are estimated to be present at 1 or 2 copies per cell (Matteau et al., 2017). In addition, their homology with the endogenous oriC region causes frequent recombination events with the host chromosome, a tendency also observed with M. florum oriC plasmids. While in some cases the integrated DNA cargo can interfere with the normal replication of the chromosome, this property can be exploited for genome engineering purposes. This was well demonstrated by the whole genome cloning (WGC) of the M. florum chromosome in the yeast S. cerevisiae (Labroussaa et al., 2016; Baby et al., 2018a) (Figure 2A). In that context, sequences enabling replication, partitioning, and selection in yeast were first introduced into the M. florum chromosome by the recombination of an oriC plasmid derivative. Following transformation in yeast, this allowed the M. florum chromosome to be replicated as a practically inert extrachromosomal element, with only minor impact on the yeast growth and cell cycle. WGC in yeast offers the opportunity to use the vast and well characterized molecular toolbox available in this model organism. For instance, the natural capacity of yeast to perform efficient homologous recombination was used to replace the duplicated oriC region resulting from the recombination of the oriC derivative plasmid by an URA3 cassette (Baby et al., 2018a). Since many Mollicutes lack efficient molecular tools to modify their genome, WGC in yeast has been performed for several species, including M. mycoides and M. genitalium (Labroussaa et al., 2019). This procedure is at the heart of the strategy used to create JCVI-syn1.0 and JCVI-syn3.0 (Gibson et al., 2010; Hutchison et al., 2016). Whole genomes cloned and engineered in yeast must however be transplanted into a suitable recipient bacterium to assess their viability, a delicate procedure known as genome transplantation (Lartigue et al., 2007; Labroussaa et al., 2019) (Figure 2A). Due to its remarkable capacity to recognize the oriC region of other Mollicutes species, M. capricolum is generally used for this task (Lartigue et al., 2003; Lartigue et al., 2007; Labroussaa et al., 2016). Following transplantation and selection, the M. capricolum genome is replaced by the donor genome, and individual transplants can be recovered for validation and characterization. While many aspects of genome transplantation are still puzzling, the phylogenetic distance between the donor and recipient bacteria is known to play a critical role in the overall efficiency of the method (Labroussaa et al., 2016). Sharing ∼92% identity on the core proteome with M. capricolum, M. florum appears to be the most phylogenetically distant organism for which the transplantation with this recipient bacterium is possible (Baby et al., 2018a). Indeed, attempts to transplant the genome of S. citri and S. floricola have failed, and genome transplantation of more distant Mollicutes species such as M. genitalium and M. hominis has never been reported, albeit their genomes have been successfully cloned in yeast (Labroussaa et al., 2016; Labroussaa et al., 2019). Apart from the phylogenetic distance, other factors such as the concentration and quality of donor genomic DNA as well as the presence of mobile genetic elements or restriction-modification systems are also known to affect the success of the procedure (Lartigue et al., 2007; Gibson et al., 2010; Labroussaa et al., 2016; Labroussaa et al., 2019). The topology of the transplanted genomes might also be important as supercoiled DNA seems to drastically increase the transformability of large DNA molecules in E. coli (Mukai et al., 2020; Yoneji et al., 2021; Fujita et al., 2022).
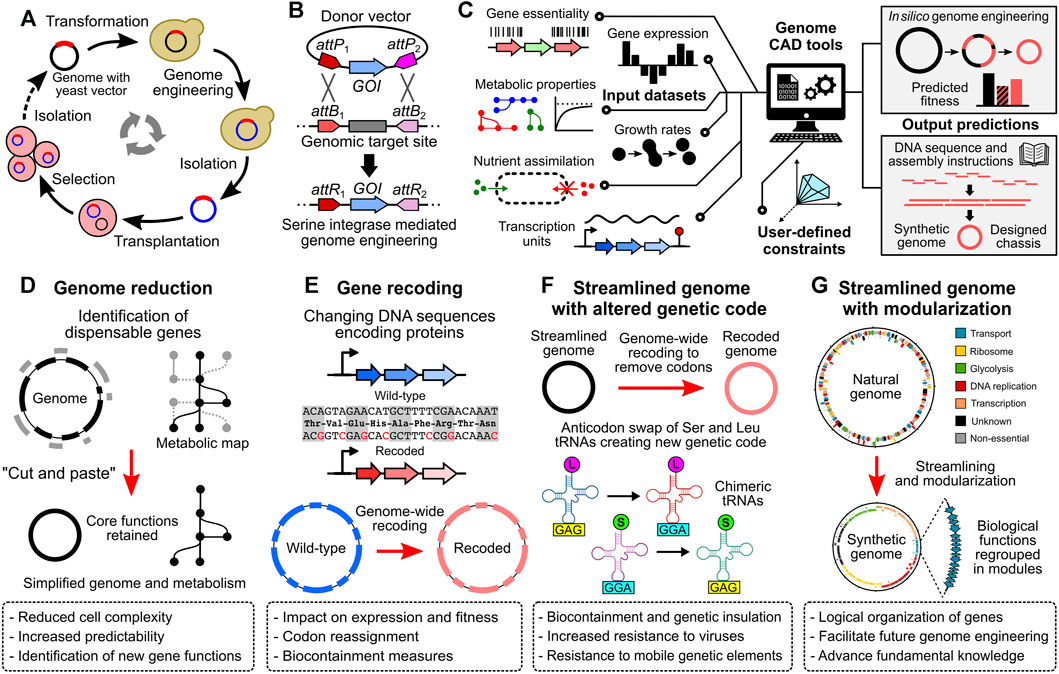
FIGURE 2. Genome engineering tools and projects to transform Mesoplasma florum into an optimized cell chassis for systems and synthetic biology. (A) Whole genome cloning and transplantation procedure. Bacterial genomes containing a yeast vector are first transformed in yeast to allow genetic modifications using the available molecular tools. Modified genomes are then carefully isolated and transplanted into a compatible recipient bacterium. Recipient cells adopt the phenotype conferred by the transplanted genome (see Lartigue et al., 2009; Labroussaa et al., 2019). (B) Serine integrase mediated genome engineering methodology. DNA fragments such as genes of interest (GOI) can be inserted or exchanged with the genome by the expression of a serine integrase. Serine integrases such as Bxb1 or PhiC31 catalyze the recombination between specific DNA sequences, namely, the attP and attB sites, resulting in attL and attR sites (attL sites not shown, see Merrick et al., 2018 for more details). (C) Integration of multiple data types by computer-aided design (CAD) tools to guide the design and optimize the synthesis, amplification, and assembly of large DNA fragments according to user-defined constraints. In silico models such as genome-scale models (GEMs) can be used to predict the fitness of designed genomes. (D) to (G) Example of synthetic genome projects. (D) Genome reduction, in which non-essential genes are removed from the genome, resulting in a reduced cell complexity and simplified metabolism. (E) Gene recoding at the genome scale. Within a given open reading frame, codons can be exchanged for synonymous ones, modifying the DNA sequence but not the corresponding amino acid sequence. (F) Streamlined genome using a swapped amino acid genetic code. By removing non-essential genes and recoding all remaining genes, specific codons can be removed from the genome, allowing anticodon swap between given tRNAs and creating an artificial genetic code. (G) Streamlined genome with gene of similar function regrouped in modules. Regrouping genes with related functions into modules can facilitate genome engineering efforts, and can be used to test specific hypotheses about gene regulation and genome organization.
What could be the next steps in M. florum research?
Expanding the available molecular toolbox should certainly be one of the key priorities to fully harness the potential of M. florum for systems and synthetic biology. Even if the transplantation of the M. florum genome is possible (Figure 2A), the very low efficiency and high variability associated with this method using M. florum constitutes an important limitation to the in-yeast genome engineering strategy. It is not rare to obtain less than 10 M. florum transplants per experiment, or even no transplant at all (Labroussaa et al., 2016; Baby et al., 2018a). Further investigations are therefore required to enable rapid and easy prototyping of the M. florum genome cloned in yeast. Finding a new compatible recipient strain phylogenetically closer to M. florum than M. capricolum could in principle improve transplantation rates. Alternatively, targeted engineering of the recipient strain could also favor the recognition and boot-up of the transplanted genome. Nevertheless, genome transplantation remains a complex and delicate procedure. Complementary approaches should therefore be developed to facilitate the genetic modification of M. florum. Methods using serine integrases (Merrick et al., 2018) to efficiently exchange or insert DNA fragments at specific positions in the genome (Figure 2B) could prove very useful for M. florum since Tn5 transposons insert randomly and current oriC plasmids tend to recombine only at the oriC region. Another option would be to adapt the well-known recombineering technique by properly expressing proteins of the λ-Red system (Datsenko and Wanner, 2000) or the GP35 recombinase, which was recently demonstrated to be functional in M. pneumoniae (Piñero-Lambea et al., 2020; Piñero-Lambea et al., 2022). This approach could even be coupled with the expression of the CRISPR-Cas9 system to further stimulate DNA recombination by cutting M. florum’s genome and counter-selecting unmodified or incorrectly repaired cells. Unlocking the CRISPR-Cas9 technology in M. florum would be a significant asset for future research on this bacterium, with a wide array of potential applications (Adli, 2018; Mariscal et al., 2018; Pickar-Oliver and Gersbach, 2019). Yet, heterologous proteins such as Cas9 must be sufficiently expressed in the host to display desired effect. On the other hand, constitutive or uncontrolled expression of many proteins is known to cause toxicity and can affect cell viability. Unfortunately, as of now not even a handful of promoters have been tested and validated on synthetic constructs introduced in M. florum (Matteau et al., 2017), none of which are inducible. Testing additional promoters -natural or synthetic- and combining them with other regulatory elements enabling strong activation or tight repression would unlock several methods (Kim et al., 2007; Breton et al., 2010; Domin et al., 2017; Etzel and Mörl, 2017; Ruegg et al., 2018; Piñero-Lambea et al., 2020). In addition, comparing these results with published transcriptional data would provide valuable information about the DNA sequences enabling strong transcription in this organism.
By increasing the molecular toolbox available in M. florum, performing large or extensive genome modifications and testing new hypotheses will become significantly easier. Combined with the most recent gene synthesis and high-throughput DNA assembly technologies (Gibson et al., 2010; Hughes and Ellington, 2017; Juhas and Ajioka, 2017; Schindler et al., 2018; Hoose et al., 2023), genome engineering projects could be undertaken (Figures 2D–G). For example, minimal genomes are powerful tools to study fundamental aspects of life, and constitute interesting cell chassis to learn genome design principles and develop promising applications in synthetic biology (Morowitz, 1984; Glass et al., 2017; Lachance et al., 2019). Their limited complexity increases predictability using modeling approaches and decreases the chance of unexpected interactions between artificial gene circuits and native host functions. Stripping the M. florum genome near its minimum would reduce the number of genes without any assigned function, and slightly decrease the costs associated with genome synthesis projects. Moreover, the comparison between a minimal M. florum genome and JCVI-syn3.0 could provide invaluable information about the different strategies used by bacteria to fulfill essential functions. Still, to enable rapid construction and testing of synthetic M. florum genomes, additional tools should be developed to integrate multiple data sources and properly guide the design as well as optimize the synthesis, amplification, and assembly of large DNA fragments (Figure 2C). With an efficient M. florum genome prototyping platform in hands, other exciting genome-wide projects could also become more realistic. Entire genome fractions could be recoded, separately or in combination with genome reduction efforts, to systematically investigate the impact of several parameters such as the GC content or the removal of internal transcription start sites (iTSSs) (Matteau et al., 2020) on gene expression and cell fitness (Figure 2E). Engineered or minimal M. florum cells will probably be sub-optimal at first, as observed with JCVI-syn3.0 and many other genome-reduced bacteria (Iwadate et al., 2011; Karcagi et al., 2016; Breuer et al., 2019; Pelletier et al., 2021; Dervyn et al., 2023). Artificial cells could next be subjected to adaptive laboratory evolution (ALE) for fine-tuning and selection of the most adapted mutants (Dragosits and Mattanovich, 2013; Sandberg et al., 2019). This strategy could be performed without adding any mutagenic compound or plasmid (Badran and Liu, 2015) given the particularly high DNA replication error rate of M. florum (Sung et al., 2012; Lynch et al., 2016). Interestingly, ALE experiments performed on JCVI-syn3A cultures led to growth rate improvements of >15%, corresponding to a doubling time of ∼80 min (Sandberg et al., 2023). The resulting M. florum mutants could be compared with ALE evolved JCVI-syn3A strains to see if they share similar mutation profiles and growth rates. Rare codons could also be systematically removed from the M. florum genome (Isaacs et al., 2011; Fredens et al., 2019), allowing codon reassignment and strict biocontainment measures. Artificial genetic codes could be developed and tested by swapping tRNA anticodons, thereby improving resistance to viruses and mobile genetic genetic elements (Zürcher et al., 2022; Nyerges et al., 2023) (Figure 2F). Genes with related functions could be regrouped into modules, reorganizing and streamlining the entire genome for engineering purposes (Hutchison et al., 2016; Coradini et al., 2020) (Figure 2G). Large genome portions could be inverted to study the importance of DNA orientation at large-scale. Every predicted transcriptional regulator could be tagged for genome-wide binding site assays, enabling high-throughput experimental determination of transcription regulation networks (Matteau and Rodrigue, 2015; Rossi et al., 2018). Protein sequences of entire pathways could be replaced by more or less phylogenetically related homologs to study protein compatibility and create chimeric genomes with enhanced properties. Guided by predictive tools such as the iJL208 GEM (Rees-Garbutt et al., 2020b; Rees-Garbutt et al., 2020a; Lachance et al., 2021), new metabolic capacities or biosynthetic pathways could be introduced by testing a large number of protein variants in parallel and finding the most optimal sequence combination for M. florum (Emanuel et al., 2017; Schubert et al., 2021). Synthetic genomics unlocks new possibilities that were simply not technically feasible not so long ago. As we move forward, the frontiers of biology will be redefined, allowing us to pursue and test hypotheses that long remained out of reach, thereby enhancing our comprehension of life at a deeper level.
Author contributions
DM: Investigation, Writing–original draft, Writing–review and editing. AD: Investigation, Writing–original draft. VB: Investigation, Writing–original draft. SR: Funding acquisition, Resources, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a Discovery grant (RGPIN-2020-06151) from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
GEM, Genome-scale model; PEG, Polyethylene glycol; oriC, Chromosomal origin of replication; WGC, Whole genome cloning; iTSS, Internal transcription start site; ALE, Adaptive laboratory evolution.
References
Adli, M. (2018). The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9, 1911. doi:10.1038/s41467-018-04252-2
Algire, M. A., Lartigue, C., Thomas, D. W., Assad-Garcia, N., Glass, J. I., and Merryman, C. (2009). New selectable marker for manipulating the simple genomes of Mycoplasma species. Antimicrob. Agents Chemother. 53, 4429–4432. doi:10.1128/AAC.00388-09
Asai, T., Zaporojets, D., Squires, C., and Squires, C. L. (1999). An Escherichia coli strain with all chromosomal rRNA operons inactivated: complete exchange of rRNA genes between bacteria. Proc. Natl. Acad. Sci. U. S. A. 96, 1971–1976. doi:10.1073/pnas.96.5.1971
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. doi:10.1038/msb4100050
Baby, V., Labroussaa, F., Brodeur, J., Matteau, D., Gourgues, G., Lartigue, C., et al. (2018a). Cloning and transplantation of the Mesoplasma florum genome. ACS Synth. Biol. 7, 209–217. doi:10.1021/acssynbio.7b00279
Baby, V., Lachance, J.-C., Gagnon, J., Lucier, J.-F., Matteau, D., Knight, T. F., et al. (2018b). Inferring the minimal genome of Mesoplasma florum by comparative genomics and transposon mutagenesis. mSystems 3, 001988–e217. doi:10.1128/mSystems.00198-17
Baby, V., Matteau, D., Brodeur, J., and Rodrigue, S. (2013). Whole genome sequencing and comparative genomics of the near-minimal bacterium Mesoplasma florum: first steps towards a simplified chassis for synthetic biology. Ottawa, Canada: Conférence Annuelle de la Société Canadienne des Microbiologistes.
Badran, A. H., and Liu, D. R. (2015). Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun. 6, 8425–8510. doi:10.1038/ncomms9425
Bakshi, S., Siryaporn, A., Goulian, M., and Weisshaar, J. C. (2012). Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol. Microbiol. 85, 21–38. doi:10.1111/j.1365-2958.2012.08081.x
Bonnet, F., Saillard, C., Vignault, J. C., Garnier, M., Carle, P., Bove, J. M., et al. (1991). Acholeplasma seiffertii sp. nov., a mollicute from plant surfaces. Int J Syst Microbiol. 40 (1), 45–49. doi:10.1099/00207713-41-1-45
Bionumbers (2015). What is the macromolecular composition of the cell. Available at: http://book.bionumbers.org/what-is-the-macromolecular-composition-of-the-cell/(Accessed March 19, 2019).
Breton, M., Sagné, E., Duret, S., Béven, L., Citti, C., and Renaudin, J. (2010). First report of a tetracycline-inducible gene expression system for mollicutes. Microbiology 156, 198–205. doi:10.1099/mic.0.034074-0
Breuer, M., Earnest, T. M., Merryman, C., Wise, K. S., Sun, L., Lynott, M. R., et al. (2019). Essential metabolism for a minimal cell. Elife 8, e36842–e36877. doi:10.7554/elife.36842
Brunner, M., and Bujard, H. (1987). Promoter recognition and promoter strength in the Escherichia coli system. EMBO J. 6, 3139–3144. doi:10.1002/j.1460-2075.1987.tb02624.x
Cameron, D. E., and Collins, J. J. (2014). Tunable protein degradation in bacteria. Nat. Biotechnol. 32, 1276–1281. doi:10.1038/nbt.3053
Chalkley, O., Purcell, O., Grierson, C., and Marucci, L. (2019). The genome design suite: enabling massive in-silico experiments to design genomes. bioRxiv, doi:10.1101/681270
Clark, T. B., Tully, J. G., Rose, D. L., Henegar, R., and Whitcomb, R. F. (1986). Acholeplasmas and similar nonsterol-requiring mollicutes from insects: missing link in microbial ecology. Curr. Microbiol. 13, 11–16. doi:10.1007/BF01568152
Coradini, A. L. V., Hull, C. B., and Ehrenreich, I. M. (2020). Building genomes to understand biology. Nat. Commun. 11, 6177–6211. doi:10.1038/s41467-020-19753-2
Cordova, C. M. M., Lartigue, C., Sirand-Pugnet, P., Renaudin, J., Cunha, R. A. F., and Blanchard, A. (2002). Identification of the origin of replication of the Mycoplasma pulmonis chromosome and its use in oriC replicative plasmids. J. Bacteriol. 184, 5426–5435. doi:10.1128/JB.184.19.5426-5435.2002
Dai, X., and Zhu, M. (2018). High osmolarity modulates bacterial cell size through reducing initiation volume in Escherichia coli. mSphere 3, 004300–e518. doi:10.1128/mSphere.00430-18
Datsenko, K. a., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97, 6640–6645. doi:10.1073/pnas.120163297
Dervyn, E., Planson, A. G., Tanaka, K., Chubukov, V., Guérin, C., Derozier, S., et al. (2023). Greedy reduction of Bacillus subtilis genome yields emergent phenotypes of high resistance to a DNA damaging agent and low evolvability. Nucleic Acids Res. 51, 2974–2992. doi:10.1093/nar/gkad145
Domin, G., Findeiß, S., Wachsmuth, M., Will, S., Stadler, P. F., and Mörl, M. (2017). Applicability of a computational design approach for synthetic riboswitches. Nucleic Acids Res. 45, 4108–4119. doi:10.1093/nar/gkw1267
Dragosits, M., and Mattanovich, D. (2013). Adaptive laboratory evolution - principles and applications for biotechnology. Microb. Cell Fact. 12, 64–17. doi:10.1186/1475-2859-12-64
Emanuel, G., Moffitt, J. R., and Zhuang, X. (2017). High-throughput, image-based screening of pooled genetic-variant libraries. Nat. Methods 14, 1159–1162. doi:10.1038/nmeth.4495
Etzel, M., and Mörl, M. (2017). Synthetic riboswitches: from plug and pray toward plug and play. Biochemistry 56, 1181–1198. doi:10.1021/acs.biochem.6b01218
Fraser, C. M., Gocayne, J. D., White, O., Adams, M. D., Clayton, R., Fleischmann, R. D., et al. (1995). The minimal gene complement of Mycoplasma genitalium. Science 270, 397–403. doi:10.1126/science.270.5235.397
Fredens, J., Wang, K., de la Torre, D., Funke, L. F. H., Robertson, W. E., Christova, Y., et al. (2019). Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518. doi:10.1038/s41586-019-1192-5
Fujita, H., Osaku, A., Sakane, Y., Yoshida, K., Yamada, K., Nara, S., et al. (2022). Enzymatic supercoiling of bacterial chromosomes facilitates genome manipulation. ACS Synth. Biol. 11, 3088–3099. doi:10.1021/acssynbio.2c00353
Gasparich, G. E., and Chih-Horng, K. (2019). Genome analysis-based union of the genus Mesoplasma with the genus Entomoplasma. Int. J. Syst. Evol. Microbiol. 69, 2735–2738. doi:10.1099/ijsem.0.003548
Gibson, D. G., Glass, J. I., Lartigue, C., Noskov, V. N., Chuang, R.-Y. Y., Algire, M. A., et al. (2010). Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329, 52–56. doi:10.1126/science.1190719
Glass, J. I., Merryman, C., Wise, K. S., Iii, C. A. H., and Smith, H. O. (2017). Minimal cells — real and imagined. Cold Spring Harb. Perspect. Biol. 1, 0238611–a23912. doi:10.1101/cshperspect.a023861
Glass, J. I. J., Assad-Garcia, N., Alperovich, N., Yooseph, S., Lewis, M. R., Maruf, M., et al. (2006). Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. U. S. A. 103, 425–430. doi:10.1073/pnas.0510013103
Goryshin, I. Y., Jendrisak, J., Hoffman, L. M., Meis, R., and Reznikoff, W. S. (2000). Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat. Biotechnol. 18, 97–100. doi:10.1038/72017
Gur, E., and Sauer, R. T. (2008). Evolution of the ssrA degradation tag in Mycoplasma: specificity switch to a different protease. Proc. Natl. Acad. Sci. U. S. A. 105, 16113–16118. doi:10.1073/pnas.0808802105
Hoose, A., Vellacott, R., Storch, M., Freemont, P. S., and Ryadnov, M. G. (2023). DNA synthesis technologies to close the gene writing gap. Nat. Rev. Chem. 7, 144–161. doi:10.1038/s41570-022-00456-9
Huang, D., Holtz, W. J., and Maharbiz, M. M. (2012). A genetic bistable switch utilizing nonlinear protein degradation. J. Biol. Eng. 6, 9–13. doi:10.1186/1754-1611-6-9
Hughes, R. A., and Ellington, A. D. (2017). Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb. Perspect. Biol. 9, a023812. doi:10.1101/cshperspect.a023812
Hutchison, C. A., Chuang, R. Y., Noskov, V. N., Assad-Garcia, N., Deerinck, T. J., Ellisman, M. H., et al. (2016). Design and synthesis of a minimal bacterial genome. Science 351, aad6253–11. doi:10.1126/science.aad6253
Hutchison, C. A., Peterson, S. N., Gill, S. R., Cline, R. T., White, O., Fraser, C. M., et al. (1999). Global transposon mutagenesis and a minimal Mycoplasma genome. Science 286, 2165–2169. doi:10.1126/science.286.5447.2165
Isaacs, F. J., Carr, P. a., Wang, H. H., Lajoie, M. J., Sterling, B., Kraal, L., et al. (2011). Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 333, 348–353. doi:10.1126/science.1205822
Ishag, H. Z. A., Xiong, Q., Liu, M., Feng, Z., and Shao, G. (2017). Development of oriC-plasmids for use in Mycoplasma hyorhinis. Sci. Rep. 7, 10596–10610. doi:10.1038/s41598-017-10519-3
Iwadate, Y., Honda, H., Sato, H., Hashimoto, M., and Kato, J. (2011). Oxidative stress sensitivity of engineered Escherichia coli cells with a reduced genome. FEMS Microbiol. Lett. 322, 25–33. doi:10.1111/j.1574-6968.2011.02331.x
Juhas, M., and Ajioka, J. W. (2017). High molecular weight DNA assembly in vivo for synthetic biology applications. Crit. Rev. Biotechnol. 37, 277–286. doi:10.3109/07388551.2016.1141394
Karas, B. J., Wise, K. S., Sun, L., Craig Venter, J., Glass, J. I., Hutchison, C. A., et al. (2014). Rescue of mutant fitness defects using in vitro reconstituted designer transposons in Mycoplasma mycoides. Front. Microbiol. 5, 369–9. doi:10.3389/fmicb.2014.00369
Karcagi, I., Draskovits, G., Umenhoffer, K., Fekete, G., Kovács, K., Méhi, O., et al. (2016). Indispensability of horizontally transferred genes and its impact on bacterial genome streamlining. Mol. Biol. Evol. 33, 1257–1269. doi:10.1093/molbev/msw009
Kim, J. N., Roth, A., and Breaker, R. R. (2007). Guanine riboswitch variants from Mesoplasma florum selectively recognize 2’-deoxyguanosine. Proc. Natl. Acad. Sci. U. S. A. 104, 16092–16097. doi:10.1073/pnas.0705884104
Krishnakumar, R., Assad-garcia, N., Benders, G. A., Phan, Q., Montague, M. G., and Glass, J. I. (2010). Targeted chromosomal knockouts in Mycoplasma pneumoniae. Appl. Environ. Microbiol. 76, 5297–5299. doi:10.1128/AEM.00024-10
Kühner, S., Van Noort, V., Betts, M. J., Leo-Madas, A., Batisse, C., Rode, M., et al. (2009). Proteome organization in a genome-reduced bacterium. Science 326, 1235–1240. doi:10.1126/science.1176343
Labroussaa, F., Baby, V., Rodrigue, S., and Lartigue, C. (2019). Whole genome transplantation: bringing natural or synthetic bacterial genomes back to life. Médecine/Sciences 35, 761–770. doi:10.1051/medsci/2019154
Labroussaa, F., Lebaudy, A., Baby, V., Gourgues, G., Matteau, D., Vashee, S., et al. (2016). Impact of donor-recipient phylogenetic distance on bacterial genome transplantation. Nucleic Acids Res. 44, 8501–8511. doi:10.1093/nar/gkw688
Lachance, J.-C., Matteau, D., Brodeur, J., Lloyd, C., Mih, N., King, Z. A., et al. (2021). Genome-scale metabolic modeling reveals key features of a minimal gene set. Mol. Syst. Biol. 17, 1–20. doi:10.15252/msb.202010099
Lachance, J.-C., Rodrigue, S., and Palsson, B. O. (2019). Synthetic Biology: minimal cells, maximal knowledge. Elife 8, 1–4. doi:10.7554/elife.45379
Lartigue, C., Blanchard, A., Renaudin, J., Thiaucourt, F., and Sirand-Pugnet, P. (2003). Host specificity of mollicutes oriC plasmids: functional analysis of replication origin. Nucleic Acids Res. 31, 6610–6618. doi:10.1093/nar/gkg848
Lartigue, C., Duret, S., Garnier, M., and Renaudin, J. (2002). New plasmid vectors for specific gene targeting in Spiroplasma citri. Plasmid 48, 149–159. doi:10.1016/S0147-619X(02)00121-X
Lartigue, C., Glass, J. I., Alperovich, N., Pieper, R., Parmar, P. P., Hutchison, C. A., et al. (2007). Genome transplantation in bacteria: changing one species to another. Science 317, 632–638. doi:10.1126/science.1144622
Lartigue, C., Vashee, S., Algire, M. A., Chuang, R. Y., Benders, G. A., Ma, L., et al. (2009). Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325, 1693–1696. doi:10.1126/science.1173759
Lloyd, C. J., Ebrahim, A., Yang, L., King, Z. A., Catoiu, E., O’Brien, E. J., et al. (2018). COBRAme: a computational framework for genome-scale models of metabolism and gene expression. PLoS Comput. Biol. 14, e1006302. doi:10.1371/journal.pcbi.1006302
Lo, W. S., Gasparich, G. E., and Kuo, C. H. (2018). Convergent evolution among ruminant-pathogenic Mycoplasma involved extensive gene content changes. Genome Biol. Evol. 10, 2130–2139. doi:10.1093/gbe/evy172
Lynch, M., Ackerman, M. S., Gout, J. F., Long, H., Sung, W., Thomas, W. K., et al. (2016). Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 17, 704–714. doi:10.1038/nrg.2016.104
Maglennon, G. A., Cook, B. S., Matthews, D., Deeney, A. S., Bossé, J. T., Langford, P. R., et al. (2013). Development of a self-replicating plasmid system for Mycoplasma hyopneumoniae. Vet. Res. 44, 63–10. doi:10.1186/1297-9716-44-63
Mariscal, A. M., Kakizawa, S., Hsu, J. Y., Tanaka, K., González-González, L., Broto, A., et al. (2018). Tuning gene activity by inducible and targeted regulation of gene expression in minimal bacterial cells. ACS Synth. Biol. 7, 1538–1552. doi:10.1021/acssynbio.8b00028
Matteau, D., Baby, V., Pelletier, S., and Rodrigue, S. (2015). A small-volume, low-cost, and versatile continuous culture device. PLoS One 10, e0133384. doi:10.1371/journal.pone.0133384
Matteau, D., Lachance, J.-C., Grenier, F., Gauthier, S., Daubenspeck, J. M., Dybvig, K., et al. (2020). Integrative characterization of the near-minimal bacterium Mesoplasma florum. Mol. Syst. Biol. 16, 98444–e9925. doi:10.15252/msb.20209844
Matteau, D., Pepin, M., Baby, V., Gauthier, S., Arango Giraldo, M., Knight, T. F., et al. (2017). Development of oriC-based plasmids for Mesoplasma florum. Appl. Environ. Microbiol. 83, 033744–e3416. doi:10.1128/AEM.03374-16
Matteau, D., and Rodrigue, S. (2015). “Precise identification of DNA-binding proteins genomic location by exonuclease coupled chromatin immunoprecipitation (ChIP-exo),” in DNA-protein interactions SE - 11 methods in molecular biology. Editors B. P. Leblanc, and S. Rodrigue (New York: Springer), 173–193. doi:10.1007/978-1-4939-2877-4_11
McCoy, R. E., Basham, H. G., Tully, J. G., and Rose, D. L. (1980). “Isolation of a new Acholeplasma from flowers in Florida,” in Third conference of the international organization for mycoplasmology. Custer, South Dakota, USA: (ICSP).
McCoy, R. E., Basham, H. G., Tully, J. G., Rose, D. L., Carle, P., Bové, J. M., et al. (1984). Acholeplasma florum, a new species isolated from plants. Int. J. Syst. Bacteriol. 34, 11–15. doi:10.1099/00207713-34-1-11
Merrick, C. A., Zhao, J., and Rosser, S. J. (2018). Serine integrases: advancing synthetic biology. ACS Synth. Biol. 7, 299–310. doi:10.1021/acssynbio.7b00308
Messer, W. (2002). The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol. Rev. 26, 355–374. doi:10.1111/j.1574-6976.2002.tb00620.x
Milo, R. (2013). What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays 35, 1050–1055. doi:10.1002/bies.201300066
Morowitz, H. J., and Tourtellotte, M. E. (1962). The smallest living cells. Sci. Am. 206, 117–126. doi:10.1038/scientificamerican0362-117
Mukai, T., Yoneji, T., Yamada, K., Fujita, H., Nara, S., and Su’etsugu, M. (2020). Overcoming the challenges of megabase-sized plasmid construction in Escherichia coli. ACS Synth. Biol. 9, 1315–1327. doi:10.1021/acssynbio.0c00008
Navas-Castillo, J., Laigret, F., Tully, J., and Bové, J.-M. (1992). Le mollicute Acholeplasma florum possède un gène du système phosphoénolpyruvate sucre-phosphotransférase et il utilise UGA comme codon tryptophane. Comptes Rendus l’Academie Des. Sci. - Ser. III 315, 43–48.
Nyerges, A., Vinke, S., Flynn, R., Owen, S. V., Rand, E. A., Budnik, B., et al. (2023). A swapped genetic code prevents viral infections and gene transfer. Nature 615, 720–727. doi:10.1038/s41586-023-05824-z
Pelletier, J. F., Sun, L., Wise, K. S., Assad-Garcia, N., Karas, B. J., Deerinck, T. J., et al. (2021). Genetic requirements for cell division in a genomically minimal cell. Cell 184, 2430–2440. doi:10.1016/j.cell.2021.03.008
Peterson, S. N., Fraser, C. M., and Claire, M. (2001). The complexity of simplicity. Genome Biol. 2, 1–2002. doi:10.1016/S0378-4754(97)00044-X
Pettersson, B., and Johansson, K.-E. (2002). “Taxonomy of mollicutes,” in Molecular biology and pathogenicity of mycoplasmas. Editors S. Razin, and R. Herrmann (New York (USA): Springer), 1–30.
Pickar-Oliver, A., and Gersbach, C. A. (2019). The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507. doi:10.1038/s41580-019-0131-5
Piñero-Lambea, C., Garcia-Ramallo, E., Martinez, S., Delgado, J., Serrano, L., and Lluch-Senar, M. (2020). Mycoplasma pneumoniae genome editing based on oligo recombineering and cas9-mediated counterselection. ACS Synth. Biol. 9, 1693–1704. doi:10.1021/acssynbio.0c00022
Piñero-Lambea, C., Garcia-Ramallo, E., Miravet-Verde, S., Burgos, R., Scarpa, M., Serrano, L., et al. (2022). SURE editing: combining oligo-recombineering and programmable insertion/deletion of selection markers to efficiently edit the Mycoplasma pneumoniae genome. Nucleic Acids Res. 50, e127. doi:10.1093/nar/gkac836
Pollack, J., Williams, M., Banzon, J., Jones, M. A., Harvey, L., and Tully, J. G. (1996). Comparative metabolism of Mesoplasma, Entomoplasma, mycoplasma, and Acholeplasma. Int. J. 46, 885–890. doi:10.1099/00207713-46-4-885
Rees-Garbutt, J., Chalkley, O., Landon, S., Purcell, O., Marucci, L., and Grierson, C. (2020a). Designing minimal genomes using whole-cell models. Nat. Commun. 11, 836–912. doi:10.1038/s41467-020-14545-0
Rees-Garbutt, J., Rightmyer, J., Karr, J. R., Grierson, C., and Marucci, L. (2020b). Furthering genome design using models and algorithms. Curr. Opin. Syst. Biol. 24, 120–126. doi:10.1016/j.coisb.2020.10.007
Rose, D. L., Kocka, J. P., Somerson, N. L., Tully, J. G., Whitcomb, R. F., Carle, P., et al. (1990). Mycoplasma lactucae sp. nov., a sterol-requiring mollicute from a plant surface. Int J Syst Bacteriol. 40 (2), 138–42. doi:10.1099/00207713-40-2-138
Rossi, M. J., Lai, W. K. M., and Pugh, B. F. (2018). Simplified ChIP-exo assays. Nat. Commun. 9, 2842–2913. doi:10.1038/s41467-018-05265-7
Ruegg, T. L., Pereira, J. H., Chen, J. C., DeGiovanni, A., Novichkov, P., Mutalik, V. K., et al. (2018). Jungle Express is a versatile repressor system for tight transcriptional control. Nat. Commun. 9, 3617–3713. doi:10.1038/s41467-018-05857-3
Saglio, P., Lhospital, M., Laflèche, D., Dupont, G., Bové, J. M., Tully, J. G., et al. (1973). Spiroplasma citri gen. and sp. n.: a mycoplasma-like organism associated with “ stubborn ” disease of Citrus. Int. J. Syst. Bacteriol. 23, 191–204. doi:10.1099/00207713-23-3-191
Sakkos, J. K., Hernandez-Ortiz, S., Osteryoung, K. W., and Ducat, D. C. (2021). Orthogonal degron system for controlled protein degradation in cyanobacteria. ACS Synth. Biol. 10, 1667–1681. doi:10.1021/acssynbio.1c00035
Sandberg, T. E., Salazar, M. J., Weng, L. L., Palsson, B. O., and Feist, A. M. (2019). The emergence of adaptive laboratory evolution as an efficient tool for biological discovery and industrial biotechnology. Metab. Eng. 56, 1–16. doi:10.1016/j.ymben.2019.08.004
Sandberg, T. E., Wise, K. S., Dalldorf, C., Szubin, R., Feist, A. M., Glass, J. I., et al. (2023). Adaptive evolution of a minimal organism with a synthetic genome. iScience 26, 107500. doi:10.1016/j.isci.2023.107500
Sapountzis, P., Zhukova, M., Hansen, L. H., Sørensen, S. J., Schiøtt, M., and Boomsma, J. J. (2015). Acromyrmex leaf-cutting ants have simple gut microbiota with nitrogen-fixing potential. Appl. Environ. Microbiol. 81, 5527–5537. doi:10.1128/AEM.00961-15
Sapountzis, P., Zhukova, M., Shik, J. Z., Schiott, M., and Boomsma, J. J. (2018). Reconstructing the functions of endosymbiotic mollicutes in fungus-growing ants. Elife 7, e39209–e39231. doi:10.7554/eLife.39209
Schindler, D., Dai, J., and Cai, Y. (2018). Synthetic genomics: a new venture to dissect genome fundamentals and engineer new functions. Curr. Opin. Chem. Biol. 46, 56–62. doi:10.1016/j.cbpa.2018.04.002
Schubert, M. G., Goodman, D. B., Wannier, T. M., Kaur, D., Farzadfard, F., Lu, T. K., et al. (2021). High-throughput functional variant screens via in vivo production of single-stranded DNA. Proc. Natl. Acad. Sci. U. S. A. 118, e2018181118. doi:10.1073/pnas.2018181118
Seto, S., and Miyata, M. (1998). Cell reproduction and morphological changes in Mycoplasma capricolum. J. Bacteriol. 180, 256–264. doi:10.1128/JB.180.2.256-264.1998
Sirand-Pugnet, P., Citti, C., Barré, A., and Blanchard, A. (2007). Evolution of mollicutes: down a bumpy road with twists and turns. Res. Microbiol. 158, 754–766. doi:10.1016/j.resmic.2007.09.007
Sleator, R. D. (2010). The story of Mycoplasma mycoides JCVI-syn1.0. Bioeng. Bugs 1, 231–232. doi:10.4161/bbug.1.4.12465
Sleator, R. D. (2016). JCVI-syn3.0 – a synthetic genome stripped bare. Bioengineered 7, 53–56. doi:10.1080/21655979.2016.1175847
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi:10.1093/bioinformatics/btu033
Su, C. J., and Baseman, J. B. (1990). Genome size of Mycoplasma genitalium. Genome Size Mycoplasma genitalium 172, 4705–4707. doi:10.1128/jb.172.8.4705-4707.1990
Sundararaj, S., Guo, A., Habibi-Nazhad, B., Rouani, M., Stothard, P., Ellison, M., et al. (2004). The CyberCell Database (CCDB): a comprehensive, self-updating, relational database to coordinate and faciliate in silico modeling of Escherichia coli. Nucleic Acids Res. 32, D293–D295. doi:10.1093/nar/gkh108
Sung, W., Ackerman, M. S., Miller, S. F., Doak, T. G., and Lynch, M. (2012). Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl. Acad. Sci. U. S. A. 109, 18488–18492. doi:10.1073/pnas.1216223109
Szydlo, K., Ignatova, Z., and Gorochowski, T. E. (2022). Improving the robustness of engineered bacteria to nutrient stress using programmed proteolysis. ACS Synth. Biol. 11, 1049–1059. doi:10.1021/acssynbio.1c00490
Tully, J., Rose, D., Whitcomb, R. F., Hackett, K. J., Clark, T. B., Henegar, R. B., et al. (1987). Characterization of some new insect-derived acholeplasmas. Isr. J. Med. Sci. 23, 699–703.
Tully, J. G. (1979). Special features of the acholeplasmas. Mycoplasmas 1, 431–449. doi:10.1016/b978-0-12-078401-1.50022-4
Tully, J. G. (1983). The Emmy Klieneberger-Nobel Award lecture. Reflections on recovery of some fastidious mollicutes with implications of the changing host patterns of these organisms. Yale J. Biol. Med. 56, 799–813.
Tully, J. G., Bove, J. M., Laigret, F., and Whitcomb, R. F. (1993). Revised taxonomy of the class mollicutes: proposed elevation of a monophyletic cluster of arthropod-associated mollicutes to ordinal rank (Entomoplasmatales ord. Nov.), with provision for familial rank to separate species with nonhelical morphology. Entom. Int. J. Syst. Bacteriol. 43, 378–385. doi:10.1099/00207713-43-2-378
Tully, J. G., Rose, D. L., Hackett, K. J., Whitcomb, R. F., Carle, P., Bove, J. M., et al. (1989). Mycoplasma ellychniae sp. nov., a sterol-requiring mollicute from the firefly beetle Ellychnia corrusca. Int. J. Syst. Bacteriol. 39, 284–289. doi:10.1099/00207713-39-3-284
Tully, J. G., Whitcomb, R. F., Hackett, K. J., Rose, D. L., Henegar, R. B., Bové, J. M., et al. (1994). Taxonomic descriptions of eight new non-sterol-requiring Mollicutes assigned to the genus Mesoplasma. Int. J. Syst. Bacteriol. 44, 685–693. doi:10.1099/00207713-44-4-685
Tully, J. G., Whitcomb, R. F., Rose, D. L., Hackett, K. J., Edward, C. L. A. R. K., Henegar, R. B., et al. (1990a). Current insight into the host diversity of acholeplasmas. Zentralblatt für Bakteriol. 20, 461–467.
Tully, J. G., Rose, D. L., McCoy, R. E., Carle, P., Bové, J. M., Whitcomb, W. G., et al. (1990b). Mycoplasma melaleucae sp. nov., a sterol-requiring mollicute from flowers of several tropical plants. Int J Syst Bacteriol. 40 (2), 143–7. doi:10.1099/00207713-40-2-143
Tully, J. G., Whitcomb, R. F., Hackett, K. J., Williamson, D. L., Laigret, F., Carle, P., et al. (1998). Entomoplasma freundtii sp. nov., a new species from a green tiger beetle (Coleoptera: Cicindelidae). Int J Syst Bacteriol. 48 (4), 1197–204. doi:10.1099/00207713-48-4-1197
Volkmer, B., and Heinemann, M. (2011). Condition-Dependent cell volume and concentration of Escherichia coli to facilitate data conversion for systems biology modeling. PLoS One 6, e23126–6. doi:10.1371/journal.pone.0023126
Whitcomb, R., and Tully, J. (1995). Revised minimum standards for description of new species of the class mollicutes (division tenericutes). J. Syst. 45, 605–612. doi:10.1099/00207713-45-3-605
Whitcomb, R. F., Tully, J. G., Rose, D. L., Stephens, E. B., Smith, A., McCoy, R. E., et al. (1982). Wall-less prokaryotes from fall flowers in Central United States and Maryland. Curr. Microbiol. 7, 285–290. doi:10.1007/bf01566864
Williamson, D. L., Tully, J. G., Rose, D. L., Hackett, K. J., Henegar, R., Carle, P., et al. (1990). Mycoplasma somnilux sp. nov., Mycoplasma luminosum sp. nov., and Mycoplasma lucivorax sp. nov., new sterol-requiring mollicutes from firefly beetles (Coleoptera: Lampyridae). Int J Syst Bacteriol. 40 (2), 160–4. doi:10.1099/00207713-40-2-160
Wodke, J. A. H., Pucha ka, J., Lluch-Senar, M., Marcos, J., Yus, E., Godinho, M., et al. (2013). Dissecting the energy metabolism in Mycoplasma pneumoniae through genome-scale metabolic modeling. Mol. Syst. Biol. 9, 653. doi:10.1038/msb.2013.6
Yoneji, T., Fujita, H., Mukai, T., and Su’etsugu, M. (2021). Grand scale genome manipulation via chromosome swapping in Escherichia coli programmed by three one megabase chromosomes. Nucleic Acids Res. 49, 8407–8418. doi:10.1093/nar/gkab298
Yus, E., Maier, T., Michalodimitrakis, K., van Noort, V., Yamada, T., Chen, W. H., et al. (2009). Impact of genome reduction on bacterial metabolism and its regulation. Science 326, 1263–1268. doi:10.1126/science.1177263
Zhou, Q., Wu, Y., Deng, J., Liu, Y., Li, J., Du, G., et al. (2023). Combinatorial metabolic engineering enables high yield production of α-arbutin from sucrose by biocatalysis. Appl. Microbiol. Biotechnol. 107, 2897–2910. doi:10.1007/s00253-023-12496-2
Keywords: Mesoplasma florum, Mollicutes, synthetic biology, systems biology, minimal genome
Citation: Matteau D, Duval A, Baby V and Rodrigue S (2024) Mesoplasma florum: a near-minimal model organism for systems and synthetic biology. Front. Genet. 15:1346707. doi: 10.3389/fgene.2024.1346707
Received: 29 November 2023; Accepted: 24 January 2024;
Published: 09 February 2024.
Edited by:
Christine Citti, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Rosario Gil, University of Valencia, SpainIsabel Gordo, Gulbenkian Institute of Science (IGC), Portugal
Copyright © 2024 Matteau, Duval, Baby and Rodrigue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sébastien Rodrigue, Sebastien.Rodrigue@usherbrooke.ca