 Qingqing Liu
Qingqing Liu Guisheng Ye1
Guisheng Ye1- 1Agriculture and Animal Husbandry College, Qinghai University, Xining, Qinghai, China
- 2College of Animal Husbandry and Veterinary Science, Qinghai University, Xining, China
Hippophae rhamnoides subsp. sinensis Rousi is a cold- and drought-tolerant pioneer species with significant economic and ecological value. Evaluating its genetic diversity and population structure is of great importance for guiding the development and utilization of resources. In this study, a total of 41,804 SSRs were generated by transcriptome sequencing of Hippophae rhamnoides subsp. sinensis Rousi. Among the different SSR motif types, mononucleotide repeats (26,972) were the most abundant, followed by trinucleotides, tetranucleotides, and pentanucleotides. 200 pairs of SSR primers were selected to detect polymorphisms, of which 15 pairs primers were selected as validated polymorphic SSRs used for genetic diversity and population structure analysis. A total of 63 alleles were identified with 15 pairs primers, with Nei’s genetic diversity index ranged from 0.27 to 0.83 (average: 0.54), and the expected heterozygosity ranged from 0.16 to 0.73 (average: 0.46). The polymorphism information content ranged from 0.23 to 0.81 (average: 0.48). Genetic structure analyses showed that the 10 populations could be broadly categorized into two groups. AMOVA denoted that genetic variations primarily originated from within the populations, with minimal differences observed between the groups, accounting for only 7% of the total genetic variation. This implies that mutation in H. rhamnoides subsp. sinensis Rousi mainly occurred within the populations. The results showed that the 10 populations of H. rhamnoides subsp. sinensis Rousi are rich in genetic diversity, with low levels of population differentiation and a high degree of gene exchange, which should be taken into consideration for the future work of germplasm resource preservation and seedling breeding.
1 Introduction
Hippophae rhamnoides subsp. sinensis Rousi (Chinese sea buckthorn) is a subspecies of H. rhamnoides L. with the largest distribution range and the longest cultivation history (Wang et al., 2014). It is widely distributed in Europe and Asia (Duan et al., 2022; Kaur et al., 2017; Mei et al., 2023; Ui Haq et al., 2021; Wang et al., 2021a). Hippophae rhamnoides subsp. sinensis has a well-developed root system with nodules, which can effectively improve the soil ecological environment and fertility (He et al., 2023). Additionally, it is a drought and cold-resistant species with a strong adaptive ability to soil, enabling it to survive and reproduce even in infertile gravel areas. Moreover, it can be utilized for water and soil conservation, windbreak, and sand fixation (Dong et al., 2020). The fruits of H. rhamnoides subsp. sinensis is rich in nutrients such as vitamins, linoleic acid, flavonoids, and various amino acids (Bal et al., 2011; Michel et al., 2011; Ghendov-Mosanu et al., 2022). Sea buckthorn oil, known as liquid gold, can effectively reduces cholesterol levels, protects the gastric mucous membrane (Trineeva et al., 2020; Pundir et al., 2021), prevents hypertension and hyperlipidemia, and promotes wound healing (Tuomasjukka et al., 2006). Therefore, the research and development of sea buckthorn can acquire excellent ecological, economic, and social value.
For a long time, due to environmental degradation and over-exploitation of natural resources, the stock of H. rhamnoides subsp. sinensis has declined dramatically. Thus, effective strategies are important to ensure the conservation and scientific utilization of H. rhamnoides subsp. sinensis (Dawson, 2011; Hu et al., 2021), and the diversity research is a prerequisite for the conservation of Chinese sea buckthorn resources. Recently, there have been a few studies on the genetic diversity of cultivated H. rhamnoides subsp. sinensis Rousi. From the morphological point of view, It found rich genetic diversity in H. rhamnoides subsp. sinensis across different regions, as evidenced by variations in morphological indexes such as leaf blade, fruit, tree height, crown width, and 100-fruit weight (Liu et al., 2023). Molecular analyses conducted by using ITS sequence analysis revealed genetic interpenetration between H. rhamnoides subsp. sinensis and other sea buckthorn subspecies, which contribute to the species diversity within H. rhamnoides subsp. sinensis Rousi (Jia et al., 2012). Additionally, Li Hongmei analyzed genetic variation in Chinese sea buckthorn through allelopathic enzymes, chromosomes, and morphology, the results showed that the degree of variation in H. rhamnoides subsp. sinensis differed between and within populations. Inter-population variation was mainly observed in traits such as 100-fruit weight, tepal shape, fruit shape, and seed thousand-grain weight, while intra-population variation among individuals mainly involved the number of thorns on 1-year-old shoots and the number of inflorescences bearing flowers (Li et al., 2003). It above research indicates the rich genetic diversity of Chinese sea buckthorn.
Transcriptome sequencing is widely used in the study of diversity research because of its wide dynamic range, sensitivity, precise, unbiased quantification of transcripts, and comprehensive coverage of all expressed sequences in a given tissue sample (Yang et al., 2018; Chakraborty et al., 2022; Liu et al., 2022). Simple-sequence repeats (SSRs), also known as microsatellites, are short tandem repeated motifs that may vary in the number of repeats at a given locus (Mahfooz et al., 2017). SSR markers have many advantages over other molecular markers, such as genetic co-dominance. They are multi-allelic, relatively abundant, widely dispersed across the genome, and easily and automatically scored (Gupta and Sharma, 2020; Yasar et al., 2020). SSR has become the best choice for DNA fingerprinting, gene mapping, variety identification, genetic diversity analysis and other applications. (Bhusan et al., 2016). Recently, SSRs have been widely employed in numerous studies on plant diversity, including research on species such as Camellia fascicularis (Bin et al., 2022), Beta vulgaris L. (Burns et al., 2010), Vanda stangeana Rchb. f. (Soneja et al., 2023), Oryza sativa L. (Li et al., 2023), or Punica granatum L. (Angelica et al., 2023). However, there is lack of the research on the genetic diversity of H. rhamnoides subsp. sinensis Rousi in Qinghai, and no published reports on the utilization of SSR markers, which makes it difficult to conduct in-depth studies on the genetic diversity of H. rhamnoides subsp. sinensis Rousi. The above studies show that Chinese sea buckthorn has rich genetic diversity. It is of great significance to study its genetic diversity. However, there is no systematic study on the development and application of ssr primers for Chinese sea buckthorn.
In this study, 15 pairs of SSR primers with clear bands, rich polymorphisms, and good repeatability were selected based on the transcriptome data of the H. rhamnoides subsp. sinensis (Ye et al., 2018). SSR molecular markers were analyzed on 150 samples from 10 districts in Qinghai Province. Our study aimed to develop polymorphic microsatellite markers and verify their polymorphism level for H. rhamnoides subsp. sinensis Rousi; and then evaluate the genetic diversity and structure among different populations using the polymorphic SSR markers from transcriptome sequencing. Our findings can provide valuable information for effective breeding and conservation strategies of H. rhamnoides subsp. sinensis Rousi germplasm resource.
2 Materials and methods
2.1 Experimental materials
A total of 150 test samples from female plants of H. rhamnoides subsp. Sinensis Rousi without diseases and pests were collected from Menyuan, Maqin, Banma, Huzhu, Qilian, Tongde, Minhe, Huangyuan, Datong, and Guinan in Qinghai Province from August to September 2020 and 2021 respectively, with distances between individuals not less than 20 m. Fresh leaves were collected in separate ziplock bags and quickly dried with allochroic silica gel. The information of the test samples is shown in Table 1 and Figure 1.
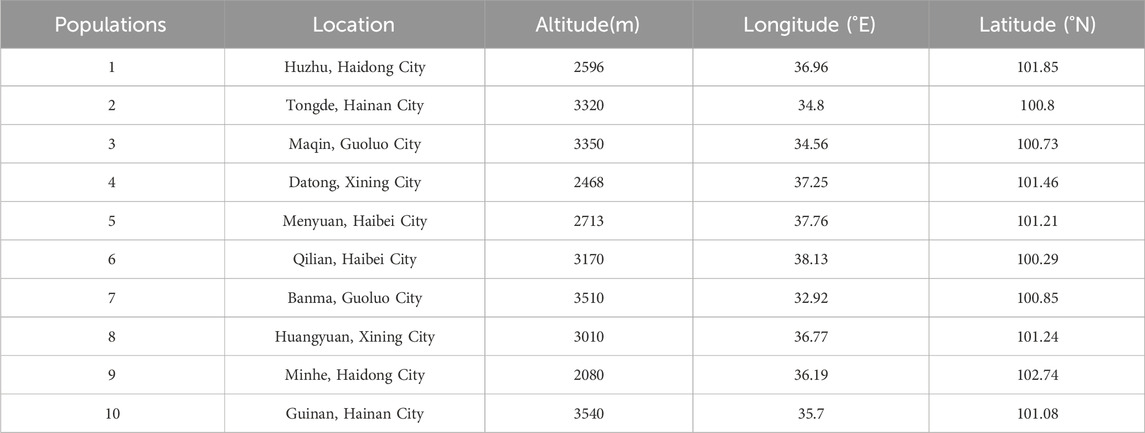
Table 1. Sample information of Hippophae rhamnoides subsp. sinensis.
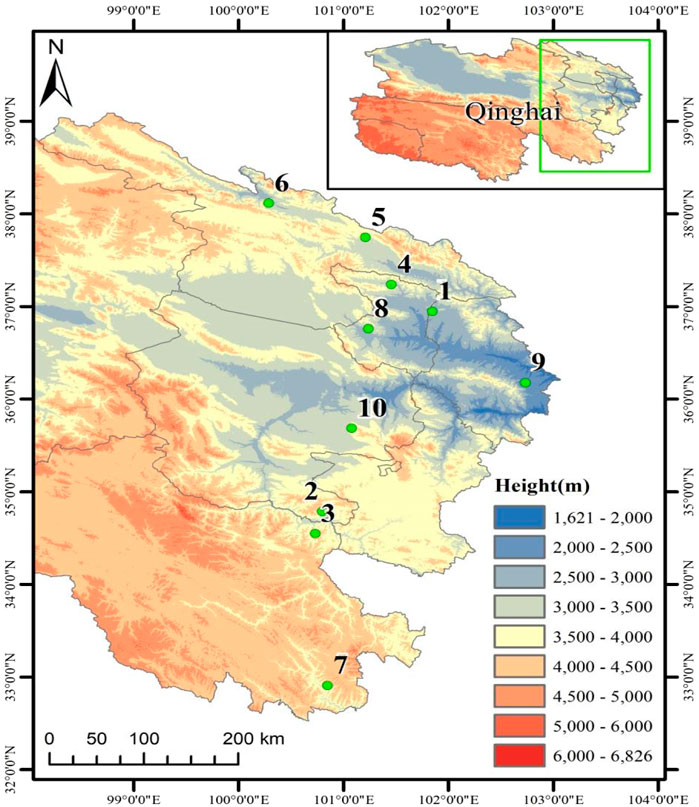
Figure 1. Sample collection map of Hippophae rhamnoides subsp. sinensis.
2.2 The sourced of transcriptome data
Transcriptome data were derived from our previous experiments, and the specific method has been written in the previously published article (Ye et al., 2018).
2.3 DNA extraction
An adequate sample were taken for genomic DNA extraction according to the instructions of the modified CTAB Plant Genomic DNA Rapid Extraction Kit. The integrity and quality of the DNA were evaluated by electrophoresis on 0.8% agarose gels, and the concentration of the DNA was determined by NanoDrop One Spectrophotometer, and then all the DNA samples were diluted to 50 ng/μL and stored at −20°C for future use.
2.4 SSR primer-blast design and analyze
Primer3 was used to screen microsatellite loci and the primers were designed based on the identified SSR loci of the whole genome H. rhamnoides subsp. Sinensis. The SSR primers obtained from the systematic screening were further evaluated using oligo7 software to filter for hairpin structures, dimers, and mismatches. The main parameters of primer design were as follows: the length of primer was 18–28 bp, the temperature difference between the upstream and downstream primers was ≤5°C, and the annealing temperature of the primer pair was 50–65°C. The proportion of G and C bases ranged from 40%–70%; the fragment length of the amplified product was expected to be between 100 and 350 bp. According to the difference in Tm value and the size of the product, 200 pairs of primers were screened and synthesized. At the initial stage of the experiment, mixed samples of H. rhamnoides subsp. sinensis DNA were used for PCR with 200 pairs of primers. The effectively amplified bands of each pair of primers were examined.
2.5 SSR-PCR reaction system and program
The optimal PCR system developed by the project team was adopted: 0.8 U Taq enzyme, 2 mmol L-1 Buffer, 1.5 mmol·L-1dNTP, 0.5 umol·L-1 primer, 25 ng of template DNA, and 20 uL of ddH2O. The PCR amplification procedure was shown in Table 2, and the PCR reaction products were identified by 6% polypropylene gel electrophoresis and then the data were statistically analyzed.

Table 2. PCR amplification procedure.
2.6 PCR amplification product detection of polymorphic primers and data analysis
Fifteen pairs of SSR primers of H. rhamnoides subsp. sinensis were selected for the marker analysis of 150 samples from 10 natural populations. Popgene32 statistics were used to calculate the number of observed alleles (Na), the number of effective alleles (Ne), Shannon’s information index (I), Observed homogeneity (Obs_Hom), Observed heterozygosity (Obs_Het), Expected homogeneity (Exp_Hom), Expected heterozygosity (Exp_Het), Diversity index Nei, genetic similarity and genetic distance. Genetic diversity indices were obtained by analyzing the genetic diversity and genetic differentiation of 15 pairs of SSR primers and 150 Chinese sea buckthorn samples by the Excel 2010 add-in GenAlEx 6.51b2 (Peakall, 2012). The analysis of molecular variance (AMOVA) between and within different populations of the H. rhamnoides subsp. sinensis was performed by GenAlEx 6.51b2. The polymorphism information content (PIC) of SSR loci of Chinese sea buckthorn was analyzed with Power Marker software, while Nei’s genetic distances were calculated among different populations. A clustering tree was constructed based on Nei’s genetic distances in MEGA X. STRUCTURE 2.2.3 software and used to analyze the genetic structure of the 10 populations of Chinese sea buckthorn. To obtain the best k value, CLUMPP software was used to merge the data, DISTRUCT software was used to obtain the structural map under the best k value, and AI software was used to obtain the final result map.
3 Results
3.1 Transcriptome assembly and sequence annotation
The raw reads produced in this study have been deposited in the Short Read Archive of the National Center for Biotechnology Information (NCBI) with accession numbers SRR7003894, SRR7003893, SRR7003896, SRR7003895, SRR7003892, and PRJNA449450. The articles published in the early stage of transcriptome assembly and gene annotation have been analyzed in detail (Ye et al., 2018). Which include GO and KEGG pathway enrichment analysis.
The Nr annotation results showed that E-value ≤0. Similarity distribution showed that more than 85% of sequences with more than 60% similarity, which indicates a high degree of confidence in these results. Similarity classifications showed that H. rhamnoides subsp. sinensis had the highest similarity with Prunus mume (16.9%) and the second highest similarity with Prunus persica (13.9%), indicating that H. rhamnoides subsp. sinensis and Prunus mume are most closely related (Figure 2).
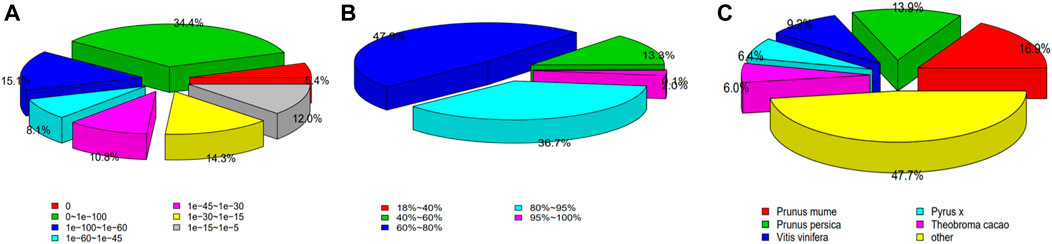
Figure 2. Nr Annotation of the Hippophae rhamnoides subsp. sinensis. (A) E-value distribution. (B) similarity distribution. (C) species classification.
3.2 Development of the SSRs based on transcriptome sequencing
141,380,000 bp in Genome discovered by MISA and 32,709 sequences contained a total of 41,804 SSR sites, of which 7,148 sequences contained 1 SSR site. The genome of H. rhamnoides subsp. sinensis. Has a rich variety of SSR nucleotide repeating motifs, and the repeating motifs have base variations ranging from 1 to 6. The mononucleotide SSR motifs were the most abundant type of repeats (26,976), accounting for 64.53% of the total SSRs, followed by the dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide SSR motifs, and the penta-type was the least abundant (Table 3; Figure 3).
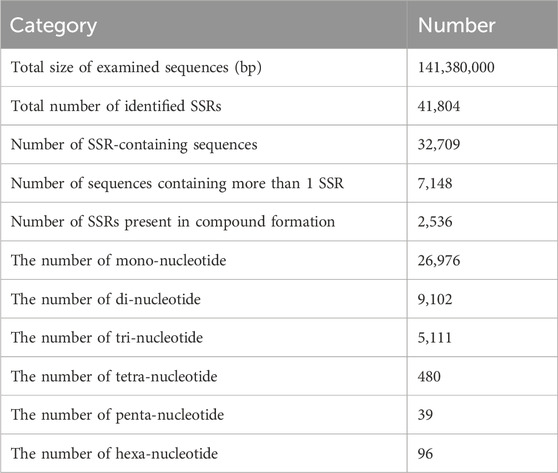
Table 3. SSR loci information in Hippophae rhamnoides subsp. sinensis.genome.
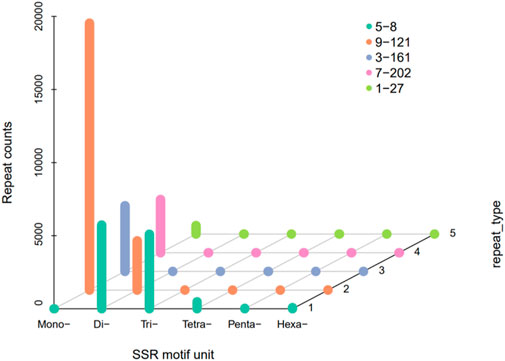
Figure 3. The number distribution of SSR motif unit in Hippophae rhamnoides subsp. sinensis. transcriptome.
According to the length of the genomic SSRs based on the number of repeat units (Table 4), the most abundant type of repeats was ten, accounting for 13,235 (31.66%), followed by eleven, six, five, and seven repeat units. Only 1,332 units presented more than 20 repeats (3.19%). The five, six, seven, ten, and eleven repeat units accounted for 65.52% of the total repeat units, which could explain the predominant diversity of SSR repeat unit types. The most abundant type of repeat motif was A/T, accounting for 26,502 of the repeats (63.40%), followed by AG/CT, AT/AT, and AAG/CTT, the other SSR motifs types (2,108) accounted for 5.04% of the repeats (Figure 4).
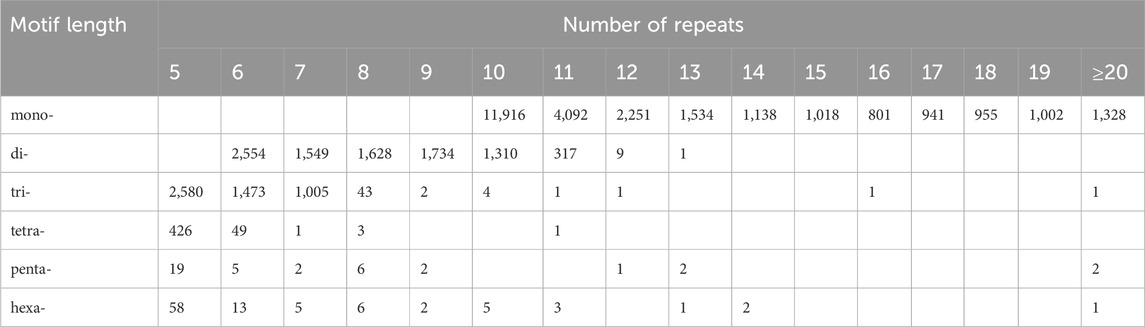
Table 4. Frequency of SSR motifs of genome-SSRs in Hippophae rhamnoides subsp. sinensis.
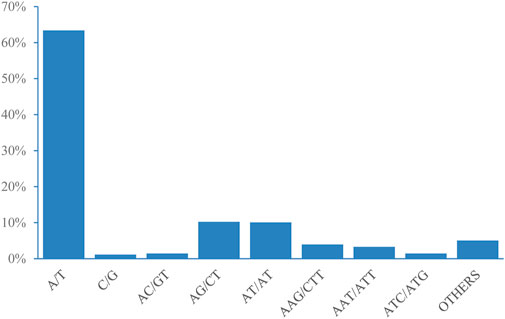
Figure 4. Frequency distribution of genome-SSRs based on motif numbers.
3.3 Polymorphism primer
From the H. rhamnoides subsp. sinensis SSR primer sequences designed by Primer3 software, 200 pairs of qualified primers were randomly selected and sent to Shanghai Sangong Bioengineering Co., Ltd. for synthesis. Based on the optimized SSR system, the synthesized primers were used to amplify the H. rhamnoides subsp. sinensis samples, finally, 90 pairs of primers were able to amplify clear bands, which were then screened by polypropylene gel electrophoresis. A total of 15 pairs of polymorphic primers were selected (Table 5).
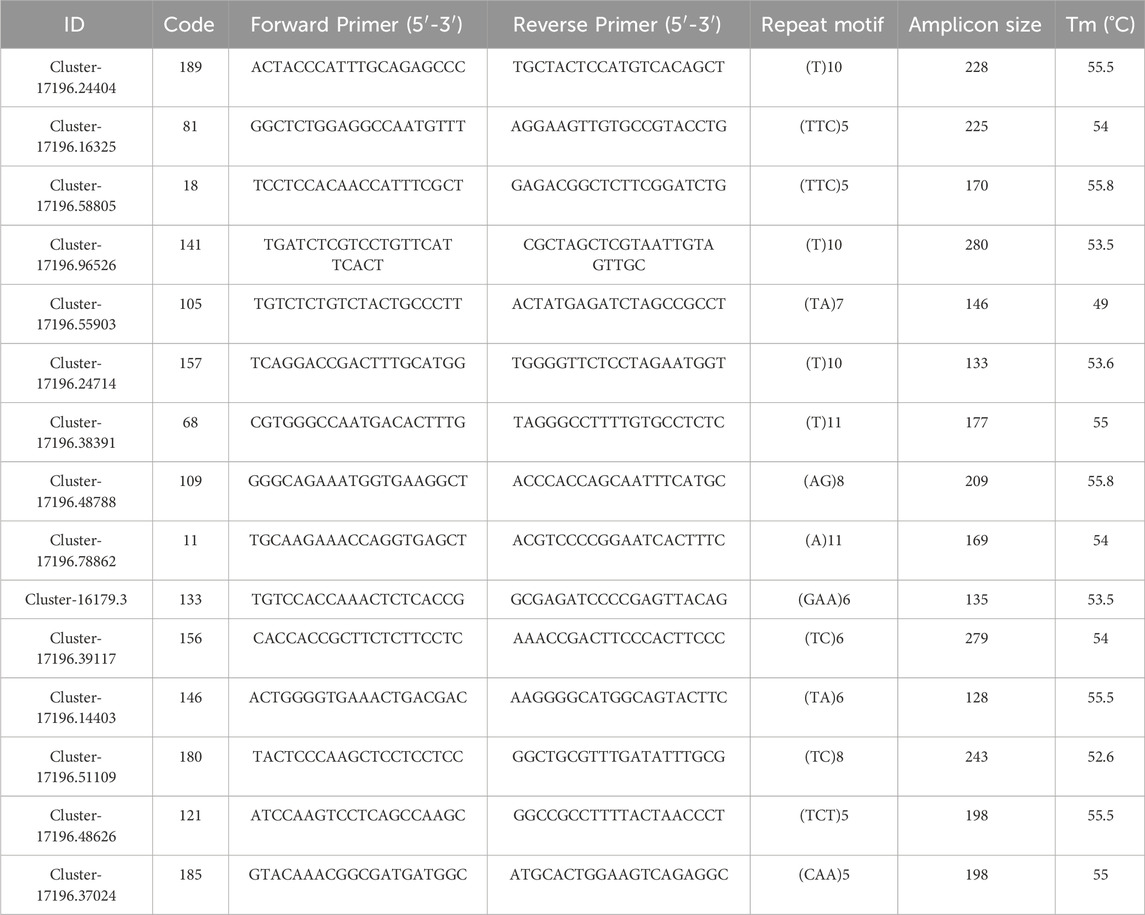
Table 5. Sequence of SSR primers.
3.4 Primer polymorphism analysis
200 pairs of primers were used to screen polymorphic primers, of which 15 pairs of primers could produce clear bands and rich polymorphisms, and then were used to detect the genetic diversity of 150 H. rhamnoides subsp. sinensis Rousi samples from 10 populations (Table 6). 63 alleles were identified, ranging from 2 to 10 (NA), among them, primer 156 amplified the most allelic sites (10), 189, 141, 68, and 11 amplified the least allelic sites (2). The Ne ranged from 1.37 to 5.99. The I varied from 0.44 to 1.95 (mean: 0.98). The Exp_Hom varied from 0.16 to 0.73 (mean: 0.46). The Obs_Hom varied from 0.01 to 0.76, (mean: 0.34). The Exp_Het varied from 0.27 to 0.84 (mean: 0.54). The Obs_Het varied from 0.24 to 0.99 (mean: 0.66). This indicates that the test material has a high degree of heterozygosity. The average number of effective alleles was 2.51, and the diversity index Nei ranged from 0.27 to 0.83 (mean:0.54). The PIC ranged from 0.23 to 0.81 (mean: 0.48), among which there were 5 pairs of primers with PIC>0.5 and 9 pairs of primers with PIC values between 0.25 and 0.5. When the PIC is greater than 0.25, it indicates that the locus is medium polymorphic. It can be seen that 93.3% of the primers had medium to high polymorphism, indicating that the SSR primers selected in this study were highly polymorphic.
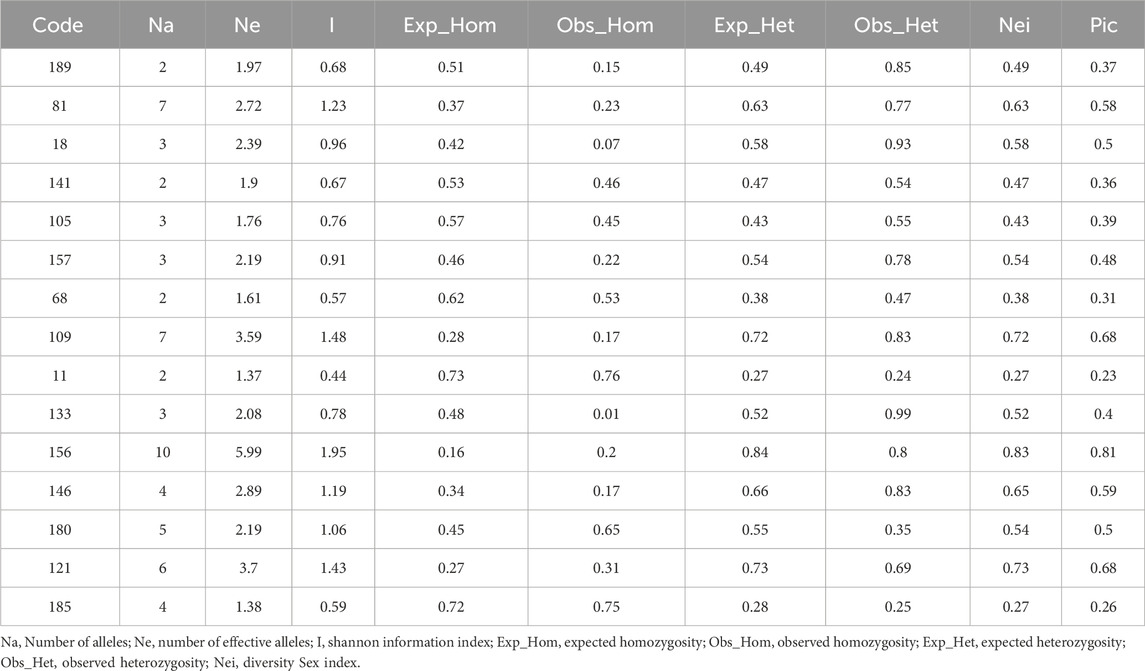
Table 6. Statistical analysis of polymorphic parameters of the 15 SSR primer.
3.5 Genetic diversity within populations
The genetic diversity of H. rhamnoides subsp. sinensis Rousi from different populations were analyzed, and all parameters of the 15 SSR loci were calculated (Table 7). The Na ranged from 2.67 to 3.40, with an average of 3.13; Ne ranged from 1.91 to 2.42, with an average of 2.25; Na and Ne indicated that there were allelic differences among Chinese sea buckthorn populations, but the genetic differences were not significant. The highest I was 0.90, the lowest was 0.66, and the average was 0.83; the He ranged from 0.40 to 0.51, with an average of 0.49; the Ho ranged from 0.58 to 0.73, with an average of 0.66; the uHe ranged from 0.42 to 0.53, with an average of 0.50; the HO was greater than the He has shown the majority of SSR loci had null alleles. Population 7 had the highest genetic diversity in all the populations (Na = 3.40, I = 0.90, He = 0.51). In the selection and breeding of H. rhamnoides subsp. sinensis Rousi, priority can be given to plants in Population 7.
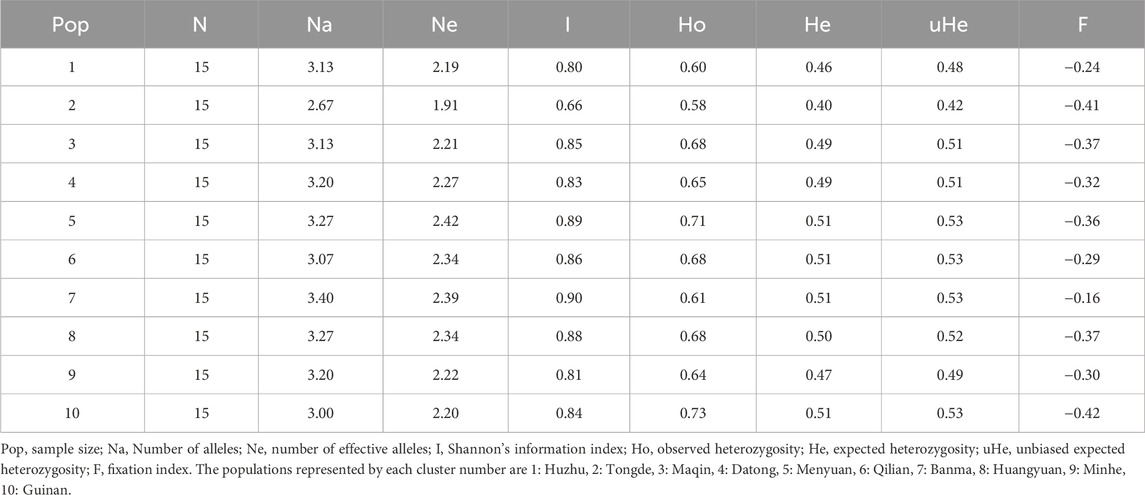
Table 7. Genetic diversity analysis of Hippophae rhamnoides.
3.6 Analysis of population genetic differentiation
Inbreeding coefficients (Fis) ranged from −0.91 to 0.24 (Table 8) with an average value less than 0, indicating that there were excessive heterozygotes in the H. rhamnoides subsp. sinensis Rousi populations. The coefficient of inbreeding (Fit) ranged from −0.90 to 0.36, the coefficient of genetic differentiation (Fst) ranged from 0.01 to 0.21, and the minimum value of gene flow (Nm) was 0.93 and the maximum value was 45.35, which indicated that gene flow was at a relatively high level among Chinese sea buckthorn populations, and that a large number of genes were exchanged among populations.
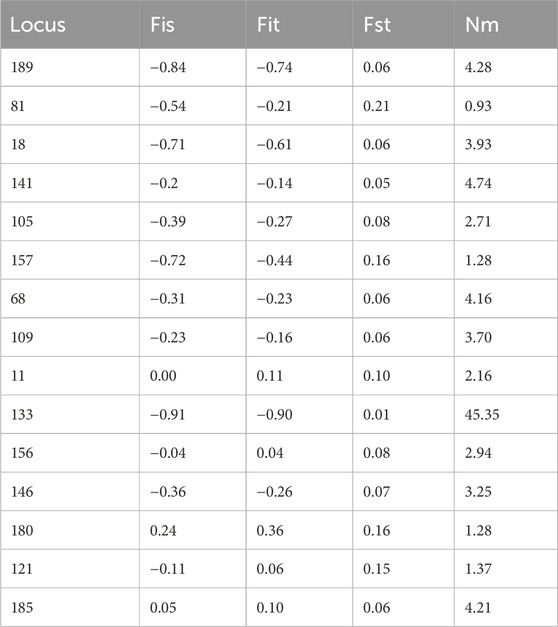
Table 8. Correlation coefficient of population genetic differentiation.
Genetic distance and genetic concordance are two important indicators used to determine the kinship between populations. A genetic similarity of zero suggests complete dissimilarity and unrelatedness between populations, while a genetic similarity of 1 indicates complete identity. The magnitude of the genetic distance directly reflects the degree of relatedness. In this study, the genetic consistency ranged from 0.80 to 0.96, and the genetic distance ranged from 0.04 to 0.22 (Table 9), indicating that the 10 populations were relatively close to each other. Among them, the genetic similarity between the population 9 and the population 5 was the highest (0.96), and the genetic distance was the smallest (0.04). It suggests that the population 9 and the population 5 are the least genetically differentiated and most closely related.
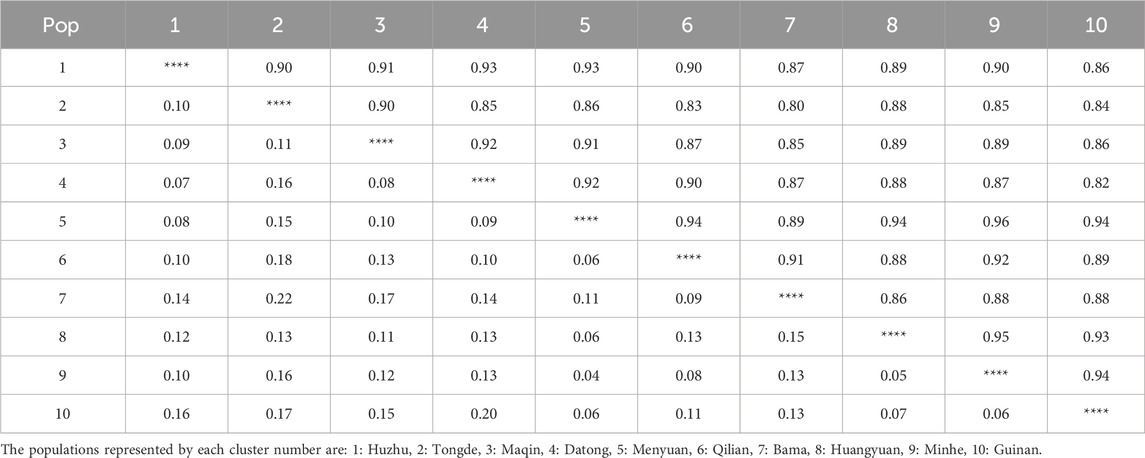
Table 9. Genetic consistency (upper right triangle) and genetic distances (lower left triangle) of Hippophae rhamnoides subsp. sinensis.
3.7 Analysis of molecular variance (AMOVA) of the settlements
The Analysis of molecular variance results are shown in Table 10. AMOVA revealed that 7% of the total variation was due to differences among populations, with the remaining 97% due to differences within populations, the genetic variation rate within the populations was much larger than that between populations, which indicated that the genetic variations of H. rhamnoides subsp. sinensis in Qinghai Province mainly occurred in the individuals of the populations. Intra-individual variation refers to genetic differences caused by heterozygous alleles, the size of which correlates with the number of individual heterozygous, the genetic diversity of individual suggesting that there is a high degree of genetic differentiation within individual, and that individual genetic variation is the main source of H. rhamnoides subsp. sinensis Rousi variation.
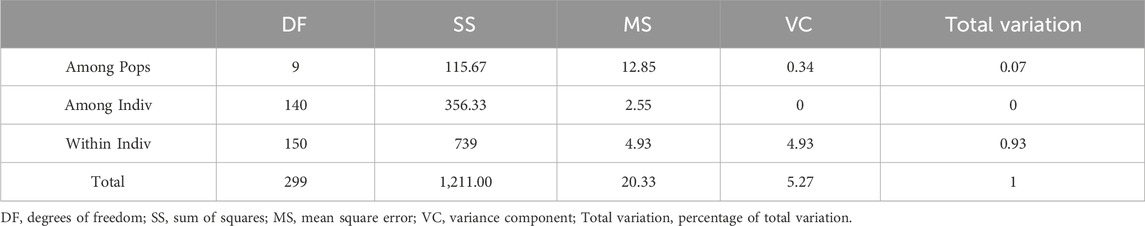
Table 10. Molecular analysis of variance within and between populations.
3.8 Population principal coordinate analysis (PCoA)
To further study the genetic similarity and evolution of different populations of H. rhamnoides subsp. sinensis and to visualize the results clearly and concisely, the GenAlEx6.51b2 software was used to analyze PCoA of 10 populations based on Nei’s genetic distance. The eigenvalues of the three principal coordinates contributing to the total variation accounted for 31.13%, of which the first principal coordinate accounted for 12.27% of the total variation, the second principal coordinate accounted for 10.64% of the total variation, and the third principal coordinate explained 8.23% of the total variation (Table 11). The PCA analysis revealed two groups of samples, of which one group included populations 1, 2, 3, and 4. Because they are more concentrated in the lower half of the plot. The other group included populations 5, 6, 7, 8, 9 and 10, which are more concentrated in the upper half of the plot (Figure 5).
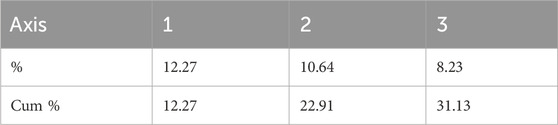
Table 11. Percentage change explained by the first three axes.
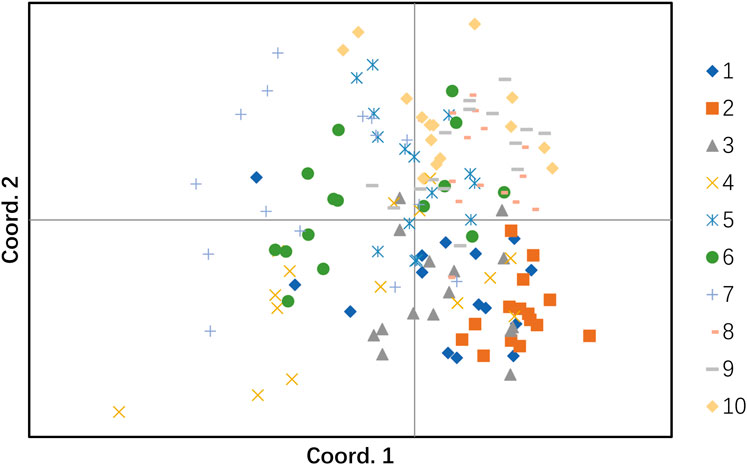
Figure 5. The principal coordinate analysis (PCoA). The cluster numbers represent the following: 1: Huzhu, 2: Tongde, 3: Maqin, 4: Datong, 5: Menyuan, 6: Qilian, 7: Banma, 8: Huangyuan, 9: Minhe, 10: Guinan.
3.9 Cluster analysis
The UPGMA clustering diagram were constructed using MEGAX software based on Nei’s genetic distance among different populations of H. rhamnoides subsp. sinensis Rousi, as shown in Figure 6. The bootstrap value of each arm are greater than 80%, indicating that the results are credible. The 10 populations were divided into two groups: populations 1, 2, 3, and 4 formed the first group, while populations 5, 6, 7, 8, 9, and 10 constituted the second group, consistent with the PCA results.
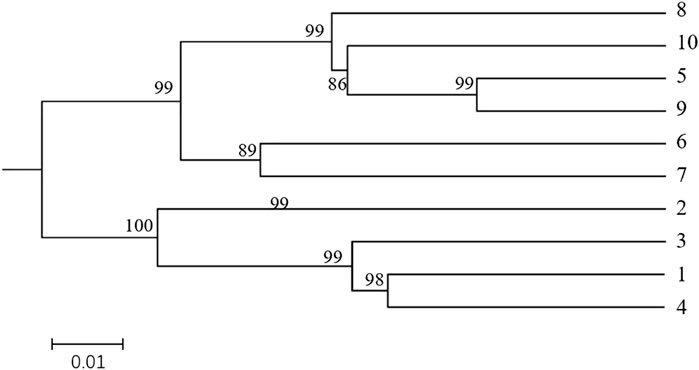
Figure 6. Dendrogram of the Hippophae rhamnoides subsp. sinensis Rousi populations constructed using UPGMA methods UPGMA methods based on matrices of pairwise Nei′s genetic distances. Note:The clusters represented by the numbers are: 1: Huzhu, 2: Tongde, 3: Maqin, 4: Datong, 5: Menyuan, 6: Qilian, 7: Banma, 8: Huangyuan, 9: Minhe, 10: Guinan.
3.10 Population genetic structure analysis
The genetic structure of 10 H. rhamnoides subsp. sinensis Rousi populations were analyzed using STRUCTURE 2.2.3 software, setting the range of the group number K values as 1 to 10, repeating the operation 10 times for each K value, and plotting the relationship between K values and ΔK. submitted to the website http://taylor0.biology.ucla.edu/structure Harvester/for analysis. The structure of H. rhamnoides subsp. sinensis Rousi germplasm resource genotypes was analyzed based on the likelihood of data [LnP(D)]. The germplasm resources were integrated into groups (K) to assess the variant frequency of each group, and the individual germplasms were reintegrated into groups based on the estimated frequencies. The number of subgroups varies with the LnP(D) values. The curve occurs between K = 1 and K = 2, and all the values are at their maximum. (Table 12, Figure 7). Therefore, the 150 accessions could be divided into two subgroups. The outputs of K = 2 were visualized through CLUMPP and DISTRUCT. Then, we visualized that all individuals are clustered into two groups, including the wild group (green) and the cultivated group (red) (Figure 8). The first group consisted of clusters from Huzhu, Tongde, Maqin, and Datong, and the second group consisted of clusters from Mengyuan, Qilian, Banma, Huangyuan, Minhe and Guinan. This is consistent with the results of MEGAX software clustering.
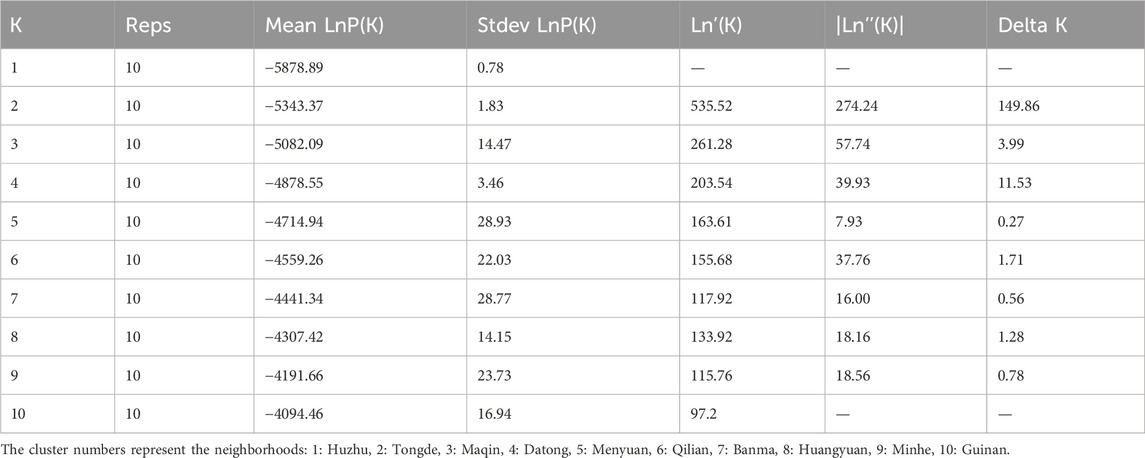
Table 12. Parameter values under different K values.
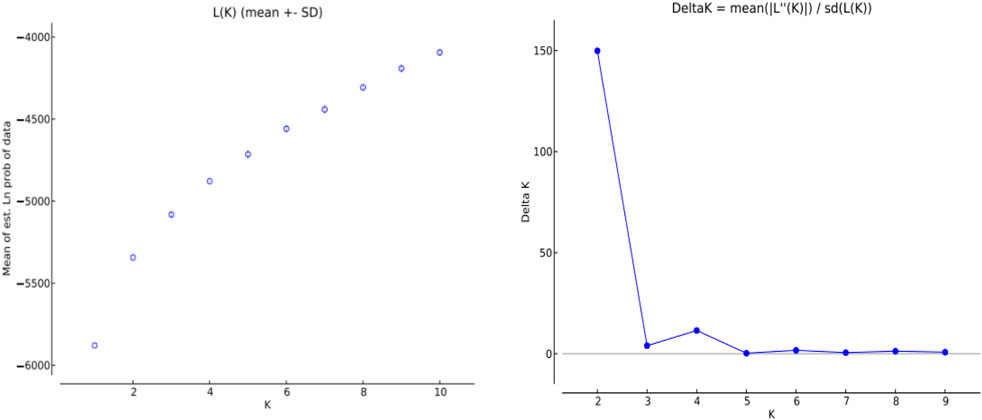
Figure 7. Variation law of ΔK value with K value.
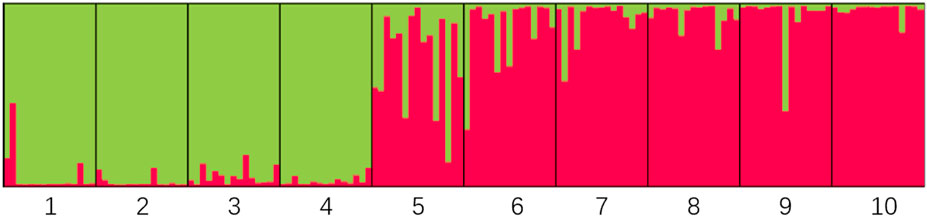
Figure 8. Structure of optimal K value. Note: The residence groups represented by each cluster number are: 1: Huzhu, 2: Tongde, 3: Maqin, 4: Datong, 5: Menyuan, 6: Qilian, 7: Banma, 8: Huangyuan, 9: Minhe, 10: Guinan.
4 Discussion
Hippophae rhamnoides subsp. sinensis is a typical plant with significant ecological and economic value. With the rapid advancement of high-throughput sequencing technology, scholars have analyzed the transcriptome of H. rhamnoides subsp. sinensis Rousi. Zhou et al. used RNA-Seq high-throughput sequencing to obtain transcriptome data from the leaves of male and female H. rhamnoides subsp. sinensis Rousi plants, identifying 33,248 microsatellite markers, which provided the basic data for gene mining and molecular labeling of Chinese sea buckthorn (Zhou et al., 2019). In this study, the transcriptunigene sequence count reached 175,425, with a total of 41,804 SSR sites identified. The SSR repeat types were abundant, and the content of short repeat units was high in the Chinese sea buckthorn transcriptom, which was consistent with the results of Wang et al. (Wang et al., 2021b). This study provides new high-throughput data for transcriptomics, which provides valuable information for germplasm conservation and breeding of H. rhamnoides subsp. sinensis Rousi. The Nr results showed that Chinese sea buckthorn had the highest sequence similarity with Prunus mume, but the SSR primers of Prunus mume can not applied to H. rhamnoides subsp. sinensis Rousi (Xue, 2021), because the SSR sequences obtained in this research had more sites and appeared more frequently. It indicates that SSRs are variable in different plants, and this situation is mainly caused by different development methods, search criteria, and the abundance of DNA sequence resources (Wei et al., 2011; Xie et al., 2023).
Currently, the development of SSR markers using transcriptome sequences has been further extended and has been widely used in population genetics, the construction of genetic linkage maps, and analysis of genetic relationships between germplasm and marker-assisted selection breeding (Gong, et al., 2016; Tulsani, et al., 2020; Tian, et al., 2021; Duan, et al., 2022a), RNA-Seq has become an effective method for analyzing transcriptome data. SSR markers have also been developed and applied by Chinese sea buckthorn RNA-seq by previous authors. Li et al. used 17 polymorphic RNA-Seq SSR primers to study the genetic variation and population genetic structure of 80 H. rhamnoides subsp. sinensis Rousi from Datong and Dongsheng, found that the genetic diversity of Chinese sea buckthorn was consistent with their origin and genetic background (Li, et al., 2019). Wang et al. found that the genetic diversity of H. rhamnoides subsp. sinensis Rousi was higher than that of Yunnan sea buckthorn by using RNA-Seq SSR primers (Wang, et al., 2021b). However, due to the late start of the application of SSR based on transcriptome sequencing in Chinese sea buckthorn, it is difficult to develop SSR primers.
Chinese sea buckthorn is wind-pollinated and dioecious, combined with the wide distribution area, the differences in ecological factors such as climate, soil and altitude in the distribution area, as well as the evolution of the population, forming a rich variation type of plants in the genus (Ma et al., 2014). Qinghai is the birthplace of seabuckthorn, it is of great significance to study the genetic variation of Chinese seabuckthorn. The ssr primers developed in this study can provide the basis for the genetic diversity of Chinese sea buckthorn in Qinghai. 15 pairs of SSR primers designed were able to amplify clear and polymorphic bands. The reason why the remaining primers could not be amplified may be: (1) There are many intronic fragments in the genomic DNA, and one or both ends of the primers designed for the SSR site are located at the intronic or exonic shear point, so the primers could not find the binding site in the genome (Gao, 2013). (2) In the initial screening of primers, PCR was carried out by uniformly annealing temperature of 55°C, which may lead to the failure of those primers with lower annealing temperature.
In this study, the genetic diversity of 10 H. rhamnoides subsp. sinensis Rousi populations were analyzed and the results showed that the number of alleles detected in SSRs was 31.34, with an average of 3.13 alleles per locus. At the population level, the Shannon information index of Chinese sea buckthorn ranged from 0.66 to 0.90, indicating high genetic diversity. Wu Q (Wu et al., 2007) analyzed the phenotypic traits of H. rhamnoides subsp. sinensis Rousi in Wutai Mountain, Shanxi Province, showed that Chinese sea buckthorn at the same altitude and habitat had a wide range of morphological traits, which were rich in genetic diversity. Tian (Tian et al., 2004) analyzed its genetic diversity by ISSR and found that H. rhamnoides subsp. sinensis Rousi was very rich in genetic diversity. The above results consistent with ourresearch results and indicate the genetic diversity was very rich in Chinese sea buckthorn form different population. It can be shown that the developed primers have a high level of polymorphism. The test material has rich genetic diversity, which can provide certain theoretical references for the conservation, development and utilization of the excellent germplasm of H. rhamnoides subsp. sinensis Rousi.
Both the clustering tree and STRUCTURE 2.2.3 software classified the 10 populations of H. rhamnoides subsp. sinensis Rousi into two categories. The first group consisting of wild populations from Huzhu, Tongde, Maqin, and Datong. The second group consisting of populations from Menyuan, Qilian, Banma, Huangyuan, Minhe, and Guinan. The genetic diversity of H. rhamnoides subsp. sinensis Rousi on both sides of the Qilian Mountains found that the geographical isolation of the Qilian Mountains led to restricted gene exchange among H. rhamnoides subsp. sinensis Rousi populations, thus forming obvious genetic differentiation (Zhang et al., 2006). The genetic variation and structure of wild populations of H. rhamnoides subsp. sinensis Rousi using cpSSR and RAPD showed varying degrees of genetic differentiation among populations from different regions (Zhao, 2007). DNA molecular markers found that H. rhamnoides subsp. sinensis Rousi has rich genetic diversity due to the influence of altitude and geographical distance, indicating that geographical isolation and gene exchange were the main factors affecting its genetic diversity. (Sun et al., 2013). However, in this study, the genetic diversity of H. rhamnoides subsp. sinensis Rousi was not significantly related to geographic distance and altitude, which may be attributed to the genetic exchange within the sampled area. It is also possible that the high genetic diversity of Chinese sea buckthorn is due to its dioecious and wind-borne characteristics, the rich genetic variation of the species may expand its distribution range and facilitate adaptation to new environments (Nybom, 2004; Wang et al., 2014).
The rate of genetic variation among the 10 H. rhamnoides subsp. sinensis Rousi populations was 7%, and the rate of genetic variation within the populations was 93%, indicating that the genetic variation of Chinese sea buckthorn mainly occurred within the populations. It may be due to the stochastic nature of plant genetic diversity caused by geographic barriers and the diversity of the environments in which they are located, plants may undergo different types of variation patterns, such as continuous variation and random variation, in response to geographic changes (Liu et al., 2020). It is also found that H. rhamnoides subsp. sinensis Rousi from different geographic populations had a low degree of differentiation, and more genetic variation originated from different individuals within the populations (Wang et al., 2021a). The investigatedion of Chinese sea buckthorn at different altitudes by using ISSR molecular markers also indicate that H. rhamnoides subsp. sinensis Rousi at different altitudes had little spatial distance but showed obvious genetic diversity and genetic differentiation among populations (Chen et al., 2004). The genetic similarity of the populations in the 10 regions of Qinghai Province was relatively high. Thus, the appropriate inclusion of H. rhamnoides subsp. sinensis Rousi populations from other provenance should be considered in the future breeding of seedlings in Qinghai Province to enrich the genetic diversity.
5 Conclusion
In this study, 41,804 SSRs were identified from transcriptome data of H. rhamnoides subsp. sinensis Rousi, and the distribution and frequency of motifs were characterized and evaluated, fifteen SSR markers were developed for H. rhamnoides subsp. sinensis Rousi with abundant polymorphisms and showed a moderate level of genetic diversity. Finally, fifteen polymorphic SSRs were selected to investigate the genetic variation and structure of ten Chinese sea buckthorn populations in Qinghai province. The findings revealed a high level of genetic diversity in H. rhamnoides subsp. sinensis Rousi. AMOVA revealed that individual genetic variation is the main source of variation of H. rhamnoides subsp. sinensis Rousi. PCoA and Genetic structure analysis revealed two different genetic groups of natural H. rhamnoides subsp. sinensis Rousi in Qinghai Province. In addition, Our findings can provide a basis for the further investigation of H. rhamnoides subsp. sinensis Rousi by quantitative trait loci mapping, association analysis, and molecular-assisted breeding. Which can also further the exchange of Chinese sea buckthorn germplasms among different areas in China and the introduction of new Chinese sea buckthorn varieties from abroad.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/search/all/?term=PRJNA449450.
Author contributions
QL: Methodology, Writing–original draft. GY: Investigation, Resources, Writing–review and editing. YM: Data curation, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National Natural Science Foundation of China (31660071), Qinghai Provincial Science and Technology Department (2024-ZJ-941), and Qinghai Province’s “Thousands of High-end Innovative Talents Program” (2022).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Angelica, G., Lucia, S. G., Ilaria, M., Giuseppe, F., and Agata, G. (2023). Datasets for genetic diversity assessment in a collection of wild and cultivated pomegranates (Punica granatum L.) by microsatellite markers. Data Brief 49, 109346. doi:10.1016/J.DIB.2023.109346
Bal, L. M., Meda, V., Naik, S. N., and Satya, S. (2011). Sea buckthorn berries: a potential source of valuable nutrients for nutraceuticals and cosmeceuticals. Food Res. Int. 44, 0–1727. doi:10.1016/j.foodres.2011.03.002
Bhusan, P. B., Young, P. S., Won, K. S., Hwang, H. J., Wang, T. H., Park, E. B., et al. (2016). Transcriptome profile of the asian giant hornet (vespa mandarinia) using illumina HiSeq 4000 sequencing: de novo assembly, functional annotation, and discovery of SSR markers. Int. J. Genomics 2016, 1–15. doi:10.1155/2016/4169587
Bin, L., Cheng, L., Jun, R. T., Ya, X. X., Zhang, H. D., Bin, B., et al. (2022). Genetic diversity analysis of Camellia fascicularis H. T. Chang based on SSR markers. J. Appl. Res. Med. Aromatic Plants 31, 1–8. doi:10.1016/J.JARMAP.2022.100404
Burns, M. J., Edwards, K. J., Newbury, H. J., Ford-Lloyd, B. V., and Baggott, C. D. (2010). Development of simple sequence repeat (SSR) markers for the assessment of gene flow and genetic diversity in pigeonpea (Cajanus cajan). Mol. Ecol. Resour. 1, 283–285. doi:10.1046/j.1471-8278.2001.00109.x
Chakraborty, D., Sharma, N., Kour, S., Sodhi, S. S., Gupta, M. K., Lee, S. J., et al. (2022). Applications of omics technology for livestock selection and improvement. Front. Genet. 13, 774113. doi:10.3389/fgene.2022.774113
Chen, Y. M., Liu, G. B., Xu, B. C., and Chen, Y. Q. (2004). Research progress and prospect of function on soil and water conservation of seabuckthorn in China. Sci. Soil Water Conservation. doi:10.3969/j.issn.1672-3007.2004.02.017
Dawson, T. P. (2011). Beyond predictions: biodiversity conservation in a changing climate. Science 332, 664. doi:10.1126/science.1200303
Dong, S. T., Chen, Y., and Gu, Q. Y. (2020). Research progress on bioactive components and functions of seabuckthorn fruit China Brewing 39: 26–32. doi:10.11882/j.issn.0254-5071.2020.02.005
Duan, B., Mu, S., Guan, Y., Li, S., Yu, Y., Liu, W., et al. (2022a). Genetic diversity and population structure of the swimming crab (portunus trituberculatus) in China seas determined by genotyping-by-sequencing (GBS). Aquaculture 555, 738233. doi:10.1016/j.aquaculture.2022.738233
Duan, B., Mu, S., Guan, Y., Liu, W., Kang, T., Cheng, Y., et al. (2022). Development of microsatellite markers based on transcriptome sequencing and evaluation of genetic diversity in swimming crab (portunus trituberculatus). Front. Genet. 13, 932173. doi:10.3389/fgene.2022.932173
Gao, L. X. (2013). Development of EST-SSR primers for mulberry. South. J. Agric. 44, 1254–1257. doi:10.3969/j:issn.2095-1191.2013.8.1254
Ghendov-Mosanu, A., Cristea, E., Patras, A., Sturza, R., Padureanu, S., Deseatnicova, O., et al. (2022). Potential application of Hippophae rhamnoides in wheat bread production. Molecules 25, 1272. doi:10.3390/molecules25061272
Gong, S., Wang, Z., Xiao, S., Lin, A., and Xie, Y. (2016). Development and verification of SSR based on transcriptome of yellow drum. Nibea albiflora. J. Jimei Univ. Nat. Sci. 21, 241–246. doi:10.19715/j.jmuzr.2016.04.001
Gupta, S, and Sharma, D. H. (2020). Genetic diversity studies on 23 hybrid rice genotypes using microsatellite (SSR) markers. Int. J. Genet. 12.5, 731–734.
He, X. H., Si, J. H., Zhu, L., Zhou, D. M., Zhao, C. Y., Jia, B., et al. (2023). Modeling habitat suitability of Hippophae rhamnoides L. using MaxEnt under climate change in China: a case study of H. r. sinensis and H. r. turkestanica. Front. For. Glob. Change 5, 1095784. doi:10.3389/ffgc.2022.1095784
Hu, J. Z., Wen, X. F., and Wang, D. J. (2021). Investigation and suggestions on the construction development and utilization of seabuckthorn resources in Xinjiang. China Soil Water Conservation: 16–18. doi:10.3969/j.issn.1000-0941.2021.09.009
Jia, D. R., Abbott, R. J., Liu, T. L., Mao, K. S., Bartish, I. V., and Liu, J. Q. (2012). Out of the Qinghai-Tibet Plateau: evidence for the origin and dispersal of Eurasian temperate plants from a phylogeographic study of Hippophae rhamnoides (Elaeagnaceae). New Phytol. 194, 1123–1133. doi:10.1111/j.1469-8137.2012.04115.x
Kaur, T., Singh, G., and Kapoor, D. N. (2017). A review of pharmacognostic, phytochemical, and pharmacological data of various species of Hippophae (Sea buckthorn). Int. J. Green Pharm. 11: S62–S75. doi:10.7897/2230-8407.0811211
Li, H., Ruan, C. J., Wang, L., Li, J. B., Guo, H., and Tian, X. J. (2019). Analysis of genetic diversity and population structure of sea buckthorn germplasm based on RNA-Seq SSR markers. Mol. Plant Breed. 17, 1567–1574. doi:10.13271/j.mpb.017.001567
Li, H. M., Lan, D. M., and He, W. X. (2003). Analysis of morphological genetic parameters in different lines of Chinese and Russian sea buckthorn. Seabuckthorn 16, 8–13.
Li, R. J., Huang, Y. L., Yang, X. S., Su, M., Xiong, H. Y., Dai, Y., et al. (2023). Genetic diversity and relationship of shanlan upland rice were revealed based on 214 upland rice SSR markers. Plants Basel, Switz. 12, 2876–1120. doi:10.3390/PLANTS12152876
Liu, Q. Q. X. J., Li, Y. Q., Ma, M. J., Cheng, C. Z., Wang, P. G., et al. (2023). Diversity analysis of phenotypic traits in wild Hippophae rhamnoides subsp. sinensis Rousi resources in Qinghai. J. Plant Genet. Resour. 24, 1057–1064. doi:10.13430/j.cnki.jpgr.20230115001
Liu, R. H., Bai, J. L., Bao, H. J., Nong, L., Zhao, J. J., et al. (2020). Variation and correlation in functional traits of main woody plants in the <i>Cyclobalanopsis glauca </i>community in the karst hills of Guilin, southwest China. southwest China 44, 828–841. doi:10.17521/cjpe.2019.0146
Liu, X., Xie, X., Liu, H., Nie, H., Ma, H., Li, D., et al. (2022). Population genomic evidence for genetic divergence in the northwest pacific ark shell (scapharca broughtonii). Aquac. Rep. 24, 101100. doi:10.1016/j.aqrep.2022.101100
Ma, Y. H., Ye, G. S., Xiang, Q. S., Gao, Y., Yang, C. J., Wei, G. L., et al. (2014). Phylogenetic relationships of seabuckthorn based on ITS sequences. J. Appl. Ecol. 25 (10), 2985–2990. doi:10.13287/j.1001-9332.20140731.009
Mahfooz, S., Singh, S. P., Mishra, N., and Mishra, A. (2017). A comparison of microsatellites in phytopathogenic Aspergillus species in order to develop markers for the assessment of genetic diversity among its isolates. Front. Microbiol. 8, 1774. doi:10.3389/fmicb.2017.01774
Mei, D., Ma, X., Fu, F., and Cao, F. (2023). Research status and development prospects of sea buckthorn (Hippophae rhamnoides L.) resources in China. Forests 14, 2461. doi:10.3390/f14122461
Michel, T., Destandau, E., and Elfakir, C. (2011). Antimicrobial, antioxidant, and phytochemical investigations of sea buckthorn (Hippophaë rhamnoides L.) organs. Planta Medica 77. doi:10.1055/s-0031-1282703
Nybom, H. (2004). Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 13, 1143–1155. doi:10.1111/j.1365-294X.2004.02141.x
Peakall, E., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinforma. Oxf. Engl. 28, 2537–2539. doi:10.1093/bioinformatics/bts460
Pundir, S., Garg, P., Dviwedi, A., Ali, A., Kapoor, V. K., Kapoor, D., et al. (2021). Ethnomedicinal uses, phytochemistry and dermatological effects of Hippophae rhamnoides L.: a review. J. Ethnopharmacol. 266, 113434. doi:10.1016/j.jep.2020.113434
Soneja, D. K., Rakesh, S., Samarjit, S. N., Julia, D. E., and Sunitibala, D. H. (2023). Intergeneric hybridization of two endangered orchids, Vanda stangeana and phalaenopsis hygrochila and molecular confirmation of hybridity using SSR and Scot markers. South Afr. J. Bot. 161, 140–150. doi:10.1016/J.SAJB.2023.07.048
Sun, K., Wang, R. X., Chen, W., Hou, Q. Z., and Li, M. (2013). Genetic diversity of the Hippophae rhamnoides subsp. sinensis Rousi subpopulation in Ziwuling. J. Lanzhou Univ. 49, 75–79. doi:10.3321/j.issn:0455-2059.2004.03.017
Tian, C., Nan, P., Shi, M. S., Chen, J., and Zhong, Y. (2004). Molecular genetic variation in Chinese populations of three subspecies of Hippophae rhamnoides. Biochem. Genet. 42: 259–267. doi:10.1023/B:BIGI.0000034430.93055.ff
Tian, Z., Chen, A., Wu, Y., Chen, S., Zhang, Y., Cao, Y., et al. (2021). Bioinformatics analysis of microsatellite sites in the RNA-sequencing of <italic&gt;Meretrix meretrix&lt;/italic&gt; 43, 160–167. doi:10.3724/sp.j.1004-2490.2021.0204
Trineeva, O. V., Rudaya, M. A., and Slivkin, A. I. (2020). The study of the carotenoid composition of the fruits of sea buckthorn different varieties by thin layer chromatography. Chem. plant raw material, 223–228. doi:10.14258/jcprm.2020016345
Tulsani, N. J., Hamid, R., Jacob, F., Umretiya, N. G., Nandha, A. K., Tomar, R. S., et al. (2020). Transcriptome landscaping for gene mining and SSR marker development in coriander (coriandrum sativum L.). Genomics 112, 1545–1553. doi:10.1016/j.ygeno.2019.09.004
Tuomasjukka, S., Kallio, H., and Forssell, P. (2006). Effect of microencapsulation of dietary oil on postprandial lipemia. J. Food Sci. 71, S225–S230. doi:10.1111/j.1365-2621.2006.tb15645.x
Ui Haq, S. A., Mir, M. A., Lone, S. M., Banoo, A., Shafi, F., et al. (2021). Explicating genetic diversity based on its characterization and determination of antioxidant potential in sea buckthorn (Hippophae spp.). Mole. Biol. Rep. 49, 5229–5240. doi:10.1007/s11033-021-06619-z
Wang, H., Liu, H., Yang, M., Bao, L., and Ge, J.(2014). Phylogeographic study of Chinese seabuckthorn (Hippophae rhamnoides subsp. sinensis Rousi) reveals two distinct haplotype groups and multiple microrefugia on the Qinghai-Tibet Plateau. Ecol. Evol., 4.4370–4379. doi:10.1002/ece3.1295
Wang, L. Y., Li, S. S., Wang, T. Y., He, C. Y., Luo, H. M., Zhang, J. G., et al. (2021a). Genomic SSR and EST-SSR markers for phylogenetic and pedigree reconstructions—a comparison in sea buckthorn. Plant Breed. 140 (1), 167–183. doi:10.1111/pbr.12889
Wang, T. Y., Xu, Y., Wang, L. Y., Zhang, J., and Gand Zeng, Y. F. (2021b). Genetic differentiation and genetic diversity of Hippophae rhamnoides subsp. sinensis Rousi and Yunnan sea buckthorn. For. Sci. Res. 34, 13–21. doi:10.13275/j.cnki.lykxyj.2021.04.002
Wei, W. L., Qi, X. Q., Wang, L. H., Zhang, Y. X., Hua, W., Li, D. H., et al. (2011). Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC genomics 12 (1), 451. doi:10.1186/1471-2164-12-451
Wu, Q., Sun, K., Zhang, H., Cheng, W., Su, X., and Chen, X. L. (2007). Phenotypic diversity of natural populations of Hippophae rhamnoides subsp. sinensis Rousi in Shanxi Province. J. Northwest Normal Univ. Nat. Sci. Ed., 78–84. doi:10.3969/j.issn.1001-988X.2007.03.020
Xie, Q., Zhang, S. Y., Jiang, L., Ding, M. Y., Liu, L. L., Wu, R. J., et al. (2023). Analysis of SSR information of olive transcriptome and development and application of molecular markers. J. Hortic. 50 (11), 2350–2364. doi:10.16420/j.issn.0513-353x.2023-0355
Xue, S. (2021). Analysis of chloroplast phylogeny and development of molecular markers in three species of Lippia. Nanjing Agric. Univ. doi:10.27244/d.cnki.gnjnu.2019.001196
Yang, W., Zheng, J., Jia, B., Wei, H., Wang, G., and Yang, F. (2018). Isolation of novel microsatellite markers and their application for genetic diversity and parentage analyses in sika deer. Gene 643, 68–73. doi:10.1016/j.gene.2017.12.007
Yasar, K., Damla, G., and Ezgi, C. (2020). Biber (capsicum annuuml.) genotiplerinin SSR markörleri ile genetik karakterizasyonu. Ege Üniversitesi Ziraat Fakültesi Derg. 57, 185–191. doi:10.20289/zfdergi.614237
Ye, G. S., Ma, Y. H., Feng, Z. P., and Zhang, X. F. (2018). Transcriptomic analysis of drought stress responses of sea buckthorn (Hippophae rhamnoidessubsp. sinensis) by RNA-Seq. PLoSONE 13 (8), e0202213. doi:10.1371/journal.pone.0202213
Zhang, H., Sun, X. W., Ma, J. Q., Chen, X. l., et al. (2006). Genetic diversity of different populations of Hippophae rhamnoides subsp. sinensis Rousi on both sides of the Qilian Mountains. Northwest J. Bot. 26, 702–706. doi:10.3321/j.issn:1000-4025.2006.04.009
Zhao, C. F. (2007). Genetic variation analysis of natural populations of sea buckthorn (Hippophae rhamnoides subsp. sinensis) in China. Available at: .http://210.75.237.14/handle/351003/2369
Keywords: Hippophae rhamnoides subsp. sinensis Rousi, SSR, primer development, genetic diversity evaluation, population genetic structure
Citation: Liu Q, Ye G and Ma Y (2024) Development and application of microsatellite markers in Hippophae rhamnoides subsp. sinensis Rousi (Hippophae rhamnoides L.) based on transcriptome sequencing. Front. Genet. 15:1373028. doi: 10.3389/fgene.2024.1373028
Received: 19 January 2024; Accepted: 11 April 2024;
Published: 09 May 2024.
Edited by:
Fatemeh Maghuly, University of Natural Resources and Life Sciences Vienna, AustriaReviewed by:
Sandip Debnath, Visva-Bharati University, IndiaAnoop Anand Malik, Indian Council of Agricultural Research, India
Copyright © 2024 Liu, Ye and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuhua Ma, qhxnmyh@163.com