 Ke Wu
Ke Wu Yuying Zhu2†
Yuying Zhu2†- 1Laboratory of Prenatal Diagnosis Center, Quzhou Maternal and Child Health Care Hospital, Quzhou, Zhejiang, China
- 2Prenatal Diagnosis Center, Quzhou Maternal and Child Health Care Hospital, Quzhou, Zhejiang, China
- 3Obstetrics Department, Quzhou Maternal and Child Health Care Hospital, Quzhou, Zhejiang, China
Silver–Russell syndrome (SRS) is a clinically and genetically heterogeneous disorder. A retrospective analysis predicted that the live birth prevalence of SRS in Estonia is 1:15,886 [Yakoreva et al., Eur J Hum Genet, 2019, 27(11), 1649–1658]. The most common causative genetic mechanism in the proband is loss of paternal methylation in the imprinted control region 1 (ICR1) at 11p15.5 chromosome. A few studies suggested that inherited or de novo loss-of-function alterations of the PLAG1 gene, including the whole-gene deletion and intragenic pathogenic variants, could cause a rare type of SRS. To date, less than 20 unrelated PLAG1-related SRS cases have been reported, and the clinical information about these cases is limited. We report the first prenatal case of SRS with 8q12 deletion (including the PLAG1 gene). The fetus presented with intrauterine growth retardation, small for gestational age, relative macrocephaly at birth, and a protruding forehead. Unlike classical SRS cases, the fetus had micrognathia and did not show body asymmetry. We hope that the literature review in this study provides new insights into genotype–phenotype relationships of PLAG1-related SRS.
Introduction
SRS is a rare and well-recognized condition presenting with intrauterine growth retardation (IUGR), small for gestational age (SGA), postnatal growth failure, relative macrocephaly at birth, protruding forehead, body asymmetry, and feeding difficulties and/or low body mass index (BMI). The first international consensus diagnostic clinical criteria and management for SRS were published in 2016 (Wakeling et al., 2017). SRS is a genetically heterogeneous disorder, caused by paternal hypomethylation at chromosome 11p15.5 (SRS1, MIM#180860), maternal uniparental disomy for chromosome 7 (SRS2, MIM#618905), paternally inherited heterozygous IGF2 variants (SRS3, MIM#616489), heterozygous PLAG1 variants (SRS4, MIM#618907), and heterozygous HMGA2 variants (SRS5, MIM#618908). PLAG1 gene-related SRS4 is quite rarely reported. Since Schinzel et al. (1994) first reported the 8q12 microdeletion-related SRS-like conditions in a 10-year-old girl and suggested certain gene variants or deletions in 8q12 may cause the clinical presentation of SRS. There are only nine documents related to SRS4 for now, including one document related to the prenatal diagnosis of SRS4. In this study, we reported a fetus with a de novo PLAG1 deletion affected with SRS4. We conducted a literature review to summarize previously reported prenatal/postnatal phenotypes and PLAG1 genotypes.
Materials and methods
Clinical features
At 23 weeks of gestation, a fetal ultrasound scan showed biparietal diameter (BPD = 55 mm) on the 13th centile, abdominal circumference (AC = 182 mm) on the 20th centile, head circumference (HC = 219 mm) on the 68th centile, and femur length (FL = 36 mm) below the 1st centile. Due to abnormal fetal biometry and oligohydramnios, the pregnant woman was referred to the prenatal diagnosis center. The couple did not have a consanguineous relationship, and their family history was not notable. Amniocentesis was performed at 25 weeks of gestation. Trio-based whole-exome sequencing (trio-WES) was recommended. The study was approved by the Medical Ethics Committee of Quzhou Maternal and Child Health Care Hospital (approval KY-2023-11), and written informed consent was obtained from the pregnant woman.
Methods
Whole-exome sequencing (WES)
Trio-WES was performed on the genomic DNA (gDNA) of the amniotic fluid sample and peripheral blood from the parents. The xGen™ Exome Research Panel v2 (designed by Integrated DNA Technologies) was used for WES. Quality control (QC) of the DNA library was performed using an Agilent 2100 Bioanalyzer system. DNA nanoball (DNB) preps of clinical samples were sequenced on an ultra-high-throughput DNBSEQ-T7 platform (MGI, Shenzhen, China) with the 150-nt paired-end strategy, following the manufacturer’s protocol. The trimmed reads were then mapped to the UCSC GRCh38 reference genome using Burrows-–Wheeler aligner (BWA) software. The alignment process was refined by local realignment and base-quality-score recalibration steps by means of the Genome Analysis Toolkit (GATK v4.2 software). CNVs were analyzed using CANoe, Convex, XHMM, and ExomeDepth software applications.
Copy-number variant (CNV) validation
All CNVs identified by trio-WES were verified by real-time quantitative PCR (qPCR), using a novel Duplex TaqMan CNV assay (Applied Biosystems, TaqMan CN early access program; TaqMan probe sequences are available on request). The assays used 20 ng of genomic DNA in a 20 μL reaction. The cycling conditions were as follows: 95°C for 10 min for initial denaturation and enzyme activation, followed by 40 cycles each of 95°C for 15 s and 60°C for 1 min. Relative quantification (RQ) was performed using CopyCaller Software (Applied Biosystems, United States), following the comparative ΔΔCT method.
Results
Genetic analysis and confirmation
Trio-WES identified a de novo deletion of 8q11.21q12.1: Seq[hg38]del(8) (q11.21q12.1) chr8:g.50402844_58939544del (Figure 1) including 32 protein-coding genes (RB1CC1, RP1, PLAG1, etc.). All detected CNVs were compared with known CNVs in the scientific literature and with those in the following publicly available databases: Database of Genomic Variants (DGV), DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources (DECIPHER), and Online Mendelian Inheritance in Man (OMIM). According to the joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) for reporting constitutional copy-number variants (Riggs et al., 2020), this CNV was a variant of uncertain significance (VUS) (3B: the number of protein-coding RefSeq genes wholly or partially included in the CNV region is 32; the score was +0.45). The CNV validation of the qPCR assay confirmed the result of trio-WES (Figure 2).
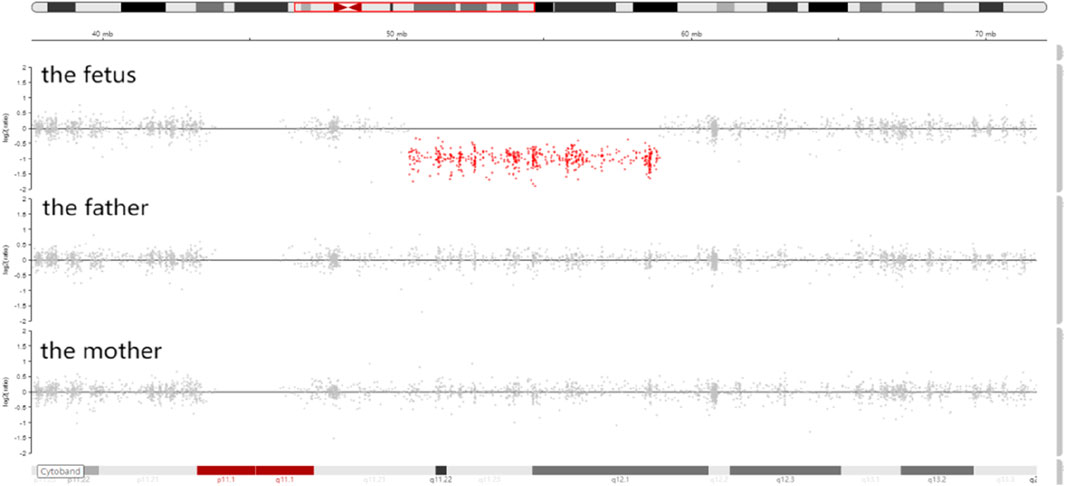
Figure 1. Integrative genomics viewer (IGV) showed that the sequencing coverage of 8q11.21q12.1 in the fetus decreased significantly (the red dots in 8q11.21q12.1 were below the zero line).
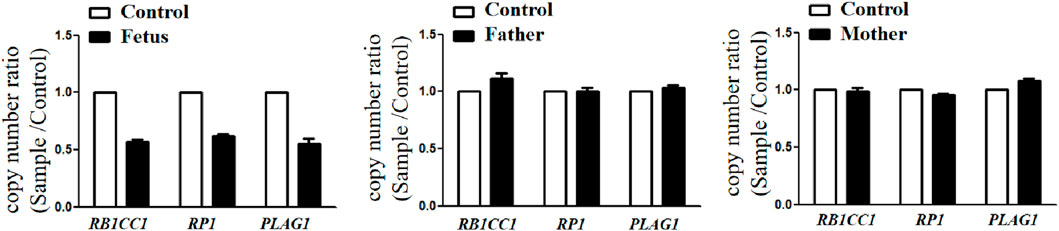
Figure 2. CNVs validation by qPCR showed that the copy number ratio (RB1CC1, RP1, and PLAG1 located in the 8q12 deletion) of the fetus was equivalent to the half that of the parents.
Pregnancy outcome
A follow-up fetal ultrasound scan at 29 weeks of gestation showed that fetal biometric parameters including BPD, AC, and FL, were all below the 1st centile except for HC on the 5th centile. The pregnant woman was offered a termination for medical reasons because the genetic tests and fetal ultrasound scan showed that the fetus was not developing as expected. Written informed consent for the fetal computed tomography (CT) scan (Figure 3) and magnetic resonance imaging (MRI) was obtained from the pregnant woman. The CT scan showed that the fetus presented with relative macrocephaly and micrognathia; the fetal trunk and extremities were symmetrical. The MRI of the fetal brain was abnormal. The fetus had a distinctive facial dysmorphism (Figure 4).
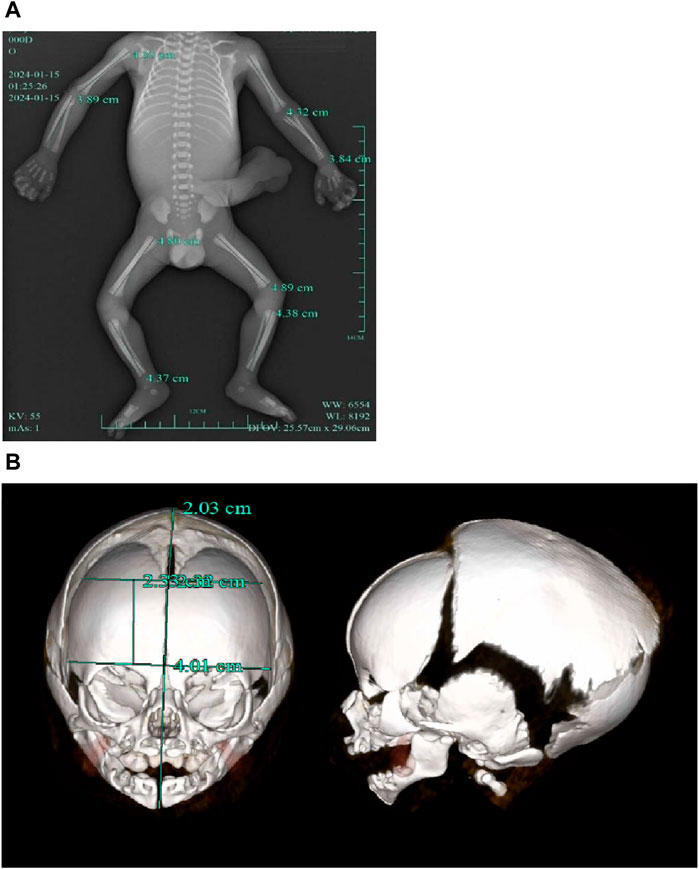
Figure 3. (A) CT scan of trunk and extremities did not show body asymmetry. (B) The head CT scan showed relative macrocephaly and micrognathia.
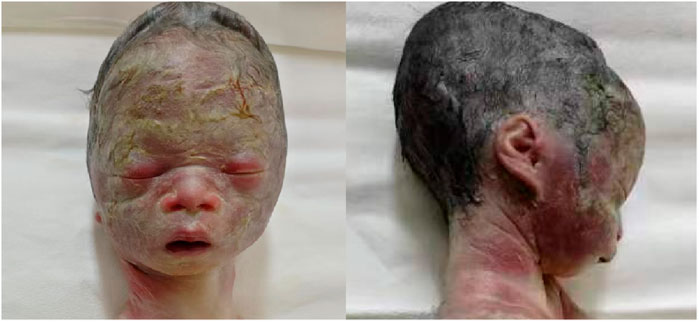
Figure 4. Fetal distinctive facial dysmorphism: prominent forehead, triangular face, orbital hypertelorism, micrognathia, long philtrum, and flat nasal bridge.
Literature review
We searched the PubMed database, the Human Gene Variant Database (HGMD), and Online Mendelian Inheritance in Man (OMIM) using “Silver–Russell syndrome” and “PLAG1” as keywords. The search time was from the establishment of the databases to 31 January 2024. Previous studies with PLAG1 aberrations and clinical manifestations were reviewed. Ten documents were retrieved. Fourteen postnatal cases with the PLAG1 gene deletion or intragenic variants and one prenatal case with a heterozygous PLAG1 gene variant have been reported (Table 1).
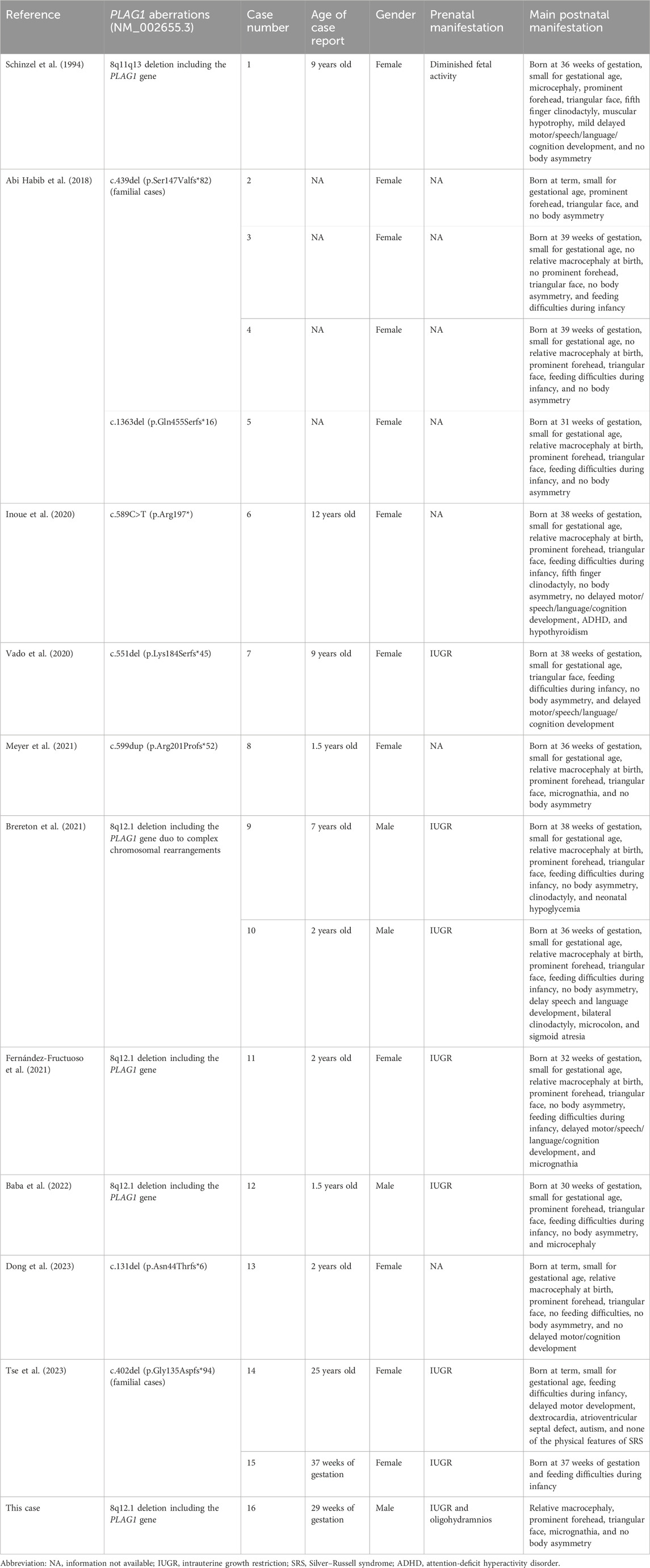
Table 1. Prenatal and postnatal clinical manifestations with PLAG1 gene aberrations.
Discussion
The PLAG1 gene, also known as “pleiomorphic adenoma gene 1,” is located on chromosome 8q21. The transcript of PLAG1 (NM_002655.3) has 5 exons, with the transcript length of 7,311 base pairs and translation length of 500 amino acids. It encodes a zinc finger protein, which is broadly expressed during fetal development (Karim et al., 2011), and plays a critical role in the oncogenic HMGA2–PLAG1–IGF2 pathway (Sélénou et al., 2022). PLAG1 acts as a transcription factor whose activation results in the upregulation of target genes (such as IGF2), leading to cell proliferation (Hensen et al., 2002). HMGA2 positively regulates IGF2 expression in a PLAG1-independent manner (Abi Habib et al., 2018). IGF2 acts as fetal growth hormone and plays a key role in possessing growth-promoting activity (Cedano Prieto et al., 2017). The loss-of-function (LoF) variants in any of these three genes in this pathway can cause a reduction in IGF2 expression and lead to SRS (Hübner et al., 2020).
To date, there are only 10 documents reporting PLAG1 gene aberrations associated with SRS4 [14 postnatal cases (case 1–14) and 1 prenatal case (case 15)]. The ratio of male to female patients was 1:4. Differing from classical SRS, all these 16 cases (including our case) did not present with body asymmetry. The most common manifestations (16/16, 100%) were small for gestational age and growth restriction. Other more common manifestations described in documents were triangular face (14/16, 87.5%), prominent forehead (12/16, 68.8%), relative macrocephaly at birth (8/16, 50.0%), and feeding difficulties during infancy (12/14, 85.7%). Some cases showed clinodactyly of the fifth finger (4/16, 25.0%), delayed development of motor/speech/language/cognition (4/14, 28.6%), and micrognathia (3/16, 18.8%). The findings of the three cases (case 1, 12, and 14) least consistent with SRS were their microcephalies and none of the physical features of SRS4. Some uncommon manifestations described in documents were heart defects, microcolon, sigmoid atresia, muscular hypotrophy, hypoglycemia, and autism. The most frequent prenatal manifestation (8/16, 50%) of SRS4 was IUGR. Other nonspecific manifestations were diminished fetal activity and oligohydramnios (our case). The deleted interval wholly or partially involves 32 protein-coding RefSeq genes. Among them, autosomal dominant genes were PLAG1, RP1 (associated with retinitis pigmentosa), LYN (associated with autoinflammatory disease), and SOX17 (associated with vesicoureteral reflux). However, the above mentioned three gene (RP1, LYN, and SOX17)-related phenotypes have not been reported in previous cases with the 8q12 deletion. We speculate that the PLAG1 gene may be the key haploinsufficient gene on the 8q12 deletion. Three types of PLAG1 gene variants have been reported: 8q21 deletion (including PLAG1) (6 cases), frameshift mutation (9 cases), and nonsense mutation (one case). The most common manifestations in patients with the PLAG1 deletion (6/6, 100%) were growth restriction, prominent forehead, and triangular face, which were not 100% frequency of occurrence in patients with other PLAG1 variants. Other more common manifestations described in patients with the PLAG1 deletion were relative macrocephaly (4/6, 66.7%), feeding difficulties during infancy (4/6, 66.7%), and clinodactyly (3/6, 50%). Differing from other types of PLAG1 variants, microcephaly only happened to patients with the PLAG1 deletion. Only one case (1/10, 10%, case 6) with other types of PLAG1 variants had fifth finger clinodactyly. The frequency of feeding difficulties during infancy in patients with other types of PLAG1 variants was 70% (7/10), which was no different from patients with the PLAG1 deletion.
One common pathogenic mechanism of these variations is haploinsufficiency. The probability of the LoF intolerance (pLI) score of the PLAG1 gene is 1.0, suggesting extremely LoF intolerance (pLI≥0.9). Frameshift and nonsense mutations can cause premature termination codons (PTCs), and then, the aberrant mRNA will be degraded by a nonsense-mediated decay (NMD) pathway, resulting in a complete loss-of-function of the truncated protein (Miller and Pearce, 2014). Although the Clinical Genome Resource (ClinGen) has not yet published curations for the PLAG1 gene, functional analysis in vitro has shown that the whole PLAG1 gene deletion could result in a reduction in IGF2 (Abi Habib et al., 2018). Recent studies found that deletion mutations within the PLAG1 gene in goats or cattle were associated with the regulation of growth traits (Wei et al., 2021), (Xu et al., 2018). The manifestations of PLAG1-knockout mice suggested that PLAG1 genes played an indispensable role in postnatal growth and fertility (Juma et al., 2016).
To sum up, we suggest that some clinical manifestations of PLAG1-related SRS4 may vary with types of PLAG1 gene aberrations. Microcephaly and clinodactyly tend to occur in patients with the PLAG1 deletion. The frequency of growth restriction, prominent forehead, triangular face, and relative macrocephaly is lower in patients with other types of PLAG1 aberrations. Most (15/16) PLAG1-related SRS4 patients presented with the classical clinical manifestations of SRS (apart from body asymmetry). Although we consider PLAG1 to be a haploinsufficiency gene, further evidence is still requisite.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethical Committee of Quzhou Maternity and Child Healthcare Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
KW: writing–original draft and writing–review and editing. YZ: data curation and writing–review and editing. QZ: writing–original draft and Writing–review and editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by grants from projects of Science and Technology of Quzhou (2023ZD084, 2023K183, and 2023K186).
Acknowledgments
The authors are grateful to the patients and their families for participation in this study and the help of all the physicians in the course of the medical treatment. They wish to thank the staffs of Chigene (Beijing) Translational Medical Research Center Co., Ltd. for assisting with whole-exome sequencing and data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abi Habib, W., Brioude, F., Edouard, T., Bennett, J. T., Lienhardt-Roussie, A., Tixier, F., et al. (2018). Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 20 (2), 250–258. doi:10.1038/gim.2017.105
Baba, N., Lengyel, A., Pinti, E., Yapici, E., Schreyer, I., Liehr, T., et al. (2022). Microdeletions in 1q21 and 8q12.1 depict two additional molecular subgroups of Silver-Russell syndrome like phenotypes. Mol. Cytogenet 15 (1), 19. Published 2022 May 13. doi:10.1186/s13039-022-00596-z
Brereton, R. E., Nickerson, S. L., Woodward, K. J., Edwards, T., Sivamoorthy, S., Ramos Vasques Walters, F., et al. (2021). Further heterogeneity in Silver-Russell syndrome: PLAG1 deletion in association with a complex chromosomal rearrangement. Am. J. Med. Genet. A 185 (10), 3136–3145. doi:10.1002/ajmg.a.62391
Cedano Prieto, D. M., Cheng, Y., Chang, C. C., Yu, J., Takada, Y. K., and Takada, Y. (2017). Direct integrin binding to insulin-like growth factor-2 through the C-domain is required for insulin-like growth factor receptor type 1 (IGF1R) signaling. PLoS One 12 (9), e0184285. doi:10.1371/journal.pone.0184285
Dong, P., Zhang, N., Zhang, Y., Liu, C. X., and Li, C. L. (2023). Clinical characterization of PLAG1-related Silver-Russell syndrome:A clinical report. Eur. J. Med. Genet. 66 (10), 104837. doi:10.1016/j.ejmg.2023.104837
Fernández-Fructuoso, J. R., De la Torre-Sandoval, C., Harbison, M. D., Chantot-Bastaraud, S., Temple, K., Lloreda-Garcia, J. M., et al. (2021). Silver Russell syndrome in a preterm girl with 8q12.1 deletion encompassing PLAG1. Clin. Dysmorphol. 30 (4), 194–196. doi:10.1097/MCD.0000000000000375
Hensen, K., Van Valckenborgh, I. C., Kas, K., Van de Ven, W. J., and Voz, M. L. (2002). The tumorigenic diversity of the three PLAG family members is associated with different DNA binding capacities. Cancer Res. 62 (5), 1510–1517.
Hübner, C. T., Meyer, R., Kenawy, A., Ambrozaityte, L., Matuleviciene, A., Kraft, F., et al. (2020). HMGA2 variants in silver-russell syndrome: homozygous and heterozygous occurrence. J. Clin. Endocrinol. Metab. 105 (7), dgaa273. doi:10.1210/clinem/dgaa273
Inoue, T., Nakamura, A., Iwahashi-Odano, M., Tanase-Nakao, K., Matsubara, K., Nishioka, J., et al. (2020). Contribution of gene mutations to Silver-Russell syndrome phenotype: multigene sequencing analysis in 92 etiology-unknown patients. Clin. Epigenetics 12 (1), 86. Published 2020 Jun 16. doi:10.1186/s13148-020-00865-x
Juma, A. R., Damdimopoulou, P. E., Grommen, S. V., Van de Ven, W. J., and De Groef, B. (2016). Emerging role of PLAG1 as a regulator of growth and reproduction. J. Endocrinol. 228 (2), R45–R56. doi:10.1530/JOE-15-0449
Karim, L., Takeda, H., Lin, L., Druet, T., Arias, J. A. C., Baurain, D., et al. (2011). Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat. Genet. 43 (5), 405–413. doi:10.1038/ng.814
Meyer, R., Begemann, M., Hübner, C. T., Dey, D., Kuechler, A., Elgizouli, M., et al. (2021). One test for all: whole exome sequencing significantly improves the diagnostic yield in growth retarded patients referred for molecular testing for Silver-Russell syndrome. Orphanet J. Rare Dis. 16 (1), 42. Published 2021 Jan 22. doi:10.1186/s13023-021-01683-x
Miller, J. N., and Pearce, D. A. (2014). Nonsense-mediated decay in genetic disease: friend or foe? Mutat. Res. Rev. Mutat. Res. 762, 52–64. Epub 2014 May 28. PMID: 25485595; PMCID: PMC4260155. doi:10.1016/j.mrrev.2014.05.001
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Schinzel, A. A., Robinson, W. P., Binkert, F., and Fanconi, A. (1994). An interstitial deletion of proximal 8q (q11-q13) in a girl with Silver-Russell syndrome-like features. Clin. Dysmorphol. 3 (1), 63–69. doi:10.1097/00019605-199401000-00009
Sélénou, C., Brioude, F., Giabicani, E., Sobrier, M. L., and Netchine, I. (2022). IGF2: development, genetic and epigenetic abnormalities. Cells 11 (12), 1886. doi:10.3390/cells11121886
Tse, W. T., Bass, C., Gurney, L., and Kinning, E. (2023). Maternally inherited autosomal dominant PLAG-1 related Silver Russell syndrome in a fetus with intra-uterine growth restriction. Prenat. Diagn 43 (6), 724–726. doi:10.1002/pd.6364
Vado, Y., Pereda, A., Llano-Rivas, I., Gorria-Redondo, N., Díez, I., and Perez de Nanclares, G. (2020). Novel variant in PLAG1 in a familial case with silver-russell syndrome suspicion. Genes (Basel) 11 (12), 1461. Published 2020 Dec 5. doi:10.3390/genes11121461
Wakeling, E. L., Brioude, F., Lokulo-Sodipe, O., O'Connell, S. M., Salem, J., Bliek, J., et al. (2017). Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat. Rev. Endocrinol. 13 (2), 105–124. doi:10.1038/nrendo.2016.138
Wei, Z., Wang, K., Wu, H., Pan, C., Chen, H., et al. (2021). Detection of 15-bp deletion mutation within PLAG1 gene and its effects on growth traits in goats. Anim. (Basel) 11 (7), 2064. Published 2021 Jul 10. doi:10.3390/ani11072064
Xu, W., He, H., Zheng, L., Xu, J. W., Lei, C. Z., Zhang, G. M., et al. (2018). Detection of 19-bp deletion within PLAG1 gene and its effect on growth traits in cattle. Gene 675, 144–149. doi:10.1016/j.gene.2018.06.041
Keywords: Silver–Russell syndrome, obstetrics department, PLAG1, loss-of-function, chromosome 8q12
Citation: Wu K, Zhu Y and Zhu Q (2024) Prenatal diagnosis of Silver–Russell syndrome with 8q12 deletion including the PLAG1 gene: a case report and review. Front. Genet. 15:1387649. doi: 10.3389/fgene.2024.1387649
Received: 18 February 2024; Accepted: 30 April 2024;
Published: 17 May 2024.
Edited by:
Yongchu Pan, Nanjing Medical University, ChinaReviewed by:
Thomas Eggermann, RWTH Aachen University, GermanyLudmila Matyakhina, GeneDx, United States
Copyright © 2024 Wu, Zhu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiumin Zhu, 15157079960@163.com
†These authors have contributed equally to this work