 Jessica Alber
Jessica Alber Kelly McGarry3
Kelly McGarry3 Richard B. Noto
Richard B. Noto Peter J. Snyder
Peter J. Snyder- 1Department of Psychiatry and Human Behavior, Warren Alpert Medical School of Brown University & Rhode Island Hospital, Providence, RI, United States
- 2Department of Neurology, Warren Alpert Medical School of Brown University, Providence, RI, United States
- 3Division of General Internal Medicine, Department of Medicine, Warren Alpert Medical School of Brown University & Rhode Island Hospital, Providence, RI, United States
- 4Department of Radiology, Warren Alpert Medical School of Brown University & Rhode Island Hospital, Providence, RI, United States
- 5Interdisciplinary Neuroscience Program, University of Rhode Island, Kingston, RI, United States
- 6Ryan Institute for Neurosciences, University of Rhode Island, Kingston, RI, United States
Background: Recent genome-wide association screening (GWAS) studies have linked Alzheimer's disease (AD) neuropathology to gene networks that regulate immune function. Kan et al. recently reported that Arg1 (an anti-inflammatory gene that codes for arginase-1) is expressed in parts of the brain associated with amyloidosis prior to the onset of neuronal loss, suggesting that chronic brain arginine deprivation promotes AD-related neuropathology. They blocked arginine catabolism in their mouse AD model by administration of eflornithine (DFMO) to juvenile animals, effectively blocking the expression of AD-related amyloid pathology as the mice aged. We report results from a single-case study in which DFMO was administered, for the first time, in an attempt to slow progression of AD in a single woman with multi-domain, amnestic MCI who was unable to tolerate an acetylcholinesterase inhibitor.
Methods: Patient C.S. is a 74-year old female with a 5-year history of cognitive decline who was placed on DFMO (500 mg b.i.d.) for 12 months, with amyloid PET scans (baseline and 12-months), APOE genotyping and neuropsychological exams at baseline, 3, 9, and 12 months.
Results: C.S. suffered continued cognitive decline over 12 months, including progressive worsening of orientation, social functions and ability to engage in IADL's. She also showed progressive decline on measures of episodic memory and executive function. Florbetapir PET imaging yielded elevated total neocortical SUVr scores at both baseline (SUVr = 1.55) and at 12 months (SUVr = 1.69).
Conclusions: We report a first attempt at using DFMO to slow AD progression. This 12-month single-case trial did not halt continued amyloidosis nor cognitive decline. Although this trial was predicated on data reported by Kan et al. (2015) showing that DFMO administered to juvenile AD-prone mice led to diminished amyloid aggregation, this attempt to treat an older mild AD patient may not be a fair test of Kan et al.'s model and results. A future trial might seek to block amyloidosis in young adults who are autosomal gene carriers for early onset AD, or perhaps in adults who are very clearly in the pre-clinical disease stage.
Trial Registration: This trial was registered as a Compassionate Use IND #128888 with the United States Food and Drug Administration (FDA).
Background
Alzheimer's disease (AD) is a neurodegenerative condition associated with the accumulation of parenchymal β-amyloid (Aβ) and hyperphosphorylated tau tangles in the neurons. The pathogenesis of AD is a complicated process that occurs as a result of a combination of downstream interactions between DNA-sequence variants and non-genetic (biological risk factors such as vascular health and psychosocial risks) contributors that act through molecular networks to affect disease risk (Schadt, 2009). The exact mechanism of AD pathogenesis remains unknown despite decades of careful research. To date, there are no preventative or disease-modifying AD therapies, despite the projected more than doubling of the incidence of AD by the year 2050 (Alzheimer's Association Strategic Plan, 2017). Currently, one dominant etiologic model of AD is the referred to as the “amyloid hypothesis” (Hardy and Allsop, 1991; Selkoe, 1991; Hardy and Higgins, 1992; Jack et al., 2013), which states that parenchymal Aβ builds up decades prior to the onset of clinical symptoms, resulting in a silent period termed preclinical AD (Sperling et al., 2011), followed by neurodegeneration and the accumulation of hyperphosphorylated tau. Therapeutic interventions now focus on this preclinical AD period, with the aim of eliminating the accumulation of parenchymal Aβ early in the pathological process of the disease.
The apolipoprotein E (APOE) gene is the AD susceptibility gene that has been most extensively researched, and accounts for an estimated 30% of the genetic variability in AD (Corder et al., 1993; Saunders et al., 1993). Despite recent developments indicating other genetic contributions to AD such as APP and TREM2 (Jonsson et al., 2012, 2013; Guerreiro et al., 2013), a large proportion of the genetic variance in AD remains unaccounted for. In addition to APOE, genome wide association studies (GWAS) have linked AD pathology to gene networks that regulate immune function and microglial expression (Kamboh et al., 2012; Zhang et al., 2013).
Most animal models of AD involve transgenic expression of mutated amyloid precursor protein (APP), which leads to parenchymal Aβ deposition but does not usually invoke tau pathology and associated neuronal cell death. Consequently, these models have been criticized as incomplete models of AD pathology (Irizarry et al., 1997; Radde et al., 2008). In a recently developed CVN-AD mouse model, immune-mediated nitric oxide (iNOS) was lowered to mimic human levels, resulting in a model that demonstrates the complete pathological course of AD, including parenchymal amyloidosis, gradual spread of hyperphosphorlyated tau, episodic memory impairment, and significant hippocampal neuronal degeneration (Colton et al., 2008, 2014; Wilcock et al., 2014). These mice are mNos2 -deficient and transgenic for the Swedish K670N/M671L vasculotropic Dutch/Iowa E693Q/D694N mutant (APPSwDI) APP (Kan et al., 2015), which prevents the expression of inducible nitric oxide synthase protein, thus lowering the level of nitric oxide production (for a detailed description of this model, see Colton et al., 2008, 2014; Wilcock et al., 2014).
Using this model, Kan et al first found that these CVN-AD mice followed the pathological progression of human AD (Kan et al., 2015). In an additional study, they found that Arg1 (an anti-inflammatory gene that codes for arginase1, an arginine utilization enzyme) was expressed in the parts of the animal brain with high levels of amyloidosis prior to the onset of neuronal degeneration and cell death, an indication that chronic brain arginine deprivation could be the cause of neuronal cell death in AD, and the subsequent cascade of AD pathology. Based on this information, they administered eflornithine (DFMO; an arginase inhibitor) to juvenile animals, which effectively blocked the expression of Aβ and other AD related pathology, immunosuppression, and spatial memory impairments. Moreover, the DFMO mice had higher arginine expression in the hippocampus and subiculum, areas that are associated with early neuronal loss in AD pathology. In other words, inhibition of abnormal arginine utilization and resultant arginine expression resulted in marked improvement in pathology and cognitive function when administered to juvenile CVN-AD mice (Kan et al., 2015).
In response to a special request by a family member of a patient, who happened to be a pediatric oncologist with clinical experience in the use of DFMO for the treatment of pediatric neuroblastomas and who also was aware of the Kan et al. (2015) publication, one of us (K.M.) applied for—and received approval for—a compassionate use Investigation New Drug (IND) application to administer DFMO to a single woman with multi-domain, amnestic mild cognitive impairment (MCI) who was unable to tolerate an acetylcholinesterase inhibitor. DFMO is an Food and Drug Administration (FDA) approved treatment of Trypanosoma brucei gambiense encephalitis (“African sleeping sickness”), has been used in clinical trials for neuroblastoma in children, and has a favorable safety and tolerability profile. It is unknown whether or not DFMO would slow or reverse amyloid pathology, or improve cognitive outcomes in a human subject with MCI.
Methods
Case Presentation
The participant (C.S.) is a 74 year old, left-handed female with 16 years of education. Her past medical history is notable for hypertension, gastric reflux, osteoarthritis, headaches, benign essential tremor, glaucoma, anxiety, and depression. Concurrent medications upon initiation of the trial were omeprazole, sertraline, trazadone, Miralax, calcium, fish oil, and baby aspirin. The researchers had access to the participants' medical records and previous neuropsychological evaluations in 2011 and 2013, which were negative for neurodegenerative disease diagnosis but noted mild cognitive changes due to anxiety and depression. Family history was positive for Alzheimer's disease in both parents, with onset in her father in the early 80s and onset in her mother in mid-80's.
The participant presented with a 5-year gradually progressive history of cognitive decline and in 2015 she received a diagnosis of multi-domain amnestic MCI due to possible Alzheimer's disease. Symptoms included disorientation to time, repetitive speech, mild word finding difficulty, difficulty following conversations, confusability, and difficulties with attention and concentration. The patient had mild, stable symptoms of depression and anxiety and had very minor dilapidation of instrumental activities of daily living (IADL's) such as driving and financial management. The patient had also become more socially withdrawn as a result of conversational difficulties and lost ~25 pounds over the 2 years preceding presentation. After diagnosis, the participant began a regimen of donepezil 5 mg, but this was discontinued due to GI problems and excessive weight loss. The patient's inability to tolerate a cholinesterase inhibitor resulted in application for a compassionate use IND for eflornithine (DFMO).
Procedure
Screening
Baseline screening assessments included medical history, physical exam, vital signs (blood pressure, pulse, temperature), and complete blood count (CBC) with differential and comprehensive metabolic panel (CMP). Prior to baseline dosing, the participant completed an amyloid PET scan and neuropsychological evaluation.
Treatment
The participant was administered eflornithine (DFMO) 500 mg (2 tabs) PO BID (morning and evening), concomitant with safe therapeutic dosing in the United States for colon cancer prevention and neuroblastoma. DFMO was prescribed by KM (primary care physician) as oral capsules, taken morning and evening with water. Dosing and medication instructions were explained verbally to the participant and her caregiver, and written instructions were also provided regarding dosing and timing of dose.
Safety Monitoring
Safety monitoring involved a CBC with differential and a CMP every 2 weeks for four results, then once a month for four results, then once every 2 months for four results. Additionally, repeat medical history, physical exam, vital signs, and blood tests were conducted once per month for 6 months, and then once every 2 months for 6 months.
Outcome Measures
Neuropsychological Evaluation
Comprehensive neuropsychological evaluation was conducted at baseline, 3, 9, and 12 months. Neuropsychological evaluation included the domains and tests described in Table 1. A clinical interview was conducted with the participant and her caregiver prior to each neuropsychological session to assess changes in medications, medical history, changes in health, functional changes, and current symptoms of depression/anxiety. Clinical interview and neuropsychological tests were administered by a post-doctoral research fellow with extensive training in neuropsychological assessment and reviewed by a licensed clinical neuropsychologist.

Table 1. Neuropsychological outcome measures used in a compassionate use IND trial of DFMO in a single case.
Primary endpoints included reported decline in instrumental activities of daily living as assessed via clinical interview, change in score on the Mini Mental Status Examination (MMSE; Folstein et al., 1975), the ADAS-Cog (Rosen et al., 1993), delayed recall on Logical Memory (Wechsler, 1997), and change in amyloid PET burden as measured by Florbetapir PET scan. Secondary endpoints included change in score on the GMLT (Snyder et al., 2005; Fredrickson et al., 2008), verbal fluency (phonemic and category fluency; Borkowski et al., 1967; Tombaugh et al., 1999), OCL (Maruff et al., 2013), OBK (Maruff et al., 2013), and ISLT (Lim et al., 2009).
Amyloid PET Imaging
Amyloid positron emission tomography (PET) imaging was conducted at baseline and after 12 months. At each scan, a 370 MBq (10 mCi ± 10%) bolus injection of 18F-florbetapir was administered intravenously. Approximately 50 min post-injection, a 20-min PET scan was performed with head computerized tomography (CT) scan for attenuation and correction purposes. Images were obtained using a 128 x 128 matrix reconstructed using iterative or row action maximization likelihood algorithms. PET standardized uptake value (SUV) data were summed and normalized to the whole cerebellum SUV, resulting in a region-to-cerbellum ratio termed SUV ratio (SUVr). SUVr calculation was performed using the MIMneuro software, with a normative database of 74 individuals (48 males, 26 females) between the ages of 18 and 50 years who met the eligibility criteria for inclusion; that is, individuals needed to be in good general health, score within normal range for standard clinical neurologic and cognitive testing, and have a negative amyloid scan on visual assessment (Clark et al., 2011). Amyloid positivity was confirmed by consensus of two board-certified radiologists who were also board certified in nuclear medicine.
Results
Results from primary and secondary outcome measures are summarized in Table 2. For cognitive reasons, the participant was unable to complete the delayed recall portion of the GMLT in the baseline visit, exam 2 (3 months), and exam 3 (9 months). Additionally, she was unable to complete the GMLT reverse recall at the baseline visit and exam 2 (3 months).
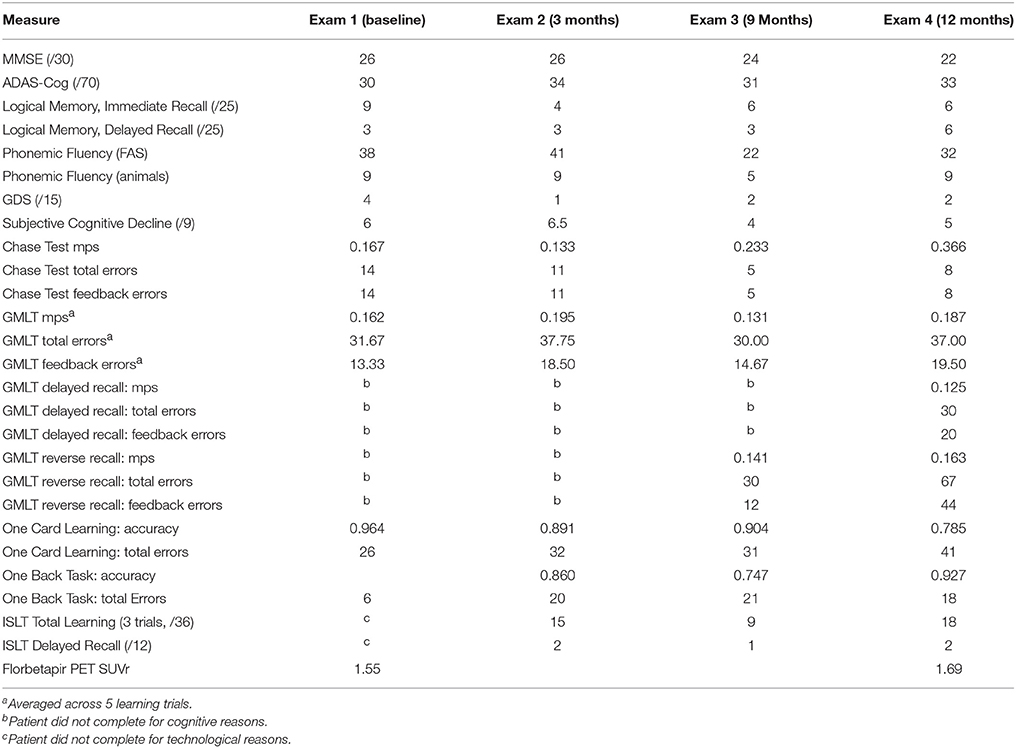
Table 2. Scores on all outcome measures for patient CS, age 74, over 12 month DFMO treatment period.
Throughout the 1 year trial period, the participant did not experience any clinical increase in depression or anxiety symptoms, assessed via self-report (GDS; Yesavage and Sheikh, 1986), caregiver report, and clinical interview. There were no significant changes in her level of subjective complaints, as measured by the Subjective Cognitive Decline scale (Gifford et al., 2015).
Primary Endpoints
The results for all four primary endpoints are summarized in Figure 1. There was no significant improvement in scores on the MMSE, ADAS-Cog, or Logical Memory delayed recall tests over the 1 year treatment period. The patient declined from an MMSE of 26/30 to an MMSE of 22/30 during this time. ADAS-Cog scores increased slightly during the treatment period, indicating cognitive decline. Learning and memory was severely impaired on the Logical Memory stories. Although the participant improved in terms of number of items recalled and percentage retained at the final visit, this was not corroborated by other memory tests, such as the word-list learning subtest of the ADAS-Cog and the ISLT.
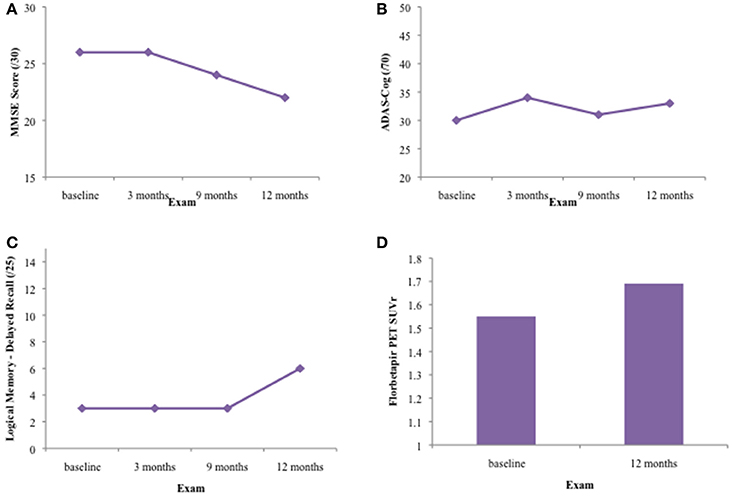
Figure 1. Results on primary outcome measures for patient CS over 12 months of DFMO therapy. (A) Results on the Mini-Mental Status Exam (MMSE)/30 at baseline, 3, 6, and 12 month exams. CS' MMSE score declined 4 points over the 12 month treatment period, from 26/30 to 22/30, indicating generalized cognitive decline. (B) Results on the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) 11—item at baseline, 3, 6, and 12 month exams. Patient CS' ADAS-Cog score increased by 3 points over the 12 month treatment period, from 30/70 to 33/70. Higher scores on this test indicate increased cognitive impairment. (C) Results on the delayed recall portion of the Logical Memory stories at baseline, 3, 6, and 12 month exams. The patient showed impaired verbal learning and episodic memory throughout the 12 month treatment period. Although the patient recalled 6/25 items at the 12 month visit, this was not concurrent with results on other episodic memory tests such as the ADAS-Cog word list learning and recall (0/10 words at delayed recall) and the ISLT (2/12 shopping items at delayed recall). (D) SUVr on Florbetapir PET scan at baseline (1.55) and after 12 month treatment period (1.69).
The patient suffered significant functional decline during the 1 year treatment period. This included complete cessation of driving and inability to prepare complex meals or perform financial management activities. Additionally, the patient and/or her caregiver reported difficulty with orientation to time and place, multi-tasking, increased confusability, and inability to complete multi-step commands. The patient is no longer able to be home independently without supervision. Given this, the family has employed a caregiver to work with the patient once per week for respite care for her spouse, who is her full-time caretaker.
Her florbetapir PET scan was positive on visual read for both the baseline and 12 month visits. Her SUVr increased from 1.55 to 1.69 during the 1 year treatment period, indicating clinically increased neocortical amyloidosis commensurate with advancing Alzheimer's disease.
Secondary Endpoints
As seen in Table 2, there were no improvements on the patient's scores on any of the secondary endpoint measures.
Efficacy Assessment
Patient CS demonstrated continued symptomatic progression, both cognitively and functionally, over the 1 year treatment period on DFMO 500 mg p.o. BID, and there was an increase in cerebral amyloidosis as measured by Florbetapir PET imaging during this time. After the 1 year treatment period, she no longer met criteria for MCI, but rather met criteria for a diagnosis of mild-moderate dementia of the Alzheimer's type.
Discussion
This report details what we believe is the first known administration of eflornithine (DFMO) as a therapeutic intended to slow mild AD progression in a single individual. The participant was a 74-year-old female with a diagnosis of multi-domain amnestic MCI, who could not tolerate cholinesterase inhibitors due to gastrointestinal irritation and subsequent weight loss. After 12 months of DFMO therapy, the participant declined on neuropsychological tests measuring global cognitive function, episodic memory, executive function, language, processing speed, and working memory. She showed no measurable improvement on the MMSE or ADAS-Cog. Additionally, the participants' cortical amyloidosis increased over the 12 month treatment period from a Florbetapir PET amyloid SUVr of 1.55 to 1.69. Functionally, she suffered decline in instrumental activities of daily living and some decline in basic activities of daily living. Taken together, these results indicate that treatment with DFMO was not effective in reducing or halting cortical amyloidosis, and did not lead to positive cognitive or functional outcomes in a patient with MCI. Over the 12 month period, the participant declined clinically and her diagnosis was changed from MCI to mild AD, commensurate with amyloid PET results, functional decline and neuropsychological test scores.
This 12 month, single-case study was predicated on a mouse model of AD (the CVN-AD model; Colton et al., 2008, 2014; Wilcock et al., 2014; Kan et al., 2015), which reduced the levels of iNOS in mice to mimic human levels, resulting in a mouse model that mirrored the course of human AD, beginning with asymptomatic amyloidosis and followed by hyperphosphorlyated tau, age-dependent hippocampal neuronal loss, and behavioral changes in episodic memory and executive function. This model showed that the pathological cascade of cortical amyloidosis in CVN-AD knockout mice is associated with increased arginine utilization, which is immune regulated, and decreased cerebral arginine bioavailability, particularly in areas affected early by AD such as the hippocampus. Additionally, they found that DFMO (a partial inhibitor of arginase, an enzyme that metabolizes arginine) administered to juvenile CVN-AD knockout mice led to reduced cortical amyloidosis and recovery of cognitive function (episodic memory) via the reduction of arginine catabolism by the enzyme arginase 1 (Arg1) and subsequent reduction of immunosuppression by cerebral microglia.
There are several potential factors contributing to the lack of efficacy of DFMO in this single case study. The primary contributor is that Kan et al. (Kan et al., 2015) administered DFMO beginning at 6–8 weeks of age in their CVN-AD knockout mice, and they had treatment dosage of 10 mg/kg via oral gavage 3 times per week for 14 weeks. CVN-AD knockout mice show Aβ accumulation at 6 weeks of age, hyperphosphorylated tau at 12 weeks of age, and behavior deficits at 24 weeks of age (Colton et al., 2014). The administration of DFMO in the CVN-AD knockout mice began concurrently with accumulation of cerebral Aβ, and the therapeutic window spanned the entire length of expected pathological changes in these juvenile mice. In our case, the participant had a Florbetapir PET SUVr of 1.55 at baseline, an indication of significant cortical amyloidosis. Visual inspection of the amyloid PET scan by a board-certified nuclear medicine specialist indicated a positive visual read, indicating increased uptake of Aβ in at least two separate neocortical areas. In our case, unlike Kan et al. (2015), the participant already had significant Aβ accumulation at baseline, which could have caused significant pathological changes resistant to change from DFMO therapy. We suspect that administration of DFMO in the very early stages of cerebral amyloidosis (preclinical AD) would have been a more accurate and reliable assessment of whether or not this therapeutic translates from mouse to human models.
There are still several unknowns regarding the mechanism by which immune suppression, particularly arginine deprivation, could prompt the initiation of the AD pathological cascade in this mouse model. We do know that there is direct evidence supporting that dysregulated arginine utilization may contribute to human AD pathology. Autopsy studies have shown an increase in arginase mRNA expression and enzymatic activity in human AD brain samples (Colton et al., 2006; Hansmannel et al., 2010; Liu et al., 2014). However, the exact cell type that is associated with increased arginine utilization and consumption remains unclear. There are several cell types capable of expressing arginase-1, and one particular cell type or a combination of these cell types could be driving arginine deprivation in AD. Kan et al. (2015) make a strong case for microglia, basing this assumption on the appearance of a particular type of microglial cell (CD11c+) very early in AD pathology that was highly correlated with AD pathology and concentrations of extracellular arginase. Complicating this story, arginine is the target of two opposing, immune regulated, enzyme systems: the iNOS pathway and the arginase pathway. These pathways co-exist, but competition between these two enzymes favors arginase in humans (Wu et al., 2009; Yi et al., 2009; Guo et al., 2012), and iNOS activity is decreased in human AD, resulting in upregulation of the arginase pathway (Colton et al., 2006; Yi et al., 2009; Liu et al., 2014). Further research elucidating the exact neurochemical mechanism by which immune arginine deprivation affects AD pathology will provide insight into next steps for potential translation to a usable therapeutic for preclinical AD patients.
More broadly, research has suggested a role for arginine in AD via interactions with atherosclerosis (Cooke and Creager, 1991; Boger et al., 1996), glucose metabolism (Schmidt et al., 1992; Smuckler et al., 2002), and neurogenesis (Ciani et al., 2006; Torroglosa et al., 2007). Further exploration of the role of arginine in these processes, which contribute to AD, will help to elucidate the peripheral effects of arginine on AD pathology.
Concluding Remarks
These unknowns notwithstanding, we have completed the first ever single case clinical trial of eflornithine (DFMO), a partial inhibitor of arginase, in a patient with multi-domain, amnestic MCI. After 12 months of DFMO treatment, the participant showed cognitive, functional, and pathological progression, and was diagnosed with mild AD. DFMO did not halt or reduce cerebral amyloidosis as measured by Florbetapir PET scan, and the participant's cognitive and functional outcomes decreased over a 12 month period at a rate commensurate with the normal course of neurodegenerative progression in AD. However, we believe that targeting immunosuppressive genes is still a reasonable scientific line of inquiry. Mouse models to date have targeted juvenile animals at the onset of cortical amyloidosis, while our participant already had a high cortical amyloid burden at onset of treatment as shown by Florbetapir PET scan.
Because DFMO is an FDA approved drug and has a favorable safety and tolerability profile, this therapeutic should be further explored as a potential target for preventative therapy in the very early stages of preclinical AD. Additionally, patients with autosomal dominant AD may be ideal targets for such therapy, as these patients are often relatively young, and age of onset tends to be in mid-late life and can be more accurately predicted.
Ethics Statement
This study was carried out in accordance with the recommendations of Institutional Review Board (IRB) at the Rhode Island Hospital (Providence, RI), and the patient described in this report provided written informed consent in accordance with the Declaration of Helsinki. This trial was registered as a Compassionate Use IND #128888 with the United States Food and Drug Administration (FDA).
Author Contributions
KM: Requested and acquired the compassionate use IND from FDA, administered medication, and completed all safety monitoring; JA and PS: Conducted all neuropsychological evaluations, analyzed all data, arranged amyloid PET scans and prepared the manuscript; RN: Analyzed amyloid PET results. All authors read and approved the final manuscript.
Funding
This case study was internally funded by PJS and the Department of Radiology, Rhode Island Hospital.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the participant, C.S., and her family, for their contributions to this study and AD research. The authors also wish to acknowledge Janice Muratori, APRN CNP, for her exceptional contributions to human subjects protection at our institution.
Abbreviations
Aβ, β-amyloid; AD, Alzheimer's disease; ADAS-Cog, Alzheimer's Disease Assessment Scale-Cognitive Subscale; ADL, activities of daily living; APOE, apolipoprotein; APP, amyloid precursor protein; Arg1, Arginase 1; BID, twice per day; CBC, complete blood count; CMP, comprehensive metabolic panel; CT, computerized tomography; DFMO, eflornithine; DNA, deoxyribonucleic acid; FDA, Food and Drug Administration; GDS, Geriatric Depression Scale; GMLT, Groton Maze Learning Test; GWAS, genome-wide association studies; IADL, instrumental activities of daily living; IND, Investigational New Drug; iNOS, immune-mediated nitric oxide; ISLT, International Shopping List Test; MCI, mild cognitive impairment; MMSE, Mini-Mental Status Exam; OBK, One-Back Task; OCL, One-Card Learning Task; PET, positron emission tomography; PO, by mouth; SCD, Subjective Cognitive Decline; SUV, standardized uptake value; SUVr, standard uptake value ratio; TREM-2, Triggering Receptor Expressed on Myeloid Cells-2.
References
Alzheimer's Association Strategic Plan (2017). Available online at: https://www.alz.org/about_us_strategic_plan.asp (Accessed June 25, 2017).
Boger, B. H., Bode-Boger, S. M., and Frolich, J. C. (1996). The L-arginine – nitric oxide pathway: role in atherosclerosis and therapeutic implications. Atherosclerosis 127, 1–11.
Borkowski, J. G., Benton, A. L., and Spreen, O. (1967). Word fluency and brain damage. Neuropsychologia 5, 135–140.
Ciani, E., Calvanese, V., Crochemore, C., Bartesaghi, R., and Contestabile, A. (2006). Proliferation of cerebellar precursor cells is negatively regulated by nitric oxide in newborn rat. J. Cell Sci. 119, 3161–3170. doi: 10.1242/jcs.03042
Clark, C. M., Schneider, J. A., Bedell, B. B., Beach, T. G., Bilker, W. G., Mintun, M. A., et al. (2011). Use of Florbetapir PET imaging amyloid-β pathology. JAMA 305, 275–283. doi: 10.1001/jama.2010.2008
Colton, C. A., Mott, R. T., Sharpe, H., Xu, Q., Van Nostrand, W. E., and Vitek, M. P. (2006). Expression profiles for macrophage alternative activation genese in AD and in mousemodels of AD. J. Neuroinflamm. 3:27. doi: 10.1186/1742-2094-3-27
Colton, C. A., Wilcock, D. M., Wink, D. A., Davis, J., Van Nostrand, W. E., and Vitek, M. P. (2008). The effects of NOS2 gene deletion on mice expression mutated human AβPP. J. Alzheimer Dis. 15, 571–587. doi: 10.3233/JAD-2008-15405
Colton, C. A., Wilson, J. G., Everhart, A., Wilcock, D. M., Puolivali, J., Heikkinen, T., et al. (2014). mNos2 deletion and human NOS2 replacement in Alzheimer disease models. J. Neuropathol. Exp. Neurol. 73, 752–769. doi: 10.1097/NEN.0000000000000094
Cooke, J. P., and Creager, A. (1991). Endothelial dysfunction in hypercholesterolemia is corrected by L-arginine. Basic Res. Cardiol. 86(Suppl. 2), 173–181.
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apoplipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923.
Folstein, M. F., Folstein, S. F., and McHugh, P. R. (1975). Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198.
Fredrickson, A., Snyder, P. J., Cromer, J., Thomas, E., Lewis, M., and Maruff, P. (2008). The use of effect sizes to characterize the nature of cognitive change in psychopharmacological studies: an example with scopolamine. Hum. Psychopharmacol. 23, 425–436. doi: 10.1002/hup.942
Gifford, K. A., Liu, D., Romano, R. R., Jones, R. N., and Jefferson, A. L. (2015). Development of a subjective cognitive decline questionnaire using item response theory: a pilot study. Alzheimers Dement. 1, 429–439. doi: 10.1016/j.dadm.2015.09.004
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guo, Z., Shao, L., Zhen, L., Du, Q., Li, P., John, B., et al. (2012). miRNA-939 regulates human inducible nitric oxide synthase posttransciptional gene expression in human hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 109, 5826–5831. doi: 10.1073/pnas.1118118109
Hansmannel, F., Sillaire, A., Kamboh, M. I., Lendon, C., Pasquier, F., Hannequin, D., Amouyel, P., et al. (2010). Is the urea cycle involved in Alzheimer's disease? J. Alzheimer Dis. 21, 1013–1021. doi: 10.3233/JAD-2010-100630
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388.
Hardy, J., and Higgins, G. A. (1992). Alzheimer's disease: the Alzheimer's cascade hypothesis. Science 256:184.
Irizarry, M. C., McNamara, M., Fedorchak, K., Hsaio, K., and Hyman, B. T. (1997). APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol. 56, 965–973.
Jack, C. R., Knopman, D. S., Jagust, W. J., Petersen, R. C., Weiner, M. W., Aisen, P. S., et al. (2013). Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/S1474-4422(12)70291-0
Jonsson, T., Atwal, K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjmosson, S., et al. (2012). A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kamboh, M. I., Demirci, F. Y., Wang, X., Minster, R. L., Carrasquillo, M. M., Pankratz, V. S., et al. (2012). Genome-wide association study of Alzheimer's disease. Transl. Psychiatr. 2:e117. doi: 10.1038/tp.2012.45
Kan, M. J., Lee, J. E., Wilson, J. G., Everhart, A. L., Brown, C. M., Hoofnagle, A. N., et al. (2015). Arginine deprivation and immune suppression in a mouse model of Alzheimer's Disease. Neurobiol dis. 35, 5969–5982. doi: 10.1523/JNEUROSCI.4668-14.2015
Lim, Y. Y., Prang, K. H., Cysique, L., Pietrzak, R., Snyder, P. J., and Maruff, P. (2009). A method for cross-cultural adaptation of a verbal memory assessment. Behav Res Methods. 41, 1190–1200. doi: 10.3758/BRM.41.4.1190
Liu, P., Fleete, M. S., Jing, Y., Collie, N. D., Curtis, M. A., Waldvogel, H. J., et al. (2014). Altered arginine metabolism in Alzheimer's disease brains. Neurobiol. Aging 35, 1992–2003. doi: 10.1016/j.neurobiolaging.2014.03.013
Maruff, P., Lim, Y. Y., Darby, D., Ellis, K. A., Pietrzak, R. H., Snyder, P. J., et al. (2013). Clincial utility of the Cogstate Brief Battery in identifying cognitive impairment in mild cognitive impairment and Alzheimer's disease. BMC Psychol. 1:30. doi: 10.1186/2050-7283-1-30
Radde, R., Duma, C., Goedert, M., and Jucker, M. (2008). The value of incomplete mouse models of Alzheimer's disease. Eur. J. Nucl. Med. Mol. Imaging 35(Suppl. 1), S70–S74. doi: 10.1007/s00259-007-0704-y
Rosen, W. G., Mohs, R. C., Davis, K. L., Ihl, R., and Weyer, G. (1993). Alzheimer's Disease Assessment Scale (ADAS). Gottingen: Hogrefe.
Saunders, A. M., Strittmatter, M. D., Schemechel, D., St. George-Hyslop, P. H., Pericak-Vance, M. D., Joo, S. H., et al. (1993). Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer's disease. Neurol. 48:1467.
Schadt, E. E. (2009). Molecular networks as sensors and drivers of common human diseases. Nature 461, 218–223. doi: 10.1038/nature08454
Schmidt, H. H., Warner, T. D., Ishii, K., Sheng, H., and Murad, F. (1992). Insulin secretion from pancreatic B cells caused by L-arginine derived nitrogen oxide. Science 255, 721–723.
Smuckler, S. R., Tang, L., Wheeler, M. B., and Salapatek, A. M. F. (2002). Exogenous nitric oxide and endogenous glucose-stimulated β-Cell nitric oxide augment insulin release. Diabetes 51, 3450–3460. doi: 10.2337/diabetes.51.12.3450
Snyder, P. J., Bednar, M. M., Cromer, J. R., Thomas, E., Lewis, M., and Maruff, P. (2005). Reversal of scopolamine-induced deficits with a single dose of donepezil, an acetylcholinesterase inhibitor. Alzheimers Dement. 1, 126–135. doi: 10.1016/j.jalz.2005.09.004
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Tombaugh, T. N., Kozakb, J., and Reesc, L. (1999). Normative data stratified by age and education for two measures of verbal fluency: FAS and animal naming. Arch. Clin. Neuropsychol. 14, 167–177.
Torroglosa, A., Murillo-Carretero, M., Romero-Grimaldi, C., Materredona, E. R., Campos-Caro, A., and Estrada, C. (2007). Nitric oxide decreases subventricular zone stem cell proliferation by inhibition of dermal growth factor receptor and phosphoinositide-3-kinase/Akt pathway. Stem Cells 25, 88–97. doi: 10.1634/stemcells.2006-0131
Wilcock, W. M., Lewis, M. R., Van Nostrand, W. E., Davis, J., Previti, M. L., and Garkholonarehe, N. (2014). Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J. Neurosci. 28, 1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008
Wu, G., Bazer, F. W., Davis, T. A., Kim, S. W., Li, P., Rhoads, M. J., et al. (2009). Arginine metabolism and nutrition in growh, health and disease. Amino Acids 37, 153–168. doi: 10.1007/s00726-008-0210-y
Yesavage, J. A., and Sheikh, J. I. (1986). Geriatric Depression Scale (GDS) recent evidence and development of a shorter version. Clin. Gerontol. 5, 165–173.
Yi, J., Horky, L. L., Friedlich, A. L., Shi, Y., Rogers, J. T., and Huang, X. (2009). L-arginine and Alzheimer's disease. Int. J. Clin. Exp. Pathol. 3, 211–238.
Keywords: mild cognitive impairment, Alzheimer's disease, case study, eflornithine (DFMO), amyloid pathology
Citation: Alber J, McGarry K, Noto RB and Snyder PJ (2018) Use of Eflornithine (DFMO) in the Treatment of Early Alzheimer's Disease: A Compassionate Use, Single-Case Study. Front. Aging Neurosci. 10:60. doi: 10.3389/fnagi.2018.00060
Received: 28 August 2017; Accepted: 20 February 2018;
Published: 06 March 2018.
Edited by:
Ashok Kumar, University of Florida, United StatesReviewed by:
Sarat C. Vatsavayai, University of California, San Francisco, United StatesWilliam Edmund Renehan, University of Rhode Island, United States
Copyright © 2018 Alber, McGarry, Noto and Snyder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Snyder, peter.j.snyder@gmail.com