
 Michael Feyder
Michael Feyder
- Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden
The basal ganglia are affected by several neuropsychiatric and neurodegenerative diseases, many of which are treated with drugs acting on the dopamine system. For instance, the loss of dopaminergic input to the striatum, which is the main pathological feature of Parkinson’s disease, is counteracted by administering the dopamine precursor, L-DOPA. Furthermore, psychotic disorders, including schizophrenia, are treated with drugs that act as antagonists at the D2-type of dopamine receptor (D2R). The use of L-DOPA and typical antipsychotic drugs, such as haloperidol, is limited by the emergence of motor side-effects, particularly after prolonged use. Striatal medium spiny neurons (MSNs) represent an ideal tool to investigate the molecular changes implicated in these conditions. MSNs receive a large glutamatergic innervation from cortex, thalamus, and limbic structures, and are controlled by dopaminergic projections originating in the midbrain. There are two large populations of striatal MSNs, which differ based on their connectivity to the output nuclei of the basal ganglia and on their ability to express dopamine D1 receptors (D1Rs) or D2Rs. Administration of L-DOPA promotes cAMP signaling and activates the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) in the D1R-expressing MSNs, which form the striatonigral, or direct pathway. Conversely, haloperidol activates the cAMP/DARPP-32 cascade in D2R-expressing MSNs, which form the striatopallidal, or indirect pathway. This review describes the effects produced on downstream effector proteins by stimulation of cAMP/DARPP-32 signaling in these two groups of MSNs. Particular emphasis is given to the regulation of the GluR1 subunit of the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate glutamate receptor, the extracellular signal-regulated protein kinases 1 and 2, focusing on functional role and potential pathological relevance.
Dysfunctions of the dopamine system are implicated in many neurodegenerative and neuropsychiatric disorders, including Parkinson’s disease (PD) and schizophrenia. The cardinal symptoms of PD (tremor, rigidity, and hypokinesia) are caused by the progressive degeneration of the midbrain dopaminergic neurons of the substantia nigra pars compacta (Hornykiewicz, 1963; Braak et al., 2003). These cells project to the dorso-lateral part of the striatum, which is the major component of the basal ganglia, a group of subcortical nuclei involved in the control of motor function. The dopamine precursor L-DOPA is widely used in the treatment of PD (Cotzias et al., 1967; Birkmayer and Hornykiewicz, 1998); however, prolonged use of this drug is complicated by the appearance of dystonic and choreic movements, or dyskinesia, which are manifested in parallel with the therapeutic, anti-akinetic action of L-DOPA (Obeso et al., 2000).
In contrast to PD, schizophrenia and psychotic disorders are treated with drugs which reduce dopaminergic transmission. In particular, conventional antipsychotic drugs, such as haloperidol, act as selective and potent antagonists at D2-type of dopamine receptors (Creese et al., 1976; Seeman et al., 1976). Even the use of these substances, however, is limited by the emergence of motor complications, generally referred to as extrapyramidal side-effects (EPS), which include tardive dyskinesia, dystonia, and parkinsonism (Miyamoto et al., 2005).
The development of motor disorders in response to administration of L-DOPA and haloperidol depends in large part on the ability of these drugs to affect transmission in the striatum, which is enriched in D1- and D2-type of dopamine receptors. The study of the effects produced in this brain region by antiparkinsonian and antipsychotic drugs is important not only to elucidate the mechanisms underlying L-DOPA-induced dyskinesia (LID) and EPS, but also to examine the impact on motor dysfunctions of aberrant signaling processes occurring in well-defined neuronal populations.
Distinct Roles of Striatal Medium Spiny Neurons in Motor Function
In the striatum, fibers originating from midbrain dopaminergic neurons innervate two populations of principal GABAergic neurons, collectively denominated medium spiny neurons (MSNs). More than 20 years ago, these two groups of neurons were proposed to form separate projection pathways, exerting opposing effects on basal ganglia output structures (i.e., substantia nigra pars reticulata and internal segment of the globus pallidus). It was suggested that the MSNs directly connected to these structures would disinhibit thalamo-cortical neurons and promote motor activity, whereas the MSNs connected to these nuclei indirectly (via external segment of the globus pallidus and subthalamic nucleus) would increase the inhibition of efferent targets within the thalamus and depress motor function (Albin et al., 1989; Alexander and Crutcher, 1990; DeLong, 1990).
Another important concept emerging during those years was that dopamine acted by stimulating the MSNs of the direct, or striatonigral pathway, and inhibiting those of the indirect, or striatopallidal pathway (Albin et al., 1989; Gerfen et al., 1990). These opposing effects were suggested to depend on the expression of dopamine D1 receptors (D1Rs) in the MSNs of the direct pathway and of dopamine D2 receptors (D2Rs) in the MSNs of the indirect pathway (Gerfen et al., 1990). The hypothesis of a contrasting action of dopamine on direct and indirect MSNs was supported by the observation that D1Rs activate, whereas D2Rs inhibit, adenylyl cyclase and cAMP synthesis (Kebabian and Calne, 1979; cf. below), thereby leading to opposite regulations of downstream effector targets and neuronal excitability.
The idea that D1Rs and D2Rs are selectively expressed in distinct groups of MSNs has gained increasing acceptance and, recently, it has been exploited to revisit and further analyze the functional properties of the MSNs of the direct and indirect pathway.
The development of techniques to engineer bacterial artificial chromosomes (BACs) has allowed the generation of mice expressing Cre recombinase, epitope-tagged proteins, and fluorescent probes in the D1R- or in the D2R-expressing MSNs of the direct and indirect pathway (Gong et al., 2003, 2007; Valjent et al., 2009). Using Cre-dependent viral delivery of channelrhodopsin-2, a light activated proton pump (Boyden et al., 2005; Kravitz et al., 2010) demonstrated that optogenetic activation of the D1R-expressing MSNs of the direct pathway promotes locomotion, whereas activation of the D2R-expressing MSNs of the indirect pathway depresses motor function.
Another study examined the effects on motor behaviors of cell-specific inactivation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), an important mediator of cAMP signaling and striatal excitability (Greengard, 2001). It was found that deletion of DARPP-32 in D1R-expressing MSNs reduced basal locomotor activity in the open-field. In contrast, inactivation of DARPP-32 in D2R-expressing MSNs increased locomotion (Bateup et al., 2010). A similar enhancement of motor function was observed following ablation of striatopallidal MSNs (Durieux et al., 2009).
Dopamine Signaling in Striatal MSNs
As previously mentioned, D1Rs and D2Rs mediate opposite regulations of cAMP signaling. In the striatum, activation of D1Rs in the direct MSNs leads to Golf-mediated stimulation of adenylyl cyclase, the enzyme responsible for the synthesis of cAMP (Zhuang et al., 2000; Herve et al., 2001). The ability of D1Rs to induce G-protein-dependent activation of cAMP signaling promotes transmission at α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA; cf. below) and N-methyl-D-aspartate (NMDA) glutamate receptors (Surmeier et al., 1995; Blank et al., 1997; Snyder et al., 1998; Liu et al., 2004). In addition, D1R-mediated activation of cAMP signaling increases L-type Ca2+ channel currents and decreases K+ channel currents (Kitai and Surmeier, 1993; Surmeier et al., 1995; Galarraga et al., 1997; Gao et al., 1997). These multiple regulations elevate the ability of sustained release of glutamate to produce depolarized up-states close to spike threshold (Wickens and Wilson, 1998), thereby promoting the excitability of striatonigral MSNs (Surmeier et al., 2007).
Activation of D2Rs in the indirect MSNs is coupled to Gi/o-mediated inhibition of adenylyl cyclase, which decreases cAMP synthesis (Kebabian and Calne, 1979; Stoof and Kebabian, 1981). This negative modulation is especially evident on the adenylyl cyclase type 5 isoform, which is preferentially expressed in striatal MSNs (Glatt and Snyder, 1993; Mons and Cooper, 1994; Robinson and Caron, 1997). D2R-mediated activation of Gi/o also leads to decreased cell excitability via G-protein-mediated activation of inwardly rectifying K+ channels (Wickman et al., 1994; Greif et al., 1995; Kuzhikandathil et al., 1998; Hopf et al., 2003). Moreover, D2Rs have been shown to regulate L-type Ca2+ currents in striatal MSNs through a G-protein- and Ca2+-dependent mechanism which does not require suppression of cAMP signaling (Hernandez-Lopez et al., 2000). The coordinated modulation of ion channels exerted by D2R via activation of Gi/o protein leads to reduced responsiveness of striatopallidal MSNs to glutamate, diminished transition of MSNs to depolarized up-states and reduced excitability (Surmeier et al., 2007).
In summary, the opposite regulation exerted by D1Rs and D2Rs on the responsiveness of MSNs to glutamate is in line with the functional model of basal ganglia described above, in which dopamine promotes motor activity via concomitant activation of direct MSNs and inhibition of indirect MSNs (Albin et al., 1989; Alexander and Crutcher, 1990; DeLong, 1990; Surmeier et al., 2007).
L-DOPA and Haloperidol in the Striatum: Focus on cAMP Signaling in MSNs
Studies performed in experimental models of PD indicate that administration of L-DOPA results in multiple changes in signal transduction, which occur specifically in the MSNs of the direct pathway. These changes depend on the loss of striatal dopamine, which results in the development of a remarkable sensitization at striatal D1Rs (Guigoni et al., 2005; Santini et al., 2008). Such sensitization potentiates the ability of L-DOPA to promote cAMP signaling, leading to stimulation of cAMP-dependent protein kinase (PKA) and phosphorylation of DARPP-32 (Picconi et al., 2003; Santini et al., 2007, 2010; Lebel et al., 2010). As discussed below, prolonged activation of this signaling cascade is implicated in the development of LID.
In contrast to L-DOPA, the actions of antipsychotic drugs (e.g., haloperidol) in the striatum are generally attributed to their ability to antagonize D2Rs on the MSNs of the indirect pathway. The primary effect of D2R blockade is the removal of the inhibition exerted by the Gi/o protein on adenylyl cyclase, leading to increased cAMP signaling. This effect, in turn, depends on basal adenylyl cyclase activity, which is maintained through the action of the neuromodulator adenosine. The MSNs of the indirect pathway express high levels of adenosine A2A receptors (A2ARs; Schiffmann et al., 1991; Fink et al., 1992), which, similarly to D1Rs, are coupled to Golf-mediated activation of adenylyl cyclase (Kull et al., 2000; Herve et al., 2001). Thus, in the MSNs of the indirect pathway, D2Rs and A2ARs exert opposite regulations on cAMP signaling. An important consequence of this functional arrangement is that blockade of A2AR-mediated transmission prevents the ability of D2R antagonists, including haloperidol, to increase cAMP signaling in indirect MSNs (Svenningsson et al., 2000; Håkansson et al., 2006; Bertran-Gonzalez et al., 2009; cf. Figure 1).
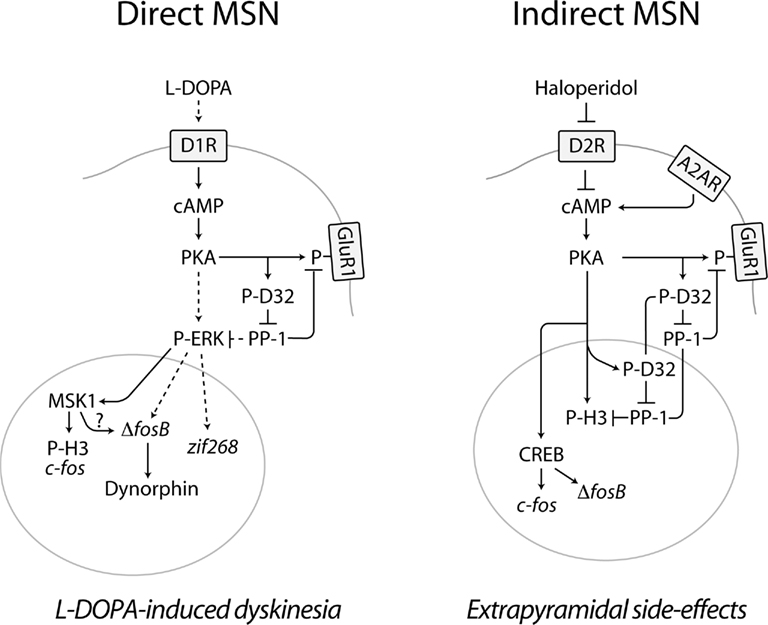
Figure 1. Diagram summarizing the effects produced by L-DOPA and haloperidol on cAMP signaling in the striatal MSNs of the direct and indirect pathway. In PD, loss of dopamine in the striatum leads to increased responsiveness of D1Rs, expressed in the direct MSNs of the striatonigral pathway. Administration of L-DOPA increases the production of dopamine and activates D1Rs, which stimulate the cAMP/PKA pathway, thereby phosphorylating DARPP-32 (D32) at Thr34 (Picconi et al., 2003; Santini et al., 2007, 2010). A similar effect is produced by haloperidol in the indirect MSNs of the striatopallidal pathway via blockade of D2Rs and disinhibition of A2AR-mediated activation of cAMP signaling (Svenningsson et al., 2000). The activation of the PKA/DARPP-32 cascade produced by L-DOPA and haloperidol increases the phosphorylation of targets located in the cytoplasm/plasma membrane, such as the GluR1 subunit of the glutamate AMPA receptor (Håkansson et al., 2006; Santini et al., 2007). In direct MSNs, L-DOPA promotes ERK signaling. This effect may be exerted via PKA/DARPP-32-mediated regulation of Ras-GRF1 (Mattingly, 1999) and/or striatal-enriched protein tyrosine phosphatase (Valjent et al., 2005), which are involved in the regulation of ERK phosphorylation. ERK-dependent activation of MSK1 results in phosphorylation of histone H3 at Ser10 and enhanced expression of c-fos (Santini et al., 2007). Activation of ERK also increases the expression of zif268 and ΔfosB, which has been associated to LID (Andersson et al., 1999; Carta et al., 2005). Increased ΔFosB, in turn, leads to enhanced levels of dynorphin (Andersson et al., 1999). In the indirect striatopallidal MSNs, the regulation of histone H3 and gene expression exerted by haloperidol occurs independently of the ERK/MSK1 cascade. Thus, histone H3 is regulated via PKA-catalyzed phosphorylation and by concomitant suppression of dephosphorylation, via DARPP-32-mediated inhibition of PP-1 (Bertran-Gonzalez et al., 2009). In addition, the ability of haloperidol to activate PKA leads to increased phosphorylation of the cAMP-response element binding protein (CREB), which may be involved in the control of Fos expression (Konradi and Heckers, 1995). The control of multiple signaling pathways exerted by L-DOPA and haloperidol in the MSNs of the direct and indirect pathway may ultimately modify the activity of these neuronal populations, leading to abnormal motor responses, including L-DOPA-induced dyskinesia and extrapyramidal side-effects. Broken lines indicate indirect effects. See text for abbreviations.
In summary, both L-DOPA and haloperidol promote cAMP-dependent signaling in striatal MSNs. The effect of L-DOPA is exerted in the MSNs of the direct pathway, via D1R-mediated activation of adenylyl cyclase. In contrast, the effect of haloperidol is exerted on the MSNs of the indirect pathway through blockade of D2Rs and disinhibition of adenylyl cyclase (Figure 1).
DARPP-32: A Mediator of the Actions of Antiparkinsonian and Antipsychotic Drugs
The ability of L-DOPA and haloperidol to promote cAMP signaling is reflected by the large increase in the phosphorylation of DARPP-32 produced by administration of these drugs. DARPP-32 is highly expressed in the MSNs of the direct and indirect pathway and plays an important role in dopamine transmission (Fienberg et al., 1998; Greengard, 2001). Activation of D1Rs in direct MSNs increases PKA-dependent phosphorylation of DARPP-32 on Thr34 (Nishi et al., 1997; Svenningsson et al., 1998). A similar effect is produced by activation of A2ARs in indirect MSNs (Svenningsson et al., 1998). Phosphorylation of Thr34 converts DARPP-32 into an inhibitor of protein phosphatase-1 (PP-1; Hemmings et al., 1984), which amplifies the effects of the cAMP/PKA cascade by reducing dephosphorylation of downstream target proteins (Fienberg et al., 1998; Greengard, 2001; cf. Figure 1).
In the dopamine depleted striatum, administration of L-DOPA increases the phosphorylation of DARPP-32 at Thr34 (Picconi et al., 2003; Santini et al., 2007, 2010; Lebel et al., 2010). In rodent and non-human primate models of PD, this effect is associated to the manifestation of abnormal involuntary movements, which are regarded as a behavioral correlate of dyskinesia (Cenci et al., 2002). In line with these observations, it has been shown that LID is attenuated in DARPP-32 knock out mice (Santini et al., 2007). Interestingly, a large reduction of LID has also been observed in mice in which DARPP-32 is specifically inactivated in the MSNs of the direct pathway, whereas deletion of DARPP-32 in indirect MSNs does not affect the development of dyskinesia (Bateup et al., 2010). These findings indicate the importance of enhanced D1R/PKA/DARPP-32 signaling in LID and identify the MSNs of the direct pathway as a key neuronal substrate implicated in this condition.
Administration of D2R antagonists, including haloperidol, promotes PKA-dependent phosphorylation of DARPP-32 (Svenningsson et al., 2000). This effect is produced via disinhibition of adenylyl cyclase, whose basal activation is maintained by A2ARs (cf. above). In support of this idea, it has been shown that both pharmacological and genetic inactivation of A2ARs prevent the increase in DARPP-32 phosphorylation induced by eticlopride, a potent D2R antagonist (Svenningsson et al., 2000). The effect of haloperidol has been examined in BAC transgenic mice overexpressing DARPP-32 with different epitope tags in D1R- or D2R-expressing MSNs (Bateup et al., 2008). In these animals, tag-specific immunoprecipitation of DARPP-32 indicates that the increase in Thr34 phosphorylation induced by haloperidol occurs specifically in the D2R-expressing MSNs of the indirect pathway (Bateup et al., 2008).
Other studies examined the role of DARPP-32 in the changes of motor activity produced by haloperidol. Blockade of D2Rs results in a state of rigid immobility, or catalepsy, which represents a surrogate marker of EPS. This form of motor depression is thought to reflect the ability of haloperidol and other D2R antagonists to activate indirect MSNs, through removal of the inhibition exerted on these neurons by D2Rs (cf. above; Albin et al., 1989; Alexander and Crutcher, 1990; DeLong, 1990; Gerfen et al., 1990). Activation of the MSNs of the indirect pathway would lead, in turn, to inhibition of thalamo-cortical neurons (Albin et al., 1989; Alexander and Crutcher, 1990; DeLong, 1990; Gerfen et al., 1990) and reduction of motor cortex activity (Parr-Brownlie and Hyland, 2005). Deletion of DARPP-32 in indirect MSNs decreases haloperidol-induced catalepsy, whereas DARPP-32 inactivation in direct MSNs only slightly affects this behavioral response (Bateup et al., 2010). These findings indicate the importance of the PKA/DARPP-32 signaling pathway in haloperidol-induced motor depression and point to the MSNs of the indirect pathway as a critical neuronal population involved in EPS.
Regulation of GluR1 Phosphorylation
Activation of the cAMP/PKA/DARPP-32 cascade increases the phosphorylation of the GluR1 subunit of the AMPA glutamate receptor (Banke et al., 2000; Snyder et al., 2000). Phosphorylation of GluR1 at the PKA site, Ser845, promotes glutamate transmission by increasing AMPA channel currents (Roche et al., 1996; Banke et al., 2000) and enhancing cell surface expression of the receptor (Mangiavacchi and Wolf, 2004). In the dopamine depleted striatum, L-DOPA enhances Ser845 phosphorylation and this effect has been associated to LID in both mice and monkeys (Santini et al., 2007, 2010). Indeed, GluR1 hyperphosphorylation may be causally linked to dyskinesia, which is currently treated with drugs that reduce glutamatergic transmission (Goetz et al., 2005).
GluR1 phosphorylation at Ser845 is also increased by administration of haloperidol (Håkansson et al., 2006). This effect depends on disinhibition of PKA in indirect MSNs, since it is abolished by pre-treatment with an A2AR antagonist (cf. above; Håkansson et al., 2006). In addition, haloperidol fails to phosphorylate GluR1 in DARPP-32 knock out mice, or in mice in which the PKA phosphorylation site of DARPP-32 (i.e., Thr34) is replaced with an alanine (T34A mutant mice; Håkansson et al., 2006). Thus, in indirect MSNs, haloperidol increases the phosphorylation of the AMPA glutamate receptor via activation of the cAMP/PKA/DARPP-32/PP-1 cascade. In view of the permissive role played by PKA-dependent phosphorylation of GluR1 on glutamatergic transmission (Roche et al., 1996; Banke et al., 2000; Mangiavacchi and Wolf, 2004), this effect of haloperidol is likely to promote the excitability of indirect MSNs, thereby leading to motor depression and, possibly, to the generation of EPS.
The activation of cAMP signaling produced by L-DOPA and haloperidol in direct and indirect MSNs is involved in the behavioral responses associated to these drugs. However, the increases in DARPP-32 and GluR1 phosphorylation discussed above are transient, whereas LID and EPS are most likely caused by long-term adaptive responses involving dysregulated gene expression.
Downstream of the cAMP/DARPP-32 Cascade I. Effects of L-DOPA in Direct MSNs
Seminal studies carried out by Gerfen et al. (1990) in 6-OHDA-lesioned rats showed that, in the striatum, dopamine deafferentation decreased the transcription of mRNA coding for substance P, a neuropeptide specifically expressed in the direct MSNs. Conversely, the 6-OHDA lesion increased the levels of mRNA coding for the neuropeptide enkephalin, which is expressed in indirect MSNs (Gerfen et al., 1990). In addition to these changes, which reflect the opposite regulation exerted by dopamine on direct and indirect MSNs (cf. above), it was found that repeated administration of a D1R agonist increased mRNA for dynorphin, specifically in direct MSNs (Gerfen et al., 1990). Later studies in the same experimental model showed that a similar increase in dynorphin expression occurs also in response to chronic administration of L-DOPA and that this effect correlates with the severity of abnormal involuntary movements (i.e., dyskinesia) produced by this drug (Andersson et al., 1999). These observations prompted the investigation of the mechanisms mediating the modifications in the expression of dynorphin and other genes potentially involved in LID.
The extracellular signal-regulated protein kinases 1 and 2 (ERK) have emerged as key signaling components involved in the control of gene expression and synaptic plasticity (Thomas and Huganir, 2004). In the striatum, ERK signaling has been implicated in the actions of addictive drugs and in corticostriatal long-term potentiation (Valjent et al., 2000, 2005; Xie et al., 2009). Stimulation of the D1R/PKA/DARPP-32 cascade leads to the phosphorylation and activation of ERK (Valjent et al., 2005; Santini et al., 2007; but see also Gerfen et al., 2008), which controls a variety of downstream effector proteins in the nucleus and cytoplasm (Thomas and Huganir, 2004). In the dopamine depleted striatum, activation of D1Rs or administration of L-DOPA increase the phosphorylation of ERK and its nuclear target, mitogen- and stress-activated kinase 1 (MSK1; Gerfen et al., 2002; Pavon et al., 2006; Santini et al., 2007, 2009; Westin et al., 2007). This, in turn, leads to phosphorylation of histone H3 at Ser10 (Santini et al., 2007, 2009; Darmopil et al., 2009), which is thought to promote transcriptional activation (Nowak and Corces, 2004).
Evidence obtained in rodent and monkey models show that activation of ERK signaling is implicated in LID (Santini et al., 2007; Schuster et al., 2008; Fasano et al., 2010). Studies in BAC transgenic mice expressing enhanced green fluorescent protein (EGFP) under the control of the promoter for either the D1R (drd1a-EGFP mice) or the D2R (drd2-EGFP mice; Santini et al., 2009) indicate that activation of the ERK/MSK1/H3 cascade occurs selectively in the D1R-containing MSNs of the direct pathway (Santini et al., 2009). Therefore, it is likely that, in these neurons, abnormal activation of PKA/DARPP-32 and ERK signaling in response to L-DOPA promotes transcriptional responses ultimately responsible for the development of dyskinesia.
Chronic administration of cocaine, a drug which promotes dopaminergic transmission, leads to the accumulation of stable, alternatively spliced isoforms of the transcription factor FosB, generally denominated as ΔFosB (Hope et al., 1994; Nestler et al., 2001). Enhanced ΔFosB levels have been also observed in the striata of rodent and non-human primate models of PD following chronic administration of L-DOPA (Andersson et al., 1999; Pavon et al., 2006; Berton et al., 2009; Fasano et al., 2010), as well as in postmortem samples from parkinsonian patients treated with L-DOPA (Tekumalla et al., 2001).
Interestingly, the ability of L-DOPA to increase ΔFosB expression correlates with the emergence of dyskinesia (Doucet et al., 1996; Andersson et al., 1999; Pavon et al., 2006; Berton et al., 2009; Fasano et al., 2010). This effect is produced in response to activation of D1Rs (Doucet et al., 1996; Darmopil et al., 2009) and is restricted to the MSNs of the direct pathway (Andersson et al., 1999; Darmopil et al., 2009), where activation of ERK is also occurring (Darmopil et al., 2009; Santini et al., 2009). Thus, in mice and monkeys, the increase in ΔFosB associated to LID is abolished by inactivation of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1), a brain specific component of the Ras-ERK signaling cascade (Fasano et al., 2010).
Several lines of evidence indicate that overexpression of ΔFosB is implicated in LID. Experiments performed in 6-OHDA-lesioned rats showed that downregulation of ΔFosB, achieved by striatal injection of a fosB antisense oligonucleotide, decreases L-DOPA-induced abnormal involuntary movements (Andersson et al., 1999). A similar effect has been recently observed in the macaque following viral-induced overexpression of ΔJunD, a dominant negative of ΔFosB (Berton et al., 2009; Cao et al., 2010). Furthermore, in the rat, LID is reduced or exacerbated by overexpression of ΔJunD and ΔFosB, respectively (Cao et al., 2010).
Taken together, the studies described above indicate that LID is produced by sensitized D1R transmission, which leads to cAMP-dependent activation of ERK/MSK1 signaling. This, in turn, results in enhanced phosphorylation of histone H3 at Ser10 and increased expression of the transcription factor ΔFosB (Figure 1). One important task for future studies will be the identification of specific sets of genes regulated by this intracellular pathway. In this regard it is interesting to note that the increase in dynorphin associated to LID has been shown to depend on the ability of chronic L-DOPA to induce ΔFosB (Andersson et al., 1999).
Downstream of the cAMP/DARPP-32 Cascade II. Effects of Haloperidol in Indirect MSNs
As discussed above, the changes in gene expression associated to LID are primarily linked to the ability of L-DOPA to act on sensitized D1Rs, leading to sequential activation of PKA/DARPP-32 and ERK signaling (Pavon et al., 2006; Santini et al., 2007, 2008; Westin et al., 2007; Lebel et al., 2010). Administration of haloperidol and other D2R antagonists is also known to induce the expression of several immediate early genes acting as transcription factors, including c-fos, fosB, and zif268 (Dragunow et al., 1990; Miller, 1990; Robertson and Fibiger, 1992; MacGibbon et al., 1994, 1995). This effect is particularly evident in the dorsal striatum and has been correlated to the propensity of antipsychotic drugs to induce EPS (Robertson and Fibiger, 1992; MacGibbon et al., 1994).
The increase in c-fos expression produced by haloperidol occurs selectively in the enkephalin-positive MSNs of the indirect pathway (Robertson et al., 1992). Recent work in drd1a- and drd2-EGFP mice has confirmed this initial observation, showing that haloperidol increases c-Fos and Zif268 specifically in D2R-expressing MSNs (Bertran-Gonzalez et al., 2008).
The control exerted by haloperidol on gene expression depends on the ability of this drug to promote cAMP signaling by antagonizing D2R-mediated transmission (cf. above). In support of this idea, it has been shown that genetic inactivation of the regulatory IIβ subunit of PKA, which is highly expressed in the striatum (Cadd and McKnight, 1989), abolishes the effect of haloperidol on c-fos expression (Adams et al., 1997). In line with this observation, intrastriatal injection of antisense oligonucleotides against the cAMP-response element binding protein, a transcription factor activated by PKA, prevents haloperidol-mediated induction of c-Fos protein (Konradi and Heckers, 1995). Other studies showed that the increase in enkephalin mRNA produced by acute and chronic administration of haloperidol is reduced in the absence of A2ARs, which are necessary to maintain normal cAMP signaling in the striatal MSNs of the indirect pathway (cf. above; Chen et al., 2001). Interestingly, the ability of haloperidol to induce catalepsy is also attenuated in both PKA- and A2AR-deficient mice (Adams et al., 1997; Chen et al., 2001), suggesting that reduced cAMP signaling in indirect MSNs may attenuate EPS.
The ability of haloperidol to promote cAMP/DARPP-32 signaling is accompanied by increased ERK phosphorylation in a small number of MSNs belonging to the indirect pathway (Bertran-Gonzalez et al., 2008, 2009). This modest effect contrasts with the relatively large increase in ERK phosphorylation detected in striatal tissue by western blotting (Pozzi et al., 2003). In fact, a considerable proportion of the increase in ERK phosphorylation produced by haloperidol is confined to the terminals of midbrain dopaminergic neurons. In support of this possibility it has been shown that, in the striatum, haloperidol increases ERK-dependent phosphorylation of tyrosine hydroxylase, whose localization is restricted to dopaminergic fibers (Håkansson et al., 2004). This effect may be secondary to the block exerted by haloperidol on inhibitory dopamine D2 autoreceptors located on cell bodies and terminals of nigrostriatal dopaminergic neurons (Håkansson et al., 2004; Fisone et al., 2007).
Administration of haloperidol results in a large increase in phosphorylation of histone H3 at Ser10, which occurs in the D2R-expressing MSNs of the indirect pathway (Bertran-Gonzalez et al., 2008, 2009). This effect differs from the more restricted and modest increase in ERK phosphorylation (cf. above; Bertran-Gonzalez et al., 2008, 2009), suggesting that ERK signaling is not involved in the phosphorylation of histone H3 produced by blockade of D2Rs. This idea is confirmed by the observation that neither pharmacological inhibition of ERK, nor genetic inactivation of MSK1, affects haloperidol-induced phosphorylation of histone H3 (Bertran-Gonzalez et al., 2009).
Several lines of evidence indicate that the ability of D2R antagonists, including antipsychotic drugs, to promote histone H3 phosphorylation at Ser10 depends exclusively on activation of the cAMP/DARPP-32 cascade. For instance, the ability of haloperidol to increase the levels of phospho-Ser10-Lys14-acetylated histone H3 is prevented by blockade of PKA (Li et al., 2004). A similar effect has been observed following genetic attenuation of the Golf protein, which couples A2ARs to activation of adenylyl cyclase (Kull et al., 2000; Herve et al., 2001), or pharmacological blockade of A2ARs (Bertran-Gonzalez et al., 2009). Finally, the ability of haloperidol to regulate histone H3 also requires PKA-dependent activation and nuclear translocation of DARPP-32 (Stipanovich et al., 2008; Bertran-Gonzalez et al., 2009). Thus, in striatopallidal MSNs, the PKA/DARPP-32 signaling pathway regulates histone H3 phosphorylation independently of the ERK/MSK1 cascade, which is generally thought to be critically involved in the generation of the nucleosomal response (Thomson et al., 1999; Figure 1).
Conclusion
The advent of sophisticated methodological approaches based on the use of BAC transgenic mice (Heintz, 2001) and optogenetics (Deisseroth, 2011) has allowed a refined analysis of the circuitries and mechanisms involved in a multitude of physiopathological conditions. In this regard, the striatal MSNs of the direct and indirect pathway have represented a particularly interesting subject of study. These neurons are the primary target of medications used in the treatment of PD and schizophrenia and are distinguishable based on the organization of their connectivity within the basal ganglia and of their ability to express distinct sets of neurotransmitter receptors (Albin et al., 1989; Gerfen et al., 1990). This latter feature has made it possible to generate transgenic mice in which the physiological properties of direct and indirect MSNs, as well as their ability to respond to a variety of psychoactive substances, can be examined by cell-specific genetic manipulations (Gong et al., 2003, 2007; Valjent et al., 2009).
This review focused on the changes in signal transduction produced in striatal MSNs by L-DOPA and haloperidol, with particular emphasis on the potential involvement of these changes in the movement disorders caused by prolonged use of these drugs. The emerging picture indicates that LID and EPS are caused by dysregulation of cAMP signaling in direct and indirect MSNs, respectively. In line with this idea, increased activity of PKA and DARPP-32 in these two groups of neurons has been implicated in L-DOPA- and haloperidol-induced motor disorders (Santini et al., 2007; Bateup et al., 2010). The activation of the PKA/DARPP-32 signaling cascade produced by administration of L-DOPA and typical antipsychotic drugs leads to chromatin modifications (e.g., histone H3 phosphorylation at Ser10) and to the expression of several immediate early genes acting as transcription factors (e.g., fos-c, fosB/ΔfosB, and zif268; Figure 1). Future studies will be necessary to examine the relative contribution of these various responses to the development and manifestation of LID and EPS. It will also be essential to identify molecular targets regulated by this complex transcriptional machinery and evaluate their possible involvement in aberrant motor behaviors.
An intriguing point emerging from recent studies concerns the existence of distinct mechanisms by which L-DOPA and haloperidol promote nucleosomal response and gene expression in striatal MSNs. In the case of L-DOPA, these effects require D1R/PKA/DARPP-32-mediated activation of ERK/MSK1 signaling. Thus, pharmacological or genetic inactivation of ERK prevents the increase in gene expression and the phosphorylation of histone H3 induced by L-DOPA and associated to LID (Santini et al., 2007; Fasano et al., 2010). In contrast, the ability of haloperidol to modify histone H3 and gene expression does not appear to involve activation of ERK signaling. This is indicated by the modest effect on ERK phosphorylation produced by haloperidol in striatal MSNs and, most importantly, by the observation that genetic inactivation of MSK1 does not affect the ability of haloperidol to phosphorylate histone H3 (Bertran-Gonzalez et al., 2009).
Taken together, the above observations indicate that cAMP-dependent regulation of long-term responses in direct and indirect MSNs engages, at least in part, different signaling pathways (Figure 1). This distinction may be exploited to design therapeutic approaches tailored to counteract motor disturbances produced by dysfunctional signaling in one or the other neuronal population. For instance, drugs interfering with ERK signaling may be particularly suited for the treatment of LID and less efficacious to counteract EPS, since haloperidol produces only a modest activation of the ERK/MSK1 cascade in indirect MSNs. Indeed, it has been shown that administration of ERK inhibitors, or genetic inactivation of Ras-GRF1, reduce LID in animal models of PD (Santini et al., 2007; Fasano et al., 2010). Further work on the characterization of signal transduction in direct and indirect MSNs will open new vistas on the functioning of the striatum in physiological and pathological conditions and provide information for the treatment of diseases caused by altered transmission in this brain region.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Gilberto Fisone was supported by Swedish Research Council grant 13482 and by the Swedish Brain Foundation. Alessandra Bonito-Oliva was supported by a fellowship from the Foundation Blanceflor Boncompagni-Ludovisi, née Bildt. Michael Feyder was supported by the National Institutes of Health – Karolinska Institutet Graduate Training Partnership Program.
References
Adams, M. R., Brandon, E. P., Chartoff, E. H., Idzerda, R. L., Dorsa, D. M., and McKnight, G. S. (1997). Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 94, 12157–12161.
Albin, R. L., Young, A. B., and Penney, J. B. (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375.
Alexander, G. E., and Crutcher, M. D. (1990). Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 13, 266–271.
Andersson, M., Hilbertson, A., and Cenci, M. A. (1999). Striatal fosB expression is causally linked with L-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol. Dis. 6, 461–474.
Banke, T. G., Bowie, D., Lee, H., Huganir, R. L., Schousboe, A., and Traynelis, S. F. (2000). Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 20, 89–102.
Bateup, H. S., Santini, E., Shen, W., Birnbaum, S., Valjent, E., Surmeier, D. J., Fisone, G., Nestler, E. J., and Greengard, P. (2010). Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc. Natl. Acad. Sci. U.S.A. 107, 14845–14850.
Bateup, H. S., Svenningsson, P., Kuroiwa, M., Gong, S., Nishi, A., Heintz, N., and Greengard, P. (2008). Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat. Neurosci. 11, 932–939.
Berton, O., Guigoni, C., Li, Q., Bioulac, B. H., Aubert, I., Gross, C. E., Dileone, R. J., Nestler, E. J., and Bezard, E. (2009). Striatal overexpression of DeltaJunD resets L-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol. Psychiatry 66, 554–561.
Bertran-Gonzalez, J., Bosch, C., Maroteaux, M., Matamales, M., Herve, D., Valjent, E., and Girault, J. A. (2008). Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J. Neurosci. 28, 5671–5685.
Bertran-Gonzalez, J., Hakansson, K., Borgkvist, A., Irinopoulou, T., Brami-Cherrier, K., Usiello, A., Greengard, P., Herve, D., Girault, J. A., Valjent, E., and Fisone, G. (2009). Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology 34, 1710–1720.
Birkmayer, W., and Hornykiewicz, O. (1998). The effect of l-3,4-dihydroxyphenylalanine (=DOPA) on akinesia in parkinsonism. Parkinsonism Relat. Disord. 4, 59–60.
Blank, T., Nijholt, I., Teichert, U., Kugler, H., Behrsing, H., Fienberg, A., Greengard, P., and Spiess, J. (1997). The phosphoprotein DARPP-32 mediates cAMP-dependent potentiation of striatal N-methyl-D-aspartate responses. Proc. Natl. Acad. Sci. U.S.A. 94, 14859–14864.
Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., and Deisseroth, K. (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268.
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211.
Cadd, G., and McKnight, G. S. (1989). Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron 3, 71–79.
Cao, X., Yasuda, T., Uthayathas, S., Watts, R. L., Mouradian, M. M., Mochizuki, H., and Papa, S. M. (2010). Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J. Neurosci. 30, 7335–7343.
Carta, A. R., Tronci, E., Pinna, A., and Morelli, M. (2005). Different responsiveness of striatonigral and striatopallidal neurons to L-DOPA after a subchronic intermittent L-DOPA treatment. Eur. J. Neurosci. 21, 1196–1204.
Cenci, A. M., Whishaw, I. Q., and Schallert, T. (2002). Animal models of neurological deficits: how relevant is the rat? Nat. Rev. Neurosci. 3, 574–579.
Chen, J.-F., Moratalla, R., Impagnatiello, F., Grandy, D. K., Cuellar, B., Rubinstein, M., Beilstein, M. A., Hackett, E., Fink, S. J., Low, M. J., Ongini, E., and Schwarzschild, M. A. (2001). The role of the D2 dopamine receptor (D2R) in A2A adenosine receptor (A2A R)-mediated behavioral and cellular responses as revealed by A2A and D2 receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 98, 1970–1975.
Cotzias, G. C., Van Woert, M. H., and Schiffer, L. M. (1967). Aromatic amino acids and modification of parkinsonism. N. Engl. J. Med. 276, 374–379.
Creese, I., Burt, D. R., and Snyder, S. H. (1976). Dopamine receptor binding predicts clinical and pharmacological potencies of anti-schizophrenic drugs. Science 192, 481–483.
Darmopil, S., Martin, A. B., De Diego, I. R., Ares, S., and Moratalla, R. (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 66, 603–613.
DeLong, M. R. (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 13, 281–285.
Doucet, J. P., Nakabeppu, Y., Bedard, P. J., Hope, B. T., Nestler, E. J., Jasmin, B. J., Chen, J. S., Iadarola, M. J., St-Jean, M., Wigle, N., Blanchet, P., Grondin, R., and Robertson, G. S. (1996). Chronic alterations in dopaminergic neurotransmission produce a persistent elevation of deltaFosB-like protein(s) in both the rodent and primate striatum. Eur. J. Neurosci. 8, 365–381.
Dragunow, M., Robertson, G. S., Faull, R. L., Robertson, H. A., and Jansen, K. (1990). D2 dopamine receptor antagonists induce fos and related proteins in rat striatal neurons. Neuroscience 37, 287–294.
Durieux, P. F., Bearzatto, B., Guiducci, S., Buch, T., Waisman, A., Zoli, M., Schiffmann, S. N., and De Kerchove d’Exaerde, A. (2009). D2R striatopallidal neurons inhibit both locomotor and drug reward processes. Nat. Neurosci. 12, 393–395.
Fasano, S., Bezard, E., D’Antoni, A., Francardo, V., Indrigo, M., Qin, L., Dovero, S., Cerovic, M., Cenci, M. A., and Brambilla, R. (2010). Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc. Natl. Acad. Sci. U.S.A. 107, 21824–21829.
Fienberg, A. A., Hiroi, N., Mermelstein, P. G., Song, W., Snyder, G. L., Nishi, A., Cheramy, A., O’Callaghan, J. P., Miller, D. B., Cole, D. G., Corbett, R., Haile, C. N., Cooper, D. C., Onn, S. P., Grace, A. A., Ouimet, C. C., White, F. J., Hyman, S. E., Surmeier, D. J., Girault, J., Nestler, E. J., and Greengard, P. (1998). DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science 281, 838–842.
Fink, J. S., Weaver, D. R., Rivkees, S. A., Peterfreund, R. A., Pollack, A. E., Adler, E. M., and Reppert, S. M. (1992). Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptor in rat striatum. Mol. Brain Res. 14, 186–195.
Fisone, G., Hakansson, K., Borgkvist, A., and Santini, E. (2007). Signaling in the basal ganglia: postsynaptic and presynaptic mechanisms. Physiol. Behav. 92, 8–14.
Galarraga, E., Hernández-López, S., Reyes, A., Barral, J., and Bargas, J. (1997). Dopamine facilitates striatal EPSPs through an L-type Ca2+ conductance. Neuroreport 8, 2183–2186.
Gao, T., Yatani, A., Dell’Acqua, M. L., Sako, H., Green, S. A., Dascal, N., Scott, J. D., and Hosey, M. M. (1997). cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19, 185–196.
Gerfen, C. R., Engber, T. M., Mahan, L. C., Susel, Z., Chase, T. N., Monsma, F. J. Jr., and Sibley, D. R. (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432.
Gerfen, C. R., Miyachi, S., Paletzki, R., and Brown, P. (2002). D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J. Neurosci. 22, 5042–5054.
Gerfen, C. R., Paletzki, R., and Worley, P. (2008). Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J. Neurosci. 28, 7113–7120.
Glatt, C. E., and Snyder, S. H. (1993). Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature 361, 536–538.
Goetz, C. G., Poewe, W., Rascol, O., and Sampaio, C. (2005). Evidence-based medical review update: pharmacological and surgical treatments of Parkinson’s disease: 2001 to 2004. Mov. Disord. 20, 523–539.
Gong, S., Doughty, M., Harbaugh, C. R., Cummins, A., Hatten, M. E., Heintz, N., and Gerfen, C. R. (2007). Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J. Neurosci. 27, 9817–9823.
Gong, S., Zheng, C., Doughty, M. L., Losos, K., Didkovsky, N., Schambra, U. B., Nowak, N. J., Joyner, A., Leblanc, G., Hatten, M. E., and Heintz, N. (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425, 917–925.
Greif, G. J., Lin, Y. J., Liu, J. C., and Freedman, J. E. (1995). Dopamine-modulated potassium channels on rat striatal neurons: specific activation and cellular expression. J. Neurosci. 15, 4533–4544.
Guigoni, C., Aubert, I., Li, Q., Gurevich, V. V., Benovic, J. L., Ferry, S., Mach, U., Stark, H., Leriche, L., Hakansson, K., Bioulac, B. H., Gross, C. E., Sokoloff, P., Fisone, G., Gurevich, E. V., Bloch, B., and Bezard, E. (2005). Pathogenesis of levodopa-induced dyskinesia: focus on D1 and D3 dopamine receptors. Parkinsonism Relat. Disord. 11(Suppl. 1), S25–S29.
Håkansson, K., Galdi, S., Hendrick, J., Snyder, G., Greengard, P., and Fisone, G. (2006). Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J. Neurochem. 96, 482–488.
Håkansson, K., Pozzi, L., Usiello, A., Haycock, J., Borrelli, E., and Fisone, G. (2004). Regulation of striatal tyrosine hydroxylase phosphorylation by acute and chronic haloperidol. Eur. J. Neurosci. 20, 1108–1112.
Heintz, N. (2001). BAC to the future: the use of bac transgenic mice for neuroscience research. Nat. Rev. Neurosci. 2, 861–870.
Hemmings, H. C. Jr., Greengard, P., Tung, H. Y., and Cohen, P. (1984). DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature 310, 503–505.
Hernandez-Lopez, S., Tkatch, T., Perez-Garci, E., Galarraga, E., Bargas, J., Hamm, H., and Surmeier, D. J. (2000). D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J. Neurosci. 20, 8987–8995.
Herve, D., Le Moine, C., Corvol, J. C., Belluscio, L., Ledent, C., Fienberg, A. A., Jaber, M., Studler, J. M., and Girault, J. A. (2001). Galpha(olf) levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J. Neurosci. 21, 4390–4399.
Hope, B. T., Nye, H. E., Kelz, M. B., Self, D. W., Iadarola, M. J., Nakabeppu, Y., Duman, R. S., and Nestler, E. J. (1994). Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron 13, 1235–1244.
Hopf, F. W., Cascini, M. G., Gordon, A. S., Diamond, I., and Bonci, A. (2003). Cooperative activation of dopamine D1 and D2 receptors increases spike firing of nucleus accumbens neurons via G-protein betagamma subunits. J. Neurosci. 23, 5079–5087.
Hornykiewicz, O. (1963). Die topische Lokalisation und das Verhalten von Noradrenalin und Dopamin (3-Hydroxytyramin) in der Substantia nigra des normalen und Parkinsonkranken Menschen. Wien. Klin. Wochenschr. 56, 426–427.
Kitai, S. T., and Surmeier, D. J. (1993). Cholinergic and dopaminergic modulation of potassium conductances in neostriatal neurons. Adv. Neurol. 60, 40–52.
Konradi, C., and Heckers, S. (1995). Haloperidol-induced Fos expression in striatum is dependent upon transcription factor cyclic AMP response element binding protein. Neuroscience 65, 1051–1061.
Kravitz, A. V., Freeze, B. S., Parker, P. R., Kay, K., Thwin, M. T., Deisseroth, K., and Kreitzer, A. C. (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626.
Kull, B., Svenningsson, P., and Fredholm, B. B. (2000). Adenosine A(2A) receptors are colocalized with and activate g(olf) in rat striatum. Mol. Pharmacol. 58, 771–777.
Kuzhikandathil, E. V., Yu, W., and Oxford, G. S. (1998). Human dopamine D3 and D2L receptors couple to inward rectifier potassium channels in mammalian cell lines. Mol. Cell. Neurosci. 12, 390–402.
Lebel, M., Chagniel, L., Bureau, G., and Cyr, M. (2010). Striatal inhibition of PKA prevents levodopa-induced behavioural and molecular changes in the hemiparkinsonian rat. Neurobiol. Dis. 38, 59–67.
Li, J., Guo, Y., Schroeder, F. A., Youngs, R. M., Schmidt, T. W., Ferris, C., Konradi, C., and Akbarian, S. (2004). Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J. Neurochem. 90, 1117–1131.
Liu, J. C., Defazio, R. A., Espinosa-Jeffrey, A., Cepeda, C., De Vellis, J., and Levine, M. S. (2004). Calcium modulates dopamine potentiation of N-methyl-D-aspartate responses: electrophysiological and imaging evidence. J. Neurosci. Res. 76, 315–322.
MacGibbon, G. A., Lawlor, P. A., Bravo, R., and Dragunow, M. (1994). Clozapine and haloperidol produce a differential pattern of immediate early gene expression in rat caudate-putamen, nucleus accumbens, lateral septum and islands of Calleja. Brain Res. Mol. Brain Res. 23, 21–32.
MacGibbon, G. A., Lawlor, P. A., Hughes, P., Young, D., and Dragunow, M. (1995). Differential expression of inducible transcription factors in basal ganglia neurons. Brain Res. Mol. Brain Res. 34, 294–302.
Mangiavacchi, S., and Wolf, M. E. (2004). D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J. Neurochem. 88, 1261–1271.
Mattingly, R. R. (1999). Phosphorylation of serine 916 of Ras-GRF1 contributes to the activation of exchange factor activity by muscarinic receptors. J. Biol. Chem. 274, 37379–37384.
Miller, J. C. (1990). Induction of c-fos mRNA expression in rat striatum by neuroleptic drugs. J. Neurochem. 54, 1453–1455.
Miyamoto, S., Duncan, G. E., Marx, C. E., and Lieberman, J. A. (2005). Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol. Psychiatry 10, 79–104.
Mons, N., and Cooper, D. M. (1994). Selective expression of one Ca(2+)-inhibitable adenylyl cyclase in dopaminergically innervated rat brain regions. Brain Res. Mol. Brain Res. 22, 236–244.
Nestler, E. J., Barrot, M., and Self, D. W. (2001). DeltaFosB: a sustained molecular switch for addiction. Proc. Natl. Acad. Sci. U.S.A. 98, 11042–11046.
Nishi, A., Snyder, G. L., and Greengard, P. (1997). Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J. Neurosci. 17, 8147–8155.
Nowak, S. J., and Corces, V. G. (2004). Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 20, 214–220.
Obeso, J. A., Olanow, C. W., and Nutt, J. G. (2000). Levodopa motor complications in Parkinson’s disease. Trends Neurosci. 23, S2–S7.
Parr-Brownlie, L. C., and Hyland, B. I. (2005). Bradykinesia induced by dopamine D2 receptor blockade is associated with reduced motor cortex activity. J. Neurosci. 25, 5700–5709.
Pavon, N., Martin, A. B., Mendialdua, A., and Moratalla, R. (2006). ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 59, 64–74.
Picconi, B., Centonze, D., Hakansson, K., Bernardi, G., Greengard, P., Fisone, G., Cenci, M. A., and Calabresi, P. (2003). Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 6, 501–506.
Pozzi, L., Hakansson, K., Usiello, A., Borgkvist, A., Lindskog, M., Greengard, P., and Fisone, G. (2003). Opposite regulation by typical and atypical anti-psychotics of ERK1/2, CREB and Elk-1 phosphorylation in mouse dorsal striatum. J. Neurochem. 86, 451–459.
Robertson, G. S., and Fibiger, H. C. (1992). Neuroleptics increase c-fos expression in the forebrain: contrasting effects of haloperidol and clozapine. Neuroscience 46, 315–328.
Robertson, G. S., Vincent, S. R., and Fibiger, H. C. (1992). D1 and D2 dopamine receptors differentially regulate c-fos expression in striatonigral and striatopallidal neurons. Neuroscience 49, 285–296.
Robinson, S. W., and Caron, M. G. (1997). Selective inhibition of adenylyl cyclase type V by the dopamine D3 receptor. Mol. Pharmacol. 52, 508–514.
Roche, K. W., O’Brien, R. J., Mammen, A. L., Bernhardt, J., and Huganir, R. L. (1996). Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16, 1179–1188.
Santini, E., Alcacer, C., Cacciatore, S., Heiman, M., Herve, D., Greengard, P., Girault, J. A., Valjent, E., and Fisone, G. (2009). L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 108, 621–633.
Santini, E., Sgambato-Faure, V., Li, Q., Savasta, M., Dovero, S., Fisone, G., and Bezard, E. (2010). Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS ONE 5, e12322.
Santini, E., Valjent, E., and Fisone, G. (2008). Parkinson’s disease: levodopa-induced dyskinesia and signal transduction. FEBS J. 275, 1392–1399.
Santini, E., Valjent, E., Usiello, A., Carta, M., Borgkvist, A., Girault, J. A., Herve, D., Greengard, P., and Fisone, G. (2007). Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 27, 6995–7005.
Schiffmann, S. N., Jacobs, O., and Vanderhaegen, J. J. (1991). Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J. Neurochem. 57, 1062–1067.
Schuster, S., Nadjar, A., Guo, J. T., Li, Q., Ittrich, C., Hengerer, B., and Bezard, E. (2008). The 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor lovastatin reduces severity of L-DOPA-induced abnormal involuntary movements in experimental Parkinson’s disease. J. Neurosci. 28, 4311–4316.
Seeman, P., Lee, T., Chau-Wong, M., and Wong, K. (1976). Anti-psychotic drug doses and neuroleptic/dopamine receptors. Nature 261, 717–719.
Snyder, G. L., Allen, P. B., Fienberg, A. A., Valle, C. G., Huganir, R. L., Nairn, A. C., and Greengard, P. (2000). Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J. Neurosci. 20, 4480–4488.
Snyder, G. L., Fienberg, A. A., Huganir, R. L., and Greengard, P. (1998). A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J. Neurosci. 18, 10297–10303.
Stipanovich, A., Valjent, E., Matamales, M., Nishi, A., Ahn, J. H., Maroteaux, M., Bertran-Gonzalez, J., Brami-Cherrier, K., Enslen, H., Corbille, A. G., Filhol, O., Nairn, A. C., Greengard, P., Herve, D., and Girault, J. A. (2008). A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature 453, 879–884.
Stoof, J. C., and Kebabian, J. W. (1981). Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature 294, 366–368.
Surmeier, D. J., Bargas, J., Hemmings, H. C. Jr., Nairn, A. C., and Greengard, P. (1995). Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14, 385–397.
Surmeier, D. J., Ding, J., Day, M., Wang, Z., and Shen, W. (2007). D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235.
Svenningsson, P., Lindskog, M., Ledent, C., Parmentier, M., Greengard, P., Fredholm, B. B., and Fisone, G. (2000). Regulation of the phosphorylation of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa in vivo by dopamine D1, dopamine D2, and adenosine A2A receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 1856–1860.
Svenningsson, P., Lindskog, M., Rognoni, F., Fredholm, B. B., Greengard, P., and Fisone, G. (1998). Activation of adenosine A2A and dopamine D1 receptors stimulates cyclic AMP-dependent phosphorylation of DARPP-32 in distinct populations of striatal projection neurons. Neuroscience 84, 223–228.
Tekumalla, P. K., Calon, F., Rahman, Z., Birdi, S., Rajput, A. H., Hornykiewicz, O., Di Paolo, T., Bedard, P. J., and Nestler, E. J. (2001). Elevated levels of DeltaFosB and RGS9 in striatum in Parkinson’s disease. Biol. Psychiatry 50, 813–816.
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183.
Thomson, S., Clayton, A. L., Hazzalin, C. A., Rose, S., Barratt, M. J., and Mahadevan, L. C. (1999). The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 18, 4779–4793.
Valjent, E., Bertran-Gonzalez, J., Herve, D., Fisone, G., and Girault, J. A. (2009). Looking BAC at striatal signaling: cell-specific analysis in new transgenic mice. Trends Neurosci. 32, 538–547.
Valjent, E., Corvol, J. C., Pages, C., Besson, M. J., Maldonado, R., and Caboche, J. (2000). Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J. Neurosci. 20, 8701–8709.
Valjent, E., Pascoli, V., Svenningsson, P., Paul, S., Enslen, H., Corvol, J. C., Stipanovich, A., Caboche, J., Lombroso, P. J., Nairn, A. C., Greengard, P., Herve, D., and Girault, J. A. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496.
Westin, J. E., Vercammen, L., Strome, E. M., Konradi, C., and Cenci, M. A. (2007). Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol. Psychiatry 62, 800–810.
Wickens, J. R., and Wilson, C. J. (1998). Regulation of action-potential firing in spiny neurons of the rat neostriatum in vivo. J. Neurophysiol. 79, 2358–2364.
Wickman, K. D., Iniguez-Lluhl, J. A., Davenport, P. A., Taussig, R., Krapivinsky, G. B., Linder, M. E., Gilman, A. G., and Clapham, D. E. (1994). Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature 368, 255–257.
Xie, G. Q., Wang, S. J., Li, J., Cui, S. Z., Zhou, R., Chen, L., and Yuan, X. R. (2009). Ethanol attenuates the HFS-induced, ERK-mediated LTP in a dose-dependent manner in rat striatum. Alcohol. Clin. Exp. Res. 33, 121–128.
Keywords: DARPP-32, dopamine D1 receptor, dopamine D2 receptor, ERK, L-DOPA, haloperidol, medium spiny neurons
Citation: Bonito-Oliva A, Feyder M and Fisone G (2011) Deciphering the actions of antiparkinsonian and antipsychotic drugs on cAMP/DARPP-32 signaling. Front. Neuroanat. 5:38. doi: 10.3389/fnana.2011.00038
Received: 11 May 2011;
Accepted: 27 June 2011;
Published online: 12 July 2011.
Edited by:
Emmanuel Valjent, Université Montpellier 1 and 2, FranceReviewed by:
Chip Gerfen, National Institutes of Health, USAMichel Cyr, Neuroscience Research Group, Canada
Copyright: © 2011 Bonito-Oliva, Feyder and Fisone. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Gilberto Fisone, Department of Neuroscience, Karolinska Institutet, Retzius väg 8, 171 77 Stockholm, Sweden. e-mail:Z2lsYmVydG8uZmlzb25lQGtpLnNl