
 Adan Hernandez1
Adan Hernandez1

- 1 Department of Psychiatry, University of Texas Southwestern Medical Center, Dallas, TX, USA
- 2 Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, TX, USA
Drug addiction is a severe neuropsychiatric disorder characterized by loss of control over motivated behavior. The need for effective treatments mandates a greater understanding of the causes and identification of new therapeutic targets for drug development. Drugs of abuse subjugate normal reward-related behavior to uncontrollable drug-seeking and -taking. Contributions of brain reward circuitry are being mapped with increasing precision. The role of synaptic plasticity in addiction and underlying molecular mechanisms contributing to the formation of the addicted state are being delineated. Thus we may now consider the role of striatal signal transduction in addiction from a more integrative neurobiological perspective. Drugs of abuse alter dopaminergic and glutamatergic neurotransmission in medium spiny neurons of the striatum. Dopamine receptors important for reward serve as principle targets of drugs abuse, which interact with glutamate receptor signaling critical for reward learning. Complex networks of intracellular signal transduction mechanisms underlying these receptors are strongly stimulated by addictive drugs. Through these mechanisms, repeated drug exposure alters functional and structural neuroplasticity, resulting in transition to the addicted biological state and behavioral outcomes that typify addiction. Ca2+ and cAMP represent key second messengers that initiate signaling cascades, which regulate synaptic strength and neuronal excitability. Protein phosphorylation and dephosphorylation are fundamental mechanisms underlying synaptic plasticity that are dysregulated by drugs of abuse. Increased understanding of the regulatory mechanisms by which protein kinases and phosphatases exert their effects during normal reward learning and the addiction process may lead to novel targets and pharmacotherapeutics with increased efficacy in promoting abstinence and decreased side effects, such as interference with natural reward, for drug addiction.
Introduction
Drug addiction is a behavioral disorder influenced by a complex interaction of genetic, environmental, and developmental factors (Crabbe, 2002; Volkow, 2005). Addictive substances include illicit drugs like the psychostimulant cocaine and the opiate heroin, as well as legally available tobacco/nicotine and alcohol (ethanol; EtOH). Prescription drugs, such as opioids, stimulants, or depressants are also increasingly being used for non-medical reasons (Compton and Volkow, 2006). Drug addiction affects not only the individual, but also society as a whole evidenced by severed interpersonal relations, failure in school or job performance, outbreaks of domestic violence and child abuse, and even the degradation of entire communities. There are few pharmacotherapeutics that exist to treat drug addiction and risk of relapse remains substantial despite sincere efforts to remain abstinent. Therefore, it is important to understand the neurobiological basis of behaviors characteristic of addiction in order to identify novel mechanisms that lead to better treatments.
Chronic drug addiction is associated with the development of pharmacological tolerance, sensitization, and dependence, and the impact of these processes is manifested in drug intake, craving, and relapse. The transition from impulsive to compulsive drug use corresponds to enhanced euphoric drug experiences, and an overshadowing of adverse consequences (Robbins et al., 2008; Torregrossa et al., 2011). These adaptations are seen clinically as reduced responding to natural rewards and a loss of control over drug intake in response to drug-associated cues (Kalivas and Volkow, 2005). Motivation to alleviate drug withdrawal-induced negative affect (i.e., stress, anxiety, depression, dysphoria) may also contribute to relapse via their negative reinforcing properties (Koob and Le Moal, 2001; Koob and Volkow, 2010).
Drugs of abuse target neural processes that normally mediate adaptive reward-based learning, but with overriding and repeated stimulation that appears to seize these neural processes (Hyman et al., 2006; Kalivas and O’brien, 2008; Milton and Everitt, 2010; Torregrossa et al., 2011). Once brain reward circuitry is activated by convergent neuronal inputs, synapses undergo functional, and structural plasticity important for learning associated with the conditioned stimulus effects of drugs of abuse (Figure 1). What drives a drug addict to uncontrollable self-destructive behavior? Pioneering behavioral psychologists emphasized the formation of stimulus–response associations as important mediators of learned behaviors. Drugs of abuse can serve as powerful rewarding, discriminative, and reinforcing stimuli. Motoric actions leading to obtaining drug are processed in tandem with environmental cues that also come to drive drug-seeking and -taking behavior. Triggers of relapse during abstinence include re-exposure to the drug itself, environmental drug-associated cues, stress, or some combination of these factors (Schmidt et al., 2005; Milton and Everitt, 2010; Weiss, 2010).
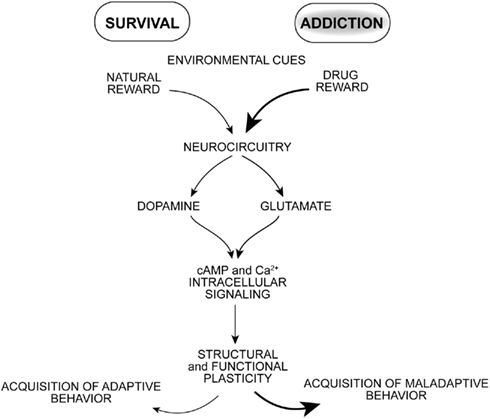
Figure 1. The pharmacological effects of drugs of abuse potently stimulate neural pathways normally mediating reward-based learning resulting in a powerful learning process and enduring changes in motivated behavior.
The striatum is a subcortical brain structure that is major target for drugs of abuse. It is comprised of ventral and dorsal subregions. The ventral striatum consists of the nucleus accumbens (NAc), olfactory tubercle, and islands of Calleja. The NAc includes shell and core subregions and plays an integral role in three dissociable aspects of psychological reward associated with addiction: “liking” (hedonic impact), “wanting” (incentive salience), and learning (predictive associations and cognition; Berridge et al., 2009). While both natural and drug rewards increase extracellular dopamine in the NAc, dopamine activity in the NAc shell does not adapt or habituate to repeated drug exposure by decreasing dopamine activity as it does with natural rewards (Wise et al., 1995a,b; Hemby et al., 1997) and repeated drug exposure induces sensitization of dopamine transmission in the NAc core (Cadoni and Di Chiara, 1999, 2000; Cadoni et al., 2000). Dopamine in the NAc shell is activated in response to primary, unconditioned, and unanticipated stimuli, for example, to an appetitive unfamiliar taste (sweet chocolate) infused intra-orally (Bassareo et al., 2002). Interestingly, food-conditioned stimuli increase dopamine in the NAc shell whereas drug-conditioned stimuli increase dopamine in the NAc core (Bassareo et al., 2011). Repeated and sustained levels of drug-induced dopamine stimulation in the shell could pathologically strengthen the acquisition (learning) of incentive stimuli predictive of drug availability through Pavlovian conditioning, while sensitization of dopamine transmission in the core might be more directly related to the initiation of behavioral activity to obtain drug reward (instrumental action). Thus, incentive learning of drug-associated cues through Pavlovian conditioning is critical for the reinforcing effects of drugs that increase the probability of future occurrences of drug-seeking and -taking behaviors (Bower and Grusec, 1964) and has been denoted as Pavlovian instrumental transfer (PIT; Ettenberg, 1990).
While reward processed in the NAc is critical for acquisition of goal-directed behaviors, these behaviors repeated over time develop into more automatic repertoires characteristic of stimulus–response learning, largely understood to involve dorsal striatal circuits (Mitchell et al., 1985; Mitchell and Hall, 1988). The dorsal striatum includes the caudate and putamen in primates, analogous to the dorsomedial and dorsolateral striatum in rodents, respectively. The transition to the addicted state appears to correspond to a recruitment of neurocircuitry from ventral to dorsal striatum (Gerdeman et al., 2003; Porrino et al., 2007; Pierce and Vanderschuren, 2010). The dorsal striatum is critical for formation of rigid or inflexible behaviors characterizing drug-seeking and -taking habits (Gerdeman et al., 2003; Pierce and Vanderschuren, 2010). This ventral to dorsal progression does not, however, negate involvement of the NAc over time. In fact, a functionally linked progression of activity from ventral to dorsal striatum in response to chronic drug exposure may invoke widespread neuroplasticity underlying cognitive and behavioral perturbations associated with drug addiction (Koob and Volkow, 2010).
Dorsal and ventral striatum are composed of medium spiny neurons (MSNs) that are predominantly γ-aminobutyric acid (GABA) projection neurons (Tepper et al., 2007). These MSNs receive dopaminergic input from afferent projections of the substantia nigra and ventral tegmental areas of the midbrain. MSNs also receive glutamatergic input from afferent projections of the prefrontal cortex, predominantly in layer 5 of the cortex.
Effects of Drugs of Abuse on Signal Transduction
Addictive substances can be generally classified into drugs that target monoamine transporters, G protein-coupled receptors (GPCRs), or ion channels and ionotropic receptors (Luscher and Ungless, 2006). Drugs of abuse mimic and surpass endogenous dopamine activation subsequent to phasic dopamine cell firing coding for salience and reward (Schultz et al., 2000). Human brain imaging studies show that drug-induced increases in striatal dopamine are associated with subjective reward (Volkow et al., 1996; Drevets et al., 2001).
All dopamine receptors are GPCRs (Neve et al., 2004). Dopamine receptors fall into two major classes: D1-like receptors (D1 and D5) and D2-like receptors (D2, D3, and D4). They are primarily distinguished via their opposing actions on adenylate cyclase (AC). D1-like receptors stimulate AC through coupling to stimulatory (Gαs) G proteins that increase cyclic adenosine 3′,5′-monophosphate (cAMP) levels, whereas D2-like receptors inhibit AC through coupling to inhibitory (Gαi) G proteins that decrease cAMP levels. Both D1 and D2 receptors contribute to aspects addiction, despite the fact that their opposing G protein-coupled functions might suggest that the two classes would interact antagonistically. One level at which this paradox resolves is the neuroanatomical circuitry. Specifically, D1 mediated effects on midbrain GABA release are direct, while D2 mediated effects are indirect. Beyond this discrepancy, interactions between D1 and D2 receptors and their circuitry during reward learning, motivated behavior, and addiction are quite complex.
While the mechanisms for the synergism between D1 and D2 receptors in mediating behavioral effects are not yet completely understood, the roles of D1- and D2-like receptors in reward can be dissociated. Either selective D1- or D2-like receptor agonists maintain intravenous drug self-administration in rats (Yokel and Wise, 1978; Self and Stein, 1992), monkeys (Woolverton et al., 1984; Weed et al., 1993), and mice (Caine et al., 2007). However, self-administration of D2-like receptor agonists requires an extensive history of self-administration with non-selective dopamine agonists like cocaine. Converse to systemic studies, intracranial self-administration studies show a requirement for concomitant D1- and D2-like receptor activation in the NAc shell but not the core subregion (Ikemoto et al., 1997). Interestingly, infusion of cocaine into the more ventral olfactory tubercle regions is even more efficacious in maintaining self-administration than in the NAc shell, and this effect is blocked by co-administration of D1- or D2-like antagonists (Ikemoto, 2003).
In contrast to voluntary drug intake modeled in self-administration procedures, compulsive drug-seeking modeled in extinction/reinstatement procedures show a predominant role for D2- but not D1-like receptors. Extinction/reinstatement procedures typically measure the level of effort an animal with prior history of drug self-administration will exert to obtain drug, as an index of craving that would precipitate relapse in humans (Wise and Rompre, 1989; Berridge and Robinson, 1998). D2-like receptor activation triggers cocaine-seeking during or after extinction of behavioral responses (Koeltzow and Vezina, 2005; Edwards et al., 2007). Conversely, D1-like receptor activation essentially has no effect, even at doses that stimulate locomotor activity comparable to D2-like receptor activation (Dias et al., 2004; Koeltzow and Vezina, 2005). However, pretreatment with a D1-like agonist blocks the ability of cocaine priming or cocaine-associated cues to induce reinstatement (Alleweireldt et al., 2003; Edwards et al., 2007), whereas, D2-like agonist pretreatment facilitates cocaine-primed reinstatement (Self et al., 1996). Thus, D2-like receptors are critical for relapse to cocaine-seeking induced by environmental cues such as cocaine-related stimuli or stress that activates the mesolimbic dopamine system (Phillips et al., 2003) and D1-like receptor tone may provide an inhibitory mechanism over cocaine-seeking via saturation of primary reward processes. However, differences between systemic and intracranial studies of D1- versus D2-like effects on drug-seeking also exist. In contrast to systemic pretreatment of a D1-like agonist, direct infusion of a D1-like agonist intra-NAc triggers cocaine-seeking (Bachtell et al., 2005; Schmidt et al., 2006). Similarly, intra-NAc infusions of D2-like agonists reinstate cocaine-seeking (Bachtell et al., 2005; Schmidt et al., 2006). Thus, dopamine effects critical for reward-related behavior can vary according to receptor subtype and systemic versus direct infusion into the NAc and either D1- or D2-like receptors have the ability to modulate the effects of the other.
Modulation of GPCR Signaling
The effects of GPCRs is modulated via transmembrane signaling pathways consisting of metabotropic cell surface receptors, G proteins, and effectors (Engelhardt and Rochais, 2007; Figure 2). In addition to G protein coupling, GPCR activation triggers a negative feedback mechanism known as desensitization (Hausdorff et al., 1990). Desensitization may be mediated via agonist-induced receptor phosphorylation by a GPCR kinase (GRK; Krupnick and Benovic, 1998). GRK-dependent GPCR phosphorylation increases receptor affinity for cytosolic arrestin protein family members. For example, the resulting phosphorylated receptor/arrestin complex prevents further coupling of the D1 receptor to its G protein, progressively reducing second messenger synthesis of cAMP. Resensitization is triggered by internalization of an uncoupled receptor to endosomal compartments, allowing protein phosphatase-dependent dephosphorylation of the receptor and recycling back to the cell surface, or degradation. Modulation of GRKs occurs via mechanisms involving Ca2+-binding proteins, phosphorylation, targeting proteins, or mechanisms governing their localizations and expressions (Penela et al., 2003). In addition to GRKs that regulate GPCR activity, several families of accessory proteins have also been discovered that control G protein signaling (Blumer and Lanier, 2003). The regulators of G protein signaling (RGS) and activators of G protein signaling (AGS) G protein modulators both are implicated in addiction.
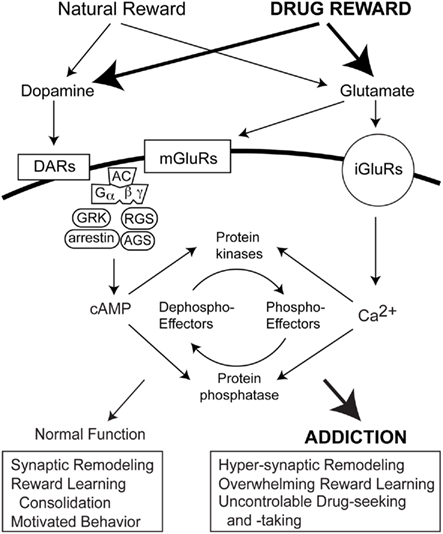
Figure 2. Signal transduction mediating reward learning and drug addiction. Dopamine and glutamate receptors are activated during rewarding experiences and exposure to addictive drugs. Both situations invoke heterotrimeric G protein-coupled receptors (GPCRs) and ionotropic glutamate receptors. In addition to G protein coupling, GRK, arrestin, RGS and AGS proteins also regulate GPCR activity to influence intracellular cAMP levels. Ionotropic receptor activation by glutamate increase intracellular Ca2+ levels. cAMP and Ca2+, in turn, affect protein kinase and phosphatase activities and the phosphorylation states of downstream effectors that modulate synaptic remodeling cognition and motivated behaviors.
RGS proteins are a large family that function as GTPase-accelerating proteins (GAP). GAP action at GTP-bound Gαi/o- or Gαq-coupled receptors increases inactivation of the Gα–GDP state and expedites recombination of the Gα and βγ signaling molecules. This allows RGS proteins to restrict G protein signal strength at steady state and negatively modulate G protein signaling, controlling potency, and efficacy of agonist effects (Garzon et al., 2008; Hooks et al., 2008; Traynor, 2010). Some RGS family members are highly enriched in striatal subregions. RGS2 is expressed in caudate putamen (Grafstein-Dunn et al., 2001; Taymans et al., 2002). RGS4 is expressed in the caudate putamen and NAc (Gold et al., 1997; Nomoto et al., 1997). The long form of RGS9 (RGS9-2) is expressed predominantly in caudate putamen, NAc, islands of Calleja, and olfactory tubercle (Thomas et al., 1998; Rahman et al., 1999). Based on striatal regional distributions, RGS2, RGS4, and especially RGS9-2 have potential in negatively regulating the effects of drugs of abuse. Correspondingly, administration of various drugs of abuse induces an up-regulation of RGS family members in striatum. RGS2, 4, and 9 have all been implicated in addiction (Burchett et al., 1998; Zachariou et al., 2003; Psifogeorgou et al., 2007; Hooks et al., 2008; Traynor, 2010). By acting at a relatively early stage of the signaling pathway, RGS promoters can affect multiple branches of D1- as well as D2-like receptor signaling to AC and its effectors and RGS proteins can function to dampen dopamine signaling in the striatum in response to various drugs of abuse.
AGS proteins constitute another family of negative G protein modulators. The AGS family contains three functionally distinct members (Blumer et al., 2005). AGS3 is the only one currently associated with addictive drug-induced neuroadaptations (Kelley and Schiltz, 2004; Bowers, 2010). AGS3 binds to and stabilizes the inactive GDP–bound Gαi conformation, preventing GDP release and, inhibiting Gαi-mediated signaling (Natochin et al., 2000). AGS3 expression increases in NAc core during protracted withdrawal (three or more weeks) from either repeated experimenter-administered injections of cocaine or 3 weeks of extinction training in rats that had self-administered cocaine (Bowers et al., 2004). Furthermore, cocaine-induced reinstatement of drug-seeking behavior was attenuated by AGS3 knockdown, but then restored by antisense washout and return of AGS3 protein. These studies suggest that AGS3 up-regulation requires protracted withdrawal from chronic drug exposure and that decreased Gαi signaling may support addiction pathogenesis.
In addition to dopamine receptors (Surmeier et al., 2007), group II metabotropic glutamate (mGlu) receptors (mGlu2/3) also modulate striatal MSNs function (Richards et al., 2005), and through these receptors cortical glutamatergic input into striatum may counter balance and potentially reverse drug-induced neuroadaptations. These receptors are Gi-coupled and are also subject to negative regulation via AGS3. Gi coupling to mGlu2/3 receptors can modulate neurotransmitter release via activation of presynaptic K+ channels, inhibition of presynaptic Ca2+ channels, or direct interference with vesicular release. Dopamine release in NAc is regulated by mGlu2/3 receptors (Karasawa et al., 2006; Xi et al., 2010). NAc mGlu2/3 receptor-G protein coupling is reduced after chronic cocaine (Xi et al., 2002; Bowers et al., 2004; Ghasemzadeh et al., 2009) or EtOH (Bowers et al., 2008). Furthermore it has been suggested that mGlu2/3 auto receptors may mediate enhanced glutamate release in NAc during protracted drug withdrawal in response to the drug itself or associated cues (Madayag et al., 2007; Lalumiere and Kalivas, 2008). Therefore, mGlu2/3 receptors are emerging as a therapeutic target to possibly reduce drug abuse relapse (Moussawi and Kalivas, 2010).
Ionotropic Mechanisms of Synaptic Plasticity
G protein-coupled receptors signaling via dopamine receptors and metabotropic glutamate receptors mediates important aspects of reward and reinstatement through regulation of excitability, which is dependent upon glutamatergic activation of ionotropic receptors. The two classes of ionotropic glutamate receptors are N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-propionate (AMPA) receptors and they contribute in important ways to addiction. For example, cocaine administration increases the NR1 NMDA and GluA1 AMPA (Figure 3) receptor subunit in the NAc (Lu et al., 2003; Hemby et al., 2005b; Schumann and Yaka, 2009) and NMDA and AMPA receptors are involved in the ability of addictive drugs to produce long-term plasticity in learning and memory circuits associated with cue–reward associations and habit formation.
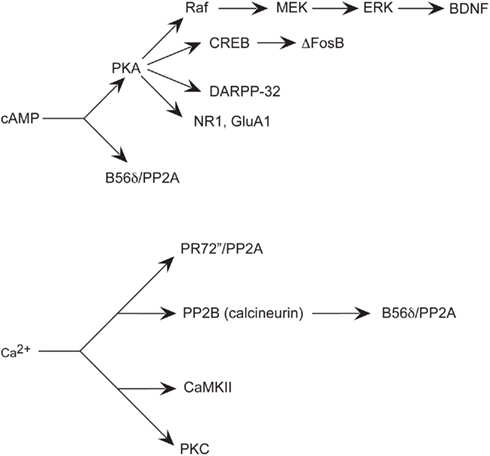
Figure 3. Simplified schematic of signaling pathways involving major effectors of cAMP and Ca2+implicated in aspects of addiction. Increased cAMP levels activate PKA, which influences multiple effectors, such as other kinases, phosphatases, transcription factors, phosphatase inhibitors, and neurotransmitter receptor subunits. Increased Ca2+ levels also affect kinases, and phosphatases.
Drugs of abuse evoke an NMDA receptor-dependent long-term potentiation (LTP) of AMPA receptor current in dopamine neurons, possibly reflecting an early memory trace in the acquisition of drug addiction (Ungless et al., 2001; Saal et al., 2003; Borgland et al., 2004; Liu et al., 2005). LTP is induced when a brief high frequency train of stimuli (a tetanus) is applied to a synaptic pathway that increases the amplitude of the excitatory postsynaptic potentials in target neurons. Long-term depression (LTD) is induced when tetanus application decreases the amplitude of the excitatory postsynaptic potentials in target neurons. Induction of LTP is associated with the formation of spines de novo and enlargement of existing spines, whereas induction of LTD is associated with contraction and retraction of spines (Nagerl et al., 2004; Okamoto et al., 2004).
Subsequent to induction of LTP, synaptic strengthening can manifest through insertion of GluA2-lacking AMPA receptors (Kauer and Malenka, 2007). The synaptic insertion of AMPA receptors may create a temporal opportunity when the acquisition of cocaine-related cues corresponds to increased synaptic plasticity (Wolf, 2010). Restricting AMPA receptor activation could reverse LTP associated with continued cocaine-seeking. AMPA receptor antagonists attenuate reinstatement of drug-seeking behavior induced by drug, cue-, or stress (Mcfarland et al., 2004; Backstrom and Hyytia, 2007; Ping et al., 2008). However, it has also been shown that degrading basal AMPA receptor function in NAc neurons is sufficient to facilitate relapse and elevating basal AMPA receptor function attenuates this behavioral effect (Bachtell et al., 2008). The role of glutamatergic mechanisms in the modulation of drug self-administration and risk of relapse is complex. Nonetheless, several non-specific glutamatergic agents have displayed potential as pharmacotherapeutics for addiction (Bowers et al., 2010).
Converse to LTP, LTD corresponds to removal of AMPA receptors from synapses (Malinow and Malenka, 2002). Persistent impairment in LTD has been associated with rigid drug-seeking behaviors resistant to modulation by environmental contingencies (Kasanetz et al., 2010). Moreover, operant cocaine self-administration attenuates LTD in both the NAc core and shell; however, LTD was abolished only in the NAc core after protracted withdrawal, suggesting long-term plasticity in the core could underlie drug-seeking behavior and relapse (Martin et al., 2006). Also, animals sensitized to repeated cocaine administration, displayed a ratio of AMPA to NMDA receptor-mediated excitatory postsynaptic currents (EPSCs) that was reduced in MSNs of the NAc shell that corresponded to decreased amplitude of miniature EPSCs and magnitude of LTD (Thomas et al., 2001). Thus aspects of both LTP and LTD in NAc subregions appear to be involved in drug-taking and -seeking behaviors.
Drug-seeking and -taking induced by exposure to cues associated with drug use imply the involvement of long-term memories, such as those induced via strong glutamatergic stimulation. At the same time, protein kinase A (PKA; see below) activation in the NAc has been shown to be necessary for reward learning in which properties of drugs become associated with environmental cues (Sutton et al., 2000; Beninger et al., 2003). These two converging observations, that glutamatergic input is elevated and necessary for drug-seeking and reward learning, and that PKA signaling invoked via activation of D1 dopamine receptors is also critical for reward associated learning, suggest that any comprehensive explanation for the biochemical mechanisms of addiction would require obligatory integration of both NMDA/AMPA receptor-dependent Ca2+ and dopamine receptor-dependent cAMP signaling cascades.
Protein Kinases, Phosphatases, and Downstream Effectors
Drugs of abuse induce dopaminergic D1 receptor activation that increases intracellular cAMP. Glutamatergic input that activates NMDA and AMPA receptors increases intracellular Ca2+ concentration. Intracellular signaling cascades involving specific kinases and phosphatases affected by cAMP and Ca2+, in turn, influence phospho- and dephospho-effectors that modulate neuroplasticity to affect behavioral outcome (Figure 2).
Cyclic AMP-Dependent Kinase (Protein Kinase A)
Under basal conditions, PKA exists as a heterotetramer composed of two regulatory and two catalytic subunits. Activation of AC through Gαs-coupled receptors causes rapid increase in intracellular cAMP. PKA is subsequently activated as cAMP binds the regulatory subunits triggering dissociation of the holoenzyme, releasing the active catalytic subunits from inhibition by the regulatory subunits (Pearce et al., 2010). There are four regulatory subunit isoforms classified as type I (RIα, RIβ) and type II (RIIα, RIIβ) and three catalytic isoforms (Cα, Cβ, Cγ). Any change in cAMP directly affects function of PKA, the most important effector for cAMP.
For example, cAMP-induced activation of PKA activates cAMP response element-binding protein (CREB; Figure 3). Induction of cAMP liberates the PKA Cα subunit that then passively diffuses into the nucleus and induces cellular gene expression by phosphorylation of CREB on Ser133 (Gonzalez and Montminy, 1989). CREB then signals transcription of genes containing a cAMP response element (CRE) in their promoter regions to regulate gene transcription (Mayr and Montminy, 2001). Both opiates and stimulants activate CREB-mediated transcription within the NAc (Cole et al., 1995; Turgeon et al., 1997; Shaw-Lutchman et al., 2002). Increased CREB in the NAc attenuates rewarding effects of cocaine (Carlezon et al., 1998) and morphine (Barrot et al., 2002) and these effects are also seen in transgenic mice with increased (Mcclung and Nestler, 2003) or decreased (Walters and Blendy, 2001) CREB. Although the CREB-regulated target genes contributing to these effects are unknown, one candidate gene in the NAc encodes preprodynorphin (Carlezon et al., 1998), the precursor gene product of the endogenous opioid peptide dynorphin, which activates the κ opioid receptor. Dynorphin is upregulated in the striatum subsequent to cocaine exposure (Cole et al., 1995) and is largely understood to contribute to the inhibition of dopamine transmission that leads to the downregulation of reward mechanisms (Shippenberg and Rea, 1997). For example, microinjection of a κ opioid receptor antagonist into the NAc of CREB overexpressing mice attenuates the inhibition of the rewarding effects of cocaine (Carlezon et al., 1998). Thus, CREB activation in the NAc may function as negative feedback to reduce sensitivity to the behavioral effects of repeated drug administration (Carlezon et al., 2005).
Another transcription factor induced in striatum by chronic drugs of abuse is ΔFosB (Figure 3). In contrast to CREB, ΔFosB appears to mediate the development of drug sensitization. ΔFosB shares homology with other Fos family transcription factors that include c-Fos, FosB, Fra1, and Fra2 (Morgan and Curran, 1995). Heterodimerization of Fos family proteins with Jun family proteins (c-Jun, JunB, or JunD) form activator protein-1 (AP-1) transcription factors. AP-1 transcription factors bind to AP-1 sites in the promoters of specific genes to regulate their transcription. Fos family proteins are highly unstable and induced rapidly and transiently in the striatum after acute drug administration. Conversely, chronic drug administration induces persistent increases inΔFosB (Mr 35–37 kD) in striatal subregions unlike other Fos family members (Chen et al., 1995, 1997; Hiroi et al., 1997). Similar to CREB, some drugs of abuse appear to induce ΔFosB selectively in dynorphin-containing MSNs of the striatum (Nye et al., 1995; Moratalla et al., 1996; Muller and Unterwald, 2005; Lee et al., 2006). ΔFosB-overexpressing mice have augmented locomotor response to acute and chronic cocaine (Kelz et al., 1999) and exhibit enhanced conditioned place preference (CPP), a Pavlovian, or classical conditioned behavior used as an indirect measure of the rewarding (or aversive) effects of drugs (Prus et al., 2009) with either cocaine (Kelz et al., 1999) or morphine (Zachariou et al., 2006). Further, ΔFosB-overexpressing mice self-administer lower doses of cocaine than do their wild-type littermates, indicating increased sensitivity to drug reinforcing effects (Colby et al., 2003). Interestingly, persistent increases in ΔFosB vary according to different striatal subregions, such as the NAc core, shell and dorsal striatum and do not differ between contingent and non-contingent drug administration procedures (Perrotti et al., 2008).
It is important to note here that volition is a critical factor in understanding the signaling mechanisms of addiction. Different outcomes between contingent versus non-contingent drug administration studies exist (Hemby et al., 2005a; Jacobs et al., 2005). While experimenter-administered drug paradigms, such as CPP and locomotor sensitization, have greatly increased understanding of neuroplasticity induced by repeated drug exposure, it has been proposed that molecular mechanisms of drug relapse may be probed with greater clinical relevance using operant drug self-administration procedures (Shaham et al., 2003; Epstein et al., 2006).
Another major substrate of the dopamine D1/cAMP/PKA pathway implicated in the mechanisms of various drugs of abuse is Dopamine- and cAMP-regulated phosphoprotein, Mr 32 kDa (DARPP-32; Svenningsson et al., 2005; Figure 3). DARPP-32 is highly enriched in striatal MSNs (Ouimet et al., 1998). DARPP-32 can serve a dual function as a protein kinase inhibitor or a protein phosphatase inhibitor, depending on the site of DARPP-32 phosphorylation. Changes in the phosphorylation state of DARPP-32, through activation of its phosphatase- or kinase-inhibitory activity, indirectly influence the phosphorylation state of other proteins and thereby mediate some of the diverse physiological effects of first messengers on cellular function. For example, activation of DARPP-32 by dopamine and cAMP can act as a positive feedback signal by inhibiting dephosphorylation of other substrates for PKA. DARPP-32 can also attenuate dephosphorylation of other substrates for other protein kinases and thereby mediate effects of first- and second-messenger systems on one another. Furthermore, DARPP-32, via phosphorylation by PKA and dephosphorylation by the Ca2+-/calmodulin-dependent phosphatase, protein phosphatase 2B (PP2B; calcineurin) can integrate physiological effects of first messengers that influence cAMP and Ca2+ systems (Figure 3). For example, dopamine activation of D1 receptors that activate the cAMP pathway phosphorylates DARPP-32, whereas NMDA or AMPA signals that activate the Ca2+pathway dephosphorylates DARPP-32.
PKA phosphorylates DARPP-32 at Thr34, rendering DARPP-32 a potent inhibitor of the multifunctional protein phosphatase 1 (PP1; Hemmings et al., 1984). Altered activity of PP1 leads to altered dephosphorylation of one of its prominent substrates: Na+, K+-ATPase. Changes in the phosphorylation state of Na+, K+-ATPase results in altered Na+transport across the cell membrane and altered membrane potential in excitable cells (Li et al., 1995). Analogous mechanisms appear to underlie modulation of Ca2+ channel activity (Surmeier et al., 1995).
Cyclin-Dependent Kinase 5
Cyclin-dependent kinase 5 (Cdk5) is another important kinase implicated in drug addiction. Cdk5 inhibition, whether by pharmacological or genetic means, potentiates both natural and cocaine reward-related behaviors. Repeated intra-NAc infusions of the Cdk5 pharmacological inhibitor roscovitine prior to cocaine injections alter both the development and expression of cocaine-induced locomotor sensitization (Taylor et al., 2007). Repeated intra-NAc infusion of roscovitine also enhances the incentive-motivational effects of cocaine as measured by responding for drug-associated cues and this effect persisted for at least 2 weeks after the last exposure of roscovitine (Taylor et al., 2007). NAc infusions of another Cdk5 inhibitor, olomoucine, produces acute, and persistent increases in “breakpoints” on a progressive ratio schedule for cocaine reinforcement (Taylor et al., 2007), an index of the degree of effort an animal will exert to obtain drug.
Cre/loxP conditional knockout systems permit temporal and spatial control of Cdk5 expression in the adult brain (Benavides et al., 2007). Mice lacking Cdk5 in adult forebrain exhibit increased locomotor-activating effects of cocaine, increased breakpoints on a progressive ratio schedule of food reinforcement and enhanced cocaine CPP corresponding to increased excitability of MSNs in the NAc, a form of functional plasticity (Benavides et al., 2007). These combined results suggest that Cdk5 acts as a homeostatic mechanism of neuronal excitability in the NAc and that Cdk5 may govern behaviors driven to obtain drug, drug-associated cues, and natural reward.
The AP-1 site in the Cdk5 gene promoter is a downstream target ofΔFosB. Chronic cocaine administration as well as overexpression of ΔFosB raises striatal Cdk5 mRNA, protein, and activity (Bibb et al., 2001). Unlike protein kinases that are activated by second messengers, Cdk5 is constitutively activated through its interactions with its cofactor p35. In this constitutively active basal state, Cdk5 appears to provide a negative tonus toward D1 dopamine receptor/cAMP/PKA signaling, through its phosphorylation of DARPP-32, for example. Cdk5 phosphorylates DARPP-32 at Thr75, rendering DARPP-32 a potent inhibitor of PKA that effectively antagonizes effects of chronic cocaine on striatal dopamine levels (Bibb et al., 1999). These data suggest that chronic cocaine induces up-regulation of Cdk5 mediated by ΔFosB that alters dopamine D1 receptor signaling. While up-regulation of Cdk5 may functionally repress hyper stimulated cAMP/PKA signaling pathways, it may also alter neuroplasticity in other ways that may contribute to addiction.
PP2A is a predominant mechanism for dephosphorylation of DARPP-32 at Thr75. Dephosphorylation of DARPP-32 at Thr34 involves primarily the Ca2+-dependent protein phosphatase calcineurin. Dephosphorylation of Thr75 primarily involves PP2A. Dephosphorylation of Thr75 is subject to control by both cAMP- and Ca2+-dependent regulatory mechanisms. The PR72 (or B′ or PPP2R3A; Figure 3) regulatory subunit of PP2A is enriched in striatum and contains two Ca2+ binding sites formed by E and F helices (EF-hands 1 and 2), of which EF-hands 1 is required for the ability of PP2A activity to be regulated by Ca2+ in vitro and in vivo (Ahn et al., 2007b). These data demonstrate that the PR72-containing form of PP2A is required for glutamate acting at AMPA and NMDA receptors to regulate Thr75 dephosphorylation. In addition, PP2A may be activated through cAMP-dependent signaling through the B56δ regulatory subunit (Figure 3), rendering it responsive to D1 receptor activation and subject to dysregulation by drugs of abuse. It has been suggested that the B56δ/AC/PP2A complex also regulates the dephosphorylation of DARPP-32 at Thr75 (Ahn et al., 2007a). As either Ca2+ or cAMP through these two regulatory subunits may activate it, PP2A represents a particularly interesting signaling molecule that may contribute to reward learning and addiction.
Ongoing studies in our laboratory suggest that Cdk5 may regulate PKA signaling through additional mechanisms. While the activation of Cdk5 is dependent upon associating with p35, this may also serve as an important regulatory target. Specifically, NMDAR-dependent Ca2+influx causes calpain to convert p35 to p25 (Wei et al., 2005; Meyer et al., 2008; Figure 4). The Cdk5/p25 holoenzyme is more soluble than Cdk5/p35 and its specificity may shift away from substrates by which it mediates attenuation of cAMP/PKA signaling. Thus, while Cdk5/p25 has been broadly implicated in neuronal injury and disease (Barnett and Bibb, 2010; Hisanaga and Endo, 2010), it may play an important role in integrating Ca2+ and cAMP signaling in localized domains within synapses invoked during reward learning. While there is growing evidence for this concept, it remains to be proven.
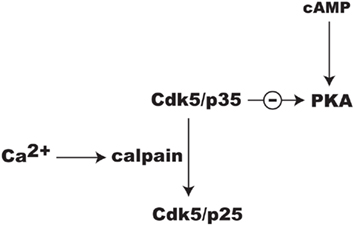
Figure 4. Possible mechanism for the integration of Ca2+ and cAMP signaling involving Cdk5 and release of its inhibition of PKA signaling by the conversion of its cofactor p35 to p25 by calpain.
Cyclin-dependent kinase 5 also modulates cocaine-induced plasticity in dendritic spine morphology, a form of structural plasticity. Rats received intra-NAc infusions of roscovitine or saline during chronic cocaine administration and Cdk5 inhibition attenuated cocaine-induced dendritic spine outgrowth in NAc core and shell 24–48 h after cocaine exposure (Norrholm et al., 2003). Cdk5 phosphorylates and inhibits myocyte enhancing factor 2 (MEF2) that increases dendritic spines in MSNs of the NAc (Pulipparacharuvil et al., 2008). Inhibition of MEF2 activity in response to cocaine can permit transcription of the cytoskeleton-associated genes, Wiskott–Aldrich syndrome protein (WASP), N-WASP, and WASP-family verprolin homologs (WAVEs) that have putative MEF binding sites in their proximal promoter regions. WAVE1 also appears to modulate spine morphogenesis in a Cdk5-dependent manner (Kim et al., 2006; Sung et al., 2008). Thus, increased activation of Cdk5 by repeated cocaine exposure via ΔFosB potentially results in modulation of WAVE activity, whereas MEF2 can modulate its expression level to mediate persistent alterations that could underlie aspects of cocaine-induced plasticity and addiction. Thus, Cdk5 could modulate functional and structural plasticity underlying cognitive inflexibility and maladaptive behaviors characteristic of addiction. The role of Cdk5 in cytoskeletal dynamics, synaptic function, and cell survival (Lopes and Agostinho, 2011) and cognition and plasticity (Angelo et al., 2006; Barnett and Bibb, 2010; Bibb et al., 2010) suggest targeting Cdk5 signaling may be a viable approach to attenuating drug-induced changes in synaptic plasticity that influence addiction behaviors.
Extracellular Signal-Regulated Kinases
Extracellular signal-regulated kinases (ERK) is also involved in the D1/cAMP/PKA signaling cascade (Figure 3; Greengard et al., 1999; Svenningsson et al., 2004). The MAPK kinases responsible for phosphorylating ERK are the ERK kinases II (MEKs). The MAPK kinase responsible for MEK activation include kinases termed Raf. Raf is activated by one of three forms of Ras that is, in turn, activated in response to many types of growth factors to their receptors, such as brain-derived neurotrophic factor (BDNF). ERKs are implicated in long-term neuroplasticity induced by various drugs of abuse (Lu et al., 2006; Girault et al., 2007; Thomas et al., 2008).
Acute cocaine injections rapidly increase ERK phosphorylation in NAc MSNs and dorsal striatum that are blocked by MEK pharmacological inhibition (Valjent et al., 2000) and cocaine-induced ERK phosphorylation is attenuated in the NAc and dorsal striatum in DARPP-32 mutant mice (Valjent et al., 2005). Increased ERK phosphorylation in the NAc has been shown with Δ9-tetrahydrocannabinol (THC; Valjent et al., 2001, 2004), D-amphetamine (Choe et al., 2002), 3,4-methylene-dioxy-methamphetamine (MDMA; Salzmann et al., 2003), morphine (Valjent et al., 2004), and nicotine (Valjent et al., 2004). Morphine-dependent mice undergoing naloxone-precipitated withdrawal show increased ERK activity in NAc but not dorsal striatum compared to controls (Li et al., 2010). Pharmacological inhibition of ERK, abolishes, or attenuates LTP induced by high frequency stimulation of population spikes in the dorsomedial striatum in rat and this effect is, in part, mediated by the ERK pathway coupling to NMDA receptors (Xie et al., 2009). Blockade of ERK phosphorylation in the NAc by direct infusion of a MEK inhibitor attenuates cocaine-induced activation of CREB (Mattson et al., 2005). Furthermore, the blockade of ERK activity corresponds to the blockade of CREB and activity of the intermediary kinase MSK1 (that directly activates CREB) in the NAc and dorsal striatum (Brami-Cherrier et al., 2005). MEK pharmacological inhibition also blocked cocaine-induced activation of the transcription factor Elk-1 and expression of Fos in the NAc and dorsal striatum (Valjent et al., 2000; Zhang et al., 2004).
Cocaine-induced CPP corresponds to increased ERK phosphorylation in the NAc and dorsal striatum (Valjent et al., 2006) and exposure to the cocaine-paired context increases ERK phosphorylation in the NAc core but not shell (Miller and Marshall, 2005). CPP corresponding to increased ERK activity in striatum has also been obtained with methamphetamine (Mizoguchi et al., 2004). Effects of MEK inhibitors in ERK1 knockout mice suggest it is the ERK2 isoform that is critical for morphine CPP (Mazzucchelli et al., 2002), cocaine CPP, and locomotor sensitization (Ferguson et al., 2006). ERK2 can inhibit ERK1 signaling, possibly by competition for phosphorylation by MEK, suggesting ERK2 is more potent than ERK1 (Valjent et al., 2005). Indeed, ERK1 and ERK2 appear to have opposing roles in drug-induced synaptic plasticity and behavior (Girault et al., 2007), thus ERK2 but not ERK1 appears to be the isoform most critical for modulating behavioral effects of drugs of abuse.
It has also been suggested that processes of memory disruption may underlie the effects of ERK blockade during CPP training on subsequent drug CPP expression (Gerdjikov et al., 2004). The modulation of ERK by drugs of abuse via dopamine and glutamate receptor interactions integrates many second messengers suggesting ERK may function in learned associations between reward and contextual cues (Girault et al., 2007). In addition to a role in classical conditioning, ERK activation is implicated in learning and performance of instrumental responding via regulation of transcription factors affecting intrinsic cellular excitability (Shiflett and Balleine, 2011a,b). ERK signaling cascades and BDNF growth factors influence glutamate transmission modulating LTP and LTD in MSNs of the striatum (Mcginty et al., 2010). Chronic EtOH intake attenuates ERK phosphorylation and LTD induction, whereas withdrawal for 1 day potentiates ERK phosphorylation and LTD induction (Cui et al., 2011). Direct infusions of BDNF into the prefrontal cortex decreases responding during extinction training and prevents cocaine-induced decreases in ERK phosphorylation in the NAc but not the dorsal striatum 1 day after drug exposure (Mcginty et al., 2010) and prevents cocaine-induced aberrations in extracellular glutamate in the NAc (Berglind et al., 2007, 2009). Thus, the ERK signaling cascade appears to mediate drug-induced synaptic plasticity linked to reward.
Ca2+/Calmodulin-Dependent Kinase II
The most prominent example of a Ca2+/calmodulin-dependent kinases (CaMK) with neuronal involvement in addiction and plasticity is CaMKII (Wayman et al., 2008). CaMKII can mediate many of the second messenger actions of Ca2+ in neurons (Figure 3). The CaMKII family consists of four subunits termed α, β, γ, and δ isoforms that are encoded by four different genes. CaMKII contains a catalytic, regulatory, and self-association domain. The regulatory domain contains (1) the autoinhibitory site that binds to and inhibits the catalytic domain while in the resting state, (2) the Ca2+/calmodulin-binding site and (3) several regulatory phosphorylation sites. Disinhibition of the autoinhibitory site occurs upon binding of Ca2+/calmodulin to the regulatory domain. CaMKIIα at Thr286 and CaMKIIβ at Thr287 undergo autophosphorylation independent of Ca2+ that switches the kinase into an autonomous activity mode (Lisman et al., 2002; Wayman et al., 2008). Due to the stimulation-sensor capability of this autophosphorylation switch, CaMKII has been proposed to act as a memory storage mechanism (Lisman et al., 2002).
The effects of psychostimulants on CaMKII activity can vary according to striatal region, drug, and dosing regimen. Acute cocaine increases CaMKII activity in the NAc (Mattson et al., 2005). Acute amphetamine increases CaMKII activity in dorsal striatum and alters the CaMKII substrates, ERK1/2, CREB, and Elk-1 (Choe and Wang, 2002) that is dependent on group I mGlu receptors (Choe and Wang, 2001). Acute methamphetamine actually reduces CaMKII in the NAc and dorsal striatum (Akiyama and Suemaru, 2000). Chronic amphetamine administration increases CaMKII phosphorylation in the striatum but not protein levels (Iwata et al., 1997). Chronic intermittent amphetamine increases striatal levels of CaMKII mRNA (Greenstein et al., 2007). Pharmacological inhibition of CaMKII activity blunts acute amphetamine-induced dopamine efflux via the dopamine transporter (DAT) in mouse midbrain dopamine cultured neurons, striatal brain slices, and in vivo (Fog et al., 2006). Acute administration of a D1-like agonist (Das et al., 1997; Dudman et al., 2003) or NMDA agonist (Das et al., 1997) increases phosphorylation of CREB that is attenuated by CaMKII pharmacological inhibitors in rat striatal neurons.
There is also cross talk between CaMKII and PKA signaling pathways. PKA may promote LTP by stabilizing and/or recycling pools of GluA1 AMPA receptors that are subsequently recruited to the synaptic membrane by Ca2+ stimulation of CaMKII (or protein kinase C PKC; see below)-driven trafficking events and GluA1 phosphorylation at Ser818 and Ser831 (Boehm et al., 2006; Guire et al., 2008). Single channel AMPA receptor activity may also be increased during LTP via PKA phosphorylation of Ser845 to increase probability of open channels and CaMKII/PKC phosphorylation of Ser831 to increase single channel conductance (Derkach et al., 2007).
Intriguingly, CaMKII has been implicated as an important negative regulator of the dopamine D3 receptor subtype that relieves the dopamine receptor-mediated inhibition on sensitized behavior (Liu et al., 2009). It was shown that D3 receptor activation inhibits AC in acute NAc slices and that NMDA receptor-mediated Ca2+ influx restores this inhibition through stimulating CaMKII-binding to the D3 receptor. Importantly, a peptide derived from the CaMKII-binding site on the D3 receptor prevented this disinhibition induced by Ca2+ influx.
Protein Kinase C
PKC comprises more than 14 isoforms subdivided into categories according to their structure, Ca2+ dependence, and lipid activators (Pearce et al., 2010). All PKC isoforms contain a catalytic domain harboring an ATP binding site and a substrate-binding site. Ca2+ in conjunction with the lipid signaling intermediate diacylglycerol (DAG) activate the classical PKCs (α, βI, βII, and γ; Figure 3). DAG but not Ca2+ activate novel PKCs (δ, ε, η, and θ). Lipids such as phosphatidic and arachidonic acid activate atypical PKCs (ζ, and ℩/λ) that are considered to be constitutively active as they require neither DAG nor Ca2+. Ca2+ and/or DAG or lipids bind to the R domain to disinhibit the C domain. PKC, similar to CaMKII, mediates multiple second messenger functions of Ca2+ to influence substrates such as receptors and ion channels.
There are numerous downstream substrates of PKCs involved in mechanisms of drugs of abuse that include neurotransmitter receptor subtypes such as α4 subunit of the nicotinic acetylcholine receptor (nAChR) phosphorylated at Ser550 (Pollock et al., 2007), dopamine D2 receptors phosphorylated at Ser229, Ser228, Thr352, Thr354, and Ser355 (Namkung and Sibley, 2004), the γ2 subunit of the GABAA receptor phosphorylated at Ser327 of the γ2 subunit of the GABAA receptor (Qi et al., 2007), and cannabinoid CB1 receptors phosphorylated at Ser317 (Garcia et al., 1998). Other PKC substrate proteins are involved in exocytosis (e.g., Ser187 of SNAP-25; Nagy et al., 2002) and synaptic plasticity (e.g., Ser36 of neurogranin and Ser41 of neuromodulin; Huang et al., 1993; Oehrlein et al., 1996) are also targeted by PKC.
PKC also phosphorylates Ser818 of the AMPA glutamate receptor subunit GluA1 that facilitates AMPA receptor externalization and LTP (Boehm et al., 2006). PKC and MAPK signaling pathways appear to be involved in AMPA receptor trafficking underlying an in vitro model of classical conditioning in pond turtles, Pseudemys scripta elegans (Zheng and Keifer, 2008). PKC activation facilitates autophosphorylated CaMKII and increased association with NMDA receptors in conjunction with NMDA postsynaptic receptor insertion that was, along with PKC-induced LTP of the AMPA receptor-mediated response, abolished by a CaMKII antagonist or by disruption of CaMKII interaction with NR2A or NR2B (Yan et al., 2011).
Due to the lack of selectivity of pharmacological inhibitors for specific PKC isoforms, targeted gene disruption in mouse models is increasingly being used to explore the precise role of PKC isoforms in behaviors related to addiction (Olive and Newton, 2010). PKCβ knockout mice have a reduced locomotor-activating effect of acute amphetamine and this isozyme is critical for effects on DAT (Chen et al., 2009). PKCγ knockout mice fail to exhibit morphine CPP (Narita et al., 2001) and show increased EtOH consumption in a two-bottle choice procedure (Bowers and Wehner, 2001). PKCε knockout mice show enhanced locomotor stimulant effects of lower doses of EtOH (Hodge et al., 1999, 2002) and potentiated rewarding and reinforcing effects of morphine (Newton et al., 2007). However, PKCε knockout mice consumed approximately 75% less EtOH and exhibit weaker EtOH preference in a two-bottle choice task compared to wildtype controls (Hodge et al., 1999). Another study showed that PKCε knockout mice exhibit reduced EtOH self-administration and attenuated alcohol deprivation effect (Olive et al., 2000). PKCε knockout mice are more sensitive to the aversive and less sensitive to the rewarding effects of EtOH in CPP (Newton and Messing, 2007). PKCε knockout mice acquire morphine intravenous self-administration at doses that do not maintain self-administration in wild-type controls (Newton et al., 2007).
These pharmacological and genetic techniques suggest a meaningful role for PKC in neuroadaptations and behavioral alterations pertinent to addiction. Indeed, the wealth of data especially on PKCε obtained in drug self-administration has led to its inclusion on a panel of markers for haplotype analysis of addiction-related genes (Hodgkinson et al., 2008). Thus, the operation of PKC isozymes and their signaling pathways provide another route to the development of pharmacotherapeutics for addiction.
Future Directions
Here we have made attempt to integrate different aspects of addiction including behavioral considerations and animal models, considerations of circuitry and its mediation of both reward and addiction learning, and have cataloged a small portion of the underlying signaling mechanism that mediate normal and addiction-related motivated behaviors. Focus on the cellular and molecular neurobiology of signal transduction has allowed the elucidation of mechanisms beyond the synaptic level that modulate addictive drug-induced functional and structural plasticity. Behavioral assays related to drug addiction that dissociates measures of relapse from reinforcement and dependence provides powerful tools to evaluate the effects of targeting specific intracellular signaling pathways. However, understanding of the molecular mechanism involved and relating them complex pathophysiological and behavioral basis of adduction is incomplete and remains a challenge. Current and future research on addiction driven toward reversing drug-induced pathology that impairs synaptic plasticity will undoubtedly include the systematic manipulation of mechanisms of protein phosphorylation.
The transition to the addicted state may initiate with a cascade of neuroadaptations from the ventral striatum to dorsal striatum. However, the delineation of drug-induced plasticity in neurocircuitry corresponding to stages of binge intoxication, withdrawal-induced negative affect and preoccupation/anticipation involves other critical brain regions that include but are not limited to the ventral tegmental area, extended amygdala, prefrontal cortex, cingulated gyrus, and hippocampus. A better understanding of the neurocircuitry altered by drugs of abuse will help form a heuristic basis in the search for genetic, molecular, and pharmacological neuroadaptations conferring risk for or protection against addiction and aid in the identification of novel targets for pharmacotherapeutics with increased therapeutic efficacy and decreased side effect liability.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a NARSAD Young Investigator Award (to Scott D. Philibin); National Institute of Drug Abuse grants DA026482, DA010460, DA008227 (to David W. Self), DA16672 (to James A. Bibb); and National Institute of Mental Health grants MH079710 and MH083711 (to James A. Bibb).
References
Ahn, J. H., Mcavoy, T., Rakhilin, S. V., Nishi, A., Greengard, P., and Nairn, A. C. (2007a). Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc. Natl. Acad. Sci. U.S.A. 104, 2979–2984.
Ahn, J. H., Sung, J. Y., Mcavoy, T., Nishi, A., Janssens, V., Goris, J., Greengard, P., and Nairn, A. C. (2007b). The B″/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc. Natl. Acad. Sci. U.S.A. 104, 9876–9881.
Akiyama, K., and Suemaru, J. (2000). Effect of acute and chronic administration of methamphetamine on calcium-calmodulin dependent protein kinase II activity in the rat brain. Ann. N. Y. Acad. Sci. 914, 263–274.
Alleweireldt, A. T., Kirschner, K. F., Blake, C. B., and Neisewander, J. L. (2003). D1-receptor drugs and cocaine-seeking behavior: investigation of receptor mediation and behavioral disruption in rats. Psychopharmacology (Berl.) 168, 109–117.
Angelo, M., Plattner, F., and Giese, K. P. (2006). Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J. Neurochem. 99, 353–370.
Bachtell, R. K., Choi, K. H., Simmons, D. L., Falcon, E., Monteggia, L. M., Neve, R. L., and Self, D. W. (2008). Role of GluR1 expression in nucleus accumbens neurons in cocaine sensitization and cocaine-seeking behavior. Eur. J. Neurosci. 27, 2229–2240.
Bachtell, R. K., Whisler, K., Karanian, D., and Self, D. W. (2005). Effects of intra-nucleus accumbens shell administration of dopamine agonists and antagonists on cocaine-taking and cocaine-seeking behaviors in the rat. Psychopharmacology (Berl.) 183, 41–53.
Backstrom, P., and Hyytia, P. (2007). Involvement of AMPA/kainate, NMDA, and mGlu5 receptors in the nucleus accumbens core in cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl.) 192, 571–580.
Barnett, D. G., and Bibb, J. A. (2010). The role of Cdk5 in cognition and neuropsychiatric and neurological pathology. Brain Res. Bull. 85, 9–13.
Barrot, M., Olivier, J. D., Perrotti, L. I., Dileone, R. J., Berton, O., Eisch, A. J., Impey, S., Storm, D. R., Neve, R. L., Yin, J. C., Zachariou, V., and Nestler, E. J. (2002). CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc. Natl. Acad. Sci. U.S.A. 99, 11435–11440.
Bassareo, V., De Luca, M. A., and Di Chiara, G. (2002). Differential expression of motivational stimulus properties by dopamine in nucleus accumbens shell versus core and prefrontal cortex. J. Neurosci. 22, 4709–4719.
Bassareo, V., Musio, P., and Di Chiara, G. (2011). Reciprocal responsiveness of nucleus accumbens shell and core dopamine to food- and drug-conditioned stimuli. Psychopharmacology (Berl.) 214, 687–697.
Benavides, D. R., Quinn, J. J., Zhong, P., Hawasli, A. H., Dileone, R. J., Kansy, J. W., Olausson, P., Yan, Z., Taylor, J. R., and Bibb, J. A. (2007). Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J. Neurosci. 27, 1967–12976.
Beninger, R. J., Nakonechny, P. L., and Savina, I. (2003). cAMP-dependent protein kinase and reward-related learning: intra-accumbens Rp-cAMPS blocks amphetamine-produced place conditioning in rats. Psychopharmacology (Berl.) 170, 23–32.
Berglind, W. J., See, R. E., Fuchs, R. A., Ghee, S. M., Whitfield, T. W. Jr., Miller, S. W., and Mcginty, J. F. (2007). A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur. J. Neurosci. 26, 757–766.
Berglind, W. J., Whitfield, T. W. Jr., Lalumiere, R. T., Kalivas, P. W., and Mcginty, J. F. (2009). A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J. Neurosci. 29, 3715–3719.
Berridge, K. C., and Robinson, T. E. (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 28, 309–369.
Berridge, K. C., Robinson, T. E., and Aldridge, J. W. (2009). Dissecting components of reward: “liking,” “wanting,” and learning. Curr. Opin. Pharmacol. 9, 65–73.
Bibb, J. A., Chen, J., Taylor, J. R., Svenningsson, P., Nishi, A., Snyder, G. L., Yan, Z., Sagawa, Z. K., Ouimet, C. C., Nairn, A. C., Nestler, E. J., and Greengard, P. (2001). Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature 410, 376–380.
Bibb, J. A., Mayford, M. R., Tsien, J. Z., and Alberini, C. M. (2010). Cognition enhancement strategies. J. Neurosci. 30, 14987–14992.
Bibb, J. A., Snyder, G. L., Nishi, A., Yan, Z., Meijer, L., Fienberg, A. A., Tsai, L. H., Kwon, Y. T., Girault, J. A., Czernik, A. J., Huganir, R. L., Hemmings, H. C. Jr., Nairn, A. C., and Greengard, P. (1999). Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature 402, 669–671.
Blumer, J. B., Cismowski, M. J., Sato, M., and Lanier, S. M. (2005). AGS proteins: receptor-independent activators of G-protein signaling. Trends Pharmacol. Sci. 26, 470–476.
Blumer, J. B., and Lanier, S. M. (2003). Accessory proteins for G protein-signaling systems: activators of G protein signaling and other nonreceptor proteins influencing the activation state of G proteins. Recept. Channels 9, 195–204.
Boehm, J., Kang, M. G., Johnson, R. C., Esteban, J., Huganir, R. L., and Malinow, R. (2006). Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 51, 213–225.
Borgland, S. L., Malenka, R. C., and Bonci, A. (2004). Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J. Neurosci. 24, 7482–7490.
Bower, G., and Grusec, T. (1964). Effect of prior pavlovian discrimination training upon learning an operant discrimination. J. Exp. Anal. Behav. 7, 401–404.
Bowers, B. J., and Wehner, J. M. (2001). Ethanol consumption and behavioral impulsivity are increased in protein kinase Cgamma null mutant mice. J. Neurosci. 21, RC180.
Bowers, M. S. (2010). Activators of G-protein signaling 3: a drug addiction molecular gateway. Behav. Pharmacol. 21, 500–513.
Bowers, M. S., Chen, B. T., and Bonci, A. (2010). AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron 67, 11–24.
Bowers, M. S., Hopf, F. W., Chou, J. K., Guillory, A. M., Chang, S. J., Janak, P. H., Bonci, A., and Diamond, I. (2008). Nucleus accumbens AGS3 expression drives ethanol seeking through G betagamma. Proc. Natl. Acad. Sci. U.S.A. 105, 12533–12538.
Bowers, M. S., Mcfarland, K., Lake, R. W., Peterson, Y. K., Lapish, C. C., Gregory, M. L., Lanier, S. M., and Kalivas, P. W. (2004). Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron 42, 269–281.
Brami-Cherrier, K., Valjent, E., Herve, D., Darragh, J., Corvol, J. C., Pages, C., Arthur, S. J., Girault, J. A., and Caboche, J. (2005). Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J. Neurosci. 25, 11444–11454.
Burchett, S. A., Volk, M. L., Bannon, M. J., and Granneman, J. G. (1998). Regulators of G protein signaling: rapid changes in mRNA abundance in response to amphetamine. J. Neurochem. 70, 2216–2219.
Cadoni, C., and Di Chiara, G. (1999). Reciprocal changes in dopamine responsiveness in the nucleus accumbens shell and core and in the dorsal caudate-putamen in rats sensitized to morphine. Neuroscience 90, 447–455.
Cadoni, C., and Di Chiara, G. (2000). Differential changes in accumbens shell and core dopamine in behavioral sensitization to nicotine. Eur. J. Pharmacol. 387, R23–R25.
Cadoni, C., Solinas, M., and Di Chiara, G. (2000). Psychostimulant sensitization: differential changes in accumbal shell and core dopamine. Eur. J. Pharmacol. 388, 69–76.
Caine, S. B., Thomsen, M., Gabriel, K. I., Berkowitz, J. S., Gold, L. H., Koob, G. F., Tonegawa, S., Zhang, J., and Xu, M. (2007). Lack of self-administration of cocaine in dopamine D1 receptor knock-out mice. J. Neurosci. 27, 13140–13150.
Carlezon, W. A. Jr., Duman, R. S., and Nestler, E. J. (2005). The many faces of CREB. Trends Neurosci. 28, 436–445.
Carlezon, W. A. Jr., Thome, J., Olson, V. G., Lane-Ladd, S. B., Brodkin, E. S., Hiroi, N., Duman, R. S., Neve, R. L., and Nestler, E. J. (1998). Regulation of cocaine reward by CREB. Science 282, 2272–2275.
Chen, J., Kelz, M. B., Hope, B. T., Nakabeppu, Y., and Nestler, E. J. (1997). Chronic Fos-related antigens: stable variants of deltaFosB induced in brain by chronic treatments. J. Neurosci. 17, 4933–4941.
Chen, J., Nye, H. E., Kelz, M. B., Hiroi, N., Nakabeppu, Y., Hope, B. T., and Nestler, E. J. (1995). Regulation of delta FosB and FosB-like proteins by electroconvulsive seizure and cocaine treatments. Mol. Pharmacol. 48, 880–889.
Chen, R., Furman, C. A., Zhang, M., Kim, M. N., Gereau, R. W. T., Leitges, M., and Gnegy, M. E. (2009). Protein kinase Cbeta is a critical regulator of dopamine transporter trafficking and regulates the behavioral response to amphetamine in mice. J. Pharmacol. Exp. Ther. 328, 912–920.
Choe, E. S., Chung, K. T., Mao, L., and Wang, J. Q. (2002). Amphetamine increases phosphorylation of extracellular signal-regulated kinase and transcription factors in the rat striatum via group I metabotropic glutamate receptors. Neuropsychopharmacology 27, 565–575.
Choe, E. S., and Wang, J. Q. (2001). Group I metabotropic glutamate receptors control phosphorylation of CREB, Elk-1 and ERK via a CaMKII-dependent pathway in rat striatum. Neurosci. Lett. 313, 129–132.
Choe, E. S., and Wang, J. Q. (2002). CaMKII regulates amphetamine-induced ERK1/2 phosphorylation in striatal neurons. Neuroreport 13, 1013–1016.
Colby, C. R., Whisler, K., Steffen, C., Nestler, E. J., and Self, D. W. (2003). Striatal cell type-specific overexpression of deltaFosB enhances incentive for cocaine. J. Neurosci. 23, 2488–2493.
Cole, R. L., Konradi, C., Douglass, J., and Hyman, S. E. (1995). Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron 14, 813–823.
Compton, W. M., and Volkow, N. D. (2006). Abuse of prescription drugs and the risk of addiction. Drug Alcohol Depend. 83(Suppl. 1), S4–S7.
Cui, S. Z., Wang, S. J., Li, J., Xie, G. Q., Zhou, R., Chen, L., and Yuan, X. R. (2011). Alteration of synaptic plasticity in rat dorsal striatum induced by chronic ethanol intake and withdrawal via ERK pathway. Acta Pharmacol. Sin. 32, 175–181.
Das, S., Grunert, M., Williams, L., and Vincent, S. R. (1997). NMDA and D1 receptors regulate the phosphorylation of CREB and the induction of c-fos in striatal neurons in primary culture. Synapse 25, 227–233.
Derkach, V. A., Oh, M. C., Guire, E. S., and Soderling, T. R. (2007). Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 8, 101–113.
Dias, C., Lachize, S., Boilet, V., Huitelec, E., and Cador, M. (2004). Differential effects of dopaminergic agents on locomotor sensitisation and on the reinstatement of cocaine-seeking and food-seeking behaviour. Psychopharmacology (Berl.) 175, 414–427.
Drevets, W. C., Gautier, C., Price, J. C., Kupfer, D. J., Kinahan, P. E., Grace, A. A., Price, J. L., and Mathis, C. A. (2001). Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol. Psychiatry 49, 81–96.
Dudman, J. T., Eaton, M. E., Rajadhyaksha, A., Macias, W., Taher, M., Barczak, A., Kameyama, K., Huganir, R., and Konradi, C. (2003). Dopamine D1 receptors mediate CREB phosphorylation via phosphorylation of the NMDA receptor at Ser897-NR1. J. Neurochem. 87, 922–934.
Edwards, S., Whisler, K. N., Fuller, D. C., Orsulak, P. J., and Self, D. W. (2007). Addiction-related alterations in D1 and D2 dopamine receptor behavioral responses following chronic cocaine self-administration. Neuropsychopharmacology 32, 354–366.
Engelhardt, S., and Rochais, F. (2007). G proteins: more than transducers of receptor-generated signals? Circ. Res. 100, 1109–1111.
Epstein, D. H., Preston, K. L., Stewart, J., and Shaham, Y. (2006). Toward a model of drug relapse: an assessment of the validity of the reinstatement procedure. Psychopharmacology (Berl.) 189, 1–16.
Ettenberg, A. (1990). Haloperidol prevents the reinstatement of amphetamine-rewarded runway responding in rats. Pharmacol. Biochem. Behav. 36, 635–638.
Ferguson, S. M., Fasano, S., Yang, P., Brambilla, R., and Robinson, T. E. (2006). Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology 31, 2660–2668.
Fog, J. U., Khoshbouei, H., Holy, M., Owens, W. A., Vaegter, C. B., Sen, N., Nikandrova, Y., Bowton, E., Mcmahon, D. G., Colbran, R. J., Daws, L. C., Sitte, H. H., Javitch, J. A., Galli, A., and Gether, U. (2006). Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron 51, 417–429.
Garcia, D. E., Brown, S., Hille, B., and Mackie, K. (1998). Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. J. Neurosci. 18, 2834–2841.
Garzon, J., Rodriguez-Munoz, M., and Sanchez-Blazquez, P. (2008). Do pharmacological approaches that prevent opioid tolerance target different elements in the same regulatory machinery? Curr. Drug Abuse Rev. 1, 222–238.
Gerdeman, G. L., Partridge, J. G., Lupica, C. R., and Lovinger, D. M. (2003). It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 26, 184–192.
Gerdjikov, T. V., Ross, G. M., and Beninger, R. J. (2004). Place preference induced by nucleus accumbens amphetamine is impaired by antagonists of ERK or p38 MAP kinases in rats. Behav. Neurosci. 118, 740–750.
Ghasemzadeh, M. B., Mueller, C., and Vasudevan, P. (2009). Behavioral sensitization to cocaine is associated with increased glutamate receptor trafficking to the postsynaptic density after extended withdrawal period. Neuroscience 159, 414–426.
Girault, J. A., Valjent, E., Caboche, J., and Herve, D. (2007). ERK2: a logical and gate critical for drug-induced plasticity? Curr. Opin. Pharmacol. 7, 77–85.
Gold, S. J., Ni, Y. G., Dohlman, H. G., and Nestler, E. J. (1997). Regulators of G-protein signaling (RGS) proteins: region-specific expression of nine subtypes in rat brain. J. Neurosci. 17, 8024–8037.
Gonzalez, G. A., and Montminy, M. R. (1989). Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59, 675–680.
Grafstein-Dunn, E., Young, K. H., Cockett, M. I., and Khawaja, X. Z. (2001). Regional distribution of regulators of G-protein signaling (RGS) 1, 2, 13, 14, 16, and GAIP messenger ribonucleic acids by in situ hybridization in rat brain. Brain Res. Mol. Brain Res. 88, 113–123.
Greengard, P., Allen, P. B., and Nairn, A. C. (1999). Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron 23, 435–447.
Greenstein, R., Novak, G., and Seeman, P. (2007). Amphetamine sensitization elevates CaMKIIbeta mRNA. Synapse 61, 827–834.
Guire, E. S., Oh, M. C., Soderling, T. R., and Derkach, V. A. (2008). Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J. Neurosci. 28, 6000–6009.
Hausdorff, W. P., Caron, M. G., and Lefkowitz, R. J. (1990). Turning off the signal: desensitization of beta-adrenergic receptor function. FASEB J. 4, 2881–2889.
Hemby, S. E., Co, C., Koves, T. R., Smith, J. E., and Dworkin, S. I. (1997). Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology (Berl.) 133, 7–16.
Hemby, S. E., Horman, B., and Tang, W. (2005a). Differential regulation of ionotropic glutamate receptor subunits following cocaine self-administration. Brain Res. 1064, 75–82.
Hemby, S. E., Tang, W., Muly, E. C., Kuhar, M. J., Howell, L., and Mash, D. C. (2005b). Cocaine-induced alterations in nucleus accumbens ionotropic glutamate receptor subunits in human and non-human primates. J. Neurochem. 95, 1785–1793.
Hemmings, H. C. Jr., Greengard, P., Tung, H. Y., and Cohen, P. (1984). DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature 310, 503–505.
Hiroi, N., Brown, J. R., Haile, C. N., Ye, H., Greenberg, M. E., and Nestler, E. J. (1997). FosB mutant mice: loss of chronic cocaine induction of Fos-related proteins and heightened sensitivity to cocaine’s psychomotor and rewarding effects. Proc. Natl. Acad. Sci. U.S.A. 94, 10397–10402.
Hisanaga, S., and Endo, R. (2010). Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J. Neurochem. 115, 1309–1321.
Hodge, C. W., Mehmert, K. K., Kelley, S. P., Mcmahon, T., Haywood, A., Olive, M. F., Wang, D., Sanchez-Perez, A. M., and Messing, R. O. (1999). Supersensitivity to allosteric GABA(A) receptor modulators and alcohol in mice lacking PKCepsilon. Nat. Neurosci. 2, 997–1002.
Hodge, C. W., Raber, J., Mcmahon, T., Walter, H., Sanchez-Perez, A. M., Olive, M. F., Mehmert, K., Morrow, A. L., and Messing, R. O. (2002). Decreased anxiety-like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase Cepsilon. J. Clin. Invest. 110, 1003–1010.
Hodgkinson, C. A., Yuan, Q., Xu, K., Shen, P. H., Heinz, E., Lobos, E. A., Binder, E. B., Cubells, J., Ehlers, C. L., Gelernter, J., Mann, J., Riley, B., Roy, A., Tabakoff, B., Todd, R. D., Zhou, Z., and Goldman, D. (2008). Addictions biology: haplotype-based analysis for 130 candidate genes on a single array. Alcohol Alcohol. 43, 505–515.
Hooks, S. B., Martemyanov, K., and Zachariou, V. (2008). A role of RGS proteins in drug addiction. Biochem. Pharmacol. 75, 76–84.
Huang, K. P., Huang, F. L., and Chen, H. C. (1993). Characterization of a 7.5-kDa protein kinase C substrate (RC3 protein, neurogranin) from rat brain. Arch. Biochem. Biophys. 305, 570–580.
Hyman, S. E., Malenka, R. C., and Nestler, E. J. (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 29, 565–598.
Ikemoto, S. (2003). Involvement of the olfactory tubercle in cocaine reward: intracranial self-administration studies. J. Neurosci. 23, 9305–9311.
Ikemoto, S., Glazier, B. S., Murphy, J. M., and Mcbride, W. J. (1997). Role of dopamine D1 and D2 receptors in the nucleus accumbens in mediating reward. J. Neurosci. 17, 8580–8587.
Iwata, S. I., Hewlett, G. H., Ferrell, S. T., Kantor, L., and Gnegy, M. E. (1997). Enhanced dopamine release and phosphorylation of synapsin I and neuromodulin in striatal synaptosomes after repeated amphetamine. J. Pharmacol. Exp. Ther. 283, 1445–1452.
Jacobs, E. H., Smit, A. B., De Vries, T. J., and Schoffelmeer, A. N. (2005). Long-term gene expression in the nucleus accumbens following heroin administration is subregion-specific and depends on the nature of drug administration. Addict. Biol. 10, 91–100.
Kalivas, P. W., and O’brien, C. (2008). Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 33, 166–180.
Kalivas, P. W., and Volkow, N. D. (2005). The neural basis of addiction: a pathology of motivation and choice. Am. J. Psychiatry 162, 1403–1413.
Karasawa, J., Yoshimizu, T., and Chaki, S. (2006). A metabotropic glutamate 2/3 receptor antagonist, MGS0039, increases extracellular dopamine levels in the nucleus accumbens shell. Neurosci. Lett. 393, 127–130.
Kasanetz, F., Deroche-Gamonet, V., Berson, N., Balado, E., Lafourcade, M., Manzoni, O., and Piazza, P. V. (2010). Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science 328, 1709–1712.
Kauer, J. A., and Malenka, R. C. (2007). Synaptic plasticity and addiction. Nat. Rev. Neurosci. 8, 844–858.
Kelley, A. E., and Schiltz, C. A. (2004). Accessories to addiction: G protein regulators play a key role in cocaine seeking and neuroplasticity. Neuron 42, 181–183.
Kelz, M. B., Chen, J., Carlezon, W. A. Jr., Whisler, K., Gilden, L., Beckmann, A. M., Steffen, C., Zhang, Y. J., Marotti, L., Self, D. W., Tkatch, T., Baranauskas, G., Surmeier, D. J., Neve, R. L., Duman, R. S., Picciotto, M. R., and Nestler, E. J. (1999). Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature 401, 272–276.
Kim, Y., Sung, J. Y., Ceglia, I., Lee, K. W., Ahn, J. H., Halford, J. M., Kim, A. M., Kwak, S. P., Park, J. B., Ho Ryu, S., Schenck, A., Bardoni, B., Scott, J. D., Nairn, A. C., and Greengard, P. (2006). Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 442, 814–817.
Koeltzow, T. E., and Vezina, P. (2005). Locomotor activity and cocaine-seeking behavior during acquisition and reinstatement of operant self-administration behavior in rats. Behav. Brain Res. 160, 250–259.
Koob, G. F., and Le Moal, M. (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24, 97–129.
Koob, G. F., and Volkow, N. D. (2010). Neurocircuitry of addiction. Neuropsychopharmacology 35, 217–238.
Krupnick, J. G., and Benovic, J. L. (1998). The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 38, 289–319.
Lalumiere, R. T., and Kalivas, P. W. (2008). Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 28, 3170–3177.
Lee, K. W., Kim, Y., Kim, A. M., Helmin, K., Nairn, A. C., and Greengard, P. (2006). Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc. Natl. Acad. Sci. U.S.A. 103, 3399–3404.
Li, D., Aperia, A., Celsi, G., Da Cruz E Silva, E. F., Greengard, P., and Meister, B. (1995). Protein phosphatase-1 in the kidney: evidence for a role in the regulation of medullary Na(+)-K(+)-ATPase. Am. J. Physiol. 269, F673–F680.
Li, T., Hou, Y., Cao, W., Yan, C. X., Chen, T., and Li, S. B. (2010). Naloxone-precipitated withdrawal enhances ERK phosphorylation in prefrontal association cortex and accumbens nucleus of morphine-dependent mice. Neurosci. Lett. 468, 348–352.
Lisman, J., Schulman, H., and Cline, H. (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190.
Liu, Q. S., Pu, L., and Poo, M. M. (2005). Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature 437, 1027–1031.
Liu, X. Y., Mao, L. M., Zhang, G. C., Papasian, C. J., Fibuch, E. E., Lan, H. X., Zhou, H. F., Xu, M., and Wang, J. Q. (2009). Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron 61, 425–438.
Lopes, J. P., and Agostinho, P. (2011). Cdk5: multitasking between physiological and pathological conditions. Prog. Neurobiol. 94, 49–63.
Lu, L., Grimm, J. W., Shaham, Y., and Hope, B. T. (2003). Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J. Neurochem. 85, 1604–1613.
Lu, L., Koya, E., Zhai, H., Hope, B. T., and Shaham, Y. (2006). Role of ERK in cocaine addiction. Trends Neurosci. 29, 695–703.
Luscher, C., and Ungless, M. A. (2006). The mechanistic classification of addictive drugs. PLoS Med. 3, e437. doi: 10.1371/journal.pmed.0030437
Madayag, A., Lobner, D., Kau, K. S., Mantsch, J. R., Abdulhameed, O., Hearing, M., Grier, M. D., and Baker, D. A. (2007). Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J. Neurosci. 27, 13968–13976.
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126.
Martin, M., Chen, B. T., Hopf, F. W., Bowers, M. S., and Bonci, A. (2006). Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat. Neurosci. 9, 868–869.
Mattson, B. J., Bossert, J. M., Simmons, D. E., Nozaki, N., Nagarkar, D., Kreuter, J. D., and Hope, B. T. (2005). Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. J. Neurochem. 95, 1481–1494.
Mayr, B., and Montminy, M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609.
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W., Welzl, H., Wolfer, D. P., Pages, G., Valverde, O., Marowsky, A., Porrazzo, A., Orban, P. C., Maldonado, R., Ehrengruber, M. U., Cestari, V., Lipp, H. P., Chapman, P. F., Pouyssegur, J., and Brambilla, R. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34, 807–820.
Mcclung, C. A., and Nestler, E. J. (2003). Regulation of gene expression and cocaine reward by CREB and deltaFosB. Nat. Neurosci. 6, 1208–1215.
Mcfarland, K., Davidge, S. B., Lapish, C. C., and Kalivas, P. W. (2004). Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J. Neurosci. 24, 1551–1560.
Mcginty, J. F., Whitfield, T. W. Jr., and Berglind, W. J. (2010). Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 1314, 183–193.
Meyer, D. A., Richer, E., Benkovic, S. A., Hayashi, K., Kansy, J. W., Hale, C. F., Moy, L. Y., Kim, Y., O’callaghan, J. P., Tsai, L. H., Greengard, P., Nairn, A. C., Cowan, C. W., Miller, D. B., Antich, P., and Bibb, J. A. (2008). Striatal dysregulation of Cdk5 alters locomotor responses to cocaine, motor learning, and dendritic morphology. Proc. Natl. Acad. Sci. U.S.A. 105, 18561–18566.
Miller, C. A., and Marshall, J. F. (2005). Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron 47, 873–884.
Milton, A. L., and Everitt, B. J. (2010). The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur. J. Neurosci. 31, 2308–2319.
Mitchell, J. A., Channell, S., and Hall, G. (1985). Response-reinforcer associations after caudate-putamen lesions in the rat: spatial discrimination and overshadowing-potentiation effects in instrumental learning. Behav. Neurosci. 99, 1074–1088.
Mitchell, J. A., and Hall, G. (1988). Learning in rats with caudate-putamen lesions: unimpaired classical conditioning and beneficial effects of redundant stimulus cues on instrumental and spatial learning deficits. Behav. Neurosci. 102, 504–514.
Mizoguchi, H., Yamada, K., Mizuno, M., Mizuno, T., Nitta, A., Noda, Y., and Nabeshima, T. (2004). Regulations of methamphetamine reward by extracellular signal-regulated kinase 1/2/ets-like gene-1 signaling pathway via the activation of dopamine receptors. Mol. Pharmacol. 65, 1293–1301.
Moratalla, R., Elibol, B., Vallejo, M., and Graybiel, A. M. (1996). Network-level changes in expression of inducible Fos-Jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron 17, 147–156.
Morgan, J. I., and Curran, T. (1995). Immediate-early genes: ten years on. Trends Neurosci. 18, 66–67.
Moussawi, K., and Kalivas, P. W. (2010). Group II metabotropic glutamate receptors (mGlu2/3) in drug addiction. Eur. J. Pharmacol. 639, 115–122.
Muller, D. L., and Unterwald, E. M. (2005). D1 dopamine receptors modulate deltaFosB induction in rat striatum after intermittent morphine administration. J. Pharmacol. Exp. Ther. 314, 148–154.
Nagerl, U. V., Eberhorn, N., Cambridge, S. B., and Bonhoeffer, T. (2004). Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44, 759–767.
Nagy, G., Matti, U., Nehring, R. B., Binz, T., Rettig, J., Neher, E., and Sorensen, J. B. (2002). Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J. Neurosci. 22, 9278–9286.
Namkung, Y., and Sibley, D. R. (2004). Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J. Biol. Chem. 279, 49533–49541.
Narita, M., Aoki, T., Ozaki, S., Yajima, Y., and Suzuki, T. (2001). Involvement of protein kinase Cgamma isoform in morphine-induced reinforcing effects. Neuroscience 103, 309–314.
Natochin, M., Lester, B., Peterson, Y. K., Bernard, M. L., Lanier, S. M., and Artemyev, N. O. (2000). AGS3 inhibits GDP dissociation from galpha subunits of the Gi family and rhodopsin-dependent activation of transducin. J. Biol. Chem. 275, 40981–40985.
Neve, K. A., Seamans, J. K., and Trantham-Davidson, H. (2004). Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 24, 165–205.
Newton, P. M., Kim, J. A., Mcgeehan, A. J., Paredes, J. P., Chu, K., Wallace, M. J., Roberts, A. J., Hodge, C. W., and Messing, R. O. (2007). Increased response to morphine in mice lacking protein kinase C epsilon. Genes Brain Behav. 6, 329–338.
Newton, P. M., and Messing, R. O. (2007). Increased sensitivity to the aversive effects of ethanol in PKCepsilon null mice revealed by place conditioning. Behav. Neurosci. 121, 439–442.
Nomoto, S., Adachi, K., Yang, L. X., Hirata, Y., Muraguchi, S., and Kiuchi, K. (1997). Distribution of RGS4 mRNA in mouse brain shown by in situ hybridization. Biochem. Biophys. Res. Commun. 241, 281–287.
Norrholm, S. D., Bibb, J. A., Nestler, E. J., Ouimet, C. C., Taylor, J. R., and Greengard, P. (2003). Cocaine-induced proliferation of dendritic spines in nucleus accumbens is dependent on the activity of cyclin-dependent kinase-5. Neuroscience 116, 19–22.
Nye, H. E., Hope, B. T., Kelz, M. B., Iadarola, M., and Nestler, E. J. (1995). Pharmacological studies of the regulation of chronic FOS-related antigen induction by cocaine in the striatum and nucleus accumbens. J. Pharmacol. Exp. Ther. 275, 1671–1680.
Oehrlein, S. A., Parker, P. J., and Herget, T. (1996). Phosphorylation of GAP-43 (growth-associated protein of 43 kDa) by conventional, novel and atypical isotypes of the protein kinase C gene family: differences between oligopeptide and polypeptide phosphorylation. Biochem. J. 317(Pt 1), 219–224.
Okamoto, K., Nagai, T., Miyawaki, A., and Hayashi, Y. (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 7, 1104–1112.
Olive, M. F., Mehmert, K. K., Messing, R. O., and Hodge, C. W. (2000). Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKCepsilon-deficient mice. Eur. J. Neurosci. 12, 4131–4140.
Olive, M. F., and Newton, P. M. (2010). Protein kinase C isozymes as regulators of sensitivity to and self-administration of drugs of abuse-studies with genetically modified mice. Behav. Pharmacol. 21, 493–499.
Ouimet, C. C., Langley-Gullion, K. C., and Greengard, P. (1998). Quantitative immunocytochemistry of DARPP-32-expressing neurons in the rat caudatoputamen. Brain Res. 808, 8–12.
Pearce, L. R., Komander, D., and Alessi, D. R. (2010). The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22.
Penela, P., Ribas, C., and Mayor, F. Jr. (2003). Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell. Signal. 15, 973–981.
Perrotti, L. I., Weaver, R. R., Robison, B., Renthal, W., Maze, I., Yazdani, S., Elmore, R. G., Knapp, D. J., Selley, D. E., Martin, B. R., Sim-Selley, L., Bachtell, R. K., Self, D. W., and Nestler, E. J. (2008). Distinct patterns of deltaFosB induction in brain by drugs of abuse. Synapse 62, 358–369.
Phillips, P. E., Stuber, G. D., Heien, M. L., Wightman, R. M., and Carelli, R. M. (2003). Subsecond dopamine release promotes cocaine seeking. Nature 422, 614–618.
Pierce, R. C., and Vanderschuren, L. J. (2010). Kicking the habit: the neural basis of ingrained behaviors in cocaine addiction. Neurosci. Biobehav. Rev. 35, 212–219.
Ping, A., Xi, J., Prasad, B. M., Wang, M. H., and Kruzich, P. J. (2008). Contributions of nucleus accumbens core and shell GluR1 containing AMPA receptors in AMPA- and cocaine-primed reinstatement of cocaine-seeking behavior. Brain Res. 1215, 173–182.
Pollock, V. V., Pastoor, T. E., and Wecker, L. (2007). Cyclic AMP-dependent protein kinase (PKA) phosphorylates Ser362 and 467 and protein kinase C phosphorylates Ser550 within the M3/M4 cytoplasmic domain of human nicotinic receptor alpha4 subunits. J. Neurochem. 103, 456–466.
Porrino, L. J., Smith, H. R., Nader, M. A., and Beveridge, T. J. (2007). The effects of cocaine: a shifting target over the course of addiction. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 1593–1600.
Prus, A. J., James, J. R., and Rosecrans, J. A. (2009). “Conditioned place preference,” in Methods of Behavior Analysis in Neuroscience, 2nd Edn., Chap. 4, ed. J. J. Buccafusco (Boca Raton, FL: CRC Press).
Psifogeorgou, K., Papakosta, P., Russo, S. J., Neve, R. L., Kardassis, D., Gold, S. J., and Zachariou, V. (2007). RGS9-2 is a negative modulator of mu-opioid receptor function. J. Neurochem. 103, 617–625.
Pulipparacharuvil, S., Renthal, W., Hale, C. F., Taniguchi, M., Xiao, G., Kumar, A., Russo, S. J., Sikder, D., Dewey, C. M., Davis, M. M., Greengard, P., Nairn, A. C., Nestler, E. J., and Cowan, C. W. (2008). Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron 59, 621–633.
Qi, Z. H., Song, M., Wallace, M. J., Wang, D., Newton, P. M., Mcmahon, T., Chou, W. H., Zhang, C., Shokat, K. M., and Messing, R. O. (2007). Protein kinase C epsilon regulates gamma-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of gamma2 subunits. J. Biol. Chem. 282, 33052–33063.
Rahman, Z., Gold, S. J., Potenza, M. N., Cowan, C. W., Ni, Y. G., He, W., Wensel, T. G., and Nestler, E. J. (1999). Cloning and characterization of RGS9-2: a striatal-enriched alternatively spliced product of the RGS9 gene. J. Neurosci. 19, 2016–2026.
Richards, G., Messer, J., Malherbe, P., Pink, R., Brockhaus, M., Stadler, H., Wichmann, J., Schaffhauser, H., and Mutel, V. (2005). Distribution and abundance of metabotropic glutamate receptor subtype 2 in rat brain revealed by [3H]LY354740 binding in vitro and quantitative radioautography: correlation with the sites of synthesis, expression, and agonist stimulation of [35S]GTPgammas binding. J. Comp. Neurol. 487, 15–27.
Robbins, T. W., Ersche, K. D., and Everitt, B. J. (2008). Drug addiction and the memory systems of the brain. Ann. N. Y. Acad. Sci. 1141, 1–21.
Saal, D., Dong, Y., Bonci, A., and Malenka, R. C. (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37, 577–582.
Salzmann, J., Marie-Claire, C., Le Guen, S., Roques, B. P., and Noble, F. (2003). Importance of ERK activation in behavioral and biochemical effects induced by MDMA in mice. Br. J. Pharmacol. 140, 831–838.
Schmidt, H. D., Anderson, S. M., Famous, K. R., Kumaresan, V., and Pierce, R. C. (2005). Anatomy and pharmacology of cocaine priming-induced reinstatement of drug seeking. Eur. J. Pharmacol. 526, 65–76.
Schmidt, H. D., Anderson, S. M., and Pierce, R. C. (2006). Stimulation of D1-like or D2 dopamine receptors in the shell, but not the core, of the nucleus accumbens reinstates cocaine-seeking behaviour in the rat. Eur. J. Neurosci. 23, 219–228.
Schultz, W., Tremblay, L., and Hollerman, J. R. (2000). Reward processing in primate orbitofrontal cortex and basal ganglia. Cereb. Cortex 10, 272–284.
Schumann, J., and Yaka, R. (2009). Prolonged withdrawal from repeated noncontingent cocaine exposure increases NMDA receptor expression and ERK activity in the nucleus accumbens. J. Neurosci. 29, 6955–6963.
Self, D. W., Barnhart, W. J., Lehman, D. A., and Nestler, E. J. (1996). Opposite modulation of cocaine-seeking behavior by D1- and D2-like dopamine receptor agonists. Science 271, 1586–1589.
Self, D. W., and Stein, L. (1992). The D1 agonists SKF 82958 and SKF 77434 are self-administered by rats. Brain Res. 582, 349–352.
Shaham, Y., Shalev, U., Lu, L., De Wit, H., and Stewart, J. (2003). The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl.) 168, 3–20.
Shaw-Lutchman, T. Z., Barrot, M., Wallace, T., Gilden, L., Zachariou, V., Impey, S., Duman, R. S., Storm, D., and Nestler, E. J. (2002). Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J. Neurosci. 22, 3663–3672.
Shiflett, M. W., and Balleine, B. W. (2011a). Contributions of ERK signaling in the striatum to instrumental learning and performance. Behav. Brain Res. 218, 240–247.
Shiflett, M. W., and Balleine, B. W. (2011b). Molecular substrates of action control in cortico-striatal circuits. Prog. Neurobiol. 95, 1–13.
Shippenberg, T. S., and Rea, W. (1997). Sensitization to the behavioral effects of cocaine: modulation by dynorphin and kappa-opioid receptor agonists. Pharmacol. Biochem. Behav. 57, 449–455.
Sung, J. Y., Engmann, O., Teylan, M. A., Nairn, A. C., Greengard, P., and Kim, Y. (2008). WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 105, 3112–3116.
Surmeier, D. J., Bargas, J., Hemmings, H. C. Jr., Nairn, A. C., and Greengard, P. (1995). Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14, 385–397.
Surmeier, D. J., Ding, J., Day, M., Wang, Z., and Shen, W. (2007). D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235.
Sutton, M. A., Mcgibney, K., and Beninger, R. J. (2000). Conditioned locomotion in rats following amphetamine infusion into the nucleus accumbens: blockade by coincident inhibition of protein kinase A. Behav. Pharmacol. 11, 365–376.
Svenningsson, P., Nairn, A. C., and Greengard, P. (2005). DARPP-32 mediates the actions of multiple drugs of abuse. AAPS J. 7, E353–E360.
Svenningsson, P., Nishi, A., Fisone, G., Girault, J. A., Nairn, A. C., and Greengard, P. (2004). DARPP-32: an integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 44, 269–296.
Taylor, J. R., Lynch, W. J., Sanchez, H., Olausson, P., Nestler, E. J., and Bibb, J. A. (2007). Inhibition of Cdk5 in the nucleus accumbens enhances the locomotor-activating and incentive-motivational effects of cocaine. Proc. Natl. Acad. Sci. U.S.A. 104, 4147–4152.
Taymans, J. M., Wintmolders, C., Te Riele, P., Jurzak, M., Groenewegen, H. J., Leysen, J. E., and Langlois, X. (2002). Detailed localization of regulator of G protein signaling 2 messenger ribonucleic acid and protein in the rat brain. Neuroscience 114, 39–53.
Tepper, J. M., Abercrombie, E. D., and Bolam, J. P. (2007). Basal ganglia macrocircuits. Prog. Brain Res. 160, 3–7.
Thomas, E. A., Danielson, P. E., and Sutcliffe, J. G. (1998). RGS9: a regulator of G-protein signalling with specific expression in rat and mouse striatum. J. Neurosci. Res. 52, 118–124.
Thomas, M. J., Beurrier, C., Bonci, A., and Malenka, R. C. (2001). Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat. Neurosci. 4, 1217–1223.
Thomas, M. J., Kalivas, P. W., and Shaham, Y. (2008). Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br. J. Pharmacol. 154, 327–342.
Torregrossa, M. M., Corlett, P. R., and Taylor, J. R. (2011). Aberrant learning and memory in addiction. Neurobiol. Learn. Mem. PMID: 21376820. [Epub ahead of print].
Traynor, J. (2010). Regulator of G protein-signaling proteins and addictive drugs. Ann. N. Y. Acad. Sci. 1187, 341–352.
Turgeon, S. M., Pollack, A. E., and Fink, J. S. (1997). Enhanced CREB phosphorylation and changes in c-Fos and FRA expression in striatum accompany amphetamine sensitization. Brain Res. 749, 120–126.
Ungless, M. A., Whistler, J. L., Malenka, R. C., and Bonci, A. (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411, 583–587.
Valjent, E., Corbille, A. G., Bertran-Gonzalez, J., Herve, D., and Girault, J. A. (2006). Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc. Natl. Acad. Sci. U.S.A. 103, 2932–2937.
Valjent, E., Corvol, J. C., Pages, C., Besson, M. J., Maldonado, R., and Caboche, J. (2000). Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J. Neurosci. 20, 8701–8709.
Valjent, E., Pages, C., Herve, D., Girault, J. A., and Caboche, J. (2004). Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur. J. Neurosci. 19, 1826–1836.
Valjent, E., Pages, C., Rogard, M., Besson, M. J., Maldonado, R., and Caboche, J. (2001). Delta 9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur. J. Neurosci. 14, 342–352.
Valjent, E., Pascoli, V., Svenningsson, P., Paul, S., Enslen, H., Corvol, J. C., Stipanovich, A., Caboche, J., Lombroso, P. J., Nairn, A. C., Greengard, P., Herve, D., and Girault, J. A. (2005). Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496.
Volkow, N. D., Wang, G. J., Fowler, J. S., Gatley, S. J., Ding, Y. S., Logan, J., Dewey, S. L., Hitzemann, R., and Lieberman, J. (1996). Relationship between psychostimulant-induced “high” and dopamine transporter occupancy. Proc. Natl. Acad. Sci. U.S.A. 93, 10388–10392.
Walters, C. L., and Blendy, J. A. (2001). Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J. Neurosci. 21, 9438–9444.
Wayman, G. A., Lee, Y. S., Tokumitsu, H., Silva, A. J., and Soderling, T. R. (2008). Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron 59, 914–931.
Weed, M. R., Vanover, K. E., and Woolverton, W. L. (1993). Reinforcing effect of the D1 dopamine agonist SKF 81297 in rhesus monkeys. Psychopharmacology (Berl.) 113, 51–52.
Wei, F. Y., Tomizawa, K., Ohshima, T., Asada, A., Saito, T., Nguyen, C., Bibb, J. A., Ishiguro, K., Kulkarni, A. B., Pant, H. C., Mikoshiba, K., Matsui, H., and Hisanaga, S. (2005). Control of cyclin-dependent kinase 5 (Cdk5) activity by glutamatergic regulation of p35 stability. J. Neurochem. 93, 502–512.
Weiss, F. (2010). “Advances in animal models of relapse for addiction research,” in Advances in the Neuroscience of Addiction, 2nd Edn., Chap. 1, eds C. M. Kuhn and G. F. Koob (Boca Raton, FL: CRC Press).
Wise, R. A., Leone, P., Rivest, R., and Leeb, K. (1995a). Elevations of nucleus accumbens dopamine and DOPAC levels during intravenous heroin self-administration. Synapse 21, 140–148.
Wise, R. A., Newton, P., Leeb, K., Burnette, B., Pocock, D., and Justice, J. B. Jr. (1995b). Fluctuations in nucleus accumbens dopamine concentration during intravenous cocaine self-administration in rats. Psychopharmacology (Berl.) 120, 10–20.
Wolf, M. E. (2010). Regulation of AMPA receptor trafficking in the nucleus accumbens by dopamine and cocaine. Neurotox. Res. 18, 393–409.
Woolverton, W. L., Goldberg, L. I., and Ginos, J. Z. (1984). Intravenous self-administration of dopamine receptor agonists by rhesus monkeys. J. Pharmacol. Exp. Ther. 230, 678–683.
Xi, Z. X., Kiyatkin, M., Li, X., Peng, X. Q., Wiggins, A., Spiller, K., Li, J., and Gardner, E. L. (2010). N-acetylaspartylglutamate (NAAG) inhibits intravenous cocaine self-administration and cocaine-enhanced brain-stimulation reward in rats. Neuropharmacology 58, 304–313.
Xi, Z. X., Ramamoorthy, S., Baker, D. A., Shen, H., Samuvel, D. J., and Kalivas, P. W. (2002). Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J. Pharmacol. Exp. Ther. 303, 608–615.
Xie, G. Q., Wang, S. J., Li, J., Cui, S. Z., Zhou, R., Chen, L., and Yuan, X. R. (2009). Ethanol attenuates the HFS-induced, ERK-mediated LTP in a dose-dependent manner in rat striatum. Alcohol. Clin. Exp. Res. 33, 121–128.
Yan, J. Z., Xu, Z., Ren, S. Q., Hu, B., Yao, W., Wang, S. H., Liu, S. Y., and Lu, W. (2011). Protein kinase C promotes N-methyl-D-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation. J. Biol. Chem. 286, 25187–25200.
Yokel, R. A., and Wise, R. A. (1978). Amphetamine- type reinforcement by dopaminergic agonists in the rat. Psychopharmacology (Berl.) 58, 289–296.
Zachariou, V., Bolanos, C. A., Selley, D. E., Theobald, D., Cassidy, M. P., Kelz, M. B., Shaw-Lutchman, T., Berton, O., Sim-Selley, L. J., Dileone, R. J., Kumar, A., and Nestler, E. J. (2006). An essential role for deltaFosB in the nucleus accumbens in morphine action. Nat. Neurosci. 9, 205–211.
Zachariou, V., Georgescu, D., Sanchez, N., Rahman, Z., Dileone, R., Berton, O., Neve, R. L., Sim-Selley, L. J., Selley, D. E., Gold, S. J., and Nestler, E. J. (2003). Essential role for RGS9 in opiate action. Proc. Natl. Acad. Sci. U.S.A. 100, 13656–13661.
Keywords: addiction, signaling, plasticity, spines, dopamine, glutamate, phosphorylation
Citation: Philibin SD, Hernandez A, Self DW and Bibb JA (2011) Striatal signal transduction and drug addiction. Front. Neuroanat. 5:60. doi: 10.3389/fnana.2011.00060
Received: 10 May 2011;
Accepted: 01 September 2011;
Published online: 20 September 2011.
Edited by:
Emmanuel Valjent, Université Montpellier 1 & 2, FranceReviewed by:
Marina Picciotto, Yale University, USABruce Thomas Hope, National Institute on Drug Abuse, USA
Copyright: © 2011 Philibin, Hernandez, Self and Bibb. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Scott D. Philibin, Laboratory of Dr. James A. Bibb, Department of Psychiatry, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9070, USA. e-mail:c2NvdHQucGhpbGliaW5AdXRzb3V0aHdlc3Rlcm4uZWR1