 Steffen Gonda1†
Steffen Gonda1† Jan Giesen1†
Jan Giesen1† Alexander Sieberath1Fabian West1Raoul Buchholz1Oliver Klatt1Tim Ziebarth2Andrea Räk1Sabine Kleinhubbert1
Alexander Sieberath1Fabian West1Raoul Buchholz1Oliver Klatt1Tim Ziebarth2Andrea Räk1Sabine Kleinhubbert1 Christian Riedel1
Christian Riedel1 Michael Hollmann3Mohammad I. K. Hamad1‡
Michael Hollmann3Mohammad I. K. Hamad1‡ Andreas Reiner2
Andreas Reiner2 Petra Wahle1*
Petra Wahle1*- 1Developmental Neurobiology, Faculty of Biology and Biotechnology, Ruhr University Bochum, Bochum, Germany
- 2Cellular Neurobiology, Faculty of Biology and Biotechnology, Ruhr University Bochum, Bochum, Germany
- 3Biochemistry I – Receptor Biochemistry, Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Bochum, Germany
NMDA receptors are important players for neuronal differentiation. We previously reported that antagonizing NMDA receptors with APV blocked the growth-promoting effects evoked by the overexpression of specific calcium-permeable or flip-spliced AMPA receptor subunits and of type I transmembrane AMPA receptor regulatory proteins which both exclusively modify apical dendritic length and branching of cortical pyramidal neurons. These findings led us to characterize the role of GluN2B and GluN2A for dendritogenesis using organotypic cultures of rat visual cortex. Antagonizing GluN2B with ifenprodil and Ro25-6981 strongly impaired basal dendritic growth of supra- and infragranular pyramidal cells at DIV 5–10, but no longer at DIV 15–20. Growth recovered after washout, and protein blots revealed an increase of synaptic GluN2B-containing receptors as indicated by a enhanced phosphorylation of the tyrosine 1472 residue. Antagonizing GluN2A with TCN201 and NVP-AAM077 was ineffective at both ages. Dendrite growth of non-pyramidal interneurons was not altered. We attempted to overexpress GluN2A and GluN2B. However, although the constructs delivered currents in HEK cells, there were neither effects on dendrite morphology nor an enhanced sensitivity to NMDA. Further, co-expressing GluN1-1a and GluN2B did not alter dendritic growth. Visualization of overexpressed, tagged GluN2 proteins was successful after immunofluorescence for the tag which delivered rather weak staining in HEK cells as well as in neurons. This suggested that the level of overexpression is too weak to modify dendrite growth. In summary, endogenous GluN2B, but not GluN2A is important for pyramidal cell basal dendritic growth during an early postnatal time window.
Introduction
Dendritic growth is a highly dynamic process (Wong and Ghosh, 2002). Elongation and outgrowth of side branches and retraction-remodeling occur within short time windows of hours and days as has been shown for tadpole tectal neurons (Cline, 2001). The synaptotropic hypothesis postulates that immature branches probe for potential afferent partners, and become stabilized when afferent presynapses form stable contacts (Vaughn, 1989; Cline and Haas, 2008). More meaningful synaptic activity for instance evoked by rearing in an enriched environment leads to an enhanced dendritic complexity (Volkmar and Greenough, 1972). Repetitive maze training enhances branching of distal apical, but not basal dendrites of layer IV and V pyramidal cells in visual cortex even of adult animals (Greenough et al., 1979), whereas deprivation of activity leads to a stunted growth. Among the underlying mediators are glutamate receptors at excitatory axodendritic synapses. Depolarization is translated into calcium currents via voltage-gated channels and NMDA receptors (NMDARs) (Konur and Ghosh, 2005), activation of G-protein signaling (Van Aelst and Cline, 2004), local protein synthesis, and the release of trophic factors acting in a paracrine or autocrine manner (McAllister, 2000; Wong and Ghosh, 2002; Wirth et al., 2003).
The three classes of ionotropic glutamate receptors, AMPA receptors (AMPARs), kainate receptors (KARs) and NMDARs, are richly expressed in developing and adult cortical neurons (Hadzic et al., 2017). All contribute to dendritic growth (Wong and Ghosh, 2002). For developing neocortical pyramidal neurons selected AMPAR subunits, most effectively as flip-spliced variants, are important as well as the KAR GluK2, and both mediate their effects with a contribution by NMDARs (Hamad et al., 2011, 2014; Jack et al., 2018). The importance of NMDARs has been shown in tadpole tectal neurons which fail to grow when inhibited with 100 μM of the non-selective NMDAR antagonist APV, whereas AMPARs and sodium channel activity were not yet important at these early stages (Rajan and Cline, 1998). Similarly, blocking NMDARs with CGP 40116 from postnatal day 1–21 in rats in vivo reduces the length of basal, but not apical dendrites of prefrontal cortical pyramidal cells (Wedzony et al., 2005). These studies relied on non-selective antagonists which do not allow to differentiate between GluN2B- and GluN2A-containing receptors; the subunits known to switch during development (Sheng et al., 1994; Traynelis et al., 2010). The recent generation of GluN2A-preferring antagonists now provide the tools to perform such a differentiation. Thus, we aimed to characterize in organotypic slice cultures the involvement of GluN2A and GluN2B for dendritic growth of cortical neurons.
Materials and Methods
Preparation of Organotypic Cultures (OTC)
Organotypic cortex cultures (OTCs) were prepared as described (Hamad et al., 2011; Jack et al., 2018) from pigmented Long-Evans rats from the in-house breeding facility with approval from the Ruhr-University Animal Research Board and the State of North Rhine-Westphalia. Briefly, visual cortex was explanted at postnatal day 0/P1 (P0, day of birth) and cut into 350 μm thick coronal slices using a McIlwain tissue chopper. Cultures were fed three times a week with semiartificial medium containing 25% adult horse serum, 25% Hank’s balanced Salt Solution, 50% Eagle’s Basal Medium, 1 mM L-glutamine (all from Life Technologies, Karlsruhe, Germany) and 0.65% D-Glucose (Merck, Darmstadt, Germany). To inhibit glial growth, 10 μl of an antimitotic cocktail consisting of uridine, cytosine-β-D-arabinofuranoside and 5-fluorodeoxyuridine (each stock 1 mM, all from Sigma; 10 μM final concentration for each) was applied at DIV 2 for 24 h.
Plasmid Transfection
OTC were gene gun-transfected as described (Hamad et al., 2011; Jack et al., 2018). All plasmids (Table 1) were prepared as endotoxin-free solutions using the EndoFree Plasmid Maxi Kit (Qiagen, Cat No./ID: 12362). Plasmid stocks were diluted to 1 μg/μl and stored at −20°C. Briefly, cartridges were prepared by coating 7 mg gold microparticles (1 μm diameter; Bio-Rad) with 10 μg of the selected plasmids. Cultures were blasted (Helios Gene Gun, Bio-Rad, Munich, Germany) with 180 psi helium pressure.
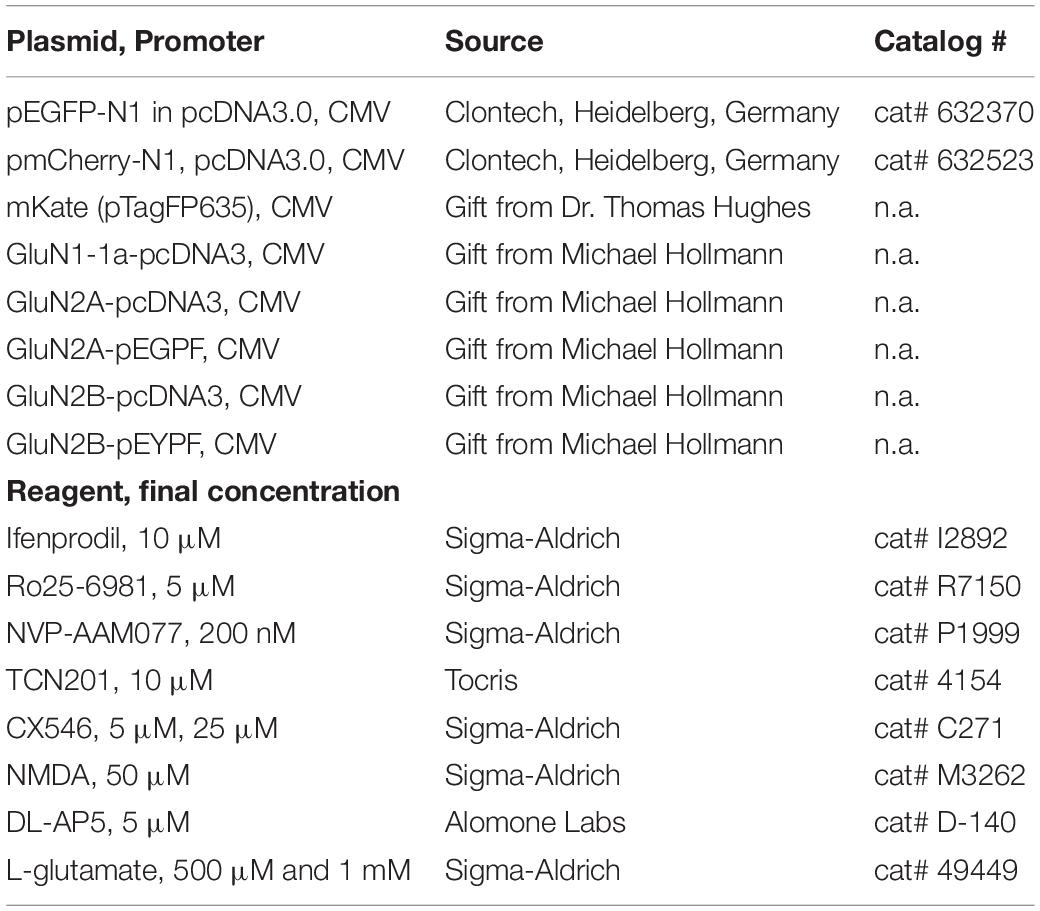
Table 1. Plasmids and NMDA receptor antagonists.
Pharmacological Treatment
The first experiment was designed as follow-up of previous work (Hamad et al., 2011). We tested if an activation of AMPARs with a cognitive enhancer, the ampakine CX546, could alter dendritic growth. For the CX546 experiment, EGFP was transfected at DIV 4 and DIV 9 with analysis at DIV 10 and DIV 20, respectively. CX546 (Sigma) was dissolved in DMSO and applied at 5 μM or 25 μM final concentration to the medium; concentrations were kept low to avoid a knockdown of glutamate receptor expression reported to occur with high concentrations (Lauterborn et al., 2003). DMSO was used as vehicle control at a concentration not exceeding 0.1% in the medium. CX546 at 30 μM has been shown to alter firing pattern in vitro (Lu et al., 2016), to restore long-term potentiation at 25 μM in a Neto1 protein null mouse hippocampus by indirectly enhancing NMDAR synaptic currents (Ng et al., 2009), and to enhance neurite growth of cultured subventricular zone progenitor cells at 5–50 μM (Schitine et al., 2012).
For the NMDAR experiments, OTC were transfected with EGFP either at DIV 4 with analysis at DIV 10, or at DIV 14 with analysis at DIV 20. In the earlier time window GluN2 receptor antagonists were applied at DIV 7 and DIV 9, and in the later time window at DIV 15 and DIV 17, always combined with a medium change. For the recovery experiment, OTC were transfected at DIV 4, treated with ifenprodil at DIV 6 and DIV 8, switched to normal medium at DIV 10, and allowed to recover until DIV 15.
We decided to study in parallel two antagonists selective for GluN2B receptors, ifenprodil [dissolved in ethanol and further diluted with H2O] and Ro25-6981 [dissolved in DMSO], and two antagonists for GluN2A, NVP-AAM077 [dissolved in H2O] and TCN201 [dissolved in DMSO] (Table 1 for concentrations in the medium). Control cultures were mock-stimulated with DMSO at the final concentration or with H2O. Biophysically, ifenprodil and Ro25-6981 have the same mechanism of action at the N-terminal domain; and for ifenprodil a 10 μM concentration has been used for the 2-day incubation of the 150–300 μm thick slice cultures although 3 μM has been shown to already elicit the maximal inhibition (Gray et al., 2011). The two selected GluN2A antagonists differ in action with NVP-AAM077 being a competitive antagonist at the glutamate binding site and TCN201 acting at the glycine cofactor binding site (Neyton and Paoletti, 2006; Traynelis et al., 2010; Edman et al., 2012; Hansen et al., 2012, 2014; Tovar et al., 2013). Subunit-specific inhibition has been shown to deliver results that differ from those obtained with APV (He et al., 2013). The use of two antagonists for each subunit should reduce the probability of false negative or false positive results, and we expected the two pairs of antagonists to deliver comparable results, which turned out to be the case.
Finally, we followed the design of our previous studies overexpressing AMPAR and KAR subunits and their accessory proteins (Hamad et al., 2011, 2014; Jack et al., 2018). We attempted to express plasmids encoding the GluN2A and GluN2B subunit together with EGFP as reporter (for morphometry), or to express plasmids encoding EGFP-tagged GluN2A and EYFP-tagged GluN2B together with mCherry as reporter (for visualization). Further, we expressed GluN1-1a alone and GluN1-1a together with GluN2B (for morphometry). Transfection was done at DIV 5 followed by staining and analysis at DIV 10.
Immunostaining
OTCs were fixed with 37°C 4% paraformaldehyde in 0.1 M phosphate buffer pH 7.4 for 2 h, rinsed, permeabilized for 30 min with 0.3% Triton X-100 in phosphate buffer, blocked with TBS/BSA solution, and incubated overnight in mouse anti-GFP antibody (1:1000; clone GSN24, Sigma-Aldrich, RRID: AB_563117). To visualize the neuronal morphology via the reporter protein, staining proceeded with biotinylated goat anti-mouse (1:1000; Dako A/S, Glostrup, Denmark, product no. E043301-2) for 3 h, followed by ABC reagent for 2 h (Vector Laboratories Inc., Burlingame, CA, United States, cat# PK-7100; RRID: AB_2336827), a HRP reaction with 3,3’-diaminobenzidine (DAB, Sigma-Aldrich, Steinheim, Germany) and H2O2. The resulting DAB product was intensified with 1% OsO4 (Sigma-Aldrich, Steinheim, Germany). Cultures were dehydrated and coverslipped in DEPEX (Sigma-Aldrich, Steinheim, Germany).
To visualize the overexpression, GluN2A-EGFP and GluN2B-EYFP tagged constructs were transfected together with mCherry fluorescence as reporter, and were stained with the mouse anti-GFP antibody developed with an Alexa 488-conjugated secondary (1:500; Thermo Fisher Scientific, cat# A-11001, RRID: AB_2534069) followed by embedding in ImmuMount or RIMS buffer.
NMDA-Induced Dendritic Injury Assay
OTC were challenged with 50 μM NMDA in the medium under live imaging followed by fixation and staining for EGFP as described above. Antagonists were preincubated to allow diffusion into the slice cultures. The time of exposure is indicated in the figure legends. The percentage of transfectants displaying dendritic beading was determined after DAB immunostaining.
Protein Blots
Lysates and blots were performed as described (Engelhardt et al., 2018; Neumann et al., 2019) with OTC from 3 preparations that were exposed to ifenprodil, and allowed to recover from ifenprodil treatment for 12h and 24h. Using membrane strips placed over the molecular weight range of the target protein of interest several proteins of distinct kDa can be assessed per lysate. Antibodies against the following proteins and epitopes were used: GAD-65/67 (mouse; clone 9A6, cat# #ADI-MSA-225-E, 1:2000; Enzo Life Science, Lörrach, Germany), phY1472 GluN2B (rabbit; 1:1000; Rockland Inc., cat# 612-401-C89; via Biomol, Hamburg, Germany), GluN2B (mouse; 1:1000; cat# 06-600, Merck Millipore, Darmstadt, Germany), GluN1 (mouse; 1:1000; NeuroMab, UC Davis, Davis, CA, United States), β-tubulin (mouse; 1:1000; cat# T5293, Sigma-Aldrich, Deisenhofen, Germany), β-actin (mouse; 1:4000; cat# 1978, Sigma-Aldrich, Deisenhofen, Germany), PSD-95 (rabbit; 1:1000; cat# 124 002, Synaptic Systems, Göttingen, Germany). Visualization was done with alkaline phosphatase-conjugated secondaries and reaction with 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (Promega, Mannheim, Germany). Blots were scanned, band intensities assessed with ImageJ followed by normalization to a housekeeping protein. Representative bands were presented together with the actin and tubulin bands.
Expression, Patch-Clamp Recordings, and Immunocytochemistry of NMDA Receptor Constructs in HEK293T Cells
Functional expression of GluN2A-EGFP and GluN2B-EYFP was verified in whole-cell patch clamp recordings. Human embryonic kidney (HEK) 293T cells were grown in DMEM with 8% FBS at 37°C and 5% CO2 on plastic coverslips. Plasmid triple transfections were carried out using polyethylenimine 25.000 with ∼0.2 μg GluN1-1a and ∼0.1 μg GluN2A or GluN2B per ml medium; 0.1 μg mKate was added as transfection marker. To reduce detrimental effects due to NMDA receptor expression the medium was supplemented with 5 mM Mg2+ or 5 μM DL-AP5 (Alomone Labs, D-140). All other chemicals were from Sigma. Whole-cell recordings were performed 24–48 h after transfection on an inverse microscope (DMi8, Leica) using an Axopatch 200B patch-clamp amplifier, a Digidata 1550 A/D converter and pClamp 10.7 software (all Molecular Devices) and a micro-manipulator (Patchstar, Scientifica). In brief, patch pipettes (3–4 MΩ resistance) were pulled from borosilicate glass and filled with internal solution containing 122 mM CsCl, 2 mM NaCl, 2 mM MgCl2, 10 mM EGTA and 10 mM HEPES, pH 7.2. The coverslips were placed in external solution containing 138 mM NaCl, 1.5 mM KCl, 2.5 mM CaCl2, 50 μM glycine and 10 mM HEPES, pH 7.3. External solution and L-glutamate (500 μM or 1 mM; Sigma, cat# 49449) in external solution were applied by means of a gravity-driven perfusion system. Recordings were performed at 22–25°C in voltage-clamp mode at a holding potential of −70 mV. Experiments were repeated minimum three times after independent transfections and baseline corrected. For the evaluation of peak and steady-state currents only recordings with leak currents <500 pA and current changes >20 pA were taken into account.
For immunocytochemistry in HEK293T cells, the expression was performed as described above, but without co-transfection of mKate. After 24–48 h, the transfected HEK293T cells were washed with DPBS and fixed with 4% PFA for 15 min. After washing the cells with DPBS, unspecific binding sites were blocked with Roti-Immunoblock (Roth) and cells permeabilized with 0.25% Triton X-100 for 45 min. Cells were washed with DPBS and incubated with mouse anti-GFP antibody (Sigma, 1:1000) overnight at 4°C. Following multiple washing steps, the cells were incubated with the secondary antibody (Alexa Fluor 568, Invitrogen, cat# A10037) for 60 min at room temperature. Finally, the coverslips were washed and mounted with Roti-Mount FluorCare DAPI. Confocal imaging was performed on a Leica SP5 using 405, 488, and 561 nm laser lines. Fluorescence overlays were prepared using ImageJ/Fiji 1.53c.
Morphological Analysis
For morphometrical analysis, immunostained neurons were reconstructed with the Neurolucida system (MicroBrightField, Inc., Williston, VT, United States) by trained observers blinded to conditions. All reconstructions were crosschecked by an observer blinded to condition for correctness and to classify the cell type. Pyramidal cells and multipolar sparsely spinous interneurons were classified by criteria of dendritic and axonal patterns. Pyramidal neurons were grouped as follows: those of layers II/III have an apical dendrite reaching into layer I, and for layers V/VI we consider those which have an apical dendrite ending in middle layers; thick and thin tufted large layer V pyramidal cells occurred too rarely and were not sampled (Hamad et al., 2014; Jack et al., 2018). Using a ZEISS Axioskop equipped with a discussion bridge we assessed with two trained observers blinded to conditions all completely stained neuronal transfectants per slice culture for symptoms of dendritic injury. We considered only completely labeled neurons with recognizable axons, because dendritic injury often starts with blebbing at distal dendrites. Data are presented as Tables and Sholl plots. Statistical analyses were done with Sigma Plot 12.3 (Systat Software, Erkrath, Germany). Non-parametric ANOVA on ranks tests with corrections for multiple testing (Dunn’s test) and/or non-parametric Mann–Whitney rank sum tests were conducted.
Results
No Effect of the Ampakine CX546 but Underdevelopment With Ifenprodil
The first experiment followed our earlier observation which showed that calcium-permeable and/or flip splice variants (with longer channel open times) of specific GluA subunits increase complexity of pyramidal cell apical, but not basal dendrites (Hamad et al., 2011). We tested if a cognitive enhancer can alter dendritic growth. The classical type 1 ampakine CX546 slows desensitization and deactivation of AMPARs, prolongs the synaptic response, and strengthens long-term potentiation (Arai and Kessler, 2007). This can increase the synthesis of BDNF which stays at somewhat higher levels for several hours and activates TrkB receptors in spines (Lauterborn et al., 2000, 2003, 2009; Montgomery et al., 2009). BDNF via TrkB promotes dendritogenesis (McAllister, 2000; Wirth et al., 2003), and chronic ampakine stimulation has been reported to prevent age-dependent dendritic pruning (Lauterborn et al., 2016). Since previous data had indicated a role of NMDARs (Hamad et al., 2011), we included a treatment with ifenprodil. However, the ampakine treatment at 5 μM from DIV 5 to DIV 10 did not alter dendritic complexity of pyramidal neurons (layers II/III tested) (Supplementary Table 1A). Further, acute wash-in of CX546 at concentrations up to 1 mM did not evoke dendritic beading in 10 DIV OTC, and there was no neuroprotection by ifenprodil (CX546: 4.3 + −1.1% neurons with dendritic beading, 227 neurons from 5 OTC; control, vehicle stimulated: 3.8 + −1.0% neurons with dendritic beading, 233 neurons from 5 OTC; ifenprodil pretreatment followed by ampakine: 3.9 + −1.3% neurons with dendritic beading, 289 neurons from 4 OTC; this is in the <10% range typical for untreated OTC; Hamad et al., 2011). The inefficiency might be due to the low concentration of endogenous AMPARs at DIV 5–10. However, a daily stimulation with CX546 at a higher concentration (25 μM final concentration in the medium, medium change every second day) in more mature cultures from DIV 10–20 also did not alter dendritic complexity of pyramidal neurons of supra- and infragranular layers (Supplementary Table 2A). Further, multipolar non-pyramidal neurons with smooth or sparsely spinous dendrites were neither altered by CX546 exposure at DIV 10 (Supplementary Table 1B) nor at DIV 20 (Supplementary Table 2B). The result is in line with the interpretation of previous studies. Potentiating AMPARs for instance by overexpressing the modulatory type II TARPs does not alter dendritic complexity whereas increasing the number of plasma membrane receptors does promote dendritic growth (Hamad et al., 2011, 2014). The unexpected result was that pyramidal neurons reconstructed from the DIV 10 ifenprodil-treated cultures showed significantly shorter and less branched basal dendrites (Supplementary Table 1A). Interneurons, however, were not affected (Supplementary Table 1B). This led us to study the role of endogenous GluN2B-containing receptors for pyramidal cell dendritic growth.
Blocking of GluN2B but Not GluN2A Prevents NMDA-Induced Dendritic Injury
We have previously reported that GluN1, GluN2A, and GluN2B are expressed in OTC at DIV 5, DIV 10, and DIV 15–20 and the expression level of the three proteins increases with age (Engelhardt et al., 2018; Neumann et al., 2019). First, we confirmed that young neurons in OTC have functional NMDARs on the cell surface. GFP-transfected OTC were challenged at DIV 10 with 50 μM NMDA to evoke dendritic beading as previously shown (Hamad et al., 2011; Jack et al., 2018). Images of live neurons were taken at 5 min before, or just before (0 min) adding NMDA and subsequently every minute for up to 10 to up to 15 min. This revealed that both, pyramidal neurons (Figures 1A–C) and multipolar sparsely spinous interneurons (Figures 1D–G) responded with dendritic beading to NMDA. Preincubation for 1 h with the GluN2B antagonist Ro25-6981 prevented dendritic beading as shown exemplarily for a multipolar neuron (Figures 1H,I). Preincubation for 1 h with the GluN2A antagonist TCN201 could not prevent dendritic beading as shown exemplarily for a pyramidal neuron (Figures 1J,K).
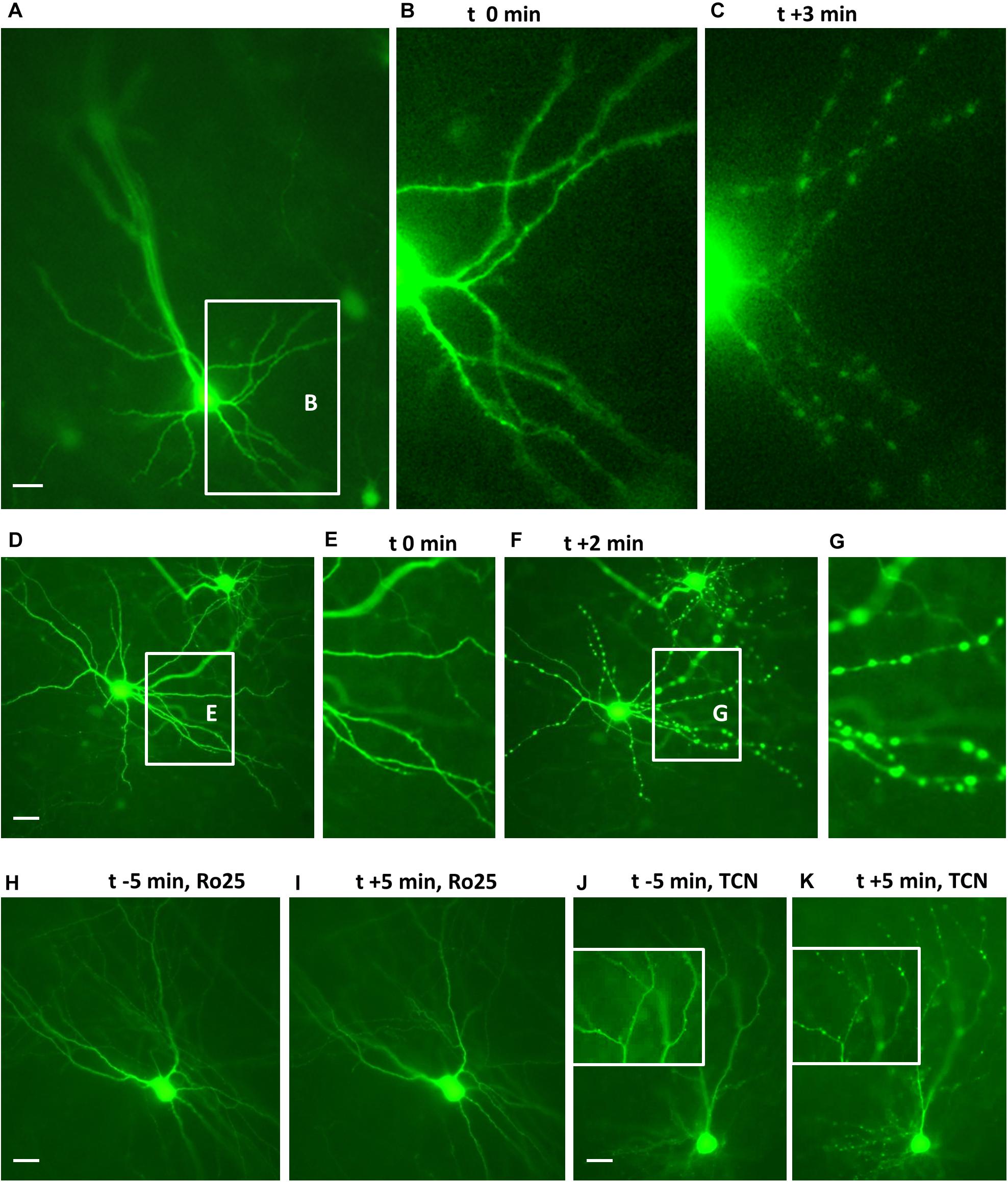
Figure 1. Endogenous GluN2 receptors are functional on DIV 10 neurons. Cultures with live EGFP-expressing neurons were challenged with 50 μM NMDA to evoke dendritic beading. (A–C) Pyramidal neuron before and 3 min (t + 3 min) after application of NMDA. The inset in (A) shows basal dendrites which are seen at higher magnification in (B,C). (D–G): Interneuron before (D,E) and 2 min after NMDA application (F,G). The insets in (D,F) show the dendrites which are at higher magnification in (E,G), respectively. (H,I): Interneuron protected from the NMDA-induced dendritic beading by preincubation with 5 μM Ro25-6981. (J,K) Pyramidal cell not protected from the NMDA-induced dendritic beading by preincubation with 10 μM TCN201; insets show details of apical dendritic branches at higher magnification before and 5 min after application of NMDA. Pial surface is to the upper left for (A–I) and to the top for (J,K), Scales: 10 μm.
To quantify the effects, the proportion of EGFP-labeled neurons with dendritic beading was determined at DIV 10 in OTC that were exposed to 50 μM NMDA for 10 min followed by fixation and staining. About 80% of the transfected pyramidal neurons (Figure 2A) and multipolar interneurons (Figure 2B) displayed swollen varicosities along apical and basal dendrites after exposure to NMDA. Pretreatment with Ro25-6981 substantially prevented dendritic beading; in contrast, preincubation with TCN201 still resulted in over 80% affected neurons of both neuron classes (Figures 2A,B). Together, these results confirm the presence of functional GluN2B-containing NMDA receptors during the early time window.
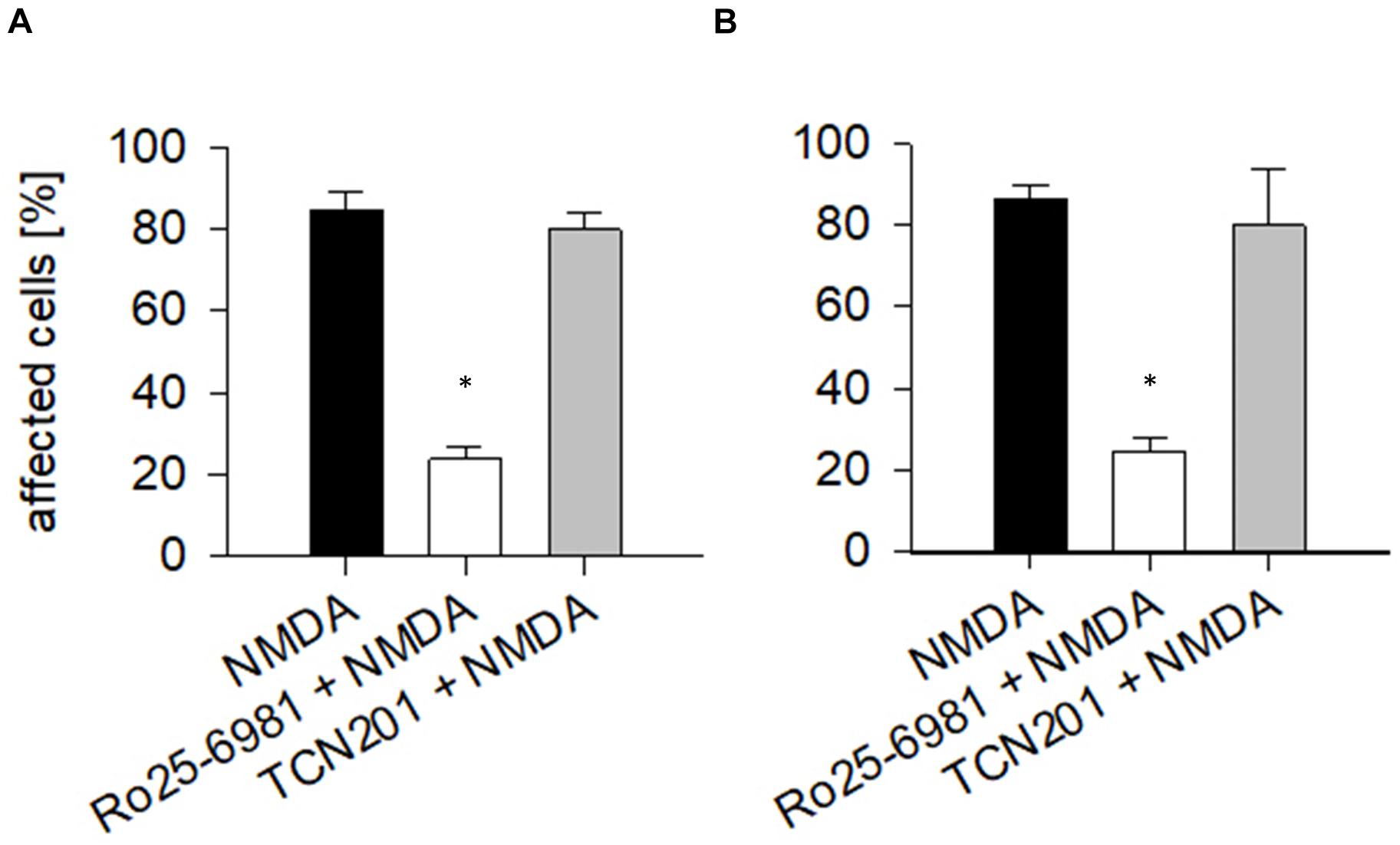
Figure 2. Endogenous NMDA receptors render neurons sensitive to NMDA. At DIV 10, the vast majority of (A) pyramidal cells and of (B) interneurons responded with dendritic beading to NMDA (50 μM) applied for 10 min. Preincubation with Ro25-6981 (5 μM) was neuroprotective (ANOVA on ranks versus control, *p < 0.05). TCN201 [10 μM] was unable to protect. Antagonists had been applied 60 min before the treatment with 50 μM NMDA, followed by immediate fixation. The average percentage of pyramidal neurons (in total 523 cells analyzed) and multipolar non-pyramidal neurons (in total 218 cells analyzed) with dendritic beading per culture was plotted as mean ± S.E.M. 6–10 OTC per condition were analyzed. Only completely stained neurons identifiable by axonal projections were assessed.
Blocking GluN2B Receptors Impairs Dendritic Growth of Young Pyramidal Neurons
Next, we quantified dendritic length and branching of pyramidal neurons of layers II/III and V/VI. All neurons had spiny dendrites and a primary axon descending toward deeper layers and white matter, giving rise to a few long-running, obliquely ascending collaterals. Since the two pairs of antagonists delivered comparable results (reported for supra- and infragranular pyramidal neurons at DIV 10 in Supplementary Table 3), we pooled the data sets for the two GluN2B antagonists (ifenprodil/Ro25-6981) and the two data sets for the GluN2A antagonists (NVP-AAM077/TCN201). The numbers of neurons and independent preparations (number of batches) are given in the Tables. Comparisons were done to the batch-internal controls which were also pooled.
The GluN2B subunit strongly influenced the formation of the basal dendritic tree of pyramidal cells in layers II/III and V/VI. Both, mean basal dendritic length and mean number of basal dendritic segments were significantly reduced after treatment with ifenprodil/Ro25-6981 (Figure 3 and Table 2A). However, the number of basal dendrites per neuron was not different between the groups (pyramidal cells of layers II/III: 4.07 ± 0.12 for EGFP control; 4.10 ± 0.16 for ifenprodil/Ro25-6981; and 4.17 ± 0.16 for NVP-AAM077/TCN201, ANOVA on ranks p = 0.76; pyramidal cells of layers V/VI: 3.87 ± 0.14 for EGFP control; 4.04 ± 0.15 for ifenprodil/Ro25-6981; and 3.5 ± 0.11 for NVP-AAM077/TCN201, ANOVA on ranks p = 0.074). This ruled out that the reduction of average basal dendritic length was caused by an increased sprouting of shorter processes. In the case of supragranular pyramidal cells the reduction of apical dendritic length and branching was only borderline (Table 2A). GluN2A seemed not to be involved in dendritic growth during the early time window since neither NVP-AAM077 nor TCN201 treatment changed the dendritic morphology. Both GluN2 subunits are expressed in cortical non-pyramidal interneurons (Hadzic et al., 2017). However, their dendritic growth seemed independent of endogenously expressed GluN2A- and GluN2B-containing NMDAR since none of the antagonists was able to alter the dendritic growth pattern (Table 2B).
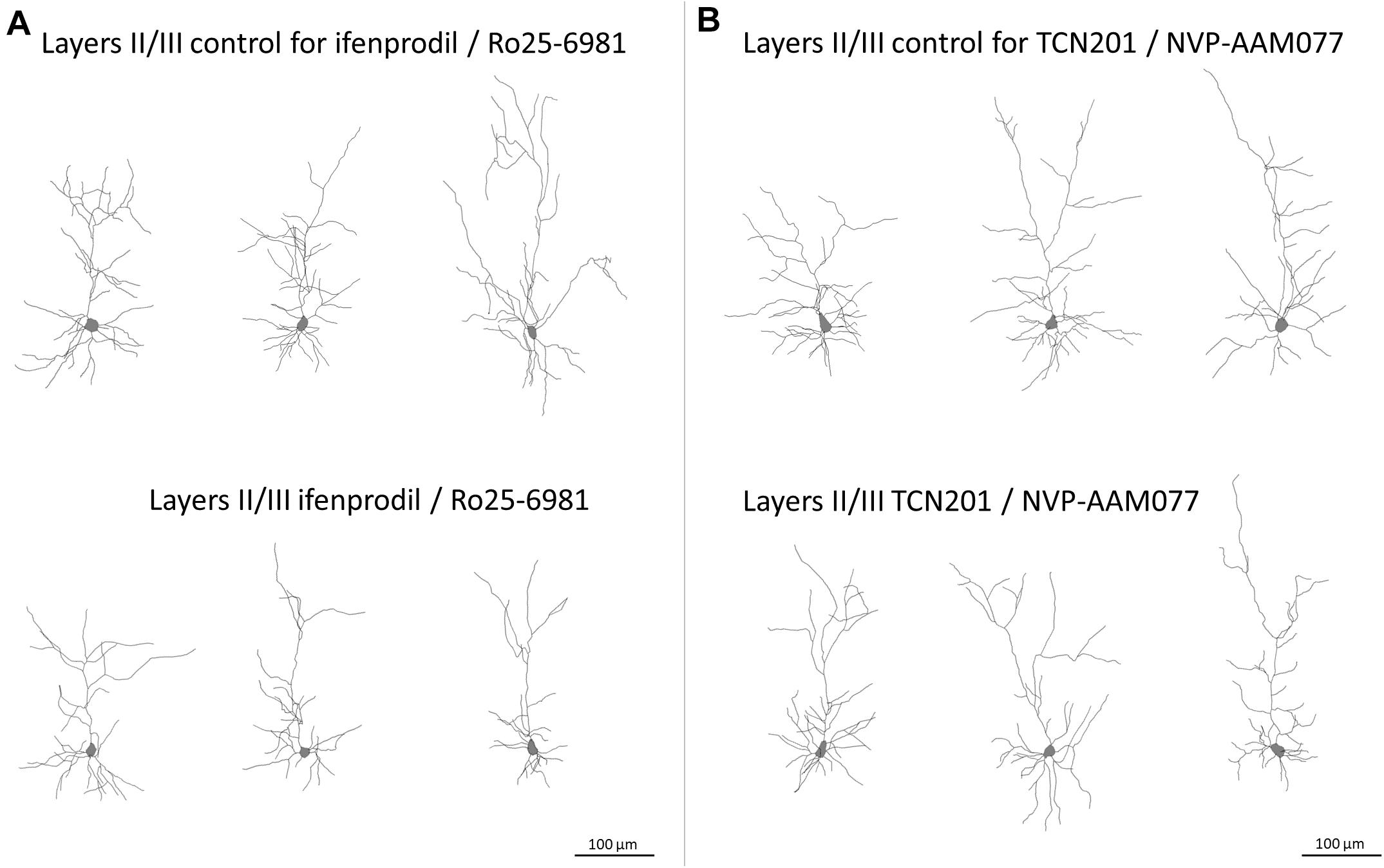
Figure 3. Skeletal drawings of representative pyramidal neurons at DIV 10. (A) Control and ifenprodil/Ro25-6981 treated pyramidal neurons of layers II/III. Note reduced dimensions of basal dendrites in the ifenprodil/Ro25-6981 treated neurons. (B) Control and TCN201/NVP-AAM077 treated pyramidal neurons of layers II/III, Scale = 100 μm.
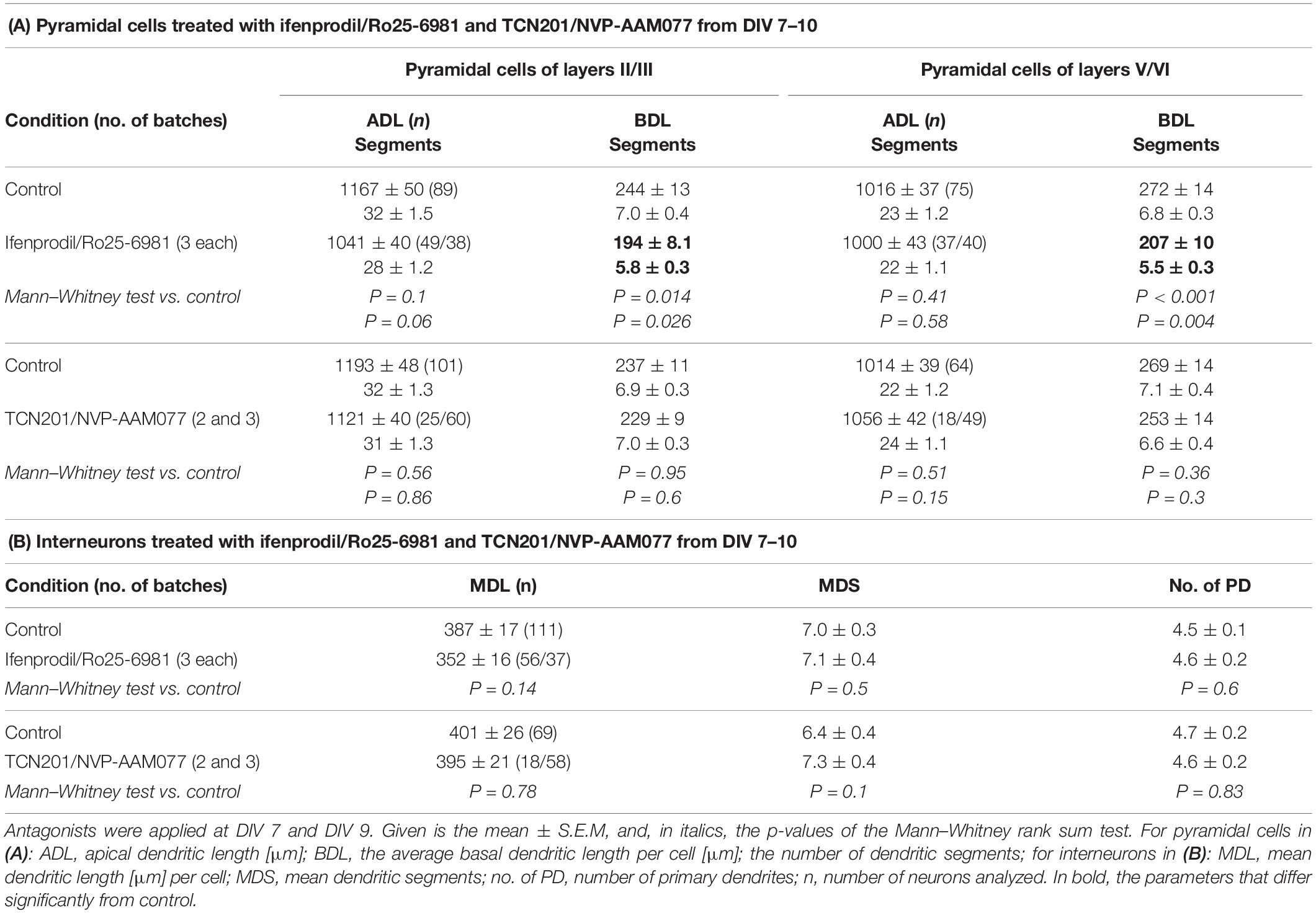
Table 2. Effects of the GluN2B and GluN2A antagonists at DIV 10.
The Sholl analyses confirmed the hypomorphy of ifenprodil/Ro25-6981 treated neurons (Figure 4). Apical dendrites of supragranular, but not infragranular pyramidal neurons seemed to have somewhat less branches (Figures 4A,B) although total intersections were not significantly different according to the ANOVA on ranks; further, apical dendrites were not different in the pilot experiment (Supplementary Table 1). The basal dendritic branching of supra- infragranular pyramidal neurons (Figures 4C,D) clearly remained below control within 50 to up to 200 μm distance from the soma suggesting a substantial reduction in dendritic branching. In contrast, the NVP-AAM077/TCN201 curves overlap with the control (Figures 4C,D).
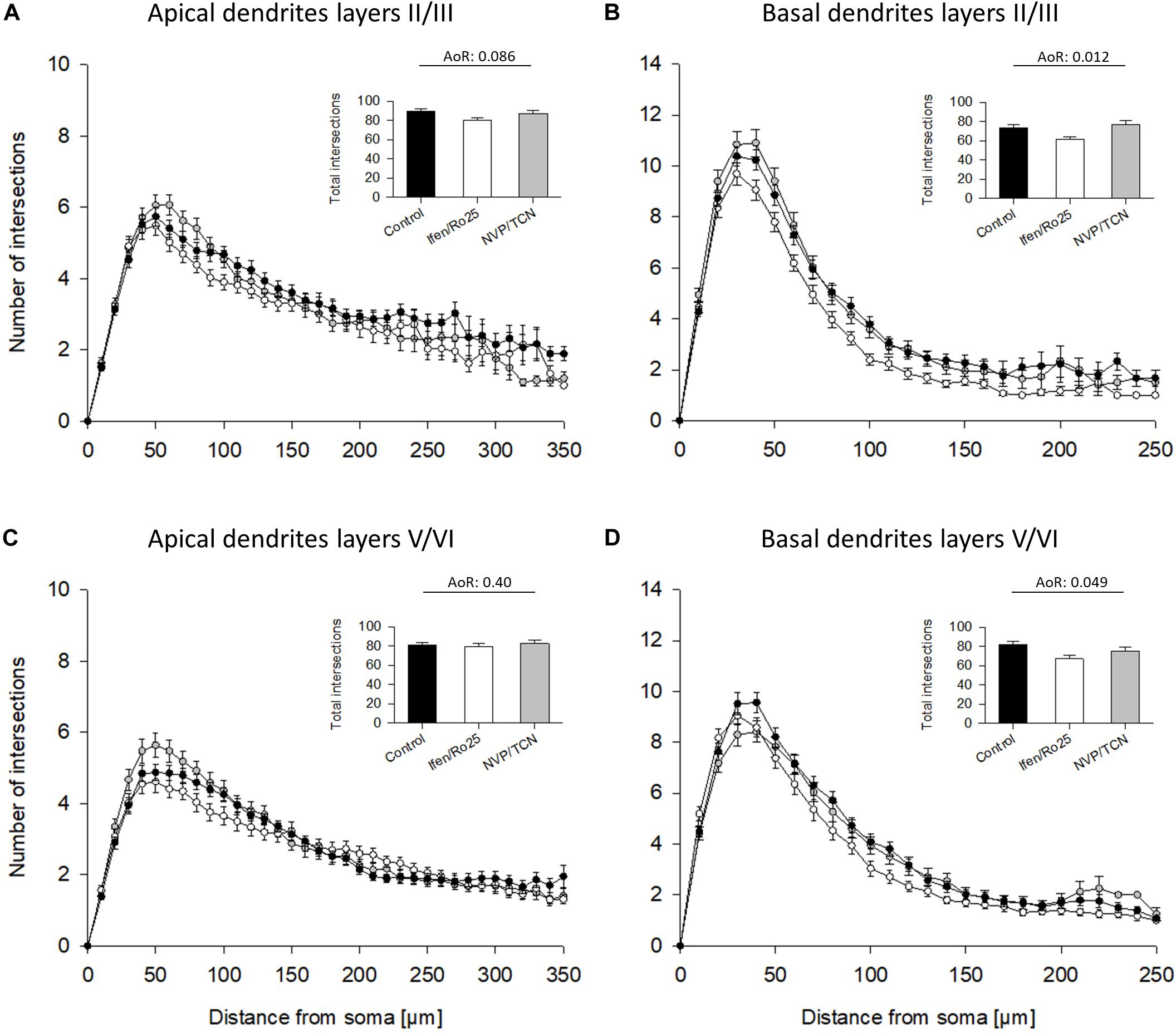
Figure 4. Sholl analyses of apical and basal dendrites of DIV 10 pyramidal cells of layers II/III and V/VI. (A) Sholl analyses of apical dendrites of pyramidal cells of layers II/III. Total intersections (mean with S.E.M.) are given in the insets. (B) Sholl analyses of basal dendrites of pyramidal cells of layers II/III showed a decrease of basal dendritic complexity in the ifenprodil/Ro25-6981 (Ifen/Ro25) treated neurons, but not NVP-AAM077/TCN201 (NVP/TCN) treated neurons between 50 and 200 μm from the soma. (C) Sholl analyses of apical dendrites of pyramidal cells of layers V/VI. (D) Sholl analyses of basal dendrites of pyramidal cells of layers V/VI showed a decrease of basal dendritic complexity in ifenprodil/Ro-6981 treated neurons, but not NVP-AAM077/TCN201 treated neurons between 50 and 150 μm distance from the soma. (B,D) ANOVA on ranks (AoR) versus control identified the significant reduction of total intersections in the ifenprodil/Ro25-6981 groups. The number of neurons analyzed per condition is given in Table 2.
The Contribution of GluN2B-Containing Receptors to Dendritogenesis Was Temporally Limited
The GluN2 receptor composition switches during development from mainly GluN2B- to mainly GluN2A-containing heterodimeric and heterotrimeric receptors (Traynelis et al., 2010) with GluN1/2A/2B triheteromers presumably representing a majority of synaptic NMDAR in juvenile and adult neocortex and hippocampus (Sheng et al., 1994; Tovar et al., 2013; Stroebel et al., 2018). The switch depends on the maturational state of the neurons and also occurs in in vitro systems (Williams et al., 1993). Therefore, we tested a blockade of GluN2A and GluN2B receptors in more differentiated neurons at DIV 15–20. Again, results of the two antagonists of each pair went into the same direction, and we therefore pooled the DIV 20 data. None of the antagonists was able to significantly alter dendritic dimensions of pyramidal cells (Table 3A) or of interneurons (Table 3B). The Sholl analysis revealed a larger scatter at the higher age group, but by and large curves overlapped the control curve and total intersections were not different from control (Figure 5). This suggested that GluN2B has lost its role as dendritic modifier by DIV 15, and that GluN2A has no measurable role in dendritic growth.
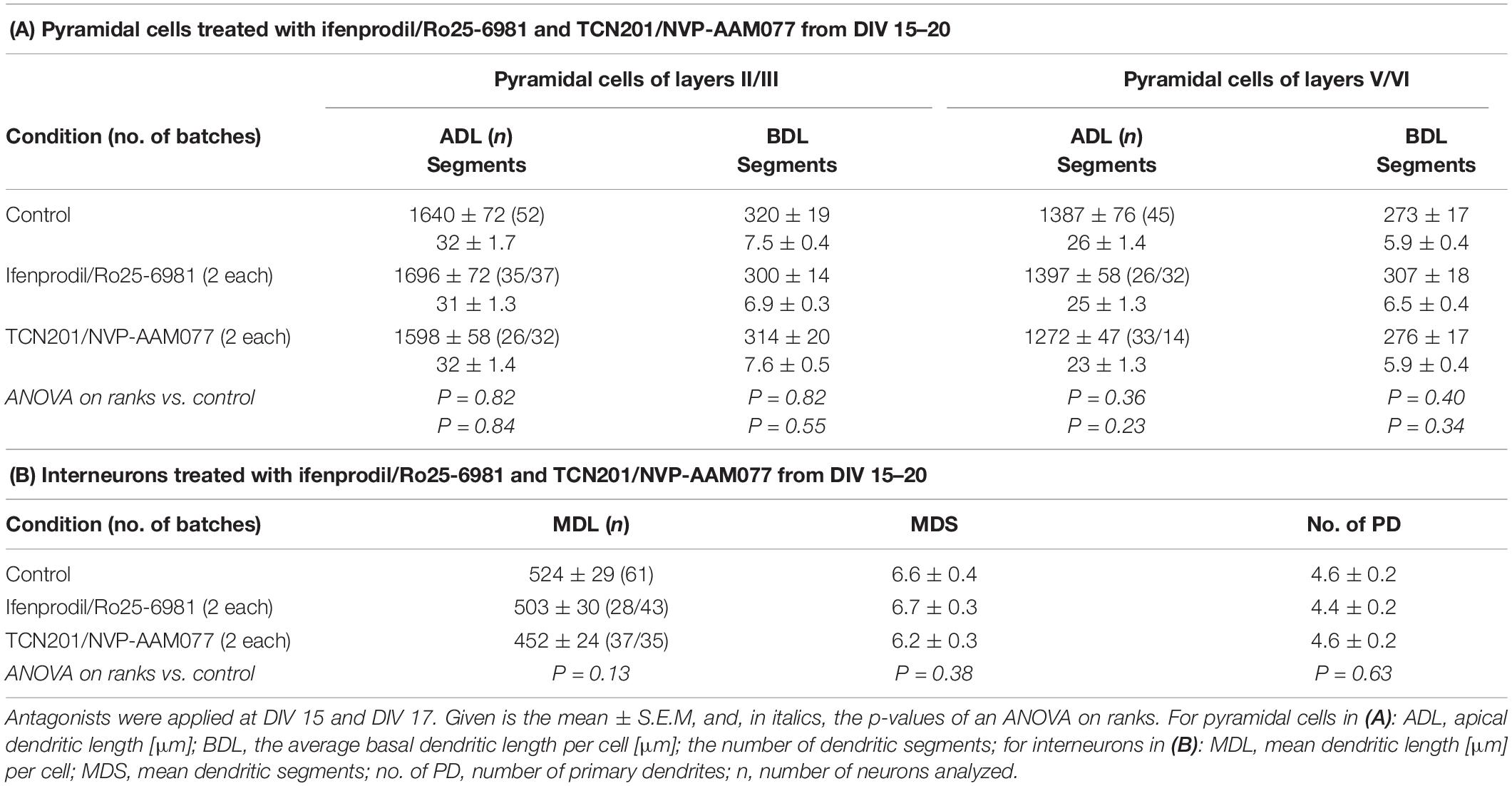
Table 3. Effects of the GluN2B and GluN2A antagonists at DIV 20.
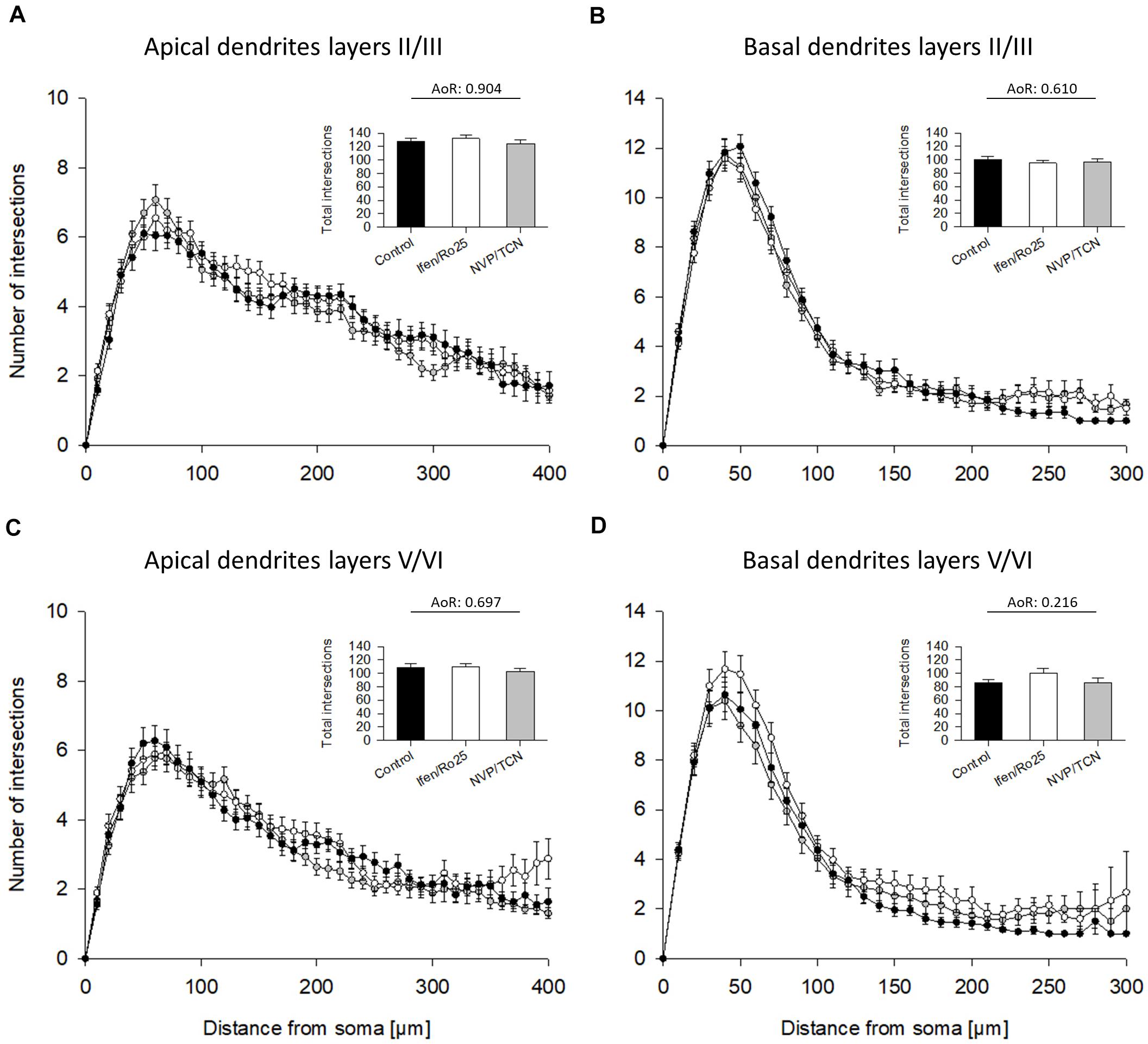
Figure 5. Sholl analyses of apical and basal dendrites of DIV 20 pyramidal cells of layers II/III and V/VI. (A) Sholl analyses of apical dendrites of pyramidal cells of layers II/III treated with ifenprodil/Ro25-6981 (Ifen/Ro25) and NVP-AAM077/TCN201 (NVP/TCN). Total intersections (mean with S.E.M.) are given in the insets. (B) Sholl analyses of basal dendrites of pyramidal cells of layers II/III. (C) Sholl analyses of apical dendrites of pyramidal cells of layers V/VI. (D) Sholl analyses of basal dendrites of pyramidal cells of layers V/VI. Note that the curves overlap with no obvious differences. ANOVA on ranks (AoR) run for the total intersections of the treatment groups versus control revealed no statistical differences. The number of neurons analyzed per condition is given in Table 3.
Basal Dendritic Growth Recovered After Ifenprodil Washout
To test if neurons are able to recover from an early antagonism of GluN2B we transfected at DIV 4, and applied ifenprodil at DIV 6 and DIV 8. Ifenprodil can be washed out, and at DIV 10 we switched the OCT to medium without antagonist until fixation, staining and reconstruction at DIV 15. Basal dendritic complexity was at the level of the mock-stimulated control cells with apical dendrites (Table 4A) and dendrites of interneurons (Table 4B) unaltered. Protein blots (Figure 6) revealed that the ifenprodil treatment caused a reduction of GAD-65/67 expression which was still lower at 12 h recovery, but had returned to control values at 24 h after washout (Table 5). Developmental GAD-65/67 expression is known to be activity-dependent with a contribution of NMDA receptors (Qin et al., 1994; Patz et al., 2003). Interestingly, although total GluN2B protein was not altered under ifenprodil and after 24 h recovery (Table 5), the phosphorylation at tyrosine 1472 was increased after 12 and 24 h recovery (Figure 6 and Table 5). The phosphorylation serves to anchor GluN2B-containing receptors at the synapse and prevent internalization (Sanz-Clemente et al., 2013). GluN1 was not altered at 12 and 24 h recovery (Table 5). PSD-95, a marker for glutamatergic synapses, were neither altered under ifenprodil nor at during recovery (Figure 6 and Table 5). This suggested that pyramidal neurons within this time window can recover basal dendritic growth.
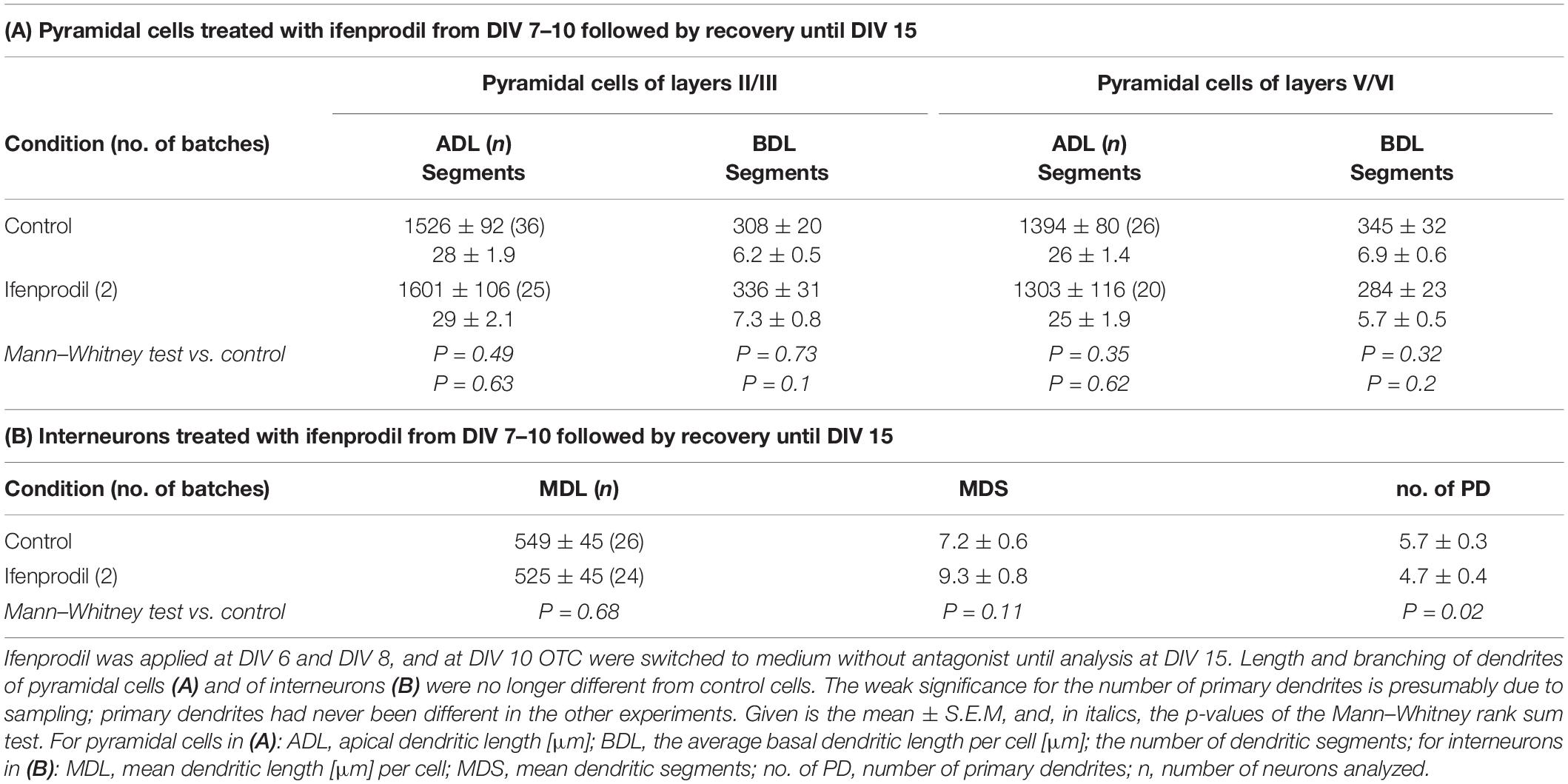
Table 4. Recovery of basal dendritic growth.

Figure 6. Higher expression of Y1472 phosphorylated GluN2B during recovery from ifenprodil treatment. The five nitrocellulose strips used to detect six proteins of different molecular weight [in kDa] derive all from one gel. Per lane, the lysate of 1 OTC (each about 1 mg wet weight) was loaded. Shown are arbitrarily paired OTC from one batch of cultures, two OTC at 12 h and two OTC at 24 h after washout of ifenprodil, and two OTC that were mock-stimulated, and harvested at the 24 h time point. Note that the level of GluN2B Y1472 phosphorylation was higher in the recovery cultures, the GAD-65 level was lower under ifenprodil and still lower after 12 h recovery; the PSD-95 levels were not affected. Tubulin and actin served as house-keeping proteins. For quantification see Table 5.

Table 5. Ifenprodil treatment decreased GAD expression and increased phosphorylation of GluN2B.
Attempts to Overexpress NMDAR Subunits Were Unsuccessful in Rat Cortical OTC
Following the strategy successfully employed for AMPAR and KAR subunits (Hamad et al., 2011; Jack et al., 2018) we attempted to overexpress GluN2B and GluN2A subunits from DIV 5–10. An elevated expression of GluN2B during development and through adulthood results in a smarter mouse (Tang et al., 1999). We hypothesized an increase in basal dendritic complexity. Unexpectedly, pyramidal neurons and multipolar interneurons transfected with either GluN2A- or GluN2B-encoding plasmids resulted in dendritic dimensions that were not different from those of EGFP-only control neurons (Table 6). Not even trends e.g., toward longer or more complex branches were seen. We questioned if the subunits were produced by the neurons. We transfected EGFP-tagged GluN2A and EYFP-tagged GluN2B into neurons in OTC, however, the tag fluorescence was barely detectable in some few transfectants. Only immunostaining with anti-EGFP antibody delivered a rather faint fluorescence in cell bodies and proximal processes of a minority of the often brilliantly mCherry-positive transfected neurons and glia cells (Figures 7A–H). Labeling appeared cytosolic and plasma membrane-associated, occluded the nucleus, and sometimes revealed intracellular aggregates (Figures 7C,C’,G,G’) which might suggest retention of the tagged proteins at the endoplasmatic reticulum.
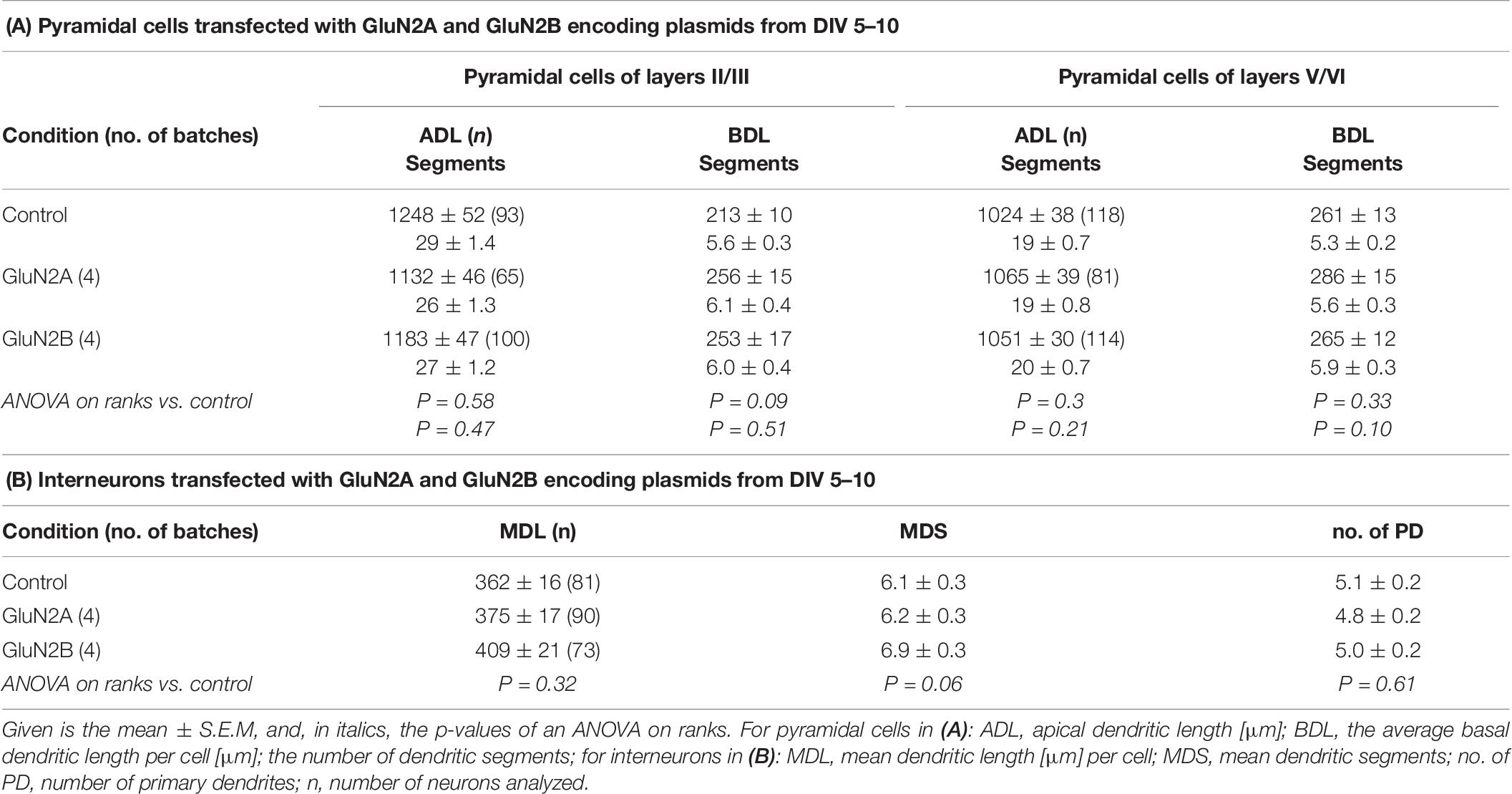
Table 6. Effects of transfection with plasmids encoding GluN2A and GluN2B.
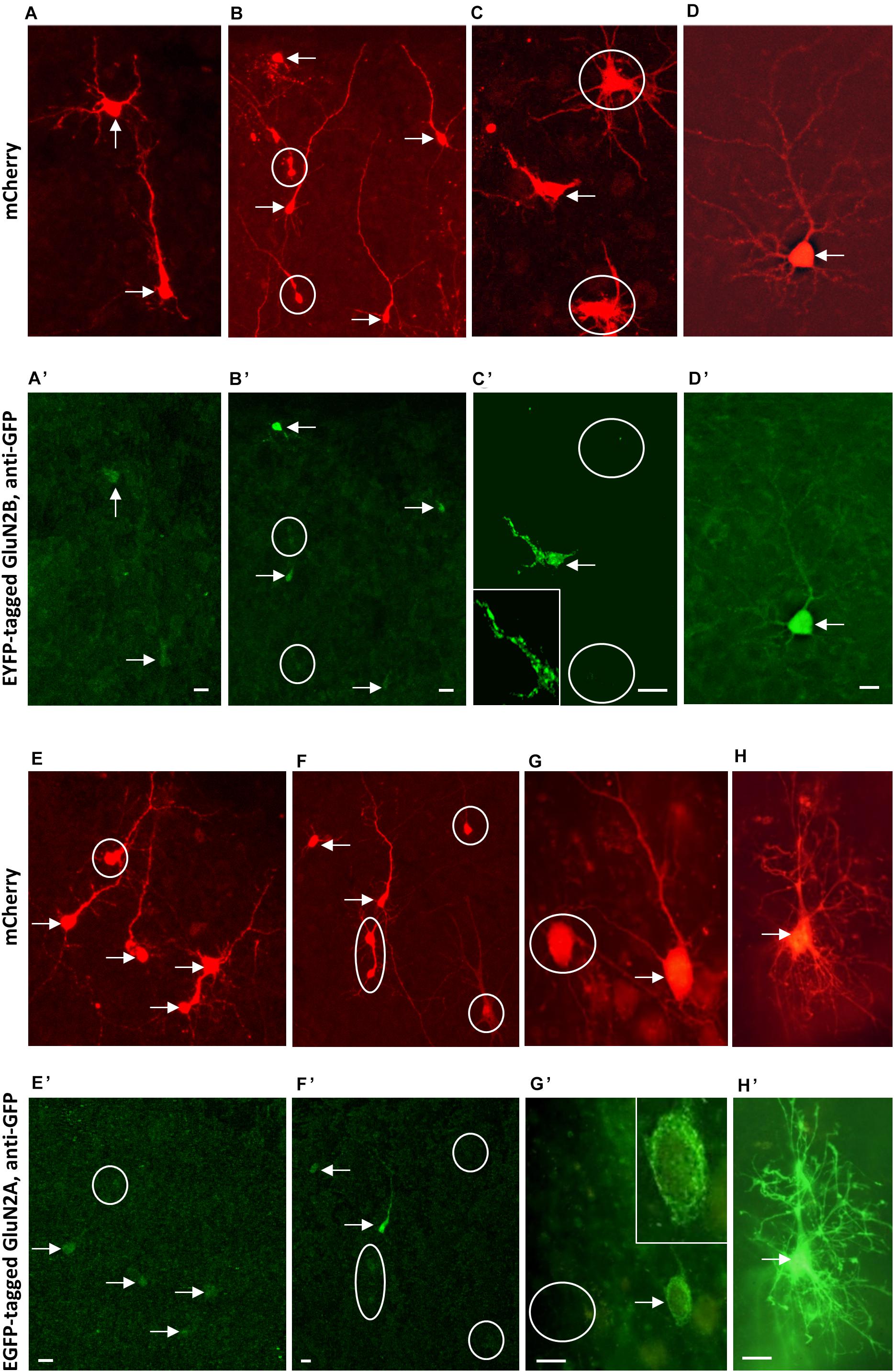
Figure 7. Immunofluorescence staining of overexpressed tagged GluN2 subunits. (A–D) Co-transfection of mCherry (mCherry fluorescence) (A–D) and EYFP-tagged GluN2B-encoding plasmids stained with mouse anti-GFP/Alexa-488 (A’–D’). (E–H): Co-transfection of mCherry (mCherry fluorescence) (E–H) and EGFP-tagged GluN2A-encoding plasmids stained with mouse anti-GFP/Alexa-488 (E’–H’). Note intense mCherry staining of immature as well as more mature neurons and the barely visible GluN2A and GluN2B signal. Intensity of the GluN2A and GluN2B immunofluorescence staining increases in the pictures arranged from left to right, and the right-most cells [the pyramidal neuron in (D,D’), and the astrocyte in (H,H’)] had in fact been the two cells we found most intensely labeled for the subunits. Note that the tag fluorescence appeared rather cytosolic, also occluding the nucleus. Double-labeled cells marked by arrows, mCherry-positive cells without GluN2 fluorescence marked by white circles. About 10 OTC transfected from two preparations for each GluN2 subunit; sequential confocal imaging. Pial surface is to the top for all pictures. Scale: 10 μm.
The overexpression of AMPAR and KAR subunits confers an elevated sensitivity toward the ligands with dendritic beading as quantitative readout (Hamad et al., 2011; Jack et al., 2018). Thus, cultures transfected with GluN2A and GluN2B, respectively, from DIV 5–10 were challenged with 50 μM NMDA for 10 min. Counting revealed that dendritic injury in pyramidal neurons and multipolar non-pyramidal neurons occurred to similar degrees that were statistically not different from the proportion of injured cells evoked in EGFP-only cultures [mean + S.E.M in (n) OTC; for pyramidal cells, control: 63.2 + 3.8% (11), GluN2A: 58.4 + 8.5% (9), GluN2B: 64.0 + 7.8% (9); ANOVA on ranks p = 0.916; for interneurons, control: 51.3 + 13.6%, GluN2A: 45.7 + 10.5%, GluN2B: 53.1 + 8.1%; ANOVA on ranks p = 0.21].
To confirm that the tagged NMDA receptor channels are functional we transfected HEK cells for immunostaining and patch-clamp recordings. At 24–48h post-transfection (together with GluN1 which is not endogenously present in HEK cells as compared to neurons) the EYFP-GluN2B and the EGFP-GluN2A tag fluorescence was barely detectable in HEK cells. As in neurons, only the immunofluorescence for the tag with anti-GFP antibodies yielded a weak to moderate staining of HEK cells along the membrane and sometimes clustered within the cytosol (Supplementary Figures 1A,B). Albeit small, currents were present. Application of 500 μM glutamate elicited typical inward currents with the inactivation being less pronounced in GluN1/GluN2B-transfected HEK cells (Supplementary Figures 2A–C). Peak currents were similar and steady-state currents were larger in GluN1/GluN2B-transfected HEK cells (Supplementary Figures 2D,E). This confirmed that the NMDA receptor constructs encode functional channels.
Finally, to test if the endogenous GluN1 expression could be a limiting factor in neurons, we co-transfected GluN1-1a together with GluN2B encoding plasmids from DIV 5-10. GluN1-1a is abundantly expressed postnatally (Laurie and Seeburg, 1994). To this end, focusing on the reconstruction of supragranular pyramidal neurons, neither GluN1-1a alone nor GluN1-1a/GluN2B expression resulted in an altered dendritic complexity (Table 7).
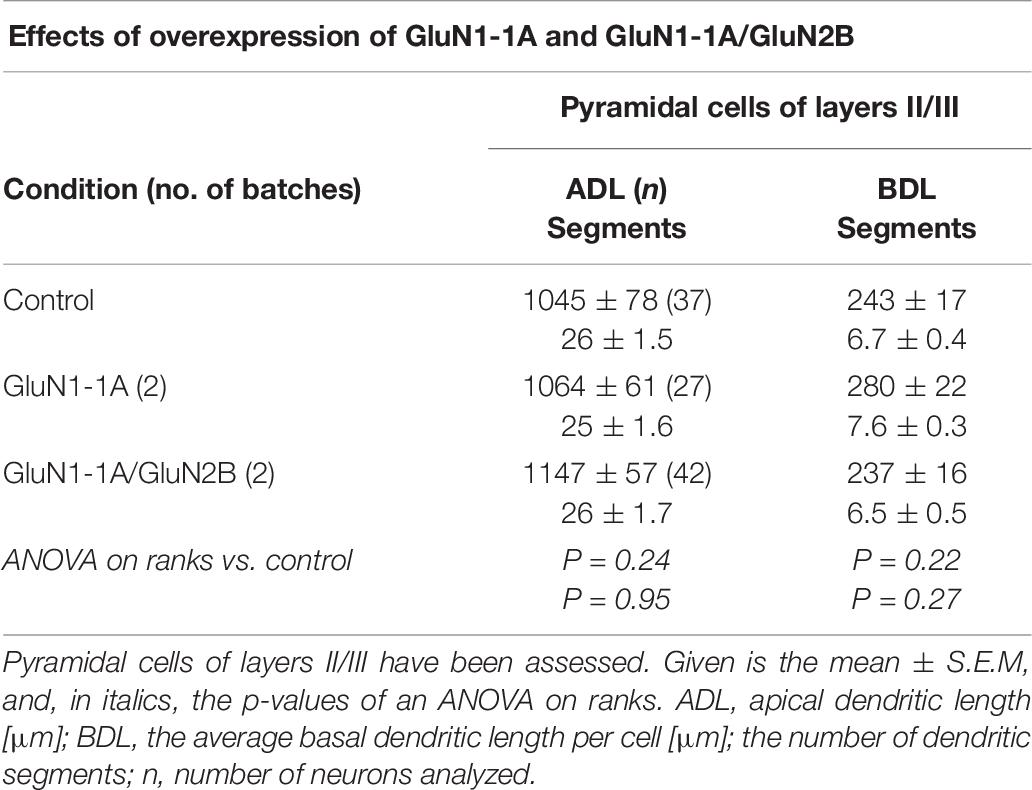
Table 7. Effects of transfection with plasmids encoding GluN1-1a and GluN1-1a/GluN2B from DIV 5–10.
Taken together, the fairly weak presence of the transfected GluN2 proteins in HEK cells and in neurons, no enhanced sensitivity of the neuronal transfectants to NMDA, and their unchanged dendritic complexity suggested that GluN2 transfections did not result in a substantially increased production of functional GluN2 proteins in spontaneously active postnatal neocortical neurons.
Discussion
The present study reports first, a class-specific effect on dendritic morphology in that only pyramidal cells are affected, second, a compartment effect with basal dendrites being affected, and third, an effect mediated by GluN2B-containing receptors that is limited to an early time window. The importance of neural activity and glutamate receptors for neuron morphology has been demonstrated in cortex and hippocampus of mammals as well as the frog optic tectum (Cline, 2001). Yet, the question of which subunits may or may not elicit certain effects, in which dendritic compartments they occur and in what developmental time windows has not been fully resolved. For instance, after blocking transmitter release by tetanus toxin injections into newborn rat hippocampus stratum oriens and stratum radiatum, CA1 pyramidal cells display less branched basal, but normal apical dendrites at day 6–10 (Groc et al., 2002). Ablation of NMDARs impairs dendritic maturation by altering the membrane excitability, a critical component for dendrite growth (Frangeul et al., 2017). Hippocampal granule cells with fragile-X-associated NMDAR hypofunction display reduced dendritic length and branching (Yau et al., 2019). Blocking NMDARs from P1-P21 by oral supplementation of the non-selective antagonist CGP 40116 results in stunted basal dendrites of adult prefrontal cortical pyramidal cells suggesting an inability to recover with time; apical dendrites had been either not altered or have managed to recover a normal morphology (Wedzony et al., 2005).
Antagonizing GluN2-containing receptors between DIV 5–10 impaired basal dendrites by slowing the growth and/or freezing the dendrites to the degree of complexity they had reached at the start of the treatment. Yet, growth resumes after ifenprodil was removed, and quickly catches up to the age-matched control level. Blots suggested that it was possibly mediated by the transient deficit of GABA-ergic inhibition as suggested by the reduction and delayed recovery of GAD-65/67 expression and by a higher synaptic GluN2B signaling as suggested by the enhanced Y1472 phosphorylation. In the presence of ifenprodil the neurons might have increased the density of synaptic GluN2B-containing receptors by homeostatic mechanisms. Synaptic currents are particularly important for dendritic growth and remodeling (Wong and Ghosh, 2002).
The specific effect on basal dendrites was surprising. GluN2A and GluN2B receptors are fairly equally distributed in apical and basal dendrites, as has been shown for layer V prefrontal cortical pyramidal cells, albeit of postnatal day 21–33 mice (Balsara et al., 2014) which are older than our neurons. Further, blocking NMDARs with APV has been shown to neutralize the growth-promoting effect delivered by certain AMPAR subunits on the apical dendrites (Hamad et al., 2011), but APV did not block basal growth in this experimental setting. Biophysical differences exist between basal and apical dendrites (Schiller et al., 2000; Milojkovic et al., 2005; Nevian et al., 2007; Major et al., 2008; Antic et al., 2010). For instance, basal dendrites receive a major fraction of intracortical excitatory synapses, NMDA spikes occur somewhat enriched in basal dendrites (Chalifoux and Carter, 2011) and evoke large calcium transients (Schiller et al., 2000) which are linearly related to the neuron’s spike activity (Hill et al., 2013). Further, excited basal dendrites have been found to elicit sustained UP states in the neuron’s cell body (Milojkovic et al., 2005; Antic et al., 2010). UP states of higher excitability are regarded as important triggers of transcriptional and translational processes required for dendritic growth. Yet, a balanced NMDAR signaling seems to be important. In hippocampal slice cultures, a 4-days chronic disinhibition with bicuculline at an age equivalent to our DIV 10 cortex cultures impairs elongation and branching of basal dendrites. This can be prevented by APV suggesting that hyperexcitability with impairment of CREB signaling (Nishimura et al., 2008) is as detrimental as hypoexcitability under GluN2B inhibition.
The temporal limitation was presumably caused by the time course of subunit expression. The developmental decrease of GluN2B protein is relative (Williams et al., 1993; Traynelis et al., 2010; Sanz-Clemente et al., 2013), and in fact the assembly of GluN1/GluN2B raises quite steadily until P20 in rat cortex as determined by immunoprecipitation (Babb et al., 2005) with surface expression of GluN2B being independent of activity. GluN2A protein increases strongly during the first postnatal week, and this proceeds also in OTC (Neumann et al., 2019). Given the presence of spontaneous activity, GluN2A efficiently replaces GluN2B in GluN2B-containing receptors at the cell surface (Barth and Malenka, 2001; Barria and Malinow, 2002). Yet, a majority of synaptic NMDAR in cortex and hippocampus seem to be GluN1/2A/2B triheteromeric receptors (Stroebel et al., 2018). Extrasynaptic NMDARs also contain GluN2B (Papouin and Oliet, 2014). Early in development dendrites have very few spines, and receptors are not necessarily clustered. What in adult neurons is an extrasynaptic NMDA receptor may in a developing neuron be a receptor at a site where a post synapse is going to form or has just been eliminated (Petralia et al., 2010). Along that line, we could trigger massive dendritic injury in pyramidal neurons in the early time window. Extrasynaptic GluN2B receptors contribute to excitotoxicity (Hardingham and Bading, 2010), but also to calcium plateau potentials (Oikonomou et al., 2012) which can influence early dendritic growth dynamics (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Kwon and Sabatini, 2011).
Growth requirements of GABA-ergic interneurons have rarely been analyzed. Of nine AMPAR subunits tested, exclusively the GluA1(Q)-flip subunit has been able to increase dendritic complexity of interneurons (Hamad et al., 2011). Further, interneuronal dendritic growth is promoted by GluK1 (Jack et al., 2018). Fast-spiking as well as regular-spiking non-pyramidal neurons express NMDAR (Hadzic et al., 2017) and undergo the subunit switch (Wang and Gao, 2009). An impairment of interneuronal dendrites has been reported in mutant Dlx1/2 mice with evidence that a reduction of GluN2B expression is the cause (Pla et al., 2018). This is in contrast to our findings that neither the block of GluN2B nor of GluN2A could alter interneuronal dendritic complexity. Opposed to GluN1/2A/2B-containing pyramidal cells, interneurons are more enriched for GluN1/2B/2D-containing receptors, and the efficacy of ifenprodil at GluN1/2B/2D receptors is much less than at GluN1/2B receptors (Yi et al., 2019). A recent study in mouse cortex has revealed that parvalbuminergic interneurons mature via tonically active GluN2C/2D-containing receptors which transform the depolarizing signaling of ambient glutamate into an increase of dendritic complexity. Interestingly, the action is temporally limited to an early postnatal time window: a pharmacological block of GluN2C/2D receptors between day 7 and 9 yields at day 21 interneurons with less dendritic branch points and reduced inhibitory activity (Hanson et al., 2019).
The attempt to overexpress GluN2B and GluN2A subunits via CMV-driven plasmids in spontaneously active cortical slice cultures did not alter dendritic geometry, not even after co-transfection with GluN1-1a. The transfectants were still present and look healthy suggesting that there was no cell death upon overexpression. Yet, staining for the tagged subunits gave only weak immunosignals of the transfectants, HEK cells and neurons. Further, although the HEK cell recordings demonstrated that the NMDA receptor constructs encode functional channels, the neuronal transfectants had no higher sensitivity to NMDA. In contrast, AMPAR subunits and TARPs overexpressed by the same strategy from plasmids with the same backbone resulted in well stained neurons, membrane labeling, and a higher sensitivity to AMPA (Hamad et al., 2011, 2014).
Results of studies employing GluN2 subunit overexpression are controversial. Transgenic GluN2B overexpressing mice display enhanced Ro25-6981 dependent LTP in frontal cortex (Cui et al., 2011), but visual cortical neurons of GluN2B-overexpressing mice display parameters of synaptic plasticity not any different from wildtype control (Philpot et al., 2001), suggestive of areal differences. Toward morphofunctional differentiation, reported effects seem to depend on timing and endogenous receptor status. Ventral spinal cord neurons dissociated from E14 rat embryos downregulate NMDARs, and undergo an enhanced dendritic growth upon transfection with GluN2B, but not GluN2A (Sepulveda et al., 2010). Hippocampal neurons richly express GluN2 receptors, and here, the knockdown of GluN2B results in a slight decrease in number of only the secondary dendrites whereas knockdown of GluN2A increases the number of such secondary branches (Sepulveda et al., 2010). In the tadpole tectum, the knockdown of GluN2B has no effect on dendritic development (Ewald et al., 2008). Overexpressing GluN2B promotes branching in dissociated hippocampal neurons at DIV 7, but no longer in older neurons (Bustos et al., 2014). A GluN2B knockdown reduces dendritic complexity (Bustos et al., 2017). Genetic ablation of GluN2B does not affect dendritic dimensions of dentate granule cells and barrel cortex layer IV spiny stellates, but the lack of GluN2B prevents the reorganization of spiny stellate dendrites toward the preferred barrel (Espinosa et al., 2009). Moreover, the effects of overexpression are sometimes surprisingly small. For instance, in tadpole tectum neurons overexpressing GluN2 receptors, recordings reveal a shift to either one of the two subunits, but total branch length and branch tip numbers remain at control level, and only a small difference of the distance between dendritic end points has been found (Ewald et al., 2008). The biological significance of such a difference remains up to debate. The amount of GluN2B at the surface is difficult to change since synaptic incorporation can neither be increased by enhancing subunit expression (Philpot et al., 2001; Barria and Malinow, 2002), nor by blocking AMPARs (Von Engelhardt et al., 2009). Our results now add to this showing that the total amount of GluN2B and GluN1 protein were not altered under ifenprodil and during recovery from ifenprodil treatment. Possibly, the overexpression-evoked surface levels of GluN2 proteins are large enough to be detectable with sensitive recordings, and this would be supported by our HEK cell recordings. However, in neurons, such currents presumably are not large enough to measurably alter the dendritic architecture. Another reason could be that we transfected wildtype GluN2B. Possibly, only the overexpression of an endocytosis-resistant E1479Q mutant (De Solis et al., 2019) might result in a GluN2B surface expression large enough to evoke morphological effects. Other mental disorder-related mutants of GluN2B have been recently discovered. For instance, GluN2B truncated at position 724 assembles with GluN1 but fails to reach the plasma membrane, and overexpressing this mutant in dissociated rat cortical pyramidal neurons results at DIV 10–14 in dramatically stunted dendritic growth (Sceniak et al., 2019). Further, a recent study of transgenic mice expressing chimeric GluN2 subunits reports that overexpression of the GluN2A with the GluN2B C-terminus, but not N-terminus or transmembrane domains, results in longer dendrites of CA1 pyramidal neurons suggesting that particular aspects of intracellular, i.e., metabotropic signaling rather than the ion conductance per se causes these effects (Keith et al., 2019). For instance, nanomolar concentrations of kainate drives dendritic growth via a metabotropic action (Jack et al., 2018), and changes of spine morphology are evoked by non-ionotropic actions of NMDARs because the changes persist in the presence of antagonists (Stein et al., 2015, 2020). The role of mutant GluN2B or non-ionotropic actions for postnatal maturation of basal dendrites remains to be unraveled. To this end, it appears as if young neocortical neurons in spontaneously active slice cultures strictly regulate the amount of functional NMDARs, possibly as a trade-off between growth and toxicity.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
All animal protocols were approved by the Ruhr University Bochum Animal Research Board and the State of North Rhine-Westphalia.
Author Contributions
MIKH, MH, ARe, and PW designed the experiments. ARä and SK prepared the cultures and solutions. SG, JG, AS, FW, RB, OK, TZ, CR, MIKH, and PW performed the experiments, reconstructions, and data management. SG, JG, MIKH, ARe, and PW interpreted the results. PW wrote the manuscript. SG, JG, OK, MIKH, ARe, and MH contributed to writing and gave feedback on the manuscript. All authors approved the final version.
Funding
This work was supported by Deutsche Forschungsgemeinschaft grants WA 541/10-1, WA 541/13-1, and RE 3101/3-1.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge support by the DFG Open Access Publication Funds of the Ruhr University Bochum.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnana.2020.571351/full#supplementary-material
References
Antic, S. D., Zhou, W. L., Moore, A. R., Short, S. M., and Ikonomu, K. D. (2010). The decade of the dendritic NMDA spike. J. Neurosci. Res. 88, 2991–3001. doi: 10.1002/jnr.22444
Arai, A. C., and Kessler, M. (2007). Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr. Drug Targets 8, 583–602. doi: 10.2174/138945007780618490
Babb, T. L., Mikuni, N., Najm, I., Wylie, C., Olive, M., Dollar, C., et al. (2005). Pre- and postnatal expressions of NMDA receptors 1 and 2B subunit proteins in the normal rat cortex. Epilepsy Res. 64, 23–30. doi: 10.1016/j.eplepsyres.2005.02.008
Balsara, R. D., Ferreira, A. N., Donahue, D. L., Castellino, F. J., and Sheets, P. L. (2014). Probing NMDA receptor GluN2A and GluN2B subunit expression and distribution in cortical neurons. Neuropharmacology 79, 542–549. doi: 10.1016/j.neuropharm.2014.01.005
Barria, A., and Malinow, R. (2002). Subunit-specific NMDA receptor trafficking to synapses. Neuron 35, 345–353. doi: 10.1016/s0896-6273(02)00776-6
Barth, A. L., and Malenka, R. C. (2001). NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat. Neurosci. 4, 235–236. doi: 10.1038/85070
Bustos, F. J., Jury, N., Martinez, P., Ampuero, E., Campos, M., Abarzúa, S., et al. (2017). NMDA receptor subunit composition controls dendritogenesis of hippocampal neurons through CAMKII, CREB-P, and H3K27ac. J. Cell. Physiol. 232, 3677–3692. doi: 10.1002/jcp.25843
Bustos, F. J., Varela-Nallar, L., Campos, M., Henriquez, B., Phillips, M., Opazo, C., et al. (2014). PSD95 suppresses dendritic arbor development in mature hippocampal neurons by occluding the clustering of NR2B-NMDA receptors. PLoS One 9:e94037. doi: 10.1371/journal.pone.0094037
Chalifoux, J. R., and Carter, A. G. (2011). Glutamate spillover promotes the generation of NMDA spikes. J. Neurosci. 31, 16435–16446. doi: 10.1523/JNEUROSCI.2777-11.2011
Cline, H. T. (2001). Dendritic arbor development and synaptogenesis. Curr. Opin. Neurobiol. 11, 118–126.
Cline, H. T., and Haas, K. (2008). The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J. Physiol. 586, 1509–1517. doi: 10.1016/s0959-4388(00)00182-3
Cui, Y., Jin, J., Zhang, X., Xu, H., Yang, L., Du, D., et al. (2011). Forebrain NR2B overexpression facilitating the prefrontal cortex long-term potentiation and enhancing working memory function in mice. PLoS One 6:e020312. doi: 10.1371/journal.pone.0020312
De Solis, C. A., Gonzalez, C. U., Galdamez, M. A., Perish, J. M., Woodard, S. W., Salinas, C. E., et al. (2019). Increasing synaptic GluN2B levels within the basal and lateral amygdala enables the modification of strong reconsolidation resistant fear memories. bioRxiv [Preprint], doi: 10.1101/537142
Edman, S., McKay, S., MacDonald, L. J., Samadi, M., Livesey, M. R., Hardingham, G. E., et al. (2012). TCN201 selectively blocks GluN2A-containing NMDARs in a GluN1 co-agonist dependent but non-competitive manner. Neuropharmacology 63, 441–449. doi: 10.1016/j.neuropharm.2012.04.027
Engelhardt, M., Hamad, M. I. K., Jack, A., Ahmed, K., König, J., Rennau, L. M., et al. (2018). Interneuron synaptopathy in developing rat cortex induced by the pro-inflammatory cytokine LIF. Exp. Neurol. 302, 169–180. doi: 10.1016/j.expneurol.2017.12.011
Engert, F., and Bonhoeffer, T. (1999). Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399, 66–70. doi: 10.1038/19978
Espinosa, J. S., Wheeler, D. G., Tsien, R. W., and Luo, L. (2009). Uncoupling dendrite growth and patterning: single cell knockout analysis of NMDA receptor 2B. Neuron 62, 205–217. doi: 10.1016/j.neuron.2009.03.006
Ewald, R. C., Van Keuren-Jensen, K. R., Aizenman, C. D., and Cline, H. T. (2008). Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J. Neurosci. 28, 850–861. doi: 10.1523/JNEUROSCI.5078-07.2008
Frangeul, L., Kehayas, V., Sanchez-Mut, J. V., Fièvre, S., Krishna-K, K., Pouchelon, G., et al. (2017). Input-dependent regulation of excitability controls dendritic maturation in somatosensory thalamocortical neurons. Nat. Commun. 8:2015. doi: 10.1038/s41467-017-02172-1
Gray, J. A., Shi, Y., Usui, H., During, M. J., Sakimura, K., and Nicoll, R. A. (2011). Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71, 1085–1101. doi: 10.1016/j.neuron.2011.08.007
Greenough, W. T., Juraska, J. M., and Volkmar, F. R. (1979). Maze training effects on dendritic branching in occipital cortex of adult rats. Behav. Neural. Biol. 26, 287–297. doi: 10.1016/s0163-1047(79)91278-0
Groc, L., Petanjek, Z., Gustafsson, B., Ben-Ari, Y., Hanse, E., and Khazipov, R. (2002). In vivo blockade of neural activity alters dendritic development of neonatal CA1 pyramidal cells. Eur. J. Neurosci. 16, 1931–1938. doi: 10.1046/j.1460-9568.2002.02264.x
Hadzic, M., Jack, A., and Wahle, P. (2017). Ionotropic glutamate receptors: which ones, when, and where in the mammalian neocortex. J. Comp. Neurol. 525, 976–1033. doi: 10.1002/cne.24103
Hamad, M. I. K., Jack, A., Lorkowski, M., Strasdeit, T., Klatt, O., Kott, S., et al. (2014). Type-I TARPs promote dendritic growth in early postnatal neocortical pyramidal cells. Development 141, 1737–1748. doi: 10.1242/dev.099697
Hamad, M. I. K., Ma-Högemeier, Z. L., Riedel, C., Conrads, C., Veitinger, T., Habijan, T., et al. (2011). Cell class-specific regulation of neocortical dendrite and spine growth by AMPA receptor splice and editing variants. Development 138, 4301–4313. doi: 10.1242/dev.071076
Hansen, K. B., Ogden, K. K., and Traynelis, S. F. (2012). Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J. Neurosci. 32, 6197–6208. doi: 10.1523/JNEUROSCI.5757-11.2012
Hansen, K. B., Ogden, K. K., Yuan, H., and Traynelis, S. F. (2014). Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81, 1084–1096. doi: 10.1016/j.neuron.2014.01.035
Hanson, E., Armbruster, M., Lau, L. A., Sommer, M. E., Klaft, Z. J., Swanger, S. A., et al. (2019). Tonic activation of GluN2C/GluN2D-containing NMDA receptors by ambient glutamate facilitates cortical interneuron maturation. J. Neurosci. 39, 3611–3626. doi: 10.1523/JNEUROSCI.1392-18.2019
Hardingham, G. E., and Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696. doi: 10.1038/nrn2911
He, S., Shao, L. R., Wang, Y., and Bausch, S. B. (2013). Synaptic and extrasynaptic plasticity in glutamatergic circuits involving dentate granule cells following chronic N-methyl-D-aspartate receptor inhibition. J. Neurophysiol. 109, 1535–1547. doi: 10.1152/jn.00667.2012
Hill, D. N., Varga, Z., Jia, H., Sakmann, B., and Konnerth, A. (2013). Multibranch activity in basal and tuft dendrites during firing of layer 5 cortical neurons in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 13618–13623. doi: 10.1073/pnas.1312599110
Jack, A., Hamad, M. I. K., Gonda, S., Gralla, S., Pahl, S., Hollmann, M., et al. (2018). Development of cortical pyramidal cell and interneuronal dendrites: a role for kainate receptor subunits and NETO1. Mol. Neurobiol. 56, 4960–4979. doi: 10.1007/s12035-018-1414-0
Keith, R. E., Azcarate, J. M., Keith, M. J., Hung, C. W., Badakhsh, M. F., and Dumas, T. C. (2019). Direct intracellular signaling by the carboxy terminus of NMDA receptor GluN2 subunits regulates dendritic morphology in hippocampal CA1 pyramidal neurons. Neuroscience 396, 138–153. doi: 10.1016/j.neuroscience.2018.11.021
Konur, S., and Ghosh, A. (2005). Calcium signaling and the control of dendritic development. Neuron 46, 401–405. doi: 10.1016/j.neuron.2005.04.022
Kwon, H. B., and Sabatini, B. L. (2011). Glutamate induces de novo growth of functional spines in developing cortex. Nature 474, 100–104. doi: 10.1038/nature09986
Laurie, D. J., and Seeburg, P. H. (1994). Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J. Neurosci. 14, 3180–3194. doi: 10.1523/JNEUROSCI.14-05-03180.1994
Lauterborn, J. C., Palmer, L. C., Jia, Y., Pham, D. T., Hou, B., Wang, W., et al. (2016). Chronic ampakine treatments stimulate dendritic growth and promote learning in middle-aged rats. J. Neurosci. 36, 1636–1646. doi: 10.1523/JNEUROSCI.3157-15.2016
Lauterborn, J. C., Pineda, E., Chen, L. Y., Ramirez, E. A., Lynch, G., and Gall, C. M. (2009). Ampakines cause sustained increases in brain-derived neurotrophic factor signaling at excitatory synapses without changes in AMPA receptor subunit expression. Neuroscience 159, 283–295. doi: 10.1016/j.neuroscience.2008.12.018
Lauterborn, J. C., Truong, G. S., Baudry, M., Bi, X., Lynch, G., and Gall, C. M. (2003). Chronic elevation of brain-derived neurotrophic factor by ampakines. J. Pharmacol. Exp. Ther. 307, 297–305. doi: 10.1124/jpet.103.053694
Lauterborn, L. C., Lynch, G., Vanderklish, P., Arai, A., and Gall, C. M. (2000). Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J. Neurosci. 20, 8–21. doi: 10.1523/JNEUROSCI.20-01-00008.2000
Lu, C., Chen, Q., Zhou, T., Bozic, D., Fu, Z., Pan, J. Q., et al. (2016). Micro-electrode array recordings reveal reductions in both excitation and inhibition in cultured cortical neuron networks lacking Shank3. Mol. Psychiatry 21, 159–168. doi: 10.1038/mp.2015.173
Major, G., Polsky, A., Denk, W., Schiller, J., and Tank, D. W. (2008). Spatiotemporally graded NMDA spike/plateau potentials in basal dendrites of neocortical pyramidal neurons. J. Neurophysiol. 99, 2584–2601. doi: 10.1152/jn.00011.2008
Maletic-Savatic, M., Malinow, R., and Svoboda, K. (1999). Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283, 1923–1927. doi: 10.1126/science.283.5409.1923
McAllister, A. K. (2000). Cellular and molecular mechanisms of dendrite growth. Cereb. Cortex 10, 963–973. doi: 10.1093/cercor/10.10.963
Milojkovic, B. A., Radojicic, M. S., and Antic, S. D. (2005). A strict correlation between dendritic and somatic plateau depolarizations in the rat prefrontal cortex pyramidal neurons. J. Neurosci. 25, 3940–3951. doi: 10.1523/JNEUROSCI.5314-04.2005
Montgomery, K. E., Kessler, M., and Arai, A. C. (2009). Modulation of agonist binding to AMPA receptors by 1-(1,4-benzodioxan-6-ylcarbonyl)piperidine (CX546): differential effects across brain regions and GluA1-4/transmembrane AMPA receptor regulatory protein combinations. J. Pharmacol. Exp. Ther. 331, 965–974. doi: 10.1124/jpet.109.158014
Neumann, J. R., Dash-Wagh, S., Jack, A., Hamad, M. I. K., Räk, A., Jüngling, K., et al. (2019). The primate-specific peptide Y-P30 promotes morphological maturation of neocortical dendritic spines. PLoS One 14:e0211151. doi: 10.1371/journal.pone.0211151
Nevian, T., Larkum, M. E., Polsky, A., and Schiller, J. (2007). Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nat. Neurosci. 10, 206–214. doi: 10.1038/nn1826
Neyton, J., and Paoletti, P. (2006). Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J. Neurosci. 26, 1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006
Ng, D., Pitcher, G. M., Szilard, R. K., Sertié, A., Kanisek, M., Clapcote, S. J., et al. (2009). Neto1 is a novel CUB-domain NMDA receptor-interacting protein required for synaptic plasticity and learning. PLoS Biol. 7:41. doi: 10.1371/journal.pbio.1000041
Nishimura, M., Owens, J., and Swann, J. W. (2008). Effects of chronic network hyperexcitability on the growth of hippocampal dendrites. Neurobiol. Dis. 29, 267–277. doi: 10.1016/j.nbd.2007.08.018
Oikonomou, K. D., Short, S. M., Rich, M. T., and Antic, S. D. (2012). Extrasynaptic glutamate receptor activation as cellular bases for dynamic range compression in pyramidal neurons. Front. Physiol. 3:334. doi: 10.3389/fphys.2012.00334
Papouin, T., and Oliet, S. H. R. (2014). Organization, control and function of extrasynaptic NMDA receptors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130601. doi: 10.1098/rstb.2013.0601
Patz, S., Wirth, M. J., Gorba, T., Klostermann, O., and Wahle, P. (2003). Neuronal activity and neurotrophic factors regulate GAD-65/67 mRNA and protein expression in organotypic cultures of rat visual cortex. Eur. J. Neurosci. 18, 1–12. doi: 10.1046/j.1460-9568.2003.02702.x
Petralia, R. S., Wang, Y. X., Hua, F., Yi, Z., Zhou, A., Ge, L., et al. (2010). Organization of NMDA receptors at extrasynaptic locations. Neuroscience 167, 68–87. doi: 10.1016/j.neuroscience.2010.01.022
Philpot, B. D., Weisberg, M. P., Ramos, M. S., Sawtell, N. B., Tang, Y. P., Tsien, J. Z., et al. (2001). Effect of transgenic overexpression of NR2B on NMDA receptor function and synaptic plasticity in visual cortex. Neuropharmacology 41, 762–770. doi: 10.1016/s0028-3908(01)00136-8
Pla, R., Stanco, A., Howard, M. A., Rubin, A. N., Vogt, D., Mortimer, N., et al. (2018). Dlx1 and Dlx2 promote interneuron GABA synthesis, synaptogenesis, and dendritogenesis. Cereb. Cortex 28, 3797–3815. doi: 10.1093/cercor/bhx241
Qin, Z. H., Zhang, S. P., and Weiss, B. (1994). Dopaminergic and glutamatergic blocking drugs differentially regulate glutamic acid decarboxylase mRNA in mouse brain. Mol. Brain Res. 21, 293–302. doi: 10.1016/0169-328x(94)90260-7
Rajan, I., and Cline, H. T. (1998). Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo. J. Neurosci. 18, 7836–7846. doi: 10.1523/JNEUROSCI.18-19-07836.1998
Sanz-Clemente, A., Nicoll, R. A., and Roche, K. W. (2013). Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19, 62–75. doi: 10.1177/1073858411435129
Sceniak, M. P., Fedder, K. N., Wang, Q., Droubi, Q., Sroubi, S., Babcock, K., et al. (2019). An autism-associated mutation in GluN2B prevents NMDA receptor trafficking and interferes with dendrite growth. J. Cell Sci. 132:jcs232892. doi: 10.1242/jcs.232892
Schiller, J., Major, G., Koester, H. J., and Schiller, Y. (2000). NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature 404, 285–289. doi: 10.1038/35005094
Schitine, C., Xapelli, S., Agasse, F., Sardà-Arroyo, L., Silva, A. P., De Melo Reis, R. A., et al. (2012). Ampakine CX546 increases proliferation and neuronal differentiation in subventricular zone stem/progenitor cell cultures. Eur. J. Neurosci. 35, 1672–1683. doi: 10.1111/j.1460-9568.2012.08072.x
Sepulveda, F. J., Bustos, F. J., Inostroza, E., Zúñiga, F. A., Neve, R. L., Montecino, M., et al. (2010). Differential roles of NMDA receptor subtypes NR2A and NR2B in dendritic branch development and requirement of RasGRF1. J. Neurophysiol. 103, 1758–1770. doi: 10.1152/jn.00823.2009
Sheng, M., Cummings, J., Roldan, L. A., Jan, Y. N., and Jan, L. Y. (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. doi: 10.1038/368144a0
Stein, I. S., Gray, J. A., and Zito, K. (2015). Non-ionotropic NMDA receptor signaling drives activity-induced dendritic spine shrinkage. J. Neurosci. 35, 12303–12308. doi: 10.1523/JNEUROSCI.4289-14.2015
Stein, I. S., Park, D. K., Flores, J. C., Jahncke, J. N., and Zito, K. (2020). Molecular mechanisms of non-ionotropic NMDA receptor signaling in dendritic spine shrinkage. J. Neurosci. 40, 3741–3750. doi: 10.1523/JNEUROSCI.0046-20.2020
Stroebel, D., Casado, M., and Paoletti, P. (2018). Triheteromeric NMDA receptors: from structure to synaptic physiology. Curr. Opin. Physiol. 2, 1–12. doi: 10.1016/j.cophys.2017.12.004
Tang, Y. P., Shimizu, E., Dube, G. R., Rampon, C., Kerchner, G. A., Zhuo, M., et al. (1999). Genetic enhancement of learning and memory in mice. Nature 401, 63–69. doi: 10.1038/4343
Tovar, K. R., McGinley, M. J., and Westbrook, G. L. (2013). Triheteromeric NMDA receptors at hippocampal synapses. J. Neurosci. 33, 9150–9160. doi: 10.1523/JNEUROSCI.0829-13.2013
Traynelis, S. F., Wollmuth, L. P., McBrain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Van Aelst, L., and Cline, H. T. (2004). Rho GTPases and activity-dependent dendrite development. Curr. Opin. Neurobiol. 14, 297–304. doi: 10.1016/j.conb.2004.05.012
Vaughn, J. E. (1989). Fine structure of synaptogenesis in the vertebrate central nervous system. Synapse 3, 255–285. doi: 10.1002/syn.890030312
Volkmar, F. R., and Greenough, W. T. (1972). Rearing complexity affects branching of dendrites in the visual cortex of the rat. Science 176, 1445–1447. doi: 10.1126/science.176.4042.1445
Von Engelhardt, J., Doganci, B., Seeburg, P. H., and Monyer, H. (2009). Synaptic NR2A- but not NR2B-containing NMDA receptors increase with blockade of ionotropic glutamate receptors. Front. Mol. Neurosci. 2:19. doi: 10.3389/neuro.02.019.2009
Wang, H. X., and Gao, W. J. (2009). Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology 34, 2028–2040. doi: 10.1038/npp.2009.20
Wedzony, K., Fijał, K., and Maækowiak, M. (2005). Alterations in the dendritic morphology of prefrontal pyramidal neurons in adult rats after blockade of NMDA receptors in the postnatal period. Brain. Res. 1062, 166–170. doi: 10.1016/j.brainres.2005.09.012
Williams, K., Russell, S. L., Shen, Y. M., and Molinoff, P. B. (1993). Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron 10, 267–278. doi: 10.1016/0896-6273(93)90317-k
Wirth, M. J., Brun, A., Grabert, J., Patz, S., and Wahle, P. (2003). Accelerated dendritic development of rat cortical pyramidal cells and interneurons after biolistic transfection with BDNF and NT4/5. Development 130, 5827–5838. doi: 10.1242/dev.00826
Wong, R. O. L., and Ghosh, A. (2002). Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci. 3, 803–812. doi: 10.1038/nrn941
Yau, S. Y., Bettio, L., Chiu, J., Chiu, C., and Christie, B. R. (2019). Fragile-X syndrome is associated with NMDA receptor hypofunction and reduced dendritic complexity in mature dentate granule cells. Front. Mol. Neurosci. 11:495. doi: 10.3389/fnmol.2018.00495
Keywords: rodent neocortex, ampakine CX546, biolistic transfection, GluN1, inhibition
Citation: Gonda S, Giesen J, Sieberath A, West F, Buchholz R, Klatt O, Ziebarth T, Räk A, Kleinhubbert S, Riedel C, Hollmann M, Hamad MIK, Reiner A and Wahle P (2020) GluN2B but Not GluN2A for Basal Dendritic Growth of Cortical Pyramidal Neurons. Front. Neuroanat. 14:571351. doi: 10.3389/fnana.2020.571351
Received: 15 June 2020; Accepted: 06 October 2020;
Published: 13 November 2020.
Edited by:
Shigeo Okabe, The University of Tokyo, JapanReviewed by:
Carlo Sala, Italian National Research Council, ItalyJohn A. Gray, University of California, Davis, United States
Copyright © 2020 Gonda, Giesen, Sieberath, West, Buchholz, Klatt, Ziebarth, Räk, Kleinhubbert, Riedel, Hollmann, Hamad, Reiner and Wahle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Wahle, cGV0cmEud2FobGVAcnVoci11bmktYm9jaHVtLmRl; d2FobGVAbmV1cm9iaW9sb2dpZS5ydWhyLXVuaS1ib2NodW0uZGU=; orcid.org/0000-0002-8710-0375
†These authors have contributed equally to this work
‡Present address: Mohammad I. K. Hamad, Faculty of Health, University Witten/Herdecke, Witten, Germany