 Sylvia J. Wowro
Sylvia J. Wowro Giang Tong
Giang Tong Jana Krech1
Jana Krech1- 1Department of Congenital Heart Disease/Pediatric Cardiology, Universitäres Herzzentrum Berlin – Medical Heart Center of Charité and German Heart Institute Berlin, Berlin, Germany
- 2Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany
Introduction: Hypothermia attenuates cerebral ischemia-induced neuronal cell death associated with neuroinflammation. The calcineurin inhibitor cyclosporin A (CsA) has been shown to be neuroprotective by minimizing activation of inflammatory pathways. Therefore, we investigated whether the combination of hypothermia and treatment with CsA has neuroprotective effects in an oxygen-glucose deprivation/reperfusion (OGD/R) injury model in neuronal and BV-2 microglia monocultures, as well as in an organotypic hippocampal slice culture (OHSC).
Methods: Murine primary neurons, BV-2 microglia, and OHSC were pretreated with CsA and exposed to 1 h OGD (0.2% O2) followed by reperfusion at normothermia (37∘C) or hypothermia (33.5∘C). Cytotoxicity was measured by lactate dehydrogenase and glutamate releases. Damage-associated molecular patterns (DAMPs) high mobility group box 1 (HMGB1), heat shock protein 70 (Hsp70), and cold-inducible RNA-binding protein (CIRBP) were detected in cultured supernatant by western blot analysis. Interleukin-6 (IL-6), Interleukin-1α and -1β (IL-1α/IL1-β), tumor necrosis factor-α (TNF-α), monocyte chemotactic protein 1 (MCP1), inducible nitric oxide synthase (iNOS), glia activation factors ionized calcium-binding adapter molecule 1 (Iba1), and transforming growth factor β1 (TGF-β1) gene expressions were analyzed by RT-qPCR.
Results: Exposure to OGD plus 10 μM CsA was sufficient to induce necrotic cell death and subsequent release of DAMPs in neurons but not BV-2 microglia. Moreover, OGD/R-induced secondary injury was also observed only in the neurons, which was not attenuated by cooling and no increased toxicity by CsA was observed. BV-2 microglia were not sensitive to OGD/R-induced injury but were susceptible to CsA-induced toxicity in a dose dependent manner, which was minimized by hypothermia. CsA attenuated IL-1β and Iba1 expressions in BV-2 microglia exposed to OGD/R. Hypothermia reduced IL-1β and iNOS expressions but induced TNF-α and Iba1 expressions in the microglia. However, these observations did not translate to the ex vivo OHCS model, as general high expressions of most cytokines investigated were observed.
Conclusion: Treatment with CsA has neurotoxic effects on primary neurons exposed to OGD but could inhibit BV-2 microglia activation. However, CsA and hypothermia treatment after ischemia/reperfusion injury results in cytotoxic neuroinflammation in the complex ex vivo OHSC.
Introduction
Therapeutic hypothermia (TH), also referred to as targeted temperature management (TTM), is a clinically established strategy for neuroprotection against ischemia/reperfusion injury. Several clinical studies in newborns and infants suffering from perinatal asphyxia and neonatal encephalopathy have demonstrated the efficacy of cooling to 33–34°C, resulting in lower mortality rates and improved neurological outcomes (Shankaran et al., 2012; Azzopardi et al., 2014). Additionally, cooling to a targeted temperature of 32–36°C has been established as a standard of care in patients after out-of-hospital cardiac arrest to reduce risk of death and improve neurological outcome (Hypothermia after Cardiac Arrest Study, 2002; Callaway et al., 2015; Nolan et al., 2015). Accumulating data also promotes hypothermia as a promising neuroprotective strategy in rodent models of traumatic brain injury (Liu et al., 2016b; Zhao et al., 2017) and stroke (Lee et al., 2016; Liu et al., 2018), but have yet to be translated to human trials. The recent Prophylactic Hypothermia Trial to Lessen Traumatic Brain Injury–Randomized Clinical Trial (POLAR-RCT) trial reported no benefits in neurological outcome at 6 months after applying early moderate hypothermia after severe traumatic brain injury (Cooper et al., 2018) and an acute ischemic stroke trial (Intravascular Cooling in the Treatment of Stroke) reported increased incidents of pneumonia and mortality in the hypothermia vs. normothermia treated group (Lyden et al., 2016).
Experimental investigations on the cytoprotective effects of hypothermia implicate a complex multi-modal response to protect from various ischemia/reperfusion injury mechanisms, including calcium influx, oxidative stress, mitochondrial dysfunction, apoptosis, excitotoxicity (neuronal death), and inflammation (Krech et al., 2017; Kurisu and Yenari, 2018), but the complete mechanism underlying hypothermia-induced neuroprotection remains to be elucidated. Additionally, these studies also did not investigate the effects of hypothermia on the injury-induced sterile inflammatory response.
In contrast to inflammation induced by pathogens, a sterile inflammatory response is induced by an acute condition, such as ischemia/reperfusion injury, in the absence of pathogens. Necrotic cells and other irritant particles are the stimuli for sterile inflammation by activating NLRP3 inflammasomes and inducing the release of interleukin-1 (IL-1), however, the complete mechanism is still not fully understood (Rock et al., 2010; Kono et al., 2014). Increasing evidence points to a sterile inflammatory response within the first few days after an ischemic insult that can exacerbate neuronal cell death (Ceulemans et al., 2010; Jin et al., 2013). Necrotic cell death due to an acute hypoxic-ischemic incident results in the release of DAMPs into the extracellular matrices, where they bind to pattern recognition receptors (e.g., TLRs) on local immunocompetent glial cells and initiate a neuroinflammatory response (Gulke et al., 2018).
CsA is an immunosuppressor used primarily in transplantation medicine to suppress the activation of T-lymphocytes (Borel et al., 1976). CsA binds to intracellular cyclophilin A and inhibits calcineurin, which regulates the immune response by modulating transcription factors activity (Clipstone and Crabtree, 1992). CsA has also been intensively discussed as a potential neuroprotectant as it inhibits the formation of the mitochondrial permeability transition pore by binding to cyclophilin D, thereby preventing mitochondrial dysfunction and apoptosis (Crompton, 1999; Osman et al., 2011; Fakharnia et al., 2017). Therefore, we investigated the anti-neuroinflammatory and neuroprotective effect of clinically established hypothermia in combination with CsA treatment in a murine primary neurons and BV-2 microglia monoculture model of simulated ischemia/reperfusion-induced injury. Additionally, we developed a complex murine OHSC simulated ischemia/reperfusion model where the tissue structures and cell interactions are preserved, and which is well suited for studying cell death and neuroprotective agents ex vivo (Li et al., 2016).
Materials and Methods
Cell Culture
BV-2 microglial cells are immortalized murine microglial cells (Blasi et al., 1990) with a phenotype functionally identical to native primary microglia (Henn et al., 2009), and were a kind gift from Prof. Ullrich (Zurich, Switzerland). BV-2 cells were cultured in high glucose Dulbecco’s Modified Eagle’s Medium supplemented with 1 mM pyruvate (Biochrom), 10% heat inactivated fetal bovine serum (Biochrom), and 100 U/ml penicillin/100 μg/ml streptomycin (Merck Millipore) in a 5% CO2 humidified atmosphere at 37°C. Cells were seeded 24 h prior to experimental start at a density of 500,000 cells in a 60 mm (21 cm2) dish (Sarstedt) pre-coated with 10 μg/ml PLL (Sigma-Aldrich).
Animals and Preparation of Primary Cultures
All animal experiments were approved and performed in accordance with the guidelines of the Charité – Universitätsmedizin Berlin, Germany, and animals were housed in a conventional animal facility (FEM, Charité – Universitätsmedizin Berlin, Germany).
Preparation of Primary Neurons
Primary neurons were prepared from embryonic day 15 (E15) C57BL/6N mice with slight modifications to the protocol previously described (Schmitt et al., 2006). Briefly, after removal of the meninges, cerebral cortices and hippocampi were digested in Hanks Balanced Salt Solution (Thermo Fisher Scientific), containing 0.2% Trypsin (Biochrom) and 100 μg/ml DNase I (Roche Diagnostics), and dissociated with a glass pipette. Cells were initially plated on 200 μg/ml PLL (Sigma-Aldrich; in 0.1 M borate buffer) pre-coated 35 mm dishes (9.2 cm2, TPP) in Minimum Essential Medium (MEM, Gibco) supplemented with 10% heat-inactivated horse serum (Biochrom), 6 g/L glucose (B. Braun), and 1 mg/ml Primocin (InvivoGen). The plating medium was changed to serum free Neurobasal®Medium (Gibco) supplemented with 1x B-27TM (Gibco), 0.5 mM L-glutamine, and 1 mg/ml Primocin. After 5 days in vitro (DIV5) medium was changed to Neurobasal® Medium supplemented with 1x B-27TM minus antioxidants (Gibco). Primary neurons were cultured at 37°C in a humidified atmosphere with 5% CO2.
Preparation of Organotypic Hippocampal Slice Cultures
Hippocampal slice cultures were prepared from C57BL/6N mice at postnatal day 3–5, with slight modifications to the protocol previously described (Schmitt et al., 2007). Briefly, mice were sacrificed by decapitation and the hippocampi were quickly removed and placed in ice cold MEM supplemented with 2.2 g/L sodium bicarbonate, 2 mM L-glutamine, and 8 mM Tris base (all Merck Millipore; pH 7.2). The hippocampi were transversely cut into 350 μm thick slices using a tissue chopper (McIlwain) and 6–8 slices were randomly distributed onto 30 mm membrane inserts with 0.4 μM pore size (Merck Millipore). The inserts were placed in a six-well culture dish with 1.3 ml culture medium, containing MEM, 20% heat-inactivated horse serum, 30 mM hepes (Biochrom), 2 mM L-glutamine, 2.2 g/L sodium bicarbonate, 1 μg/ml insulin (Insuman Rapid), 2.3 g/L glucose, 0.1 mg/ml Primocin, and 88 μg/ml vitamin C (Rotexmedica) at pH 7.2. Slices were cultured at 37°C in a humidified atmosphere with 5% CO2 for 14 days and medium was changed 1 day after preparation and every second to third day thereafter.
Oxygen-Glucose Deprivation and Reperfusion (OGD/R)
Ischemia was simulated in vitro by incubation for 1 h in pre-equilibrated glucose and serum free medium at 0.2% O2, 5% CO2, and 94.8% N2 in a CO2 incubator (Binder). Control cells were maintained in glucose containing medium at Normoxia for 1 h (21% O2). OGD exposure in primary neurons (DIV7) and OHSC (DIV14) was conducted in glucose-free Neurobasal®-A Medium and in BV-2 microglia in glucose-free DMEM. After OGD, reperfusion was simulated by restoration of glucose, serum and 21% O2 for 24 h in all groups in appropriate complete medium.
Time-Temperature Protocol
An experimental time-temperature protocol is illustrated in Figure 1. All cultures were exposed to OGD (0.2% O2) at normothermic temperature (37°C) for 1 h, followed by either normothermic (37°C) or moderate hypothermic (33.5°C) simulated reperfusion (21% O2) for 24 h. Control groups were maintained under normoxic and normothermic conditions (21% O2 and 37°C) for the duration of the experiment.
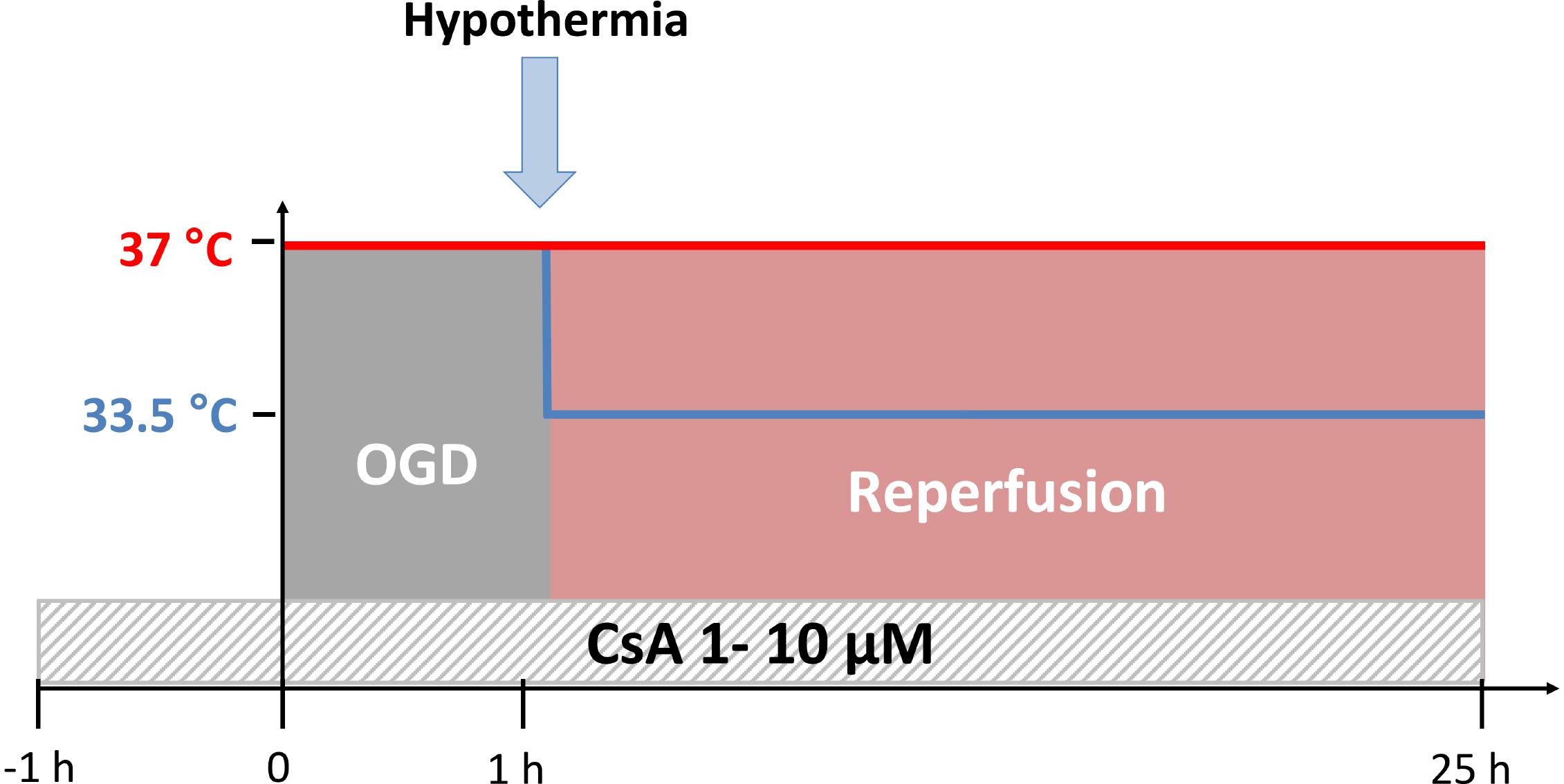
Figure 1. Experimental time-temperature protocol. Cyclosporin A (CsA) was applied 1 h before the simulated ischemia phase by oxygen-glucose-deprivation (OGD at 0.2% O2 in glucose-depleted medium) and maintained after the reperfusion phase for the duration of experiment. Exposure to OGD for 1 h was performed at 37°C, followed by reperfusion (OGD/R) for 24 h at 37°C (normothermia) or 33.5°C (hypothermia).
Cyclosporin A Treatment
Cyclosporin A was purchased from Cayman (#12088). A 20 mM stock solution dissolved in ethanol (EtOH) was aliquoted and stored at −20°C. Working CsA dilutions (1 or 10 μM) in appropriate culture medium were prepared freshly and used to pre-treat cultures at 37°C and 21% O2 for 1 h prior to exposure to OGD and maintained throughout the duration of the experiment. Control cells were incubated with equal amount EtOH solvent.
Assessment of Lactate Dehydrogenase (LDH) Release
Cell death was assessed by LDH released into the cultured supernatant using a colorimetric Cytotoxicity Detection Kit (Roche Diagnostics) according to the manufacturer’s instructions. Briefly, cultured supernatants were separated from cells by centrifugation and mixed with reagents (1:1) in a 96-well plate. Extinction was measured at 490 nm minus 630 nm using a microtiter plate reader (Thermo Fisher Multiskan Ascent). LDH release is expressed as a percentage of total LDH content, as determined from lysed normoxic control cells using a freeze/thaw method at −80°C.
Assessment of Glutamate Release
Glutamate release into the cultured supernatant was measured using the Glutamat-GloTM Assay (Promega) according to the manufacturer’s instruction. Briefly, cell free supernatant was incubated with glutamate detection reagent (1:1) in a 96-well plate and luminescence signal was measured using a microplate reader (Tecan Infinite®M200 Pro). Glutamate concentration was extrapolated from a concentration curve and expressed in μM.
Assessment of Propidium Iodide (PI) Staining in the OHSCs
Cell death in the OHSCs was assessed by PI staining. OHSCs were incubated with 4 μg/ml solution of PI (Sigma-Aldrich, Germany) for the last 30 min of the reperfusion phase. After staining, samples were washed twice with PBS and documented using a Keyence BZ-9000 inverted microscope.
Extracellular Proteins Isolation and Western Blot Analysis
Supernatants were collected after 1 h of OGD or Normoxia and extracellular proteins were isolated by trichloroacetic acid precipitation. Briefly, supernatants were incubated with 20% trichloroacetic acid (VWR) for 30 min on ice, and then centrifuged at 16,000 × g for 20 min. The precipitated proteins were washed with ice cold aceton and dissolved in RIPA buffer. Samples were incubated with Pierce Lane Marker Reducing Sample Buffer (Thermo Scientific) at 95°C for 5 min and subjected to a 15% SDS polyacryl gel electrophoresis. Proteins were transferred onto a polyvinylidene fluoride membrane (PALL Life Sciences) overnight at 30 V using a tank blot procedure (Bio-Rad Laboratories). The membrane was blocked for 1 h at room temperature using 5% bovine albumin fraction V (Carl Roth) for Hsp70 and HMGB1 or 5% dry milk (Applied Biosystems) for CIRBP in TBS + 0.1% Tween 20. Primary antibodies against Hsp70 (1:1000, Cell Signaling Technology, Cat#4872) HMGB1 (1:1000, Chondrex, Cat#7028), and CIRBP (1:1000, Abclonal, Cat#A6080) were diluted in blocking solution and blots were incubated overnight at 4°C. HRP-conjugated secondary antibodies (anti-rabbit IgG, 1:20,000 Dianova) were incubated on the blots for 1 h at room temperature. We used Dura Super Signal West (Thermo Fisher Scientific) to visualize protein expression and ChemiDocTM Imaging Systems and Image LabTM Software (Bio-Rad) for densitometry analysis.
RNA Isolation and Semi-Quantitative Real-Time qPCR
Total RNA from BV-2 microglia and primary neurons was isolated by acidic phenol/chloroform extraction using RNA PureTM (Peqlab) and RNA from OHSC was isolated by using the GenUPTM Total RNA Kit (Biotechrabbit) followed by genomic DNA digestion using Turbo DNA-freeTM Kit (Ambion) according to manufacturers’ instructions, respectively. RNA concentration and purity was determined by spectrophotometric measurements at 260 nm and 280 nm using a Nanodrop 2000 (Nanodrop) and agarose gel electrophoresis. cDNA was transcribed from 1 μg total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) in a thermal cycler (PTC200, MJ Research). Expression of target genes and the endogenous control, GAPDH, was assessed by real-time qPCR using the TaqMan®Gene Expression Assays (summarized in Table 1) and StepOnePlusTM Real-Time PCR System (Applied Biosystems) according to manufacturer’s recommendations. Reactions with no templates and RNA control were included as negative controls. Relative quantification of gene expression was normalized to the housekeeping gene GAPDH, using the 2−ΔΔct method, and illustrated as fold change (Livak and Schmittgen, 2001).

Table 1. TaqMan®Gene Expression Assays.
Statistical Analysis
Experimental data from at least four independent experiments were analyzed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, United States). Values are presented as box-and-whiskers plot (box from 25th to 75th percentile and whisker min to max). Comparisons between experimental groups were made using one-way ANOVA followed by the Tukey post-test for multiple comparisons and p < 0.05 was considered to be significant.
Results
Cyclosporin A is an immunosuppressant medication that has been reported to exhibit neuroprotective properties in multiple experimental models (Trumbeckaite et al., 2013). Therefore, we investigated the potential neuroprotective effect of pre-treatment with CsA (1 and 10 μM) in a simulated ischemia/reperfusion-induced injury model (1 h OGD followed by 24 h restoration of oxygen, glucose, and serum) in either a monoculture of murine primary neurons (DIV7) prepared from E15 mice, BV-2 microglia, or in an OHSC (DIV14) prepared from early postnatal (P3–5) mice that retains the cytoarchitecture and synaptic circuits of the hippocampus. Additionally, moderate hypothermia (33.5°C) was applied post-OGD to both, the in vitro and ex vivo models, to investigate any potential additive neuroprotective effects of the combined treatment.
Cytotoxicity in Primary Neuron and Microglia Monocultures
We observed that initial exposure to OGD for 1 h did not significantly induce necrotic cell death compared to normoxic control in the primary neuronal and BV-2 microglial cell cultures, as measured by LDH release (Figures 2A,B). However, treatment with 10 μM CsA under both normoxic and OGD conditions resulted in measurable toxicity in the primary neurons, as observed by significantly increased LDH release compared to Normoxia control (2.8-fold and 4.1-fold increase, respectively), while 1 μM CsA was not toxic (Figure 2A). Furthermore, BV-2 microglia were resistant to CsA-induced toxicity, as no significant increases in LDH releases were measurable after exposure to 1 h OGD and treatment with 1 and 10 μM CsA (Figure 2B).
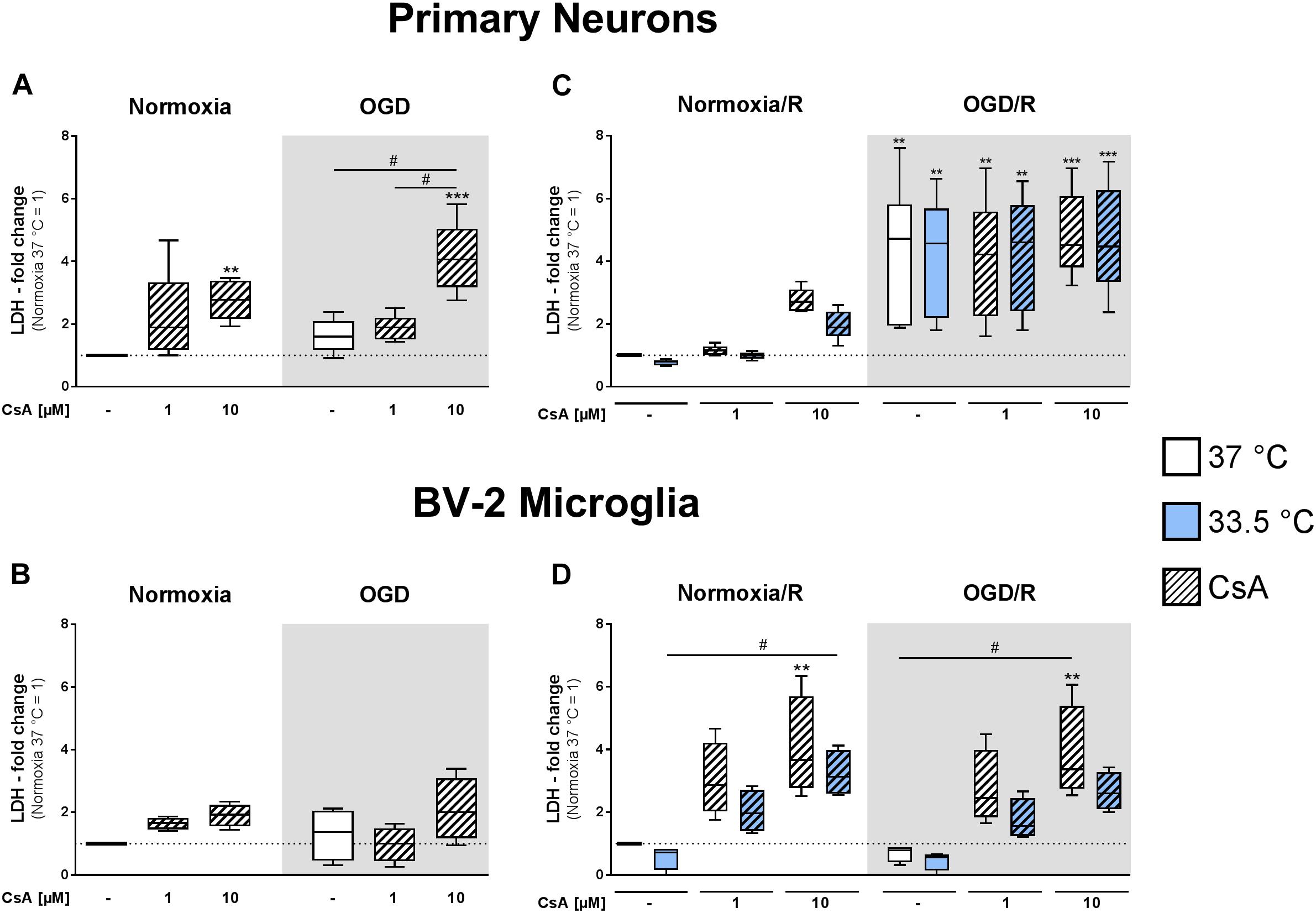
Figure 2. Lactate dehydrogenase releases were measured from primary neurons (A) and BV-2 microglia cells (B) in the experimental control group exposed to 1 h normoxia (21% O2 in glucose-containing medium) and simulated ischemia group exposed to 1 h oxygen-glucose deprivation (OGD at 0.2% O2 in glucose-depleted medium) at 37°C, and also after reperfusion from primary neurons (C) and BV-2 microglia cells (D) exposed to 1 h normoxia + 24 h reperfusion with complete medium (Normoxia/R) or 1 h OGD + 24 h reperfusion (OGD/R) at either 37 or 33.5°C. All CsA groups were pre-treated 1 h before experimental start with either 1 or 10 μM CsA at 37°C. Data from 4 to 6 individual experiments are presented as box-and-whiskers plot (box from 25th to 75th percentile and whisker min to max). Statistical analysis were conducted using one-way ANOVA followed by the Tukey post hoc test; ∗∗p < 0.01, ∗∗∗p < 0.001 compared to Normoxia at 37°C, and #p < 0.05 for group comparison were considered significant.
Exposure to 1 h OGD followed by 24 h reperfusion (OGD/R) resulted in a significantly higher LDH release (4.4-fold increase relative to Normoxia/R control) in the primary neurons (Figure 2C). Treatment with 1 and 10 μM CsA had no additive cytotoxic effect and necrotic cell death was not attenuated by cooling. In contrast, BV-2 microglia exposed to OGD/R showed no observable necrotic cell death, but treatment with CsA was observed to be toxic in a dose dependent manner under both Normoxia/R and OGD/R conditions, as measurable by an incremental increase in LDH release that was significant with 10 μM CsA (Normoxia/R: 4.0-fold; OGD/R: 3.8-fold) (Figure 2D). Treatment with hypothermia resulted in only slight decreases in LDH releases that did not reach significancy under all test conditions.
Interestingly, longer exposure to CsA [1 h (Figure 2B) vs. 1+24 h (Figure 2D)] induced higher LDH release in BV-2 microglia, which was not observable in the neuronal cultures. BV-2 exposed to 1 μM CsA under normoxic conditions resulted in an increase of LDH release from 1.6-fold to 3.0-fold, and 10 μM CsA resulted in an increase from 1.9-fold to 4.0-fold, respectively (Figures 2B,D).
Cytotoxicity in Organotypic Hippocampal Slice Cultures
Organotypic hippocampal slice culture are a suitable model for studying cell death and neuroprotective agents ex vivo, as the complex cellular structures and cell interactions are preserved. Therefore, we further investigated the cytotoxic effects of exposure to 1 h OGD and treatment with 10 μM CsA in OHSCs prepared from early postnatal (P3–5) mice and cultivated for 14 days (DIV14). Necrotic cell death was assessed by the release of LDH and glutamate, a neuronal excitotoxicity marker. We did not observe a significant increase in LDH release after 1 h exposure to OGD or treatment with 10 μM CsA under normoxic condition alone. However, treatment with 10 μM CsA under OGD condition resulted in an increase in LDH release that was significantly higher than for both treatments with normoxic CsA and OGD alone (Figure 3A). Interestingly, we observed significantly higher glutamate release in the OHSC exposed to 1 h OGD compared to the normoxic control (3.9 vs. 0.8 μM, respectively) (Figure 3B). Significantly higher release of glutamate was also observed in OHSC treated with 10 μM CsA under OGD condition compared to normoxic control (6.42 vs. 0.83 μM, respectively), confirming our LDH assessment indicating necrosis in the OHSCs.
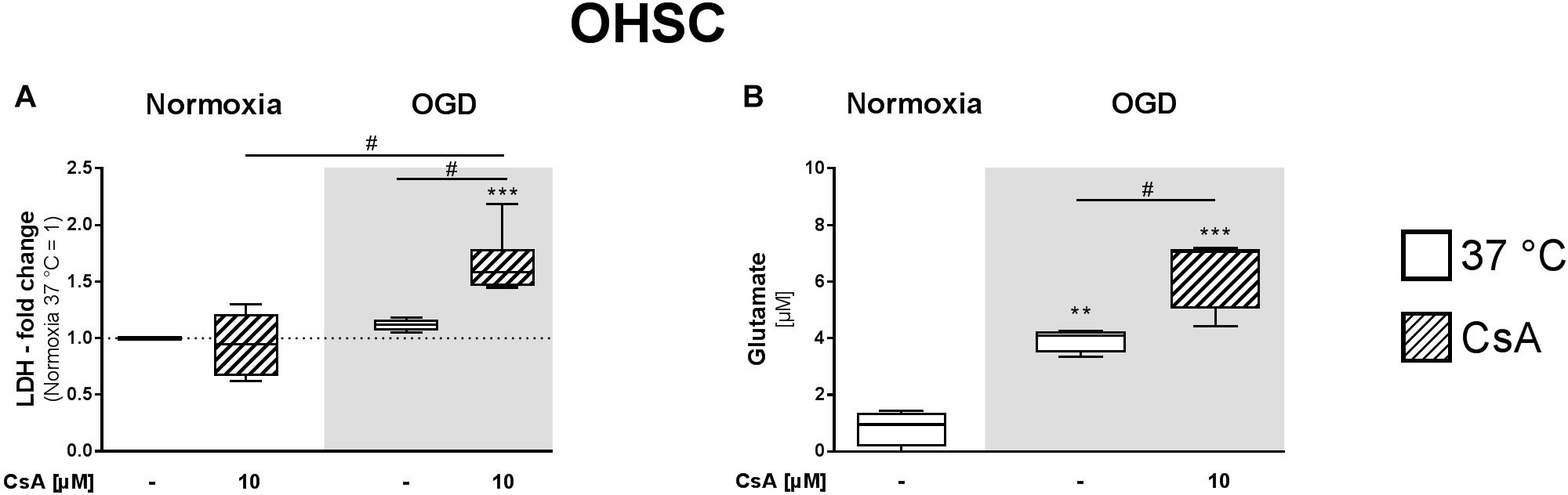
Figure 3. (A) LDH and (B) glutamate releases from OHSCs were assessed from the cultured supernatants of the experimental control group exposed to 1 h normoxia (21% O2 in glucose-containing medium) and simulated ischemia group exposed to 1 h oxygen-glucose deprivation (OGD at 0.2% O2 in glucose-depleted medium) at 37°C. All CsA containing groups were pre-treated 1 h before experimental start with 10 μM CsA at 37°C. Data from 4 to 5 individual experiments are presented as box-and-whiskers plot (box from 25th to 75th percentile and whisker min to max). Statistical analysis were conducted using one-way ANOVA followed by the Tukey post hoc test; ∗∗p < 0.01, ∗∗∗p < 0.001 compared to Normoxia at 37°C, and #p < 0.05 for group comparison were considered significant.
Due to high serum concentration in the reperfusion medium, LDH and glutamate levels were not detectable after OGD/R-induced injury. Therefore, we performed PI staining to assess necrotic cell death in the OHSC after reperfusion (Figure 4). Exposure of the OHSCs to OGD/R at 37°C resulted in a marked increase in PI positive cells, as compared to Normoxia/R at 37°C. OHSCs exposed to OGD/R at 33.5°C resulted in less observable PI positive cells. OHSCs exposed to Normoxia/R +10 μM CsA at 37°C also showed a marginal increased in PI positive cells that dramatically increased when CsA was applied in combination with OGD/R at 37°C. No observable differences in PI positive cells were seen between OGD/R and OGD/R+CsA slices incubated at 37°C, nor with OGD/R+CsA slices incubated at 33.5°C.
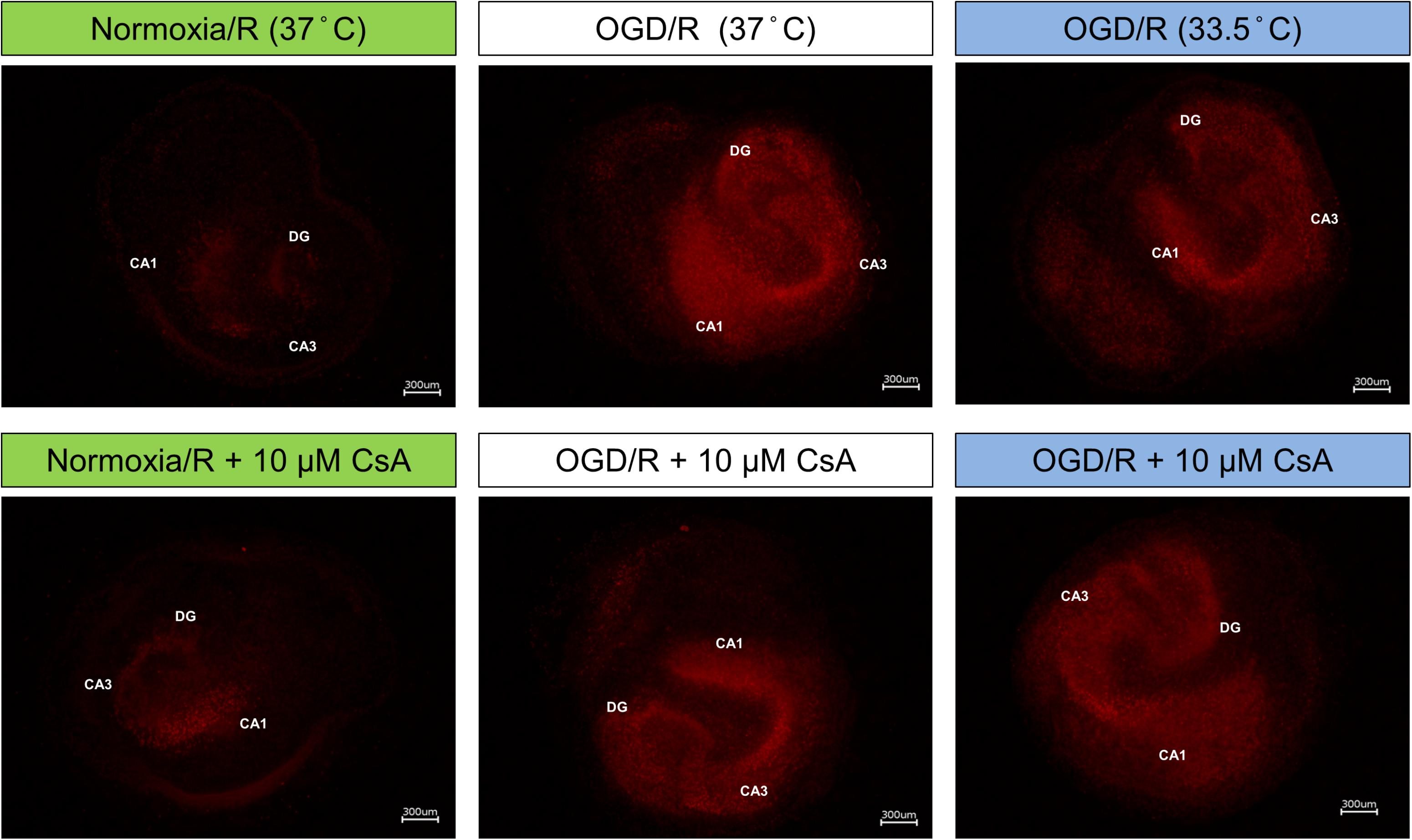
Figure 4. Propidium iodide (PI) staining; OHSCs in the experimental control group (Normoxia/R) were exposed to 1 h normoxia (21% O2 in glucose-containing medium) + 24 h reperfusion (21% O2 in glucose-containing medium) and simulated ischemia groups were exposed to 1 h oxygen-glucose deprivation (OGD at 0.2% O2 in glucose-depleted medium) + 24 h reperfusion (OGD/R at 21% O2 in glucose-containing medium) at either 37 or 33.5°C. All cyclosporin A (CsA) containing groups were pre-treated 1 h before experimental start with 10 μM CsA at 37°C and maintained throughout the duration of the experiment. Images are shown at 40× magnification. CA, Cornu Ammonis; DG, Dentate Gyrus.
Release of DAMPs
Soluble inflammatory mediators are released from the necrotic brain tissue. Therefore, we isolated extracellular proteins from cell-cultured supernatants to investigate the release of inflammation inducing DAMPs from primary neurons and BV-2 microglia exposed to 1 h OGD. Correlating with the LDH measurements, western blot analysis showed significantly higher releases of HMGB1 and HSP70 in primary neurons treated with 10 μM CsA under OGD conditions, in comparison to normoxic control, as well as to all other OGD treated groups (Figures 5A,B). Additionally, we observed a trend toward a higher release of CIRBP in neurons treated with 10 μM CsA under OGD conditions (Figure 5C). Also in correlation to LDH release, no significant increases in extracellular HMGB1 and HSP70 were observed in non-necrotic BV-2 microglia exposed to OGD and treated with 10 μM CsA (Figures 5D,E). Interestingly, we observed elevated but non-significant DAMPs releases in all OGD damaged BV-2 cells in comparison to Normoxia control, which was not seen in the primary neurons. Extracellular CIRBP was not detectable in the BV-2 microglia cultured supernatant. Furthermore, none of the investigated DAMPs were detectable in the cultured supernatant of the OHSCs.
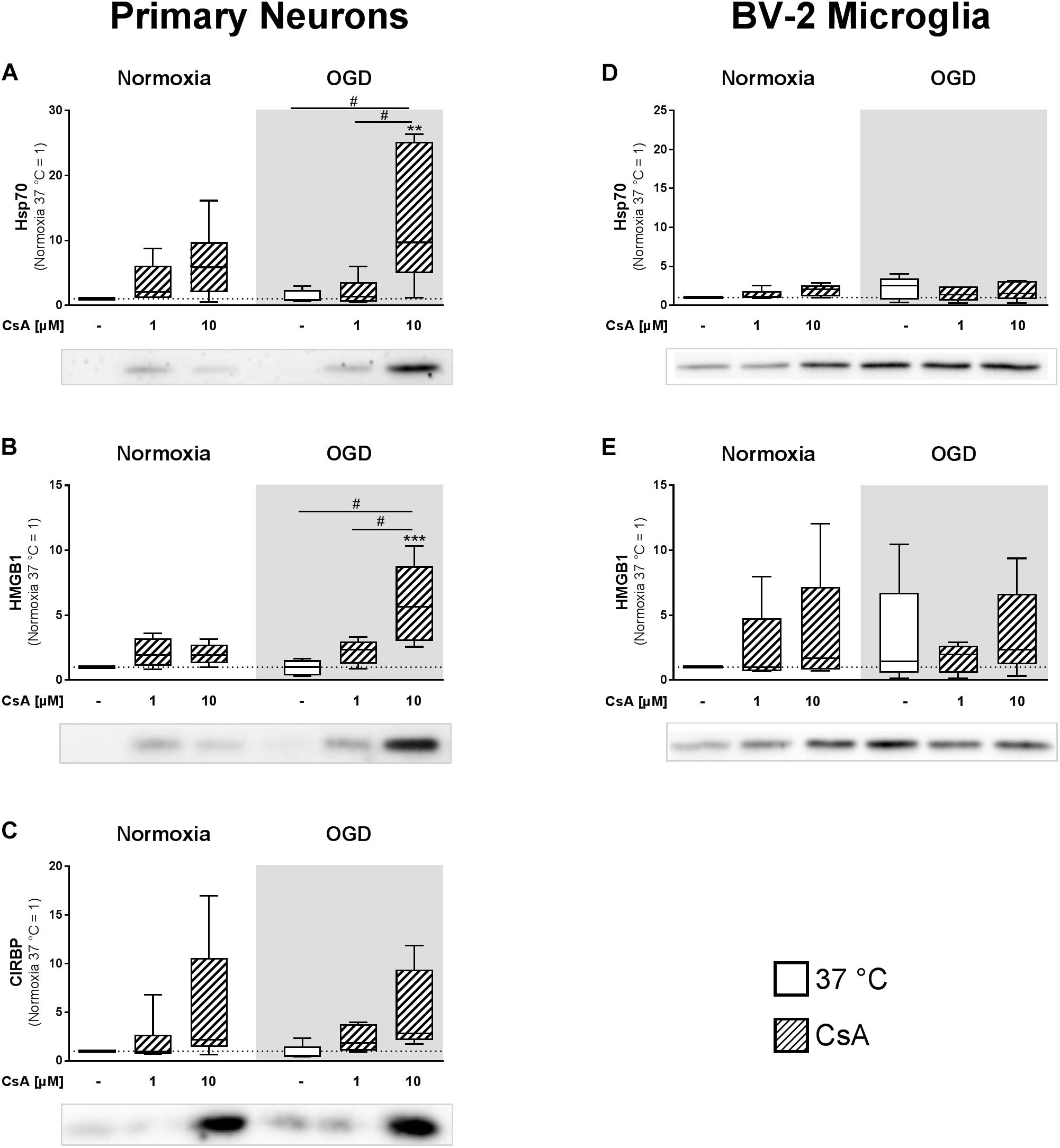
Figure 5. Western Blot analysis was used to assess extracellular DAMPs, (A,D) heat shock protein 70 (Hsp70), (B,E) high mobility group box 1 (HMGB1), and (C) cold-inducible RNA-binding protein (CIRBP) released into the cultured supernatant from primary neurons and BV-2 microglial cells. The experimental control group was exposed to 1 h normoxia (21% O2 in glucose-containing medium) and simulated ischemia group was exposed to 1 h oxygen-glucose deprivation (OGD at 0.2% O2 in glucose-depleted medium) at 37°C. Quantitative densitometric analysis from 5 to 6 individual experiments is presented as box-and-whiskers plot (box from 25th to 75th percentile and whisker min to max), along with the representative immunoblots. Statistical analysis were conducted using one-way ANOVA followed by the Tukey post hoc test; ∗∗p < 0.01, ∗∗∗p < 0.001 compared to normoxia at 37°C, and #p < 0.05 for group comparison were considered significant.
Cytokine and Chemokine Expression in BV-2 Microglia, Primary Neurons, and OHSC
Based on our observation of increased DAMPs release from necrotic primary neurons, we next investigated the inflammatory response in the immune competent BV-2 microglia, in primary neurons, and OHSC after exposure to OGD/R and treatment with CsA where necrosis was detectable in neurons and OHSC. Additionally, we also investigated the effect of cooling (33.5°C) applied during the reperfusion phase on inflammatory cytokines and chemokines gene expressions.
Cytokine Expressions in BV-2 Microglia and Primary Neurons
We observed a significant increase in IL-6 expression in the BV-2 microglia treated with 10 μM CsA under both Normoxia/R and OGD/R conditions, which correlated with the observed necrotic cell death (Figure 6A). Cooling significantly reduced IL-6 expression in CsA treated Normoxia/R control and in weaker extend in the OGD/R injury group. Similar increased IL-6 expression was also observed in the primary neurons treated with 10 μM CsA (Figure 6I). Interestingly, TNF-α expression was significantly induced by cooling under both Normoxia/R and OGD/R conditions in BV-2 microglia, which was attenuated by treatment with CsA (Figure 6B). Additionally, cooling and CsA alone and in combination significantly inhibited IL-1β expression under both Normoxia/R and OGD/R conditions (Figure 6C), but had no significant effect on IL-1α expression in BV-2 microglia (Figure 6D). TNF-α, IL-1β, and IL-1α expressions were under detection limit in the primary neurons.
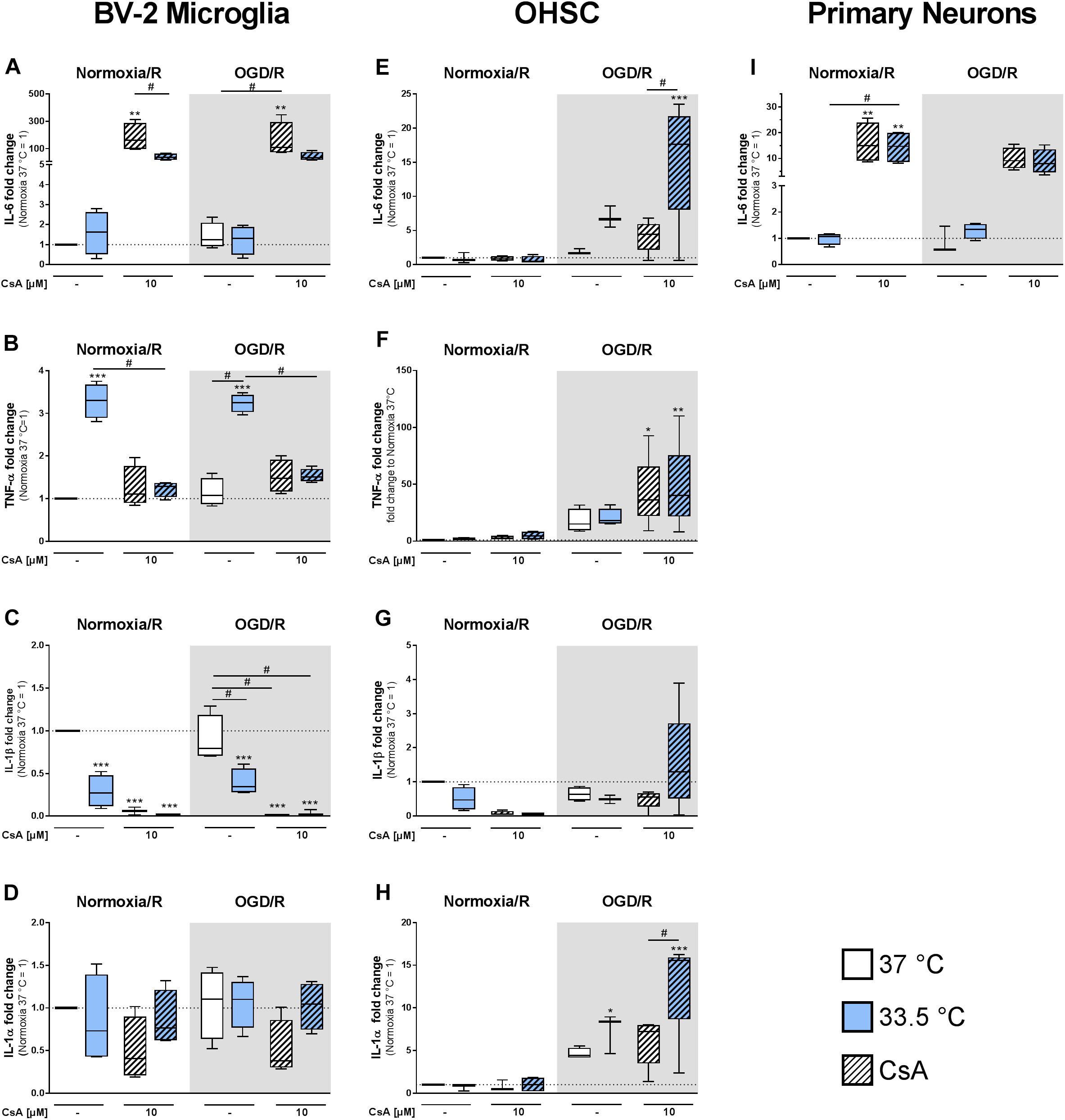
Figure 6. RT-qPCR was used to assess gene expressions in BV-2 microglia, OHSCs, and primary neurons for inflammatory (A,E,I) IL-6, (B,F) TNF-α, (C,G) IL-1β, and (D,H) IL-1α after exposure to Normoxia/R (1 h normoxia + 24 h reperfusion at 21% O2 in glucose-containing medium) or OGD/R (1 h OGD + 24 h reperfusion at 0.2% O2 in glucose-depleted medium) at either 37 or 33.5°C. All CsA containing groups were pre-treated with 10 μM CsA at 37°C. Data from 4 to 5 individual experiments are presented as box-and-whiskers plot (box from 25th to 75th percentile and whisker min to max). Statistical analysis were conducted using one-way ANOVA followed by the Tukey post hoc test; ∗p < 0.01, ∗∗p < 0.01, ∗∗∗p < 0.001 compared to Normoxia/R at 37°C, and #p < 0.05 for group comparison were considered significant.
Cytokine Expressions in the OHSC Model
We next investigated if exposure to OGD/R injury and treatment with CsA had a similar cytokine expression regulation in the OHSC model as observed in the BV-2 microglia. In general, inflammatory cytokines expressions in the OHSC model did not reflect what we observed in the BV-2 microglia monoculture. Interestingly, the combined treatment of 10 μM CsA, OGD/R, and cooling resulted in the greatest general increase in cytokines expressions. Under these combined conditions, IL-6 expression was significantly higher than Normoxia/R control and OGD/R+CsA, indicating a profound effect of cooling on IL-6 expression (Figure 6E). TNF-α expression was significantly higher than Normoxia/R control in the OGD/R+CsA treated group with no observable effect of cooling (Figure 6F). IL-1β expression was not suppressed as observed in the BV-2 microglia (Figure 6G), and IL-1α expression appears to be more induced by cooling under both OGD/R and OGD/R+CsA treatments compared to 37°C Normoxia/R group (Figure 6H).
Chemokine and Growth Factor Expressions in BV-2 Microglia and Primary Neurons
Furthermore, we investigated the expression of targets regulating migration and activation of microglia and monocytes. In BV-2 microglia we observed a trend toward hypothermia-induced MCP1 expression, whereas treatment with CsA rather reduced MCP1 expression in comparison to Normoxia/R control (Figure 7A). In contrast to BV-2 microglia, we found an increased MCP1 expression in primary neurons treated with 10 μM CsA, which was even higher at 33.5°C in the OGD/R injured group (Figure 7I).
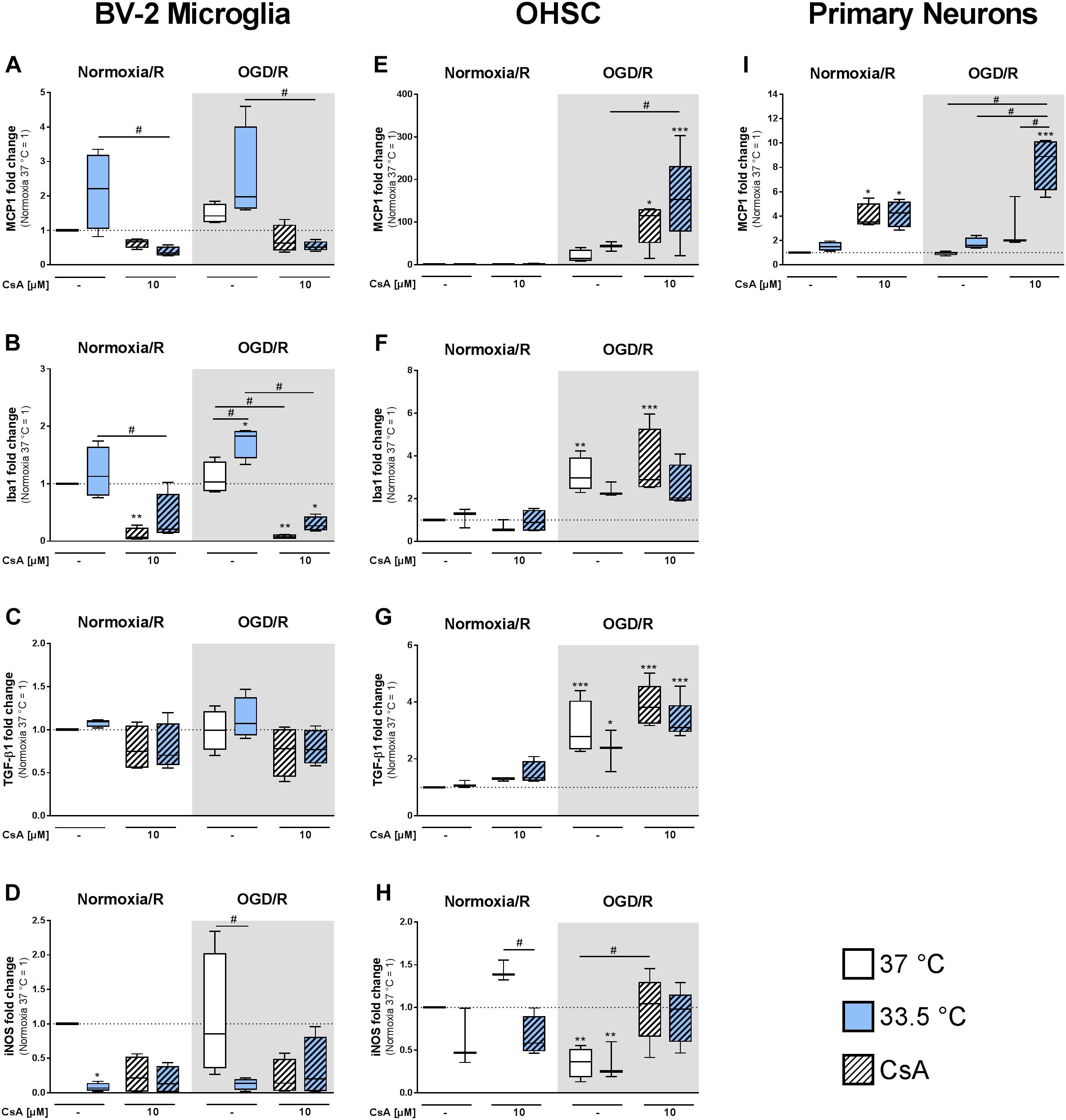
Figure 7. RT-qPCR was used to assess gene expressions in the BV-2 microglia, OHSCs, and primary neurons for (A,E,I) MCP1, (B,F) Iba1, (C,G) TGF-β1, and (D,H) iNOS after exposure to Normoxia/R (1 h normoxia + 24 h reperfusion at 21% O2 in glucose-containing medium) or OGD/R (1 h OGD + 24 h reperfusion at 0.2% O2 in glucose-depleted medium) at either 37 or 33.5°C. All CsA containing groups were pre-treated with 10 μM CsA at 37°C. Data from 4 to 5 individual experiments are presented as box-and-whiskers plot (box from 25th to 75th percentile and whisker min to max). Statistical analysis were conducted using one-way ANOVA followed by the Tukey post hoc test; ∗p < 0.01, ∗∗p < 0.01, ∗∗∗p < 0.001 compared to Normoxia/R at 37°C, and #p < 0.05 for group comparison were considered significant.
Activation of microglia is associated with increased Iba1 expression (Hovens et al., 2014). Similar to MCP1, we found Iba1 expression significantly induced in the cooled OGD/R injured group, while treatment with CsA significantly decreased Iba1 expression (Figure 7B). The expression of growth factor TGF-β1 was unchanged in all BV-2 microglia groups (Figure 7C). Additionally, we found iNOS expression to be significantly suppressed by cooling under Normoxia/R, and to a less extend by OGD/R at 33.5°C and by CsA treatment (Figure 7D). Iba1, TGF-β1, and iNOS expressions were under detection limit in the primary neurons.
Chemokine and Growth Factor Expressions in OHSC
In the OHSC model we observed a strong increase in chemokine MCP1 expression in OGD/R and OGD/R+CsA compared to Normoxia/R control (Figure 7E). A similar response was also found in primary neurons (Figure 7I). Iba1 gene expression was induced in OGD/R injury alone and in combination with CsA (Figure 7F). Comparable to Iba1, the expression of growth factor TGF-β1 was increasingly expressed in OGD/R injured slices, while cooling and CsA treatment had no additional effect (Figure 7G). Interestingly, iNOS expression was significantly decreased by OGD/R-induced injury and increased to Normoxia/R level by OGD/R+CsA (Figure 7H).
Discussion
Neuroinflammation plays a central role in the pathogenesis of ischemic brain injury (Bhalala et al., 2014; Hagberg et al., 2015). Therefore, we evaluated the anti-inflammatory and neuroprotective effect of clinically discussed hypothermia in combination with CsA immunosuppressant treatment on OGD/R injured brain cells. In our study treatment with hypothermia and CsA caused an alteration in the inflammatory pathways. While pro-inflammatory IL-1β and iNOS expressions are effectually suppressed in BV-2 microglia and OHSC, expression of TNF-α, IL-1α, MCP1, Iba1, and TGF-β1 are increased in OGD/R injured OHSC. We found that primary neurons undergo cell death by OGD/R-induced injury and by CsA treatment, suggesting a complex activation of inflammatory pathways through the involvement of a secondary damage mechanism in complex slice culture. Our findings demonstrate that CsA, a highly specific inhibitor of calcineurin, affects the survival of primary neurons and modulate the inflammatory response in microglial cultures, as well as in the OHSC.
Cytotoxicity-Induced DAMPs Release
Primary neurons pre-treated with 10 μM CsA followed by 1 h exposure to OGD undergo necrotic cell death, as indicated by increased LDH release. Additionally, increased amounts of DAMPs, namely Hsp70, HMGB1, and CIRBP were detected in the cultured supernatants. Hsp70 and HMGB1 are classical DAMPs and have been extensively investigated (Asea et al., 2000; Muhammad et al., 2008), whereas the inflammation-inducing DAMP property of extracellular CIRBP is a recent observation that has been shown to perpetuate the inflammatory response in hemorrhagic shock and sepsis patients (Qiang et al., 2013). Although both, intracellular Hsp70 and CIRBP, have been shown to have desirable anti-apoptotic properties, their presence in the extracellular matrix has been shown to initiate inflammatory responses (Liao et al., 2017; Kim et al., 2018). Their affinity to bind to surface receptors, including RAGE, toll-like receptor 2 (TLR2), and 4 (TLR4) on adjacent cells leads to the activation of the nuclear factor “kappa-light-chain-enhancer” of activated B-cells (NF-κB) pathway and transcription of inflammatory cytokines, such as TNF-α, IL-1β, and IL-6 (Johnson and Fleshner, 2006; Andersson and Tracey, 2011; Zhou et al., 2014). In comparison to neurons, BV-2 microglia were more vulnerable to OGD-induced release of HMGB1 and Hsp70, which did not reach significance, but were less vulnerable to OGD+CsA-induced cytotoxicity, which resulted in no additional DAMPs release.
Organotypic hippocampal slice culture exposed to 1 h of OGD had a significant increased release of glutamate, but no increase in LDH release was measureable. Under non-pathological conditions glutamate in the extracellular space is cleared by astrocytes via glutamate transporters, but under ischemic conditions the uptake is reversed due to ion gradients changes, resulting in the release of glutamate to the extracellular space. High concentration of glutamate can cause extensive neuronal injury and subsequently trigger cell death (Rossi et al., 2000; Nishizawa, 2001; Zhang et al., 2019). The addition of CsA during the OGD phase resulted in an increase in LDH release and an additional increase of glutamate in the cultured supernatant, presumably by necrotic cells, as we also observed CsA-induced cell death in both microglial and neuronal mono-cultures. Our findings are in line with an in vitro study of primary mixed neurons-astrocytes culture showing CsA-induced cytotoxicity only in the neurons, while the astrocytes were unharmed (Kaminska et al., 2001).
Reperfusion-induced injury occurs when restoration of energy and oxygen after an OGD phase is achieved, causing alterations in multiple pathways affecting inflammation, redox-system, and many other signaling pathways (Lopez-Neblina et al., 2005; Mizuma and Yenari, 2017). In line with previous findings describing a higher resistance of microglia than neurons to OGD/R-induced injury, we also observed a marked increase in necrosis in primary neurons but absent in BV-2 microglia after exposure to OGD/R (Goldberg and Choi, 1993; Lim et al., 2006; Li et al., 2007). Surprisingly, neither treatment with moderate hypothermia for 24 h, nor CsA protected the neurons from OGD/R-induced cell death. Conversely, treatment with CsA resulted in BV-2 microglia cell death in a concentration dependent manner, which could be partially attenuated by cooling. In the ex vivo OHSCs, we observed a dramatic increase in PI positive cells after exposure to OGD/R at 37°C, which was decreased by cooling to 33.5°C. Similar exposure to OGD/R at 37°C also resulted in significant cell death in the primary neuronal culture. However, treatment with CsA during OGD/R had no observable protective effect in the slice cultures independent of temperature, as similar intensities in PI staining were also observed in the slices exposed to OGD/R alone. This is in contrast to Liu et al. (2016a), who reported a protective effect of CsA on the mitochondria which was augmented by hypothermia in an in vivo rat cardiac arrest model. Unfortunately, quantification of DAMPs release in the reperfusion phase was not possible by western blot analysis due to the high serum concentration in the cultured medium, and warrants further investigation.
Hypothermia
To date, it is widely accepted that multiple pathways and mechanisms are involved in the protective effect of hypothermia (Kurisu and Yenari, 2018). Preclinical studies in vitro and in vivo suggest that anti-inflammatory mechanisms induced by hypothermia play an important role (Lee et al., 2016). Exposure of BV-2 microglia to hypothermia for 24 h had variable effects on known pro-inflammatory mediators, resulting in increased TNF-α and MCP1 expressions and decreased IL-1β and iNOS expressions under both Normoxia/R and OGD/R conditions. While increased iNOS and IL-1β levels are generally associated with neurotoxic effects, TNF-α also exhibits neuroprotective properties (Kawabori and Yenari, 2015). Lambertsen et al. (2009) showed in an in vivo cerebral ischemia model that microglia derived TNF-α enhances neuronal survival. Other ex vivo OHSC studies showed that deep hypothermia induces TNF-α secretion, resulting in neurite outgrowths (Schmitt et al., 2010). Nevertheless, neurotoxic effects by high levels of TNF-α in the ischemic brain injury are also well documented (Sriram and O’Callaghan, 2007). Also the expression of the chemokines MCP1 leads to recruitment and migration of leukocyte from the periphery to the site of injury. MCP1 is upregulated in models of cerebral ischemia and inhibition of MCP1 can decrease brain injury (Chen et al., 2003).
When the same OGD/R injury protocol was applied to the OHSC model ex vivo, neither significant induction of TNF-α and MCP1 expressions, nor a significant inhibition of IL-1β by cooling was observed. The heterogeneity of cells in the OHSC, with 5–15% of microglial cells in the hippocampus (Lawson et al., 1990) may explain the variability.
Cyclosporin A
Cyclosporin A is a cyclophilin binding substance whose primary immunosuppressive function is to inhibit calcineurin, a Ca2+/calmodulin dependent protein phosphatase. Calcineurin is ubiquitously expressed in most tissues, but at particularly high concentrations in the brain. In glia cells calcineurin plays a global role in neuroinflammation, as it interacts and modulates multiple transcription factors, including NFAT, NF-κB, and AP1, which are associated with cytokines expression (Furman and Norris, 2014).
In the BV-2 microglia, cooling suppressed IL-1β and iNOS expressions, but also induced TNF-α and MCP1 expressions, which was attenuated by CsA treatment. Increased pro-inflammatory iNOS expression has been observed to result in the production of high amounts of NO, which attributes to brain cytotoxicity and promotes ischemic cell death (Amantea et al., 2009; Terpolilli et al., 2012). Zawadzka et al. (2012) observed that CsA inhibits iNOS expression in an in vitro model of lipopolysaccharide stimulated microglia, by interfering with the MAPK and NF-κB signaling pathways. Our findings indicate that NF-κB driven cytokines expressions are also inhibited by CsA treatment. This is not surprising as calcineurin has been shown to be indirectly involved in the activation of NF-κB by degrading IκBα, as well as NFATs regulated cytokine gene expressions by coupling with AP1 or NF-κB, amongst others (Macian et al., 2001; Sama et al., 2008; Palkowitsch et al., 2011). Multiple isoforms of NFAT that are calcineurin activated transcription factors have been shown to be expressed by microglia and can be specifically inhibited by the NFAT inhibitor VIVIT, resulting in decreased secretions of TNF-α and MCP1 (Nagamoto-Combs and Combs, 2010; Rojanathammanee et al., 2015). Additionally, microglia activation marker, Iba1, expression was also inhibited by CsA. In contrast to reduced MCP1 expression observed in the BV-2 microglia, CsA induces expression of MCP1 in the primary neuronal cultures under both Normoxia/R and OGD/R conditions. This is not surprising, as neurons have been observed to be capable of expressing chemokines, including MCP1, during the early phase after ischemia in an in vivo model of focal cerebral ischemia (Che et al., 2001).
Interestingly, treatment with CsA upregulated IL-6 expression, but had no measurable effect on TGF-β1 and IL-1α expressions in the BV-2 microglia. IL-6 was also the only cytokine we observed to be expressed under OGD/R+CsA treatment in primary neurons. IL-6 can be secreted by both immune (microglia) and non-immune (neurons) cells and function as a neurotrophic factor and inhibitor of neuronal death (Loddick et al., 1998). At the same time IL-6 is involved in the pathological progression of several inflammatory diseases (Rothaug et al., 2016).
Altogether, CsA exhibits an immunosuppressive effect in a BV-2 microglia model, but it also had a toxic effect at the same concentration in a murine dissociated neuronal culture. The response to OGD/R-induced injury in combination with CsA and hypothermia treatment was also investigated in a more complex ex vivo OHSC model, which shows a different pattern of response to simulated ischemia than the individual primary neuronal cultures and BV-2 microglia. We did not observe significant increased induction of the inflammatory pathways investigated in the OGD/R-induced injured slices, but surprisingly increases in TNF-α, IL-1α, IL-6, and MCP1 expressions were observed when 10 μM CsA was additionally introduced, which was not observed in the Normoxia/R control treated with CsA nor in the BV-2 microglia under OGD/R+CsA conditions. As astrocytes are the most abundant glial cell type in the brain capable of participating in the immune response (Becerra-Calixto and Cardona-Gomez, 2017), the observed increase in inflammatory response in our ex vivo OHSC model could be considered to be astrocytes driven. Increased MCP1 expression has been observed in hippocampal astrocytes after in vivo transient global ischemia (Sakurai-Yamashita et al., 2006), which can trigger the adaptive immunity response in the inflamed CNS (Farina et al., 2007). Additionally, in vitro studies with primary astrocytes showed longer exposure to OGD (24 h) resulted in increased TNF-α and IL-1β secretions, which was attenuated by CsA treatment (Gabryel et al., 2004). Moreover, in vivo studies of transient middle cerebral artery occlusion in a rat model found decreased TNF-α secretion after treatment with CsA containing nanoliposomes compared to non-treated animals (Partoazar et al., 2017). This is in contrast to our findings in the OHSC treated with OGD/R+CsA, indicating that the putative protective effect of CsA may be dominated by or overlaid with other pro-inflammatory mediators.
Other studies showed DAMPs released from necrotic neurons in hippocampal slices or treated with exogenous HMGB1 lead to induced expression of inflammatory cytokines TNF-α and IL-1β via TLR4 activation in microglia cells (Zou and Crews, 2014). As we found LDH and glutamate secretions are increased in OGD+CsA-induced damaged OHSC, it is likely that various DAMPs are released from necrotic neurons, thereby activating neighboring microglia and astrocytes to drive the secondary inflammatory process after OGD/R-induced injury. While CsA mediated suppression of microglia activation, it is not sufficient to reduce the overall inflammatory response in the complex OHSC. To our knowledge, the effect of CsA on the inflammatory response in OGD/R-induced OHSC injury has not been investigated, as most research studies have investigated the amelioration of cell death by CsA treatment, and protection of the mitochondria from OGD/R-induced opening of the mitochondrial permeability transition pores (Kawakami, 2013; Trumbeckaite et al., 2013; Yu et al., 2013).
An inflammatory response in the brain can be both beneficial and harmful, depending on the type and amount of cytokines expressed. An increase in Iba1 expression is associated with microglia activation (Hovens et al., 2014). TGF-β1 is mainly expressed by these activated microglial cells and is associated with a reduction of neuronal cell death and decreased infarct size after cerebral ischemia (Meyers and Kessler, 2017). We observed both Iba1 and TGF-β1 expressions to be increased in OGD/R injured slices, whereas other cytokines were not significantly altered. Our findings indicate a beneficial activation of inflammatory processes after exposure to OGD/R in the OHSC model, but it becomes highly neuroinflammatory when CsA is additionally applied.
We acknowledge several limitations of our study. First, we only assessed cytokine and chemokine mRNA expressions and not the secreted form. Therefore, any conclusion concerning inflammation should be approached with caution, as intracellular and secreted levels may differ. Second, quantifiable necrosis in the hippocampal slice cultures as well as any subsequent DAMPs release after reperfusion was not possible due to high serum concentrations in the cultured media. Therefore, further studies are needed to investigate the impact of DAMPs associated sterile inflammation in CNS cells. Finally, our protocol focused on ischemia sensitive neurons and immunocompetent microglia. As astrocytes constitute a large population of cells in the brain and have an important role in neuroprotection, further studies investigating the effects of CsA and cooling on OGD/R-induced injury in astrocytes are warranted.
Conclusion
We found CsA treatment to be effective in suppressing inflammation in a pure microglia culture after OGD/R-induced injury, but causes necrotic cell death in primary neurons. In the complex ex vivo slice culture CsA treatment lead to an exacerbated immune response, which was not diminished by hypothermia but instead potentiated an additive effect leading to an increase in neuroinflammation. Based on our findings, the combination of cooling and CsA treatment can hereby not be considered as neuroprotective. In contrast to other studies describing the neuroprotective effects of CsA and hypothermia, we observed the induction of neuroinflammation to the combined treatment in a complex OHSC model.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
This study was carried out in accordance with the guidelines of the Charité – Universitätsmedizin Berlin, Germany and the national ethic principles (registration no. T0044/08).
Author Contributions
SW, GT, and KS designed the experiments. SW conducted the experiments and analyzed the data. SW, GT, KS, NR, JK, and FB participated in the discussion of the results. SW prepared the manuscript. GT, JK, NR, and KS reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This study was financially supported by the Stiftung KinderHerz, Essen, Germany (Grant Number 2511-3-14-015). We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité – Universitätsmedizin Berlin.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Nora Künzel for her excellent technical assistance.
Abbreviations
AP1, activator protein 1; CIRBP, cold-inducible RNA-binding protein; CNS, central nervous system; CsA, cyclosporin A; DAMP, damage-associated molecular pattern; DIV, days in vitro; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HMGB1, high mobility group box 1; Hsp70, heat shock protein 70; iNOS, inducible nitric oxide synthase; Iba1, ionized calcium-binding adapter molecule 1; IL-6, interleukin-6; IL-1α, interleukin-1α; IL-1β, interleukin-1β; LDH, lactate dehydrogenase; MAPK, mitogen-activated protein kinase; MCP1, monocyte chemotactic protein 1; NFAT, nuclear factor of activated T-cells; OGD, oxygen-glucose-deprivation; OGD/R, OGD and reperfusion; OHSC, organotypic hippocampal slice culture; PI, propidium iodide; PLL, poly-L-lysin; RAGE, receptor for advanced glycation endproducts; RT-qPCR, reverse transcription quantitative polymerase chain reaction; TGF-β1, transforming growth factor-β1; TH, therapeutic hypothermia; TLR, toll-like receptor; TNF-α, tumor necrosis factor-α; TTM, targeted temperature management.
References
Amantea, D., Nappi, G., Bernardi, G., Bagetta, G., and Corasaniti, M. T. (2009). Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J. 276, 13–26. doi: 10.1111/j.1742-4658.2008.06766.x
Andersson, U., and Tracey, K. J. (2011). HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162. doi: 10.1146/annurev-immunol-030409-101323
Asea, A., Kraeft, S. K., Kurt-Jones, E. A., Stevenson, M. A., Chen, L. B., Finberg, R. W., et al. (2000). HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6, 435–442. doi: 10.1038/74697
Azzopardi, D., Strohm, B., Marlow, N., Brocklehurst, P., Deierl, A., Eddama, O., et al. (2014). Effects of hypothermia for perinatal asphyxia on childhood outcomes. N. Engl. J. Med. 371, 140–149. doi: 10.1056/NEJMoa1315788
Becerra-Calixto, A., and Cardona-Gomez, G. P. (2017). The role of astrocytes in neuroprotection after brain stroke: potential in cell therapy. Front. Mol. Neurosci. 10:88. doi: 10.3389/fnmol.2017.00088
Bhalala, U. S., Koehler, R. C., and Kannan, S. (2014). Neuroinflammation and neuroimmune dysregulation after acute hypoxic-ischemic injury of developing brain. Front. Pediatr. 2:144. doi: 10.3389/fped.2014.00144
Blasi, E., Barluzzi, R., Bocchini, V., Mazzolla, R., and Bistoni, F. (1990). Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 27, 229–237. doi: 10.1016/0165-5728(90)90073-v
Borel, J. F., Feurer, C., Gubler, H. U., and Stahelin, H. (1976). Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions 6, 468–475. doi: 10.1007/bf01973261
Callaway, C. W., Donnino, M. W., Fink, E. L., Geocadin, R. G., Golan, E., Kern, K. B., et al. (2015). Part 8: post-cardiac arrest care: 2015 american heart association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 132, S465–S482.
Ceulemans, A. G., Zgavc, T., Kooijman, R., Hachimi-Idrissi, S., Sarre, S., and Michotte, Y. (2010). The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J. Neuroinflammation 7:74. doi: 10.1186/1742-2094-7-74
Che, X., Ye, W., Panga, L., Wu, D. C., and Yang, G. Y. (2001). Monocyte chemoattractant protein-1 expressed in neurons and astrocytes during focal ischemia in mice. Brain Res. 902, 171–177. doi: 10.1016/s0006-8993(01)02328-9
Chen, Y., Hallenbeck, J. M., Ruetzler, C., Bol, D., Thomas, K., Berman, N. E., et al. (2003). Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J. Cereb. Blood Flow Metab. 23, 748–755. doi: 10.1097/01.wcb.0000071885.63724.20
Clipstone, N. A., and Crabtree, G. R. (1992). Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357, 695–697. doi: 10.1038/357695a0
Cooper, D. J., Nichol, A. D., Bailey, M., Bernard, S., Cameron, P. A., Pili-Floury, S., et al. (2018). Effect of early sustained prophylactic hypothermia on neurologic outcomes among patients with severe traumatic brain injury: the POLAR randomized clinical trial. JAMA 320, 2211–2220. doi: 10.1001/jama.2018.17075
Crompton, M. (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341(Pt 2), 233–249. doi: 10.1042/bj3410233
Fakharnia, F., Khodagholi, F., Dargahi, L., and Ahmadiani, A. (2017). Prevention of cyclophilin D-Mediated mPTP opening using cyclosporine-A alleviates the elevation of necroptosis, autophagy and apoptosis-related markers following global cerebral ischemia-reperfusion. J. Mol. Neurosci. 61, 52–60. doi: 10.1007/s12031-016-0843-3
Farina, C., Aloisi, F., and Meinl, E. (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol. 28, 138–145. doi: 10.1016/j.it.2007.01.005
Furman, J. L., and Norris, C. M. (2014). Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 11:158. doi: 10.1186/s12974-014-0158-7
Gabryel, B., Labuzek, K., Malecki, A., and Herman, Z. S. (2004). Immunophilin ligands decrease release of pro-inflammatory cytokines (IL-1beta, TNF-alpha and IL-2 in rat astrocyte cultures exposed to simulated ischemia in vitro. Pol. J. Pharmacol. 56, 129–136.
Goldberg, M. P., and Choi, D. W. (1993). Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci. 13, 3510–3524. doi: 10.1523/jneurosci.13-08-03510.1993
Gulke, E., Gelderblom, M., and Magnus, T. (2018). Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 11:1756286418774254. doi: 10.1177/1756286418774254
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Henn, A., Lund, S., Hedtjarn, M., Schrattenholz, A., Porzgen, P., and Leist, M. (2009). The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26, 83–94. doi: 10.14573/altex.2009.2.83
Hovens, I. B., Nyakas, C., and Schoemaker, R. G. (2014). A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: cell body to cell size ratio. Neuroimmunol. Neuroinflammation 1, 82–88.
Hypothermia after Cardiac Arrest Study (2002). Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N. Engl. J. Med. 346, 549–556. doi: 10.1056/nejmoa012689
Jin, R., Liu, L., Zhang, S., Nanda, A., and Li, G. (2013). Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 6, 834–851. doi: 10.1007/s12265-013-9508-6
Johnson, J. D., and Fleshner, M. (2006). Releasing signals, secretory pathways, and immune function of endogenous extracellular heat shock protein 72. J. Leukoc. Biol. 79, 425–434. doi: 10.1189/jlb.0905523
Kaminska, B., Figiel, I., Pyrzynska, B., Czajkowski, R., and Mosieniak, G. (2001). Treatment of hippocampal neurons with cyclosporin A results in calcium overload and apoptosis which are independent on NMDA receptor activation. Br. J. Pharmacol. 133, 997–1004. doi: 10.1038/sj.bjp.0704177
Kawabori, M., and Yenari, M. A. (2015). Inflammatory responses in brain ischemia. Curr. Med. Chem. 22, 1258–1277. doi: 10.2174/0929867322666150209154036
Kawakami, M. (2013). Molecular dissection of cyclosporin a’s neuroprotective effect reveals potential therapeutics for ischemic brain injury. Brain Sci. 3, 1325–1356. doi: 10.3390/brainsci3031325
Kim, J. Y., Han, Y., Lee, J. E., and Yenari, M. A. (2018). The 70-kDa heat shock protein (Hsp70) as a therapeutic target for stroke. Expert Opin. Ther. Targets 22, 191–199. doi: 10.1080/14728222.2018.1439477
Kono, H., Onda, A., and Yanagida, T. (2014). Molecular determinants of sterile inflammation. Curr. Opin. Immunol. 26, 147–156. doi: 10.1016/j.coi.2013.12.004
Krech, J., Tong, G., Wowro, S., Walker, C., Rosenthal, L. M., Berger, F., et al. (2017). Moderate therapeutic hypothermia induces multimodal protective effects in oxygen-glucose deprivation/reperfusion injured cardiomyocytes. Mitochondrion 35, 1–10. doi: 10.1016/j.mito.2017.04.001
Kurisu, K., and Yenari, M. A. (2018). Therapeutic hypothermia for ischemic stroke; pathophysiology and future promise. Neuropharmacology 134, 302–309. doi: 10.1016/j.neuropharm.2017.08.025
Lambertsen, K. L., Clausen, B. H., Babcock, A. A., Gregersen, R., Fenger, C., Nielsen, H. H., et al. (2009). Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J. Neurosci. 29, 1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009
Lawson, L. J., Perry, V. H., Dri, P., and Gordon, S. (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170. doi: 10.1016/0306-4522(90)90229-w
Lee, J. H., Wei, Z. Z., Cao, W., Won, S., Gu, X., Winter, M., et al. (2016). Regulation of therapeutic hypothermia on inflammatory cytokines, microglia polarization, migration and functional recovery after ischemic stroke in mice. Neurobiol. Dis. 96, 248–260. doi: 10.1016/j.nbd.2016.09.013
Li, D., Shao, Z., Vanden Hoek, T. L., and Brorson, J. R. (2007). Reperfusion accelerates acute neuronal death induced by simulated ischemia. Exp. Neurol. 206, 280–287. doi: 10.1016/j.expneurol.2007.05.017
Li, Q., Han, X., and Wang, J. (2016). Organotypic hippocampal slices as models for stroke and traumatic brain injury. Mol. Neurobiol. 53, 4226–4237. doi: 10.1007/s12035-015-9362-4
Liao, Y., Tong, L., Tang, L., and Wu, S. (2017). The role of cold-inducible RNA binding protein in cell stress response. Int. J. Cancer 141, 2164–2173. doi: 10.1002/ijc.30833
Lim, J. H., Lee, J. C., Lee, Y. H., Choi, I. Y., Oh, Y. K., Kim, H. S., et al. (2006). Simvastatin prevents oxygen and glucose deprivation/reoxygenation-induced death of cortical neurons by reducing the production and toxicity of 4-hydroxy-2E-nonenal. J. Neurochem. 97, 140–150. doi: 10.1111/j.1471-4159.2006.03715.x
Liu, J., Wang, Y., Zhuang, Q., Chen, M., Wang, Y., Hou, L., et al. (2016a). Protective effects of cyclosporine A and hypothermia on neuronal mitochondria in a rat asphyxial cardiac arrest model. Am. J. Emerg. Med. 34, 1080–1085. doi: 10.1016/j.ajem.2016.02.066
Liu, T., Zhao, D. X., Cui, H., Chen, L., Bao, Y. H., Wang, Y., et al. (2016b). Therapeutic hypothermia attenuates tissue damage and cytokine expression after traumatic brain injury by inhibiting necroptosis in the rat. Sci. Rep. 6:24547. doi: 10.1038/srep24547
Liu, L., Liu, X., Wang, R., Yan, F., Luo, Y., Chandra, A., et al. (2018). Mild focal hypothermia regulates the dynamic polarization of microglia after ischemic stroke in mice. Neurol. Res. 40, 508–515. doi: 10.1080/01616412.2018.1454090
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Loddick, S. A., Turnbull, A. V., and Rothwell, N. J. (1998). Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 18, 176–179. doi: 10.1097/00004647-199802000-00008
Lopez-Neblina, F., Toledo, A. H., and Toledo-Pereyra, L. H. (2005). Molecular biology of apoptosis in ischemia and reperfusion. J. Invest. Surg. 18, 335–350. doi: 10.1080/08941930500328862
Lyden, P., Hemmen, T., Grotta, J., Rapp, K., Ernstrom, K., Rzesiewicz, T., et al. (2016). Results of the ICTuS 2 Trial (intravascular cooling in the treatment of stroke 2). Stroke 47, 2888–2895.
Macian, F., Lopez-Rodriguez, C., and Rao, A. (2001). Partners in transcription: NFAT and AP-1. Oncogene 20, 2476–2489. doi: 10.1038/sj.onc.1204386
Meyers, E. A., and Kessler, J. A. (2017). TGF-beta family signaling in neural and neuronal differentiation. development, and function. Cold Spring Harb Perspect. Biol. 9:a022244. doi: 10.1101/cshperspect.a022244
Mizuma, A., and Yenari, M. A. (2017). Anti-inflammatory targets for the treatment of reperfusion injury in stroke. Front. Neurol. 8:467. doi: 10.3389/fneur.2017.00467
Muhammad, S., Barakat, W., Stoyanov, S., Murikinati, S., Yang, H., Tracey, K. J., et al. (2008). The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 28, 12023–12031. doi: 10.1523/JNEUROSCI.2435-08.2008
Nagamoto-Combs, K., and Combs, C. K. (2010). Microglial phenotype is regulated by activity of the transcription factor. NFAT (nuclear factor of activated T cells). J. Neurosci. 30, 9641–9646. doi: 10.1523/JNEUROSCI.0828-10.2010
Nishizawa, Y. (2001). Glutamate release and neuronal damage in ischemia. Life Sci. 69, 369–381. doi: 10.1016/s0024-3205(01)01142-0
Nolan, J. P., Soar, J., Cariou, A., Cronberg, T., Moulaert, V. R., Deakin, C. D., et al. (2015). European resuscitation council and european society of intensive care medicine 2015 guidelines for post-resuscitation care. Intensive Care Med. 41, 2039–2056. doi: 10.1007/s00134-015-4051-3
Osman, M. M., Lulic, D., Glover, L., Stahl, C. E., Lau, T., Van Loveren, H., et al. (2011). Cyclosporine-A as a neuroprotective agent against stroke: its translation from laboratory research to clinical application. Neuropeptides 45, 359–368. doi: 10.1016/j.npep.2011.04.002
Palkowitsch, L., Marienfeld, U., Brunner, C., Eitelhuber, A., Krappmann, D., and Marienfeld, R. B. (2011). The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J. Biol. Chem. 286, 7522–7534. doi: 10.1074/jbc.M110.155895
Partoazar, A., Nasoohi, S., Rezayat, S. M., Gilani, K., Mehr, S. E., Amani, A., et al. (2017). Nanoliposome containing cyclosporine A reduced neuroinflammation responses and improved neurological activities in cerebral ischemia/reperfusion in rat. Fundam. Clin. Pharmacol. 31, 185–193. doi: 10.1111/fcp.12244
Qiang, X., Yang, W. L., Wu, R., Zhou, M., Jacob, A., Dong, W., et al. (2013). Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 19, 1489–1495. doi: 10.1038/nm.3368
Rock, K. L., Latz, E., Ontiveros, F., and Kono, H. (2010). The sterile inflammatory response. Annu. Rev. Immunol. 28, 321–342. doi: 10.1146/annurev-immunol-030409-101311
Rojanathammanee, L., Floden, A. M., Manocha, G. D., and Combs, C. K. (2015). Attenuation of microglial activation in a mouse model of Alzheimer’s disease via NFAT inhibition. J. Neuroinflammation 12:42. doi: 10.1186/s12974-015-0255-2
Rossi, D. J., Oshima, T., and Attwell, D. (2000). Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321. doi: 10.1038/35002090
Rothaug, M., Becker-Pauly, C., and Rose-John, S. (2016). The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta 1863, 1218–1227. doi: 10.1016/j.bbamcr.2016.03.018
Sakurai-Yamashita, Y., Shigematsu, K., Yamashita, K., and Niwa, M. (2006). Expression of MCP-1 in the hippocampus of SHRSP with ischemia-related delayed neuronal death. Cell. Mol. Neurobiol. 26, 823–831.
Sama, M. A., Mathis, D. M., Furman, J. L., Abdul, H. M., Artiushin, I. A., Kraner, S. D., et al. (2008). Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J. Biol. Chem. 283, 21953–21964. doi: 10.1074/jbc.M800148200
Schmitt, K. R., Boato, F., Diestel, A., Hechler, D., Kruglov, A., Berger, F., et al. (2010). Hypothermia-induced neurite outgrowth is mediated by tumor necrosis factor-alpha. Brain Pathol. 20, 771–779. doi: 10.1111/j.1750-3639.2009.00358.x
Schmitt, K. R., Kern, C., Berger, F., Ullrich, O., Hendrix, S., and Abdul-Khaliq, H. (2006). Methylprednisolone attenuates hypothermia- and rewarming-induced cytotoxicity and IL-6 release in isolated primary astrocytes, neurons and BV-2 microglia cells. Neurosci. Lett. 404, 309–314. doi: 10.1016/j.neulet.2006.05.064
Schmitt, K. R., Kern, C., Lange, P. E., Berger, F., Abdul-Khaliq, H., and Hendrix, S. (2007). S100B modulates IL-6 release and cytotoxicity from hypothermic brain cells and inhibits hypothermia-induced axonal outgrowth. Neurosci. Res. 59, 68–73. doi: 10.1016/j.neures.2007.05.011
Shankaran, S., Pappas, A., Mcdonald, S. A., Vohr, B. R., Hintz, S. R., Yolton, K., et al. (2012). Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 366, 2085–2092. doi: 10.1056/NEJMoa1112066
Sriram, K., and O’Callaghan, J. P. (2007). Divergent roles for tumor necrosis factor-alpha in the brain. J. Neuroimmune Pharmacol. 2, 140–153. doi: 10.1007/s11481-007-9070-6
Terpolilli, N. A., Moskowitz, M. A., and Plesnila, N. (2012). Nitric oxide: considerations for the treatment of ischemic stroke. J. Cereb. Blood Flow Metab. 32, 1332–1346. doi: 10.1038/jcbfm.2012.12
Trumbeckaite, S., Gizatullina, Z., Arandarcikaite, O., Rohnert, P., Vielhaber, S., Malesevic, M., et al. (2013). Oxygen glucose deprivation causes mitochondrial dysfunction in cultivated rat hippocampal slices: protective effects of CsA, its immunosuppressive congener [D-Ser](8)CsA, the novel non-immunosuppressive cyclosporin derivative Cs9, and the NMDA receptor antagonist MK 801. Mitochondrion 13, 539–547. doi: 10.1016/j.mito.2012.07.110
Yu, Z., Liu, N., Li, Y., Xu, J., and Wang, X. (2013). Neuroglobin overexpression inhibits oxygen-glucose deprivation-induced mitochondrial permeability transition pore opening in primary cultured mouse cortical neurons. Neurobiol. Dis. 56, 95–103. doi: 10.1016/j.nbd.2013.04.015
Zawadzka, M., Dabrowski, M., Gozdz, A., Szadujkis, B., Sliwa, M., Lipko, M., et al. (2012). Early steps of microglial activation are directly affected by neuroprotectant FK506 in both in vitro inflammation and in rat model of stroke. J. Mol. Med. 90, 1459–1471. doi: 10.1007/s00109-012-0925-9
Zhang, L. N., Hao, L., Guo, Y. S., Wang, H. Y., Li, L. L., Liu, L. Z., et al. (2019). Are glutamate transporters neuroprotective or neurodegenerative during cerebral ischemia? J. Mol. Med. 97, 281–289. doi: 10.1007/s00109-019-01745-5
Zhao, W. Y., Chen, S. B., Wang, J. J., Xu, C., Zhao, M. L., Dong, H. J., et al. (2017). Establishment of an ideal time window model in hypothermic-targeted temperature management after traumatic brain injury in rats. Brain Res. 1669, 141–149. doi: 10.1016/j.brainres.2017.06.006
Zhou, M., Yang, W. L., Ji, Y., Qiang, X., and Wang, P. (2014). Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim. Biophys. Acta 1840, 2253–2261. doi: 10.1016/j.bbagen.2014.02.027
Keywords: cyclosporin A, hypothermia, oxygen-glucose deprivation/reperfusion, organotypic hippocampal slice culture, BV-2 microglia, primary neuron, inflammation, DAMPs
Citation: Wowro SJ, Tong G, Krech J, Rolfs N, Berger F and Schmitt KRL (2019) Combined Cyclosporin A and Hypothermia Treatment Inhibits Activation of BV-2 Microglia but Induces an Inflammatory Response in an Ischemia/Reperfusion Hippocampal Slice Culture Model. Front. Cell. Neurosci. 13:273. doi: 10.3389/fncel.2019.00273
Received: 27 March 2019; Accepted: 05 June 2019;
Published: 25 June 2019.
Edited by:
Xin Qi, Case Western Reserve University, United StatesReviewed by:
Peter Racay, Comenius University, SlovakiaSharon DeMorrow, The University of Texas at Austin, United States
Copyright © 2019 Wowro, Tong, Krech, Rolfs, Berger and Schmitt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvia J. Wowro, Sylvia.wowro@charite.de