 Adam S. Deardorff
Adam S. Deardorff Shannon H. Romer
Shannon H. Romer Patrick M. Sonner
Patrick M. Sonner Robert E. W. Fyffe
Robert E. W. Fyffe- Boonshoft School of Medicine, Department of Neuroscience, Cell Biology and Physiology, Wright State University, Dayton, OH, USA
C-boutons are important cholinergic modulatory loci for state-dependent alterations in motoneuron firing rate. m2 receptors are concentrated postsynaptic to C-boutons, and m2 receptor activation increases motoneuron excitability by reducing the action potential afterhyperpolarization. Here, using an intensive review of the current literature as well as data from our laboratory, we illustrate that C-bouton postsynaptic sites comprise a unique structural/functional domain containing appropriate cellular machinery (a “signaling ensemble”) for cholinergic regulation of outward K+ currents. Moreover, synaptic reorganization at these critical sites has been observed in a variety of pathologic states. Yet despite recent advances, there are still great challenges for understanding the role of C-bouton regulation and dysregulation in human health and disease. The development of new therapeutic interventions for devastating neurological conditions will rely on a complete understanding of the molecular mechanisms that underlie these complex synapses. Therefore, to close this review, we propose a comprehensive hypothetical mechanism for the cholinergic modification of α-MN excitability at C-bouton synapses, based on findings in several well-characterized neuronal systems.
Introduction
The neuromuscular system provides rapid and coordinated force generation, whereby the number and firing rate of recruited motor units are systematically adjusted to meet environmental demands (Monster and Chan, 1977; Henneman and Mendell, 1981; Clamann, 1993; Cope and Sokoloff, 1999). Indeed, the elegant simplicity with which animals navigate their environment relies on neural circuitry that is inherently modifiable, and the ability to perform a variety of motor tasks while responding quickly to unexpected perturbations and threats is essential for individual survival (Ladle et al., 2007; Miri et al., 2013). Control of α-MN repetitive firing properties is a therefore highly conserved and critical adaption of mammalian and non-mammalian species alike, and identifying the responsible spinal circuits has been of essential importance in our understanding of neuromuscular function and dysfunction (Miles and Sillar, 2011).
For more than 50 years, a particular class of synapse in the spinal cord ventral horn – the C-bouton – has generated sustained interest among α-MN anatomists and physiologists. Unambiguous identification of these conspicuously large cholinergic synaptic contacts and the characteristic postsynaptic SSC for which they are named has prompted numerous investigations into their distribution, source, function, and pathology. Yet despite the detailed morphologic and physiologic information generated by many neuroscientists, it is humbling to consider (a) the incrementally slow trajectory by which our understanding of this enigmatic synapse has grown and (b) that as yet there is no definitive and fully functional hypothesis regarding their distribution, their postsynaptic subcellular machinery, their contribution to motor control and behavior, and their regulation/dysregulation in health and disease.
Recently, we have learned the most elementary effect of C-boutons on α-MN f-I gain during static intracellular current injection occurs via dramatic reductions in the strength of the action potential AHP (Miles et al., 2007), which is mediated by postsynaptic small conductance Ca2+-activated K+ (SK) channels (Deardorff et al., 2013). However, the mystery of the C-bouton and its cholinergic effects on MN biophysical properties and integrative capabilities is by no means solved, as has been suggested (Frank, 2009). Using an isolated spinal cord preparation, Miles et al. (2007) demonstrate a putative role for C-boutons in ensuring appropriate levels of motor output during drug induced fictive locomotion. But complexity arises upon behavioral assessment of adult mice with selective genetic inactivation of C-bouton synaptic inputs, which during locomotion exhibit normal flexor–extensor alternation and normal EMG amplitude. Motor deficits in these mice primarily manifest during high-output tasks such as swimming (Zagoraiou et al., 2009). These data convincingly implicate C-boutons in the task-dependent regulation of α-MN excitability via reduction of outward K+ currents, but questions remain regarding (a) the functional impact of C-bouton input during different behaviors, (b) the manner in which C-bouton activity is modulated to match motor demands, (c) the expression of abnormal force generation as well as spasticity, rigidity, or tremor as a consequence of C-bouton dysfunction, and (d) the mechanism of interaction between underlying acetylcholine receptors (AChRs) and K+ channels.
To aid in the development of new in vivo and in vitro experimental strategies to answer these and related questions, this review details our current understanding of the cellular, synaptic, and genetic properties that underlie C-bouton function and proposes a hitherto unexplored mechanism for the cholinergic modification of α-MN excitability. It should be noted that the title of this review is intended to reflect and pay homage to the many dedicated and careful neuroscientists who have undertaken MN synaptological investigations over the years. This review will therefore also provide historical perspective on the foundational advances in our understanding of this complex and elusive, yet important, synapse. Neuroscientists have spent 50+ years at the C-bouton swimming against the tide. Significant progress has been slow and hard fought. And though we are a long way from shore, we must remember – as our murine colleagues have demonstrated – without C-boutons we cannot swim at all.
The C-Bouton Signaling Ensemble: A Contemporary View of a Classic Synapse
We are riding the crest of a wave. With the turn of the century and the application of advanced morphologic analyses, cellular neurophysiology, and selective genetic perturbations, we have built a decidedly robust picture of C-bouton form and function. C-boutons are an essential piece of an integrated control system set to regulate α-MN activity through a complex anatomical substrate: a signaling ensemble (Figures 1 and 2) precisely organized for highly nuanced orchestration of somatic K+ currents.
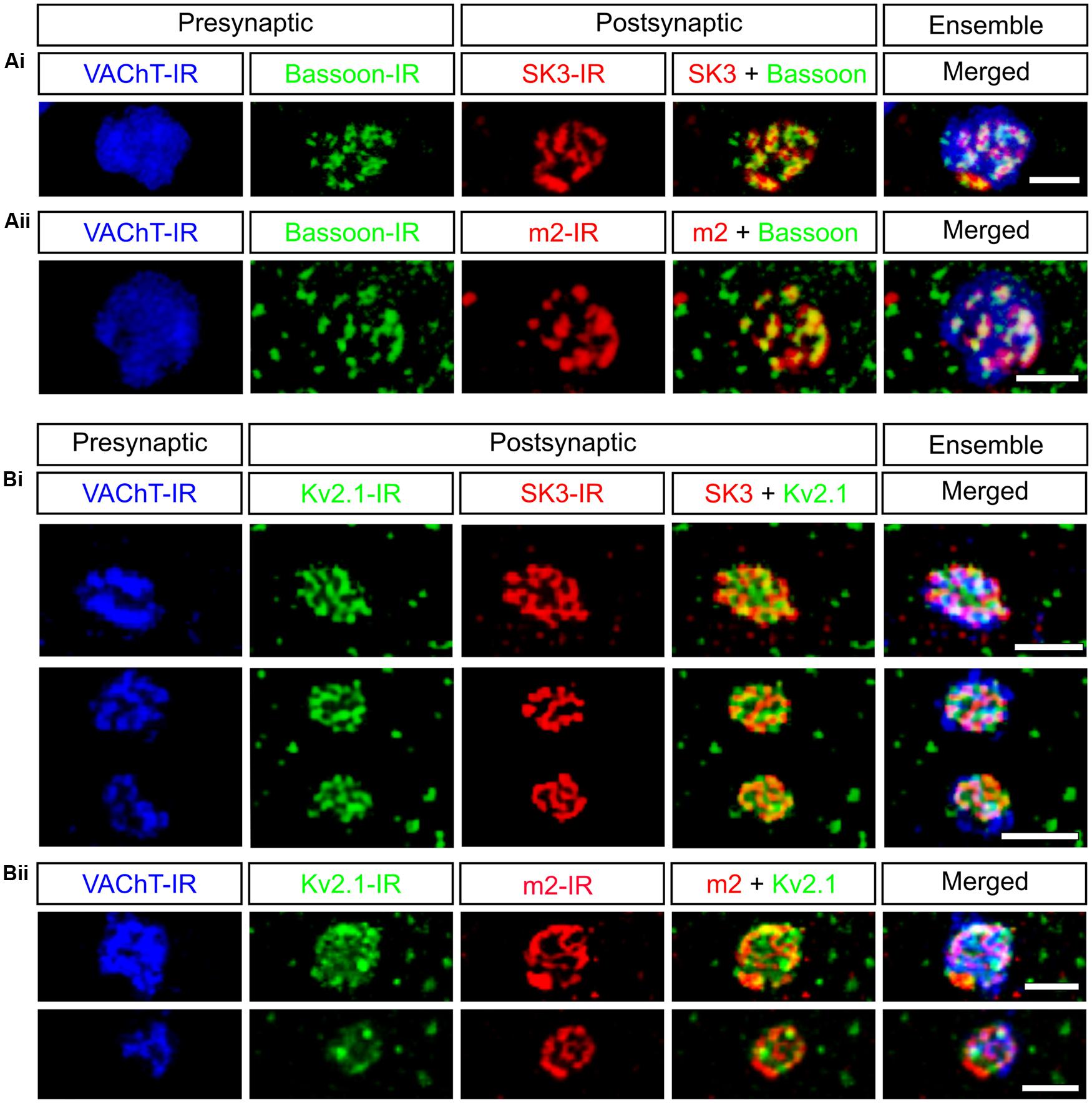
FIGURE 1. C-bouton synaptic sites contain a complex signaling ensemble. Presynaptic bassoon-IR and postsynaptic SK3-IR and m2-IR share a striking subsynaptic fenestrated appearance within the C-bouton. All images are small confocal stacks (3 × 1 μm Z-stacks) of en face C-boutons, indicated with VAChT-IR (blue), on rat lumbar α-motoneurons. (A) Presynaptic active zone protein bassoon (green) is aligned with postsynaptic ion channels SK3 (Ai, red) and m2 receptors (Aii, red). (B) Kv2.1-IR (green) intercalates with SK3-IR (Bi, red) and m2-IR (Bii), “filling in” the C-bouton postsynaptic membrane. Scale bars are 2.0 μm.
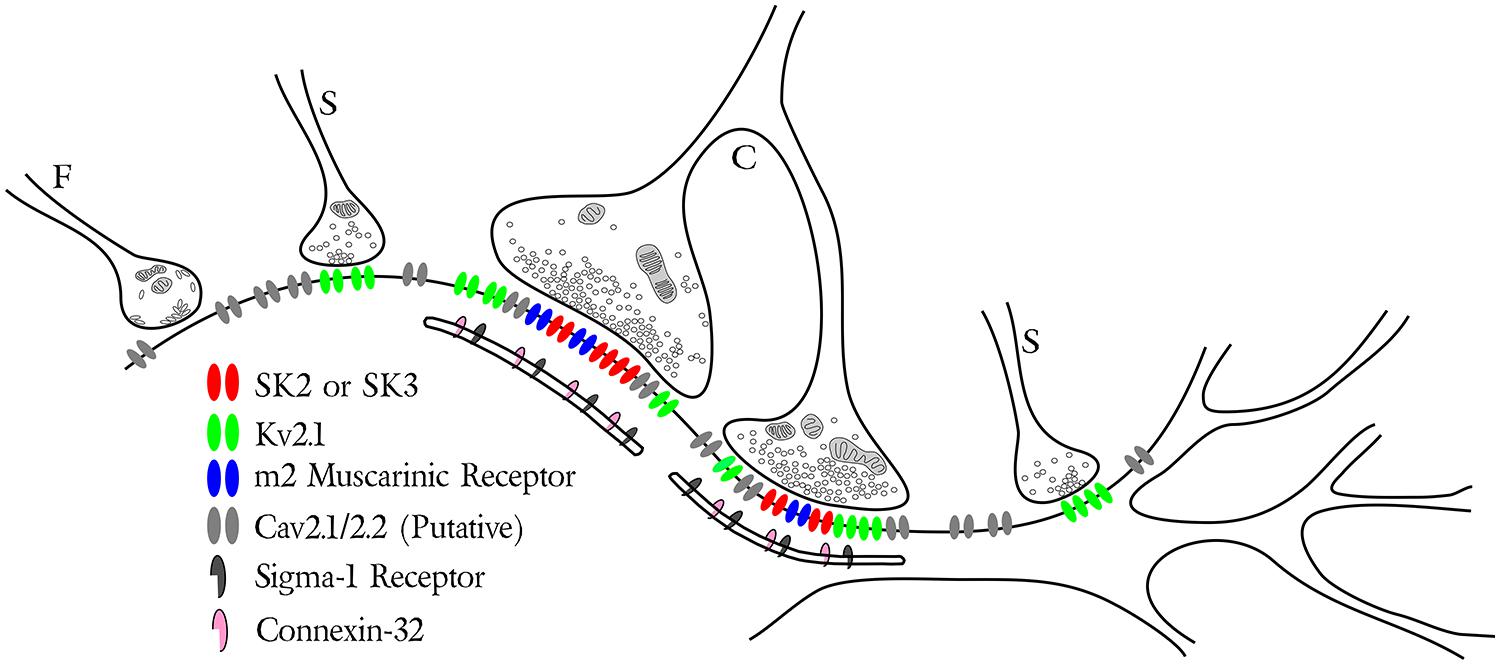
FIGURE 2. Synaptic distribution of specific ion channels and receptors on soma and proximal dendrites of motoneurons. The schematic illustrates three types of motoneuron presynaptic boutons including the glycinergic/GABAergic F-type, glutamatergic S-type and cholinergic C-type with its associated postsynaptic subsurface cistern. Note the specific localization of m2 muscarinic receptors (blue) with SK channels (red) and Kv2.1 channels (green) postsynaptic to the C-bouton. Small Kv2.1 clusters are also found postsynaptic to some S-type synapses (see Muennich and Fyffe, 2004). The P/Q- and N-type Ca2+ channels Cav2.1/2.2 (light gray) are illustrated throughout the membrane, although the precise subcellular localization of this channel is currently unknown. Both connexin 32 (pink) and the sigma-1 receptor (dark gray) are specifically associated with the C-bouton subsurface cistern.
Precise Anatomical Localization and Organization of Signaling Components: An Ensemble of Apposed Proteins and Mosaic Membrane Domains
C-type synaptic sites comprise three closely apposed membranous domains (Figure 2), spanning a breadth of <25 nm, and across which the distribution of synaptic and signaling proteins are precisely regulated. Clear and consistent immunohistochemical data demonstrate membrane clusters of α-MN Kv2.1 channels, SK2/3 channels, and m2 receptors directly apposing C-bouton presynaptic terminals (Skinner et al., 1999; Hellstrom et al., 2003; Muennich and Fyffe, 2004; Wilson et al., 2004; Deardorff et al., 2013). When visualized under high resolution, these SK2/3 channel and m2 receptor clusters are composed of an intricate, non-uniform aggregation of smaller “threadlike” structures that are woven together and closely approximate/appose C-bouton pre-synaptic vesicle release sites, which are enriched with bassoon (A. S. Deardorff, S. H. Romer, R. E. W. Fyffe, unpublished; see Figure 1). Beneath the postsynaptic membrane, in α-MN SSCs, the gap junction protein connexin32 shows a similar threadlike distribution pattern (Yamamoto et al., 1990, 1991; Zampieri et al., 2014), indicating that connexin32, SK channels/m2 receptors, and transmitter release machinery are precisely aligned across the three membranous domains. Kv2.1 channels appear to “fill in” the remaining postsynaptic α-MN membrane surface not occupied by SK channels or m2 receptors. The demarcated postsynaptic area, therefore, is a highly structured and mosaic domain of interdigitating clusters of Kv2.1 channels and co-localized SK2/3 channels and m2 receptors. The orderly, stacked apposition of proteins on the cisternal, postsynaptic, and presynaptic membranes as well as the spatial interdigitation of distinct channel and receptor clusters demonstrates a coordinated and specific signaling organization across all membranous domains at C-bouton synaptic sites.
Additional Signaling Components
Additional studies have revealed, to varying levels of specificity, other signaling components that characterize the C-bouton ensemble. Certain elements, although identified within one or another membranous or cytoplasmic domain, are not well defined in regard to specific subdomain organization nor anatomic relation to other molecular components. In this category, C-bouton synaptic terminals express a range of exocytotic proteins consistent with those necessary for fast transmitter release (Hellstrom et al., 1999), are highly associated with presynaptic P2X7 purinergic receptor immunoreactivity (~90% of C-boutons; Deng and Fyffe, 2004), and may also express presynaptic nicotinic acetylcholine receptors (nAChRs; Khan et al., 2003). In addition, the α-MN SSC is highly enriched with S1Rs (Mavlyutov et al., 2010), and with closely associated neuregulin-1 (NG1) immunoreactivity (Gallart-Palau et al., 2014). Indole-N-methyl transferase (INMT), an enzyme that converts tryptamine into the S1R ligand dimethytryptamine (DMT), is also present in close proximity to S1Rs at C-bouton postsynaptic sites (Mavlyutov et al., 2012), but the extent to which S1Rs, themselves, are diffusely distributed within the entire cisternal membrane or co-localize/interdigitate with the well-characterized connexin32 immunoreactivity is not described.
The subcellular organization of Ca2+ sources necessary for SK channel activation also remains poorly characterized. However, α-MN SK2/3 channels require high voltage activated (HVA) N- and P/Q-type Ca2+ currents to generate the AHP (Viana et al., 1993; Umemiya and Berger, 1994; Bayliss et al., 1995; Li and Bennett, 2007), and SK channels typically couple to their Ca2+ source(s) by <200 nm (Fakler and Adelman, 2008; Jones and Stuart, 2013). Internally, SSCs may amplify or shape these Ca2+ signals via RyRs or connexin32, as they do in other cell types (see discussion Section “Subsurface Cisternae and the Generation of an Isolated Ca2+ Signal”). We, therefore, expect some proportion of HVA Ca2+ channels and RyRs to localize to the C-bouton postsynaptic membrane and/or to the associated SSC (Figure 2). In support, Wilson et al. (2004) provide evidence that P/Q-type Ca2+ channels are diffusely spread throughout the α-MN somatic membrane. By inference, some proportion must then appose C-boutons. The presence of N-type Ca2+ channels on α-MNs, however, has only been demonstrated physiologically (Carlin et al., 2000; Wilson et al., 2004).
The Crest of a Wave
The unique aggregation of cytoplasmic and membrane bound pre- and postsynaptic proteins that constitute the C-bouton signaling ensemble provides mechanistic insight into the cholinergic modulation of α-MN firing rate and has advanced new research at a comparatively faster pace than that of many other α-MN synaptic inputs. While uncertainties still confound our arrival at a “simple” molecular mechanism governing C-bouton synaptic function, experiments in other cell systems can help push us forward against the tide. Further exploration of this complex synapse is clearly necessitated. However, we must first review other salient features of the C-bouton system.
Moments and Milestones: Ultrastructure
Pioneering EM investigations (Wyckoff and Young, 1956) provided accurate anatomical description and categorization of the structurally diverse presynaptic terminals contacting spinal α-MNs, and in general, most authors still conform to the descriptive abbreviations (S-, F-, C-, T-, and M-Boutons) introduced by Bodian (1966a,b) and Conradi (1969a). (An additional bouton type, the P bouton, makes presynaptic connections with specific excitatory boutons in contact with the MN surface and may form triadic arrangements; Conradi, 1969a; Fyffe and Light, 1984). Those boutons Conradi classified as “C-type” are defined by and named for a signature 10–15 nm thick postsynaptic SSC (“C-type” for cistern): a broad, flat disc of smooth endoplasmic reticulum juxtaposed a mere 5–8 nm below the postsynaptic membrane and spanning the length of the apposing presynaptic terminal (Figure 3; Conradi, 1969a). The SSC is continuous with several lamellae of rough endoplasmic reticulum oriented in parallel with the cell membrane and frequently observed alongside free ribosomal rosettes in the subcisternal cytoplasm (Figure 3). Across a particularly narrow synaptic cleft (3–8 nm; see discussion Davidoff and Irintchev, 1986), the C-boutons themselves contain a dense cytoplasmic matrix of glycogen particles and neurofilaments tightly packed with 25–55 nm (diameter) clear spherical/pleomorphic vesicles, abundant mitochondria, and occasionally a small number of large dense core vesicles intermingled therein (Figure 3; Bodian, 1966a,b; Conradi, 1969a; McLaughlin, 1972b; Hamos and King, 1980). Notably, several authors (Rosenbluth, 1962; Bodian, 1966a,b; Charlton and Gray, 1966; Van Harreveld and Khattab, 1967) identified these unique and prominent boutons prior to Conradi’s (1969a) classic and thorough description of their synaptic ultrastructure – which remains the gold standard for their identification.
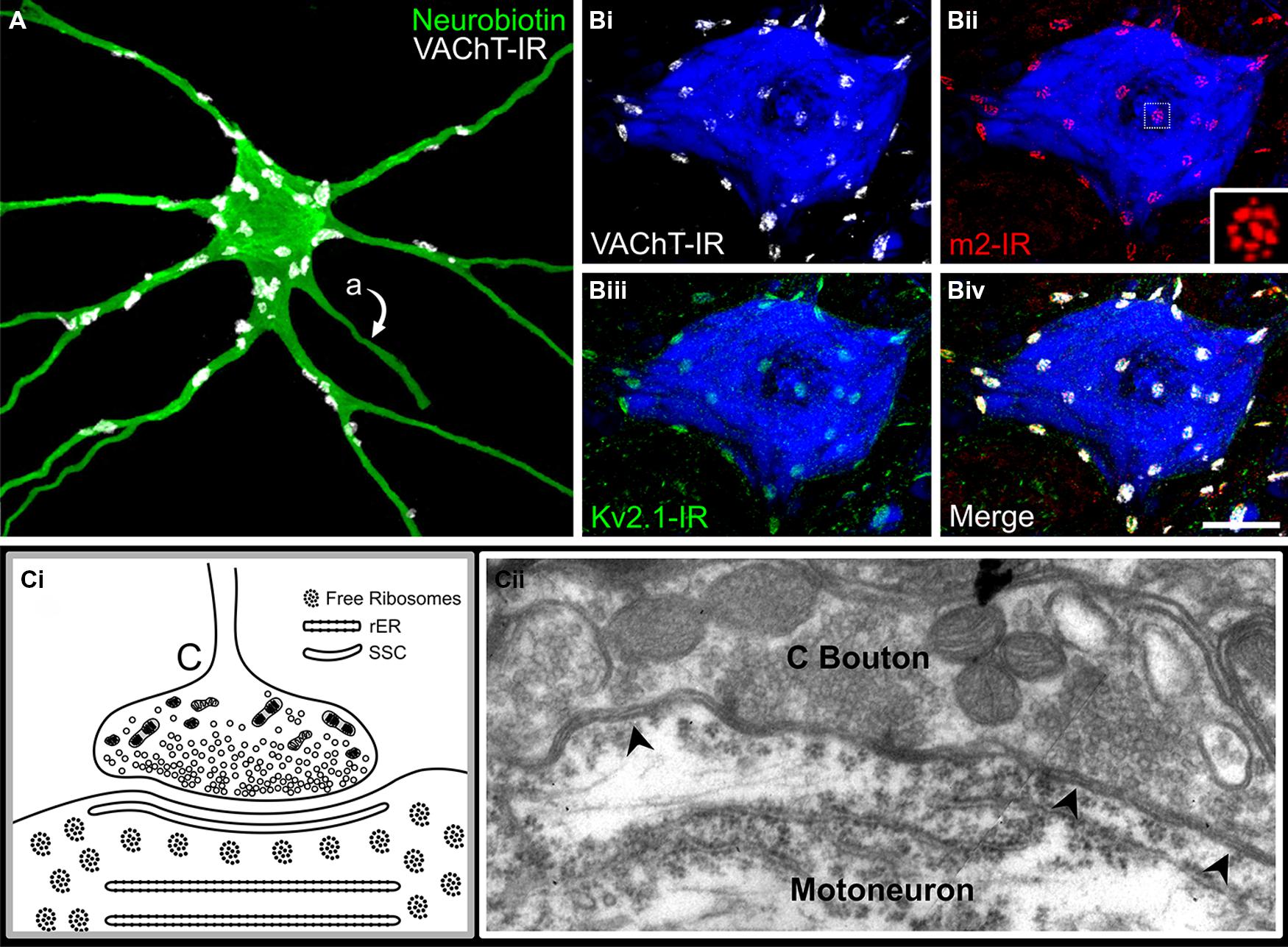
FIGURE 3. The C-bouton synapse on mammalian α-motoneurons. (A) C-bouton synapses on intracellularly labeled and reconstructed adult rat lumbar α-MN are revealed by VAChT-IR (white). Large C-boutons densely innervate the soma and proximal dendrites of α-MNs but are absent from more distal locations. Also note that C-boutons are not located on motoneuron axons (indicated by “a”). (B) C-boutons, indicated by VAChT-IR (Bi,iv, white), are presynaptic to the muscarinic m2 receptor (Bii,iv, red) and large Kv2.1 clusters (Biii,iv, green). Note that m2 receptor immunoreactivity on the α-MN soma and proximal dendrites localize exclusively to C-bouton postsynaptic sites. (Bii) Inset shows subsynaptic fenestrated distribution of m2-IR. Images are confocal stacks of 12 × 1 μm Z-stacks with nissl stain (blue) to label adult rat neuronal somata. Scale bar is 20 μm. (C) Diagrammatic representation and electron micrograph of C-bouton ultrastructure in an adult rat. (Ci) Diagram illustrates densely packed, clear spherical or pleomorphic vesicles and abundant mitochondria. Closely apposed to the postsynaptic membrane is a 10–15 nm wide subsurface cistern (SSC) that is continuous with several lamellae of underlying rough endoplasmic reticulum (rER). Free ribosomal rosettes are typically visible in the subsynaptic region. (Cii) Electron micrograph of C-bouton synapse on an α-MN soma. Arrowheads indicate a SSC extending the entire appositional length of the bouton. Note key features present in electron micrograph illustrated in diagram (Ci).
C-boutons are among the largest of α-MN somatic and proximal dendritic synaptic inputs, ranging in size from 3 to 8 μm in the cat (Conradi, 1969a; McLaughlin, 1972b; Conradi et al., 1979a), 3–6 μm in the primate (Bodian, 1966a,b), 3–5 μm in the opossum (Hamos and King, 1980), 3–6 μm in the human (Pullen, 1992), and 1–8 μm in the rodent (Alvarez et al., 1999). But despite their conspicuous size, they lack quintessential active zone ultrastructure, i.e., pronounced paramembraneous densities and associated pools of readily releasable vesicles (Bodian, 1966a,b; Conradi, 1969a; McLaughlin, 1972b; Bernstein and Bernstein, 1976), prompting early speculation that vesicle release occurs across the entire synaptic interface (McLaughlin, 1972b). However, small presynaptic dense projections and local vesicle aggregations have been subsequently described (Hamos and King, 1980; Connaughton et al., 1986; Davidoff and Irintchev, 1986), and are particularly pronounced in non-osmicated tissue stained with E-PTA (Pullen, 1988a) or uranyl acetate and lead citrate (Schroder, 1979). These observations are commonly accepted evidence for specific synaptic vesicle release sites. Supporting this notion, C-boutons express discrete punctae of the active zone specific protein bassoon rather than diffuse expression throughout the presynaptic membrane (A. S. Deardorff, S. H. Romer, R. E. W. Fyffe, unpublished; see Figure 1). Moreover, bassoon immunoreactivity precisely overlies postsynaptic SK channels and m2 receptors even though traditional postsynaptic densities are not typically observed under EM. The physiologic advantage of this characteristically atypical and peculiarly subtle active zone architecture, however, is not yet fully understood, and may be further complicated by interspecies variability (see Pullen, 1988a).
C-boutons are ubiquitous and highly specific to somatic α-MNs and have been identified on α-MN somata and proximal dendrites in all mammalian species studied thus far (see Yamamoto et al., 1991 for references). Detailed analyses of γ-MNs (Lagerback, 1985; Lagerback et al., 1986; Destombes et al., 1992), autonomic MNs (Mawe et al., 1986; Leedy et al., 1988), spinal interneurons (Johnson and Sears, 1988), and Renshaw cells (Lagerback and Ronnevi, 1982; Alvarez and Fyffe, 2007) confirm these cells lack C-type synaptic inputs. C-boutons, when properly identified (see discussion Section “Moments and Milestones: Transmitter Content”), are thus a useful anatomical criterion to distinguish somatic α-MNs in the brain and spinal cord (Conradi, 1969a; Pullen, 1988b; Deng and Fyffe, 2004; Muennich and Fyffe, 2004; Deardorff et al., 2013). Although there have been no extensive three-dimensional analyses of the total number of C-boutons per α-MN, our, and other, observations suggest on the order of 30–70 such contacts per cell (McLaughlin, 1972b; Hamos and King, 1980; Brannstrom, 1993; Brannstrom and Kellerth, 1998), and in general, there are a greater number of C-bouton synaptic contacts on large α-MNs innervating fast twitch muscle fibers, with this difference not simply due to the larger available somatic/dendritic surface area (Conradi et al., 1979a,b; Kellerth et al., 1979, 1983; Hellstrom et al., 2003). It should be noted, the features of C-boutons present on somatic α-MNs in ocular motor nuclei vary from those in the spinal cord and other brainstem motor nuclei. Specifically, C-boutons have been ultrastructurally identified (Tredici et al., 1976) and α-MN SSCs express connexin32 (Yamamoto et al., 1991), but no large VAChT-IR synaptic contacts (Hellstrom et al., 2003) nor m2 receptors are present (Vilaro et al., 1992; Hellstrom et al., 2003).
Moments and Milestones: Transmitter Content
Correlative light-electron microscopic analysis of ChAT-IR confirmed C-boutons are cholinergic (Houser et al., 1983; Connaughton et al., 1986; Li et al., 1995), a suggestion first made decades prior with ultrastructural acetylcholinesterase (AChE) histochemistry (Lewis and Shute, 1966), which alone is not sufficient for cholinergic classification (Fibiger, 1982; Satoh et al., 1983; Sakamoto et al., 1985; Davidoff and Irintchev, 1986; Nagy et al., 1993). In support, VAChT is highly associated with small clear synaptic vesicles in the C-bouton presynaptic terminal (Gilmor et al., 1996) and there is a strong association throughout the brainstem and spinal cord between large ChAT-IR synaptic boutons on α-MNs and SSCs immunolabeled for connexin32 (Nagy et al., 1993). Immunoreactivity for the cholinergic markers ChAT or VAChT, combined with anatomical criteria such as bouton size and location, therefore makes C-boutons easily identifiable in adult/neonatal histologic sections (Figure 3; Barber et al., 1984; Phelps et al., 1984; Nagy et al., 1993; Hellstrom et al., 2003; Wilson et al., 2004; Zagoraiou et al., 2009; Alvarez et al., 2011; Deardorff et al., 2013). However, this approach should be applied with caution, as a small subset of cholinergic S-type terminals arising from recurrent α-MN axon collaterals and contacting α-MN somata may approximate C-boutons in size (Cullheim et al., 1977; Lagerback et al., 1981). Definitive confirmation of C-bouton phenotype requires ultrastructural verification of the C-bouton specific “cisternal signature” or alternatively – when systematically surveying an adequate sample of α-MNs under EM is unrealistic – light level co-localization of cholinergic makers with C-bouton specific pre- and/or postsynaptic proteins (see Section “The C-bouton Signaling Ensemble: A Contemporary View of a Classic Synapse”).
Moments and Milestones: Dissecting the C-Bouton Circuitry
Unlike so many α-MN synaptic inputs, for which the neurons of origin are identifiable anatomically and physiologically (Jankowska and Lindstrom, 1972; Jankowska and Roberts, 1972a,b; Brown et al., 1981; Brown, 1983; Fyffe, 1991a,b; Burke and Glenn, 1996; Bui et al., 2003), the neuronal source of C-boutons has been elusive. Early in vivo lesion studies demonstrated that C-boutons do not degenerate following dorsal root section (Conradi, 1969b; McLaughlin, 1972a; Bodian, 1975); spinal cord hemisection/transection (McLaughlin, 1972c; Bodian, 1975; Pullen and Sears, 1978, 1983), or cortical ablation (Bodian, 1975). Neither are they labeled by injection of retrograde tracers into dorsal roots (Ralston and Ralston, 1979), nor intracellular staining of Ia afferents (Brown and Fyffe, 1978; Conradi et al., 1983; Fyffe and Light, 1984), Ib afferents (Brown and Fyffe, 1979), group II afferents (Fyffe, 1979), hair follicle afferents (Maxwell et al., 1982), or axons innervating cutaneous mechanoreceptors (Brown et al., 1978, 1980, 1981; Bannatyne et al., 1984; Maxwell et al., 1984). Similarly, intracellular labeling of α-MNs showed C-boutons do not arise from α-MN axon collaterals (Lagerback et al., 1981), which is corroborated by differential protein expression in C-type synapses versus cholinergic terminals in the Renshaw cell area (see Section “The C-bouton Signaling Ensemble: A Contemporary View of a Classic Synapse;” Hellstrom et al., 1999; Deng and Fyffe, 2004).
Though these data collectively indicate the intraspinal derivation of C-boutons, no investigator to date has intracellularly labeled a cholinergic spinal interneuron and traced its axon to an α-MN C-type synaptic contact in vivo or in vitro; the definitive test for synaptic connectivity. Advanced molecular labeling techniques, however, have very convincingly demonstrated that C-boutons arise from cholinergic V0-embryonic (V0C) interneurons identifiable transcriptionally and phenotypically by the expression of the V0-specific homeobox protein Dbx1, the paired-like homeodomain transcription factor Pitx2, and the cholinergic proteins ChAT or VAChT (Miles et al., 2007; Zagoraiou et al., 2009). (For complete information on V0 cell ontogeny, we refer the reader to studies by Moran-Rivard et al. (2001), Pierani et al. (2001), and Lanuza et al. (2004) as well as the review by Arber (2012)). V0C interneurons correspond to a known population of cholinergic partition cells (Barber et al., 1984; Phelps et al., 1984; Arvidsson et al., 1997) located lateral to the central canal in Rexed’s lamina X and medial lamina VII (Miles et al., 2007; Zagoraiou et al., 2009). They can be subdivided into ipsilateral and bilateral projecting subpopulations and span several segments rostral and caudal to their innervated motor pools (Stepien et al., 2010). Cholinergic partition cells, C-type synaptic boutons, and the “signaling ensemble” appear early in postnatal development, and are well established by approximately 1 month of age (Phelps et al., 1984; Wetts and Vaughn, 2001; Wilson et al., 2004).
The specific placement of V0C interneurons within segmental spinal circuitry is not fully characterized [see preliminary circuit diagrams in Zagoraiou et al. (2009) and Witts et al. (2014)]. Preliminary analysis of V0C connectivity demonstrates V0C interneurons receive synaptic input from several sources, including descending serotonergic pathways, local and/or descending VGluT2 projections, inhibitory interneurons (e.g., V2b cells), lamina II/III nociceptive interneurons, and non-proprioceptive primary mechanosensors (Zagoraiou et al., 2009; Witts et al., 2014; Zampieri et al., 2014; Zhang et al., 2014). Each V0C cell sends divergent axonal projections to several α-MNs of the same or functionally equivalent motor pools and avoids α-MNs innervating antagonist muscles (Stepien et al., 2010). Numerous en passant synaptic varicosities arising from a single V0C axon contact the soma and proximal dendrites of a one or more α-MNs, which in turn receive convergent input from several V0C cells (Stepien et al., 2010). Although the precise levels of convergence/divergence are unknown, this pattern of connectivity establishes a large number of release sites from each presynaptic axon onto the α-MN, likely reflecting a high probability of transmitter release and contributing to a high safety factor for strong cholinergic neuromodulation (e.g., Walmsley et al., 1998).
Recent work shows V0C interneurons also project numerous small synaptic contacts onto V1-derived IaINs (Siembab et al., 2010). These synapses are morphologically dissimilar to C-boutons (Siembab et al., 2010), and their postsynaptic effects are currently unknown. Still, it is intriguing to consider that V0C interneurons project to the only two neuronal types (α-MNS and IaINs) in the ventral horn known to receive both recurrent inhibition and group Ia excitatory drive. Whether V0C interneurons, like Renshaw cells, send parallel projections to α-MNs and the “corresponding” IaINs (i.e., those with the same Ia connections; Hultborn et al., 1971a,b,c) has yet to be elucidated. Nevertheless, these data provide further insight into segmental motor circuitry and prompt new questions into both circuit function and synaptic specificity of the V0C neuronal class.
Moments and Milestones: AHP, SK, and Motor Unit Type
Early in vivo use of the SK channel blocker, apamin, established that SK channels are uniquely responsible for generating α-MN AHP currents (Zhang and Krnjevic, 1987). In vitro investigation subsequently confirmed these findings (Viana et al., 1993; Lape and Nistri, 2000), and showed that α-MN SK currents are reduced following m2 receptor activation at C-bouton synaptic sites (Lape and Nistri, 2000; Miles et al., 2007). Consistent with these electrophysiological data, our lab has recently shown that not only are SK channels highly enriched in the C-bouton postsynaptic membrane (Deardorff et al., 2013), but individual α-MNs express a variable complement of SK2 and SK3 channel isoforms consistent with observed co-variability in α-MN size and AHP duration (Deardorff et al., 2013). In the rodent, all α-MNs express SK2, but SK3 expression is markedly heterogeneous and cell-type-specific (Figure 4) varying in intensity from negligible (<2× background) to modest (2 to 3× background) to strong (>3× background) between individual α-MNs in a single tissue section. SK3 channels, which have a longer deactivation time constant than SK2 (Xia et al., 1998), are only expressed (with SK2) at C-bouton postsynaptic sites in smaller α-MNs with longer duration/larger amplitude AHPs (Figure 5). Conversely, larger α-MNs with significantly shorter duration/smaller amplitude AHPs express only SK2 (with little or no SK3-IR; Figure 5).
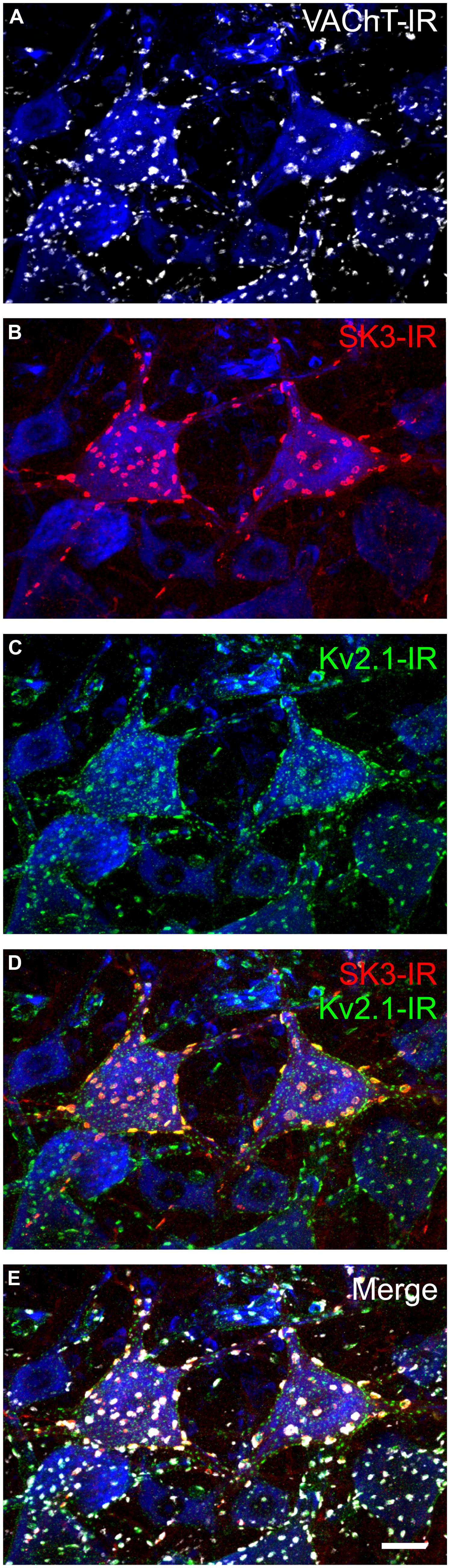
FIGURE 4. The potassium ion channel SK3 is part of the C-bouton signaling ensemble in a subset of α-motoneurons. Images are confocal stacks of 26 × 1 μm Z-stacks with nissl stain (blue) to label rat lumbar neuronal somata. Scale bar is 20 μm. (A) VAChT-IR (white) C-boutons form synapses onto all rat lumbar α-MNs on the soma and proximal dendrites. (B) SK3-IR (red) located within surface membrane of a subset of α-MNs in large distinct clusters. In rodents, SK3 channels, having slower intrinsic activation and deactivation kinetics than SK2 channels (Xia et al., 1998), are preferentially expressed in small, presumably S-type, α-MNs with long duration and large amplitude mAHP currents (Deardorff et al., 2013). (C) Large and small Kv2.1-IR (green) clusters are located within the surface membrane of all α-MNs. (D,E) The large SK3-IR and Kv2.1-IR clusters colocalize within the surface membrane of α-MNs and are apposed to VAChT-IR C-boutons.
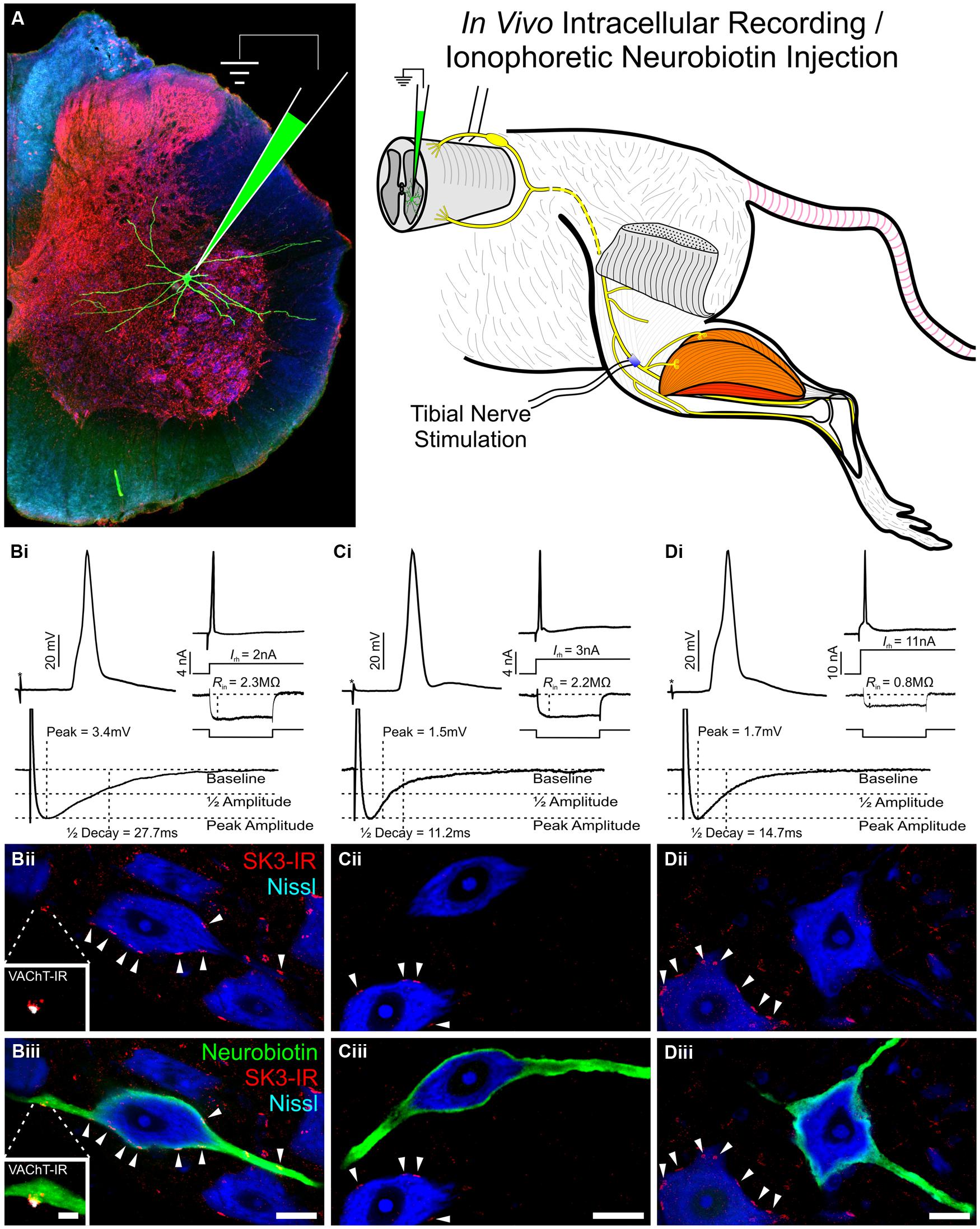
FIGURE 5. Subset of rat lumbar α-motoneurons with SK3-IR have significantly longer AHP 1/2 decay time and increased amplitude. Data shown is review of previous study reported by Deardorff et al. (2013). (A) Diagrammatic representation of experimental paradigms. In an adult in vivo rat preparation, tibial α-MNs, identified by antidromic activation of the tibial nerve, were penetrated with a sharp recording electrode. Neuronal electrical properties were recorded and neurons were filled with neurobiotin (green) for post hoc identification. Spinal cord tissue was harvested and processed for SK3-IR. (B–D) Neuronal electrical properties are of α-MNs depicted in micrographs below. Asterisk (*) denotes stimulus artifact. Micrographs are single optical confocal sections through the soma of intracellularly labeled α-MNs (green) processed for SK3-IR (red) and the general neuronal stain nissl (Blue). Scale bars are 20 μm. (B) SK3-IR (+) (Bii and Biii arrowheads) α-MNs have long duration and large amplitude AHP, low rheobase, and high input resistance. Micrograph insets show VAChT-IR (White) C-bouton in apposition to an SK3-IR (+) cluster. Inset scale bar is 5 μm. (C,D) SK3-IR (-) α-MNs have short duration and small amplitude AHPs. However, even among these SK3-IR (-) cells, rheobase and input resistance show high variance along the continuum of α-MN properties. Please note the nearby SK3-IR (+) cells (C,Dii,iii arrowheads).
SK3-expressing α-MNs share other physiological properties predictive of S-type MNs (i.e., slower conduction velocity, lower rheobase, and higher input resistance; Deardorff et al., 2013). SK3-IR within the signaling ensemble can therefore provide “brush stroke” differentiation of rodent α-MNs along their physiological spectrum, and is a useful tool for histologic analysis of α-MN subtypes in development and disease (Brownstone and Magown, 2013). Altogether these data strongly indicate that the relative proportion of SK2/SK3 isoforms and the channel cluster size and density regulates AHP duration and amplitude, and the variability of these proportions accounts, in part, for the fact that AHP properties are continuous variables across a population of α-MNs (Deardorff et al., 2013). SK channel expression may, therefore, explain the “speed match” between AHP duration of a given α-MN and the contractile speed of its innervated muscle fibers (Bakels and Kernell, 1993; Gardiner, 1993). However, critical additional factors include the source and amplitude of the necessary Ca2+ signal, the coupling of these signals to the SK channels and, potentially, the presence/absence of Ih currents (Gustafsson and Pinter, 1985). Nevertheless, differential SK channel expression at the C-bouton undoubtedly contributes to α-MN input–output gain across the spectrum of α-MN subtypes by regulating AHP properties.
Swimming Forward: A Mechanism for Cholinergic Modulation
We return now to the crest of our wave. The constancy of form and the intricacy of protein expression imply a fundamental logic to C-bouton organization and engagement during motor activity. Here, we assert the signaling ensemble is built around an organizing principle (i.e., the SSC) that allows for the generation of isolated Ca2+ signals at multiple sites on the soma. From this starting point, our intent here is to swim forward toward the synthesis of a comprehensive mechanistic hypothesis for the cholinergic modulation of α-MN firing rate. We base our rationale in the now recognized functional requirement for C-boutons in “swimming” (Zagoraiou et al., 2009), in the observation that cholinergic C-bouton function is not required for regular locomotion (Zagoraiou et al., 2009), and in the probable interactions of the key components of the C-bouton signaling ensemble (Figures 1 and 2), most of which are known to generate, regulate, or be regulated by local intracellular Ca2+.
Although C-boutons may boost recruitment gain, as proposed elsewhere (Zagoraiou et al., 2009; Brownstone and Magown, 2013), we propose that the cholinergic modulation produced by C-boutons is highly task-dependent and will be maximal only during the moderate to strong physiological drive necessary for high-output motor tasks like swimming (Zagoraiou et al., 2009; Figure 6). The mechanism we suggest accounts for the minimal appreciable requirements and effects observed during conditions of low and/or transient drive, which are appropriate for spinal reflexes and/or low-output tasks such as walking (Zagoraiou et al., 2009; Figures 6Ai,Bi). We extend this notion further to conditions of extremely powerful physiological (or pathological) drive, during which time any effects of C-bouton activity on firing rate are negated by the molecular dynamics and kinetics of the respective m2 receptors and SK/Kv channels (Figures 6Aiv,Biv). That is, while the cumulative, combined effects of these isolated Ca2+ signals on specific AHP and delayed rectifier K+ currents are likely to be quite significant throughout the α-MN activity spectrum, the functional impact of the C-bouton circuitry is only observed when imposed upon a restricted window of moderate to strong excitatory drive. We believe our synthesis, which is primarily based on interpretation of disparate datasets, will promote testable hypotheses. Elements of this synthetic approach are considered in the following sections.
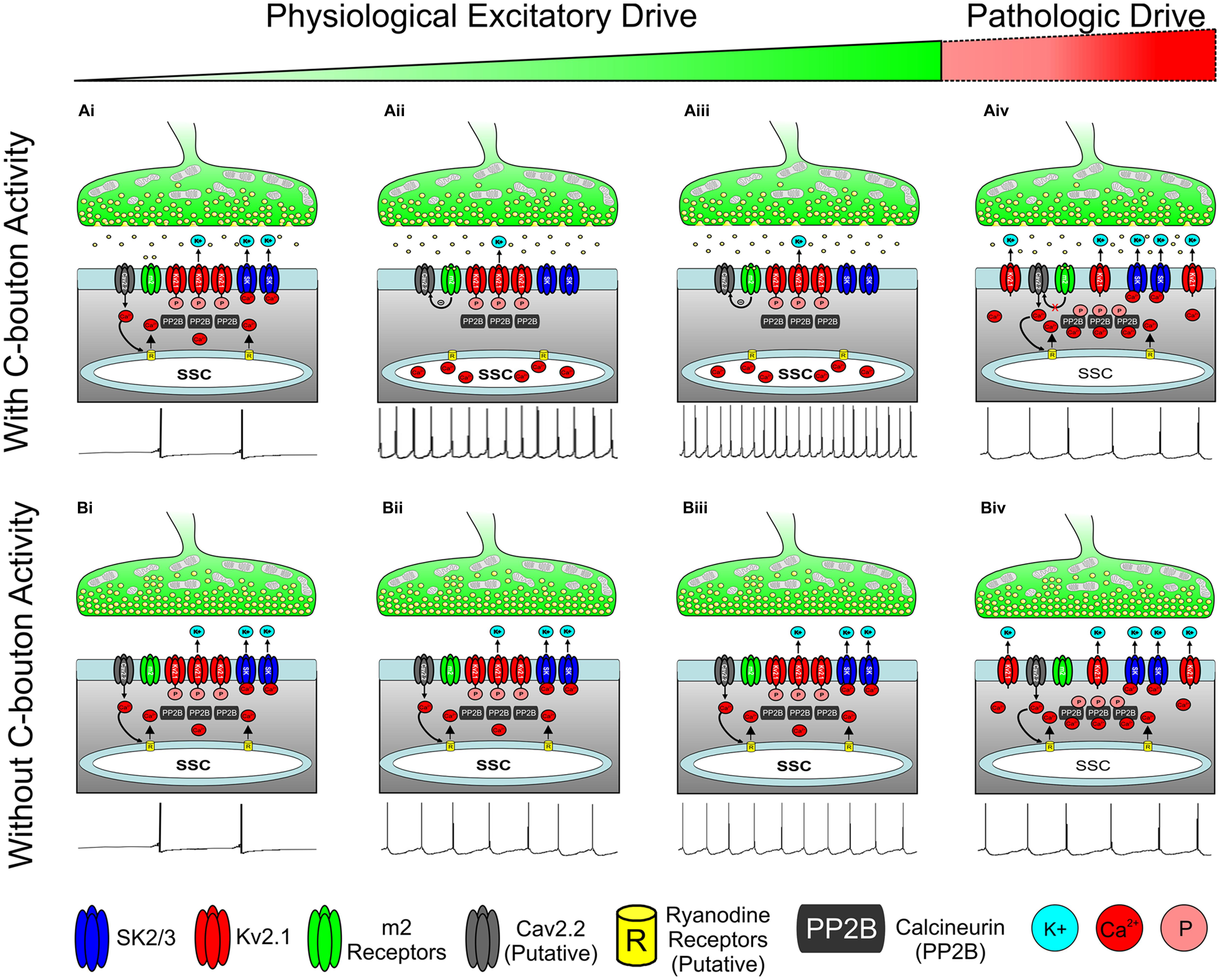
FIGURE 6. Hypothesis for state dependent regulation of motoneuron activity through the C-Bouton signaling ensemble. (A) C-boutons increase motoneuron firing frequency along a widow of the α-MN activity spectrum. (Ai) With low or transient physiological drive, m2 activation is not likely to mediate an effect on AHP duration or firing rate. (Aii,iii) As excitatory drive increases, persistent m2 receptor activation inhibits local CaV channels through a Gi/Go coupled pathway, preventing both the SK channel activation and Kv2.1 dephosphorylation. Thus, outward K+ current is reduced and neuronal firing rate is increased (relative to Bii and Biii) as illustrated with spike train below. (Aiv) m2-mediated effects on CaV channels are negated by prolonged or repeated membrane depolarization (Hille, 1994) as may occur during extremely strong or pathologic excitatory drive. Here, Ca2+ influx through N-type calcium channels activates SK channels to generate AHP and to dephosphorylate Kv2.1 to increase outward K+ current and reduce firing frequency, as illustrated with spike train below. (Bi–iii) As excitatory drive increases without C-bouton activity, the N-type Ca2+ influx activates SK channels to generate AHP. Thus, the outward K+ current maintains a lower firing frequency than in corresponding images in A. Spike trains illustrated below. (Biv) As in (Aiv), during prolonged or pathologic excitatory drive, N-type Ca2+ influx results in both SK channel activation and Kv2.1 dephosphorylation, thereby increasing outward K+ current and homeostatically decreasing firing rate, illustrated with spike train below. All spike trains depicted in this figure are added for illustrative purposes only and do not represent electrophysiological recordings or computer simulations.
Subsurface Cisternae and the Generation of an Isolated Ca2+ Signal
It is widely accepted that neuronal SSCs function as an intracellular Ca2+ store with multiple roles in Ca2+ homeostasis and mobilization (see Yamamoto et al., 1991 and Fuchs et al., 2014 for references). Indeed, Henkart et al. (1976) proposed that SSCs “are designed to release Ca2+ into the cytoplasm with whatever further effects this might produce.” SSCs serve also as a physical diffusion barrier that spatially and functionally restricts this Ca2+ signal from those originating in other cellular compartments and, during increased cellular activity, act as a Ca2+ sink to rapidly absorb and shuttle free Ca2+ from the cisternal microdomain (Yamamoto et al., 1990, 1991; Fuchs et al., 2014). Ca2+ release by RyR-rich SSCs serve, in part, to activate nearby SK channels in cochlear hair cells, which share some synaptic similarities with C-boutons (Evans et al., 2000; Lioudyno et al., 2004; Grant et al., 2006), and in sympathetic ganglion cells (Akita and Kuba, 2000). RyR release of Ca2+ may also result in an increase in nearby Kv2.1 channel conductances, via Ca2+-dependent dephosphorylation pathways, as it does in hippocampal and cortical pyramidal cells (Du et al., 1998; Antonucci et al., 2001; Misonou et al., 2005). Moreover, vesicles observed budding from the cytoplasmic surface of SSCs in cochlear hair cells and α-MNs are thought to be involved in removal of excess free Ca2+ from the subsynaptic cytoplasm (Yamamoto et al., 1991; Fuchs et al., 2014). In light of these factors, the SSC itself is highly indicative that the functional regulation of the C-bouton signaling ensemble (which includes SK and Kv2.1 channels) occurs through precise control of an isolated Ca2+ microdomain, the mechanistic underpinnings of which are considered below.
Involvement of the Signaling Ensemble with the Isolated Ca2+ Signal
The unique aggregation of cellular elements at C-bouton synaptic sites and their coordinated regulation by and/or of the isolated Ca2+ signal enables exquisite control over α-MN K+ currents. Consider first the generation of the α-MN AHP. Membrane bound N- and P/Q-type Ca2+ currents necessary for α-MN SK channel activation (see “Additional Signaling Components”) generate this Ca2+ signal, which is isolated and shaped by the SSC. The AHP currents influence repetitive discharge properties of α-MNs, in part, via reductions in the variability in the interspike interval, the slope of the f-I relation, and the maximal rate of primary-range firing (Kernell, 2006; Brownstone and Magown, 2013).
A primary effect of m2 receptor activation by C-bouton synapses is a reduction of the AHP (Lape and Nistri, 2000; Miles et al., 2007). Though their signaling pathway(s) in α-MNs are undefined, m2 receptors typically exert their effects by inhibiting N-type Ca2+ channels, as observed in sympathetic ganglion (Hille, 1994; Herlitze et al., 1996; Shapiro et al., 1999), cortical pyramidal (Stewart et al., 1999), neostriatal (Howe and Surmeier, 1995), and basal forebrain neurons (Allen and Brown, 1993). Ca2+ influx through these channels is required for activation of SK channels and dictates the number of SK channels that open. N-type channel blockade by m2 receptors is usually mediated by Gi/o protein coupled βγ subunits, which cause a depolarizing shift in the voltage dependence of channel activation (Hille, 1994; Herlitze et al., 1996; Ikeda, 1996; Jeong and Ikeda, 1999; Shapiro et al., 1999) and is negated by strong or repeated membrane depolarization (Hille, 1994).
The m2/cholinergic effect exerted by active C-boutons is quite simple and intuitive at this level: preventing N-type Ca2+ influx (which is largely triggered by synaptically evoked action potentials) from activating SK channels during moderate to strong physiologic drive of the MNs (Figures 6Aii,iii,Bii,iii). This would be consistent with observed reduction of the AHP and enhanced α-MN excitability when m2 receptors are, presumably, activated during swimming or other tasks requiring high motor output (e.g., Figures 6Aii,Aiii; Miles et al., 2007; Zagoraiou et al., 2009). This “upstream” mechanism of AHP modulation will have a minimal appreciable effect on individual AHPs and α-MN firing rate during low levels of physiologic drive causing transient or “subprimary” range firing (Manuel et al., 2009; Turkin et al., 2010), due to the physiological triggering of SK channel activation by a short duration, suprathreshold stimulus (i.e., an action potential) occurring at intervals that may be longer than the duration of the AHP itself (Figures 6Ai,Bi). This may account for observations that C-bouton function is not required for regular locomotion (Zagoraiou et al., 2009). Moreover during powerful and/or pathologic excitatory drive the m2-mediated diminution of N-type channel activity is negated (Hille, 1994), resulting in a break of the m2 generated effect and an increase in AHP size (Figures 6Aiv,Biv).
At high levels of excitatory drive we must also consider the results of modulation of other components of the signaling ensemble. Although the m2 mediated effect on AHP is significant in a particular physiological range, the whole microdomain has an important role in setting α-MN firing rate. With this in mind, the Ca2+ dependent generation of the AHP and its regulation by m2 receptor activation is one part of a coordinated series of molecular events that occur at the C-bouton, but is reliant on the complex interplay of other components in the signaling ensemble. For example, as excitatory drive increases how does the combinatorial contribution of SK and/or Kv2.1 change in the presence or absence of cholinergic input?
In the highly clustered configuration (typically) observed in hippocampal and cortical pyramidal cells, and α-MNs, Kv2.1 channels are phosphorylated and have a high activation and deactivation threshold and slow kinetics (Murakoshi et al., 1997; Misonou et al., 2004, 2005; Surmeier and Foehring, 2004; Mohapatra and Trimmer, 2006; Misonou, 2010). Interestingly, some investigators have postulated that clustered Kv2.1 channels serve primarily non-conducting functions (O’Connell et al., 2010; Fox et al., 2013); for the purposes of this discussion we will consider a more traditional role for the channels in αMNs. Importantly, upon prolonged/pathologic excitatory drive, Ca2+/calcineurin dependent dephosphorylation pathways (Figures 6Aiv,Biv) rapidly decluster Kv2.1 while simultaneously lowering its activation and deactivation threshold and accelerating its kinetics (Surmeier and Foehring, 2004; Park et al., 2006; Mohapatra et al., 2009). In α-MNs, prolonged excitatory drive causes rapid Kv2.1 channel declustering (Romer et al., 2014) by a Ca2+/calcineurin dependent mechanism (S. H. Romer, A. S. Deardorff, R. E. W. Fyffe, unpublished), though corresponding alterations in channel kinetics are uncharacterized.
Data from other cell types shows clustered Kv2 channels maintain steady state firing by regulating membrane potential during the interspike interval (Johnston et al., 2008; Guan et al., 2013; Liu and Bean, 2014), while declustered/dephosphorylated Kv2 channels serve to homeostatically lower firing rate (Surmeier and Foehring, 2004; Park et al., 2006; Mohapatra et al., 2009). In this way, Kv2 channels may increase or decrease cell excitability depending on the kinetics of channel activation (Liu and Bean, 2014). Brownstone et al. (2011) propose C-bouton activity during fictive locomotion (Miles et al., 2007; Zagoraiou et al., 2009) may contribute to steady state firing rates via the regulation of Kv2.1 phosphorylation and clustering. This is consistent with our hypothesis that m2-mediated inhibition of HVA-Ca2+ current prevents the activation of Ca2+/calcineurin dependent dephosphorylation pathways and thus maintains Kv2.1 clustering. However, if prolonged/pathologic excitatory drive causes large changes in intracellular Ca2+ sufficient to allow diffusion of Ca2+ from neighboring compartments, there would be rapid Kv2.1 channel declustering (Romer et al., 2014) by a Ca2+/calcineurin dependent mechanism (S. H. Romer, A. S. Deardorff, R. E. W. Fyffe, unpublished), negating the influence of C-boutons.
Several other components of this complex signaling ensemble likely serve to fine tune the efficacy of neuromodulation. Presynaptic nAChRs and P2X7 receptors may provide an additional regulatory mechanism for synaptic transmission, particularly if ATP is co-released with ACh as it is at other central and peripheral cholinergic synapses (Burnstock et al., 1997), and cisternal S1Rs are known to reduce the sensitivity of m2 receptors to ACh (Walker and Bourguignon, 1990; Kim et al., 2010). Altogether, we suggest the C-bouton signaling ensemble is a highly integrated system, organized around an anatomically segregated Ca2+ microdomain, for precise and nuanced regulation of cell firing. Moreover, it has a built-in fail-safe mechanism against excitotoxicity, in that this strategically organized ensemble can both be driven by, or override, the synaptic circuitry of the C-bouton.
An Alternative Mechanism
Others have suggested, based on muscarine’s minimal effect on global α-MN Ca2+ currents, that m2 receptor activation results in the direct blockade of α-MN SK channels (Miles et al., 2007; Witts et al., 2014). In support of their view, the direct phosphorylation of SK channels by protein kinase A (PKA) and casein kinase 2 (CK2) can, respectively, cause channel internalization (Kohler et al., 1996; Ren et al., 2006; Fakler and Adelman, 2008; Faber, 2009) and reduced Ca2+ sensitivity (Bildl et al., 2004; Allen et al., 2007). Moreover, neurotransmitter-initiated signaling cascades have been shown to modulate SK channel gating through CK2- or protein kinase C (PKC)-mediated phosphorylation (Maingret et al., 2008; Buchanan et al., 2010; Giessel and Sabatini, 2010). Although m2 receptors typically inhibit protein kinase activity, they can activate phosphorylation pathways in smooth muscle (Zhou et al., 2003). Therefore it is possible the direct phosphorylation of SK channels by protein kinases could provide an alternate mechanism through which m2 receptors reduce the AHP in α-MNs.
However, evidence that N- and P/Q-type Ca2+ channels are diffusely distributed throughout the α-MN somatic membrane (Wilson et al., 2004), and that α-MN SSCs function as Ca2+ diffusion barriers indicates that m2 receptor activation need only inhibit those α-MN CaV channels located within or very near to the C-bouton postsynaptic membrane to exert an effect on the AHP. In this case, m2 influence over the signaling ensemble would be masked in studies of global Ca2+ currents. The activation of CK2- or PKC-mediated phosphorylation would also be a novel finding for neuronal m2 receptors, necessitating future studies characterizing this undescribed signaling pathway. Moreover, such a mechanism would act as a binary switch, turning on and off AHP when necessary and not requiring an elaborate signaling ensemble nor the SSC. Our hypothesis, however, of a signaling ensemble organized around fine control of a Ca2+ micro-signaling domain is capable of highly nuanced and graded modulation of outward K+ current.
C-Boutons in Human Health and Disease
Dynamic reorganization of C-boutons and components of the postsynaptic signaling ensemble has been noted in a variety of pathologic conditions and in conditions of altered excitability (Saxena et al., 2013; Romer et al., 2014; Witts et al., 2014). The bulk of the data has thus far been obtained in animal models, and there is no consensus on whether C-bouton plasticity in these conditions is compensatory or pathologic. In part, the uncertainty results from the diversity of disease/injury models that affect C-boutons and the complexity of the signaling ensemble.
Analysis of effects on C-bouton structure in models of amyotrophic lateral sclerosis (ALS), spinal cord injury, and peripheral nerve injury demonstrate diverse and sometimes conflicting reports. In ALS, there has been interest in potential neuroprotective roles for C-boutons and this view is bolstered by studies that show an early increase in C-bouton size (Pullen and Athanasiou, 2009; Herron and Miles, 2012; Saxena et al., 2013); however, diminished C-bouton and V0c interneuronal ChAT/VAChT content (Nagao et al., 1998; Casas et al., 2013) and S1R expression (Casas et al., 2013; see Witts et al., 2014) have also been observed in similar murine models of the disease. The structural changes in animal models may also reflect a propensity for C-bouton reorganization to occur first in larger, less excitable, and more vulnerable α-MNs (Saxena et al., 2013), and the changes may be more pronounced in males (Herron and Miles, 2012). There is minimal data from autopsied human spinal cord from ALS patients, mostly from late stages of the disease, showing continued presence of C-boutons on degeneration-resistant sphinteric α-MNs (Pullen, 1992). Additionally, the duration of the AHP in human MNs is possibly related to disease progression (i.e., an initial shortening followed by prolongation; Piotrkiewicz and Hausmanowa-Petrusewicz, 2011).
C-bouton organization is affected by both spinal cord and peripheral nerve injury, which generally appear to cause transient or persistent loss of and/or disconnection of C-boutons from α-MNs and changes in expression and localization of SK, HCN, and Kv2.1 channels (Kerns and Hinsman, 1973; Sumner, 1975; Alvarez et al., 2011; Romer et al., 2012, 2014). These specific changes may account for some, but not all, of the physiological changes that have been observed (Kuno et al., 1974a,b; Cope et al., 1986; Bichler et al., 2007a,b; Bullinger et al., 2011; Prather et al., 2011), including altered post-spike AHP duration and repetitive firing properties (Kuno et al., 1974a; Gustafsson and Pinter, 1984).
The significance of C-bouton plasticity remains uncertain. After injury, the specific loss or disconnection could lead to postsynaptic receptors (m2) becoming constitutively active, analogous to observations made of the serotoninergic system (Fouad et al., 2010; Kong et al., 2010, 2011; Murray et al., 2010, 2011; Hultborn et al., 2013), but this has not been explored. Given the high vulnerability of large, F-type α-MNS in ALS, it would be interesting to determine if the graded expression of SK channel isoforms will promote new testable hypotheses regarding disease pathogenesis and C-bouton mediated compensatory adjustments (Brownstone and Magown, 2013; Deardorff et al., 2013).
Conclusion
Multiple neuromodulatory systems and a myriad of ion channels are available for the task dependent regulation of MN excitability. The serotonergic system, for example, originates in the brainstem raphe nucleus, provides extensive synaptic input onto α-MN dendrites (Alvarez et al., 1998) and is strongly linked to both behavioral and pathologic alterations of persistent inward Ca2+ currents (Li and Bennett, 2003, 2007; Heckmann et al., 2005; Brownstone, 2006; Heckman et al., 2008; Norton et al., 2008; Powers et al., 2008). While numerous studies have focused on inward current modulation, the state dependent regulation of α-MN outward current has only recently been investigated (see Manuel et al., 2012). New evidence has shown that a cholinergic modulatory system originating from spinal interneurons (V0C interneurons), and contributing dense synaptic coverage to α-MN somata, modulates the strength of motor output via reductions in α-MN outward K+ current (Miles et al., 2007; Zagoraiou et al., 2009). It is interesting to consider that while serotonin increases MN excitability by amplifying inward current, acetylcholine does so by reducing outward current. The dynamic interplay of these two different, but rather synergistic, systems endows the CNS with remarkable control over MN output, and the interaction between the AHP and L-type Ca2+ currents responsible for PIC may be a critical factor in regulating α-MN firing properties (Manuel et al., 2014).
Here, we illustrate large, cholinergic presynaptic terminals, termed C-boutons (Conradi, 1969a), are important modulatory loci for state-dependent alterations in MN repetitive firing, largely mediating their effects through a unique and highly specialized signaling ensemble organized for the state-dependent regulation of outward K+ currents. To effectively manipulate signal transduction at C-bouton synaptic sites may be critical in the development of new therapeutic interventions for a variety of devastating neurological conditions. However, advances in patient care will first require a complete understanding of both the transduction mechanisms, as well as which cases (if any) C-bouton synaptic reorganization and/or alterations in α-MN AHP (and other intrinsic α-MN properties) contribute to disease pathology or, alternatively, maintain α-MN viability.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Melissa A. Bautista for contributions in Figures 2 and 3. We thank Dr. Timothy Cope and Paul Nardelli for the use of their laboratory for in vivo electrophysiology and intracellular fills. We thank Dr. Francisco Alvarez for his helpful discussion in C-bouton synaptic structure. In addition, Mark Rich, Kathrin Engisch, and David Ladle for their insights during the preparation of this manuscript. This work was funded by National Institute of Neurological Disorders and Stroke Program Project Grant P01-NS-057228.
Abbreviations
AHP, afterhyperpolarization; ChAT, choline acetyltransferase; IaIN, Ia inhibitory interneuron; IR, immunoreactivity; m2 receptor, type 2 muscarinic acetylcholine receptor; MN, motoneuron; RyR, ryanodine receptors; S1R, sigma-1 receptors; SK, small conductance calcium-activated potassium channel; SSC, subsurface cistern; VAChT, vesicular acetylcholine transporter; VGluT, vesicular glutamate transporter.
References
Akita, T., and Kuba, K. (2000). Functional triads consisting of ryanodine receptors, Ca(2+) channels, and Ca(2+)-activated K(+) channels in bullfrog sympathetic neurons. Plastic modulation of action potential. J. Gen. Physiol. 116, 697–720. doi: 10.1085/jgp.116.5.697
Allen, D., Fakler, B., Maylie, J., and Adelman, J. P. (2007). Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J. Neurosci. 27, 2369–2376. doi: 10.1523/JNEUROSCI.3565-06.2007
Allen, T. G., and Brown, D. A. (1993). M2 muscarinic receptor-mediated inhibition of the Ca2+ current in rat magnocellular cholinergic basal forebrain neurones. J. Physiol. 466, 173–189.
Alvarez, F. J., Dewey, D. E., McMillin, P., and Fyffe, R. E. (1999). Distribution of cholinergic contacts on Renshaw cells in the rat spinal cord: a light microscopic study. J. Physiol. 515(Pt 3), 787–797. doi: 10.1111/j.1469-7793.1999.787ab.x
Alvarez, F. J., and Fyffe, R. E. (2007). The continuing case for the Renshaw cell. J. Physiol. 584, 31–45. doi: 10.1113/jphysiol.2007.136200
Alvarez, F. J., Pearson, J. C., Harrington, D., Dewey, D., Torbeck, L., and Fyffe, R. E. (1998). Distribution of 5-hydroxytryptamine-immunoreactive boutons on alpha-motoneurons in the lumbar spinal cord of adult cats. J. Comp. Neurol. 393, 69–83. doi: 10.1002/(SICI)1096-9861(19980330)393:1<69::AID-CNE7>3.0.CO;2-O
Alvarez, F. J., Titus-Mitchell, H. E., Bullinger, K. L., Kraszpulski, M., Nardelli, P., and Cope, T. C. (2011). Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. J. Neurophysiol. 106, 2450–2470. doi: 10.1152/jn.01095.2010
Antonucci, D. E., Lim, S. T., Vassanelli, S., and Trimmer, J. S. (2001). Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 108, 69–81. doi: 10.1016/S0306-4522(01)00476-6
Arber, S. (2012). Motor circuits in action: specification, connectivity, and function. Neuron 74, 975–989. doi: 10.1016/j.neuron.2012.05.011
Arvidsson, U., Riedl, M., Elde, R., and Meister, B. (1997). Vesicular acetylcholine transporter (VAChT) protein: a novel and unique marker for cholinergic neurons in the central and peripheral nervous systems. J. Comp. Neurol. 378, 454–467. doi: 10.1002/(SICI)1096-9861(19970224)378:4<454::AID-CNE2>3.0.CO;2-1
Bakels, R., and Kernell, D. (1993). Matching between motoneurone and muscle unit properties in rat medial gastrocnemius. J. Physiol. 463, 307–324.
Bannatyne, B. A., Maxwell, D. J., Fyffe, R. E., and Brown, A. G. (1984). Fine structure of primary afferent axon terminals of slowly adapting cutaneous receptors in the cat. Q. J. Exp. Physiol. 69, 547–557.
Barber, R. P., Phelps, P. E., Houser, C. R., Crawford, G. D., Salvaterra, P. M., and Vaughn, J. E. (1984). The morphology and distribution of neurons containing choline acetyltransferase in the adult rat spinal cord: an immunocytochemical study. J. Comp. Neurol. 229, 329–346. doi: 10.1002/cne.902290305
Bayliss, D. A., Umemiya, M., and Berger, A. J. (1995). Inhibition of N- and P-type calcium currents and the after-hyperpolarization in rat motoneurones by serotonin. J. Physiol. 485(Pt 3), 635–647.
Bernstein, J. J., and Bernstein, M. E. (1976). Ventral horn synaptology in the rat. J. Neurocytol. 5, 109–123. doi: 10.1007/BF01176185
Bichler, E. K., Carrasco, D. I., Rich, M. M., Cope, T. C., and Pinter, M. J. (2007a). Rat motoneuron properties recover following reinnervation in the absence of muscle activity and evoked acetylcholine release. J. Physiol. 585, 47–56. doi: 10.1113/jphysiol.2007.135541
Bichler, E. K., Nakanishi, S. T., Wang, Q. B., Pinter, M. J., Rich, M. M., and Cope, T. C. (2007b). Enhanced transmission at a spinal synapse triggered in vivo by an injury signal independent of altered synaptic activity. J. Neurosci. 27, 12851–12859. doi: 10.1523/JNEUROSCI.1997-07.2007
Bildl, W., Strassmaier, T., Thurm, H., Andersen, J., Eble, S., Oliver, D.,et al. (2004). Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron 43, 847–858. doi: 10.1016/j.neuron.2004.08.033
Bodian, D. (1966a). Electron microscopy: two major synaptic types on spinal motoneurons. Science 151, 1093–1094. doi: 10.1126/science.151.3714.1093
Bodian, D. (1966b). Synaptic types on spinal motoneurons: electron microscopic study. Bull. Johns Hopkins Hosp. 119, 16–46.
Bodian, D. (1975). Origin of specific synaptic types in the motoneuron neuropil of the monkey. J. Comp. Neurol. 159, 225–243. doi: 10.1002/cne.901590205
Brannstrom, T. (1993). Quantitative synaptology of functionally different types of cat medial gastrocnemius alpha-motoneurons. J. Comp. Neurol. 330, 439–454. doi: 10.1002/cne.903300311
Brannstrom, T., and Kellerth, J. O. (1998). Changes in synaptology of adult cat spinal alpha-motoneurons after axotomy. Exp. Brain Res. 118, 1–13. doi: 10.1007/s002210050249
Brown, A. G., and Fyffe, R. E. (1978). The morphology of group Ia afferent fibre collaterals in the spinal cord of the cat. J. Physiol. 274, 111–127.
Brown, A. G., and Fyffe, R. E. (1979). The morphology of group Ib afferent fibre collaterals in the spinal cord of the cat. J. Physiol. 296, 215–226.
Brown, A. G., Fyffe, R. E., and Noble, R. (1980). Projections from Pacinian corpuscles and rapidly adapting mechanoreceptors of glabrous skin to the cat’s spinal cord. J. Physiol. 307, 385–400.
Brown, A. G., Fyffe, R. E., Rose, P. K., and Snow, P. J. (1981). Spinal cord collaterals from axons of type II slowly adapting units in the cat. J. Physiol. 316, 469–480.
Brown, A. G., Rose, P. K., and Snow, P. J. (1978). Morphology and organization of axon collaterals from afferent fibres of slowly adapting type I units in cat spinal cord. J. Physiol. 277, 15–27.
Brownstone, R. M. (2006). Beginning at the end: repetitive firing properties in the final common pathway. Prog. Neurobiol. 78, 156–172. doi: 10.1016/j.pneurobio.2006.04.002
Brownstone, R. M., Krawitz, S., and Jordan, L. M. (2011). Reversal of the late phase of spike frequency adaptation in cat spinal motoneurons during fictive locomotion. J. Neurophysiol. 105, 1045–1050. doi: 10.1152/jn.00411.2010
Brownstone, R. M., and Magown, P. (2013). Matchmaking: SK channels, C-boutons and motor units. J. Physiol. 591, 747–748. doi: 10.1113/jphysiol.2012.249573
Buchanan, K. A., Petrovic, M. M., Chamberlain, S. E., Marrion, N. V., and Mellor, J. R. (2010). Facilitation of long-term potentiation by muscarinic M(1) receptors is mediated by inhibition of SK channels. Neuron 68, 948–963. doi: 10.1016/j.neuron.2010.11.018
Bui, T. V., Cushing, S., Dewey, D., Fyffe, R. E., and Rose, P. K. (2003). Comparison of the morphological and electrotonic properties of Renshaw cells, Ia inhibitory interneurons, and motoneurons in the cat. J. Neurophysiol. 90, 2900–2918. doi: 10.1152/jn.00533.2003
Bullinger, K. L., Nardelli, P., Pinter, M. J., Alvarez, F. J., and Cope, T. C. (2011). Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. II. Loss of functional connectivity with motoneurons. J. Neurophysiol. 106, 2471–2485. doi: 10.1152/jn.01097.2010
Burke, R. E., and Glenn, L. L. (1996). Horseradish peroxidase study of the spatial and electrotonic distribution of group Ia synapses on type-identified ankle extensor motoneurons in the cat. J. Comp. Neurol. 372, 465–485. doi: 10.1002/(SICI)1096-9861(19960826)372:3<465::AID-CNE9>3.0.CO;2-0
Burnstock, G., Campbell, G., Satchell, D., and Smythe, A. (1997). Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. 1970. Br. J. Pharmacol. 120, 337–357; discussion 334–336. doi: 10.1111/j.1476-5381.1997.tb06815.x
Carlin, K. P., Jiang, Z., and Brownstone, R. M. (2000). Characterization of calcium currents in functionally mature mouse spinal motoneurons. Eur. J. Neurosci. 12, 1624–1634. doi: 10.1046/j.1460-9568.2000.00050.x
Casas, C., Herrando-Grabulosa, M., Manzano, R., Mancuso, R., Osta, R., and Navarro, X. (2013). Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 3, 145–158. doi: 10.1002/brb3.104
Charlton, B. T., and Gray, E. G. (1966). Comparative electron microscopy of synapses in the vertebrate spinal cord. J. Cell Sci. 1, 67–80.
Clamann, H. P. (1993). Motor unit recruitment and the gradation of muscle force. Phys. Ther. 73, 830–843.
Connaughton, M., Priestley, J. V., Sofroniew, M. V., Eckenstein, F., and Cuello, A. C. (1986). Inputs to motoneurones in the hypoglossal nucleus of the rat: light and electron microscopic immunocytochemistry for choline acetyltransferase, substance P and enkephalins using monoclonal antibodies. Neuroscience 17, 205–224. doi: 10.1016/0306-4522(86)90237-X
Conradi, S. (1969a). Ultrastructure and distribution of neuronal and glial elements on the motoneuron surface in the lumbosacral spinal cord of the adult cat. Acta Physiol. Scand. Suppl. 332, 5–48.
Conradi, S. (1969b). Ultrastructure of dorsal root boutons on lumbosacral motoneurons of the adult cat, as revealed by dorsal root section. Acta Physiol. Scand. Suppl. 332, 85–115.
Conradi, S., Cullheim, S., Gollvik, L., and Kellerth, J. O. (1983). Electron microscopic observations on the synaptic contacts of group Ia muscle spindle afferents in the cat lumbosacral spinal cord. Brain Res. 265, 31–39. doi: 10.1016/0006-8993(83)91330-6
Conradi, S., Kellerth, J. O., and Berthold, C. H. (1979a). Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons. II. A method for the description of architecture and synaptology of the cell body and proximal dendritic segments. J. Comp. Neurol. 184, 741–754. doi: 10.1002/cne.901840407
Conradi, S., Kellerth, J. O., Berthold, C. H., and Hammarberg, C. (1979b). Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons. IV. Motoneurons innervating slow-twitch (type S) units of the soleus muscle. J. Comp. Neurol. 184, 769–782. doi: 10.1002/cne.901840409
Cope, T. C., Bodine, S. C., Fournier, M., and Edgerton, V. R. (1986). Soleus motor units in chronic spinal transected cats: physiological and morphological alterations. J. Neurophysiol. 55, 1202–1220.
Cope, T. C., and Sokoloff, A. J. (1999). Orderly recruitment tested across muscle boundaries. Prog. Brain Res. 123, 177–190. doi: 10.1016/S0079-6123(08)62855-1
Cullheim, S., Kellerth, J. O., and Conradi, S. (1977). Evidence for direct synaptic interconnections between cat spinal alpha-motoneurons via the recurrent axon collaterals: a morphological study using intracellular injection of horseradish peroxidase. Brain Res. 132, 1–10. doi: 10.1016/0006-8993(77)90702-8
Davidoff, M. S., and Irintchev, A. P. (1986). Acetylcholinesterase activity and type C synapses in the hypoglossal, facial and spinal-cord motor nuclei of rats. An electron-microscope study. Histochemistry 84, 515–524. doi: 10.1007/BF00482985
Deardorff, A. S., Romer, S. H., Deng, Z., Bullinger, K. L., Nardelli, P., Cope, T. C.,et al. (2013). Expression of postsynaptic Ca2+-activated K+ (SK) channels at C-bouton synapses in mammalian lumbar-motoneurons. J. Physiol. 591, 875–897. doi: 10.1113/jphysiol.2012.240879
Deng, Z., and Fyffe, R. E. (2004). Expression of P2X7 receptor immunoreactivity in distinct subsets of synaptic terminals in the ventral horn of rat lumbar spinal cord. Brain Res. 1020, 53–61. doi: 10.1016/j.brainres.2004.06.014
Destombes, J., Horcholle-Bossavit, G., Thiesson, D., and Jami, L. (1992). Alpha and gamma motoneurons in the peroneal nuclei of the cat spinal cord: an ultrastructural study. J. Comp. Neurol. 317, 79–90. doi: 10.1002/cne.903170106
Du, J., Tao-Cheng, J. H., Zerfas, P., and Mcbain, C. J. (1998). The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 84, 37–48. doi: 10.1016/S0306-4522(97)00519-8
Evans, M. G., Lagostena, L., Darbon, P., and Mammano, F. (2000). Cholinergic control of membrane conductance and intracellular free Ca2+ in outer hair cells of the guinea pig cochlea. Cell Calcium 28, 195–203. doi: 10.1054/ceca.2000.0145
Faber, E. S. (2009). Functions and modulation of neuronal SK channels. Cell Biochem. Biophys. 55, 127–139. doi: 10.1007/s12013-009-9062-7
Fakler, B., and Adelman, J. P. (2008). Control of K(Ca) channels by calcium nano/microdomains. Neuron 59, 873–881. doi: 10.1016/j.neuron.2008.09.001
Fibiger, H. C. (1982). The organization and some projections of cholinergic neurons of the mammalian forebrain. Brain Res. 257, 327–388. doi: 10.1016/0165-0173(82)90011-X
Fouad, K., Rank, M. M., Vavrek, R., Murray, K. C., Sanelli, L., and Bennett, D. J. (2010). Locomotion after spinal cord injury depends on constitutive activity in serotonin receptors. J. Neurophysiol. 104, 2975–2984. doi: 10.1152/jn.00499.2010
Fox, P. D., Loftus, R. J., and Tamkun, M. M. (2013). Regulation of Kv2.1 K(+) conductance by cell surface channel density. J. Neurosci. 33, 1259–1270. doi: 10.1523/JNEUROSCI.3008-12.2013
Frank, E. (2009). A new class of spinal interneurons: the origin and function of C boutons is solved. Neuron 64, 593–595. doi: 10.1016/j.neuron.2009.11.030
Fuchs, P. A., Lehar, M., and Hiel, H. (2014). Ultrastructure of cisternal synapses on outer hair cells of the mouse cochlea. J. Comp. Neurol. 522, 717–729. doi: 10.1002/cne.23478
Fyffe, R. E. (1979). The morphology of group II muscle afferent fibre collaterals [proceedings]. J. Physiol. 296, 39P–40P.
Fyffe, R. E. (1991a). Glycine-like immunoreactivity in synaptic boutons of identified inhibitory interneurons in the mammalian spinal cord. Brain Res. 547, 175–179. doi: 10.1016/0006-8993(91)90590-R
Fyffe, R. E. (1991b). Spatial distribution of recurrent inhibitory synapses on spinal motoneurons in the cat. J. Neurophysiol. 65, 1134–1149.
Fyffe, R. E., and Light, A. R. (1984). The ultrastructure of group Ia afferent fiber synapses in the lumbosacral spinal cord of the cat. Brain Res. 300, 201–209. doi: 10.1016/0006-8993(84)90831-X
Gallart-Palau, X., Tarabal, O., Casanovas, A., Sabado, J., Correa, F. J., Hereu, M.,et al. (2014). Neuregulin-1 is concentrated in the postsynaptic subsurface cistern of C-bouton inputs to alpha-motoneurons and altered during motoneuron diseases. FASEB J. 28, 3618–3632. doi: 10.1096/fj.13-248583
Gardiner, P. F. (1993). Physiological properties of motoneurons innervating different muscle unit types in rat gastrocnemius. J. Neurophysiol. 69, 1160–1170.
Giessel, A. J., and Sabatini, B. L. (2010). M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron 68, 936–947. doi: 10.1016/j.neuron.2010.09.004
Gilmor, M. L., Nash, N. R., Roghani, A., Edwards, R. H., Yi, H., Hersch, S. M.,et al. (1996). Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. J. Neurosci. 16, 2179–2190.
Grant, L., Slapnick, S., Kennedy, H., and Hackney, C. (2006). Ryanodine receptor localisation in the mammalian cochlea: an ultrastructural study. Hear. Res. 219, 101–109. doi: 10.1016/j.heares.2006.06.002
Guan, D., Armstrong, W. E., and Foehring, R. C. (2013). Kv2 channels regulate firing rate in pyramidal neurons from rat sensorimotor cortex. J. Physiol. 591, 4807–4825. doi: 10.1113/jphysiol.2013.257253
Gustafsson, B., and Pinter, M. J. (1984). Effects of axotomy on the distribution of passive electrical properties of cat motoneurones. J. Physiol. 356, 433–442.
Gustafsson, B., and Pinter, M. J. (1985). Factors determining the variation of the afterhyperpolarization duration in cat lumbar alpha-motoneurones. Brain Res. 326, 392–395. doi: 10.1016/0006-8993(85)90053-8
Hamos, J. E., and King, J. S. (1980). The synaptic organization of the motor nucleus of the trigeminal nerve in the opossum. J. Comp. Neurol. 194, 441–463. doi: 10.1002/cne.901940210
Heckman, C. J., Hyngstrom, A. S., and Johnson, M. D. (2008). Active properties of motoneurone dendrites: diffuse descending neuromodulation, focused local inhibition. J. Physiol. 586, 1225–1231. doi: 10.1113/jphysiol.2007.145078
Heckmann, C. J., Gorassini, M. A., and Bennett, D. J. (2005). Persistent inward currents in motoneuron dendrites: implications for motor output. Muscle Nerve 31, 135–156. doi: 10.1002/mus.20261
Hellstrom, J., Arvidsson, U., Elde, R., Cullheim, S., and Meister, B. (1999). Differential expression of nerve terminal protein isoforms in VAChT-containing varicosities of the spinal cord ventral horn. J. Comp. Neurol. 411, 578–590. doi: 10.1002/(SICI)1096-9861(19990906)411:4<578::AID-CNE4>3.0.CO;2-L
Hellstrom, J., Oliveira, A. L., Meister, B., and Cullheim, S. (2003). Large cholinergic nerve terminals on subsets of motoneurons and their relation to muscarinic receptor type 2. J. Comp. Neurol. 460, 476–486. doi: 10.1002/cne.10648
Henkart, M., Landis, D. M., and Reese, T. S. (1976). Similarity of junctions between plasma membranes and endoplasmic reticulum in muscle and neurons. J. Cell Biol. 70, 338–347. doi: 10.1083/jcb.70.2.338
Henneman, E., and Mendell, L. M. (1981). “Functional organization of the motoneuron pool and its inputs,” in Handbook of Physiology, ed. V. E. Brooks (Bethesda, MD: American Physiological Society), 423–507.
Herlitze, S., Garcia, D. E., Mackie, K., Hille, B., Scheuer, T., and Catterall, W. A. (1996). Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 380, 258–262. doi: 10.1038/380258a0
Herron, L. R., and Miles, G. B. (2012). Gender-specific perturbations in modulatory inputs to motoneurons in a mouse model of amyotrophic lateral sclerosis. Neuroscience 226, 313–323. doi: 10.1016/j.neuroscience.2012.09.031
Hille, B. (1994). Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 17, 531–536. doi: 10.1016/0166-2236(94)90157-0
Houser, C. R., Crawford, G. D., Barber, R. P., Salvaterra, P. M., and Vaughn, J. E. (1983). Organization and morphological characteristics of cholinergic neurons: an immunocytochemical study with a monoclonal antibody to choline acetyltransferase. Brain Res. 266, 97–119. doi: 10.1016/0006-8993(83)91312-4
Howe, A. R., and Surmeier, D. J. (1995). Muscarinic receptors modulate N-, P-, and L-type Ca2+ currents in rat striatal neurons through parallel pathways. J. Neurosci. 15, 458–469.
Hultborn, H., Jankowska, E., and Lindstrom, S. (1971a). Recurrent inhibition from motor axon collaterals of transmission in the Ia inhibitory pathway to motoneurones. J. Physiol. 215, 591–612.
Hultborn, H., Jankowska, E., and Lindstrom, S. (1971b). Recurrent inhibition of interneurones monosynaptically activated from group Ia afferents. J. Physiol. 215, 613–636.
Hultborn, H., Jankowska, E., Lindstrom, S., and Roberts, W. (1971c). Neuronal pathway of the recurrent facilitation of motoneurones. J. Physiol. 218, 495–514.
Hultborn, H., Zhang, M., and Meehan, C. F. (2013). Control and role of plateau potential properties in the spinal cord. Curr. Pharm. Des. 19, 4357–4370. doi: 10.2174/1381612811319240004
Ikeda, S. R. (1996). Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 380, 255–258. doi: 10.1038/380255a0
Jankowska, E., and Lindstrom, S. (1972). Morphology of interneurones mediating Ia reciprocal inhibition of motoneurones in the spinal cord of the cat. J. Physiol. 226, 805–823.
Jankowska, E., and Roberts, W. J. (1972a). An electrophysiological demonstration of the axonal projections of single spinal interneurones in the cat. J. Physiol. 222, 597–622.
Jankowska, E., and Roberts, W. J. (1972b). Synaptic actions of single interneurones mediating reciprocal Ia inhibition of motoneurones. J. Physiol. 222, 623–642.
Jeong, S. W., and Ikeda, S. R. (1999). Sequestration of G-protein beta gamma subunits by different G-protein alpha subunits blocks voltage-dependent modulation of Ca2+ channels in rat sympathetic neurons. J. Neurosci. 19, 4755–4761.
Johnson, I. P., and Sears, T. A. (1988). Ultrastructure of interneurons within motor nuclei of the thoracic region of the spinal cord of the adult cat. J. Anat. 161, 171–185.
Johnston, J., Griffin, S. J., Baker, C., Skrzypiec, A., Chernova, T., and Forsythe, I. D. (2008). Initial segment Kv2.2 channels mediate a slow delayed rectifier and maintain high frequency action potential firing in medial nucleus of the trapezoid body neurons. J. Physiol. 586, 3493–3509. doi: 10.1113/jphysiol.2008.153734
Jones, S. L., and Stuart, G. J. (2013). Different calcium sources control somatic versus dendritic SK channel activation during action potentials. J. Neurosci. 33, 19396–19405. doi: 10.1523/JNEUROSCI.2073-13.2013
Kellerth, J. O., Berthold, C. H., and Conradi, S. (1979). Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons. III. Motoneurons innervating fast-twitch (type FR) units of the gastrocnemius muscle. J. Comp. Neurol. 184, 755–767. doi: 10.1002/cne.901840408
Kellerth, J. O., Conradi, S., and Berthold, C. H. (1983). Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons: V. Motoneurons innverating fast-twitch (type FF) units of the gastrocnemius muscle. J. Comp. Neurol. 214, 451–458. doi: 10.1002/cne.902140409
Kernell, D. (2006). The Motoneurone and Its Muscle Fibres. Oxford: Oxford University Press. doi: 10.1093/acprof:oso/9780198526551.001.0001
Kerns, J. M., and Hinsman, E. J. (1973). Neuroglial response to sciatic neurectomy. II. Electron microscopy. J. Comp. Neurol. 151, 255–280. doi: 10.1002/cne.901510304
Khan, I., Osaka, H., Stanislaus, S., Calvo, R. M., Deerinck, T., Yaksh, T. L.,et al. (2003). Nicotinic acetylcholine receptor distribution in relation to spinal neurotransmission pathways. J. Comp. Neurol. 467, 44–59. doi: 10.1002/cne.10913
Kim, F. J., Kovalyshyn, I., Burgman, M., Neilan, C., Chien, C. C., and Pasternak, G. W. (2010). Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol. Pharmacol. 77, 695–703. doi: 10.1124/mol.109.057083
Kohler, M., Hirschberg, B., Bond, C. T., Kinzie, J. M., Marrion, N. V., Maylie, J.,et al. (1996). Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273, 1709–1714. doi: 10.1126/science.273.5282.1709
Kong, X. Y., Wienecke, J., Chen, M., Hultborn, H., and Zhang, M. (2011). The time course of serotonin 2A receptor expression after spinal transection of rats: an immunohistochemical study. Neuroscience 177, 114–126. doi: 10.1016/j.neuroscience.2010.12.062
Kong, X. Y., Wienecke, J., Hultborn, H., and Zhang, M. (2010). Robust upregulation of serotonin 2A receptors after chronic spinal transection of rats: an immunohistochemical study. Brain Res. 1320, 60–68. doi: 10.1016/j.brainres.2010.01.030
Kuno, M., Miyata, Y., and Munoz-Martinez, E. J. (1974a). Differential reaction of fast and slow alpha-motoneurones to axotomy. J. Physiol. 240, 725–739.
Kuno, M., Miyata, Y., and Munoz-Martinez, E. J. (1974b). Properties of fast and slow alpha motoneurones following motor reinnervation. J. Physiol. 242, 273–288.
Ladle, D. R., Pecho-Vrieseling, E., and Arber, S. (2007). Assembly of motor circuits in the spinal cord: driven to function by genetic and experience-dependent mechanisms. Neuron 56, 270–283. doi: 10.1016/j.neuron.2007.09.026
Lagerback, P. A. (1985). An ultrastructural study of cat lumbosacral gamma-motoneurons after retrograde labelling with horseradish peroxidase. J. Comp. Neurol. 240, 256–264. doi: 10.1002/cne.902400304
Lagerback, P. A., Cullheim, S., and Ulfhake, B. (1986). Electron microscopic observations on the synaptology of cat sciatic gamma-motoneurons after intracellular staining with horseradish peroxidase. Neurosci. Lett. 70, 23–27. doi: 10.1016/0304-3940(86)90431-3
Lagerback, P. A., and Ronnevi, L. O. (1982). An ultrastructural study of serially sectioned Renshaw cells. II. Synaptic types. Brain Res. 246, 181–192. doi: 10.1016/0006-8993(82)91166-0
Lagerback, P. A., Ronnevi, L. O., Cullheim, S., and Kellerth, J. O. (1981). An ultrastructural study of the synaptic contacts of alpha-motoneurone axon collaterals. I. Contacts in lamina IX and with identified alpha-motoneurone dendrites in lamina VII. Brain Res. 207, 247–266. doi: 10.1016/0006-8993(81)90363-2
Lanuza, G. M., Gosgnach, S., Pierani, A., Jessell, T. M., and Goulding, M. (2004). Genetic identification of spinal interneurons that coordinate left–right locomotor activity necessary for walking movements. Neuron 42, 375–386. doi: 10.1016/S0896-6273(04)00249-1
Lape, R., and Nistri, A. (2000). Current and voltage clamp studies of the spike medium afterhyperpolarization of hypoglossal motoneurons in a rat brain stem slice preparation. J. Neurophysiol. 83, 2987–2995.
Leedy, M. G., Bresnahan, J. C., Mawe, G. M., and Beattie, M. S. (1988). Differences in synaptic inputs to preganglionic neurons in the dorsal and lateral band subdivisions of the cat sacral parasympathetic nucleus. J. Comp. Neurol. 268, 84–90. doi: 10.1002/cne.902680109
Lewis, P. R., and Shute, C. C. (1966). The distribution of cholinesterase in cholinergic neurons demonstrated with the electron microscope. J. Cell Sci. 1, 381–390.
Li, W., Ochalski, P. A., Brimijoin, S., Jordan, L. M., and Nagy, J. I. (1995). C-terminals on motoneurons: electron microscope localization of cholinergic markers in adult rats and antibody-induced depletion in neonates. Neuroscience 65, 879–891. doi: 10.1016/0306-4522(94)00511-3
Li, X., and Bennett, D. J. (2007). Apamin-sensitive calcium-activated potassium currents (SK) are activated by persistent calcium currents in rat motoneurons. J. Neurophysiol. 97, 3314–3330. doi: 10.1152/jn.01068.2006
Li, Y., and Bennett, D. J. (2003). Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J. Neurophysiol. 90, 857–869. doi: 10.1152/jn.00236.2003
Lioudyno, M., Hiel, H., Kong, J. H., Katz, E., Waldman, E., Parameshwaran-Iyer, S.,et al. (2004). A “synaptoplasmic cistern” mediates rapid inhibition of cochlear hair cells. J. Neurosci. 24, 11160–11164. doi: 10.1523/JNEUROSCI.3674-04.2004
Liu, P. W., and Bean, B. P. (2014). Kv2 Channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J. Neurosci. 34, 4991–5002. doi: 10.1523/JNEUROSCI.1925-13.2014
Maingret, F., Coste, B., Hao, J., Giamarchi, A., Allen, D., Crest, M.,et al. (2008). Neurotransmitter modulation of small-conductance Ca2+-activated K+ channels by regulation of Ca2+ gating. Neuron 59, 439–449. doi: 10.1016/j.neuron.2008.05.026
Manuel, M., Iglesias, C., Donnet, M., Leroy, F., Heckman, C. J., and Zytnicki, D. (2009). Fast kinetics, high-frequency oscillations, and subprimary firing range in adult mouse spinal motoneurons. J. Neurosci. 29, 11246–11256. doi: 10.1523/JNEUROSCI.3260-09.2009
Manuel, M., Li, Y., Elbasiouny, S. M., Murray, K., Griener, A., Heckman, C. J.,et al. (2012). NMDA induces persistent inward and outward currents that cause rhythmic bursting in adult rodent motoneurons. J. Neurophysiol. 108, 2991–2998. doi: 10.1152/jn.00518.2012
Manuel, M., Zytnicki, D., and Meunier, C. (2014). The dendritic location of the L-type current and its deactivation by the somatic AHP current both contribute to firing bistability in motoneurons. Front. Comput. Neurosci. 8:4. doi: 10.3389/fncom.2014.00004
Mavlyutov, T. A., Epstein, M. L., Andersen, K. A., Ziskind-Conhaim, L., and Ruoho, A. E. (2010). The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience 167, 247–255. doi: 10.1016/j.neuroscience.2010.02.022
Mavlyutov, T. A., Epstein, M. L., Liu, P., Verbny, Y. I., Ziskind-Conhaim, L., and Ruoho, A. E. (2012). Development of the sigma-1 receptor in C-terminals of motoneurons and colocalization with the N,N′-dimethyltryptamine forming enzyme, indole-N-methyl transferase. Neuroscience 206, 60–68. doi: 10.1016/j.neuroscience.2011.12.040
Mawe, G. M., Bresnahan, J. C., and Beattie, M. S. (1986). A light and electron microscopic analysis of the sacral parasympathetic nucleus after labelling primary afferent and efferent elements with HRP. J. Comp. Neurol. 250, 33–57. doi: 10.1002/cne.902500104
Maxwell, D. J., Bannatyne, B. A., Fyffe, R. E., and Brown, A. G. (1982). Ultrastructure of hair follicle afferent fibre terminations in the spinal cord of the cat. J. Neurocytol. 11, 571–582. doi: 10.1007/BF01262425
Maxwell, D. J., Bannatyne, B. A., Fyffe, R. E., and Brown, A. G. (1984). Fine structure of primary afferent axon terminals projecting from rapidly adapting mechanoreceptors of the toe and foot pads of the cat. Q. J. Exp. Physiol. 69, 381–392.
McLaughlin, B. J. (1972a). Dorsal root projections to the motor nuclei in the cat spinal cord. J. Comp. Neurol. 144, 461–474. doi: 10.1002/cne.901440405
McLaughlin, B. J. (1972b). The fine structure of neurons and synapses in the motor nuclei of the cat spinal cord. J. Comp. Neurol. 144, 429–460. doi: 10.1002/cne.901440404
McLaughlin, B. J. (1972c). Propriospinal and supraspinal projections to the motor nuclei in the cat spinal cord. J. Comp. Neurol. 144, 475–500. doi: 10.1002/cne.901440406
Miles, G. B., Hartley, R., Todd, A. J., and Brownstone, R. M. (2007). Spinal cholinergic interneurons regulate the excitability of motoneurons during locomotion. Proc. Natl. Acad. Sci. U.S.A. 104, 2448–2453. doi: 10.1073/pnas.0611134104
Miles, G. B., and Sillar, K. T. (2011). Neuromodulation of vertebrate locomotor control networks. Physiology (Bethesda) 26, 393–411. doi: 10.1152/physiol.00013.2011
Miri, A., Azim, E., and Jessell, T. M. (2013). Edging toward entelechy in motor control. Neuron 80, 827–834. doi: 10.1016/j.neuron.2013.10.049
Misonou, H. (2010). Homeostatic regulation of neuronal excitability by K(+) channels in normal and diseased brains. Neuroscientist 16, 51–64. doi: 10.1177/1073858409341085
Misonou, H., Mohapatra, D. P., Menegola, M., and Trimmer, J. S. (2005). Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J. Neurosci. 25, 11184–11193. doi: 10.1523/JNEUROSCI.3370-05.2005
Misonou, H., Mohapatra, D. P., Park, E. W., Leung, V., Zhen, D., Misonou, K.,et al. (2004). Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 7, 711–718. doi: 10.1038/nn1260
Mohapatra, D. P., Misonou, H., Pan, S. J., Held, J. E., Surmeier, D. J., and Trimmer, J. S. (2009). Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin) 3, 46–56. doi: 10.4161/chan.3.1.7655
Mohapatra, D. P., and Trimmer, J. S. (2006). The Kv2.1 C terminus can autonomously transfer Kv2.1-like phosphorylation-dependent localization, voltage-dependent gating, and muscarinic modulation to diverse Kv channels. J. Neurosci. 26, 685–695. doi: 10.1523/JNEUROSCI.4620-05.2006
Monster, A. W., and Chan, H. (1977). Isometric force production by motor units of extensor digitorum communis muscle in man. J. Neurophysiol. 40, 1432–1443.
Moran-Rivard, L., Kagawa, T., Saueressig, H., Gross, M. K., Burrill, J., and Goulding, M. (2001). Evx1 is a postmitotic determinant of v0 interneuron identity in the spinal cord. Neuron 29, 385–399. doi: 10.1016/S0896-6273(01)00213-6
Muennich, E. A., and Fyffe, R. E. (2004). Focal aggregation of voltage-gated, Kv2.1 subunit-containing, potassium channels at synaptic sites in rat spinal motoneurones. J. Physiol. 554, 673–685. doi: 10.1113/jphysiol.2003.056192
Murakoshi, H., Shi, G., Scannevin, R. H., and Trimmer, J. S. (1997). Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol. Pharmacol. 52, 821–828. doi: 10.1124/mol.52.5.821
Murray, K. C., Nakae, A., Stephens, M. J., Rank, M., D’amico, J., Harvey, P. J.,et al. (2010). Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT2C receptors. Nat. Med. 16, 694–700. doi: 10.1038/nm.2160
Murray, K. C., Stephens, M. J., Ballou, E. W., Heckman, C. J., and Bennett, D. J. (2011). Motoneuron excitability and muscle spasms are regulated by 5-HT2B and 5-HT2C receptor activity. J. Neurophysiol. 105, 731–748. doi: 10.1152/jn.00774.2010
Nagao, M., Misawa, H., Kato, S., and Hirai, S. (1998). Loss of cholinergic synapses on the spinal motor neurons of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 57, 329–333. doi: 10.1097/00005072-199804000-00004
Nagy, J. I., Yamamoto, T., and Jordan, L. M. (1993). Evidence for the cholinergic nature of C-terminals associated with subsurface cisterns in alpha-motoneurons of rat. Synapse 15, 17–32. doi: 10.1002/syn.890150103
Norton, J. A., Bennett, D. J., Knash, M. E., Murray, K. C., and Gorassini, M. A. (2008). Changes in sensory-evoked synaptic activation of motoneurons after spinal cord injury in man. Brain 131, 1478–1491. doi: 10.1093/brain/awn050
O’Connell, K. M., Loftus, R., and Tamkun, M. M. (2010). Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc. Natl. Acad. Sci. U.S.A. 107, 12351–12356. doi: 10.1073/pnas.1003028107
Park, K. S., Mohapatra, D. P., Misonou, H., and Trimmer, J. S. (2006). Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science 313, 976–979. doi: 10.1126/science.1124254
Phelps, P. E., Barber, R. P., Houser, C. R., Crawford, G. D., Salvaterra, P. M., and Vaughn, J. E. (1984). Postnatal development of neurons containing choline acetyltransferase in rat spinal cord: an immunocytochemical study. J. Comp. Neurol. 229, 347–361. doi: 10.1002/cne.902290306
Pierani, A., Moran-Rivard, L., Sunshine, M. J., Littman, D. R., Goulding, M., and Jessell, T. M. (2001). Control of interneuron fate in the developing spinal cord by the progenitor homeodomain protein Dbx1. Neuron 29, 367–384. doi: 10.1016/S0896-6273(01)00212-4
Piotrkiewicz, M., and Hausmanowa-Petrusewicz, I. (2011). Motoneuron afterhyperpolarisation duration in amyotrophic lateral sclerosis. J. Physiol. 589, 2745–2754. doi: 10.1113/jphysiol.2011.204891
Powers, R. K., Nardelli, P., and Cope, T. C. (2008). Estimation of the contribution of intrinsic currents to motoneuron firing based on paired motoneuron discharge records in the decerebrate cat. J. Neurophysiol. 100, 292–303. doi: 10.1152/jn.90296.2008
Prather, J. F., Nardelli, P., Nakanishi, S. T., Ross, K. T., Nichols, T. R., Pinter, M. J.,et al. (2011). Recovery of proprioceptive feedback from nerve crush. J. Physiol. 589, 4935–4947. doi: 10.1113/jphysiol.2011.210518
Pullen, A. H. (1988a). Feline ‘C’-type terminals possess synaptic sites associated with a hypolemmal cistern and Nissl body. Neurosci. Lett. 84, 143–148. doi: 10.1016/0304-3940(88)90398-9
Pullen, A. H. (1988b). Quantitative synaptology of feline motoneurones to external anal sphincter muscle. J. Comp. Neurol. 269, 414–424. doi: 10.1002/cne.902690308
Pullen, A. H. (1992). Presynaptic terminal loss from alpha-motoneurones following the retrograde axonal transport of diphtheria toxin. Acta Neuropathol. 83, 488–498. doi: 10.1007/BF00310025
Pullen, A. H., and Athanasiou, D. (2009). Increase in presynaptic territory of C-terminals on lumbar motoneurons of G93A SOD1 mice during disease progression. Eur. J. Neurosci. 29, 551–561. doi: 10.1111/j.1460-9568.2008.06602.x
Pullen, A. H., and Sears, T. A. (1978). Modification of “C” synapses following partial central deafferentation of thoracic motoneurones. Brain Res. 145, 141–146. doi: 10.1016/0006-8993(78)90802-8
Pullen, A. H., and Sears, T. A. (1983). Trophism between C-type axon terminals and thoracic motoneurones in the cat. J. Physiol. 337, 373–388.
Ralston, H. J., and Ralston, D. D. (1979). Identification of dorsal root synaptic terminals on monkey ventral horn cells by electron microscopic autoradiography. J. Neurocytol. 8, 151–166. doi: 10.1007/BF01175558
Ren, Y., Barnwell, L. F., Alexander, J. C., Lubin, F. D., Adelman, J. P., Pfaffinger, P. J.,et al. (2006). Regulation of surface localization of the small conductance Ca2+-activated potassium channel, Sk2, through direct phosphorylation by cAMP-dependent protein kinase. J. Biol. Chem. 281, 11769–11779. doi: 10.1074/jbc.M513125200
Romer, S., Deardorff, A., Ahmed, S., Schwieterman, R., and Fyffe, R. (2012). “Alterations in the ion channels SK3, HCN-1 and Kv2.1 in rodent lumbar alpha-motoneurons following peripheral axotomy,” in Society for Neuroscience: 42nd Annual Meeting, New Orleans, LA.
Romer, S. H., Dominguez, K. M., Gelpi, M. W., Deardorff, A. S., Tracy, R. C., and Fyffe, R. E. (2014). Redistribution of Kv2.1 ion channels on spinal motoneurons following peripheral nerve injury. Brain Res. 1547, 1–15. doi: 10.1016/j.brainres.2013.12.012
Rosenbluth, J. (1962). Subsurface cisterns and their relationship to the neuronal plasma membrane. J. Cell Biol. 13, 405–421. doi: 10.1083/jcb.13.3.405
Sakamoto, H., Ohsato, K., and Atsumi, S. (1985). Acetylcholinesterase activity at the synapses of presynaptic boutons with presumed alpha-motoneurons in chicken ventral horn. Light- and electron-microscopic studies. Histochemistry 83, 291–298. doi: 10.1007/BF00684373
Satoh, K., Armstrong, D. M., and Fibiger, H. C. (1983). A comparison of the distribution of central cholinergic neurons as demonstrated by acetylcholinesterase pharmacohistochemistry and choline acetyltransferase immunohistochemistry. Brain Res. Bull. 11, 693–720. doi: 10.1016/0361-9230(83)90013-8
Saxena, S., Roselli, F., Singh, K., Leptien, K., Julien, J. P., Gros-Louis, F.,et al. (2013). Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96. doi: 10.1016/j.neuron.2013.07.027
Schroder, H. D. (1979). Paramembranous densities of ‘C’ terminal-motoneuron synapses in the spinal cord of the rat. J. Neurocytol. 8, 47–52. doi: 10.1007/BF01206457
Shapiro, M. S., Loose, M. D., Hamilton, S. E., Nathanson, N. M., Gomeza, J., Wess, J.,et al. (1999). Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca(2+) channels by using knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 10899–10904. doi: 10.1073/pnas.96.19.10899
Siembab, V. C., Smith, C. A., Zagoraiou, L., Berrocal, M. C., Mentis, G. Z., and Alvarez, F. J. (2010). Target selection of proprioceptive and motor axon synapses on neonatal V1-derived Ia inhibitory interneurons and Renshaw cells. J. Comp. Neurol. 518, 4675–4701. doi: 10.1002/cne.22441
Skinner, H. J., Girling, K. J., Whitehurst, A., and Nathanson, M. H. (1999). Influence of metoclopramide on plasma cholinesterase and duration of action of mivacurium. Br. J. Anaesth. 82, 542–545. doi: 10.1093/bja/82.4.542
Stepien, A. E., Tripodi, M., and Arber, S. (2010). Monosynaptic rabies virus reveals premotor network organization and synaptic specificity of cholinergic partition cells. Neuron 68, 456–472. doi: 10.1016/j.neuron.2010.10.019
Stewart, A. E., Yan, Z., Surmeier, D. J., and Foehring, R. C. (1999). Muscarine modulates Ca2+ channel currents in rat sensorimotor pyramidal cells via two distinct pathways. J. Neurophysiol. 81, 72–84.
Sumner, B. E. (1975). A quantitative study of subsurface cisterns and their relationships in normal and axotomized hypoglossal neurones. Exp. Brain Res. 22, 175–183. doi: 10.1007/BF00237687
Surmeier, D. J., and Foehring, R. (2004). A mechanism for homeostatic plasticity. Nat. Neurosci. 7, 691–692. doi: 10.1038/nn0704-691
Tredici, G., Pizzini, G., and Milanesi, S. (1976). The ultrastructure of the nucleus of the oculomotor nerve (somatic efferent portion) of the cat. Anat. Embryol. (Berl.) 149, 323–346. doi: 10.1007/BF00315448
Turkin, V. V., O’Neill, D., Jung, R., Iarkov, A., and Hamm, T. M. (2010). Characteristics and organization of discharge properties in rat hindlimb motoneurons. J. Neurophysiol. 104, 1549–1565. doi: 10.1152/jn.00379.2010
Umemiya, M., and Berger, A. J. (1994). Properties and function of low- and high-voltage-activated Ca2+ channels in hypoglossal motoneurons. J. Neurosci. 14, 5652–5660.
Van Harreveld, A., and Khattab, F. I. (1967). Electron microscopy of asphyxiated spinal cords of cats. J. Neuropathol. Exp. Neurol. 26, 521–536. doi: 10.1097/00005072-196710000-00001
Viana, F., Bayliss, D. A., and Berger, A. J. (1993). Multiple potassium conductances and their role in action potential repolarization and repetitive firing behavior of neonatal rat hypoglossal motoneurons. J. Neurophysiol. 69, 2150–2163.
Vilaro, M. T., Wiederhold, K. H., Palacios, J. M., and Mengod, G. (1992). Muscarinic M2 receptor mRNA expression and receptor binding in cholinergic and non-cholinergic cells in the rat brain: a correlative study using in situ hybridization histochemistry and receptor autoradiography. Neuroscience 47, 367–393. doi: 10.1016/0306-4522(92)90253-X
Walker, G., and Bourguignon, L. Y. (1990). Membrane-associated 41-kDa GTP-binding protein in collagen-induced platelet activation. FASEB J. 4, 2925–2933.
Walmsley, B., Alvarez, F. J., and Fyffe, R. E. (1998). Diversity of structure and function at mammalian central synapses. Trends Neurosci. 21, 81–88. doi: 10.1016/S0166-2236(97)01170-3
Wetts, R., and Vaughn, J. E. (2001). Development of cholinergic terminals around rat spinal motor neurons and their potential relationship to developmental cell death. J. Comp. Neurol. 435, 171–183. doi: 10.1002/cne.1200
Wilson, J. M., Rempel, J., and Brownstone, R. M. (2004). Postnatal development of cholinergic synapses on mouse spinal motoneurons. J. Comp. Neurol. 474, 13–23. doi: 10.1002/cne.20089
Witts, E. C., Zagoraiou, L., and Miles, G. B. (2014). Anatomy and function of cholinergic C bouton inputs to motor neurons. J. Anat. 224, 52–60. doi: 10.1111/joa.12063
Wyckoff, R. W., and Young, J. Z. (1956). The motorneuron surface. Proc. R. Soc. Lond. B Biol. Sci. 144, 440–450. doi: 10.1098/rspb.1956.0002
Xia, X. M., Fakler, B., Rivard, A., Wayman, G., Johnson-Pais, T., Keen, J. E.,et al. (1998). Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507. doi: 10.1038/26758
Yamamoto, T., Hertzberg, E. L., and Nagy, J. I. (1990). Epitopes of gap junctional proteins localized to neuronal subsurface cisterns. Brain Res. 527, 135–139. doi: 10.1016/0006-8993(90)91071-N
Yamamoto, T., Hertzberg, E. L., and Nagy, J. I. (1991). Subsurface cisterns in alpha-motoneurons of the rat and cat: immunohistochemical detection with antibodies against connexin32. Synapse 8, 119–136. doi: 10.1002/syn.890080206
Zagoraiou, L., Akay, T., Martin, J. F., Brownstone, R. M., Jessell, T. M., and Miles, G. B. (2009). A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron 64, 645–662. doi: 10.1016/j.neuron.2009.10.017
Zampieri, N., Jessell, T. M., and Murray, A. J. (2014). Mapping sensory circuits by anterograde transsynaptic transfer of recombinant rabies virus. Neuron 81, 766–778. doi: 10.1016/j.neuron.2013.12.033
Zhang, J., Lanuza, G. M., Britz, O., Wang, Z., Siembab, V. C., Zhang, Y.,et al. (2014). V1 and v2b interneurons secure the alternating flexor–extensor motor activity mice require for limbed locomotion. Neuron 82, 138–150. doi: 10.1016/j.neuron.2014.02.013
Zhang, L., and Krnjevic, K. (1987). Apamin depresses selectively the after-hyperpolarization of cat spinal motoneurons. Neurosci. Lett. 74, 58–62. doi: 10.1016/0304-3940(87)90051-6
Keywords: SK, Kv2.1, α-motoneuron, acetylcholine, C-boutons, afterhyperpolarization, subsurface cistern
Citation: Deardorff AS, Romer SH, Sonner PM and Fyffe REW (2014) Swimming against the tide: investigations of the C-bouton synapse. Front. Neural Circuits 8:106. doi: 10.3389/fncir.2014.00106
Received: 30 April 2014; Paper pending published: 23 June 2014;
Accepted: 17 August 2014; Published online: 18 September 2014.
Edited by:
Hans Hultborn, University of Copenhagen, DenmarkReviewed by:
C. J. Heckman, Northwestern University, USAClaire Francesca Meehan, University of Copenhagen, Denmark
Copyright © 2014 Deardorff, Romer, Sonner and Fyffe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert E. W. Fyffe, Boonshoft School of Medicine, Department of Neuroscience, Cell Biology and Physiology, Wright State University, 202A University Hall, 3640 Col. Glenn Highway, Dayton, OH 45435, USA e-mail:cm9iZXJ0LmZ5ZmZlQHdyaWdodC5lZHU=
†Adam S. Deardorff and Shannon H. Romer have contributed equally to this work.