 Hung-Yun Lin1,2
Hung-Yun Lin1,2 Faith B. Davis1
Faith B. Davis1 Mary K. Luidens1,3 Shaker A. Mousa1,2 James H. Cao1,2
Mary K. Luidens1,3 Shaker A. Mousa1,2 James H. Cao1,2 Min Zhou1,4
Min Zhou1,4 Paul J. Davis1,2,3*
Paul J. Davis1,2,3*- 1 Ordway Signal Transduction, Albany, NY, USA
- 2 Albany College of Pharmacy and Health Sciences, Albany, NY, USA
- 3 Albany Medical College, Albany, NY, USA
- 4 Ohio State University College of Medicine, Columbus, OH, USA
The pathophysiology of brain damage that is common to ischemia–reperfusion injury and brain trauma include disodered neuronal and glial cell energetics, intracellular acidosis, calcium toxicity, extracellular excitotoxic glutamate accumulation, and dysfunction of the cytoskeleton and endoplasmic reticulum. The principal thyroid hormones, 3,5,3′-triiodo-L-thyronine (T3) and L-thyroxine (T4), have non-genomic and genomic actions that are relevant to repair of certain features of the pathophysiology of brain damage. The hormone can non-genomically repair intracellular H+ accumulation by stimulation of the Na+/H+ exchanger and can support desirably low [Ca2+]i.c. by activation of plasma membrane Ca2+–ATPase. Thyroid hormone non-genomically stimulates astrocyte glutamate uptake, an action that protects both glial cells and neurons. The hormone supports the integrity of the microfilament cytoskeleton by its effect on actin. Several proteins linked to thyroid hormone action are also neuroprotective. For example, the hormone stimulates expression of the seladin-1 gene whose gene product is anti-apoptotic and is potentially protective in the setting of neurodegeneration. Transthyretin (TTR) is a serum transport protein for T4 that is important to blood–brain barrier transfer of the hormone and TTR also has been found to be neuroprotective in the setting of ischemia. Finally, the interesting thyronamine derivatives of T4 have been shown to protect against ischemic brain damage through their ability to induce hypothermia in the intact organism. Thus, thyroid hormone or hormone derivatives have experimental promise as neuroprotective agents.
Introduction
The critical contributions of thyroid hormone to development of the nervous system (Farwell et al., 2006; Bernal, 2007), to mature and immature brain glial cell (Siegrist-Kaiser et al., 1990; Farwell et al., 1995, 2006), and neuronal function (Yonkers and Ribera, 2008; Zhou et al., 2011) and to behavior (Davis et al., 2010) are well-known. While hypothyroidism has been reported by some observers to be neuroprotective in the setting of ischemia (Shuaib et al., 1994a; Alevizaki et al., 2006), repeated administration of L-thyroxine (T4) has also been shown to protect against brain ischemia (Rami and Krieglstein, 1992) and thyroid hormone is known to support the integrity of brain vasculature (Schlenker et al., 2008). Iodothyronines may support by non-genomic or genomic molecular mechanisms support the activities of plasma membrane ion pumps or channels that are important to normal excitable cell function. These pumps that are stimulated by 3, 5, 3′-triiodo-L-thyronine (T3) or, in some cases, by T4 include the Na+/H+ exchanger (Incerpi et al., 1998), Na, K-ATPase (Lei et al., 2004, 2006), and Ca2+-ATPases (Davis et al., 1983a,b; Mylotte et al., 1985) and the channels include the inward rectifier K+ channel (Sakaguchi et al., 1996) and the Na+ current (Yonkers and Ribera, 2008; Zhou et al., 2011). A contribution to normalizing intracellular pH in the setting of excess H+ accumulation (Incerpi et al., 1998) that occurs with ischemia/acidosis is obviously a desirable action of the hormone, as would be stimulation of Na, K-ATPase that would maintain normal [Na+]i.c. in the face of enhanced proton-for-Na+ exchange. Thyroid hormone can also increase glial cell glutamate uptake (Mendes-de-Aguiar et al., 2008), thus protecting neurons and astrocytes against the toxicity of glutamate at the cell surface.
In this review, we will briefly describe the molecular basis for several actions of thyroid hormone analogs that can now be interpreted as neuroprotective. Most of these actions are non-genomic in mechanism, that is, they do not rely primarily on the interaction of T3 with nuclear thyroid hormone receptors (TR proteins) and consequent specific gene transcription (Cheng et al., 2010). The hormones are T3, T4, and the thyronamines (Piehl et al., 2011). We will also comment on the apparently neuroprotective action of a serum thyroid hormone transport protein, transthyretin (TTR; Santos et al., 2010).
Glutamate Neurotoxicity and Thyroid Hormone
Increases in extraneuronal glutamate neurotransmitter concentration stimulate nerve cell glutamate receptors and can result in excitotoxicity and neuronal death (Danbolt, 2001; Mendes-de-Aguiar et al., 2008). Via several high affinity glutamate-specific transporters, astrocytes take up glutamate from the extracellular matrix environment and are a defense against neuronal excitotoxicity. Within the cell, glutamate supports several metabolic cascades, including energy production and antioxidant (glutathione) functions. The brain damage that results from ischemia is mediated to an important degree by excessive glutamate release from neurons and glial cells (see review by Mongin, 2007), resulting in excessive depolarization and accumulation of cytoplasmic Ca2+. In addition to glutamate and calcium toxicity, other factors contributing to neuronal damage and death in ischemia include free radical and nitric oxide accumulations and dysfunction of mitochondria and endoplasmic reticulum (Guo et al., 2009).
Against this background, studies by Mendes-de-Aguiar et al. (2008) reported that T3 at 10−8 M eliminated the “gliotoxic” effect of glutamate on cultured cerebellar astrocytes from newborn rats and ensured viability of astrocytes and of neurons co-cultured with the astrocytes. The mechanism involved in these observations included increased astrocyte uptake of glutamate that was attributable to increased cellular abundance of GLT-1 and GLAST mRNAs and protein. GLT-1 and GLAST are the principal plasma membrane glutamate transporters of astrocytes. Losi et al. (2008) showed that T3 protected rat hippocampal neurons against glutamate toxicity by a non-genomic mechanism). Thus, the defense against glutamate toxicity in neurons and glial cells that is imposed by T3 may be both genomic and non-genomic in mechanism. The existence of seamless interfaces between thyroid hormone-initiated non-genomic and genomic actions has recently been appreciated (Lin et al., 2009; Cheng et al., 2010; Davis et al., 2011).
Given the contributions of excessive glutamate accumulation extraneuronally to ischemic brain injury, brain concussive injury (Yi and Hazell, 2006; Jennings et al., 2008), heavy metal toxicity (Struzyriska, 2009), and a variety of other brain damage syndromes, there is a rationale for investigating the feasibility of modulating glutamate toxicity with thyroid hormone analogs. Depending upon the pathological setting and model, the scheduling of hormone administration may be acute or subacute. It may be useful to quantitate the proportion of the hormonal effect in models that is non-genomic in initiation and thus subject to management with T3 that has been reformulated to act at the cell surface (Lin et al., 2011).
Hypothyroidism and Neuroprotection; Thyroid Hormone Administration and Neuroprotection
Because both hypothyroidism and thyroid hormone administration have been reported to be protective in the setting of brain ischemia, it is important to specify the experimental conditions under which these actions have been described. Experimental hypothyroidism (Shuaib et al., 1994a; Rastogi et al., 2006) and subclinical hypothyroidism (Alevizaki et al., 2006; Akhoundi et al., 2011) may result in apparently improved neurological outcomes from brain ischemia, that is, preservation of function. The experimental hypothyroidism observations have been collected in several animal models of transient cerebral ischemia in which the state of established hypothyroidism has either reduced neuronal death (Shuaib et al., 1994a; Rastogi et al., 2006) or has delayed, but did not prevent, neuronal death (Lee et al., 2010). The rationales for an apparently protective effect of thyroid hypofunction established prior to neurological insult include decreased metabolic demands of nerve cells, decreased oxidative stress (Rastogi et al., 2006), decreased excitatory glutamate production (Shuaib et al., 1994b), and, possibly, decreased body temperature. Decreased glutamate production per se can be viewed as neuroprotective in the sense of attenuated excitotoxicity (see below), however, it may not be desirable when it impairs antioxidant glutathione production (Mendes-de-Aguiar et al., 2008). It is presently impractical clinically to induce hypothyroidism acutely, although, as will be described below, the thyronamine thyroid hormone analogs can induce hypothermia quickly and neuroprotection may be afforded by this measure.
Reduction of hippocampal neuronal damage from ischemia with repeated daily T4 administration was shown in the rat 20 years ago by Rami and Krieglstein (1992), with approximately a 50% increase in neuronal density attributable to hormone treatment. Subsequently, it was shown that experimental hyperthyroidism that is established before focal ischemia may increase neuronal insult in the rat (Rastogi et al., 2008). Thus, any neuronal rescue that may occur with thyroid hormone administration must avoid induction of hypermetabolism. The molecular basis for an acute effect of thyroid hormone isoforms on post-ischemic neuronal survival may involve on genomic actions of the hormone on intracellular pH, Na+, and Ca2+, as suggested above. T3 has been shown to stimulate Na+/H+ (NHE1) exchanger/transporter activity in excitable cells and to enhance recovery of intracellular pH after an acid load (Incerpi et al., 1998). The mechanism of increased exchanger activity includes thyroid hormone-dependent activation of mitogen-activated protein kinase (MAPK; D’Arezzo et al., 2004). The ischemic neuron has accumulated H+ and thus enhanced exchanger pump activity is desirable from the standpoint of pH. The theoretical price that is paid for increased Na+/H+ activity is the import of Na+, the consequent possibility of reverse Na+/Ca2+ exchange (NSCE) and an undesirable increase in [Ca2+]i.c.. However, T3 also stimulates Na, K-ATPase activity (Lei et al., 2004, 2006), and increases the number of pump subunits inserted in the plasma membrane. This effect acts to reduce [Na+]i.c.. Finally, a number of studies have shown iodothyronines activate Ca2+-ATPase activity (Davis et al., 1983a; Mylotte et al., 1985) by a calmodulin-dependent mechanism (Davis et al., 1983b); this non-genomic effect contributes to modulation of any increase in intracellular Ca2+ that may occur.
Given that iodothyronines can increase certain membrane ion pump activities, it seems paradoxical that overall neuronal excitability – spike number and action potentials – might be decreased by the hormone. We have already described the inhibitory effect of T3 on glutamate excitotoxicity that affects neurons and appears to be mediated by glial cells. In addition, Cao et al. (2011) have recently reported a non-genomic effect of T4 to decrease the excitability of pyramidal neurons of prefrontal cortex slices. While this is potentially an energy-conserving mechanism at a time of hypoxic stress, the observation requires confirmation and studies of mechanism, since thyroid hormone is known increase certain membrane ion currents (Sakaguchi et al., 1996; Yonkers and Ribera, 2008; Zhou et al., 2011).
In summary, the decreased metabolic demands of the hypothyroid brain may reduce the impact of ischemia. However, acute imposition of hypothyroidism in the clinical context of CNS ischemia is impractical. Protection of intraneuronal or glial pH, and [Ca2+]i.c. and suppression of glutamate neuroexcitation (see below) with a short-term, non-hyperthyroid increase in iodothyronines appears to deserve further experimental evaluation in euthyroid animal models of ischemia/stroke. Certain of the neuroprotective actions of thyroid hormone are depicted in Figure 1.
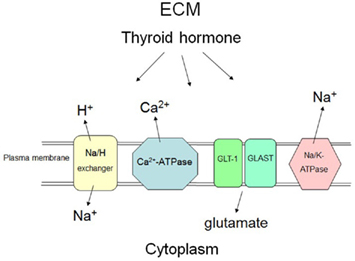
Figure 1. Selected mechanisms at the plasma membrane by which thyroid hormone may non-genomically or genomically have protective effects on neurons and glial cells in the setting of acute brain ischemia. In response to thyroid hormone (T3), astrocytes and neurons may take up glutamate from the extracellular apace via increased activity of specific transporters (GLAST and GLT-1), reducing neuroexcitation fostered by glutamate at the neuronal surface. The hormone may also increase the number of glutamate transporters in the plasma membrane. Thyroid hormone (T3) increases activity of the Na/H exchanger, reducing the intracellular acid load that results from ischemia. Sodium pump (Na, K-ATPase) activity is enhanced by T3 and there is increased expression of the Na, K-ATPase gene in response to iodothyronines. Export of Na+ by the pump is relevant because of the increased activity of the Na/H exchanger results in import of Na+. An increase in [Na+]i.c. results in an undesirable increase in cell uptake of Ca2+ by sodium–calcium exchange (not shown in the figure). Enhanced activity of the calcium pump (Ca2+-ATPase) caused by thyroid hormone (T4 and T3) also protects against intracellular accumulation of Ca2+. ECM, extracellular matrix.
Seladin-1 and Amyloid-β Protein Precursor and Thyroid Hormone
The SELective AD Indicator-1 (seladin-1) gene codes for a novel protective factor that has been seen to be deficient in Alzheimer’s disease (AD; Greeve et al., 2000; Peri and Serio, 2008). Among several effects, the protein inhibits pro-apoptotic caspase 3. The seladin-1 protein also has enzymatic activity as a hydroxysterol reductase that converts desmosterol to cholesterol. Thyroid hormone, particularly T3, promotes the expression of seladin-1 in neuronal precursor cells, such as human mesenchymal stem cells and fetal neuroepithelial cells (Benvenuti et al., 2008). Thus, the actions of thyroid hormone on the seladin-1 axis are relevant to brain cell development/differentiation and appear to protect neuron precursor cells from programmed cell death. The critical contributions of thyroid hormone to embryonic-fetal brain development are very well-recognized and now may include contributions from seladin-1 gene expression. The optimistic possibility may also exist that seladin-1 is a factor that supports a pool of multipotent CNS cells beyond the period of brain development and that might be accessed in the settings of certain brain disorders.
In addition to stimulating expression of seladin-1, T3 is known to suppress expression of the β-amyloid precursor protein (APP) gene in neuroblastoma cells in vitro (Belandia et al., 1998). The hormone had been reported earlier by the same group to have complex and variable effects on APP gene splicing (Latasa et al., 1998). Tan and Vasan (2009) have recently reviewed a substantial literature on thyroid function in the setting of AD and have speculated that suboptimal iodothyronine concentrations in the CNS may support AβPP accumulation and the products thereof, It is not clear whether the amyloid-β-peptide and amyloid-β-protein are etiologic or secondary factors in a complex pattern of pathogenesis in AD. However, the possibility that thyroid hormone has a neuroprotective effect in the context of neurodegeneration would appear to warrant additional experimental consideration.
Such a consideration also raises the issue of transport of thyroid hormone isoforms from blood into brain tissue. At least in part, such intercompartmental transport involves TTR, a serum transport protein for T4 and for tetraiodothyroacetic acid (tetrac); TTR is discussed below as a possible neuroprotective factor in ischemia. In addition, and emphasizing the complexity of the relationship of TTR to neuroprotection and neurodegenertation, TTR has been reported to have apparently contradictory actions in the mouse model of AD, in that it may enhance cleavage of the Aβ-peptide (Costa et al. (2006)) and accelerate vascular deposition of Aβ protein (Wati et al., 2009). This controversy requires resolution.
Transthyretin in Brain Ischemia
Transthyretin, is a blood and CSF protein that contributes to maintaining intravascular pools of T4 and of retinol. As noted above, TTR is also responsible for facilitating access of thyroid hormone to brain. TTR is secreted by the liver and by choroid plexus. It is involved in amyloid deposition. Because brain TTR gene expression is increased in response to injury/ischemia, Santos et al. (2010) recently searched for a possible contribution of TTR to neuroprotection. The investigators were surprised to find that the brain of the TTR null mouse did not exhibit increased vulnerability to ischemic damage. However, when a mouse heterozygous for heat-shock transcription factor 1 (HSF1) – and thus exhibiting a somewhat impaired stress response was rendered TTR−/−, middle cerebral artery occlusion induced a profoundly severe injury in terms of volume of brain cortex infarct, cerebral edema, and white blood cell immigration. The increase in severity of damage was expressed relative to that in the TTR+/+ HSF1+/− control animals. Interestingly, TTR, itself, was distributed throughout the area of infarction in the TTR−/− HSF1+/− mouse brain. Thus, TTR can be neuroprotective, but this is apparent only when a second stress or defense factor is impaired and such is the case in the HSF1+/− mouse brain in the setting of ischemia. In these studies, it was CSF-source TTR that was important to neuroprotection, since inhibition by RNAi of liver TTR production did not affect the model.
The results emphasize that there may be redundancy in neuroprotective responses that are based on proteins such as TTR and HSF1 and this has implications for experimental design. The study does not provide insight into possible contributions of thyroid hormone-binding or retinol-binding (or retinol-binding globulin) to the behavior of TTR in the neuroprotective response. This issue is of some relevance, given the preceding discussion of neuroprotective actions of thyroid hormone isoforms, themselves, on local accumulations of glutamate or on intracellular pH of glial cells or neurons.
Thyronamines, Hypothermia, and Neuroprotection
Thyronamines are thyroid hormone derivatives whose side chain has been decarboxylated. Two thyronamines – the completely deiodinated product (T0AM) and a derivative that retains the 3-iodine (T1AM) – have been exploited for several experimental purposes by Scanlan and colleagues (Piehl et al., 2011) and are of particular interest with regard to brain protection. The compounds occur naturally in small quantities and are ligands for a G protein-coupled receptor (TAAR1), rather than for TRs. One of their remarkable effects is that of lowering body temperature without inducing homeostatic responses. Because of the known protective effect of hypothermia on brain damage associated with ischemia, the thyronamines have been tested by Doyle et al. (2007) against stroke injury. Administered intraperitoneally 1 h after middle cerebral artery occlusion, the agents promptly lowered mouse body temperature to 31°C from 37°C. The thyronamines significantly reduced brain infarct volume at 24 h by about 33%. The specificity of the neuroprotective impact of hormone analog-induced hypothermia was shown by prevention of the neuronal salvage by maintaining animal body temperature at 37°C during administration of thyronamines. The induction of hypothermia by the thyronamines is presumed to be mediated by the TAAR1 receptor and the nerve cell salvage is thought to reflect decreased metabolic rate and lowered free radical generation.
The apparent effectiveness of the agents in ischemia-induced brain injury does not exclude consideration of the thyronamines for application to other models of CNS injury, for example, concussive trauma and global anoxic damage at the time of cardiac arrest. It is clear that systemic administration of the thyronamines permits their trespass of the blood–brain barrier, but it is not known by what precise mechanism the barrier is crossed. The thyronamines do not appear to gain access to the interior of cells by the principal thyroid hormone uptake system, monocarboyxlate transporter 8 (MCT8; Kinne et al., 2010).
Possibly Protective Effects of T4 on Glial Cell Cytoskeleton and Intracellular Protein Trafficking
Thyroid hormone, notably T4, has important actions on the actin cytoskeleton that were first described in glial cells in 1990 (Siegrist-Kaiser et al., 1990). Interestingly, reverse T3, but not T3, also stabilizes the cytoskeleton (Farwell et al., 2006). Under the direction of thyroid hormone, soluble actin is converted to F-actin in a variety of cells, a function that stabilizes microfilaments, enabling cell motility, and the interaction of cells with extracellular proteins. The latter include laminins that support adhesion to the basal lamina (basement membrane) and growth factors that are neuroprotective, such as basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF; Hicks et al., 1998). Dissolution of the cytoskeleton is one phase of ischemic neuronal cell death (Lipton, 1999) and thus, thyroid hormone may be a factor that desirably stabilizes cell and tissue structure in the face of ischemic challenge to structure. Iodothyronines have been shown to enhance activities of EGF (Lin et al., 1999) and bFGF (Davis et al., 2004).
The cytoskeleton is also important to cell protein trafficking, a process whose maintenance is imperative in response to ischemia/hypoxia. Thyroid hormone has very substantial effects on the intracellular trafficking of kinases, nuclear hormone receptors (cytoplasm-to-nucleus), and a variety of transactivators (Cao et al., 2009; Davis et al., 2011). The energetics of the hypoxic cell may be impaired and this may limit the activation of MAPK and phosphatidylinositol 3-kinase by iodothyronines (Lin et al., 2009; Davis et al., 2011) that is critical to trafficking.
Conclusion
Thyroid hormone has a broad spectrum of actions on the nervous system. These relate to brain development and morphogenesis and to a variety of brain functions and to behaviors of the intact organism. The molecular basis for such actions include modulation of expression of specific genes, regulation of energetics in the mitochondrion and a variety of non-genomic actions on ion transport systems, on the cytoskeleton, intracellular protein trafficking, on specific kinase activation and specific protein phosphorylation, and on cell migration and angiogenesis; these multiple actions have been reviewed by Cheng et al. (2010) and Davis et al. (2011). That thyroid hormone may be neuroprotective has been explored in this review. What is attractive about the apparently neuroprotective effect of T3 and T4 is that they coherently affect multiple defense mechanisms that can be invoked in brain damage settings. The possibility has also been discussed that thyroid hormone-relevant proteins such as TTR may be defensive in the setting of ischemia/reperfusion injury. Finally, the concept that thyroid hormone analogs such as the thyronamines are relevant to neuroprotection should encourage exploration of additional forms of the hormone for desirable effects on the nervous system at risk for injury.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are the beneficiaries of the support of a research endowment established by M. Frank and Margaret D. Rudy.
References
Akhoundi, F. H., Ghorbani, A., Soltani, A., and Meysamie, A. (2011). Favorable functional out-comes in acute ischemic stroke patients with subclinical hypothyroidism. Neurology 77, 349–354.
Alevizaki, M., Synetou, M., Xynos, K., Alevizaki, C. C., and Vemmos, K. N. (2006). Hypothyroidism as a protective factor in acute stroke patients. Clin. Endocrinol. (Oxf) 65, 369–372.
Belandia, B., Latasa, M. J., Villa, A., and Pascual, A. (1998). Thyroid hormone negatively regulates the transcriptional activity of the β-amyloid precursor protein gene. J. Biol. Chem. 273, 30366–30371.
Benvenuti, S., Luciani, P., Cellai, I., Deledda, C., Baglioni, S., Saccardi, R., Urbani, S., Francini, F., Squecco, R., Giuliani, C., Vennelli, G. B., Serio, M., Pinchera, A., and Peri, A. (2008). Thyroid hormone promote cell differentiation and up-regulate the expression of the seladin-1 gene in in vitro models of human neuronal precursors. Exp. Neurol. 197, 437–446.
Bernal, J. (2007). Thyroid hormone receptors in brain development and function. Nat. Clin. Pract. Endocrinol. Metab. 3, 249–259.
Cao, H. J., Lin, H. Y., Luidens, M. K., Davis, F. B., and Davis, P. J. (2009). Cytoplasm-to-nucleus shuttling of thyroid hormone receptor β1 (TRβ1) is directed from a plasma membrane integrin receptor by thyroid hormone. Endocr. Res. 34, 31–42.
Cao, H. J., Pan, J., Lin, H. Y., Davis, F. B., Zhou, M., and Davis, P. J. (2011). L-Thyroxine attenuates pyramidal neuron excitability in rat acute prefrontal cortex slices. Immunol. Endocr. Metab. Agents Med. Chem. 11, 152–156.
Cheng, S. Y., Leonard, J. L., and Davis, P. J. (2010). Molecular aspects of thyroid hormone actions. Endocr. Rev. 31, 139–170.
Costa, R., Goncalves, A., Saraiva, M. J., and Cardoso, I. (2006). Transthyretin binding to A-β peptide – impact on A-β fibrillogenesis and toxicity. FEBS Lett. 582, 936–942.
D’Arezzo, S., Incerpi, S., Davis, F. B., Acconcia, F., Marino, M., Farias, R. N., and Davis, P. J. (2004). Rapid nongenomic effects of 3, 5, 3′-triiodo-L-thyronine on the intracellular pH of L-6 myoblasts are mediated by intracellular calcium mobilization and kinase pathways. Endocrinology 145, 5694–5703.
Davis, F. B., Cody, V., Davis, P. J., Borzynski, L. J., and Blas, S. D. (1983a). Stimulation by thyroid hormone analogues of red blood cell Ca2+-ATPase activity in vitro. Correlations between structure, and biological activity in a human cell system. J. Biol. Chem. 258, 12373–12377.
Davis, F. B., Davis, P. J., and Blas, S. D. (1983b). Role of calmodulin in thyroid hormone stimulation in vitro of human erythrocyte Ca2+-ATPase activity. J. Clin. Invest. 71, 579–586.
Davis, F. B., Mousa, S. A., O’Connor, L., Mohamed, S., Lin, H. Y., Cao, H. J., and Davis, P. J. (2004). Proangiogenic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Circ. Res. 94, 1500–1506.
Davis, P. J., Davis, F. B., Mousa, S. A., Luidens, M. K., and Lin, H.-Y. (2011). Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu. Rev. Pharmacol. Toxicol. 51, 99–115.
Davis, P. J., Zhou, M., Davis, F. B., Lansing, L., Mousa, S. A., and Lin, H. Y. (2010). Mini-review: cell surface receptor for thyroid hormone and nongenomic regulation of ion fluxes in excitable cells. Physiol. Behav. 99, 237–239.
Doyle, K. P., Suchland, K. L., Ciesielski, T. M., Lessov, N. S., Grandy, D. K., Scanlan, T. S., and Stenzel-Poore, M. P. (2007). Novel thyroxine derivatives, thyronamine and 3-iodo-thyronamine, induce transient hypothermia and marked neuroprotection against stroke injury. Stroke 38, 2569–2576.
Farwell, A., Dubord-Tomasetti, S. A., Pietrzykowski, A. Z., and Leonard, J. L. (2006). Dynamic nongenomic actions of thyroid hormone in the developing brain. Endocrinology 147, 2567–2574.
Farwell, A., Tranter, M. P., and Leonard, J. L. (1995). Thyroxine-dependent regulation of integrin-laminin interactions in astrocytes. Endocrinology 136, 3909–3915.
Greeve, I., Hermans-Borgmayer, I., Brellinger, C., Kasper, D., Gomez-Isla, T., Behl, C., Levkau, B., and Nilsch, R. M. (2000). The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. J. Neurosci. 20, 7345–7352.
Guo, Z. H., Li, F., and Wang, W. Z. (2009). The mechanisms of brain ischemic insult and poten tial protective interventions. Neurosci. Bull. 25, 139–152.
Hicks, D., Heidinger, V., Mohand-Said, S., Sahel, J., and Dreyfus, H. (1998). Growth factors and gangliosides as neuroprotective agents in excitotoxicity and ischemia. Gen. Pharmacol. 30, 265–273.
Incerpi, S., Luly, P., De Vito, P., and Farias, R. N. (1998). Short-term effects of thyroid hormone on the Na/H antiport in L-6 myoblasts: high molecular specificity for 3, 3′, 5-triiodo-L-thyronine. Endocrinology 140, 683–689.
Jennings, J. S., Gerber, A. M., and Vallano, M. L. (2008). Pharmacological strategies for neuro protection in traumatic brain injury. Mini Rev. Med. Chem. 8, 689–701.
Kinne, A., Kleinau, G., Hoefig, C. S., Gruters, A., Köhrle, J., Kruase, G., and Schweizer, G. (2010). Essential molecular determinants for thyroid hormone transport and first structural implications for monocarboxylate transporter 8. J. Biol. Chem. 285, 28054–28063.
Latasa, M. J., Belandia, B., and Pascual, A. (1998). Thyroid hormones regulate β-amyloid gene splicing and protein secretion in neuroblastoma cells. Endocrinology 139, 2692–2698.
Lee, C. H., Yoo, K. Y., Hwang, I. K., Choi, J. H., Park, O. K., Li, H., Kang, I. J., Kwon, Y. G., Kim, Y. M., and Won, M. H. (2010). Hypothyroid state does not protect but delays neuronal death in the hippocampal CA1 region following transient cerebral ischemia: focus on oxidative stress and gliosis. J. Neurosci. Res. 88, 2661–2668.
Lei, J., Mariash, C. N., and Ingbar, D. H. (2004). 3, 3′, 5-Triiodo-L-thyronine regulation of Na, K-ATPase activity and cell surface expression in alveolar epithelial cells is Src kinase and phosphoinositide 3-kinase-dependent. J. Biol. Chem. 279, 47589–47600.
Lei, J., Mariash, C. N., and Ingbar, D. H. (2006). T3 increases Na, K-ATPase activity via a MAPK/ERK1/2-dependent pathway in rat adult alveolar cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L749–L754.
Lin, H. Y., Cody, V., Davis, F. B., Hercbergs, A. A., Luidens, M. K., Mousa, S. A., and Davis, P. J. (2011). Identification and function of the plasma membrane receptor for thyroid hormone analogues. Discov. Med. 11, 337–347.
Lin, H. Y., Shih, A., Davis, F. B., and Davis, P. J. (1999). Thyroid hormone promotes the phosphorylation of STAT3 and potentiates the action of epidermal growth factor in cultured cells. Biochem. J. 338, 427–432.
Lin, H. Y., Sun, M., Tang, H. Y., Simone, T. M., Wu, Y. H., Grandis, J. R., Cao, H. J., Davis, P. J., and Davis, F. B. (2009). L-Thyroxine vs. 3, 5, 3′-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am. J. Physiol. Cell Physiol. 296, C980–C991.
Losi, G., Garzon, G., and Puia, G. (2008). Nongenomic regulation of glutamatergic neurotrans mission in hippocampus by thyroid hormones. Neuroscience 151, 155–163.
Mendes-de-Aguiar, C. B., Alchini, R., Decker, H., Alvarez-Silva, M., Tasca, C. I., and Trentin, A. G. (2008). Thyroid hormone increases astrocyte glutamate uptake and protects astrocytes and neurons against glutamate toxicity. J. Neurosci. Res. 86, 3117–3125.
Mongin, A. A. (2007). Disruption of ionic and cell volume homeostasis in cerebral ischemia: the perfect storm. Pathophysiology 14, 183–193.
Mylotte, K. M., Cody, V., Davis, P. J., Davis, F. B., Blas, S. D., and Schoenl, M. (1985). Milrinone and thyroid hormone stimulate myocardial membrane Ca2+-ATPase activity and share structural homologies. Proc. Natl. Acad. Sci. U.S.A. 82, 7974–7978.
Peri, A., and Serio, M. (2008). Neuroprotective effects of the Alzheimer’s disease-related gene seladin-1. J. Mol. Endocrinol. 41, 251–261.
Piehl, S., Hoefig, C. S., Scanlan, T. S., and Kohrle, J. (2011). Thyronamines – past, present and future. Endocr. Rev. 32, 64–80.
Rami, A., and Krieglstein, J. (1992). Thyroxine attenuates hippocampal neuronal damage caused by ischemia in the rat. Life Sci. 50, 645–650.
Rastogi, L., Godbole, M. M., Ray, M., Rathore, P., Pradhan, S., Gupta, S. K., and Pandey, C. M. (2006). Reduction in oxidative stress and cell death explains hypothyroidism-induced neuroprotection subsequent to ischemia/reperfusion insult. Exp. Neurol. 200, 290–300.
Rastogi, L., Gupta, S., and Godbole, M. M. (2008). Pathophysiological basis for thyrotoxicosis as an aggravating factor in post-ischemic brain injury in rats. J. Endocrinol. 196, 335–341.
Sakaguchi, Y., Cui, G., and Sen, L. (1996). Acute effects of thyroid hormone on inward rectifier potassium channel currents in guinea pig ventricular myocytes. Endocrinology 137, 4744–4751.
Santos, S. D., Lambertsen, K. L., Clausen, B. H., Akinc, A., Alvarez, R., Finsen, B., and Saraiva, M. J. (2010). CSF transthyretin neuroprotection in a mouse model of brain ischemia. J. Neurochem. 115, 1434–1444.
Schlenker, E. H., Hora, M., Liu, Y., Redetzke, R. A., Morkin, E., and Gerdes, A. M. (2008). Effects of thyroidectomy, T4, and DITPA replacement on brain blood vessel density in adult rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1504–R1509.
Shuaib, A., Ijaz, S., Mazagri, R., Kalra, J., Hemmings, S., Senthilsvlvan, A., and Crosby, N. (1994a). Hypothyroidism protects the brain during transient forebrain ischemia in gerbils. Exp. Neurol. 127, 119–125.
Shuaib, A., Ijaz, S., Hemmings, S., Galazka, P., Ishaqzay, R., Liu, L., Ravindran, J., and Miyashita, H. (1994b). Decreased glutamate release during hypothyroidism may contribute to protection in cerebral ischemia. Exp. Neurol. 128, 260–265.
Siegrist-Kaiser, C. A., Juge-Aubry, C., Tranter, M. P., Ekenbarger, D. M., and Leonard, J. L. (1990). Thyroxine-dependent modulation of actin polymerization in cultured astrocytes. A novel extranuclear action of thyroid hormone. J. Biol. Chem. 265, 5296–5302.
Struzyriska, L. (2009). A glutamatergic component of lead toxicity in adult brain: the role of astrocytic glutamate transporters. Neurochem. Int. 55, 151–156.
Tan, Z. S., and Vasan, R. S. (2009). Thyroid function and Alzheimer’s disease. J. Alzheimers Dis. 16, 503–507.
Wati, H., Kawarabayashi, T., Matsubara, E., Kasai, A., Hirasawa, T., Kubota, T., Harigaya, Y., Shoji, M., and Maeda, S. (2009). Transthyretin accelerates vascular Aβ deposition in a mouse model of Alzheimer’s disease. Brain Pathol. 19, 48–57.
Yi, J. H., and Hazell, A. S. (2006). Excitotoxic mechanisms and the role of astrocyte glutamate transporters in traumatic brain injury. Neurochem. Int. 48, 394–403.
Yonkers, M. A., and Ribera, A. B. (2008). Sensory sodium current requires nongenomic actions of thyroid hormone during development. J. Neurophysiol. 100, 2719–2725.
Keywords: thyroid hormone, thyronamines, sodium-proton exchanger, calcium ATPase, seladin-1, transthyretin, ischemia–reperfusion injury
Citation: Lin H-Y, Davis FB, Luidens MK, Mousa SA, Cao JH, Zhou M and Davis PJ (2011) Molecular basis for certain neuroprotective effects of thyroid hormone. Front. Mol. Neurosci. 4:29. doi: 10.3389/fnmol.2011.00029
Received: 29 July 2011;
Paper pending published: 12 August 2011;
Accepted: 19 September 2011;
Published online: 14 October 2011.
Edited by:
Isabel Varela-Nieto, Consejo Superior Investigaciones Científicas, SpainReviewed by:
Beatriz Morte, Centro de Investigación Biomédica en Red de Enfermedades Raras, SpainAndréa Gonçalves Trentin, Universidade Federal de Santa Catarina, Brazil
Copyright: © 2011 Lin, Davis, Luidens, Mousa, Cao, Zhou and Davis. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Paul J. Davis, Ordway Signal Transduction, 150 New Scotland Avenue, Albany, NY 12208, USA. e-mail:cGRhdmlzQG9yZHdheXJlc2VhcmNoLm9yZw==