 Ariadna Recasens
Ariadna Recasens Celine Perier
Celine Perier Carolyn M. Sue1,2*
Carolyn M. Sue1,2*- 1Department of Neurogenetics, Kolling Institute, The Royal North Shore Hospital, Northern Sydney Local Health District, St. Leonards, NSW, Australia
- 2Northern Clinical School, Sydney Medical School, University of Sydney, Sydney, NSW, Australia
- 3Neurodegenerative Disease Laboratory, Vall d’Hebron Research Institute and Centre for Networked Biomedical Research on Neurodegenerative Diseases (CIBERNED), Barcelona, Spain
Growing evidence suggests that increased levels of α-synuclein might contribute to the pathogenesis of Parkinson’s disease (PD) and therefore, it is crucial to understand the mechanisms underlying α-synuclein expression. Recently, microRNAs (miRNAs) have emerged as key regulators of gene expression involved in several diseases such as PD and other neurodegenerative disorders. A systematic literature search was performed here to identify microRNAs that directly or indirectly impact in α-synuclein expression/accumulation and describe its mechanism of action. A total of 27 studies were incorporated in the review article showing evidences that six microRNAs directly bind and regulate α-synuclein expression while several miRNAs impact on α-synuclein expression indirectly by targeting other genes. In turn, α-synuclein overexpression also impacts miRNAs expression, indicating the complex network between miRNAs and α-synuclein. From the current knowledge on the central role of α-synuclein in PD pathogenesis/progression, miRNAs are likely to play a crucial role at different stages of PD and might potentially be considered as new PD therapeutic approaches.
Introduction
Growing evidence suggests that increased levels of α-synuclein are toxic and may contribute to the pathogenesis of Parkinson’s disease (PD). Supporting evidence includes: (i) duplications and triplication of the α-synuclein gene cause dominantly inherited PD, with a dose-correlation of α-synuclein load to the PD phenotype (Singleton et al., 2003; Ibáñez et al., 2004, 2009; Ahn et al., 2008; Ross et al., 2008); (ii) polymorphisms in α-synuclein promoters are associated with increased PD risk by enhancing α-synuclein expression (Chiba-Falek and Nussbaum, 2001; Touchman et al., 2001; Maraganore et al., 2006); (iii) increased α-synuclein mRNA levels are found in surviving dopaminergic (DA) neurons in the substantia nigra (SN) of idiopathic PD patients (Gründemann et al., 2008); (iv) induced pluripotent stem cells (iPS) from PD patients exhibited α-synuclein accumulation (Nguyen et al., 2011; Sánchez-Danés et al., 2012; Mazzulli et al., 2016); (v) α-synuclein is up-regulated in several in vivo PD models including 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mice and monkeys (Vila et al., 2000, 2001; Purisai et al., 2005); and (vi) overexpression of human wild-type and A53T mutant α-synuclein in rats and monkeys induced nigrostriatal degeneration (Kirik et al., 2002, 2003). In addition to PD, α-synuclein plays a key role in and other synucleinopathies such as dementia with Lewy bodies (DLB) and multiple system atrophy (MSA; Tagliafierro and Chiba-Falek, 2016).
MicroRNAs (miRNAs) are endogenous 17–24 base-pair (bp) single-stranded non-coding RNAs that have recently emerged as a key regulators of gene expression. Biogenesis of miRNAs, which are encoded within the genome as independent genomic transcription units or as introns of protein-coding genes, required a multi-step process that takes place in the nucleus and the cytoplasm (Figure 1; Meister and Tuschl, 2004; Ameres and Zamore, 2013; Catalanotto et al., 2016). First, miRNAs are transcribed in the nucleus by the RNA polymerase II as long primary miRNAs (pri-miRNAs) which are converted by the RNAse III enzyme Drosha into 60 bp stem-loop structures called pre-miRNAs. The pre-miRNAs are subsequently exported to the cytoplasm by the Exportin 5 via a nuclear pore. Once in the cytoplasm, the pre-miRNAs are further processed by a second RNase II enzyme called Dicer into a ~22nt miRNA-miRNA complex intermediate. Then, the RNA-duplex binds to an Argonaute (AGO) protein and one of the strands is removed resulting in the mature RNA-induced silencing complex (RISC). Finally, the RISC will bind to complementary mRNA sequence (seed matches) and repress their expression by: (i) translational repression via blocking translational initiation, poly(A) tail shortening or recruiting translation blockers; (ii) mRNA decay; or (iii) direct cleavage of target mRNAs by RISC (Ameres and Zamore, 2013). Via regulation of target genes, miRNAs are involved in several biological process including cell proliferation, differentiation, apoptosis, development, angiogenesis and immune response (Huang et al., 2011; Tüfekci et al., 2014a) and therefore, miRNAs dysregulation is associated with the pathogenesis of several human disease such as cancer, diabetes, autoimmune diseases, neurological disorders, diabetes and cardiovascular disease (Tüfekci et al., 2014b).
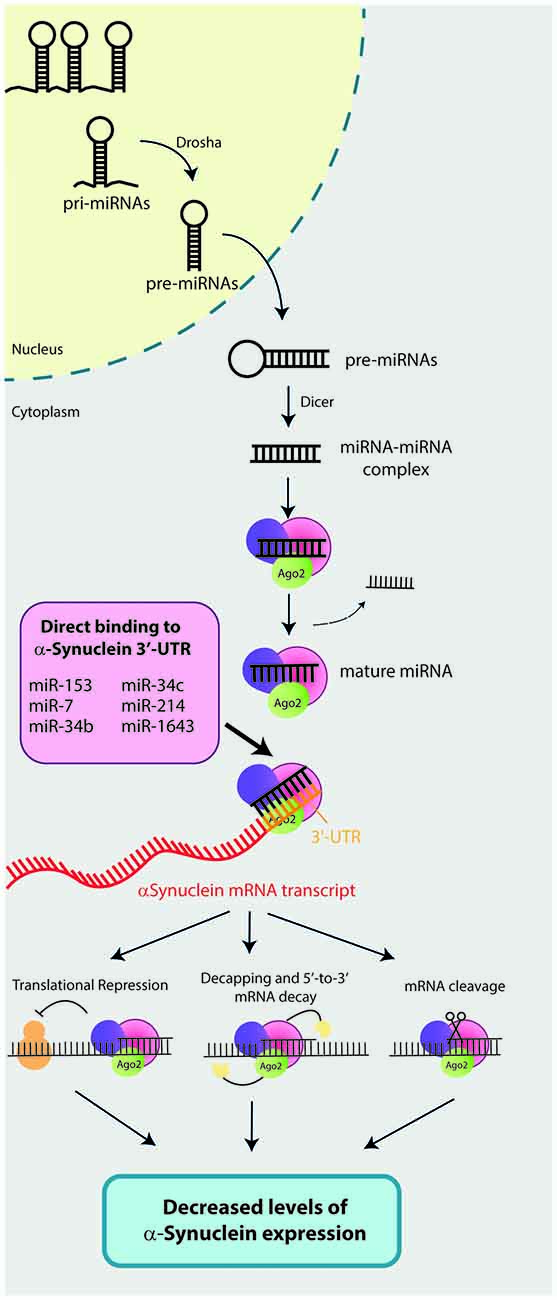
Figure 1. Mechanisms of action of microRNAs (miRNAs) that directly bind and regulate α-synuclein expression. Biogenesis of miRNAs required a multi-step process that takes place in the nucleus and the cytoplasm of the cells. First, miRNAs are transcribed in the nucleus by the RNA polymerase II as long primary miRNAs (pri-miRNAs). Then, a RNAse III enzyme called Drosha converts pri-miRNAs into 60 base-pair (bp) stem-loop structures (pre-miRNAs). Pre-miRNAs are subsequently exported to the cytoplasm by the Exportin 5, where a second RNase II enzyme called Dicer will process pre-miRNAs into a ~22nt miRNA-miRNA complex. The RNA-duplex binds to an Argonaute (AGO) protein and one of the strands is removed resulting in the mature RNA-induced silencing complex (RISC). Finally, RISC binds to complementary mRNA sequence (seed matches) and represses its expression by: (i) translational repression; (ii) mRNA decapping and decay; or (iii) direct cleavage of mRNAs target by RISC. To date, six miRNAs have been described to directly bind the 3′-untranslational region (UTR) of the α-synuclein mRNA transcript and repress its expression. These six miRNAs are: miR-7, miR-153, miR-34b, miR-34c, miR-214 and miR-1643.
Considering the importance of modulating α-synuclein levels in PD and other related disorders, the objective of this study is to review miRNAs that impact directly or indirectly in α-synuclein expression and describe their mechanisms of action.
Materials and Methods
Registration
Following PRISMA recommendations (Liberati et al., 2009; PRISMA Checklist available in Supplementary Materials), the systematic review was registered in The Joanna Bridge Institute (JBI) website with date 27th July 2016. Registration details are described in Supplementary Table 1.
Eligibility Criteria
All the studies selected for the review satisfied the PICOS selection criteria detailed in Supplementary Methods. No language or publication date restrictions were imposed.
Information Sources and Search
Three different database were used in this review article: PubMed, Scopus and Web of Science. The last search was run on 25th May 2016. No supplementary approaches were used to identify additional studies. Duplicated records were removed. The full electronic search strategy for each database is described in Table 1.

Table 1. Full electronic search strategy for each database used in the review article.
Study Selection
First, an over-inclusive screening by titles and abstracts was done to identify potential relevant studies. At this stage, irrelevant records, reviews, abstracts, editorials, letters, comments, perspective, reports, opinion and book chapter were removed. Full-text articles from the candidate studies were read and a second screening was done accordingly to the exclusion criteria detailed in Supplementary Methods.
Data Extraction
All the included studies were divided into two groups: (i) overexpressing studies: studies using α-synuclein overexpressing models (OEM); and (ii) standard studies. Relevant information from all included studies was extracted using two different extraction datasheets, depending on the category of the article (overexpressing vs. standard). See Supplementary Methods for detailed data extraction.
Results
Initially, a total of 353 publications were identified using three databases: Pubmed, Scopus and Web of Science (Figure 2). After duplicates removal, a total of 223 studies were screened by title and abstract and 61 potential relevant studies were selected for full-text review. A second screening was performed and 34 studies were discarded according to the following exclusion criteria: five were single nucleotide polymorphism (SNP) association studies with PD, another five were miRNA expression profile studies in PD patients, 12 studies did not investigate the impact on α-synuclein expression, two studies did not describe the impact of miRNAs expression, and 10 studies did not correlate miRNA and α-synuclein expression. Finally, 27 studies were included in the present review article, from which 12 investigations studied the effect of miRNA that directly bind and regulates α-synuclein expression, 10 studies focused on miRNAs that indirectly impact on α-synuclein expression and five studies used α-synuclein overexpressing in vivo models (Table 2).
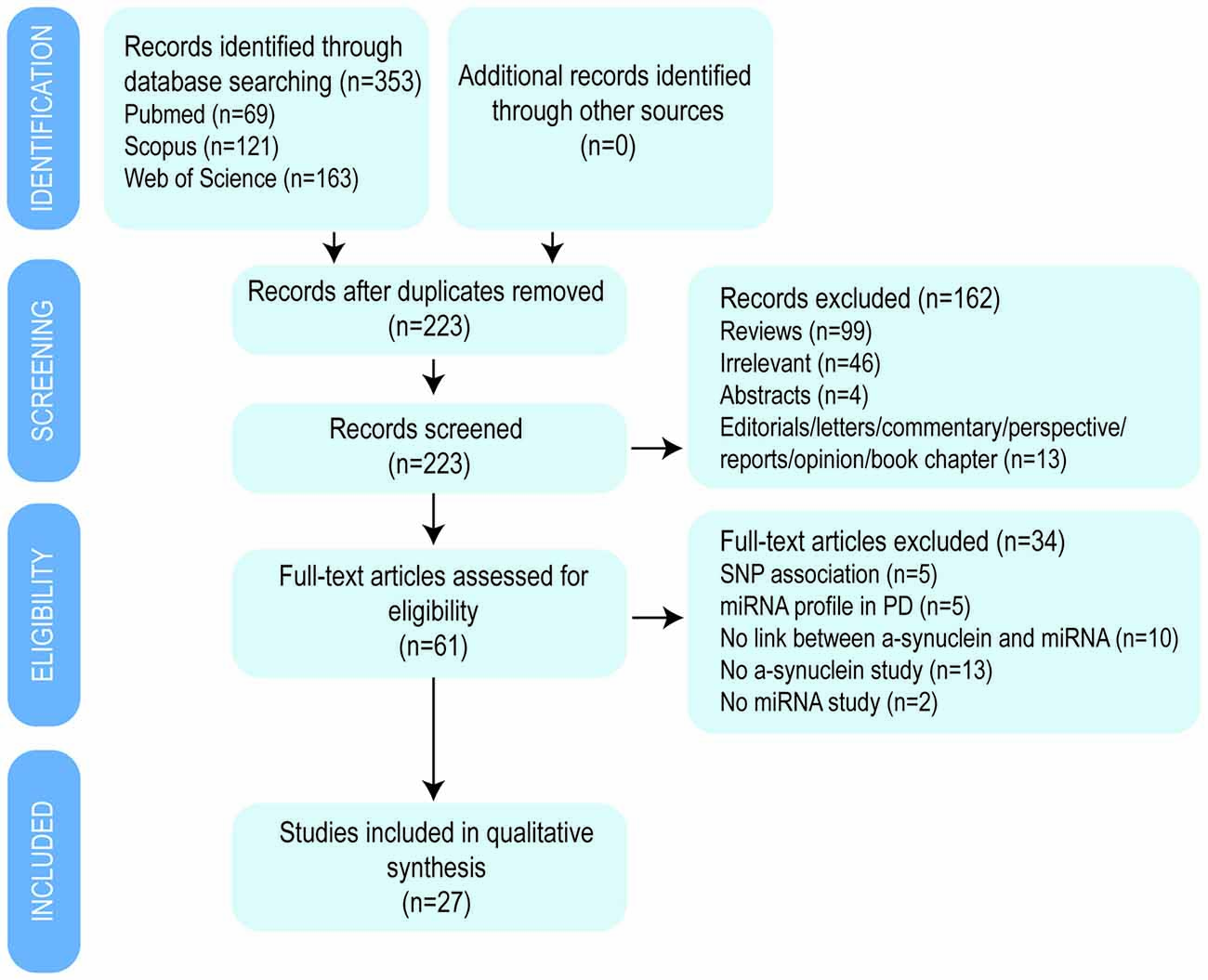
Figure 2. Flow diagram of the selection process by which the studies were included in the review article.

Table 2. Summary of studies included in the review article.
miRNAs that Directly Bind 3′-UTR α-Synuclein and Negatively Regulate α-Synuclein Expression
The results derived from all included studies demonstrated that a total of six miRNAs (miR-7, miR-153, miR-34b, miR-34c, miR-214 and miR-1643) directly bind to the 3′-untranslated region (UTR) of the α-synuclein mRNA transcript and negatively regulate its expression (Figure 1).
miR-7
Seven studies demonstrated the impact of miR-7 in α-synuclein expression (Junn et al., 2009; Doxakis, 2010; Choi et al., 2014; Fragkouli and Doxakis, 2014; Latreille et al., 2014; Fan et al., 2015; Zhou et al., 2016). From these studies, a total of three confirmed the direct binding of miR-7 to α-synuclein 3′-UTR sequence using luciferase reporter assays in three different in vitro models (SH-SY5Y, HEK293T and primary neurons; Supplementary Table 2). The specificity of the binding site was confirmed by introducing mutations in the α-synuclein 3′-UTR sequence that blocked the effect of miR-7 in the luciferase activity assay. The predicted binding site of miR-7 within the α-synuclein gene required to repress its expression is located at bases 119–217 of the α-synuclein 3′-UTR (Junn et al., 2009; Doxakis, 2010).
The direct effect of miR-7 in α-synuclein expression was first reported by Junn et al. (2009). In particular, transfection with 40 nM of premiR-7-2 in HEK293T cells resulted in a reduction of α-synuclein expression both at protein and mRNA levels. On the other hand, treatment with miR-7 inhibitors significantly increased the levels of α-synuclein protein in SH-SY5Y cells. The direct impact of miR-7 in α-synuclein expression was reproduced by Doxakis (2010) using both HEK293T cells and murine primary neurons.
One of the studies was focused on the role of miR-7 in pancreatic β-cell function (Latreille et al., 2014) and generated a miR-7 conditional knockout mice using Cre/Lox system (miR7a2fl/fl mice) which developed diabetes due to impaired insulin secretion and β cell differentiation. The direct impact of miR-7 in α-synuclein expression was confirmed in MIN6 cells and pancreatic islets obtained from miR7a2fl/fl mice. In particular, adenovirus-miR7a-mediated overexpression in MIN6 cells resulted in a reduction of α-synuclein transcript levels, while exposure to miR-7a inhibitors increased α-synuclein mRNA and protein levels. In addition, α-synuclein levels were increased in miR7a2fl/fl pancreatic islets. Interestingly, miR-7 played a role in insulin secretion by repressing the expression of α-synuclein which in turn modulated the granule fusion with the plasma membrane. These results are in line with the previous observation that α-synuclein, whose exact function still remains unknown, plays a role in neurotransmitter release via regulating the pool of vesicles available in the synaptic bouton and its fusion with the plasma membrane (Murphy et al., 2000; Cabin et al., 2002; Fernández-Chacón et al., 2004; Chandra et al., 2005; Larsen et al., 2006; Mazzulli et al., 2016).
The neuroprotective effect of miR-7 has been assessed under different conditions (Junn et al., 2009; Choi et al., 2014; Fragkouli and Doxakis, 2014; Fan et al., 2015). One of the studies investigated the protective effect of miR-7 in N20Y cells overexpressing mutant A53T α-synuclein challenged with hydrogen peroxide (H2O2). Notably, the presence of miR-7 reduced H2O2-induced cell death in A53T α-synuclein mutant cells (Junn et al., 2009). Additional protective effects of miR-7 against the MPTP-active metabolite 1-methyl-4-phenylpyridinium (MPP+) in vitro was investigated in two studies (Choi et al., 2014; Fragkouli and Doxakis, 2014). Both of them demonstrated that overexpression of miR-7 significantly increased cell viability after MPP+ treatment in SH-SY5Y cells, ReNcell VM cells and mouse primary neurons. One of the studies suggested that the protective effect of miR-7 against MPP+ is independent of α-synuclein repression, since knocking down α-synuclein in SH-SY5Y cells did not impact on miR-7-enhanced cell viability. This study rather suggested that miR-7 protected against MPP+-induced cell death by directly targeting the expression of RelA, a component of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) consequently relieving NF-κB suppression (Choi et al., 2014). On the other hand, Fragkouli and Doxakis (2014) suggested that miR-7 protects against MPP+-induced cell death by activating the mTOR pathway. Relevant to this context, SH-SY5Y cells treated with MPP+ and subchronic MPTP administration in mice resulted in a significant reduction of miR-7 expression in both models (Junn et al., 2009; Choi et al., 2014; Fragkouli and Doxakis, 2014). Two studies were focused on the protective effect of miR-7 against A53T mutant α-synuclein-induced toxicity (Fan et al., 2015; Zhou et al., 2016). Both studies concluded that miR-7 protects against PD-like degeneration by directly targeting nod-like receptor protein 3 (Nlrp3) expression and therefore modulating NLRP3 inflammasome activation. The protective effect of miR-7 in vivo was also assessed in the MPTP mice model (Zhou et al., 2016), in which the injection of miR-7 mimics into wild type mice treated with subacute MPTP dose rescued the loss of tyrosine hydroxylase (TH)-positive neuron number in the SN and dramatically inhibited Ionized calcium binding adaptor molecule 1 (Iba1) microglial activation via supressing NLRP3 inflammasome-mediated neuroinflammation. Further supporting the concept that miR-7 regulates α-synuclein expression in vivo, Song et al. (2012) reported that schizophrenia-like transgenic mice overexpressing heme oxygenas-1 (HO-1) protein in astrocytes exhibited decreased levels of miR-7 and increased α-synuclein levels in the SN/ventral tegmental area (VTA) at 48-weeks of age compare to control animals.
miR-153
A total of four studies investigated the impact of miR-153 in α-synuclein expression and its protective effect (Doxakis, 2010; Kim et al., 2013; Fragkouli and Doxakis, 2014; Lim and Song, 2014). Two studies investigated the combined effects of both miR-7, miR-153 and the combination of miR-7/153 (Doxakis, 2010; Fragkouli and Doxakis, 2014). One study predicted the binding site of miR-153 within the α-synuclein gene in the 442–448 bases of the α-synuclein 3′-UTR (Doxakis, 2010). The specificity of the predicted binding site for miR-153 was confirmed in vitro (Doxakis, 2010; Kim et al., 2013; Lim and Song, 2014) using luciferase assays and introducing mutations in the α-synuclein 3′-UTR. The direct effect of miR-153 in α-synuclein expression has now been studied in HEK293T cells. Cotransfection with an α-synuclein plasmid containing the 3′-UTR and miR-153 significantly reduced α-synuclein levels both at protein and mRNA level (Doxakis, 2010). The protective effect of miR-153 was also studied in embryonic murine neurons treated with MPP+. As reported with miR-7, overexpression of miR-153 in primary cortical neurons attenuated MPP+-induced neurotoxicity by upregulating the mTOR pathway (Fragkouli and Doxakis, 2014).
miR-34b and miR-34c
One study demonstrated that miR-34b and miR-34c directly targeted α-synuclein expression (Kabaria et al., 2015). Computational algorithms were used to predict two miR-34b and one miR-34c binding sites in the 3′-UTR of α-synuclein mRNA (Kabaria et al., 2015): miR-34b binding site #1: located between bases 528–549; miR-34b binding site #2: between bases 732–754; and miR-34c binding site: between bases 1149–1171. These bindings sites were verified by cotransfecting SH-SY5Y cells with a plasmid construct expressing α-synuclein 3′-UTR with miR-34b or miR-34c. Interestingly, the introduction of a polymorphic variation (rs10024743) which lies within the target site 1 of miR-34b significantly decreased the impact of miR-34b in the luciferase activity. As a consequence of the direct binding between miR-34b/miR-34c and α-synuclein, overexpression of miR-34b or miR-34c in SH-SY5Y cells resulted in significant reduction in α-synuclein mRNA and protein levels. Interestingly, miR-34b and miR-34c did not repress β-synuclein, but rather increased its expression by up to 2.3-fold. Moreover, inhibition of miR-34b and miR-34c increased α-synuclein mRNA and protein level as well as the formation of α-synuclein-containing aggregates in DA neurons.
miR-214
Only one study has demonstrated the direct impact of miR-214 in α-synuclein expression. Using luciferase assays in SH-SY5Y cells, miR-214 has been shown to directly target the α-synuclein 3′-UTR. In addition, miR-214 overexpression reduced α-synuclein expression both at mRNA and protein levels, while downregulation of miR-214 increased not only α-synuclein expression (mRNA and protein) but also the number of α-synuclein-aggregates in cells (Wang et al., 2015). This work also investigated whether the regulation of α-synuclein by miR-214 was the mechanism underlying the neuroprotective effect of Resveratrol. First, they showed that Resveratrol could ameliorate MPP+/MPTP-induced cell death both in vitro and in vivo. Interestingly, miR-214 inhibitors reversed the neuroprotective effect of resveratrol treatment in MPP+/MPTP models.
miR-1643
One study found that miR-1643 is a direct regulator of α-synuclein expression (Lim and Song, 2014). Luciferase assay in 293TF cells confirmed the direct binding of miR-1643 to α-synuclein 3′-UTR sequence.
miRNAs that Indirectly Impact on α-Synuclein Expression Without Binding to α-Synuclein 3′-UTR Sequence
In addition to miRNAs that directly bind and regulate α-synuclein protein, several miRNAs have been reported to indirectly regulate α-synuclein levels by targeting the expression of other genes. There are five different studies that report miRNAs that directly impact on proteolytic pathways and result in α-synuclein accumulation (Figure 3; Alvarez-Erviti et al., 2013; Decressac et al., 2013; Li et al., 2014; Zhang and Cheng, 2014; Su et al., 2016). In addition, miR-133b (Niu et al., 2016) and miR-433 (Wang et al., 2008; Schmitt et al., 2012) have been reported to impact on α-synuclein by directly targeting Ras homolog gene family, member A (RhoA) and fibroblast growth factor 20 (FGF20), respectively (Figure 4).
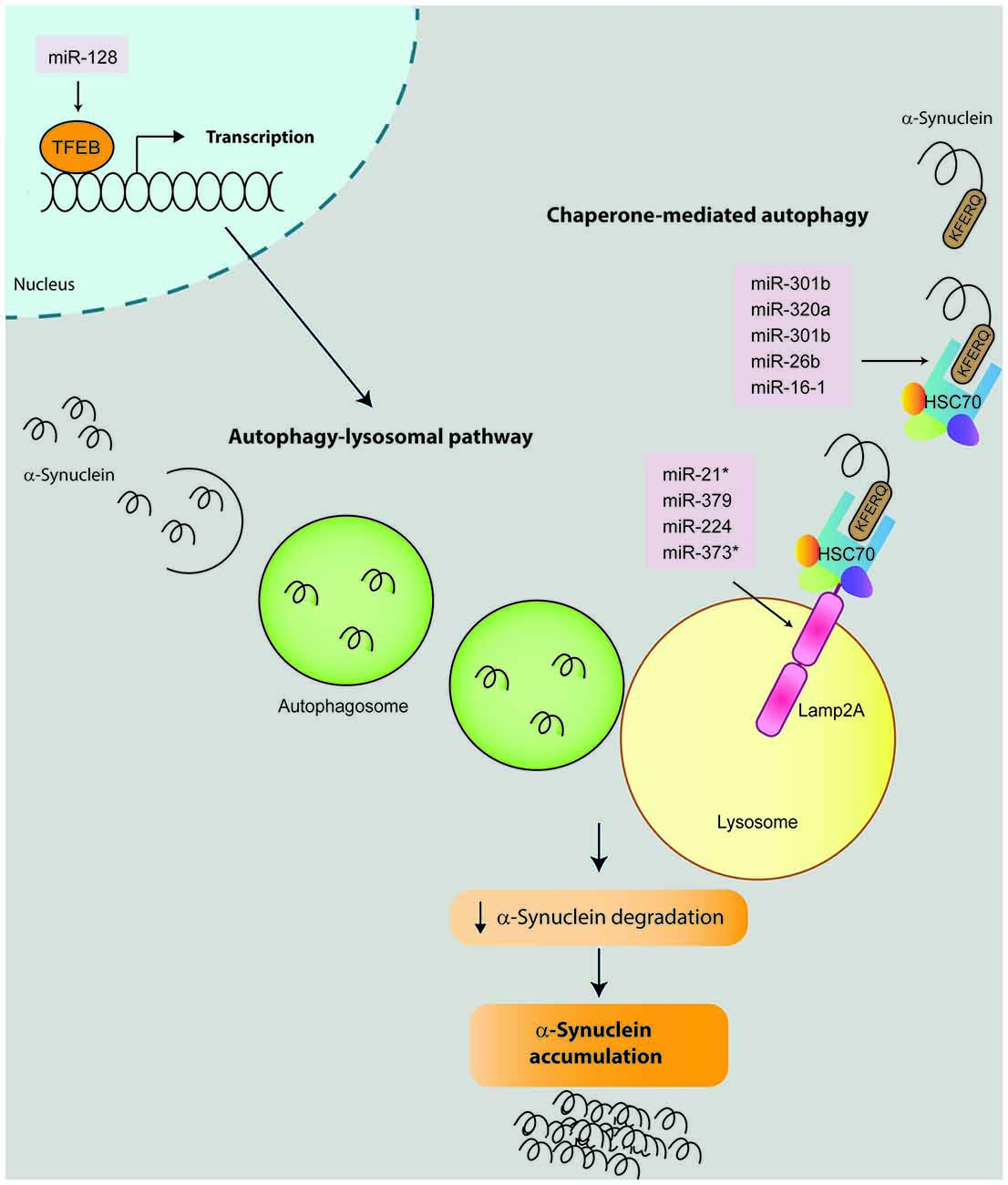
Figure 3. miRNAs that impact on α-synuclein expression by modulating proteolytic degradation pathways. α-Synuclein can be degraded by several proteolytic pathways including chaperone-mediated autophagy (CMA) and autophagy-lysosomal pathway (ALP). During the CMA, the KFREQ-like sequence of the α-synuclein protein is recognized by a chaperone complex which includes the Heat shock protein 70 (Hsc70). This complex is guided to the lysosomes and recognized by the Lysosome-associated membrane protein 2 (Lamp2A), which in turn translocate the α-synuclein into the lysosome where it is finally degraded by hydrolytic enzymes. To date, nine microRNAs have been described to modulate the CMA pathway and impact on α-synuclein degradation by directly binding and repressing the expression of Hsc70 (miR-301b, miR-26b, miR-320a, miR-106a and miR-16-1) or Lamp2a (miR-21*, miR-379, miR-373* and miR-224). For ALP degradation, α-synuclein is firstly enclosed into an autophagosome. Then the autophagosome is guided and fused with a lysosome where α-synuclein is finally degraded. In this context, miRNA-128 activates transcription factor EB (TFEB) which has been demonstrated to promote the transcription of genes involved in ALP pathway. Therefore miRNA-repression of Hsc7, Lamp2a or TFEB result in alterations in the α-synuclein degradation and its consequent accumulation.
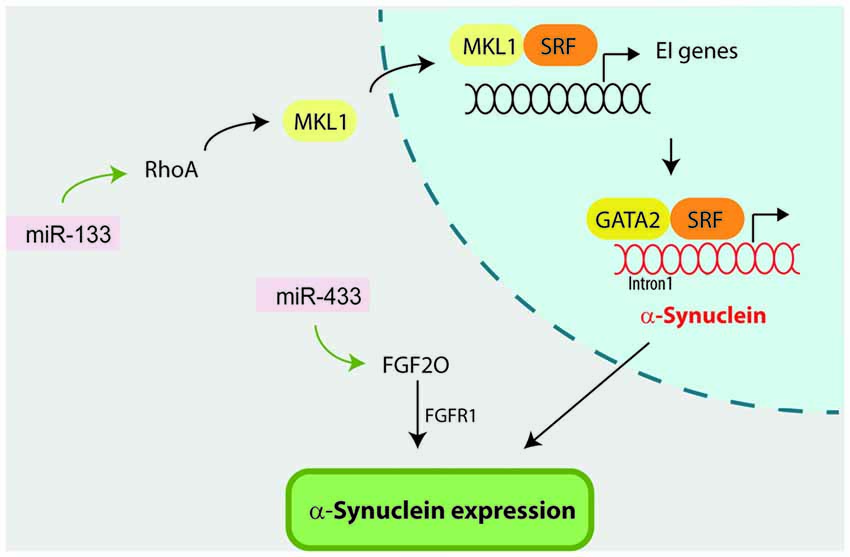
Figure 4. miR-133 and miR-433 indirectly influence α-synuclein by targeting Ras homolog gene family, member A (RhoA) and fibroblast growth factor 20 (FGF20) respectively. On one hand, miR-133 targets and regulates RhoA expression which has been previously reported to regulate α-synuclein expression. In this context, RhoA first activates megakaryoblastic leukemia 1 (MKL-1) factor, which in turn activates serum response element (SRF) transcription factor. MKL-1 and SRF activation promote the transcription of early immediate (EI) genes. Finally SRF forms a multiprotein complex with GATA-2 factor which regulates α-synuclein expression via occupancy at the intron-1. On the other hand, miR-433 directly targets FGF20, which has been suggested to directly regulate α-synuclein expression through the FGF-receptor 1 (FGFR1).
miRNAs, Proteolytic System and α-Synuclein
α-Synuclein turnover predominantly involves chaperone-mediated autophagy (CMA). Therefore, alterations in CMA result in pathological α-synuclein accumulation. Four studies have investigated how miRNA regulation of CMA influences α-synuclein accumulation (Alvarez-Erviti et al., 2013; Li et al., 2014; Zhang and Cheng, 2014; Su et al., 2016). Firstly, Alvarez-Erviti et al. (2013) transfected SH-SY5Y cells overexpressing α-synuclein with seven miRNAs that directly bind and negatively regulate two key proteins involved in CMA: Lysosome-associated membrane protein 2 (Lamp2a, hsa-miR-21*; hsa-miR-224; hsa-miR-373*; and hsa-miR-379) and Heat shock protein 70 (Hsc70, hsa-miR-26b: hsa-miR-106a*; and hsa-miR-301b). In addition to the expected reduction in Lamp2a and Hsc70 gene expression, transfection with the seven candidate miRNAs significantly increased α-synuclein protein levels. Notably, only two of them (miR-106a* and miR-301b) caused a significant decrease in α-synuclein mRNA levels. Interestingly, miR-106a* was predicted to target the 3′-UTR of α-synuclein although the direct binding has not yet been confirmed.
The impact of miR-21 on Lamp2a and α-synuclein aggregation was confirmed by a second study using SH-SY5Y cells. Cells transfected with miR-21 mimics exhibited decreased levels of Lamp2a both at protein and mRNA levels, and increased α-synuclein only at the protein level. On the other hand, SH-SY5Y cells treated with miR-21 inhibitors displayed increased levels of Lamp2a (protein and mRNA) and decreased α-synuclein levels. This study also suggested that geniposide had a neuroprotective effect against MPP+/MPTP by inhibiting α-synuclein expression through the miR-21/Lamp2a axis (Su et al., 2016). In relation to Hsc70, two studies added miR-320 (Li et al., 2014) and miR-16-1(Zhang and Cheng, 2014) as direct regulators of Hsc70 expression, which negatively downregulated Hsc70 expression promoting α-synuclein aggregation in SH-SY5Y cells overexpressing α-synuclein, without affecting α-synuclein mRNA levels. Interestingly, miR-16 is a member of the miR-15/107 superfamily, a miRNA family highly dysregulated in Alzheimer’s disease (AD; Parsi et al., 2015). In this context, a preclinical study aimed to evaluate members of the superfamily miR-15/107 as potential drugs for AD, discovered that the brainstem of mice treated with a miR-16 mimic exhibited decreased α-synuclein protein levels (Parsi et al., 2015). This result was confirmed in HT22 cells, whereby overexpression of miR-16 downregulated α-synuclein protein levels (Parsi et al., 2015).
Another proteolytic pathway related with α-synuclein induced-toxicity is the autophagy-lysosomal pathway (ALP). The impact of ALP-associated miRNAs in α-synuclein expression was studied by Decressac et al. (2013) using rat midbrain overexpressing human wild-type α-synuclein. First, they demonstrated that α-synuclein toxicity is linked to impairment of the transcription factor EB (TFEB), a master regulator of the ALP controlled by mTOR signaling. In this context, AAV-mediated overexpression of miR-128 (which directly targeted TFEB) increased the formation of α-synuclein oligomers and the number of α-synuclein-positive axonal swellings, which resulted in α-synuclein-induced toxicity as revealed by a significant loss of nigral DA neurons, striatal innervation and DA levels, as well as development of motor deficits at 8 weeks after vector injection.
miR-133b
RhoA is a Rho family member that acts downstream of Rho-associated kinase (ROCK) and is a major regulator of the morphological events during apoptosis and neurite extension (Katoh et al., 1998; Shi and Wei, 2007). The fact that miR-133b was previously shown to promote neurite outgrowth and enhance neural function recovery after spinal cord injury and stroke by targeting RhoA (Liu et al., 2009; Yu et al., 2011; Xin et al., 2013), prompted Niu et al. (2016) to investigate the potential neuroprotective effect of miR-133b in the MPP+ model. In this scenario, Niu et al. (2016) reported that MPP+ treatment reduced miR-133b levels, increased RhoA expression and reduced neurite length in PC2 cells and rat DA neuron. Overexpression of miR-133b reversed the negative impact of MPP+ in neurite length and decreased RhoA protein level, although it had no impact on RhoA mRNA levels. Interestingly, ectopic expression of miR-133b in PC2 cells and primary neurons downregulated α-synuclein mRNA levels, both under baseline and MPP+ conditions. The authors attributed α-synuclein downregulation to miR-133 inhibition of RhoA, although this pathway has not been experimentally confirmed. Supporting this idea it has been previously reported that RhoA can directly modulate α-synuclein expression by activating the serum response element (SRF) transcription factor and GATA-2 transcription factor which regulates α-synuclein expression via occupancy at the intron-1 (Scherzer et al., 2008; Zhou et al., 2011).
miR-433
The discovery that SNPs located within the miR-433 binding sites in FGF20 gene were associated with PD (van der Walt et al., 2004; Haghnejad et al., 2015) triggered two studies to investigate the potential impact of miR-433 in α-synuclein expression (Wang et al., 2008; Schmitt et al., 2012). Wang et al. (2008) first demonstrated that miR-433 directly targets the FGF20 mRNA transcript and negatively regulates FGF20 protein translation. They showed that when SH-SY5Y cells were treated with the miR-433-target FGF20, α-synuclein protein levels were significantly increased compared to control cells. The authors suggest that FGF20 might regulate α-synuclein expression via FGF-receptor 1 (FGFR1), as it was previously demonstrated for FGF2 (Ohmachi et al., 2003; Rideout et al., 2003). Supporting this hypothesis, miR-433 did not bind to the α-synuclein 3′-UTR as reported with luciferase assays in Neuro2A and SK-N-SH cells (Schmitt et al., 2012).
Synuclein-Induced Changes in miRNAs Expression
In total, six studies report in vivo overexpressing α-synuclein models to investigate the impact of α-synuclein in miRNA expression (Figure 5; Table 2 and Supplementary Table 3; Gillardon et al., 2008; Asikainen et al., 2010; Ubhi et al., 2014; Kong et al., 2015; Schafferer et al., 2016; Thome et al., 2016). Four out of six studies used mice models (Gillardon et al., 2008; Ubhi et al., 2014; Schafferer et al., 2016; Thome et al., 2016), one was performed in Caenorhabditis elegans (Asikainen et al., 2010) and one in a Drosophila model (Kong et al., 2015). In all the species, overexpression of α-synuclein dysregulated several miRNAs (results summarized in Figure 5 and Supplementary Table 3).
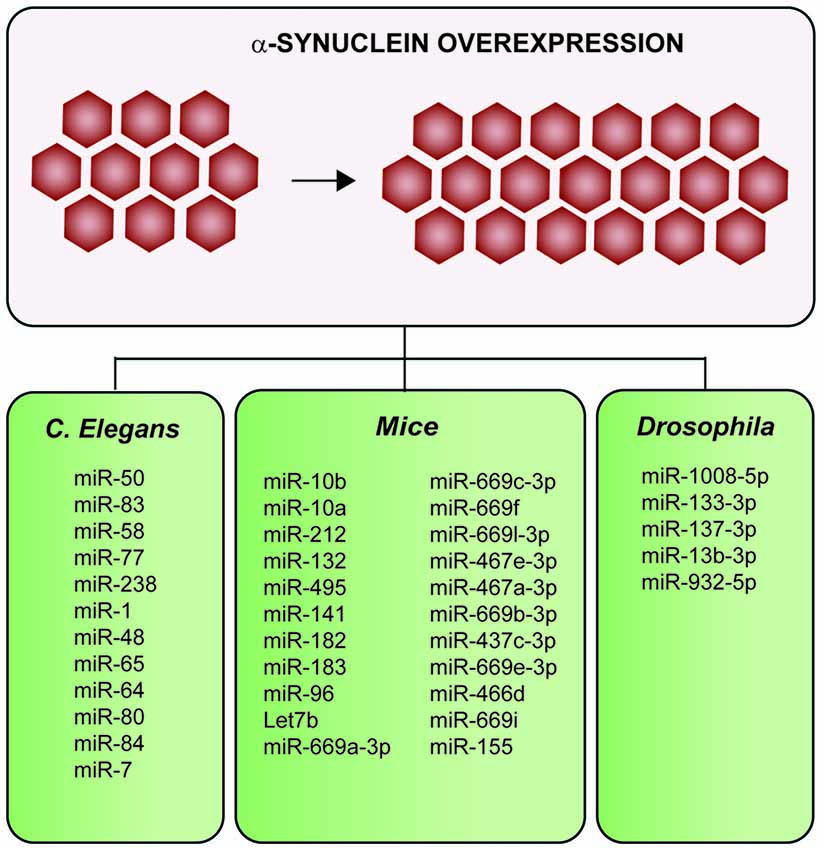
Figure 5. Summary of the impacts on the miRNAs profile after α-synuclein-overexpression in multiple in vivo models. Overexpression of α-synuclein induces alterations of several miRNAs in mice, Drosophila and Caenorhabditis elegans (C. elegans).
One of the three mice model studies investigated alterations in the miRNA profile of early-symptomatic α-synuclein (A30P)-transgenic mice using microfluidic chip technology (LC Sciences, Houston, TX, USA; Gillardon et al., 2008). The study of 266 unique mature mouse miRNA probes using μParaflo microfluidic chip (#MRA-1002) revealed that five microRNAs were downregulated in early-symptomatic α-synuclein (A30P)-transgenic mice: mmu-miR-10b, mmu-miR-10a, mmu-miR-212, mmu-miR-132 and mmu-miR-495. Two out of three mice studies were focused on MSA mice models overexpressing α-synuclein in oligodendroglial cells (Ubhi et al., 2014; Schafferer et al., 2016). Of these two MSA models, one was a comparative study of frontal cortex from several neurodegenerative transgenic mice models: (i) two different models of MSA in which α-synuclein was expressed under the control of oligodendrocyte-specific MPG promoter (lines MBP1- hαsyn and MBP29-hαsyn, medium and high αsyn expression respectively); (ii) DLB/PD; (iii) AD; and (iv) tauopathy (Ubhi et al., 2014). The study revealed that 55 out of 88 microRNAs analyzed were dysregulated in both MSA models (MBP1 and MBP29) compared to non-transgenic animals, and five of these genes were disease specific (Supplementary Table 3). Surprisingly, the DLB/PD model expressing human α-synuclein under the control of the mThy1.2. promoter did not show significant differences. The second MSA study analyzed the striatum and SN in premotor MSA models overexpressing oligodendroglial α-synuclein in the third postnatal month using microarrays (Schafferer et al., 2016). The results showed that 33 miRNAs were dysregulated in the striatum and 59 miRNAs in the SN compared to control groups. Particularly, the miRNA family miR-437 was significantly enriched (p < 0.0001) among the up-regulated miRNAs (Figure 4 and Supplementary Table 3).
Neuroinflammation and miR-155
Considering the growing evidence that neuroinflammation plays a key role in the pathogenesis and progression of PD, Thome et al. (2016) investigated the impact of miR-155 expression, one of the key microRNA modulators of neuroinflammation, in the α-synuclein transgenic mice. Interestingly, adenovirus-mediated overexpression of α-synuclein (AAV2-Syn) enhanced the expression of miR-155 in the SN of α-synuclein-overexpressing mice compared to control (30% increment 2 weeks after transduction) and induced a 29.7 ± 6.6% loss of TH positive neurons in the SN 6 months after transduction. Reactive microgliosis markers Major Histocompatibility Complex Class II (MHCII) and CD68 were also increased in the AAV2-Syn transgenic mice. Interestingly, genetic deletion of miR-155 prevented the increments of MHCII and CD68 and markedly attenuated the TH positive neuronal loss in the SN of AAV2-syn transgenic mice. These results were confirmed in vitro using primary microglial murine cells. The authors first showed that microglial cells treated with fibrils of human wild-type α-synuclein exhibited increased levels of MHCII and inducible nitric oxide synthase (iNOS), while monomeric α-synuclein did not activate the inflammatory response. On the other hand, α-synuclein fibrils did not activate the inflammatory process in microglial cells derived from miR-155−/− mice. However, miR-155 mimic treatment restored the inflammatory activity in miR-155−/− microglial cells.
Discussion
miRNAs both regulate and are regulated by α-synuclein expression, indicating the complex network between miRNAs and α-synuclein.
miRNAs Regulate α-Synuclein Expression
Growing evidence indicates that increased levels of α-synuclein are toxic and may initiate a deleterious cascade of events leading to neuronal death in PD. However, the cause that triggers α-synuclein upregulation in PD is only understood in a small percentage of patients with duplications/triplications in the α-synuclein gene or SNPs in the α-synuclein promoter. miRNAs impact on α-synuclein expression raises the hypothesis that dysregulated miRNAs in PD patients are responsible for α-synuclein upregulation and/or accumulation. Supporting this idea, several studies have demonstrated that PD patients exhibited dysregulated miRNAs in brain (Kim et al., 2007; Cardo et al., 2013; Miñones-Moyano et al., 2013; Briggs et al., 2015; Hoss et al., 2016), blood (Margis et al., 2011; Martins et al., 2011; Khoo et al., 2012; Botta-Orfila et al., 2014; Burgos et al., 2014), cerebrospinal fluid (CSF; Burgos et al., 2014; Gui et al., 2015; Hossein-Nezhad et al., 2016) and medulla (Liao et al., 2013).
Synuclein-Induced Changes in miRNAs Expression
α-synuclein overexpression impacts on miRNAs expression. Recent studies suggest that changes in miRNA expression can be directly linked to the pathophysiology of several diseases. Therefore, it is possible that the early α-synuclein overexpression linked to PD patients induces a stable miRNAs deregulation which can be the beginning of a process of neuronal death and the subsequent development of PD (Eacker et al., 2009). Supporting this idea, changes in miRNAs expression as a consequence of cellular damage and brain injury can be detected in the CSF and in the blood plasma/serum (Moldovan et al., 2014).
miRNAs as Potential Therapeutic Opportunity in PD
Similar to other neurodegenerative diseases, there is still no treatment available that stops or halts the progression of PD; and symptomatic treatments are the only option for PD patients. In this context, a large proportion of therapeutic approaches under development are aimed to reduce α-synuclein expression levels.
Targeting miRNAs seems to be a potential therapeutic opportunity for PD. Indeed, multiple α-synuclein-targeting miRNAs (miR-7, miR-153, miR-214 and miR-133b) have displayed protective effects against the PD-like-induced toxins MPP+/MPTP. Strikingly, their effects are normally attributed to α-synuclein-independent mechanisms; for example, miR-7 may exert its protective effect by activating RelA, Nlpr3 and mTOR pathways. However, each miRNAs is unique and displays its own protective/deleterious effect: overexpression of miR-7 or miR-155 induced a protective effect in MPP+/MPTP models and α-synuclein-induced toxicity, while overexpression of miR-128 targeting TFEB exacerbated α-synuclein-induced toxicity in mice. Nonetheless, when considering miRNAs as therapeutic opportunities, one have to keep in mind that each miRNA can target various mRNA transcripts, rendering difficult to target a specific molecular way.
In addition to miRNAs that impact on α-synuclein expression, other miRNAs might play essential roles in the pathogenesis of PD. As an example, the list of miRNAs that target other PD-related genes such as LRKK2, Parkin and Pink becomes longer every year. Several reviews are available that illustrate the complex interplay of miRNAs in PD (Salta and De Strooper, 2012; Ma et al., 2013; Majidinia et al., 2016; Xie and Chen, 2016).
Conclusion
This review article highlights that miRNAs regulate and are subject to regulation by α-synuclein. Considering the central role of α-synuclein in PD pathogenesis/progression, miRNAs are likely to play a crucial role at different stages of PD and might potentially be used in the future in new PD therapeutic approaches.
Author Contributions
AR: conception and design of the work; acquisition, analysis and interpretation of data for the work; drafting, final approval of the version to be published and agreement to be accountable for all aspects of the work. CP: substantial contributions to the design of the work; drafting the work, revising it critically for important intellectual content; final approval of the version to be published and agreement to be accountable for all aspects of the work. CMS: substantial contributions to the conception of the work; revising it critically for important intellectual content; final approval of the version to be published and agreement to be accountable for all aspects of the work.
Funding
This study was supported by the Sydney Medical School Foundation (University of Sydney).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank V. Garcia Cardenas (University Technology of Sydney) for her technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnmol.2016.00128/full#supplementary-material
References
Ahn, T. B., Kim, S. Y., Kim, J. Y., Park, S. S., Lee, D. S., Min, H. J., et al. (2008).α-synuclein gene duplication is present in sporadic Parkinson disease. Neurology 70, 43–49. doi: 10.1212/01.wnl.0000271080.53272.c7
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Rodriguez-Oroz, M. C., Obeso, J. A., and Cooper, J. M. (2013). Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson’s disease. Cell Death Dis. 4:e545. doi: 10.1038/cddis.2013.73
Ameres, S. L., and Zamore, P. D. (2013). Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 14, 475–488. doi: 10.1038/nrm3611
Asikainen, S., Rudgalvyte, M., Heikkinen, L., Louhiranta, K., Lakso, M., Wong, G., et al. (2010). Global microRNA expression profiling of Caenorhabditis elegans Parkinson’s disease models. J. Mol. Neurosci. 41, 210–218. doi: 10.1007/s12031-009-9325-1
Botta-Orfila, T., Morató, X., Compta, Y., Jose Lozano, J., Falgàs, N., Valldeoriola, F., et al. (2014). Identification of blood serum micro-RNAs associated with idiopathic and LRRK2 Parkinson’s disease. J. Neurosci. Res. 92, 1071–1077. doi: 10.1002/jnr.23377
Briggs, C. E., Wang, Y., Kong, B., Woo, T.-U. W., Iyer, L. K., and Sonntag, K. C. (2015). Midbrain dopamine neurons in Parkinson’s disease exhibit a dysregulated miRNA and target-gene network. Brain Res. 1618, 111–121. doi: 10.1016/j.brainres.2015.05.021
Burgos, K., Malenica, I., Metpally, R., Courtright, A., Rakela, B., Beach, T., et al. (2014). Profiles of extracellular miRNA in cerebrospinal fluid and serum from patients with Alzheimer’s and Parkinson’s diseases correlate with disease status and features of pathology. PLoS One 9:e94839. doi: 10.1371/journal.pone.0094839
Cabin, D. E., Shimazu, K., Murphy, D., Cole, N. B., Gottschalk, W., McIlwain, K. L., et al. (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 22, 8797–8807.
Cardo, L. F., Coto, E., de Mena, L., Ribacoba, R., Moris, G., Menéndez, M., et al. (2013). Profile of microRNAs in the plasma of Parkinson’s disease patients and healthy controls. J. Neurol. 260, 1420–1422. doi: 10.1007/s00415-013-6900-8
Catalanotto, C., Cogoni, C., and Zardo, G. (2016). MicroRNA in control of gene expression: an overview of nuclear functions. Int. J. Mol. Sci. 17:1712. doi: 10.3390/ijms17101712
Chandra, S., Gallardo, G., Fernández-Chacón, R., Schlüter, O. M., and Südhof, T. C. (2005). α-synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chiba-Falek, O., and Nussbaum, R. L. (2001). Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 10, 3101–3109. doi: 10.1093/hmg/10.26.3101
Choi, D. C., Chae, Y. J., Kabaria, S., Chaudhuri, A. D., Jain, M. R., Li, H., et al. (2014). MicroRNA-7 protects against 1-methyl-4-phenylpyridinium-induced cell death by targeting RelA. J. Neurosci. 34, 12725–12737. doi: 10.1523/JNEUROSCI.0985-14.2014
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Bjorklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. U S A 110, E1817–E1826. doi: 10.1073/pnas.1305623110
Doxakis, E. (2010). Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 285, 12726–12734. doi: 10.1074/jbc.m109.086827
Eacker, S. M., Dawson, T. M., and Dawson, V. L. (2009). Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 10, 837–841. doi: 10.1038/nrn2726
Fan, Z., Lu, M., Qiao, C., Zhou, Y., Ding, J.-H., and Hu, G. (2015). MicroRNA-7 enhances subventricular zone neurogenesis by inhibiting NLRP3/caspase-1 axis in adult neural stem cells. Mol. Neurobiol. 53, 7057–7069 doi: 10.1007/s12035-015-9620-5
Fernández-Chacón, R., Wölfel, M., Nishimune, H., Tabares, L., Schmitz, F., Castellano-Muñoz, M., et al. (2004). The synaptic vesicle protein CSP α prevents presynaptic degeneration. Neuron 42, 237–251. doi: 10.1016/s0896-6273(04)00190-4
Fragkouli, A., and Doxakis, E. (2014). miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Front. Cell. Neurosci. 8:182. doi: 10.3389/fncel.2014.00182
Gillardon, F., Mack, M., Rist, W., Schnack, C., Lenter, M., Hildebrandt, T., et al. (2008). MicroRNA and proteome expression profiling in early-symptomatic α-synuclein(A30P)-transgenic mice. Proteomics Clin. Appl. 2, 697–705. doi: 10.1002/prca.200780025
Gründemann, J., Schlaudraff, F., Haeckel, O., and Liss, B. (2008). Elevated α-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic Acids Res. 36:e38. doi: 10.1093/nar/gkn084
Gui, Y., Liu, H., Zhang, L., Lv, W., and Hu, X. (2015). Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 6, 37043–37053. doi: 10.18632/oncotarget.6158
Haghnejad, L., Emamalizadeh, B., Jamshidi, J., Bidoki, A. Z., Ghaedi, H., Ahmadi, E., et al. (2015). Variation in the miRNA-433 binding site of FGF20 is a risk factor for Parkinson’s disease in Iranian population. J. Neurol. Sci. 355, 72–74. doi: 10.1016/j.jns.2015.05.020
Hoss, A. G., Labadorf, A., Beach, T. G., Latourelle, J. C., and Myers, R. H. (2016). microRNA profiles in Parkinson’s disease prefrontal cortex. Front. Aging Neurosci. 8:36. doi: 10.3389/fnagi.2016.00036
Hossein-Nezhad, A., Fatemi, R. P., Ahmad, R., Peskind, E. R., Zabetian, C. P., Hu, S. C., et al. (2016). Transcriptomic profiling of extracellular RNAs present in cerebrospinal fluid identifies differentially expressed transcripts in Parkinson’s disease. J. Parkinsons Dis. 6, 109–117. doi: 10.3233/jpd-150737
Huang, Y., Shen, X. J., Zou, Q., Wang, S. P., Tang, S. M., and Zhang, G. Z. (2011). Biological functions of microRNAs: a review. J. Physiol. Biochem. 67, 129–139. doi: 10.1007/s13105-010-0050-6
Ibáñez, P., Bonnet, A.-M., Débarges, B., Lohmann, E., Tison, F., Pollak, P., et al. (2004). Causal relation between α-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171. doi: 10.1016/s0140-6736(04)17104-3
Ibáñez, P., Lesage, S., Janin, S., Lohmann, E., Durif, F., Destée, A., et al. (2009). α-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype and mechanisms. Arch. Neurol. 66, 102–108. doi: 10.1001/archneurol.2008.555
Junn, E., Lee, K.-W., Byeong, S. J., Chan, T. W., Im, J.-Y., and Mouradian, M. M. (2009). Repression of α-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. U S A 106, 13052–13057. doi: 10.1073/pnas.0906277106
Kabaria, S., Choi, D. C., Chaudhuri, A. D., Mouradian, M. M., and Junn, E. (2015). Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease. FEBS Lett. 589, 319–325. doi: 10.1016/j.febslet.2014.12.014
Katoh, H., Aoki, J., Ichikawa, A., and Negishi, M. (1998). p160 RhoA-binding kinase ROKalpha induces neurite retraction. J. Biol. Chem. 273, 2489–2492. doi: 10.1074/jbc.273.5.2489
Khoo, S. K., Petillo, D., Kang, U. J., Resau, J. H., Berryhill, B., Linder, J., et al. (2012). Plasma-based circulating MicroRNA biomarkers for Parkinson’s disease. J. Parkinsons Dis. 2, 321–331. doi: 10.3233/JPD-012144
Kim, H. J., Park, G., Jeon, B. S., Park, W. Y., and Kim, Y. E. (2013). A mir-153 binding site variation in SNCA in a patient with Parkinson’s disease. Mov. Disord. 28, 1755–1756. doi: 10.1002/mds.25505
Kim, J., Inoue, K., Ishii, J., Vanti, W. B., Voronov, S. V., Murchison, E., et al. (2007). A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317, 1220–1224. doi: 10.1126/science.1140481
Kirik, D., Annett, L. E., Burger, C., Muzyczka, N., Mandel, R. J., and Bjorklund, A. (2003). Nigrostriatal α-synucleinopathy induced by viral vector-mediated overexpression of human α-synuclein: a new primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 100, 2884–2889. doi: 10.1073/pnas.0536383100
Kirik, D., Rosenblad, C., Burger, C., Lundberg, C., Johansen, T. E., Muzyczka, N., et al. (2002). Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J. Neurosci. 22, 2780–2791.
Kong, Y., Liang, X., Liu, L., Zhang, D., Wan, C., Gan, Z., et al. (2015). High throughput sequencing identifies MicroRNAs mediating α-synuclein toxicity by targeting neuroactive-ligand receptor interaction pathway in early stage of drosophila Parkinson’s disease model. PLoS One 10:e0137432. doi: 10.1371/journal.pone.0137432
Larsen, K. E., Schmitz, Y., Troyer, M. D., Mosharov, E., Dietrich, P., Quazi, A. Z., et al. (2006). Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922. doi: 10.1523/jneurosci.3821-06.2006
Latreille, M., Hausser, J., Stützer, I., Zhang, Q., Hastoy, B., Gargani, S., et al. (2014). MicroRNA-7a regulates pancreatic β cell function. J. Clin. Invest. 124, 2722–2735. doi: 10.1172/JCI73066
Li, G., Yang, H., Zhu, D., Huang, H., Liu, G., and Lun, P. (2014). Targeted suppression of chaperone-mediated autophagy by miR-320a promotes α-synuclein aggregation. Int. J. Mol. Sci. 15, 15845–15857. doi: 10.3390/ijms150915845
Liao, X.-Y., Wang, W.-W., Yang, Z.-H., Wang, J., Lin, H., Wang, Q. S., et al. (2013). Microarray analysis of transcriptome of medulla identifies potential biomarkers for Parkinson’s disease. Int. J. Genomics 2013:606919. doi: 10.1155/2013/606919
Liberati, A., Altman, D. G., Tetzlaff, J., Mulrow, C., Gøtzsche, P. C., Ioannidis, J. P., et al. (2009). The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J. Clin. Epidemiol. 62, e1–e34. doi: 10.1016/j.jclinepi.2009.06.006
Lim, W., and Song, G. (2014). Identification of novel regulatory genes in development of the avian reproductive tracts. PLoS One 9:e96175. doi: 10.1371/journal.pone.0096175
Liu, N. K., Wang, X. F., Lu, Q. B., and Xu, X. M. (2009). Altered microRNA expression following traumatic spinal cord injury. Exp. Neurol. 219, 424–429. doi: 10.1016/j.expneurol.2009.06.015
Ma, L., Wei, L., Wu, F., Hu, Z., Liu, Z., and Yuan, W. (2013). Advances with microRNAs in Parkinson’s disease research. Drug Des. Devel. Ther. 7, 1103–1113. doi: 10.2147/DDDT.S48500
Majidinia, M., Mihanfar, A., Rahbarghazi, R., Nourazarian, A., Bagca, B., and Avci, C. B. (2016). The roles of non-coding RNAs in Parkinson’s disease. Mol. Biol. Rep. 43, 1193–1204. doi: 10.1007/s11033-016-4054-3
Maraganore, D. M., de Andrade, M., Elbaz, A., Farrer, M. J., Ioannidis, J. P., Krüger, R., et al. (2006). Collaborative analysis of α-synuclein gene promoter variability and Parkinson disease. JAMA 296, 661–670. doi: 10.1001/jama.296.6.661
Margis, R., Margis, R., and Rieder, C. R. M. (2011). Identification of blood microRNAs associated to Parkinsonis disease. J. Biotechnol. 152, 96–101. doi: 10.1016/j.jbiotec.2011.01.023
Martins, M., Rosa, A., Guedes, L. C., Fonseca, B. V., Gotovac, K., Violante, S., et al. (2011). Convergence of miRNA expression profiling, α-synuclein interacton and GWAS in Parkinson’s disease. PLoS One 6:e25443. doi: 10.1371/journal.pone.0025443
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016). Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient Midbrain neurons. J. Neurosci. 36, 7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016
Meister, G., and Tuschl, T. (2004). Mechanisms of gene silencing by double-stranded RNA. Nature 431, 343–349. doi: 10.1038/nature02873
Miñones-Moyano, E., Friedläender, M. R., Pallares, J., Kagerbauer, B., Porta, S., Escaramís, G., et al. (2013). Upregulation of a small vault RNA (svtRNA2–1a) is an early event in parkinson disease and induces neuronal dysfunction. RNA Biol. 10, 1093–1106. doi: 10.4161/rna.24813
Moldovan, L., Batte, K. E., Trgovcich, J., Wisler, J., Marsh, C. B., and Piper, M. (2014). Methodological challenges in utilizing miRNAs as circulating biomarkers. J. Cell. Mol. Med. 18, 371–390. doi: 10.1111/jcmm.12236
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., and Lee, V. M. (2000). Synucleins are developmentally expressed and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220.
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Niu, M., Xu, R., Wang, J., Hou, B., and Xie, A. (2016). MiR-133b ameliorates axon degeneration induced by MPP+ via targeting RhoA. Neuroscience 325, 39–49. doi: 10.1016/j.neuroscience.2016.03.042
Ohmachi, S., Mikami, T., Konishi, M., Miyake, A., and Itoh, N. (2003). Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J. Neurosci. Res. 72, 436–443. doi: 10.1002/jnr.10592
Parsi, S., Smith, P. Y., Goupil, C., Dorval, V., and Hébert, S. S. (2015). Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for Alzheimer’s disease. Mol. Ther. Nucleic Acids 4:e256. doi: 10.1038/mtna.2015.33
Purisai, M. G., McCormack, A. L., Langston, W. J., Johnston, L. C., and Di Monte, D. A. (2005). α-synuclein expression in the substantia nigra of MPTP-lesioned non-human primates. Neurobiol. Dis. 20, 898–906. doi: 10.1016/j.nbd.2005.05.028
Rideout, H. J., Dietrich, P., Savalle, M., Dauer, W. T., and Stefanis, L. (2003). Regulation of α-synuclein by bFGF in cultured ventral midbrain dopaminergic neurons. J. Neurochem. 84, 803–813. doi: 10.1046/j.1471-4159.2003.01574.x
Ross, O. A., Braithwaite, A. T., Skipper, L. M., Kachergus, J., Hulihan, M. M., Middleton, F. A., et al. (2008). Genomic investigation of α-synuclein multiplication and parkinsonism. Ann. Neurol. 63, 743–750. doi: 10.1002/ana.21380
Salta, E., and De Strooper, B. (2012). Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol. 11, 189–200. doi: 10.1016/s1474-4422(11)70286-1
Sánchez-Danés, A., Richaud-Patin, Y., Carballo-Carbajal, I., Jimenáz-Delgado, S., Caig, C., Mora, S., et al. (2012). Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 4, 380–395. doi: 10.1002/emmm.201200215
Schafferer, S., Khurana, R., Refolo, V., Venezia, S., Sturm, E., Piatti, P., et al. (2016). Changes in the miRNA-mRNA regulatory network precede motor symptoms in a mouse model of multiple system atrophy: clinical implications. PLoS One 11:e0150705. doi: 10.1371/journal.pone.0150705
Scherzer, C. R., Grass, J. A., Liao, Z., Pepivani, I., Zheng, B., Eklund, A. C., et al. (2008). GATA transcription factors directly regulate the Parkinson’s disease-linked gene α-synuclein. Proc. Natl. Acad. Sci. U S A 105, 10907–10912. doi: 10.1073/pnas.0802437105
Schmitt, I., Wüellner, U., van Rooyen, J. P., Khazneh, H., Becker, J., Volk, A., et al. (2012). Variants in the 3 ’ UTR of SNCA do not affect miRNA-433 binding and α-synuclein expression. Eur. J. Hum. Genet. 20, 1265–1269. doi: 10.1038/ejhg.2012.84
Shi, J., and Wei, L. (2007). Rho kinase in the regulation of cell death and survival. Arch. Immunol. Ther. Exp. (Warsz) 55, 61–75. doi: 10.1007/s00005-007-0009-7
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). α-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Song, W., Zukor, H., Lin, S. H., Hascalovici, J., Liberman, A., Tavitian, A., et al. (2012). Schizophrenia-like features in transgenic mice overexpressing human HO-1 in the astrocytic compartment. J. Neurosci. 32, 10841–10853. doi: 10.1523/JNEUROSCI.6469-11.2012
Su, C., Yang, X., and Lou, J. (2016). Geniposide reduces α-synuclein by blocking microRNA-21/lysosome-associated membrane protein 2A interaction in Parkinson disease models. Brain Res. 1644, 98–106. doi: 10.1016/j.brainres.2016.05.011
Tagliafierro, L., and Chiba-Falek, O. (2016). Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics 17, 145–157. doi: 10.1007/s10048-016-0478-0
Thome, A. D., Harms, A. S., Volpicelli-Daley, L. A., and Standaert, D. G. (2016). microRNA-155 regulates α-synuclein-induced inflammatory responses in models of Parkinson disease. J. Neurosci. 36, 2383–2390. doi: 10.1523/JNEUROSCI.3900-15.2016
Touchman, J. W., Dehejia, A., Chiba-Falek, O., Cabin, D. E., Schwartz, J. R., Orrison, B. M., et al. (2001). Human and mouse α-synuclein genes: comparative genomic sequence analysis and identification of a novel gene regulatory element. Genome Res. 11, 78–86. doi: 10.1101/gr.165801
Tüfekci, K. U., Meuwissen, R. L., and Genç, S. (2014a). The role of microRNAs in biological processes. Methods Mol. Biol. 1107, 15–31. doi: 10.1007/978-1-62703-748-8_2
Tüfekci, K. U., Oner, M. G., Meuwissen, R. L., and Genç, Ş. (2014b). The role of microRNAs in human diseases. Methods Mol. Biol. 1107, 33–50. doi: 10.1007/978-1-62703-748-8_3
Ubhi, K., Rockenstein, E., Kragh, C., Inglis, C., Spencer, B., Michael, S., et al. (2014). Widespread microRNA dysregulation in multiple system atrophy–disease-related alteration in miR-96. Eur. J. Neurosci. 39, 1026–1041. doi: 10.1111/ejn.12444
van der Walt, J. M., Noureddine, M. A., Kittappa, R., Hauser, M. A., Scott, W. K., McKay, R., et al. (2004). Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am. J. Hum. Genet. 74, 1121–1127. doi: 10.1086/421052
Vila, M., Jackson-Lewis, V., Vukosavic, S., Djaldetti, R., Liberatore, G., Offen, D., et al. (2001). Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U S A 98, 2837–2842. doi: 10.1073/pnas.051633998
Vila, M., Vukosavic, S., Jackson-Lewis, V., Neystat, M., Jakowec, M., and Przedborski, S. (2000). Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 74, 721–729. doi: 10.1046/j.1471-4159.2000.740721.x
Wang, G., van der Walt, J. M., Mayhew, G., Li, Y. J., Züchner, S., Scott, W. K., et al. (2008). Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of α-synuclein. Am. J. Hum. Genet. 82, 283–289. doi: 10.1016/j.ajhg.2007.09.021
Wang, Z. H., Zhang, J. L., Duan, Y. L., Zhang, Q. S., Li, G. F., and Zheng, D. L. (2015). MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting α-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed. Pharmacother. 74, 252–256. doi: 10.1016/j.biopha.2015.08.025
Xie, Y., and Chen, Y. (2016). microRNAs: emerging targets regulating oxidative stress in the models of Parkinson’s disease. Front. Neurosci. 10:298. doi: 10.3389/fnins.2016.00298
Xin, H., Li, Y., Liu, Z., Wang, X., Shang, X., Cui, Y., et al. (2013). MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells 31, 2737–2746. doi: 10.1002/stem.1409
Yu, Y. M., Gibbs, K. M., Davila, J., Campbell, N., Sung, S., Todorova, T. I., et al. (2011). MicroRNA miR-133b is essential for functional recovery after spinal cord injury in adult zebrafish. Eur. J. Neurosci. 33, 1587–1597. doi: 10.1111/j.1460-9568.2011.07643.x
Zhang, Z., and Cheng, Y. (2014). miR-16–1 promotes the aberrant α-synuclein accumulation in parkinson disease via targeting heat shock protein 70. Scientific World Journal 2014:938348. doi: 10.1155/2014/938348
Zhou, Z., Kim, J., Insolera, R., Peng, X., Fink, D. J., and Mata, M. (2011). Rho GTPase regulation of α-synuclein and VMAT2: implications for pathogenesis of Parkinson’s disease. Mol. Cell. Neurosci. 48, 29–37. doi: 10.1016/j.mcn.2011.06.002
Keywords: Parkinson’s disease, α-synuclein, microRNA, alpha-synuclein, gene expression, miRNA, gene regulation
Citation: Recasens A, Perier C and Sue CM (2016) Role of microRNAs in the Regulation of α-Synuclein Expression: A Systematic Review. Front. Mol. Neurosci. 9:128. doi: 10.3389/fnmol.2016.00128
Received: 10 October 2016; Accepted: 07 November 2016;
Published: 21 November 2016.
Edited by:
Jiajie Diao, University of Cincinnati, USAReviewed by:
Hong Qing, Beijing Institute of Technology, ChinaJun Wan, Hong Kong University of Science and Technology, China
Copyright © 2016 Recasens, Perier and Sue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolyn M. Sue, carolyn.sue@sydney.edu.au