 Shaily Malik
Shaily Malik Martin Theis
Martin Theis Eliseo A. Eugenin
Eliseo A. Eugenin- 1Public Health Research Institute (PHRI), Newark, NJ, United States
- 2Department of Microbiology, Biochemistry, and Molecular Genetics, Rutgers New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, NJ, United States
- 3Institute of Cellular Neurosciences, University of Bonn, Bonn, Germany
Human immunodeficiency virus-1 (HIV-1) infection compromises the central nervous system (CNS) in a significant number of infected individuals, resulting in neurological dysfunction that ranges from minor cognitive deficits to frank dementia. While macrophages/microglia are the predominant CNS cells infected by HIV, our laboratory and others have shown that HIV-infected astrocytes, although present in relatively low numbers with minimal to undetectable viral replication, play key role in NeuroAIDS pathogenesis. Our laboratory has identified that HIV “hijacks” connexin (Cx) containing channels, such as gap junctions (GJs) and hemichannels (HCs), to spread toxicity and apoptosis to uninfected cells even in the absence of active viral replication. In this study, using a murine model with an astrocyte-directed deletion of Cx43 gene (hGFAP-cre Cx43fl/fl) and control Cx43fl/fl mice, we examined whether few HIV-infected human astrocytoma cells (U87-CD4-CCR5), microinjected into the mouse cortex, can spread toxicity and apoptosis through GJ-mediated mechanisms, into the mouse cells, which are resistant to HIV infection. In the control Cx43fl/fl mice, microinjection of HIV-infected U87-CD4-CCR5 cells led to apoptosis in 84.28 ± 6.38% of mouse brain cells around the site of microinjection, whereas hGFAP-cre Cx43fl/fl mice exhibited minimal apoptosis (2.78 ± 1.55%). However, simultaneous injection of GJ blocker, 18α-glycyrrhetinic acid, and Cx43 blocking peptide along with microinjection of HIV-infected cells prevented apoptosis in Cx43fl/fl mice, demonstrating the Cx43 is essential for HIV-induced bystander toxicity. In conclusion, our findings demonstrate that Cx43 expression, and formation of GJs is essential for bystander apoptosis during HIV infection. These findings reveal novel potential therapeutic targets to reduce astrocyte-mediated bystander toxicity in HIV-infected individuals because despite low to undetectable viral replication in the CNS, Cx channels hijacked by HIV amplify viral neuropathogenesis.
Introduction
As reported by UNAIDS in 2015, approximately 36 million people are infected with human immunodeficiency virus-1 (HIV-1) worldwide (UNAIDS, 2016). In addition to immune compromise, HIV infection causes a wide spectrum of HIV-associated neurocognitive disorders (HAND) which range from mild cognitive disease to dementia, even in the context of successful anti-retroviral therapy (ART) (Anthony et al., 2005; Antinori et al., 2007; Heaton et al., 2010). HIV infection of the central nervous system (CNS) occurs within weeks of systemic infection, when circulating HIV-infected monocytes/macrophages enter the brain, and infect CNS-resident microglia, macrophages, and a small population of astrocytes (Wiley et al., 1999; Churchill et al., 2006). In the CNS, HIV-infected cells release a plethora of host and viral molecules (e.g., chemokines/cytokines and viral proteins, respectively), which promote neuroinflammation, even during ART when HIV replication is undetectable (Clifford, 2017). Interestingly, upon interruption of the ART, the virus re-emerges, suggesting existence of long-lasting viral reservoirs in HIV-infected individuals (Chun et al., 1999). Furthermore, despite peripheral control of HIV replication by ART, 50–60% of HIV-infected individuals have some degree of cognitive disease (Heaton et al., 2010), suggesting that HIV uses unknown replication-independent mechanisms to amplify damage within the CNS. Work from our laboratory has demonstrated that one of the mechanisms of CNS damage is elicited by HIV-infected astrocytes, which, while representing a small fraction of all astrocytes, act as a source of toxic, pro-apoptotic signals that spread to neighboring uninfected cells via connexin43 (Cx43) containing channels, gap junctions (GJ) and unopposed hemichannels (uHC), causing “bystander apoptosis” despite absence of viral replication (Eugenin and Berman, 2007, 2013; Eugenin et al., 2011; Orellana et al., 2014).
Gap junctions connect the cytoplasm of two adjoining cells enabling direct exchange of cytoplasmic products between the connected cells. Each GJ channel is formed from the docking of two hemichannels (HCs), and each HC comprises of a hexameric assembly of connexin (Cx) protein subunits (Bennett et al., 2003). Recently, our laboratory has identified that uHCs open under HIV-infected conditions enabling the release of intracellular factors into the extracellular milieu (Orellana et al., 2014). Both types of channels, GJs and uHCs, can pass molecules up to 1.2 kDa in size, including second messengers, ions, ATP, prostaglandins, small peptides, and RNA (Saez et al., 2003). Previously, we have demonstrated that HIV-infected astrocytes “hijack” GJs and uHCs to spread toxic, pro-apoptotic, and pro-inflammatory molecules, such as inositol triphosphate (IP3), Ca2+, glutamate, ATP, and prostaglandin E2 (PGE2), to vast areas of the CNS, into uninfected cells eliciting neuronal and glial compromise (Eugenin and Berman, 2007, 2013; Eugenin et al., 2011). Interestingly, in contrast to the uninfected cells, HIV-infected astrocytes were resistant to pro-apoptotic signals, and exhibited extended survival due to reduced mitochondrial function, IP3 receptor sensitivity, calcium response, and apoptosome formation (Eugenin and Berman, 2013). Importantly, all of these mechanisms were independent of HIV replication, and occur even in the presence of successful ART. We have also demonstrated that most of these mechanisms operate in vivo (i.e., in human and monkey brain tissue samples), underscoring the importance of glial cells and Cx43 containing channels in the pathogenesis of NeuroAIDS (Berman et al., 2016).
In the present study, we microinjected HIV-infected U87-CD4-CCR5 cells into the cortex of Cx43-expressing (Cx43fl/fl) and Cx43-deficient (hGFAP-cre Cx43fl/fl) mice, and assessed bystander apoptosis in the mouse cells. We demonstrate that Cx43 in astrocytes is essential for amplifying bystander apoptosis from few HIV-infected astrocytes to neighboring mouse cells which do not support viral replication. The mechanism of bystander apoptosis was dependent not only on Cx43 but also on IP3 receptor activation and intracellular calcium. Thus, we propose that HIV “hijacks” Cx43 containing channels even in the absence of HIV replication to amplify damage within the CNS.
Materials and Methods
Materials
U87-CD4-CCR5 cells, HIVADA, and HIV-1 p24 antibody were obtained from NIH AIDS Reagent program (Germantown, MD, United States). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, trypsin-EDTA, secondary antibody conjugated to FITC, and prolong gold anti-fade reagent with DAPI were purchased from Thermo Fisher Scientific (Waltham, MA, United States). The in situ cell death detection kit, TUNEL was purchased from Roche (Mannheim, Germany). BAPTA-AM, 18α-glycyrrhetinic acid (AGA), and isoflurane were procured from Sigma–Aldrich (St. Louis, MO, United States). Xestospongin C (XeC) was purchased from Tocris (Bristol, United Kingdom). Cx43 blocking peptide and scrambled (Scr) peptide were obtained from PeproTech (Rocky Hill, NJ, United States).
Methods
Cx43 Conditional Knockout Mice
Mice with astrocyte-specific deletion of Cx43 were generated by interbreeding of Cx43fl mice (Theis et al., 2001), in which the Cx43 coding region is flanked by loxP sites, with mice carrying a cre re-combinase transgene under the human glial fibrillary acidic protein (GFAP) promoter, hGFAP-cre (Zhuo et al., 2001) (Figure 1A). In astrocyte cultures from hGFAP-cre Cx43fl/fl mice, at least 90% of the cells displayed loss of Cx43 expression after 4 weeks in culture (Theis et al., 2003). Similar results of loss of Cx43 expression (approximately 90%) were observed for our animals using Western blotting (data not shown). For the present study, we used five adult male hGFAP-cre Cx43fl/fl mice, and as controls, four adult male Cx43fl/fl mice.
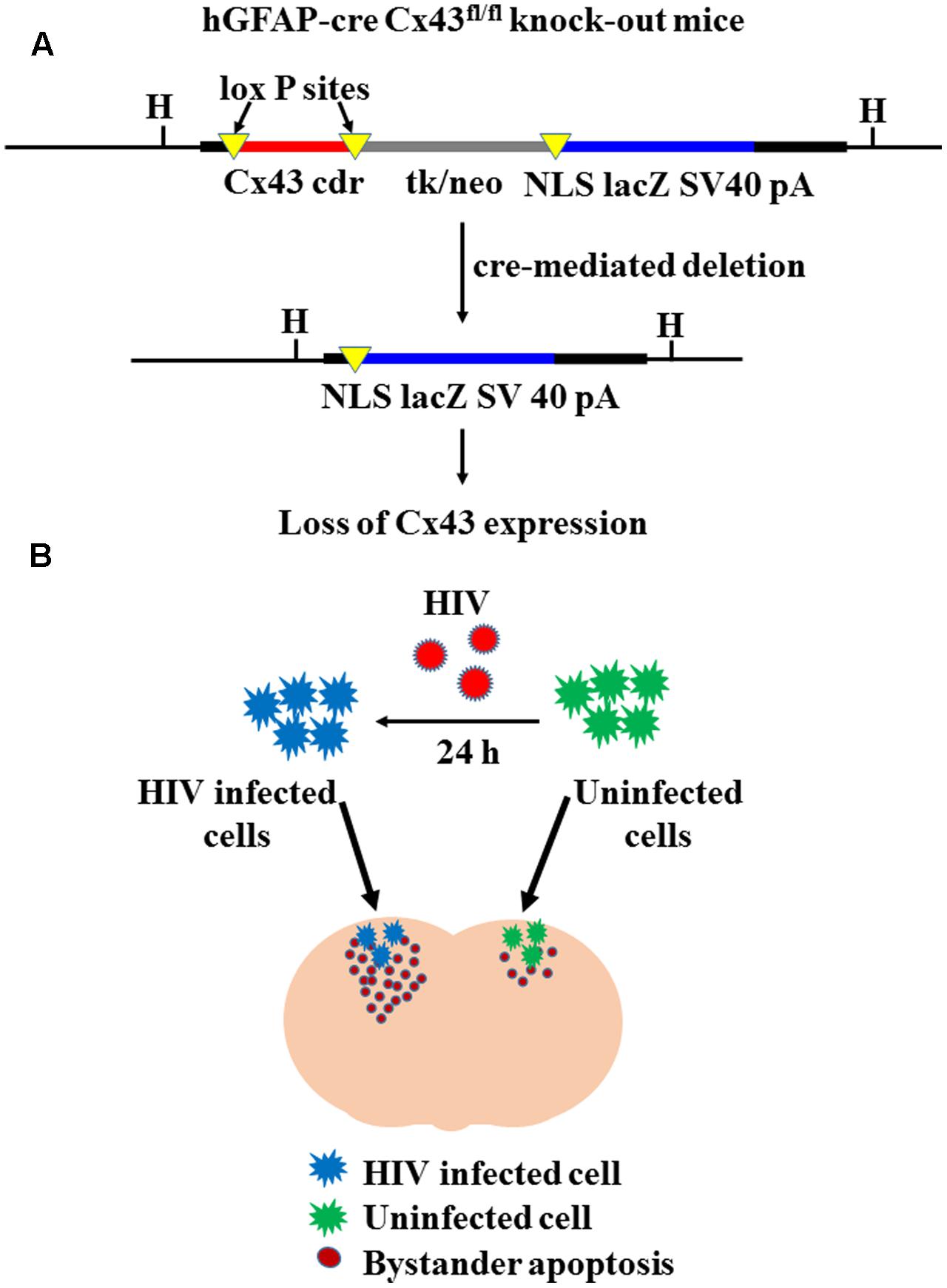
FIGURE 1. Cre-mediated deletion of Cx43 expression. (A) Represents the concept of cre-mediated deletion of floxed DNA at the Cx43 locus, leading to expression of lacZ in the cells that display Cx43 gene activity. Since cre transgene is under the human GFAP promoter (hGFAP-cre), Cx43 is specifically deleted in astrocytes. Cx43 cdr: Cx43 coding region; tk/neo: herpes simplex virus thymidine-kinase/neomycin phosphotransferase; NLS lacZ: lacZ reporter gene with nuclear localization signal; SV40 pA: SV40 polyadenylation signal; H: HindIII restriction site [Adapted from Theis et al. (2003)]. (B) Represents experimental strategy of microinjecting HIV-infected/uninfected human U87-CD4-CCR5 cells in Cx43fl/fl and hGFAP-cre Cx43fl/fl mice. Human U87-CD4-CCR5 astrocytoma cells were infected with HIVADA (50 ng/ml) for 24 h, and then microinjected into cortices of Cx43fl/fl and hGFAP-cre Cx43fl/fl mice. After 48 h of microinjection, extent of bystander toxicity was analyzed in the mouse cortex by detecting apoptosis with TUNEL assay. In the contra-lateral hemisphere, mice were injected with uninfected U87-CD4-CCR5 cells, which served as control for microinjection of HIV-infected cells.
HIV Infection of U87-CD4-CCR5 Cells
Human astrocytoma cells transfected with CD4 and CCR5, U87-CD4-CCR5, were obtained from NIH AIDS Reagent Program (Germantown, MD, United States). CD4 and CCR5 transfection enables the U87 cells to achieve 90–95% infection with HIV, and is widely used to study HIV infection in astrocytes (Bjorndal et al., 1997; Orellana et al., 2014). U87-CD4-CCR5 cells were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Prior to microinjection, U87-CD4-CCR5 cells were infected with HIVADA (50 ng/ml) for 24 h (Figure 1B). The cells were washed once with the media to remove any free virus. The cells were then re-suspended in phosphate buffered saline (PBS) at a density of 3,000 cells/0.5 μl for microinjection.
Microinjection of U87-CD4-CCR5 Cells in Mouse Cortex
For microinjection, four adult male Cx43fl/fl mice and five adult male hGFAP-cre Cx43fl/fl mice, 6 weeks of age were used in the study. The mice were anesthetized by isoflurane inhalation for the procedure (nose cone providing continuous 1.5% isoflurane). The microinjection of U87-CD4-CCR5 cells was performed as described in approved IACUC protocol 12016D015 (Rutgers University, NJ, United States). Once anesthetized, the top of the animal’s head was shaved, and sterilized with a betadine pad. A 0.5 cm incision was made, and the skin was folded back to expose the top of the cranium. A 0.01 cm hole was made into the top of the cranium using a small drill exposing the mouse brain. In this opening, a Hamilton Neuros syringe was used to inject 3,000 HIV-infected U87-CD4-CCR5 astrocytoma cells in total a volume of 0.5 μl, at four different sites in the gray matter of the somatosensory cortex (S1). The syringe needle had a fixed length (0.1 cm) to ensure that the injection is made to the cortex. After the injection, the skin was sutured using tissue adhesive. As a control, the same animal was injected with uninfected U87-CD4-CCR5 cells in a similar manner in the contralateral hemisphere, to negate the effects of host immune response on the results. The mice were kept under continuous observation after the procedure. For experiments with chemical and peptide blockers, the following concentrations were used: 18α-glycyrrhetinic acid (AGA): 35 μM; Cx43 blocking peptide: 100 μM; Scr peptide: 100 μM; Xestospongin C: 10 μM; BAPTA-AM: 5 μM. As per the experiment, either inhibitors or peptides were added to the cell mix (3,000 cells/0.5 μl) prior to microinjection. All these blockers were tested in different cell lines and models to assure specificity, concentration and toxicity (Eugenin and Berman, 2007, 2013). As a control, the same animal was injected with either HIV-infected or uninfected U87-CD4-CCR5 cells without the inhibitor/peptide in the contralateral hemisphere. After 48 h of microinjection, the mice were sacrificed by cervical dislocation and brains were harvested. We concluded our experiments 48 h post-microinjection because bystander apoptosis was evident at that time, and to reduce pain and suffering of the animals.
Immunohistochemistry and Apoptosis Assay
Harvested mice brains were fixed in 4% paraformaldehyde for 24 h and then transferred to 30% sucrose solution at 4°C. After 48 h, the brains were sectioned into 10-μm-thick coronal sections. For each animal, 20–40 sections were analyzed to reconstruct the injected area and to quantify bystander killing. Apoptosis was detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay as described previously (Eugenin and Berman, 2013). Briefly, tissue sections were incubated in TUNEL reaction mixture (Enzyme solution:Label solution 1:9) at 37°C for 1 h, washed three times with PBS, and proceeded for immunohistochemistry. The tissue sections were permeabilized with 0.01% Triton × 100, washed three times with PBS, and then incubated in blocking solution (50 μM EDTA, 1% fish gelatin, 1% bovine serum albumin, 2% human serum, and 2% horse serum) for 2 h at room temperature (RT). The samples were then incubated with HIV-1 p24 antibody (1:50), overnight at 4°C. Then, the tissue samples were washed several times with PBS at RT, and incubated with the appropriate secondary antibody conjugated to FITC for 2 h at RT. Following several washes with PBS, the tissue sections were mounted with prolong gold anti-fade reagent with DAPI. The tissue samples were examined by Leica SP2 confocal microscope (Leica, Germany), and analyzed by Adobe Photoshop, NIH ImageJ, and Nikon NIS Elements software. For calculating the percentage of apoptotic cells around the site of microinjection, a circular area of 200 μm radius from the site of microinjection was assessed for the presence of TUNEL-positive nuclei. Percentage of apoptotic cells was calculated by counting the total number of TUNEL-positive nuclei versus the total number of nuclei stained with DAPI in the circular area of 200 μm radius from the site of microinjection.
Statistical Analysis
All data are presented as mean ± standard deviation (SD). Each animal was microinjected at four sites, and in the figures, one dot represents one microinjection. All statistical analysis have been performed using one-way ANOVA. The p-values < 0.05 were considered significant.
Results
Microinjection of HIV-Infected Human Astrocytoma Cells into the Brain of Cx43 Expressing Mouse Induces Bystander Apoptosis in Mouse Cells
To examine bystander apoptosis in the absence of HIV infection, we microinjected 3000 U87-CD4-CCR5 cells into the cortex of mice containing astrocyte-specific deletion of Cx43, hGFAP-cre Cx43fl/fl, and control, Cx43fl/fl mice (with normal expression of Cx43) (Figure 1B). The U87-CD4-CCR5 cells were infected with HIVADA (50 ng/ml) for 24 h, and based on HIV-p24 immunostaining, approximately 90–95% cells were infected after the first round of replication (data not shown). Specifically, 3000 HIV-infected U87-CD4-CCR5 cells were microinjected at four sites in the somatosensory cortex (S1) of hGFAP-cre Cx43fl/fl and Cx43fl/fl mice. As a control, equal number of uninfected U87-CD4-CCR5 cells was microinjected into the contralateral hemisphere of the same animal. After 48 h of microinjection, the animals were sacrificed, and the brains were sectioned and analyzed for apoptosis.
Microinjection of human HIV-infected astrocytoma cells into cortex of Cx43fl/fl mice led to significant apoptosis, and approximately 84.28 ± 6.38% host cells were apoptotic in the region surrounding the site of microinjection [Figures 2A(Upper panel), 2B]. In contrast, microinjection of HIV-infected human astrocytes into hGFAP-cre Cx43fl/fl mice resulted in minimal apoptosis (2.78 ± 1.55%) around the site of microinjection [Figures 2A(Lower panel), 2B]. Microinjection of uninfected human astrocytoma cells resulted in minimal apoptosis in Cx43fl/fl (3.72 ± 1.44%) as well as in hGFAP-cre Cx43fl/fl (3.23 ± 1.53%) mice at the site of microinjection (Figure 2B and Table 1). Microinjection of cell free virus (50 ng/ml) did not altered the levels of apoptosis (data not shown). Since HIV does not infect mouse cells as the viral envelope glycoprotein, gp120, does not engage mouse CD4 receptors (Browning et al., 1997), we propose that the microinjected human astrocytoma cells form GJs with resident mouse brain cells (Supplementary Figure S1 and Supplementary Data), and the cytoplasmic apoptotic factors generated in HIV-infected U87-CD4-CCR5 cells diffuse into uninfected host cells via Cx43 GJs and lead to their apoptosis.
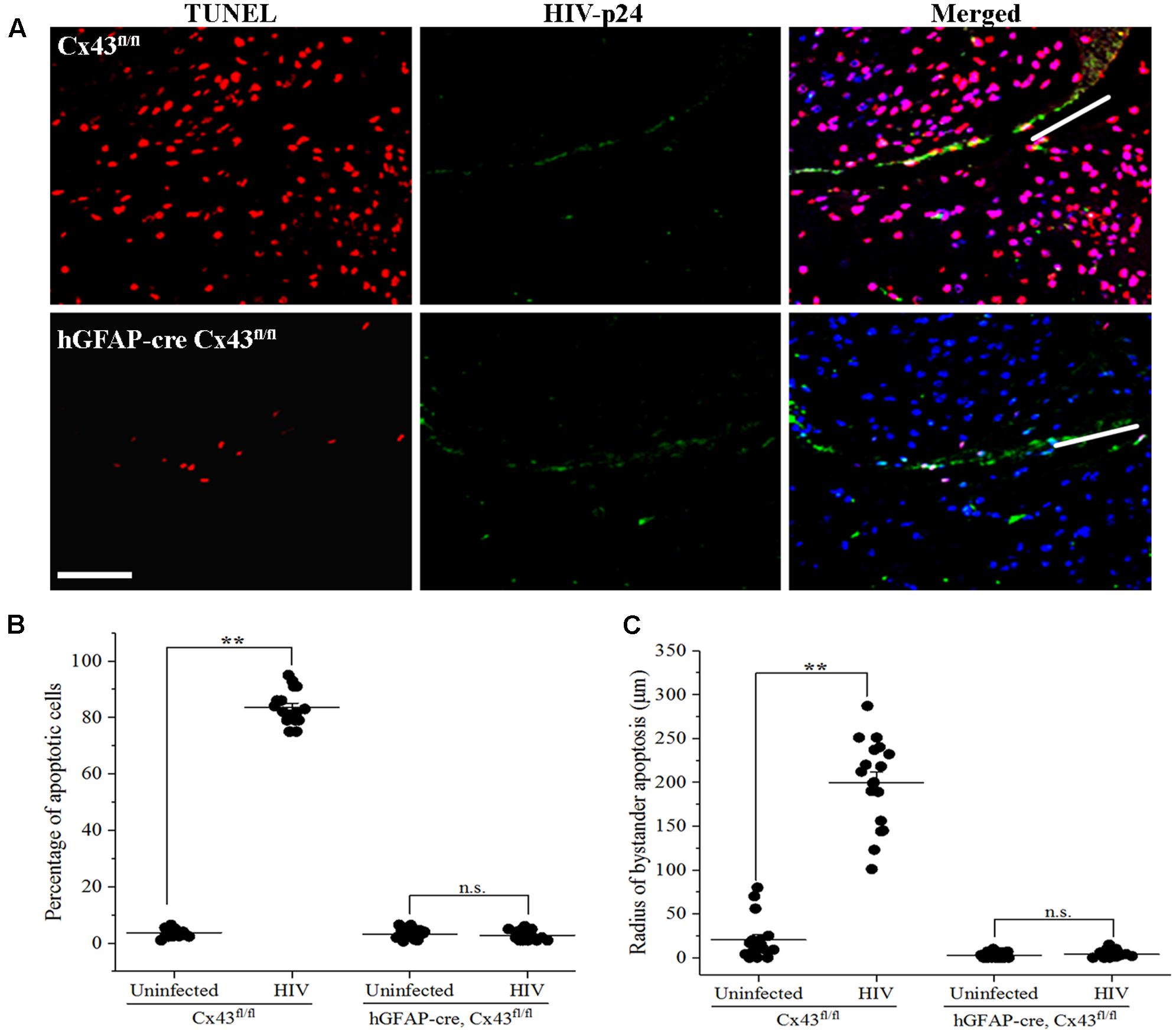
FIGURE 2. Microinjection of HIV-infected cells induces bystander apoptosis in mice cortex. Human U87-CD4-CCR5 cells infected with HIVADA (50 ng/ml) were microinjected into the cortices of Cx43fl/fl as well as hGFAP-cre Cx43fl/fl mice, and after 48 h, animals were sacrificed and brain sections were analyzed for apoptosis by TUNEL assay. (A) Representative images showing apoptosis (TUNEL, red) upon microinjection of HIV-infected astrocytes in Cx43fl/fl as well as hGFAP-cre Cx43fl/fl mice. HIV-p24 has been labeled with FITC (green), and cell nuclei with DAPI (blue). White lines in the merged images show the direction of microinjection. Scale bar denotes 75 μm. (B) Graphical representation of the extent of bystander apoptosis observed in a circular area of 200 μm around the site of microinjection in Cx43fl/fl and hGFAP-cre Cx43fl/fl mice. Microinjection of HIV-infected cells led to significant apoptosis in Cx43fl/fl mice as GJs were formed between the microinjected human cells and murine host cells, which resulted in the diffusion of apoptotic factors generated in a few HIV-infected cells to host cells leading to their apoptosis. However, hGFAP-cre Cx43fl/fl mice displayed minimal apoptosis even after microinjection of HIV-infected cells, possibly due to the absence of Cx43 GJs and HCs in these animals. (C) Represents the radius of bystander apoptosis which is the maximum distance of apoptotic cells from the site of microinjection of either HIV-infected or uninfected cells. The Cx43fl/fl mice where HIV-infected cells were microinjected exhibit significantly larger radius of bystander apoptosis as compared to Cx43fl/fl mice that received uninfected cells or hGFAP-cre Cx43fl/fl mice that received either HIV-infected or uninfected cells. Each dot in the graphs (B,C) represents a microinjection. Each animal was microinjected at four sites; hence, in total, there were 16 microinjections for Cx43fl/fl mice and 20 microinjections for hGFAP-cre Cx43fl/fl mice. ∗∗p < 0.005; n.s. – not significant.
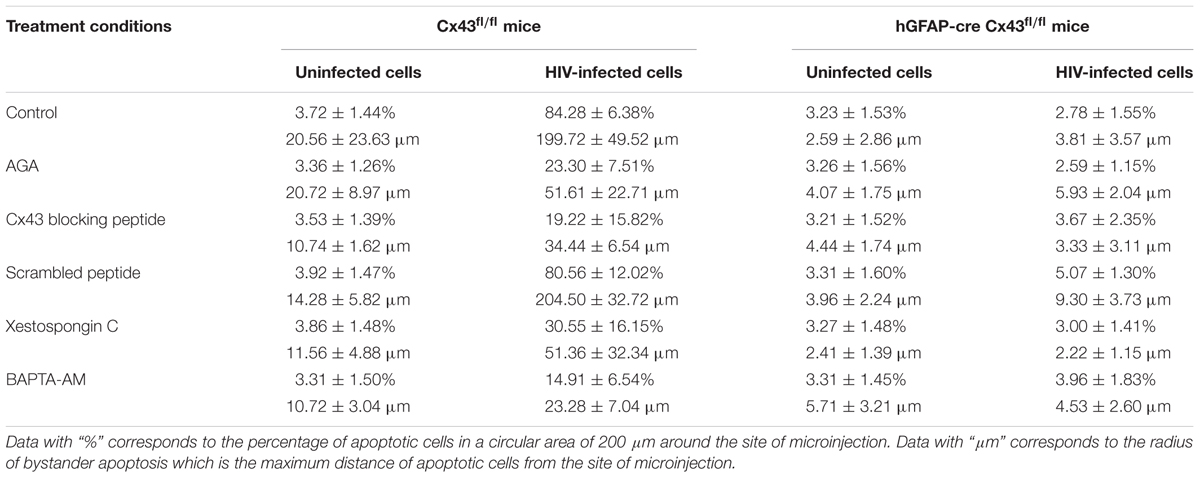
TABLE 1. Bystander apoptosis in Cx43fl/fl and hGFAP-cre Cx43fl/fl mice upon microinjection of either uninfected or HIV-infected U87-CD4-CCR5 cells under various treatment conditions.
To quantify the degree of damage, we measured the radius of bystander apoptosis, as determined by the presence of apoptotic cells in a circular area from the site of microinjection of HIV-infected/uninfected cells. As shown in Figure 2C, Cx43fl/fl mice expressing Cx43 GJ channels and HCs had amplification of apoptosis as upon microinjection of HIV-infected astrocytoma cells, TUNEL-positive cells were observed up to a distance of 199.72 ± 49.52 μm from the site of microinjection as opposed to 20.56 ± 23.63 μm when uninfected U87-CD4-CCR5 cells were microinjected (Table 1). However, microinjection of HIV-infected cells into hGFAP-cre Cx43fl/fl mice resulted in minimal cell death (Figure 2B), and apoptotic cells were observed only up to a distance of 3.81 ± 3.57 μm from the site of microinjection, exhibiting much smaller radius of bystander apoptosis as compared to Cx43fl/fl mice (Figure 2C). It is important to mention here that even microinjection of uninfected cells in hGFAP-cre Cx43fl/fl mice had significantly smaller radius of toxicity (2.59 ± 2.86 μm) as compared to Cx43fl/fl mice (20.56 ± 23.63 μm) (Figure 2C), which underscores the role of Cx43 channels in mediating the spread of toxic factors from damaged/pro-apoptotic cells to healthy cells during any pathology. Hence, we propose that the amplification of apoptosis was dependent on the microinjection of HIV-infected cells as well as on the presence of Cx43 GJ channels and HCs to propagate apoptotic stimuli from few HIV-infected cells to the uninfected cells.
Functional GJs/HCs Are Required for Propagation of Apoptotic Factors from HIV-Infected Cells to Uninfected Cells
To further characterize the role of Cx43 containing GJs and HCs in mediating HIV-induced bystander apoptosis, inhibitor for GJ-mediated communication, 18α-glycyrrhetinic acid, AGA (35 μM), and Cx43 blocking peptide (100 μM), which blocks both Cx43 GJs and HCs, were used in the study. A Scr peptide was also used in the study as a negative control for Cx43 blocking peptide. At the time of microinjection, either AGA or Cx43 blocking peptide or Scr peptide was administered along with HIV-infected/uninfected U87-CD4-CCR5 cells into the cortices of hGFAP-cre Cx43fl/fl and Cx43fl/fl mice. After 48 h, the animals were sacrificed, and the brains were sectioned and analyzed for apoptosis.
As shown in Figure 3A, microinjection of HIV-infected U87-CD4-CCR5 cells into cortices of Cx43fl/fl mice led to significant apoptosis around the site of microinjection (84.28 ± 6.38%). However, injection of AGA along with HIV-infected cells prevented apoptosis as these animals exhibited significantly lesser percentage of apoptotic cells (23.30 ± 7.51%), implicating the role of GJs in mediating HIV-induced bystander apoptosis. Similarly, simultaneous injection of Cx43 blocking peptide averted apoptosis (19.22 ± 15.82% apoptotic cells) upon microinjection of HIV-infected cells in the Cx43fl/fl mice (Figure 3B). The control animals (Cx43fl/fl) where Scr peptide was injected along with HIV-infected cells exhibited 80.56 ± 12.02% apoptotic cells around the site of microinjection, confirming the role of Cx43 GJs and HCs in mediating HIV-induced bystander apoptosis. As expected, microinjecting HIV-infected U87-CD4-CCR5 cells into hGFAP-cre Cx43fl/fl mice cortices either in presence or absence of AGA/Cx43 blocking peptide did not alter the percentage of apoptotic cells due to the lack of Cx43 GJs and HCs (Figures 3A,B). These results indicate that Cx43 channels, GJs as well as HCs, are required for bystander killing of uninfected cells surrounding HIV-infected astrocytes.
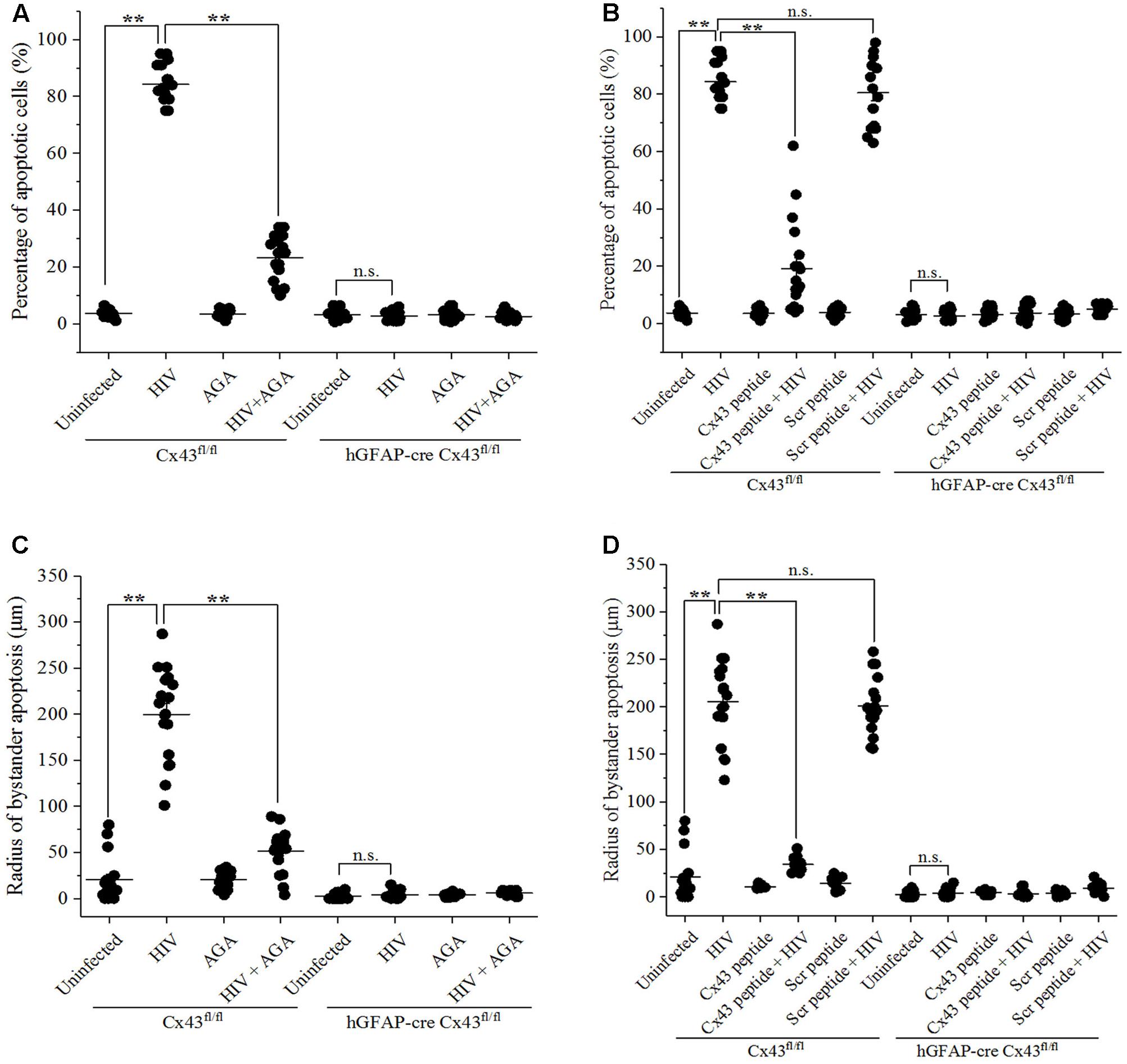
FIGURE 3. Blocking Cx43 prevents bystander toxicity induced by HIV-infected cells in the mouse brain. Human U87-CD4-CCR5 cells infected with HIVADA (50 ng/ml) were microinjected into the cortices of Cx43fl/fl as well as hGFAP-cre Cx43fl/fl mice, and simultaneously either GJ blocker, AGA (35 μM), or Cx43 blocking peptide (100 μM) was injected to inhibit GJ- and HC-mediated communication. After 48 h, animals were sacrificed, and brain sections were analyzed for apoptosis by TUNEL assay. (A,B) Blocking GJ-mediated communication by AGA (A), and blocking GJ- as well as Cx43 HC-mediated crosstalk by Cx43 blocking peptide (B) led to prevention of apoptosis in Cx43fl/fl mice signifying the role of Cx43 in mediating HIV-induced bystander toxicity. However, hGFAP-cre Cx43fl/fl mice had no effect on apoptotic cell death upon injection of either AGA (A) or Cx43 blocking peptide (B) due to the absence of Cx43 GJs and HCs. (C,D) Cx43fl/fl mice where either AGA (C) or Cx43 blocking peptide (D) was administered along with microinjection of HIV-infected cells, also had significantly smaller radii of bystander apoptosis as compared to Cx43fl/fl mice with active GJ and HC crosstalk, implying the role of Cx43 GJs and HCs in bystander apoptosis. ∗∗p < 0.005; n.s. – not significant.
Similarly, the radius of bystander apoptosis from the site of microinjection of HIV-infected cells was significantly smaller when either AGA (51.61 ± 22.71 μm) (Figure 3C) or Cx43 blocking peptide (34.44 ± 6.54 μm) (Figure 3D) was injected along with HIV-infected cells in Cx43fl/fl mice as opposed to when the HIV-infected cells were microinjected alone (199.72 ± 49.52 μm). However, in case of hGFAP-cre Cx43fl/fl mice, the addition of neither AGA (Figure 3C) nor Cx43 blocking peptide (Figure 3D) during microinjection of HIV-infected/uninfected cells had any significant difference in the radius of bystander apoptosis due to the absence of Cx43 channels. These observations reiterate the significance of Cx43 GJs and HCs in mediating bystander apoptosis of uninfected cells during HIV pathogenesis.
Bystander Apoptosis in Mice Microinjected with HIV-Infected Human Astrocytes Involves Inositol Triphosphate Receptor Activation and Increase in [Ca2+]i
Bystander apoptosis during HIV infection in astrocytes has been reported to involve cytochrome c, IP3 receptors (IP3Rs), and Ca2+ release (Eugenin and Berman, 2013). To determine whether bystander apoptosis in mice cortices upon microinjection of HIV-infected cells involved IP3Rs and Ca2+ signaling, Xestospongin C (XeC), an IP3R antagonist, and BAPTA-AM, a cell-permeant Ca2+ chelator, were used in the study. At the time of microinjection, either XeC (10 μM) or BAPTA-AM (5 μM) was administered along with HIV-infected/uninfected U87-CD4-CCR5 cells into the cortices of hGFAP-cre Cx43fl/fl and Cx43fl/fl mice. After 48 h, animals were sacrificed and brain sections were analyzed for apoptosis by TUNEL assay. As shown in Figure 4A, inhibition of IP3R-mediated Ca2+ release by XeC resulted in significantly lesser percentage of apoptotic cells (30.55 ± 16.55%) in Cx43fl/fl mice after microinjection of HIV-infected cells as opposed to control conditions (84.28 ± 6.38%). Morover, abolishing increase in intracellular Ca2+ levels by addition of BAPTA-AM prevented bystander apoptosis as only 14.91 ± 6.54% cells were apoptotic after microinjection of HIV-infected cells in Cx43fl/fl mice (Figure 4C). However, use of either XeC or BAPTA-AM during microinjection of HIV-infected U87-CD4-CCR5 cells into hGFAP-cre Cx43fl/fl mice cortices had no significant difference on the extent of bystander apoptosis as these animals do not express Cx43 GJs and HCs (Figures 4A,C). These results suggest that HIV-induced bystander apoptosis involves IP3R activation and increase in intracellular Ca2+ levels.
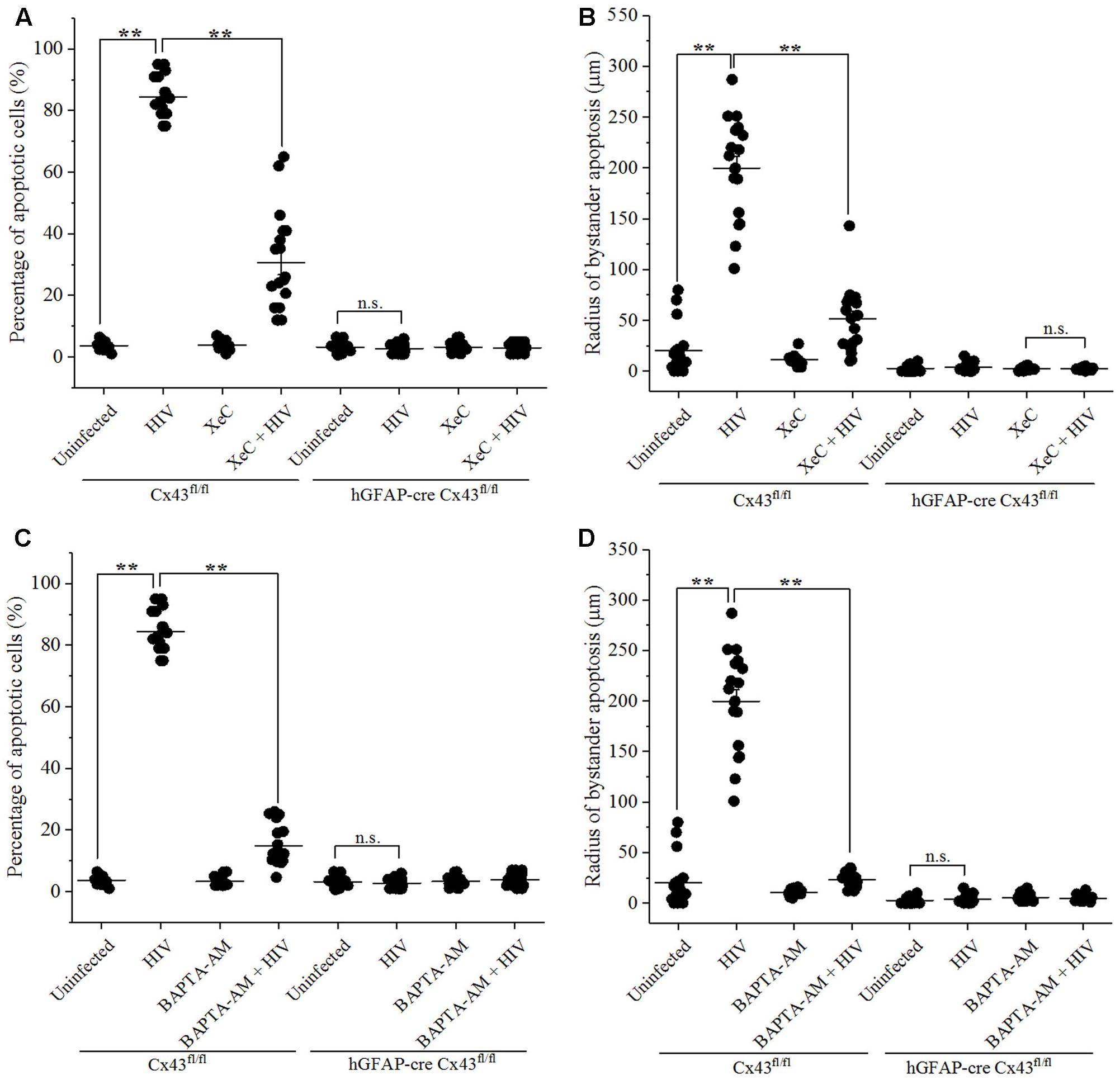
FIGURE 4. Bystander apoptosis induced by HIV-infected astrocytes microinjected in mice brain involves IP3Rs and Ca2+ release. Human U87-CD4-CCR5 cells infected with HIVADA (50 ng/ml) were microinjected into the cortices of Cx43fl/fl as well as hGFAP-cre Cx43fl/fl mice, and simultaneously either IP3R blocker, XeC (10 μM), or intracellular Ca2+ chelator, BAPTA-AM (100 μM), was administered to assess Ca2+ dependency. After 48 h, animals were sacrificed, and the brain sections were analyzed for apoptosis by TUNEL assay. (A,B) Bystander apoptosis induced by microinjected HIV-infected cells in Cx43fl/fl mice involved IP3R-mediated Ca2+ release, since inhibition of IP3R activation by XeC prevented bystander toxicity as assessed by percentage of apoptotic cells (A) as well as by radius of bystander apoptosis from the site of microinjection (B). (C,D) Administration of BAPTA-AM along with microinjection of HIV-infected cells in Cx43fl/fl mice also averted toxicity by regulating [Ca2+]i levels leading to lesser percentage of apoptotic cells (C) and smaller radius of bystander apoptosis (D) as compared to control condition. However, in case of hGFAP-cre Cx43fl/fl mice, use of either XeC (A,B) or BAPTA-AM (C,D) had no significant effect as these animals did not exhibit bystander apoptosis due to lack of Cx43 GJs and HCs. ∗∗p < 0.005; n.s. – not significant.
Analysis of the radius of bystander apoptosis from the site of microinjection of HIV-infected/uninfected cells revealed similar observations. The use of XeC and BAPTA-AM restricted the presence of apoptotic cells to 51.36 ± 32.34 μm (Figure 4B) and 23.28 ± 7.04 μm (Figure 4D), respectively, from the site of microinjection in Cx43fl/fl mice, whereas the control mice exhibited much larger radius of bystander apoptosis (199.72 ± 49.52 μm) when HIV-infected astrocytes were microinjected without IP3R inhibitor and Ca2+ chelator. hGFAP-cre Cx43fl/fl mice did not exhibit any significant differences in terms of radius of bystander apoptosis upon microinjection of HIV-infected cells with either XeC (Figure 4B) or BAPTA-AM (Figure 4D). These results stregthen our observations that Cx43 GJs and HCs mediate HIV-induced bystander toxicity which involves activation of IP3Rs and elevation of intracellular Ca2+ levels.
Discussion
In the current scenario of combined ART, HIV/AIDS has transformed from a life-threatening disease to a chronic disorder that compromises the surviving HIV-infected individuals with accelerated aging, immune reconstitution inflammatory syndrome, ART-associated toxicities, and persistence of the virus in latent hard-to-detect viral reservoirs in anatomical sites including the brain (Johnson and Nath, 2011; Nath and Clements, 2011). Although the prevalence of HIV-associated dementia has declined considerably in the post-ART era, the increasing pervasiveness of the milder forms of neurocognitive impairment still poses a threat to the complete eradication of the virus from the infected individuals (Chan et al., 2016).
Though the extent of HIV infection in the brain is limited to perivascular macrophages, microglia, and a small percentage of astrocytes, the magnitude of HIV neuropathogenesis suggests the involvement of certain host intercellular communication systems that amplify HIV pathology (Malik and Eugenin, 2016). Our laboratory and others have demonstrated that HIV infection targets host intercellular communication systems including GJs, HCs, tunneling nanotubes, and microvesicles/exosomes leading to widespread inflammation (Konadu et al., 2015), bystander apoptosis of uninfected cells (Eugenin et al., 2011), and assist the virus in evading host immune response (Kadiu et al., 2012). Several pathological conditions such as ischemia, stroke and traumatic brain injury, as well as viral and bacterial infections lead to down-regulation of Cx expression and hence GJ-mediated communication (Eugenin et al., 2012). However, during HIV infection, GJ-mediated communication is maintained in astrocytes which enables the diffusion of toxic/apoptotic signals from the few HIV-infected cells to gap-junctionally coupled uninfected cells, thereby amplifying HIV neuropathogenesis (Eugenin and Berman, 2007, 2013; Eugenin et al., 2011).
Our previous studies on HIV-induced bystander toxicity have revealed that HIV-infected astrocytes exhibit mitochondrial dysfunction which leads to an uncontrolled release of cytochrome c into the cytoplasm resulting in apoptosis of surrounding uninfected cells (Eugenin and Berman, 2013). We have demonstrated that second messengers, IP3 and Ca2+, generated in HIV-infected astrocytes as a consequence of mitochondrial dysfunction, diffuse into the surrounding uninfected cells via GJs, leading to their apoptosis since blocking either GJ crosstalk or cytochrome c/IP3 signaling abolished bystander apoptosis (Eugenin and Berman, 2013). Since the major Cx in astrocytes is Cx43, we wanted to critically examine the role of Cx43 in mediating HIV-induced bystander apoptosis.
HIV encounters dual restriction during infection and replication in the murine cells. First, the interaction between viral envelope protein, gp120, and murine surface receptors, CD4 and CCR5, does not lead to infection in the mouse cells (Browning et al., 1997). Second, HIV transactivating protein (Tat) does not bind to mouse cyclin-T1 and hence cannot activate viral transcription in the murine cells (Garber et al., 1998). Hence, murine models are relevant to study HIV-induced bystander damage in brain cells due to their inability to support HIV infection and replication. To decipher the role of astrocytic Cx43, we used a murine model with an astrocyte-directed deletion of Cx43 gene, hGFAP-cre Cx43fl/fl. This mouse model has been extremely useful in studies dealing with astrocyte-specific deletion of Cx43 (Frisch et al., 2003; Theis et al., 2003, 2004; Sridharan et al., 2007), especially because systemic deletion of Cx43 causes early postnatal death (Reaume et al., 1995).
In this study, we have demonstrated that HIV-infected human astrocytoma U87-CD4-CCR5 cells microinjected into the mouse brain, form GJs with resident mouse brain cells, and the apoptotic factors generated in these HIV-infected cells diffuse into uninfected host cells via Cx43 GJs, and lead to their apoptosis. Our results clearly signify the role of Cx43 GJs and HCs in mediating the spread of toxic factors from damaged/pro-apoptotic cells to healthy cells during HIV pathogenesis, as even without supporting HIV infection/replication, mouse brain cells undergo apoptosis owing to the presence of Cx43 protein. The involvement of Cx43 GJs and HCs in bystander apoptosis in the context of HIV is supported by our results from Cx43-deficient hGFAP-cre Cx43fl/fl mouse model, as well as from experiments using GJ blocker and Cx43 blocking peptide in Cx43fl/fl mouse model. Normally, Cx43 is down-regulated in viral infections, but CNS HIV infection is different because HIV uses Cx43 to spread toxicity from few HIV-infected cells to uninfected cells including neurons, astrocytes, and endothelial cells. Thus, Cx43 is used by the non-replicating virus to survive in the host.
Cx43 is the principle Cx in astrocytes, and plays an important role in neuroglia interactions (Malik and Eugenin, 2017). Cx43 knockout animals have been shown to exhibit impaired neuronal plasticity (Han et al., 2014), altered glutamatergic synaptic activity (Chever et al., 2014), and neurodevelopmental defects (Wiencken-Barger et al., 2007). However, limiting GJ-mediated communication during stroke (Wu et al., 2015), ischemia (Contreras et al., 2004), and oxidative stress (Blanc et al., 1998) has been documented to reduce neuronal apoptosis. Moreover, certain GJ blockers have been shown to exhibit neuroprotective as well as cardioprotective effects, and hence partially blocking gap-junctional communication could be favorable in certain pathological events (Herve and Sarrouilhe, 2005; Schulz et al., 2015).
Our study provides significant insights into the role of Cx43 in HIV-induced bystander apoptosis, and is highly relevant since even a low rate of HIV infection in astrocytes represents a significant viral reservoir which could reactivate and repopulate the periphery during ART withdrawal (Li et al., 2016). Since astrocytes form extensive intercellular GJ networks that amplify HIV neuropathogenesis several folds, astrocytes warrant extensive examination to limit the increasing rates of cognitive disease in HIV-infected individuals.
Author Contributions
SM performed experiments with in vitro microinjection, analyzed all data, and wrote the paper. MT designed the study. EAE designed the study, performed all experiments with animal microinjection, and wrote the paper.
Funding
This work was funded by National Institute of Mental Health (Grant No. MH096625), National Institute of Neurological Disorders and Stroke (Grant No. NS105584), and Public Health Research Institute funding to EAE.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2017.00404/full#supplementary-material
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anthony, I. C., Ramage, S. N., Carnie, F. W., Simmonds, P., and Bell, J. E. (2005). Influence of HAART on HIV-related CNS disease and neuroinflammation. J. Neuropathol. Exp. Neurol. 64, 529–536. doi: 10.1093/jnen/64.6.529
Antinori, A., Arendt, G., Becker, J. T., Brew, B. J., Byrd, D. A., Cherner, M., et al. (2007). Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69, 1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b
Bennett, M. V., Contreras, J. E., Bukauskas, F. F., and Saez, J. C. (2003). New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 26, 610–617. doi: 10.1016/j.tins.2003.09.008
Berman, J. W., Carvallo, L., Buckner, C. M., Luers, A., Prevedel, L., Bennett, M. V., et al. (2016). HIV-tat alters Connexin43 expression and trafficking in human astrocytes: role in NeuroAIDS. J. Neuroinflammation 13:54. doi: 10.1186/s12974-016-0510-1
Bjorndal, A., Deng, H., Jansson, M., Fiore, J. R., Colognesi, C., Karlsson, A., et al. (1997). Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71, 7478–7487.
Blanc, E. M., Bruce-Keller, A. J., and Mattson, M. P. (1998). Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. J. Neurochem. 70, 958–970. doi: 10.1046/j.1471-4159.1998.70030958.x
Browning, J., Horner, J. W., Pettoello-Mantovani, M., Raker, C., Yurasov, S., DePinho, R. A., et al. (1997). Mice transgenic for human CD4 and CCR5 are susceptible to HIV infection. Proc. Natl. Acad. Sci. U.S.A. 94, 14637–14641. doi: 10.1073/pnas.94.26.14637
Chan, P., Hellmuth, J., Spudich, S., and Valcour, V. (2016). Cognitive impairment and persistent CNS injury in treated HIV. Curr. HIV/AIDS Rep. 13, 209–217. doi: 10.1007/s11904-016-0319-7
Chever, O., Pannasch, U., Ezan, P., and Rouach, N. (2014). Astroglial connexin 43 sustains glutamatergic synaptic efficacy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130596. doi: 10.1098/rstb.2013.0596
Chun, T. W., Davey, R. T. Jr., Engel, D., Lane, H. C., and Fauci, A. S. (1999). Re-emergence of HIV after stopping therapy. Nature 401, 874–875. doi: 10.1038/44755
Churchill, M. J., Gorry, P. R., Cowley, D., Lal, L., Sonza, S., Purcell, D. F., et al. (2006). Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 12, 146–152. doi: 10.1080/13550280600748946
Clifford, D. B. (2017). HIV-associated neurocognitive disorder. Curr. Opin. Infect. Dis. 30, 117–122. doi: 10.1097/QCO.0000000000000328
Contreras, J. E., Sanchez, H. A., Veliz, L. P., Bukauskas, F. F., Bennett, M. V., and Saez, J. C. (2004). Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res. Brain Res. Rev. 47, 290–303. doi: 10.1016/j.brainresrev.2004.08.002
Eugenin, E. A., Basilio, D., Saez, J. C., Orellana, J. A., Raine, C. S., Bukauskas, F., et al. (2012). The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J. Neuroimmune Pharmacol. 7, 499–518. doi: 10.1007/s11481-012-9352-5
Eugenin, E. A., and Berman, J. W. (2007). Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J. Neurosci. 27, 12844–12850. doi: 10.1523/JNEUROSCI.4154-07.2007
Eugenin, E. A., and Berman, J. W. (2013). Cytochrome C dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. J. Neurochem. 127, 644–651. doi: 10.1111/jnc.12443
Eugenin, E. A., Clements, J. E., Zink, M. C., and Berman, J. W. (2011). Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 31, 9456–9465. doi: 10.1523/JNEUROSCI.1460-11.2011
Frisch, C., Theis, M., De Souza Silva, M. A., Dere, E., Sohl, G., Teubner, B., et al. (2003). Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur. J. Neurosci. 18, 2313–2318. doi: 10.1046/j.1460-9568.2003.02971.x
Garber, M. E., Wei, P., KewalRamani, V. N., Mayall, T. P., Herrmann, C. H., Rice, A. P., et al. (1998). The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 12, 3512–3527. doi: 10.1101/gad.12.22.3512
Han, Y., Yu, H. X., Sun, M. L., Wang, Y., Xi, W., and Yu, Y. Q. (2014). Astrocyte-restricted disruption of connexin-43 impairs neuronal plasticity in mouse barrel cortex. Eur. J. Neurosci. 39, 35–45. doi: 10.1111/ejn.12394
Heaton, R. K., Clifford, D. B., Franklin, D. R. Jr., Woods, S. P., Ake, C., Vaida, F., et al. (2010). HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: charter study. Neurology 75, 2087–2096. doi: 10.1212/WNL.0b013e318200d727
Herve, J. C., and Sarrouilhe, D. (2005). Connexin-made channels as pharmacological targets. Curr. Pharm. Des. 11, 1941–1958. doi: 10.2174/1381612054021060
Johnson, T., and Nath, A. (2011). Immune reconstitution inflammatory syndrome and the central nervous system. Curr. Opin. Neurol. 24, 284–290. doi: 10.1097/WCO.0b013e328346be57
Kadiu, I., Narayanasamy, P., Dash, P. K., Zhang, W., and Gendelman, H. E. (2012). Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J. Immunol. 189, 744–754. doi: 10.4049/jimmunol.1102244
Konadu, K. A., Chu, J., Huang, M. B., Amancha, P. K., Armstrong, W., Powell, M. D., et al. (2015). Association of cytokines with exosomes in the plasma of HIV-1-seropositive individuals. J. Infect. Dis. 211, 1712–1716. doi: 10.1093/infdis/jiu676
Li, G. H., Henderson, L., and Nath, A. (2016). Astrocytes as an HIV reservoir: mechanism of HIV infection. Curr. HIV Res. 14, 373–381. doi: 10.2174/1570162X14666161006121455
Malik, S., and Eugenin, E. A. (2016). Mechanisms of HIV neuropathogenesis: role of cellular communication systems. Curr. HIV Res. 14, 400–411. doi: 10.2174/1570162X14666160324124558
Malik, S., and Eugenin, E. A. (2017). Role of connexin and pannexin containing channels in HIV infection and NeuroAIDS. Neurosci. Lett. doi: 10.1016/j.neulet.2017.09.005 [Epub ahead of print].
Nath, A., and Clements, J. E. (2011). Eradication of HIV from the brain: reasons for pause. AIDS 25, 577–580. doi: 10.1097/QAD.0b013e3283437d2f
Orellana, J. A., Saez, J. C., Bennett, M. V., Berman, J. W., Morgello, S., and Eugenin, E. A. (2014). HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J. Neurochem. 128, 752–763. doi: 10.1111/jnc.12492
Reaume, A. G., de Sousa, P. A., Kulkarni, S., Langille, B. L., Zhu, D., Davies, T. C., et al. (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831–1834. doi: 10.1126/science.7892609
Saez, J. C., Contreras, J. E., Bukauskas, F. F., Retamal, M. A., and Bennett, M. V. (2003). Gap junction hemichannels in astrocytes of the CNS. Acta Physiol. Scand. 179, 9–22. doi: 10.1046/j.1365-201X.2003.01196.x
Schulz, R., Gorge, P. M., Gorbe, A., Ferdinandy, P., Lampe, P. D., and Leybaert, L. (2015). Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol. Ther. 153, 90–106. doi: 10.1016/j.pharmthera.2015.06.005
Sridharan, S., Simon, L., Meling, D. D., Cyr, D. G., Gutstein, D. E., Fishman, G. I., et al. (2007). Proliferation of adult sertoli cells following conditional knockout of the gap junctional protein GJA1 (connexin 43) in mice. Biol. Reprod. 76, 804–812. doi: 10.1095/biolreprod.106.059212
Theis, M., de Wit, C., Schlaeger, T. M., Eckardt, D., Kruger, O., Doring, B., et al. (2001). Endothelium-specific replacement of the connexin43 coding region by a lacZ reporter gene. Genesis 29, 1–13. doi: 10.1002/1526-968X(200101)29:1<1::AID-GENE1000>3.0.CO;2-0
Theis, M., Jauch, R., Zhuo, L., Speidel, D., Wallraff, A., Doring, B., et al. (2003). Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J. Neurosci. 23, 766–776.
Theis, M., Speidel, D., and Willecke, K. (2004). Astrocyte cultures from conditional connexin43-deficient mice. Glia 46, 130–141. doi: 10.1002/glia.10350
Wiencken-Barger, A. E., Djukic, B., Casper, K. B., and McCarthy, K. D. (2007). A role for connexin43 during neurodevelopment. Glia 55, 675–686. doi: 10.1002/glia.20484
Wiley, C. A., Achim, C. L., Christopherson, C., Kidane, Y., Kwok, S., Masliah, E., et al. (1999). HIV mediates a productive infection of the brain. AIDS 13, 2055–2059. doi: 10.1097/00002030-199910220-00007
Wu, L. Y., Yu, X. L., and Feng, L. Y. (2015). Connexin 43 stabilizes astrocytes in a stroke-like milieu to facilitate neuronal recovery. Acta Pharmacol. Sin. 36, 928–938. doi: 10.1038/aps.2015.39
Keywords: NeuroAIDS, HIV, connexin43, astrocyte, apoptosis
Citation: Malik S, Theis M and Eugenin EA (2017) Connexin43 Containing Gap Junction Channels Facilitate HIV Bystander Toxicity: Implications in NeuroHIV. Front. Mol. Neurosci. 10:404. doi: 10.3389/fnmol.2017.00404
Received: 01 August 2017; Accepted: 21 November 2017;
Published: 05 December 2017.
Edited by:
Sheriar Hormuzdi, University of Dundee, United KingdomReviewed by:
Olimpia Meucci, Drexel University, United StatesAndrew MacLean, Tulane University School of Medicine, United States
Copyright © 2017 Malik, Theis and Eugenin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eliseo A. Eugenin, eliseo.eugenin@rutgers.edu