 James Z. Curlin1,2†
James Z. Curlin1,2† Kimberly Schmitt1†
Kimberly Schmitt1† Leila Remling-Mulder1
Leila Remling-Mulder1 Ryan V. Moriarty3John J. Baczenas3Kelly Goff4Shelby O'Connor3
Ryan V. Moriarty3John J. Baczenas3Kelly Goff4Shelby O'Connor3 Mark Stenglein1
Mark Stenglein1 Preston A. Marx4,5
Preston A. Marx4,5 Ramesh Akkina1*
Ramesh Akkina1*- 1Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, CO, United States
- 2Antiviral Discovery, Evaluation and Application Research (ADEAR) Training Program, Department of Medicine, University of Colorado, Aurora, CO, United States
- 3Department of Pathology and Laboratory Medicine, School of Medicine and Public Health, University of Wisconsin, Madison, WI, United States
- 4Department of Tropical Medicine, School of Public Health and Tropical Medicine, Tulane University, New Orleans, LA, United States
- 5Tulane National Primate Research Center, Covington, LA, United States
Simian immunodeficiency virus native to sooty mangabeys (SIVsm) is believed to have given rise to HIV-2 through cross-species transmission and evolution in the human. SIVmac239 and SIVB670, pathogenic to macaques, and SIVhu, isolated from an accidental human infection, also have origins in SIVsm. With their common ancestral lineage as that of HIV-2 from the progenitor SIVsm, but with different passage history in different hosts, they provide a unique opportunity to evaluate cross-species transmission to a new host and their adaptation/evolution both in terms of potential genetic and phenotypic changes. Using humanized mice with a transplanted human system, we evaluated in vivo replication kinetics, CD4+ T cell dynamics and genetic adaptive changes during serial passage with a goal to understand their evolution under human selective immune pressure. All the three viruses readily infected hu-mice causing chronic viremia. While SIVmac and SIVB670 caused CD4+ T cell depletion during sequential passaging, SIVhu with a deletion in nef gene was found to be less pathogenic. Deep sequencing of the genomes of these viruses isolated at different times revealed numerous adaptive mutations of significance that increased in frequency during sequential passages. The ability of these viruses to infect and replicate in humanized mice provides a new small animal model to study SIVs in vivo in addition to more expensive macaques. Since SIVmac and related viruses have been indispensable in many areas of HIV pathogenesis, therapeutics and cure research, availability of this small animal hu-mouse model that is susceptible to both SIV and HIV viruses is likely to open novel avenues of investigation for comparative studies using the same host.
Introduction
The causative agents of AIDS, both HIV-1 and HIV-2, remain entrenched in the human population and continue their onslaught in the ongoing global health crisis. While HIV-1 receives the most attention due to its wider global distribution and prominence, studies on HIV-2 and its relatives have been relatively sparse and infrequent. Ten HIV-2 strains currently exist whose origins are traced back to a series of independent cross-species transmissions of simian immunodeficiency viruses (SIVsm) native to sooty mangabeys (1, 2). Several previous studies have sought to understand the genetic adaptations that were necessary for SIVsm to transition into HIV-2 (3, 4). Interestingly, besides human infections, SIVsm crossed the species barrier infecting additional non-human primate (NHP) species generating strains such as SIVmac and SIVB670 as well as other SIVsm derivatives such as SIVhu (5–10).
So far, over 40 other types of non-human primate derived SIVs have been identified, some with zoonotic potential. Thus, it is important to understand the nature of adaptations that these viruses undergo when transmitted to divergent host species (10). The SIVmac239 virus is a unique immunodeficiency causing virus resulting from an accidental transmission between two different NHP species, in this case from sooty mangabeys to rhesus macaques (5). While SIVsm is relatively harmless to its native host, SIVmac is highly pathogenic to rhesus macaques (9). The ability of SIVmac239 to cause the rapid onset of AIDS-like symptoms in rhesus macaques has led to SIVmac239 and its derivatives becoming important surrogate lentiviruses commonly used for understanding HIV pathogenesis (7, 8). Since the majority of endogenous SIVs do not cause AIDS-like symptoms in their native hosts (6, 11), SIVmac239 also serves as a useful tool for understanding the genetic adaptive changes contributing to the increased virological fitness and mobility between host species. Similar to SIVmac239, SIVB670 is widely used in non-human primate models to study lentivirus pathology (12, 13). SIVB670 is a derivative of SIVsm that was originally isolated from rhesus macaques that had been inoculated with cutaneous lepromatous leprosy lesion tissue homogenates from SIVsm infected sooty mangabeys. Infected macaque lymphoid tissues were then co-cultured with a human T cell line to isolate the virus SIVB670 (6, 14). This virus has been commonly used in HIV research due to its aggressive pathology and immunosuppression (15, 16).
In the context of rare accidental human laboratory exposures to SIVs, a laboratory worker in 1992 became exposed to SIVB670. This subject became virus positive with a productive infection and was carefully monitored for several years (17, 18). While the subject never developed AIDS-like symptoms, viral reactive antibodies were detected and an SIV was isolated from the subject's PBMC. This viral isolate was termed SIVhu denoting its isolation from a human. Genetic analysis of the viral isolate revealed a small 4 bp deletion in the nef gene that is absent in the parental SIVB670 strain resulting in a premature stop codon leading to a truncated non-functional protein (18). Due to its replication in the human and isolation from this source as well as its parental origin from SIVB670, SIVhu virus may represent an evolutionary intermediate variant between SIVsm and HIV-2. In essence, SIVmac239, SIVhu, SIVB670 and HIV-2, all have a common origin in the prototype SIVsm virus. Study of these viruses in a common in vivo setting is likely to provide interesting data in terms of their relative replicative ability and viral adaptation in a new host environment.
Host tropism of viruses and their adaptation to a new host are dictated by a complex interplay between various host and viral factors. Inherent differences exist between humans and non-human primates in terms of host factors and their respective immune environments thus restricting the cross-species transmission of lentiviruses in several ways. In addition to subtle differences in the CD4 and chemokine receptors necessary for viral entry, a number of host restriction factors play a role. Among some of these are tetherin, APOBEC3G (apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G) and TRIM5α (tripartite motif 5α protein), which are all involved in blocking viral production at various points in the viral replication cycle (19–23). While host restriction factors in human cells help impede SIV replication, they allow HIV replication and the reverse is true in simian cells (24–26). Many of the accessory proteins in HIV such as Vif, Vpx, Vpr, Vpu, and Nef are all well-known for their ability to counteract human host restriction factors (19, 27–32). Thus, counteracting restriction factors successfully is a key aspect for cross species viral transmissions.
A suitable and affordable model is necessary to effectively evaluate the initial transmission and gradual adaptation of these viruses. Non-human primate models (NHPs) have historically been important models to study HIV pathogenesis and curative strategies. However, to determine the genetic changes in SIVs that gave rise to HIV, an alternative model that can permit repeated SIV exposures to a human immune system is needed. Humanized mice, which are mice that have been xenografted with human hematopoietic stem cells (HSC), can provide a surrogative human immune environment to delineate the important questions on both SIV and HIV infection process and viral pathogenesis (3, 4, 33–39). Commonly used humanized mice models are hu-HSC and BLT mouse models. BLT mice are constructed by the transplantation of liver/thymus fragments surgically engrafted under the kidney capsule, while hu-HSC mice are derived through the direct injection of HSC (40–44). Both mouse models are capable of producing human T cells, B cells, macrophages, and dendritic cells de novo. Both the models permit HIV infection due to the presence of full immune cell repertoire and have been used widely to investigate areas such as HIV pathogenesis, transmission, and latency (33, 35, 44–47). We previously used the hu-mouse model as a human surrogate system to dissect various aspects of the initial cross-species transmission of SIV chimpanzee virus (SIVcpz) to the human giving rise to HIV-1 and the SIVsm progenitor virus in giving rise to HIV-2 (3, 4, 39, 48–50). These studies provided important insights into the important genetic changes these progenitor viruses had to undergo to quickly adapt to the human host and also shed light on the gradual genetic changes that arose as the viruses were serially passaged multiple times using this system.
The existence of three related viruses namely SIVmac239, SIVhu, SIVB670 that arose in different circumstances, but with a common origin in SIVsm which is a progenitor to HIV-2, provided a unique opportunity to evaluate their ability to cause a potential human infection in a human surrogate hu-mouse model and to ask important questions in an experimental setting. Here we determined if these viruses can infect humanized mice to simulate cross species transmission and serially passaged them in vivo to identify viral adaptive phenotypic and genetic changes with the goal to identify potential common adaptive changes between these three viruses. These studies also opened a new avenue to develop a cost-effective small animal model that can permit in vivo studies on SIVmac239 which is widely studied in more expensive NHP models. Our results show that all three viruses can productively infect humanized mice, cause chronic viremia and affect CD4+ T cell dynamics. Furthermore, evaluation of these serially passaged viruses using this system mimics the necessary viral adaptation needed during the initial phases of viral encroachment into the human population to reveal important mutations among these three viruses.
Results
SIVmac239, SIVB670, and SIVhu Are All Capable of Productive and Chronic Infection in Humanized Mice
To determine if SIVmac239, SIVB670, and SIVhu can establish productive infection, cohorts of five hu-HSC mice were inoculated with one of the three viruses and serially passaged for a total of three generations. Within 1 week of inoculation, SIVmac239 RNA was detected in the plasma using qRT-PCR in all five infected hu-HSC mice demonstrating that SIVmac239 can infect human cells in vivo (Figure 1A). During the first generation, the plasma viral loads continued to climb gradually until ~64 days post-inoculation peaking at over 3.4 × 104 RNA copies/mL. This first generation proved successful chronic infection of human immune cells in hu-HSC mice by SIVmac239. After the virus was serially passaged into a second generation of hu-mice, the viral loads displayed a remarkably similar pattern to the first generation and were detected within 1 week. Interestingly, several individual mice in the third generation displayed drastically higher peak plasma viral loads than the others, reaching 1 × 106 RNA copies/mL within 21 days post-inoculation, suggesting that the virus had fundamentally adapted to human immune cells displaying increased viral fitness in these hu-mice (Figure 1A).
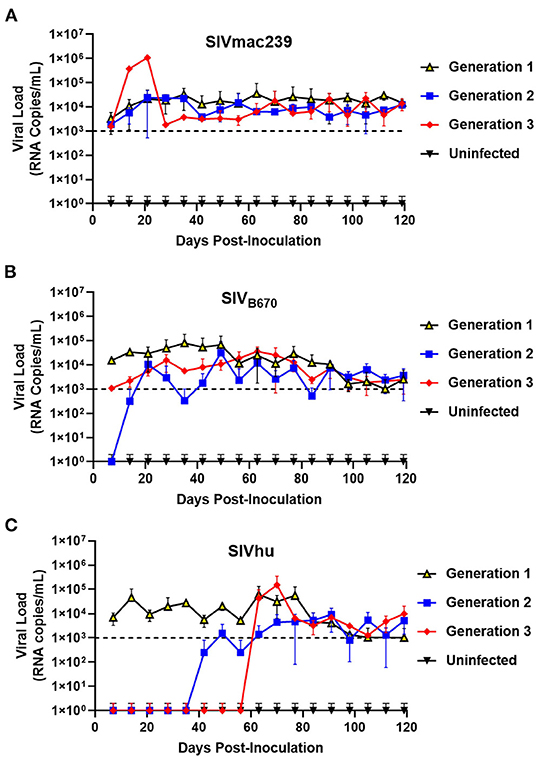
Figure 1. SIVmac239, SIVB670, and SIVhu can successfully initiate productive infection of hu-mice and are capable of serial transmission. (A) SIVmac239, (B) SIVB670, and (C) SIVhu plasma viral loads during three generations of serial passage. Plasma viral loads were assessed using qRT-PCR on a weekly basis for the duration of each passage. The limit of detection was 1 × 103 RNA copies/mL.
SIVB670 displayed a pattern of infection similar to that of SIVmac239. Following the initial inoculation, the virus was detected within the first week (1 × 104 RNA copies/mL) (Figure 1B). The viral loads gradually increased peaking (8 × 104 RNA copies/mL) around 35 days post-inoculation, at which point the viral loads showed a steady but persistent decline until the end of the first generation. In contrast, the second generation was not detected above the limit of detection until ~21 days post-inoculation and the viral loads peaked (3 × 104 RNA copies/mL) lower than the first generation at around 49 days post-inoculation. Interestingly, the plasma viral loads of the third generation were initially over a log lower than those observed in the first generation. However, the third generation showed drastically less fluctuation than the second generation peaking (3.64 × 104 RNA copies/mL) by 63 days post-inoculation.
Within the first generation of infection, SIVhu was initially detectable in the plasma by the first week and remained within a log of that before peaking (6 × 104 RNA copies/mL) around 63 days post-inoculation (Figure 1C). Following this viral peak, the viral loads rapidly dropped to around the limit of detection until the end of the generation. This demonstrated that SIVhu could readily infect human immune cells leading to productive viremia. After serial passaging, the virus struggled to replicate, and did not become detectable in the second generation until around 49 days post-inoculation. Similarly, the third generation was a continuation of the trend seen in the second generation, with the virus remaining undetected until around 63 days post-inoculation before shortly peaking (1.5 × 105 RNA copies/mL). No positive viral loads were detected in any of the uninfected control mice from these studies (Figure 1).
SIVmac239 and SIVB670 Infection Results in CD4+ T Cell Decline, but SIVhu Infection Does Not
One of the hallmarks of HIV infection in humans as well as hu-mice is the depletion of CD4+ T cell lymphocytes during the course of infection (33, 41, 44, 51, 52). In the first generation, SIVmac239 displayed a slight decline in CD4+ T cell levels within the first 100 days post-inoculation (Figure 2A). This resulted in no real net decrease of CD4+ T cell levels. In contrast, the second generation showed an immediate decline in CD4+ T cell levels through 70 days post-inoculation, while the third generation displayed a rapid decline in CD4+ T cells beginning around 28 days post-inoculation as a result of the rapid increase in plasma viral loads. By the end of the third generation, there was a statistically significant difference in the CD4+ T cell decline when compared to the uninfected control hu-mice similar to HIV-1 infection (two-tailed, Student's t-test, p < 0.01) demonstrating that serially passaging SIVmac239 results in a more rapid CD4+ T cell depletion in the humanized mouse model (33, 41, 44, 51, 52).
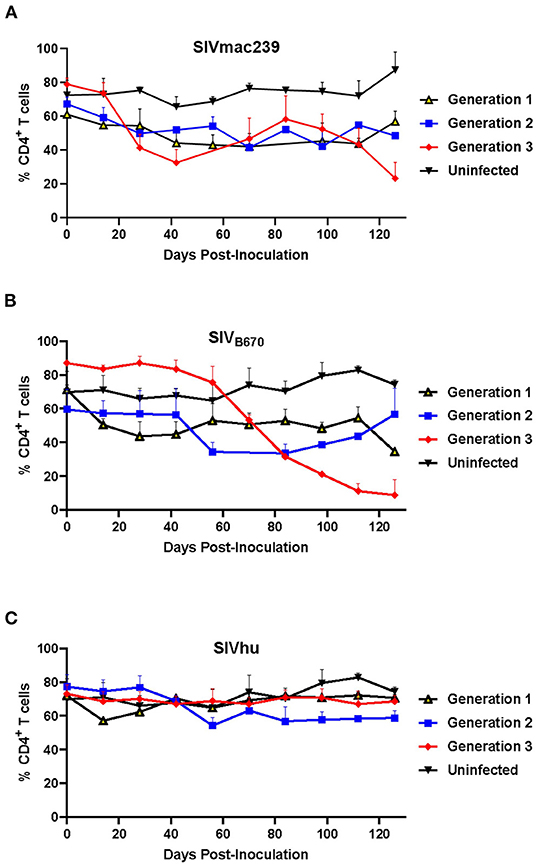
Figure 2. CD4+ T cells levels during three generations of serial passage of SIVmac239, SIVB670, and SIVhu in humanized mice. (A) SIVmac239, (B) SIVB670, and (C) SIVhu CD4+ T cell levels over three generations. CD4+ T cell levels were monitored bimonthly using flow cytometry. The percent of CD4+ T cells is presented relative to that of CD45+/CD3+ cell populations. Statistically significant CD4+ T cell depletion was seen in the third generation of SIVmac239 (p < 0.01) and SIVB670 (p < 0.05) infected hu-HSC mice relative to the uninfected control (two-tailed Student's t-test).
The first generation of SIVB670 displayed a drastic decline in CD4+ T cells compared to the controls within 14 days post-inoculation (Figure 2B). In contrast, the CD4+ T cell levels in the second generation remained initially stable through 42 days post-inoculation, followed by a decrease in CD4+ T cell levels. By the third generation, SIVB670 displayed a very consistent and gradual decline in CD4+ T cell levels until 56 days post-inoculation, followed by significantly rapid CD4+ T cell decline relative to the uninfected controls (p < 0.05). This data suggests that SIVB670 showed enhanced CD4+ T cell depletion in the humanized mouse model indicative of increased viral fitness to human immune cells by the third generation.
Flow cytometric analysis showed that there was little to no significant CD4+ T cell decline across any of the three generations of serial passaging among mice infected with SIVhu. In fact, the CD4+ T cell levels remained relatively consistent across each generation (Figure 2C). Altogether, this data demonstrates that the truncated Nef protein in SIVhu may delay viral replication kinetics in human immune cells.
Evolutionary Genetic Adaptations Arise During Sequential Passaging of SIVmac239, SIVB670, and SIVhu
In addition to the in vivo pathogenicity of these viruses, adaptation was assessed by identifying non-synonymous mutations that rose in frequency over time that could change the phenotype of the virus. Therefore, we used Illumina-based deep sequencing on plasma viral RNA from two mice (replicates) per generation collected at early, mid, and late timepoints across three serial passages. Paired end reads of the viruses were mapped to the consensus sequence of the starting viral stock. While there were numerous mutations that arose briefly and disappeared at later timepoints, the following criteria were used to identify truly significant variants: (1) Mutations must be non-synonymous and present in the coding sequence (CDS), (2) The mutations must be present in at least four of the data sets which means each variant is present beyond a single replicate in a single generation, (3) Mutations must increase in viral frequency over the subsequent generations, and (4) The frequency of the mutation in the viral population must average at least 50% at the last sequenced timepoint. Eight mutations were found in SIVmac239, 32 mutations were found in SIVB670, and 11 mutations were found in SIVhu that matched these criteria by the end of the third generation (Figure 3, Tables 1–3).
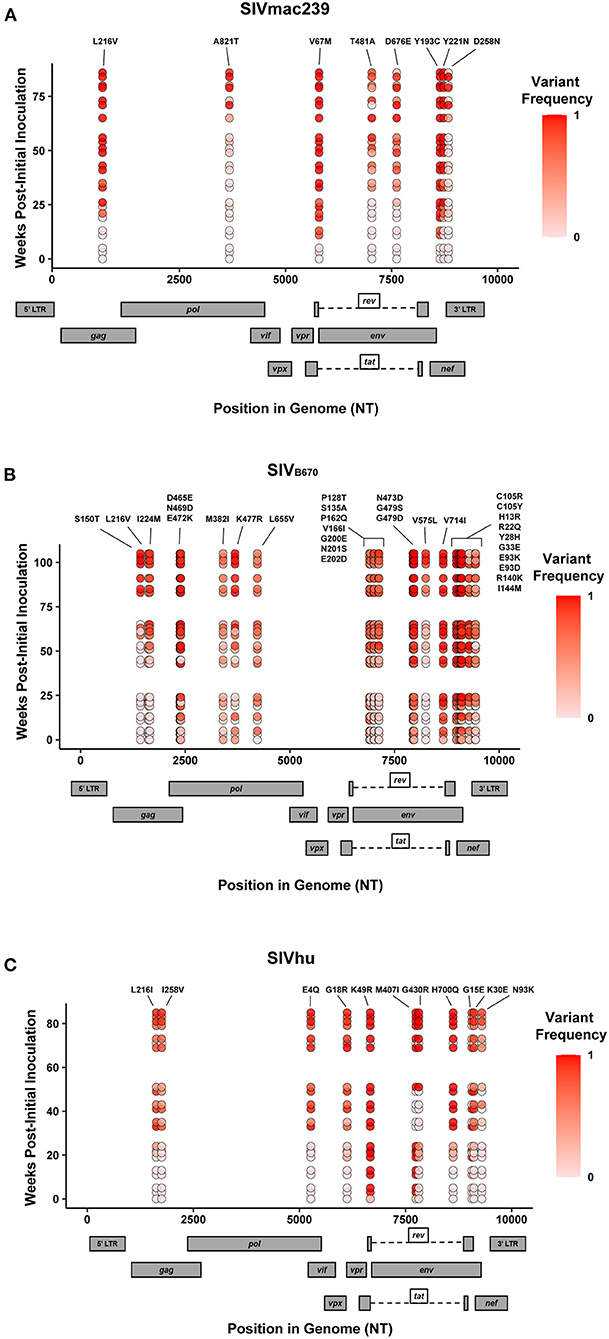
Figure 3. Single nucleotide polymorphisms showing an increase in population frequency over three serial passages. Viral RNAs isolated from two infected hu-mice per viral strain per generation at early, middle, and late timepoints were sequenced. The non-synonymous mutations identified were present in the CDS, had an average endpoint frequency of at least 50%, and were found in at least four timepoints. (A) SIVmac239, (B) SIVB670, and (C) SIVhu variants are plotted based on their position in the genome along the x-axis. The y-axis location denotes the time of collection weeks post-initial infection. Variant population frequency is denoted by the shading color scale for each point. Week 0 corresponds to the frequency in the viral stock pre-inoculation, with the second and third passages being plotted beginning at week 30 and 60, respectively. For each time point, replicates are offset vertically from each other. Both the residue changes and number for each position are listed above their respective locations.
Table 1. Amino acid substitutions accumulated during three passages of SIVmac239 in hu-mice.
Non-synonymous mutations were identified in SIVmac239 after three generations of passage across a number of genes, with the largest concentration in env and nef (Figure 3A, Table 1). Of these mutations, Env D676E and Nef D258N had individual replicate samples in the third generation that had persistently low frequencies, while all the other identified mutations were near 100% population frequency in both replicates. SIVB670 had by far the largest number of qualifying mutations, with a large number of mutations found in gag, pol, env, and nef (Figure 3B, Table 2). Interestingly, though a number of these mutations did increase to a frequency of nearly 100%, quite a few mutations were still at lower frequencies, albeit they had still increased in frequency relative to the stock virus. SIVhu had fewer qualifying mutations than SIVB670, with a relatively even distribution that was slightly weighted toward the 5′ end of the genome with env and nef having the most mutations (Figure 3C, Table 3). Interestingly, all of the mutations found in env were found to be at 100% frequency in the viral population of both replicates of the third generation. One mutation in particular at residue Gag 216, was present in all three viral strains, though the residue change was L → I in SIVhu and L → V in SIVB670 and SIVmac239, which may suggest some shared function in adaptation.
Table 2. Amino acid substitutions accumulated during three passages of SIVB670 in hu-mice.
Table 3. Amino acid substitutions accumulated during three passages of SIVhu in hu-mice.
Discussion
Here we described a unique human surrogate experimental setting that permitted the evaluation of in vivo infection features of three different SIVsm-derived viruses, namely SIVmac239, SIVB670, and SIVhu in humanized mice. This system enables the simulation of cross-species viral transmission into the human and viral evolution under this selective pressure. Serial in vivo passaging of these viruses in hu-mice allowed for the identification of the genetic changes that potentially occur during the early stages of viral adaptation to humans. In this study, hu-mice mice were exposed to SIVmac239, SIVhu, and SIVB670 by i/p injection to first ascertain their potential for infection of human immune cells in vivo. Viruses isolated from the subsequent serial passages at different stages of infection were subjected to sequence analysis to identify and ascertain if any common changes could be found among these viruses given their shared ancestry. These studies also recapitulated some key aspects of cross-species transmission that we described previously in humanized mice that centered on SIVcpz and SIVsm to shed light on the origins of HIV-1 and HIV-2 from these respective progenitor viruses (3, 4, 48, 50). Of the key observations from this present study, one was that both SIVmac239 and SIVB670 were readily able to infect hu-mice (Figures 1A,B). Viremia was detected within a week following the initial inoculation and persisted for both the viruses beyond 120 days. Successful sequential serial passaging of these viruses in hu-mice imply that these viruses have the capacity for maintaining human-to-human transmission and can evolve further to increased viral fitness.
CD4+ helper T cell loss was evaluated for these viruses across the three in vivo viral passages because this feature is a central hallmark of immunodeficiency and viral virulence (Figure 2). While the CD4+ T cell levels remained steady during chronic infection in passages 1 and 2 of SIVmac239 and SIVB670, a progressive and more rapid loss of these cells was observed during the 3rd passage indicating increased viral adaptation to these cells (Figures 2A,B). SIVhu also displayed productive infection upon the initial i/p inoculation followed by chronic viremia in during the first passage (Figure 1C). However, the SIVhu viral loads were lower compared to SIVmac239 and SIVB670 and it took longer to establish detectable viremia in the 2nd and 3rd passages. Also, in contrast to SIVmac239 and SIVB670, no discernable CD4+ T cell loss could be detected compared to uninfected control mice across all the 3 passages of SIVhu (Figure 2C). The lower replicative capacity of the SIVhu virus is likely due to the defective nature of this virus isolate harboring a deletion in the nef gene (see below).
Sequential serial passaging of each of these viruses for 3 generations in different cohorts of hu-mice allowed the original virus to adapt to the human immune cell environment in vivo and accumulate genetic mutations potentially conferring fitness (Figure 3, Tables 1–3). Overall, relative to SIVmac239 and SIVhu, SIVB670 accumulated the largest number of non-synonymous mutations with corresponding amino acid changes throughout the viral genome (Figure 3B, Table 2). The majority of these changes were found in the 3′ end of the viral genome similar to that found in previous studies on SIVsm and SIVcpz in hu-mouse studies (3, 37, 48–50). Interestingly, one of the most striking features amongst the three passaged viruses was the mutation at residue 216 in Gag that appeared within all three different strains of viruses in a highly conserved part of the genome (26, 53). This residue is part of the gag gene that encodes the capsid protein and is located within the cyclophilin binding loop. Furthermore, it is possible that this residue is involved in TRIM5α antagonism and escape, which is an important barrier to overcome for successful cross-species transmission (54, 55). The dramatic increases in the frequency of viral populations bearing this mutation further supports its potential role in conferring improved fitness (56–58).
While functional studies are needed to fully understand the biological significance of these mutations, their occurrence in known motifs involved in binding interactions provide some potential clues. For example, the amino acid changes at Gag 465 and 469 in SIVB670 are located within a dileucine-motif shown to be responsible for particle association and uptake of Vpx and Vpr in SIVmac (59, 60). This interaction between the DXAXXLL motif and Vpr allows Vpr to be diverted away from the proteasome-degradative pathway, while deletion of this particular motif was shown to result in a total lack of incorporation of Vpx in SIVmac (60). The fact that this motif altered by the D465E mutation had no marked effect on pathogenicity for SIVB670 is notable. It could be due to the fact that the LXXLF motif at the C terminus of the p6gag region remained intact, and that they share some overlap in functionality (60). Just as compelling is that the Gag N469D mutation brings SIVB670 closer to amino acid sequences found in both SIVsmE041 and SIVmac239, which were already aspartic acid residues at this particular location (53). Very few notable mutations were identified in vif, vpr, and vpx in all three viruses studied. However, the SIVhu Vpr G18R residue change is immediately adjacent to a previously described DCAF-1 binding motif canonically found in both Vpr and Vpx which has been implicated in G2 arrest and SAMHD1 degradation, respectively (29).
Both env and nef genes displayed a large number of mutations in all three viruses that increased in frequency over time and may ultimately have functional implications regarding virus-host restriction factor interactions. The Env V67M mutation identified in SIVmac239 increased from 0% frequency in the starting stock virus to 100% by the end of the third passage. This mutation was previously shown to increase viral infectivity (61). The mechanism for this increased infectivity was not tied to neutralizing antibody escape, but was implicated in conferring macrophage tropism (62). Amino acid changes P128T and S135A of SIVB670 in the Env are located within the V1 hypervariable loop region implicated in neutralizing antibody susceptibility (63, 64). Since proline residues contributing to high degree of conformational rigidity are less frequent in protein binding sites, its substitution for a threonine could suggest a shift in functionality for this region of the protein (65). Other mutations such as Env G430R in SIVhu occurred in the V4 loop and could potentially affect neutralization. Additional mutations that were identified, but did not meet the previously described criteria, such as Env I421K, R425G, and Q428R all occurred in the V4 loop of SIVB670 and could be involved in neutralizing antibody escape due to undergoing variation early in infection (66–68). The R425G substitution, at approximately the equivalent P421 residue in SIVmac239, has been directly implicated in neutralizing antibody escape and increased infectivity when mutated into P421Q in SIVmac239 (61). Env N201S, and R481Q in SIVB670 and T481A in SIVmac239 occur within N-linked glycosylation motifs of the V2 and V5 loops. The effect of N-linked glycosylation motifs in the envelope proteins of HIV and SIV on antibody neutralization have been well-characterized. These motifs allow a glycan shield to form, which protects exposed epitopes from being bound by neutralizing antibodies (69, 70). The Rev-response element (RRE) is a critical binding motif necessary for the shuttling of transcripts from the nucleus to the cytoplasm. It is possible that Env V575L in SIVB670, which falls within the RRE could be advantageously affecting this interaction, given that this mutation increased in frequency to make up nearly 90% of the viral population by the third generation.
While smaller in size than env, the nef gene accumulated a higher number of non-synonymous mutations. However, given that APOBEC3 deaminase activity begins on the 5′ end of the viral genome, it is possible that a restriction factor like APOBEC3 could play a role in the abundance of non-synonymous mutations occurring in nef though this requires further analysis (71). Similarly, to mutations seen in other genes, many of the substitutions in nef occur near previously characterized binding and interaction sites. For example, the Nef residue 93 changes that occur in both SIVhu and SIVB670 occur within a known PACS-1 binding site (72). The Nef Y193C substitution identified in SIVmac239 has a low evolutionary probability (Supplementary Figure 1) (73). This residue is found within a ExxxL (L/M) (di-leucine) motif in the α4 region of Nef, which is implicated as an AP interaction site involved in the downregulation of CD3 and CD4 (74). Additionally, this exact residue has also been implicated in tetherin antagonism (75). At a similar location in the stock viral sequences of SIVsmE041 and SIVB670 this residue is a cysteine. In HIV-2 and SIVmac, it has been shown that the di-leucine motif is flexible enough to form an alpha helix by binding to a hydrophobic crevice between α2 and α3 with Glu 190 and Leu 194 (74). Change in amino acid residue 193 to a cysteine could have a drastic impact on the ability of the di-leucine motif to bind to the hydrophobic crevice. Furthermore, Nef I144M from SIVB670 is located within the α3 region of Nef and is adjacent to a number of residues that have been shown to interact with the amino acid residues of the di-leucine sorting motif (76). However, this substitution is more biochemically favored than the others and may have less of an impact on binding for this region. The Nef Y221N substitution in SIVmac is located at the end of α5 helix and is a highly disfavored residue change based on the PAM1 matrix score (Supplementary Figure 1A). It is also adjacent to residue Y223 and Y226, which are involved in MHC-I downregulation and be functionally disrupted through substitutions (77). In SIVB670 residue Nef Y28H occurs at a conserved N-proximal tyrosine residue that has been found to be part of an adapter protein (AP) binding site that may be involved in mediating endocytosis (72, 78). Nef K30E in SIVhu also occurs at this same AP binding site.
Despite being the only strain of SIV isolated directly from a human subject, SIVhu displayed the lowest overall potential to gain increased fitness to the human among the three viruses tested during the serial passages. While the first generation showed viral loads comparable to the other two viruses, a clear decline in PVL and prolonged delay in establishing detectable viremia during subsequent passages suggests that SIVhu is slow to adapt to human immune cells (Figure 1C). Furthermore, the lack of significant CD4+ T cell decline indicates that SIVhu is not yet capable of producing AIDS-like symptoms in humanized mice after three passages (Figure 2C). However, the increased peaks and general upward trend of the third generation of plasma viral loads suggest that SIVhu is still adapting, albeit slowly. The noted difficulty that SIVhu displays could be at least partially attributed to the documented frameshift deletion in nef that encodes a truncated protein (18). Nef has been shown to be involved in downregulating the CD4 and MHC class I molecules and helps to mitigate cell-mediated cytotoxicity (79–82). Additionally, Nef is involved in counteracting the action of host restriction factors such as tetherin and SERINC3/5 and is therefore critical for the virus to properly function (27, 30, 83). No restoring mutations meeting our selection criteria were present near the frameshift or premature truncation, which indicates that after three generations of serial passaging, the nef gene had not reverted, which is supported by the consistently low plasma viral loads.
In summary, the above data showed for the first time that SIVmac239 and SIVB670 both with origins in NHP macaques can infect hu-mice and that cross-species transmission studies simulating human infection are feasible in this system for future investigations of zoonosis centered on lentiviruses. We also established that SIVmac239 and SIVsm related viruses can cause chronic infection leading to CD4+ T cell loss in hu-mice, thus mimicking key aspects of SIV pathogenesis. This has practical implications as this small animal model will permit novel studies in vivo using these primate lentiviruses that continue to play an important role in HIV pathogenesis and therapeutic studies. With regards to evolution, adaptive changes seen in all three viruses in the hu-mouse point to both unique and consensus changes in the viral genomes. Whether these evolving viruses resemble HIV-2 after prolonged and additional passages in vivo remains to be investigated and is something that we hope to explore in the future.
Materials and Methods
Cell Culture
The 293T (ATCC CRL-3216) and the TZM-bl (ARP-8129) reporter cell lines were maintained with DMEM media that contained 1% L-glutamine, 10% heat inactivated fetal bovine serum (HI FBS), and 1% antibiotic-antimycotic mix (Thermo Fisher Scientific, Waltham, MA, United States). CEMx174 (ARP-272) cells were maintained with RPMI media that contained 10% HI FBS and 2 × antibiotic-antimycotic mix (Thermo Fisher Scientific, Waltham, MA, United States). Whole blood filter packs were obtained from the Garth Englund Blood Center of Fort Collins, CO, United States. Mononuclear cells were isolated by Ficoll-Paque density centrifugation. PBMC were maintained in RPMI media with 1 × antibiotic/antimycotic mix (Thermo Fisher Scientific, Waltham, MA, United States), 10% heat-inactivated fetal bovine serum, and 20 ng/mL IL-2 (R&D Systems, Inc., Minneapolis, MN, United States). For viral propagation, PBMC were CD8-depleted by positive selection and stimulated with 100 ng/mL of anti-CD3 and anti-CD28 soluble antibody (Miltenyi Biotec Inc., Auburn, CA, United States) for 48 h.
Generation of Hu-HSC and BLT Mice
All mice used in these studies were cared for in the Colorado State University Painter Animal Center. Fetal liver-derived human CD34+ cells obtained from Advanced Bioscience Resources (ABR, Alameda CA), were isolated, column purified (Miltenyi Biotec, San Diego, CA), cultured and assessed for purity using flow cytometry (3, 4, 39, 48, 84, 85). To create humanized hematopoietic stem cell (hu-HSC) mice, neonatal (1–4 day old) Balb/c Rag1−/−γc−/− or Balb/c Rag2−/−γc−/− mouse pups were sublethally irradiated (350 rads) prior to intrahepatic injection of 0.5–1 × 106 CD34+ cells (3, 34, 86). For humanized bone marrow, liver, thymus (BLT) mice, adult Balb/c Rag1−/−γc−/− or Balb/c Rag2−/−γc−/− mice were surgically engrafted with a combination of human fetal liver and thymic tissue under the kidney capsule. This was followed by a tail-vein injection of autologous CD34+ human hematopoietic stem cells from the same source as the liver and thymic tissue (40, 41, 44). Ten to twelve weeks after engraftment, peripheral blood was collected via tail vein puncture and used to assess human immune cell engraftment. Red blood cells were lysed with the Whole Blood Erythrocyte Lysing Kit (R & D Systems, Minneapolis, MN) according to the manufacturer's instructions. The fractioned white blood cells were stained for flow cytometry using fluorophore conjugated hCD45-FITC, hCD3-PE, and hCD4-PE/Cy5 (BD Pharmingen, San Jose, CA).
SIVmac239, SIVhu, and SIVB670 Viral Stock Preparation and in vivo Infection
The SIVmac239 molecular clone plasmid was transfected into 293T cells, and the viral supernatant was concentrated using ultracentrifugation as described previously (39, 50). A small aliquot from ultracentrifugation was set aside for a functional titer in TZM-bl reporter cells as previously described (50, 87, 88). SIVhu and SIVB670 viral supernatants were obtained from the NIH AIDS Reagent Program and were cultured according to their recommended guidelines (6, 18, 89). Cohorts of hu-HSC and/or BLT mice with high human hematopoietic engraftment levels were inoculated with ~200 μL of viral supernatant via intraperitoneal (i/p) injection with one of the respective progenitor viruses (SIVmac239: 1 × 105.5 TCID50/mL, SIVhu: 2 × 108 TCID50/mL, SIVB670: 5.78 × 107 TCID50/mL).
Virus Propagation and Serial Passaging of SIVmac239, SIVhu, and SIVB670
At 24 weeks post-infection, the viremic mice that showed the highest plasma viral loads were euthanized to propagate the virus from the first passage to the next. Whole blood was collected via cardiac puncture for PBMC and the spleen, lymph nodes and bone marrow were collected as previously described (50). Leukocyte fractions of the cells were collected by Ficoll-Paque density centrifugation and seeded at a density of 2–3 × 106 cells/mL. These cells were then activated for roughly 48 h using 100 ng/mL of anti-hCD3 and anti-hCD28 soluble antibody (Miltenyi Biotec Inc., Auburn, CA). To enhance viral infection of the next cohort of hu-mice, these cells were co-cultured for 48 h with freshly isolated splenocytes obtained from the new hu-mouse cohort used for serial passage. These cultured cells together with culture supernatants containing virus were then intraperitoneally inoculated into the next batch of hu-mice.
Plasma Viral Load Determination by qRT-PCR
During each passage, peripheral blood was collected on a weekly basis to assess plasma viral loads (PVL). Viral RNA from the plasma was extracted with the E.Z.N.A. Viral RNA kit as outlined by the manufacturer's recommendations (OMEGA bio-tek, Norcross, GA). This RNA was then used to determine PVL using qRT-PCR using the iScript One-Step RT-PCR kit with SYBR green (Bio Rad, Hercules, CA). Primers were designed for SIVmac239 based on the ltr sequence (GenBank accession: M33262.1), while SIVhu and SIVB670 used primers designed for a conserved region of the ltr sequence in SIVsmE041 (GenBank accession: HM059825.1). The primers used for PCR were as follows: 1. SIVmac239: forward 5′-GCAGGTAAGTGCAACACAAA-3′ and reverse 5′-CCTGACAAGACGGAGTTTCT-3′, Tm = 54°C and 2. SIVB670 and SIVhu: forward 5′-CCACAAAGGGGATGTTATGGGG-3′ and reverse 5′-AACCTCCCAGGGCTCAATCT-3′, Tm = 60°C. These primers were used with the following cycling reactions for qRT-PCR quantification: 50°C for 10 min, 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 54 or 60°C for 30 s using a Bio Rad C1000 Thermo Cycler and CFX96 Real-Time System (Bio Rad, Hercules, CA). The standard curve was determined using a series of 10-fold dilutions of viral SIVmac239, and SIVsmE041 ltr at a known concentration. The limit of detection was 1,000 copies/mL.
CD4+ T Cell Level Determination
Peripheral blood was collected bi-monthly to assess human CD4+ T cell engraftment levels using flow cytometry. Briefly, 5 μL of FcγR-block (Jackson ImmunoResearch Laboratories, Inc. West Grove, PA) was added to the blood for 5 min. Each blood sample was then stained with fluorophore conjugated hCD45-FITC, hCD3-PE and hCD4-PE/Cy5 (BD Pharmingen, San Jose, CA) for roughly 30 min. Erythrocytes were lysed with the Whole Blood Erythrocyte Lysing kit based on the manufacturer's guidelines (R&D Systems, Minneapolis, MN). Antibody-stained cells were then fixed in 1% paraformaldehyde/PBS and passed through a 0.45 μm filter. Samples were run on the BD Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA). CD4+ T cell levels were assessed as a percentage of the CD3+/CD45+ cell population. Flow cytometry data was analyzed using the FlowJo v10.0.7 software package (FlowJo LLC, Ashland, OR). A two-tailed Student's t-test was used to determine CD4+ T cell decline between the uninfected and infected mouse groups as indicated in the figure legend.
Amplicon and Illumina-Based Deep Sequencing Preparation
To identify potential adaptive mutations, viral RNA from two mice per passage at multiple timepoints from across the duration of the passage, i.e., 3, 11, 19, and 23 weeks post-inoculation, etc. as well as the starting stock viruses were used for sequencing. Briefly, cDNA was synthesized from the viral RNA with a SuperScript IV kit according to the manufacturer's guidelines (Invitrogen, Carlsbad, CA). Two multiplexed inter-overlapping primer pools that span the coding region of the viral genomes based on the SIVmac239 reference sequence (GenBank accession: M33262.1) and an independently sequenced SIVhu reference sequence for SIVhu and SIVB670 were designed using Primal Scheme (https://primal.zibraproject.org) (90). These primer pools were then amplified using viral cDNA and Q5 Hot Start High Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA) as described by Quick et al., to produce roughly 400 bp overlapping amplicons of the viral genomes (Supplementary Tables 1, 2). Agencourt AMPure XP (Beckman Coulter Life Sciences, Indianapolis, IN) magnetic beads were used to purify the amplicons, which were further prepped for Illumina-based deep sequencing with the TruSeq Nano DNA HT Library Preparation Kit according to the manufacturer's guidelines (Illumina, San Diego, CA). Sequencing of the amplicon library was performed on a MiSeq Illumina desktop sequencer (Invitrogen, Carlsbad, CA) in the Pathogen Sequencing Services unit at the Wisconsin National Primate Research Center.
Calculation of Non-synonymous Single Nucleotide Polymorphism Frequencies
The deep sequencing reads were prepared for analysis by filtering out low quality reads, and by trimming the ~30 bp primer sequences and adapter sequences off of the reads with cutadapt software v1.9.1 (91). Filtered reads were aligned to the stock virus sequence with bowtie2 software v2.2.5 (92). The resulting BAM format output was used as the input to call variants with lofreq software v2.1.2 (93). Variants identified here had >100 read depth and at least 1% frequency. Genome plots were generated using R and ggplot2 (ISBN: 0387981403). R scripts used to graph the plots can be found at https://github.com/stenglein-lab/viral_variant_explorer.
Data Availability Statement
The raw data supporting the conclusions of this article are available on the sequence read archive (SRA) (Accession numbers: SRR17194541–SRR17194610).
Ethics Statement
The animal study was reviewed and approved by Colorado State University IACUC.
Author Contributions
RA, JC, KS, MS, PM, and SO'C are responsible for the design and conduct of the project. LR-M, KG, JB, and RM provided technical assistance. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH, USA Grant R01 AI123234 to RA and in part by NIH, USA Grant R01 AI120021 to RA. In addition to the NIH grants listed above to RA, these experiments were further supported by the National Center for Research Resources, and the Office of Research Infrastructure Programs (ORIP) of the NIH through the grant OD011104 at the Tulane National Primate Research Center. Sequencing resources were provided through the NIH Grant P51OD011106 and the Wisconsin National Primate Research Center. Computational and sequencing analysis support was provided through the NIH/NCATS Colorado CTSA Grant Number UL1 TR002535. Partial support was also provided by the ADEAR Training Program NIH Grant T32AI150547.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: SIV B670 Virus from Dr. Michael Murphey-Corb, NIH: SIVhu Virus from Dr. Thomas Folks, NIH: 174xCEM Cells, ARP-272, contributed by Dr. Peter Cresswell, NIH: TZM-bl Cells, ARP-8129, contributed by Dr. John C. Kappes and Dr. Xiaoyun Wu. The 293T (ATCC CRL-3216) cells were obtained from the American Type Culture Collection (ATCC). Some of the data in this manuscript was part of a thesis by Curlin, J (94).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2021.813606/full#supplementary-material
References
1. Sharp PM, Hahn BH. Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med. (2011) 1:a006841. doi: 10.1101/cshperspect.a006841
2. Ayouba A, Akoua-Koffi C, Calvignac-Spencer S, Esteban A, Locatelli S, Li H, et al. Evidence for continuing cross-species transmission of SIVsmm to humans: characterization of a new HIV-2 lineage in rural Cote d'Ivoire. AIDS. (2013) 27:2488–91. doi: 10.1097/01.aids.0000432443.22684.50
3. Schmitt K, Mohan Kumar D, Curlin J, Remling-Mulder L, Stenglein M, O'Connor S, et al. Modeling the evolution of SIV sooty mangabey progenitor virus towards HIV-2 using humanized mice. Virology. (2017) 510:175–84. doi: 10.1016/j.virol.2017.07.005
4. Schmitt K, Curlin J, Kumar DM, Remling-Mulder L, Feely S, Stenglein M, et al. SIV progenitor evolution toward HIV: a humanized mouse surrogate model for SIVsm adaptation toward HIV-2. J Med Primatol. (2018) 47:298–301. doi: 10.1111/jmp.12380
5. Daniel MD, Letvin NL, King NW, Kannagi M, Sehgal PK, Hunt RD, et al. Isolation of T-cell tropic HTLV-III-like retrovirus from macaques. Science. (1985) 228:1201–4. doi: 10.1126/science.3159089
6. Murphey-Corb M, Martin LN, Rangan SR, Baskin GB, Gormus BJ, Wolf RH, et al. Isolation of an HTLV-III-related retrovirus from macaques with simian AIDS and its possible origin in asymptomatic mangabeys. Nature. (1986) 321:435–7. doi: 10.1038/321435a0
7. Naidu YM, Kestler HW III, Li Y, Butler CV, Silva DP, Schmidt DK, et al. Characterization of infectious molecular clones of simian immunodeficiency virus (SIVmac) and human immunodeficiency virus type 2: persistent infection of rhesus monkeys with molecularly cloned SIVmac. J Virol. (1988) 62:4691–6. doi: 10.1128/jvi.62.12.4691-4696.1988
8. Kestler H, Kodama T, Ringler D, Marthas M, Pedersen N, Lackner A, et al. Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science. (1990) 248:1109–12. doi: 10.1126/science.2160735
9. Apetrei C, Kaur A, Lerche NW, Metzger M, Pandrea I, Hardcastle J, et al. Molecular epidemiology of simian immunodeficiency virus SIVsm in U.S. primate centers unravels the origin of SIVmac and SIVstm. J Virol. (2005) 79:8991–9005. doi: 10.1128/JVI.79.14.8991-9005.2005
10. Bell SM, Bedford T. Modern-day SIV viral diversity generated by extensive recombination and cross-species transmission. PLoS Pathog. (2017) 13:e1006466. doi: 10.1371/journal.ppat.1006466
11. Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science. (2012) 335:1188–93. doi: 10.1126/science.1217550
12. Andrieu JM, Lu W. A 30-year journey of trial and error towards a tolerogenic AIDS vaccine. Arch Virol. (2018) 163:2025–31. doi: 10.1007/s00705-018-3936-1
13. Moretti S, Virtuoso S, Sernicola L, Farcomeni S, Maggiorella MT, Borsetti A. Advances in SIV/SHIV non-human primate models of neuroAIDS. Pathogens. (2021) 10:1018. doi: 10.3390/pathogens10081018
14. Wolf RH, Gormus BJ, Martin LN, Baskin GB, Walsh GP, Meyers WM, et al. Experimental leprosy in three species of monkeys. Science. (1985) 227:529–31. doi: 10.1126/science.3917577
15. Zink WE, Zheng J, Persidsky Y, Poluektova L, Gendelman HE. The neuropathogenesis of HIV-1 infection. FEMS Immunol Med Microbiol. (1999) 26:233–41. doi: 10.1111/j.1574-695X.1999.tb01394.x
16. Dorsey JL, Mangus LM, Hauer P, Ebenezer GJ, Queen SE, Laast VA, et al. Persistent peripheral nervous system damage in simian immunodeficiency virus-infected macaques receiving antiretroviral therapy. J Neuropathol Exp Neurol. (2015) 74:1053–60. doi: 10.1097/NEN.0000000000000249
17. Khabbaz RF, Rowe T, Murphey-Corb M, Heneine WM, Schable CA, George JR, et al. Simian immunodeficiency virus needlestick accident in a laboratory worker. Lancet. (1992) 340:271–3. doi: 10.1016/0140-6736(92)92358-M
18. Khabbaz RF, Heneine W, George JR, Parekh B, Rowe T, Woods T, et al. Brief report: infection of a laboratory worker with simian immunodeficiency virus. N Engl J Med. (1994) 330:172–7. doi: 10.1056/NEJM199401203300304
19. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. (2002) 418:646–50. doi: 10.1038/nature00939
20. Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. (2004) 430:569–73. doi: 10.1038/nature02777
21. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. (2004) 427:848–53. doi: 10.1038/nature02343
22. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. (2008) 451:425–30. doi: 10.1038/nature06553
23. Le Tortorec A, Neil SJ. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J Virol. (2009) 83:11966–78. doi: 10.1128/JVI.01515-09
24. Sauter D, Hue S, Petit SJ, Plantier JC, Towers GJ, Kirchhoff F, et al. HIV-1 Group P is unable to antagonize human tetherin by Vpu, Env or Nef. Retrovirology. (2011) 8:103. doi: 10.1186/1742-4690-8-103
25. Zhang X, Zhou T, Yang J, Lin Y, Shi J, Zhang X, et al. Identification of SERINC5-001 as the predominant spliced isoform for HIV-1 restriction. J Virol. (2017) 91:e00137–17. doi: 10.1128/JVI.00137-17
26. Sauter D, Kirchhoff F. Key viral adaptations preceding the AIDS pandemic. Cell Host Microbe. (2019) 25:27–38. doi: 10.1016/j.chom.2018.12.002
27. Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, et al. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. (2009) 5:e1000429. doi: 10.1371/journal.ppat.1000429
28. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. (2011) 474:654–7. doi: 10.1038/nature10117
29. Wei W, Guo H, Han X, Liu X, Zhou X, Zhang W, et al. A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cell Microbiol. (2012) 14:1745–56. doi: 10.1111/j.1462-5822.2012.01835.x
30. Usami Y, Wu Y, Gottlinger HG. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature. (2015) 526:218–23. doi: 10.1038/nature15400
31. Lubow J, Collins KL. Vpr Is a VIP: HIV Vpr and infected macrophages promote viral pathogenesis. Viruses. (2020) 12:809. doi: 10.3390/v12080809
32. Lubow J, Virgilio MC, Merlino M, Collins DR, Mashiba M, Peterson BG, et al. Mannose receptor is an HIV restriction factor counteracted by Vpr in macrophages. Elife. (2020) 9:e51035. doi: 10.7554/eLife.51035.sa2
33. Berges BK, Wheat WH, Palmer BE, Connick E, Akkina R. HIV-1 infection and CD4 T cell depletion in the humanized Rag2-/-gamma c-/- (RAG-hu) mouse model. Retrovirology. (2006) 3:76. doi: 10.1186/1742-4690-3-76
34. Berges BK, Akkina SR, Folkvord JM, Connick E, Akkina R. Mucosal transmission of R5 and X4 tropic HIV-1 via vaginal and rectal routes in humanized Rag2-/- gammac -/- (RAG-hu) mice. Virology. (2008) 373:342–51. doi: 10.1016/j.virol.2007.11.020
35. Berges BK, Akkina SR, Remling L, Akkina R. Humanized Rag2(-/-)gammac(-/-) (RAG-hu) mice can sustain long-term chronic HIV-1 infection lasting more than a year. Virology. (2010) 397:100–3. doi: 10.1016/j.virol.2009.10.034
36. Yuan Z, Kang G, Ma F, Lu W, Fan W, Fennessey CM, et al. Recapitulating cross-species transmission of simian immunodeficiency virus SIVcpz to humans by using humanized BLT mice. J Virol. (2016) 90:7728–39. doi: 10.1128/JVI.00860-16
37. Sato K, Misawa N, Takeuchi JS, Kobayashi T, Izumi T, Aso H, et al. Experimental adaptive evolution of simian immunodeficiency virus SIVcpz to pandemic human immunodeficiency virus type 1 by using a humanized mouse model. J Virol. (2018) 92:e01905–17. doi: 10.1128/JVI.01905-17
38. Yuan Z, Kang G, Daharsh L, Fan W, Li Q. SIVcpz closely related to the ancestral HIV-1 is less or non-pathogenic to humans in a hu-BLT mouse model. Emerg Microbes Infect. (2018) 7:59. doi: 10.1038/s41426-018-0062-9
39. Curlin J, Schmitt K, Remling-Mulder L, Moriarty R, Stenglein M, O'Connor S, et al. SIVcpz cross-species transmission and viral evolution toward HIV-1 in a humanized mouse model. J Med Primatol. (2020) 49:40–3. doi: 10.1111/jmp.12440
40. Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. (2006) 108:487–92. doi: 10.1182/blood-2005-11-4388
42. Garcia S, Freitas AA. Humanized mice: current states and perspectives. Immunol Lett. (2012) 146:1–7. doi: 10.1016/j.imlet.2012.03.009
43. Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. (2012) 12:786–98. doi: 10.1038/nri3311
44. Akkina R. New generation humanized mice for virus research: comparative aspects and future prospects. Virology. (2013) 435:14–28. doi: 10.1016/j.virol.2012.10.007
45. Neff CP, Ndolo T, Tandon A, Habu Y, Akkina R. Oral pre-exposure prophylaxis by anti-retrovirals raltegravir and maraviroc protects against HIV-1 vaginal transmission in a humanized mouse model. PLoS ONE. (2010) 5:e15257. doi: 10.1371/journal.pone.0015257
46. Choudhary SK, Archin NM, Cheema M, Dahl NP, Garcia JV, Margolis DM. Latent HIV-1 infection of resting CD4(+) T cells in the humanized Rag2(-)/(-) gammac(-)/(-) mouse. J Virol. (2012) 86:114–20. doi: 10.1128/JVI.05590-11
47. Charlins P, Schmitt K, Remling-Mulder L, Hogan LE, Hanhauser E, Hobbs KS, et al. A humanized mouse-based HIV-1 viral outgrowth assay with higher sensitivity than in vitro qVOA in detecting latently infected cells from individuals on ART with undetectable viral loads. Virology. (2017) 507:135–9. doi: 10.1016/j.virol.2017.04.011
48. Curlin J, Schmitt K, Remling-Mulder L, Moriarty R, Goff K, O'Connor S, et al. Evolution of SIVsm in humanized mice towards HIV-2. J Med Primatol. (2020) 49:280–3. doi: 10.1111/jmp.12486
49. Schmitt K, Curlin J, Remling-Mulder L, Moriarty R, Goff K, O'Connor S, et al. Mimicking SIV chimpanzee viral evolution toward HIV-1 during cross-species transmission. J Med Primatol. (2020) 49:284–7. doi: 10.1111/jmp.12485
50. Schmitt K, Curlin J, Remling-Mulder L, Moriarty R, Goff K, O'Connor S, et al. Cross-species transmission and evolution of SIV chimpanzee progenitor viruses toward HIV-1 in humanized mice. Front Microbiol. (2020) 11:1889. doi: 10.3389/fmicb.2020.01889
51. Aldrovandi GM, Feuer G, Gao L, Jamieson B, Kristeva M, Chen IS, et al. The SCID-hu mouse as a model for HIV-1 infection. Nature. (1993) 363:732–6. doi: 10.1038/363732a0
52. Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, Kurrer MO, et al. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci USA. (2006) 103:15951–6. doi: 10.1073/pnas.0604493103
53. Foley BL, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, ert al. HIV Sequence Compendium 2018. Los Alamos, New Mexico: Los Alamos National Laboratory, Theoretical Biology and Biophysics (2018).
54. Bukovsky AA, Weimann A, Accola MA, Gottlinger HG. Transfer of the HIV-1 cyclophilin-binding site to simian immunodeficiency virus from Macaca mulatta can confer both cyclosporin sensitivity and cyclosporin dependence. Proc Natl Acad Sci USA. (1997) 94:10943–8. doi: 10.1073/pnas.94.20.10943
55. Wu F, Kirmaier A, Goeken R, Ourmanov I, Hall L, Morgan JS, et al. TRIM5 alpha drives SIVsmm evolution in rhesus macaques. PLoS Pathog. (2013) 9:e1003577. doi: 10.1371/journal.ppat.1003577
56. Sanjuan R, Moya A, Elena SF. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc Natl Acad Sci USA. (2004) 101:8396–401. doi: 10.1073/pnas.0400146101
57. Cuevas JM, Domingo-Calap P, Sanjuan R. The fitness effects of synonymous mutations in DNA and RNA viruses. Mol Biol Evol. (2012) 29:17–20. doi: 10.1093/molbev/msr179
58. Acevedo A, Brodsky L, Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. (2014) 505:686–90. doi: 10.1038/nature12861
59. Pancio HA, Ratner L. Human immunodeficiency virus type 2 Vpx-Gag interaction. J Virol. (1998) 72:5271–5. doi: 10.1128/JVI.72.6.5271-5275.1998
60. Accola MA, Bukovsky AA, Jones MS, Gottlinger HG. A conserved dileucine-containing motif in p6(gag) governs the particle association of Vpx and Vpr of simian immunodeficiency viruses SIV(mac) and SIV(agm). J Virol. (1999) 73:9992–9. doi: 10.1128/JVI.73.12.9992-9999.1999
61. Sato S, Yuste E, Lauer WA, Chang EH, Morgan JS, Bixby JG, et al. Potent antibody-mediated neutralization and evolution of antigenic escape variants of simian immunodeficiency virus strain SIVmac239 in vivo. J Virol. (2008) 82:9739–52. doi: 10.1128/JVI.00871-08
62. Mori K, Ringler DJ, Kodama T, Desrosiers RC. Complex determinants of macrophage tropism in env of simian immunodeficiency virus. J Virol. (1992) 66:2067–75. doi: 10.1128/jvi.66.4.2067-2075.1992
63. Johnson WE, Morgan J, Reitter J, Puffer BA, Czajak S, Doms RW, et al. A replication-competent, neutralization-sensitive variant of simian immunodeficiency virus lacking 100 amino acids of envelope. J Virol. (2002) 76:2075–86. doi: 10.1128/jvi.76.5.2075-2086.2002
64. Saunders CJ, McCaffrey RA, Zharkikh I, Kraft Z, Malenbaum SE, Burke B, et al. The V1, V2, and V3 regions of the human immunodeficiency virus type 1 envelope differentially affect the viral phenotype in an isolate-dependent manner. J Virol. (2005) 79:9069–80. doi: 10.1128/JVI.79.14.9069-9080.2005
65. Betts M, Russell R. Amino acid properties and consequences of substitutions. In: Barnes M, editor. Bioinformatics for Geneticists. Hoboken: John Wiley & Sons (2003). p. 289−316.
66. Castro E, Belair M, Rizzardi GP, Bart PA, Pantaleo G, Graziosi C. Independent evolution of hypervariable regions of HIV-1 gp120: V4 as a swarm of N-Linked glycosylation variants. AIDS Res Hum Retroviruses. (2008) 24:106–13. doi: 10.1089/aid.2007.0139
67. Moore PL, Gray ES, Choge IA, Ranchobe N, Mlisana K, Abdool Karim SS, et al. The c3-v4 region is a major target of autologous neutralizing antibodies in human immunodeficiency virus type 1 subtype C infection. J Virol. (2008) 82:1860–9. doi: 10.1128/JVI.02187-07
68. Dieltjens T, Loots N, Vereecken K, Grupping K, Heyndrickx L, Bottieau E, et al. HIV type 1 subtype A envelope genetic evolution in a slow progressing individual with consistent broadly neutralizing antibodies. AIDS Res Hum Retroviruses. (2009) 25:1165–9. doi: 10.1089/aid.2008.0161
69. Chackerian B, Rudensey LM, Overbaugh J. Specific N-linked and O-linked glycosylation modifications in the envelope V1 domain of simian immunodeficiency virus variants that evolve in the host alter recognition by neutralizing antibodies. J Virol. (1997) 71:7719–27. doi: 10.1128/jvi.71.10.7719-7727.1997
70. Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, et al. Antibody neutralization and escape by HIV-1. Nature. (2003) 422:307–12. doi: 10.1038/nature01470
71. Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3' –> 5' on single-stranded DNA. Nat Struct Mol Biol. (2006) 13:392–9. doi: 10.1038/nsmb1086
72. Heusinger E, Deppe K, Sette P, Krapp C, Kmiec D, Kluge SF, et al. (2018). Preadaptation of simian immunodeficiency virus SIVsmm facilitated env-mediated counteraction of human tetherin by human immunodeficiency virus type 2. J Virol 92:e00276–18. doi: 10.1128/JVI.00276-18
73. Dayhoff MO. Atlas of Protein Sequence and Structure. Washington, DC: Georgetown University Medical Center (1972).
74. Manrique S, Sauter D, Horenkamp FA, Lulf S, Yu H, Hotter D, et al. Endocytic sorting motif interactions involved in Nef-mediated downmodulation of CD4 and CD3. Nat Commun. (2017) 8:442. doi: 10.1038/s41467-017-00481-z
75. Serra-Moreno R, Zimmermann K, Stern LJ, Evans DT. Tetherin/BST-2 antagonism by Nef depends on a direct physical interaction between Nef and tetherin, and on clathrin-mediated endocytosis. PLoS Pathog. (2013) 9:e1003487. doi: 10.1371/journal.ppat.1003487
76. Hirao K, Andrews S, Kuroki K, Kusaka H, Tadokoro T, Kita S, et al. Structure of HIV-2 nef reveals features distinct from HIV-1 involved in immune regulation. iScience. (2020) 23:100758. doi: 10.1016/j.isci.2019.100758
77. Swigut T, Iafrate AJ, Muench J, Kirchhoff F, Skowronski J. Simian and human immunodeficiency virus Nef proteins use different surfaces to downregulate class I major histocompatibility complex antigen expression. J Virol. (2000) 74:5691–701. doi: 10.1128/JVI.74.12.5691-5701.2000
78. Rowell JF, Stanhope PE, Siliciano RF. Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J Immunol. (1995) 155:473–88.
79. Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell. (1994) 76:853–64. doi: 10.1016/0092-8674(94)90360-3
80. Rhee SS, Marsh JW. Human immunodeficiency virus type 1 Nef-induced down-modulation of CD4 is due to rapid internalization and degradation of surface CD4. J Virol. (1994) 68:5156–63. doi: 10.1128/jvi.68.8.5156-5163.1994
81. Chaudhuri R, Lindwasser OW, Smith WJ, Hurley JH, Bonifacino JS. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J Virol. (2007) 81:3877–90. doi: 10.1128/JVI.02725-06
82. Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol. (2014) 88:2633–44. doi: 10.1128/JVI.03230-13
83. Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. (2009) 6:54–67. doi: 10.1016/j.chom.2009.05.008
84. Akkina RK, Rosenblatt JD, Campbell AG, Chen IS, Zack JA. Modeling human lymphoid precursor cell gene therapy in the SCID-hu mouse. Blood. (1994) 84:1393–8. doi: 10.1182/blood.V84.5.1393.1393
85. Bai J, Gorantla S, Banda N, Cagnon L, Rossi J, Akkina R. Characterization of anti-CCR5 ribozyme-transduced CD34+ hematopoietic progenitor cells in vitro and in a SCID-hu mouse model in vivo. Mol Ther. (2000) 1:244–54. doi: 10.1006/mthe.2000.0038
86. Veselinovic M, Charlins P, Akkina R. Modeling HIV-1 mucosal transmission and prevention in humanized mice. Methods Mol Biol. (2016) 1354:203–20. doi: 10.1007/978-1-4939-3046-3_14
87. Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, et al. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. (2000) 74:8358–67. doi: 10.1128/JVI.74.18.8358-8367.2000
88. Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, et al. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. (2002) 46:1896–905. doi: 10.1128/AAC.46.6.1896-1905.2002
89. Baskin GB, Murphey-Corb M, Watson EA, Martin LN. Necropsy findings in rhesus monkeys experimentally infected with cultured simian immunodeficiency virus (SIV)/delta. Vet Pathol. (1988) 25:456–67. doi: 10.1177/030098588802500609
90. Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for MinION and illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. (2017) 12:1261–76. doi: 10.1038/nprot.2017.066
91. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. (2011) 17:10–12. doi: 10.14806/ej.17.1.200
92. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. (2012) 9:357–9. doi: 10.1038/nmeth.1923
93. Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, et al. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. (2012) 40:11189–201. doi: 10.1093/nar/gks918
Keywords: HIV origins and evolution from SIV, SIV cross-species transmission, a dual purpose humanized mouse model for HIV and SIV research, SIVmac and SIVB670 pathogenesis and evolution in humanized mice, viral adaptive changes and evolution, SIV evolution into HIV-2, origin of human pathogens in NHP, origin of human pandemic viruses
Citation: Curlin JZ, Schmitt K, Remling-Mulder L, Moriarty RV, Baczenas JJ, Goff K, O'Connor S, Stenglein M, Marx PA and Akkina R (2021) In vivo Infection Dynamics and Human Adaptive Changes of SIVsm-Derived Viral Siblings SIVmac239, SIVB670, and SIVhu in Humanized Mice as a Paralog of HIV-2 Genesis. Front. Virol. 1:813606. doi: 10.3389/fviro.2021.813606
Received: 12 November 2021; Accepted: 13 December 2021;
Published: 31 December 2021.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Scott Kitchen, University of California, Los Angeles, United StatesFatah Kashanchi, George Mason University, United States
Vicente Planelles, The University of Utah, United States
Copyright © 2021 Curlin, Schmitt, Remling-Mulder, Moriarty, Baczenas, Goff, O'Connor, Stenglein, Marx and Akkina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramesh Akkina, YWtraW5hQGNvbG9zdGF0ZS5lZHU=
†These authors have contributed equally to this work