 Takaaki Koma1
Takaaki Koma1 Tokifumi Odaka2
Tokifumi Odaka2 Sung-Il Lee3
Sung-Il Lee3 Naoya Doi1
Naoya Doi1 Tomoyuki Kondo1
Tomoyuki Kondo1 Kazu Okuma2Jun-Ichi Fujisawa2*
Kazu Okuma2Jun-Ichi Fujisawa2* Akio Adachi1,2*
Akio Adachi1,2* Masako Nomaguchi1*
Masako Nomaguchi1*- 1Department of Microbiology, Graduate School of Medicine, Tokushima University, Tokushima, Tokushima, Japan
- 2Department of Microbiology, Kansai Medical University, Hirakata, Osaka, Japan
- 3Institute of Biomedical Science, Kansai Medical University, Hirakata, Osaka, Japan
Animal models are essential for basic and clinical research on virus diseases. Humanized mice (mice reconstituted with human hematopoietic cells) have been effectively used for various virus studies as small animal models. Studies on human-tropic HIV-1 have also been performed using different humanized mouse models. Various humanized mice have been generated using distinct mouse strains and engraftment methods. These different techniques affect the reconstitution of human hematopoietic cells in individual mice, and in turn the HIV-1 replication in vivo. In this report, we describe the details of the generation method of humanized mice, i.e., severely immunodeficient mice (NSG mice) transplanted with human CD133-positive cells via intra-bone marrow injection (IBMI). It has been shown that the CD133-positive cells are highly capable to generate CD34-positive cells in vivo and IBMI is an excellent methodology for lymphoid and myeloid cell repopulation. In humanized mice transplanted with CD133-positive cells into the bone marrow, human lymphocytes were increased 3 months after the transplantation and a steady increase in CD4-positive cells was observed until 6–8 months after the transplantation. In order to test the utility of our system, CXCR4-tropic and CCR5-tropic HIV-1 clones were intraperitoneally inoculated into the resultant humanized mice 6–8 months after the transplantation. Upon inoculation at the same dose of viruses, the plasma viral load in CCR5-tropic HIV-1-inoculated mice peaked earlier than that in CXCR4-tropic HIV-1-inoculated mice (2–3 weeks vs 5–10 weeks post-inoculation). While a rapid decrease in CD4-positive cells was observed at the peak or prior to the peak of viremia for CXCR4-tropic HIV-1-inoculated mice, CD4-positive cells were gradually decreased in CCR5-tropic HIV-1-inoculated mice. Upon inoculation at the same dose of viruses, a Nef-deleted R5-tropic HIV-1 exhibited retarded growth kinetics in the inoculated mice compared to the parental virus (around 8 weeks vs 2–3 weeks post-inoculation), which appears to reflect the decrease in replication potential in primary cells. Taken all together, in addition to the humanized mice reported so far, our humanized mice generated by transplanting CD133-positive cells with the IBMI method would be an appropriate prototype model for understanding HIV-1 biology in vivo.
1 Introduction
Animal models are essential to understand the nature of viral infectious diseases including viral replication and pathogenesis in individuals (1–7). Accumulating the knowledge/data gained from these model studies has led to progress in basic research on the biology of viruses, pathogenesis, and clinical research including the development of vaccines and antivirals.
HIV-1 replicates well in humans and causes disease only in humans. This narrow tropism of HIV-1 has hampered the establishment of animal models that reflect the pathophysiological states of HIV-1-infected humans. Macaques infected with SIVs related to HIV-1 or with SIV-based simian-tropic HIV-1 (SHIV) are used as animal models that mimic HIV-1/human infection (8–11). Although non-human primate models would be the best in terms of mimicking HIV-1-infected humans, there are generally insurmountable essential issues, i.e., the lack of pathogenic macaque-tropic HIV-1 and the high cost to maintain experimental macaques and facilities. On the one hand, humanized mouse models (mice reconstituted with human hematopoietic cells) have been commonly utilized as in vivo models for HIV-1 infection, despite several serious limitations such as incomplete immune responses (12, 13). These are divided into two groups: BLT (bone marrow-liver-thymus)- and non-BLT-humanized mouse models. BLT humanized mice are generated through the implantation of human fetal liver and thymus tissues followed by the transplantation of CD34+ cells from the same donor into the bone marrow. BLT-humanized mice are excellent models for the establishment of mucosal HIV-1 infection and cellular immune responses, whereas there are several issues such as graft versus host disease. Also, human fetal materials are not usually available to many researchers (12, 13). Thus, non-BLT mice have been accepted by many researchers and widely used for HIV-1 research. To generate non-BLT humanized mice, human hematopoietic stem cells (HSCs) are transplanted into severely immunodeficient mice and thus human hematopoietic cells are reconstituted within individuals. Non-BLT humanized mice are generated in several ways using different materials, as follows (Figure 1) (12, 13).
1) Severely immunodeficient mouse strains: non-obese diabetic (NOD), NOD.Cg-PrkdcscidIl2rgtm1Sug (NOG), NOD.Cg-PrkdcscidIl2rgtm1Wjl (NSG), NOD.Cg-Rag1tm1MoMIl2rgtm1Wjl (NRG), BALB/c/Rag2null/IL2Rnull (DKO/BRG), NOD/SCID/Jak3null (NOJ)
2) CD34+ or CD133+ HSCs: derived from umbilical cord blood (UCB) or fetal liver
3) HSCs transplantation methods: transplantation into the livers of newborns or intravenous injection into adult mice
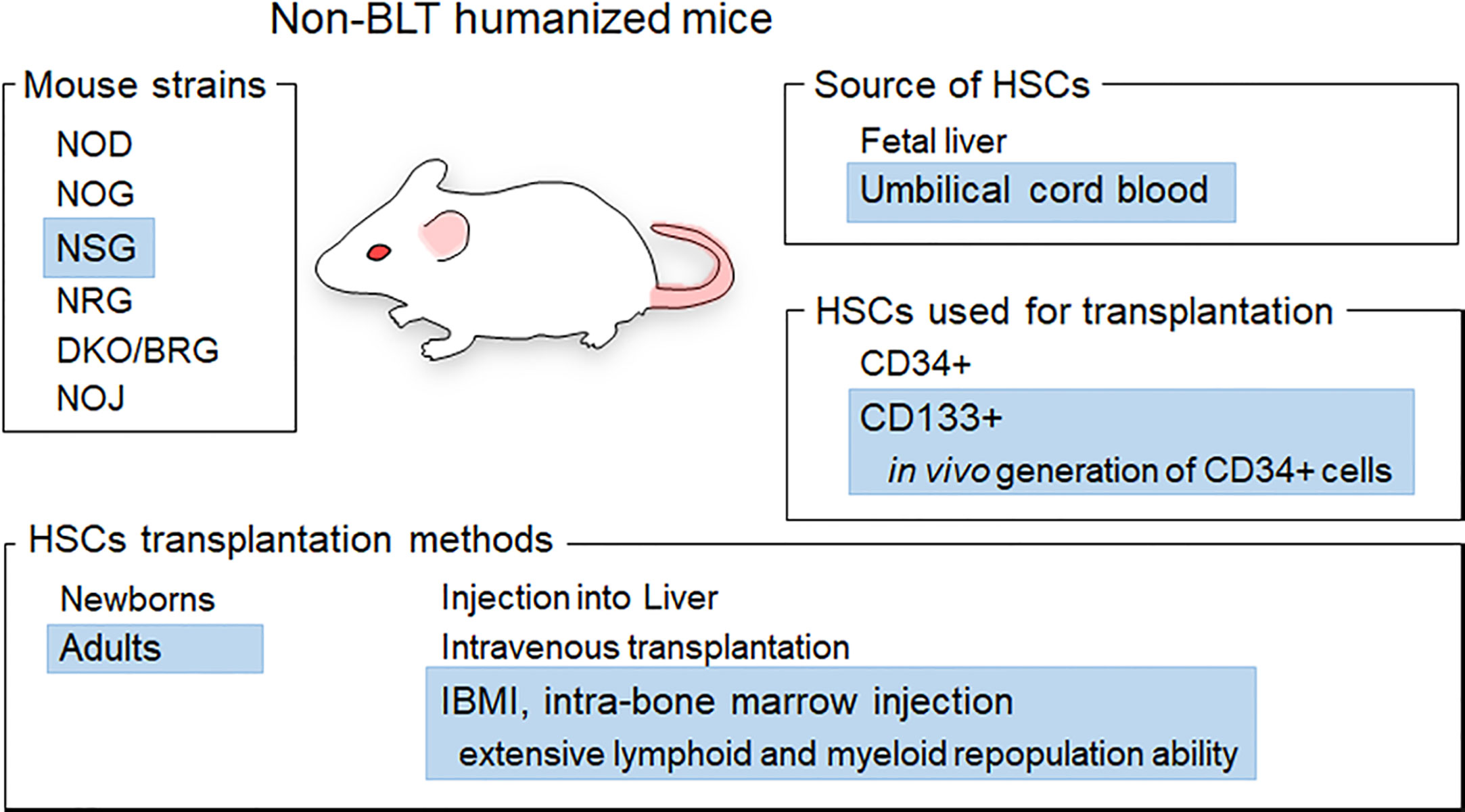
Figure 1 Generation of non-BLT humanized mice. Various materials and methods used to generate non-BLT humanized mice are presented. Blue highlights show the materials and methods used in this study. The mice used in this study were female adult NSG mice based on the efficiency of hematopoietic stem cell (HSC) engraftment (14, 15). CD133+ cells from umbilical cord blood were chosen as HSCs for transplantation because CD133+ cells exhibit in vivo generation ability of CD34+ cells and a slightly higher plasticity relative to CD34+ cells (16, 17). The IBMI method is a way to directly inject HSCs into the bone marrow, which is one of the sites important for the reconstitution of immune cells in mice (18–20). Transplantation of CD133+ HSCs into mice using the IBMI method is expected to facilitate the generation of CD34+ cells and the reconstitution of lymphoid and myeloid cells (21, 22).
These materials/methods influence the reconstitution and differentiation of human cells in individual mice, and thus can affect the successful generation of humanized mice and HIV-1 replication in vivo in the resultant humanized mice. Nevertheless, studies using non-BLT humanized mice have led to a better understanding of HIV-1 basic and clinical biology, including in vivo activity/function of viral accessory proteins, relationship between viral tropism and in vivo replication, and virus adaptation (23–32).
Our purpose in this study is to provide details of procedures to generate non-BLT humanized mice so as to be available for many researchers, and present some examples where our humanized mice can be used in HIV-1 infection experiments. Essentially, CD133+ cells purified from UCB are transplanted into the bone marrow of NSG mice using the intra-bone marrow injection (IBMI) method. This type of humanized mice is abbreviated as huNSGs/IBMI-CD133 hereafter. Female NSG mice were chosen in this study because HSC engraftment in female mice is better and faster than that in male mice and NSG as well as NOG are the best recipients for HSCs among various mouse strains including NRG and DKO/BRG (14, 15) (Figure 1). From a practical point of view, NSG mice are available earlier after birth (at 4 weeks vs. after 5 weeks), are more inexpensive, and tend to be heavier than NOG mice, thus being better recipients for HSCs. CD133 is a marker of undifferentiated stem cells derived from UCB (21, 22, 33, 34). UCB-derived CD133+ cells have a high self-renewing capability and long-term reconstituting ability in the bone marrow. The transplantation of CD133+ cells using IBMI has been shown to induce in vivo generation of CD34+ cells and extensive lymphoid and myeloid cell repopulation (21, 22, 35). Here, we show that huNSG/IBMI-CD133 is an appropriate model for HIV-1 infection, equally useful as previously reported non-BLT humanized mouse models. In huNSGs/IBMI-CD133, stable CCR5 (R5) -tropic HIV-1 replication and CD4+ cell number can be maintained over a period of about 1 year from transplantation. For CXCR4 (X4) -tropic HIV-1 infection, a rapid CD4+ cell depletion was observed in individuals. R5-tropic Nef-negative HIV-1 exhibited a lower replication potential than that of parental R5-tropic HIV-1 in peripheral blood mononuclear cells (PBMCs), and similar retarded growth kinetics for Nef-negative HIV-1 was observed in huNSGs/IBMI-CD133. Our huNSGs/IBMI-CD133 would be one of the prototypes of non-BLT humanized mice. Further improvement, such as the introduction of human cytokine and/or human leukocyte antigen, is required to develop a more suitable model that mimics HIV-1-infected human individuals.
2 Materials and methods
2.1 Generation of huNSGs/IBMI-CD133
The outline to generate huNSGs/IBMI-CD133 is shown in Figure 2. All animal experiments were performed in accordance with the guidelines established by the animal research committee of Tokushima University. All huNSGs/IBMI-CD133 used in this study are listed in Table 1.
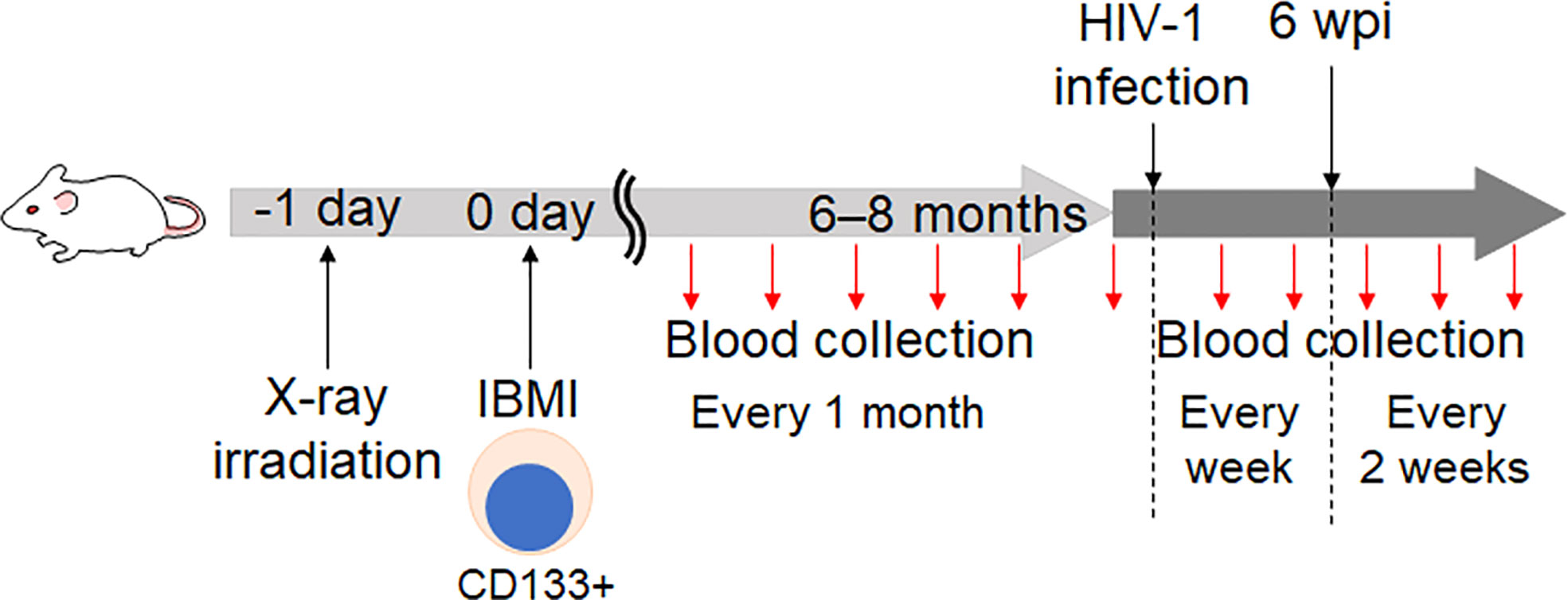
Figure 2 Schedule for the HIV-1 infection experiments using huNSGs/IBMI-CD133. NSG mice were irradiated with X-ray one day before the HSC-transplantation. CD133+ cells prepared from UCB were transplanted into NSG mice using the IBMI method (21, 22, 35). The detailed procedures of the IBMI method are shown in Figure 3 and the Supplemental Movie. The resultant mice were designated huNSGs/IBMI-CD133. At 6 to 8 months after the transplantation, virus samples were infected into huNSGs/IBMI-CD133. To monitor the reconstitution of human hematopoietic cells and the HIV-1 growth kinetics, peripheral blood samples were periodically collected.
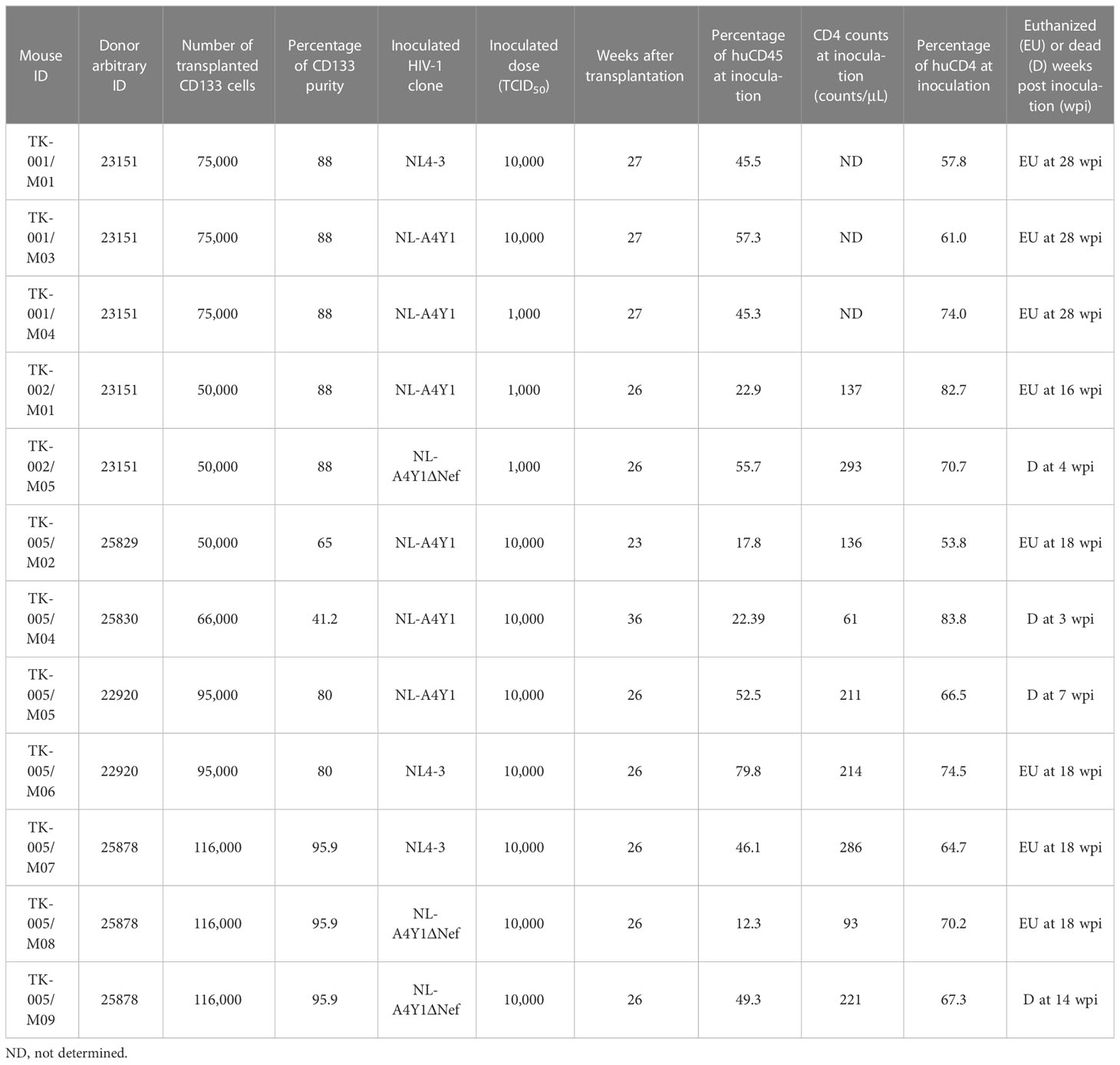
Table 1 HuNSGs/IBMI-CD133 used for HIV-1 infection in this study.
Preparation for CD133+ cells CD133+ cells from umbilical cord blood cells contain both CD34+ and CD34- cell populations, and thus are more primitive cells (36, 37). It has been shown that CD133 can be a marker for CD34+ and CD34- cells (33, 34). It is difficult to judge whether CD133+ selection has a relevant advantage over CD34+ selection (16, 38–40). It appears that there is not a large difference in the cell expansion including CD34+ cells and the differentiation of human lymphocyte cells in an individual mouse, although CD133+ cells have been suggested to display slightly higher plasticity relative to CD34+ cells (16, 17). Based on these previous studies, we decided to use CD133+ cells in our experiments because of the possibility of better cell differentiation and primitivity of CD133+ cells. CD133+ cells for transplantation were isolated from UCB, obtained from the Japanese Red Cross Kinki Cord Blood Bank (Osaka, Japan) for research purposes, similarly to as described previously (35). Around 100–150 mL of UCB with CPDA (citrate-phosphate-dextrose-adenine) was diluted with PBS (-) containing 2 mM EDTA up to 200 mL. After incubation for 15 min at room temperature, the diluted UCB samples were centrifuged at 500 × g for 15 min at 20°C (slow acceleration and deceleration). Buffy coat samples were collected in one tube, PBS (-) containing 2 mM EDTA was added to make a total volume of 30 mL, and the samples were layered on 20 mL of Ficoll-Paque PLUS (GE Healthcare, USA). After centrifugation at 500 × g for 40 min at 20°C (slow acceleration and deceleration), a layer containing mononuclear cells was collected, PBS (-) containing 2 mM EDTA was added to make a total volume of 50 mL, and centrifuged at 400 × g for 10 min at 20°C (slow acceleration and deceleration). The mononuclear cells were washed twice by the addition of 30 mL of PBS (-) containing 2 mM EDTA and centrifugation at 200 × g for 10 min at 20°C. The cells (around 15–30 × 107) were resuspended in 500 μL of CELLBANKER 1 (ZENOGEN PHARMA, Japan) and stored at -80°C until use. From our experience, the cells could be stored for at least 6 months to 1 year. Subsequent preparation of CD133+ cells was performed on the day of transplantation into mice. The stored mononuclear cells were immediately thawed in a water bath at 37°C and transferred to a conical tube. Around 10 mL of the thawing medium [RPMI-1640 containing 20% heat-inactivated (hi) FBS, 100 U/mL Heparin Sodium MOCHIDA (MOCHIDA PHARMACEUTICAL, Japan) and 100 U/mL DNase I (FUJIFILM Wako Pure Chemical, Japan)] was slowly added to the mononuclear cells. After centrifugation at 500 × g for 5 min at 4°C, RPMI-1640 containing 2% hi FBS was added to the mononuclear cells and then centrifuged at 500 × g for 5 min at 4°C to wash the cells. The mononuclear cells were resuspended in 350 μL of PBS (-), and CD133+ cells were isolated from the mononuclear cells using a CD133 MicroBead Kit (Miltenyi Biotec, Germany) in accordance with the manufacturer’s instruction.
IBMI protocol Female 5-week-old NSG mice were purchased from The Jackson Laboratory Japan, Inc. (Kanagawa, Japan). The mice were maintained under germ-free sterilized conditions. The NSG mice were irradiated with 2.5 Gy radiation one day before transplantation, and were injected with CD133+ cells (more than 5.0 x 104) using the IBMI method, as described previously (21, 22, 35) (Figures 2, 3; Supplemental Movie). The mice were anesthetized using medetomidine hydrochloride (0.75 mg/kg, Nihon Zenyaku Kogyo, Japan), midazolam (4.0 mg/kg, Sando, Japan), and butorphanol tartrate (5.0 mg/kg, Meiji Seika Pharma, Japan). After shaving the leg hairs of the anesthetized mice (Figure 3A), to make a hole to transplant CD133+ cells into the bone marrow, the position of the joint was checked (Figure 3B) and the shaved leg was disinfected with 70% ethanol (Figure 3C). The tibia was punctured while rotating a 27G × 3/4” needle (Terumo, Japan) (Figure 3D). If the needle enters bone marrow properly, the needle does not move or sway even if the hand is released. The needle was then slowly removed from the tibia without moving the hole. To transplant CD133+ cells into the bone marrow, cells were resuspended in RPMI1640 without FBS and drawn into a syringe with a needle (MS-50 micro syringe, Ito Corporation, Japan) (Figure 3E). After insertion of the syringe into the pre-punctured hole, the cells (5 μL/mouse) were slowly injected into the bone marrow over about 1 min while rotating the handle of the syringe. About 30 sec after transferring the cells, the syringe was slowly pulled out. The hole was pinched with fingers for around 30 sec, and disinfected with iodine (Figure 3F). The mice were administered an anesthetic antagonist (antisedan, Nippon Zenyaku Kogyo, Japan) and returned to the cage. Since the teeth of mice transplanted with huCD133+ cells often grow longer preventing proper feeding, caution should be taken to observe their teeth and cut them if necessary.
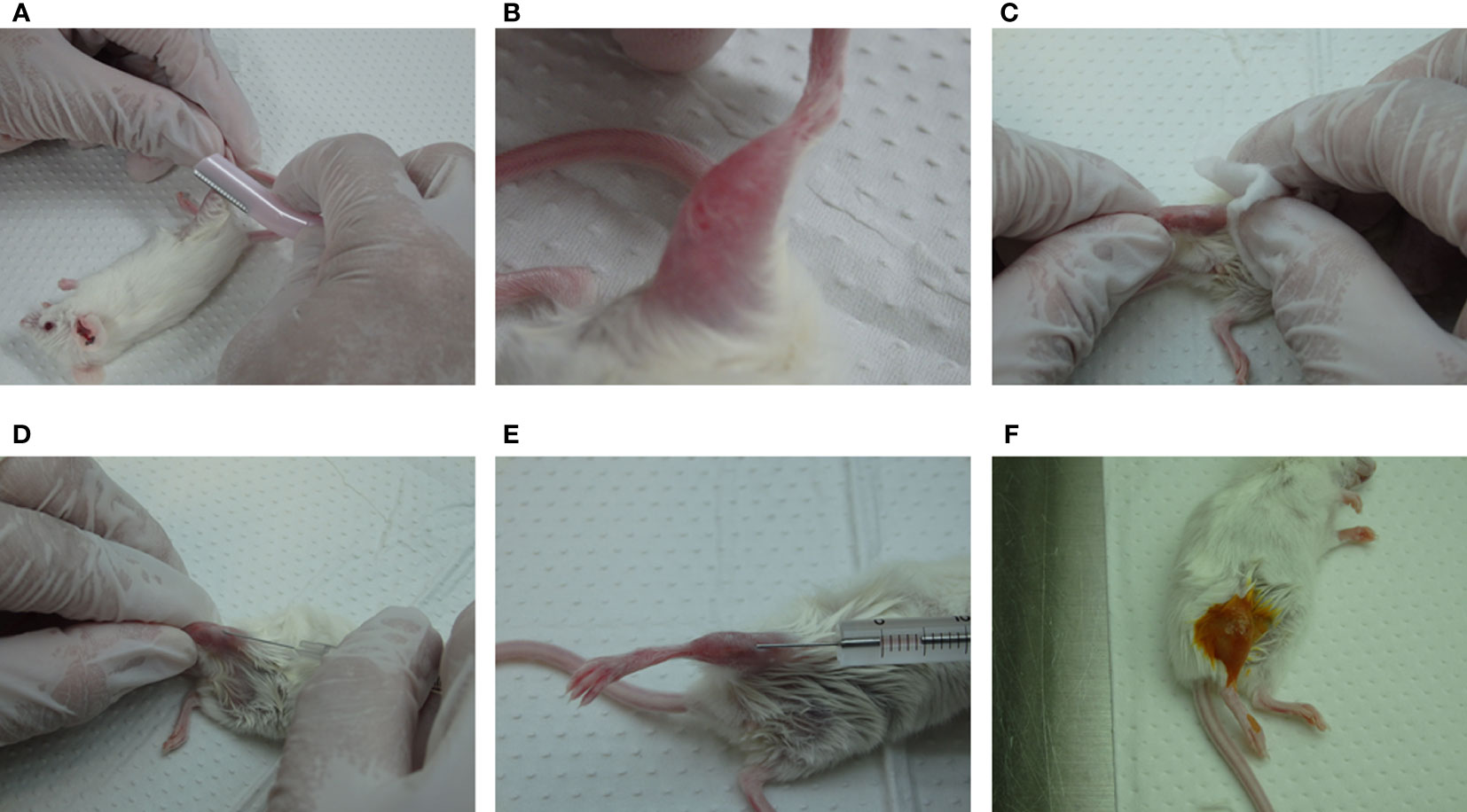
Figure 3 Outline of the IBMI method. (A, B) Shaving of the leg hairs of anesthetized mice. (C) Disinfection of the shaved leg with 70% ethanol. (D, E) Puncture of the bone marrow with a needle, 27G ×3/4" (Terumo, Japan), followed by the transplantation of CD133+ cells with a MS-50 micro syringe (Ito Corporation, Japan). (F) Treatment of the legs with iodine solution. See the Supplemental movie for further details.
2.2 Flow cytometry analysis (FACS)
As indicated in Figure 2, peripheral blood samples of huNSGs/IBMI-CD133 were periodically collected from the tail vein into EDTA-treated hematocrit glass capillary tubes (Marienfeld Superior, Lauda-Königshofen, Germany). When necessary, samples were treated with β-propiolactone and ultraviolet irradiation to inactivate viruses, as previously described (41). Briefly, 10 μL of whole blood samples were mixed with 1 uL each of 1M Tris-HCl (pH 7.50) and 10% β-Propiolactone, and then exposed to UV light (spectral power 254nm, 39W) (Atlantic Ultraviolet, USA) for 10 min at 30 cm distance. To stain cell surface markers, PE anti-mouse CD45 [30-F11], APC/Cy7 anti-human CD45 [HI30], APC anti-human CD3 [HIT3a], PerCP/Cy5.5 anti-human CD8a [HIT8a], FITC anti-human CD4 [RPA-T4], and PE/Cy7 anti-human CD19 [HIB19] (Biolegend, USA) antibodies were used. Whole blood samples (10 μL) were mixed with 50 μL of PBS containing 3% hi FBS and appropriate antibodies (0.5 μL each), and incubated at room temperature for 30 min. To determine absolute cell numbers in the blood samples, AccuCount Ultra Rainbow Fluorescent Particles (ACRFP-100-3, Spherotech, USA) were used in accordance with the manufacturer’s instruction. Before FACS analysis, erythrocytes in the samples were lysed by treating with RBC lysis buffer (144 mM NH4Cl and 17 mM Tris [pH 7.65]) for at least 5 min at room temperature. The stained cells were analyzed using BD FACSVerse and FACSuite software (Becton Dickinson, USA).
2.3 HIV-clones
HIV-1 proviral clones NL4-3 (42) and its R5-tropic derivative NL-A4Y1 were used in this study. NL-A4Y1 clone was generated by introducing the env gene of gtu+A4CI1 clone (43), derived from a clinical isolate of subtype B, into the corresponding region of NL4-3 through overlapping PCR. NL-A4Y1-ΔNef clone, a Nef-negative frameshift mutant, was constructed using the XhoI site in the nef gene, as described previously (44, 45).
2.4 Cell cultures
A human monolayer cell line 293T (46) and a luciferase reporter cell line TZM-bl (47, 48) were maintained in Eagles’s minimal essential medium (EMEM) containing 10% hi FBS. A human lymphocyte cell line MT4/CCR5 (MT4 cells stably expressing CCR5) was maintained in RPMI-1640 medium containing 10% hi FBS and 200 μg/mL hygromycin (Merck, Germany). Human PBMCs were purchased from Cellular Technology Ltd. (USA). Cryopreserved PBMCs were thawed in accordance with the manufacturer’s instruction and used for virus replication assays.
2.5 Virus replication assays in cells
Virus stocks for infection were prepared from 293T cells transfected with proviral clones by the calcium-phosphate coprecipitation method, as described previously (42, 49). Virion-associated reverse transcriptase (RT) activity was measured as described previously (50, 51). Prior to infection, PBMCs were stimulated with RPMI 1640 medium containing 10% hi FBS, 50 units/ml of recombinant human IL-2 (Bio-Rad, USA), and 2 μg/ml of phytohemagglutinin L (PHA-L) (Roche Diagnostics GmbH, Germany) for 3 days. MT4/R5 and stimulated PBMCs were spin-infected (52) with equal amounts of viruses. Infected PBMCs were cultured in the presence of IL-2 throughout the experiment. Viral growth was monitored by RT activity in the culture supernatants.
2.6 HIV-1 infection to huNSGs/IBMI-CD133
Virus stocks were prepared as described above. The titer of input viruses was determined by calculating TCID50 using infectivity to TZM-bl cells (53). Briefly, virus stocks with 10-fold serial dilution were inoculated into TZM-bl cells, and the cells were lysed and subjected to luciferase assays on day 2 post-inoculation (Promega, USA). TCID50 values were calculated by the Reed-Muench method. Virus stocks were diluted with EMEM or saline, and the diluted viruses (a total volume of 100 μL) were inoculated intraperitoneally into anesthetized IBMI-huNSG mice 6–8 months after the cell transplantation. Peripheral blood samples (around 100 μL) were periodically collected from the tails of mice (Figure 2) and used to monitor virus replication and absolute CD4 cell numbers. Virus replication was assessed by measuring viral RNA copy numbers in the plasma samples. Viral RNA genome in the plasma sample (25 μL) was automatically extracted using QIAamp Viral RNA Mini Kit (QIAGEN, Netherlands) by QIAcube (QIAGEN, Netherlands), and subjected to real-time RT-PCR analysis. The RT-PCR reaction was performed using a specific primer set qPCR Set3 F (caggccatatcacctagaactt)/qPCR Set3 R (gggtggctccttctgataat) and QuantiFast SYBR Green RT-PCR Kit (QIAGEN) in accordance with the manufacturer’s instruction. The molecular clone pNL4-3 (42) digested with EcoRI was used as a standard to quantify viral RNA copy numbers.
3 Results
3.1 Generation of huNSGs/IBMI-CD133 for HIV-1 infection experiments
The time schedule for the HIV-1 infection experiments using huNSGs/IBMI-CD133 is shown in Figure 2. NSG mice were transplanted with UCB-derived CD133+ cells by IBMI on the next day after X-ray irradiation (Figure 3; Supplemental Movie) (Table 1). Blood samples were collected periodically to monitor the development of human hematopoietic cells by FACS (Figure 4A). Three months after the transplantation, the human leukocyte population (CD45+ cells) was around 30% of the whole lymphocyte cell population and increased to approximately 40% at 6–8 months post-transplantation. The percentage of human B cells (CD19+ cells) was higher than that of human T cells (CD3+ cells) at 3 months post-transplantation (more than 80% vs less than 10%, respectively) (Figure 4B). While the percentage of human B cells in the human lymphocyte population was gradually decreased, the percentage of human T cells was significantly increased (~50%) up to a similar level to that of human B cells at 6–8 months after the transplantation (Figure 4B). This result is in good agreement with previous reports that human B cells develop earlier than human T cells in non-BLT humanized mice (17, 54). The percentage of CD4+ cells in the human T cell population was higher than that of CD8+ cells at 6–8 months post-transplantation (Figure 4C) and the absolute number of CD4+ cells was increased up to around 200 cells/uL blood. Consistent with a previous report (35), stable T cell numbers and B-cell-to-T-cell ratio were maintained in huNSGs/IBMI-CD133 for a long time (6–8 months after the transplantation).
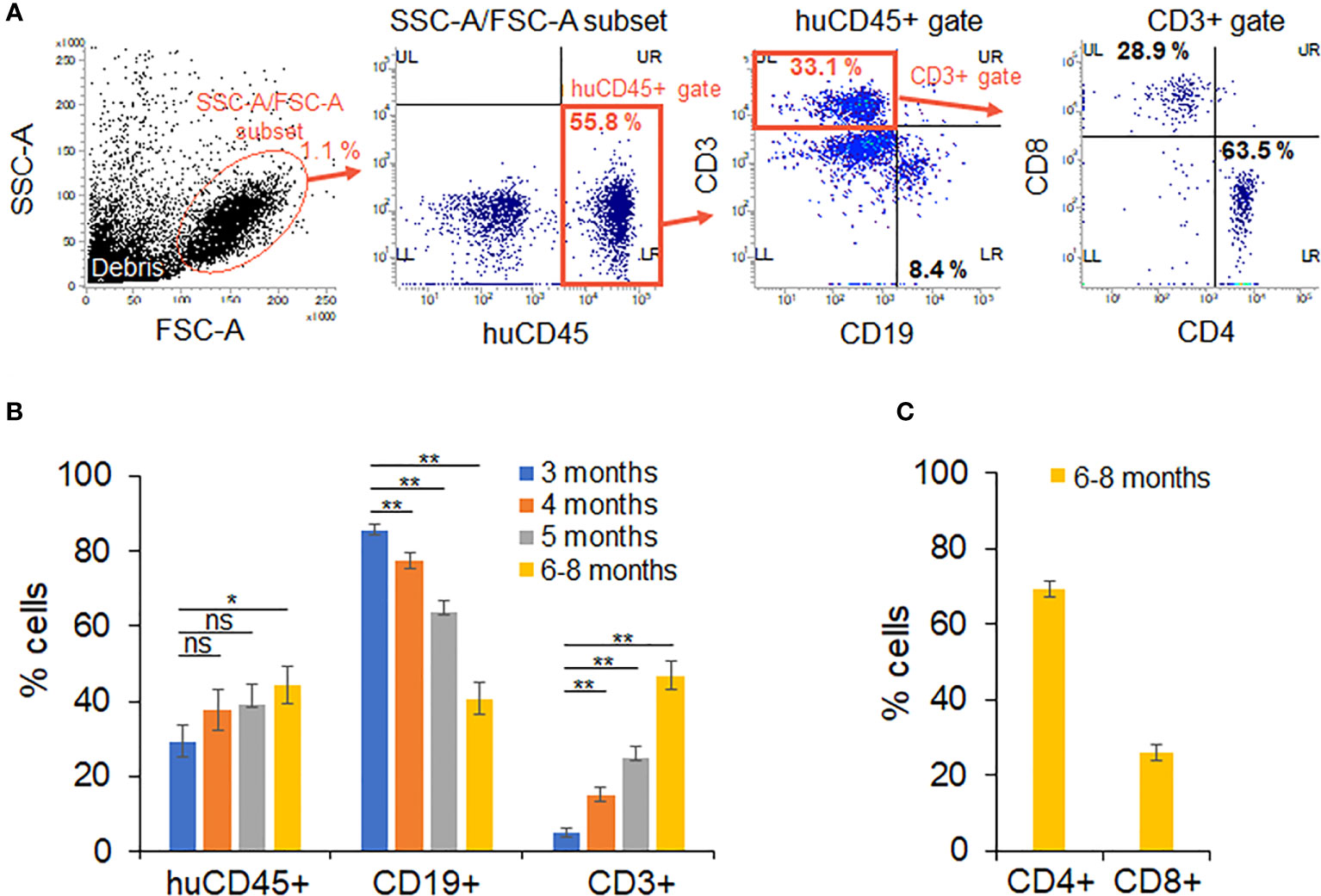
Figure 4 Analysis on the development of human hematopoietic cells in huNSGs/IBMI-CD133. The percentage of human hematopoietic cells in peripheral blood samples was assessed by flow cytometry analysis on months 3 (n=14–15), 4 (n=13–15), 5 (n=13–15), and 6–8 (n=11–14) post-transplantation. Mouse groups transplanted with CD133+ cells derived from different UCB are shown in Table 1. (A) Sequential gating to identify various cell populations. (B) The percentage of human leukocytes (huCD45) in the SSC-A/FSC-A subset, T cells (CD3)/B cells (CD19) in the huCD45-positive cell population, and (C) CD4+ and CD8+ cells within the human CD3-positive cell populations are presented with the averages and standard errors. Significance relative to the 3-month samples was determined by Welch’s t test. ns, not significant; *, P < 0.05; **, P < 0.01.
3.2 HIV-1 replication in huNSGs/IBMI-CD133
Using twelve huNSGs/IBMI-CD133 (Table 1), we assessed HIV-1 replication in individual mice. X4-tropic HIV-1 (NL4-3), R5-tropic HIV-1 (NL-A4Y1), and its nef-deletion mutant (NL-A4Y1ΔNef) were used for the infection experiments in this study. The NL-A4Y1 clone carries an env gene derived from a clinical isolate (subtype B). HuNSGs/IBMI-CD133 6–8 months after the transplantation were used for HIV-1 infection, because the percentage of T cells in the human lymphocyte population was higher than that of B cells and the CD4+ cell number was stably maintained (Figure 4). Virus samples prepared from 293T cells transfected with proviral clones were intraperitoneally inoculated into huNSGs/IBMI-CD133. Blood samples were periodically collected, and the HIV-1 replication and the CD4+ cell number were measured using quantitative RT-PCR and FACS, respectively. Of the twelve huNSGs/IBMI-CD133 infected, three died in a short period of time, within 7 weeks, from rapid increases in CD4+ cells (n=2) or HIV-1 (n=1) (Table 1). In our experience, approximately 6% of mice transplanted with CD133+ cells have died of unidentified causes after transplantation, possibly because of some infections and/or stresses caused by transplantation such as irradiation, anesthesia, blood collection, or graft versus host disease. As shown in Figure 5, the X4-tropic HIV-1 (NL4-3) viremia peaked at 5 to 10 weeks post-inoculation accompanied by a rapid reduction in the CD4+ cell number upon or just after the peak. On the other hand, upon the inoculation of the same dose of viruses (10,000TCID50), the plasma viral load with R5-tropic HIV-1 (NL-A4Y1) reached a peak at 2 to 3 weeks post-inoculation (Figures 6A, B). The viremia was maintained 4 to 6 months after the virus-inoculation with a concomitant gradual decrease in the CD4+ cell number, repeatedly increasing and decreasing (Figures 6A, C). Viral replication kinetics in mice inoculated with a lower dose of viruses (1,000TCID50) were similar to those in mice inoculated with a 10,000TCID50 dose of viruses (Figure 6A). The peak viremia and replication kinetics in the plasma of huNSGs/IBMI-CD133 individuals were not related to the percentage of human leukocyte reconstitution and the CD4+ cell number at the HIV-1 inoculation time point (Table 1; Figures 5, 6). Since p24-positive cells have been detected in lymphoid organs (23, 24), various sources of viruses may affect the plasma viral load. Considering the stable maintenance of the CD4+ cell number for at least 10 months after transplantation (our unpublished results) and the gradual decrease in the CD4+ cell number associated with fluctuations in viral growth (Figure 6), it is reasonably assumed that this decrease was not due to a spontaneous decrease in CD4+ cells in infected huNSGs/IBMI-CD133. These results show that HIV-1 replicates and persists well for a long time in huNSGs/IBMI-CD133.
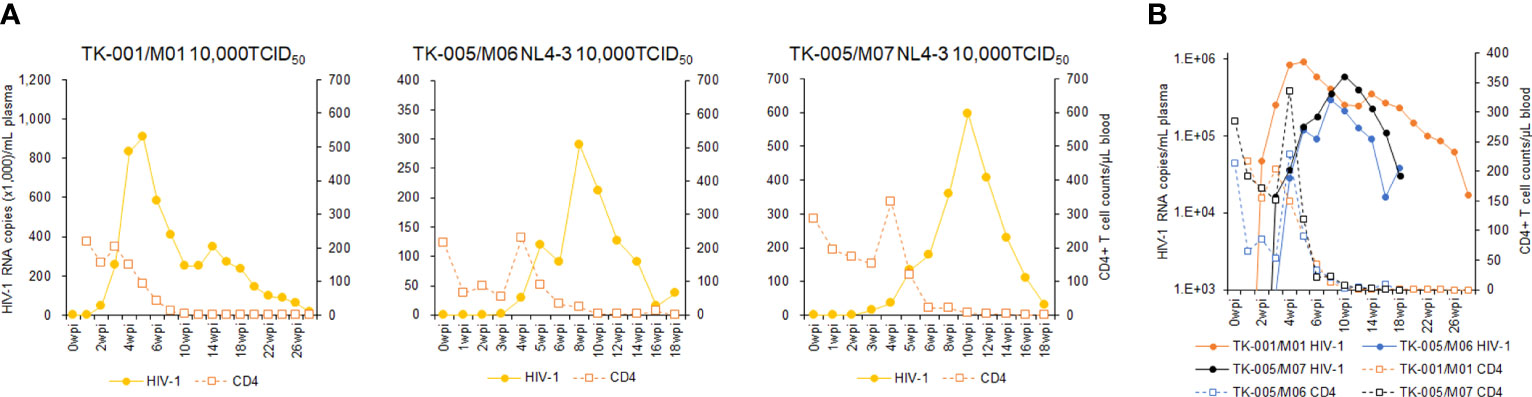
Figure 5 Analyses of viral replication and CD4+ cell counts in huNSGs/IBMI-CD133 infected with X4-tropic NL4-3. HuNSGs/IBMI-CD133, 6–8 months after the transplantation, were anesthetized and intraperitoneally inoculated with NL4-3 (see Table 1 for summary). Peripheral blood samples were collected periodically as indicated in the graphs. Virus replication in plasma samples was assessed by real-time RT-PCR analysis and absolute CD4+ cell numbers in the blood samples were determined by flow cytometry analysis. (A) The actual number of HIV-1 RNA copies and the absolute number of CD4+ cells in each individual. (B) Comparison of the logarithm of the RNA copy number and the absolute number of CD4+ cells among virus-inoculated mice.
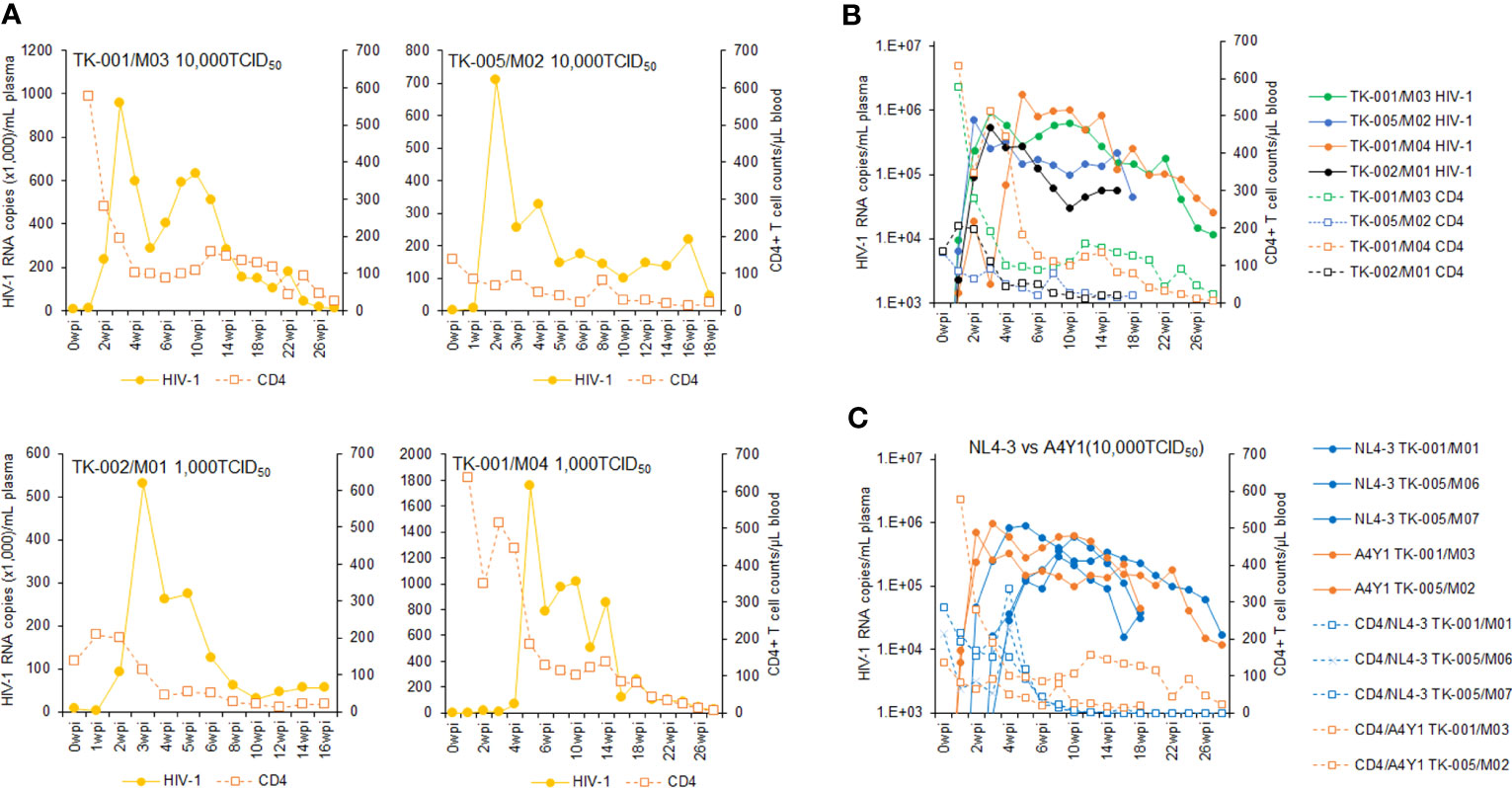
Figure 6 Analyses of viral replication and CD4+ cell counts in huNSGs/IBMI-CD133 infected with R5-tropic NL-A4Y1. HuNSGs/IBMI-CD133, 6–8 months after the transplantation, were anesthetized and intraperitoneally inoculated with NL-A4Y1 (see Table 1 for summary). Peripheral blood samples were collected periodically as indicated in the graphs. Virus replication in the plasma samples was assessed by real-time RT-PCR analysis and absolute CD4+ cell numbers in the blood samples were determined by flow cytometry analysis. (A) The actual number of HIV-1 RNA copies and the absolute number of CD4+ cells in each individual. (B) Comparison of the logarithm of the RNA copy number and the absolute number of CD4+ cells among virus-inoculated mice. (C) Comparison of the logarithm of the RNA copy number and the absolute number of CD4+ cells between X4-tropic NL4-3- and R5-tropic NL-A4Y1-inoculated mice.
3.3 Effect of nef deletion on the growth kinetics of NL-A4Y1 in PBMCs and huNSGs/IBMI-CD133
It has been shown that Nef-deleted or -inactivated HIV-1 displays an attenuated phenotype in humanized BLT mice, such as the decrease in plasma viral load and the weak decline in CD4+ cells (55–57). Using our hu/NSGs/IBMI-CD133 model, we investigated the effect of Nef deletion on HIV-1 replication in vivo. Our Nef-negative R5-tropic HIV-1 (NL-A4Y1ΔNef) was newly constructed in this study by introducing a frameshift mutation into the viral nef gene. First, we compared the growth ability of NL-A4Y1ΔNef and its parental NL-A4Y1 in human lymphocyte cells. As shown in Figure 7, these two clones appeared to exhibit similar growth kinetics in an MT4/R5 cell line, although NL-A4Y1ΔNef reached a peak slightly later than NL-A4Y1. Nef-deleted HIV-1 mutants have been reported to be attenuated in their ability to replicate in stimulated PBMCs compared to those of their parental clones (58, 59). To examine the effect of Nef-deletion on the viral replication ability, virus samples (NL-A4Y1 and NL-A4Y1ΔNef) were inoculated into human PBMCs stimulated with PHA-L. In two different preparations of PBMCs (PBMC1 and PBMC2 in Figure 7), NL-A4Y1ΔNef grew poorly compared to NL-A4Y1. The degree of decrease in the viral replication ability of the Nef-deleted clone appeared to be different in different cell types (MT4/R5 cells vs PBMCs) and in distinct preparations of PBMCs (PBMC1 vs PBMC2). This may be due to the difference in cellular physiological states and/or some specific cellular factors such as CD4 and SERINC3/5 (60, 61). Next, NL-A4Y1ΔNef virus samples were intraperitoneally inoculated into huNSGs/IBMI-CD133. As shown in Figure 8, the plasma viral load by the Nef-deleted clone reached a peak later than 8 weeks post-inoculation. In huNSGs/IBMI-CD133 inoculated with the same dose of viruses (10,000TCID50), NL-A4Y1ΔNef virus exhibited delayed growth kinetics compared to that for the parental NL-A4Y1 (Figures 6A, 8A, C). This may reflect the lowered replication ability of Nef-deleted HIV-1 in PBMCs, although further experiments need to be done. The activity/function of Nef in HIV-1 replication in vivo has been investigated using humanized BLT and non-BLT mice (55–57, 62). HuNSGs/IBMI-CD133 also may be useful for investigating the activity/function of the four accessory proteins of HIV-1 in vivo.
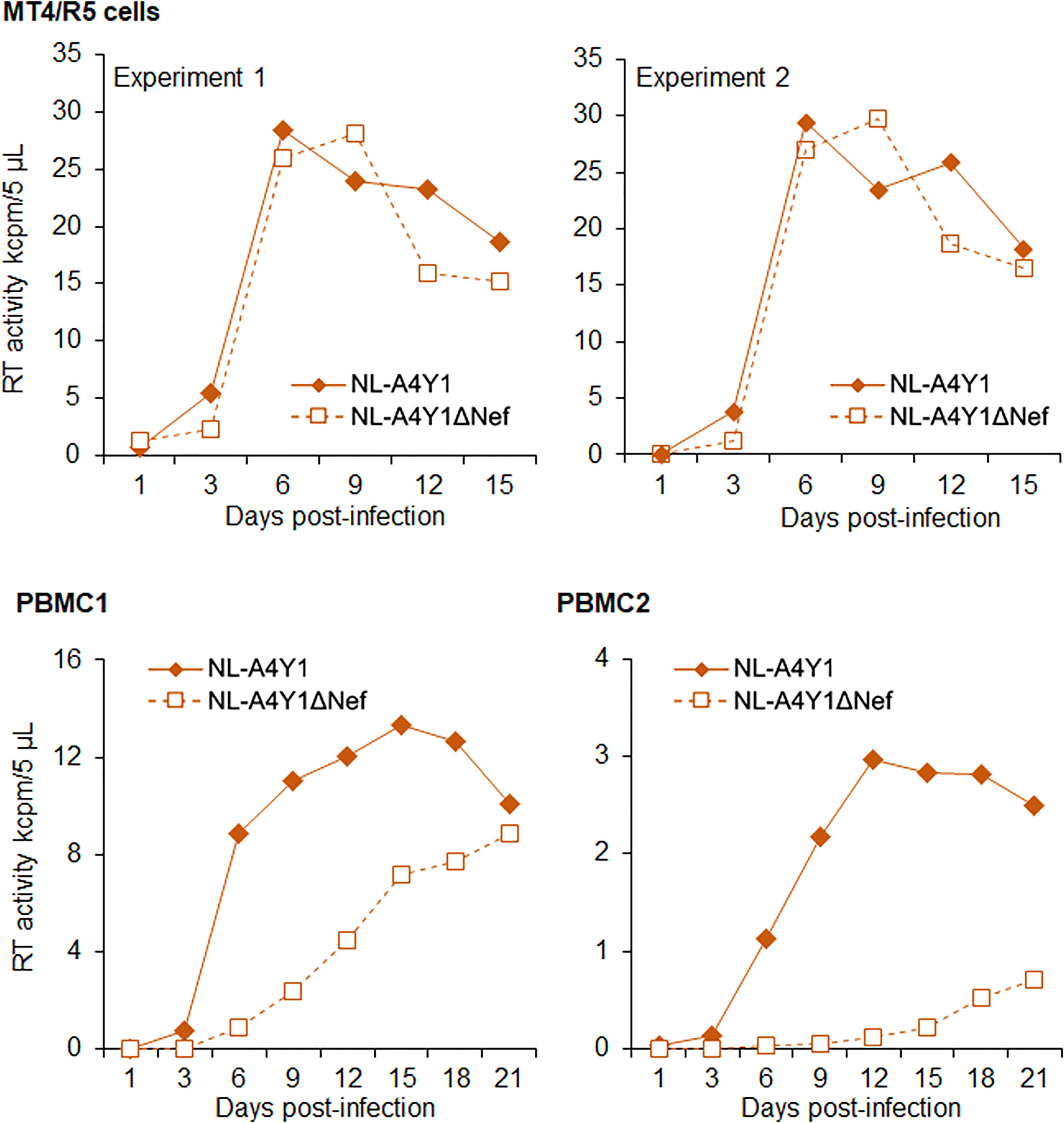
Figure 7 Growth kinetics of R5-tropic NL-A4Y1 and its Nef-negative mutant (NL-A4Y1ΔNef) clones. Viruses were prepared from 293T cells transfected with the indicated clones. MT4/R5 cells (105) and stimulated PBMCs (106) with IL-2/PHA-L were spin-infected with equal amounts of viruses (2.0 x 105 and 2.5 x 104 RT units, respectively). Virus replication was monitored by RT production in the culture supernatants. Different preparations of PBMCs were used for the experiments, as shown. For MT4/R5 cells, representative data from two independent experiments are shown.
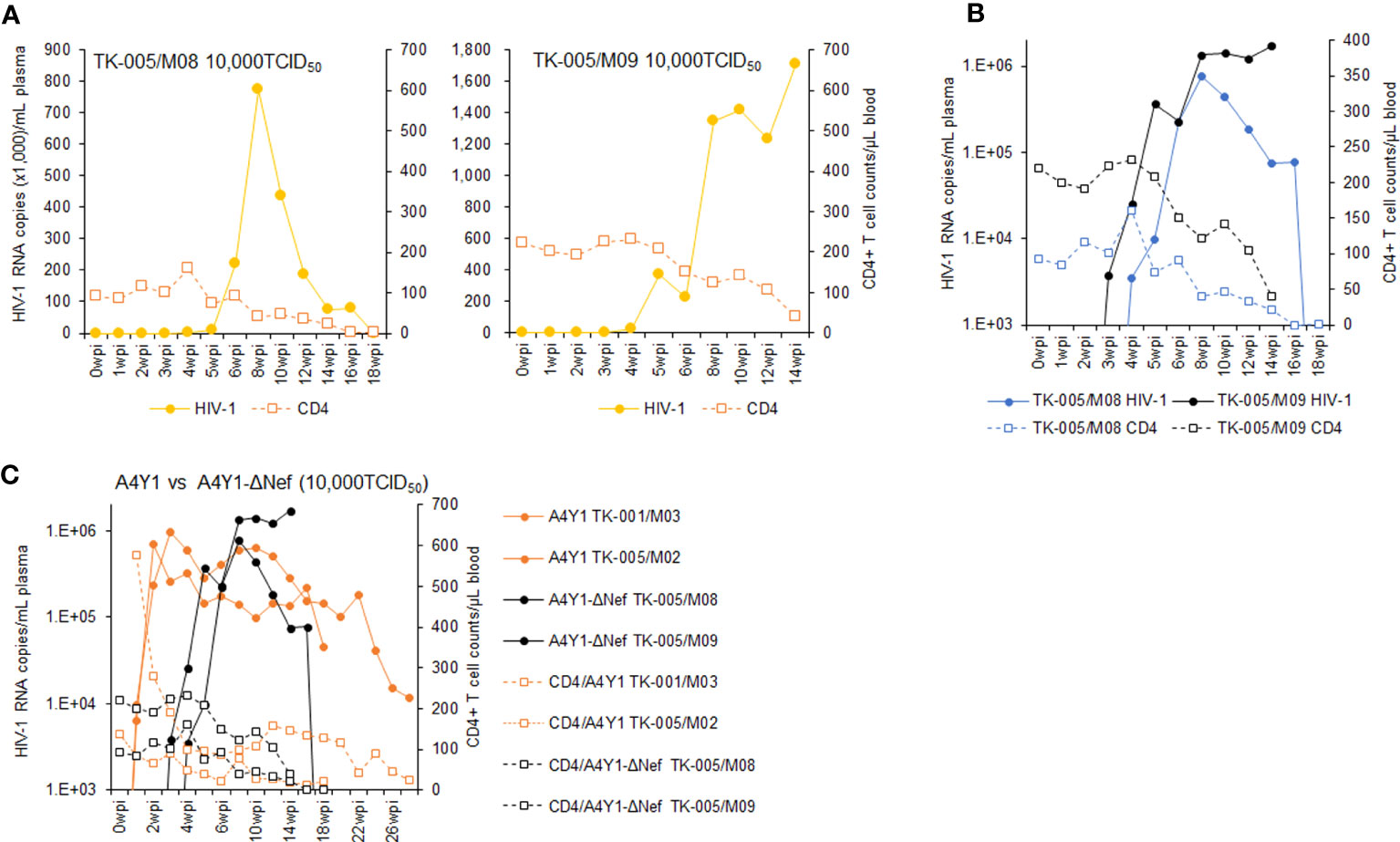
Figure 8 Analyses of viral replication and CD4+ cell counts in huNSGs/IBMI-CD133 infected with R5-tropic NL-A4Y1ΔNef. HuNSGs/IBMI-CD133, 6–8 months after the transplantation, were anesthetized and intraperitoneally inoculated with NL-A4Y1ΔNef (see Table 1 for summary). Peripheral blood samples were collected periodically as indicated in the graphs. Virus replication in the plasma samples was assessed by real-time RT-PCR analysis and absolute CD4+ cell numbers in the blood samples were determined by flow cytometry analysis. (A) The actual number of HIV-1 RNA copies and the absolute number of CD4+ cells in each individual. (B) Comparison of the logarithm of the RNA copy number and the absolute number of CD4+ cells among virus-inoculated mice. (C) Comparison of the logarithm of the RNA copy number and the absolute number of CD4+ cells between R5-tropic NL-A4Y1- and its mutant NL-A4Y1ΔNef-inoculated mice.
4 Discussion
Small animal models including humanized mice have contributed greatly to the progress of basic and clinical research on viruses. Animal models are essential for studies on viral diseases, especially on those caused by strictly human-tropic viruses such as HIV-1. Enormous effort has been expended to establish humanized mice that mimic HIV-1-infected humans (1–7). Of humanized mice, various non-BLT humanized mice have been constructed by using different materials and methods including distinct mouse strains and engraftment methods (Figure 1). These differences in materials/methods can alter the reconstitution of human hematopoietic cells and the supply of CD4+ cells, and consequently can affect HIV-1 replication in vivo among non-BLT humanized mice generated. In this report, we described the method to generate humanized mice (huNSGs/IBMI-CD133) by transplanting UCB-derived CD133+ cells through IBMI (Figures 2, 3, Supplemental Movie). HuNSGs/IBMI-CD133 showed a steady increase in CD4+ T cells from 3 to 8 months after the CD133-transplantation (Figure 4) and survived for almost one year, except for three mice that died shortly after HIV-1 inoculation (Table 1). In our huNSGs/IBMI-CD133, both X4- and R5-tropic HIV-1 clones replicated well (Figures 5, 6, 8). At the same inoculation dose of viruses, the plasma viral load of R5-tropic NL-A4Y1 virus peaked early (2 to 3 weeks) after the virus inoculation and was sustained long-term (4 to 6 months) accompanied by a gradual decrease in the CD4+ cell number, whereas X4-tropic NL4-3 peaked viremia around 5 to 10 weeks post-inoculation along with the rapid depletion of CD4+ cells. Although a small number of humanized mice were tested in this study, these results may support the pervious results obtained using non-BLT humanized mice infected with X4- and R5-tropic HIV-1 clones (23-25, 32). In total, our huNSGs/IBMI-CD133 are comparable to the other humanized mice reported so far and are expected to be capable of long-term reconstitution of human CD4+ cells by transplantation of CD133+ cells using the IBMI method. Similar to other non-BLT humanized mice, our huNSGs/IBMI-CD133 also require the improvement of immune responses through some modifications such as the transgenic expression of cytokines (M-CSF, GM-CSF, IL-3, etc.) and human leukocyte antigen molecules (12, 13, 15).
The bone marrow is one of the important sites that reconstitute immune cells including T cells and B cells in various types of non-humanized mice (18–20). Transplantation of CD133+ cells into the bone marrow with IBMI is involved in facilitating the abilities to differentiate into CD34+ cells and to reconstitute lymphoid and myeloid cells (21, 22). Therefore, it is conceivable that after R5-tropic HIV-1 inoculation, CD4+ and CD8+ cells were stably maintained for 6 to 8 months (almost one year after CD133+ cells transplantation) in huNSGs/IBMI-CD133. Sumide et al. examined the differentiation/cell repopulation ability of CD133+ cells in the bone marrow, spleen, and thymus in NOG mice transplanted with CD133+ cells using IBMI. They have reported that CD133+ cells have multi-lineage differentiation capabilities, such as CD19+ B-lymphoid and CD33+/CD11b+ myeloid, and that CD56+ NK cells in the spleen and CD3+ and CD4/CD8 single/double-positive cells in the thymus were also detected. (34). BRG mice transplanted with CD133+ cells showed the engraftment of human CD45+ cells in the bone marrow, spleen, thymus, and mesenteric lymph node, and the repopulation of human CD3/CD45 and CD19/CD45 in peripheral blood (17). It has been shown that in non-BLT humanized mice, there is a higher percentage of CXCR4 expressing cells but a lower percentage of CCR5 within the CD4+ T cell population in lymphoid organs and peripheral blood (23, 54). It can be assumed that our huNSGs/IBMI-CD133 individuals probably express CXCR4 and CCR5 on CD4+ T cells similarly to as reported previously, since X4- and R5-HIV-1 viruses replicate well in vivo. Further experiments are required for the human cell differentiations and the sub-populations of CD4+ T cells in peripheral blood and lymphoid organs in our huNSGs/IBMI-CD133 model. It has been reported that HTLV-I-specific CD8+ cytotoxic T lymphocytes and IgG antibodies were induced upon HTLV-I infection in CD133+ cells-transplanted humanized NOG mice (35). In this study, we did not observe the induction of HIV-1-specific IgG antibodies in huNSGs/IBMI-CD133 (data not shown). This may be due to differences in the immune response (the lack of CD4 helper function and B cell maturation), virus species (HTLV-I vs HIV-1), the timing of infection after transplantation (4–5 months vs 6–8 months), mouse strain (NOG vs NSG), and/or individuals used for the experiments.
Various animal models including humanized mice and non-human primates have inherent advantages and disadvantages (5, 9). Researchers have to choose which animal model is suitable for achieving the goals of their studies. For non-BLT mice, further genetic and technical modifications are necessary to improve the immune response and immune cell distribution to secondary lymphoid tissues and so on. Approaches to overcome these issues, including the introduction of human cytokines and human leukocyte antigens into mice, have been extensively made (12, 13, 15). While our IBMI-CD133 method can afford the recipient mice a weak acquired immunity against pathogenic human retrovirus HTLV-1 (35), it also requires the improvement of HIV-1-specific immune response through these modifications (our unpublished data). The generation of non-BLT humanized mice that induce sufficient immune responses and mimic HIV-1-infected humans allows us to study cell-cell interactions in lymphoid tissues and virus-host interactions. Such studies would surely contribute to a deeper understanding of HIV-1 replication in vivo and of its pathogenesis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was reviewed and approved by The animal research committee of Tokushima University.
Author contributions
TaK, J-IF, AA, and MN designed the research project. TO and S-IL guided the generation of humanized mice. TaK, ND, and MN performed the experiments. All authors (TaK, TO, S-IL, ND, ToK, KO, J-IF, AA, and MN) analyzed and discussed the results. AA and MN wrote the manuscript. All authors approved submission. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by JSPS KAKENHI Grant Numbers 21K07042 to MN, 22K07102 to TaK, 21K08491 to ND, and 20K18484 to ToK, and by a grant from the Takeda Science Foundation to TaK.
Acknowledgments
We thank Yayoi Shono (Tokushima University) for experimental assistance. We also thank Kazuko Yoshida (Tokushima University), Kyoko Inui (Tokushima University), and Fumie Nishina (Kansai Medical University) for editorial and administrative assistance.
The TZM-bl cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc. We appreciate the Support Center for Advanced Medical Sciences, Tokushima University Graduate School of Biomedical Sciences, for experimental facilities and technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
The authors TaK, AA, and MN declared that they were editorial board members of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2023.1192184/full#supplementary-material
References
1. Banadyga L, Dolan MA, Ebihara H. Rodent-adapted filoviruses and the molecular basis of pathogenesis. J Mol Biol (2016) 428:3449–66. doi: 10.1016/j.jmb.2016.05.008
2. Pena LJ, Miranda Guarines K, Duarte Silva AJ, Sales Leal LR, Mendes Félix D, Silva A, et al. In vivo and in vivo models for studying zika virus biology. J Gen Virol (2018) 99:1529–50. doi: 10.1099/jgv.0.001153
3. Chen RE, Diamond MS. Dengue mouse models for evaluating pathogenesis and countermeasures. Curr Opin Virol (2020) 43:50–8. doi: 10.1016/j.coviro.2020.09.001
4. Cleary SJ, Pitchford SC, Amison RT, Carrington R, Robaina Cabrera CL, Magnen M, et al. Animal models of mechanisms of SARS-CoV-2 infection and COVID-19 pathology. Br J Pharmacol (2020) 177:4851–65. doi: 10.1111/bph.15143
5. Nguyen TQ, Rollon R, Choi YK. Animal models for influenza research: strengths and weaknesses. Viruses (2021) 13:1011. doi: 10.3390/v13061011
6. Caceres CJ, Seibert B, Cargnin Faccin F, Cardenas-Garcia S, Rajao DS, Perez DR. Influenza antivirals and animal models. FEBS Open Bio (2022) 12:1142–65. doi: 10.1002/2211-5463.13416
7. Fan C, Wu Y, Rui X, Yang Y, Ling C, Liu S, et al. Animal models for COVID-19: advances, gaps and perspectives. Signal Transduct Target Ther (2022) 7:220. doi: 10.1038/s41392-022-01087-8
8. Nomaguchi M, Doi N, Kamada K, Adachi A. Species barrier of HIV-1 and its jumping by virus engineering. Rev Med Virol (2008) 18:261–75. doi: 10.1002/rmv.576
9. Hatziioannou T, Evans DT. Animal models for HIV/AIDS research. Nat Rev Microbiol (2012) 10:852–67. doi: 10.1038/nrmicro2911
10. Nishimura Y, Martin MA. Of mice, macaques, and men: broadly neutralizing antibody immunotherapy for HIV-1. Cell Host Microbe (2017) 22:207–16. doi: 10.1016/j.chom.2017.07.010
11. Thippeshappa R, Kimata JT, Kaushal D. Toward a macaque model of HIV-1 infection: roadblocks, progress, and future strategies. Front Microbiol (2020) 11:882. doi: 10.3389/fmicb.2020.00882
12. Gillgrass A, Wessels JM, Yang JX, Kaushic C. Advances in humanized mouse models to improve understanding of HIV-1 pathogenesis and immune responses. Front Immunol (2021) 11:617516. doi: 10.3389/fimmu.2020.617516
13. Terahara K, Iwabuchi R, Tsunetsugu-Yokota Y. Perspectives on non-BLT humanized mouse models for studying HIV pathogenesis and therapy. Viruses (2021) 13:776. doi: 10.3390/v13050776
14. Ito R, Takahashi T, Katano I, Ito M. Current advances in humanized mouse models. Cell Mol Immunol (2012) 9:208–14. doi: 10.1038/cmi.2012.2
15. Baroncini L, Bredl S, Nicole KP, Speck RF. The humanized mouse model: what added value does it offer for HIV research? Pathogens (2023) 12:608. doi: 10.3390/pathogens12040608
16. Freund D, Oswald J, Feldmann S, Ehninger G, Corbeil D, Bornhäuser M. Comparative analysis of proliferative potential and clonogenicity of MACS-immunomagnetic isolated CD34+ and CD133+ blood stem cells derived from a single donor. Cell Prolif (2006) 39:325–32. doi: 10.1111/j.1365-2184.2006.00386.x
17. De Giovanni C, Nicoletti G, Landuzzi L, Romani F, Croci S, Palladini A, et al. Human responses against HER-2-positive cancer cells in human immune system-engrafted mice. Br J Cancer (2012) 107:1302–9. doi: 10.1038/bjc.2012.394
18. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood (2002) 100:3175–82. doi: 10.1182/blood-2001-12-0207
19. Okada S, Harada H, Ito T, Saito T, Suzu S. Early development of human hematopoietic and acquired immune systems in new born NOD/Scid/Jak3null mice intrahepatic engrafted with cord blood-derived CD34 + cells. Int J Hematol (2008) 88:476–82. doi: 10.1007/s12185-008-0215-z
20. Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, et al. Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rγKO mice. J Immunol (2012) 188:6145–55. doi: 10.4049/jimmunol.1103660
21. Kushida T, Inaba M, Hisha H, Ichioka N, Esumi T, Ogawa R, et al. Intra-bone marrow injection of allogeneic bone marrow cells: a powerful new strategy for treatment of intractable autoimmune diseases in MRL/lpr mice. Blood (20010) 97:3292–9. doi: 10.1182/blood.v97.10.3292
22. Wang J, Kimura T, Asada R, Harada S, Yokota S, Kawamoto Y, et al. SCID-repopulating cell activity of human cord blood-derived CD34- cells assured by intra-bone marrow injection. Blood (2003) 101:2924–31. doi: 10.1182/blood-2002-09-2782
23. Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, Kurrer MO, et al. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci USA (2006) 103:15951–6. doi: 10.1073/pnas.0604493103
24. Nie C, Sato K, Misawa N, Kitayama H, Fujino H, Hiramatsu H. Selective infection of CD4+ effector memory T lymphocytes leads to preferential depletion of memory T lymphocytes in R5 HIV-1-infected humanized NOD/SCID/IL-2Rgammanull mice. Virology (2009) 394:64–72. doi: 10.1016/j.virol.2009.08.011
25. Berges BK, Akkina SR, Remling L, Akkina R. Humanized Rag2(-/-)gammac(-/-) (RAG-hu) mice can sustain long-term chronic HIV-1 infection lasting more than a year. Virology (2010) 397:100–3. doi: 10.1016/j.virol.2009.10.034
26. Ince WL, Zhang L, Jiang Q, Arrildt K, Su L, Swanstrom R. Evolution of the HIV-1 env gene in the Rag2-/- gammaC-/- humanized mouse model. J Virol (2010) 84:2740–52. doi: 10.1128/JVI.02180-09
27. Akkina R, Berges BK, Palmer BE, Remling L, Neff CP, Kuruvilla J, et al. Humanized Rag1-/- γc-/- mice support multilineage hematopoiesis and are susceptible to HIV-1 infection via systemic and vaginal routes. PloS One (2011) 6:e20169. doi: 10.1371/journal.pone.0020169
28. Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, et al. HIV Therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature (2012) 492:118–22. doi: 10.1038/nature11604
29. Dave VP, Hajjar F, Dieng MM, Haddad É., Cohen É.AEfficient BST2 antagonism by Vpu is critical for early HIV-1 dissemination in humanized mice. Retrovirology (2013) 10:128doi: 10.1186/1742-4690-10-128
30. Horwitz JA, Halper-Stromberg A, Mouquet H, Gitlin AD, Tretiakova A, Eisenreich TR, et al. HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc Natl Acad Sci USA (2013) 110:16538–43. doi: 10.1073/pnas.1315295110
31. Halper-Stromberg A, Lu CL, Klein F, Horwitz JA, Bournazos S, Nogueira L, et al. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell (2014) 158:989–99. doi: 10.1016/j.cell.2014.07.043
32. Terahara K, Ishige M, Ikeno S, Okada S, Kobayashi-Ishihara M, Ato M, et al. Humanized mice dually challenged with R5 and X4 HIV-1 show preferential R5 viremia and restricted X4 infection of CCR5(+)CD4(+) T cells. Microbes Infect (2015) 17:378–86. doi: 10.1016/j.micinf.2015.02.002
33. Takahashi M, Matsuoka Y, Sumide K, Nakatsuka R, Fujioka T, Kohno H, et al. CD133 is a positive marker for a distinct class of primitive human cord blood-derived CD34-negative hematopoietic stem cells. Leukemia (2014) 28:1308–15. doi: 10.1038/leu.2013.326
34. Sumide K, Matsuoka Y, Kawamura H, Nakatsuka R, Fujioka T, Asano H, et al. A revised road map for the commitment of human cord blood CD34-negative hematopoietic stem cells. Nat Commun (2018) 9:2202. doi: 10.1038/s41467-018-04441-z
35. Tezuka K, Xun R, Tei M, Ueno T, Tanaka M, Takenouchi N, et al. An animal model of adult T-cell leukemia: humanized mice with HTLV-1-specific immunity. Blood (2014) 123:346–55. doi: 10.1182/blood-2013-06-508861
36. Gallacher L, Murdoch B, Wu DM, Karanu FN, Keeney M, Bhatia M. Isolation and characterization of human CD34(-)Lin(-) and CD34(+)Lin(-) hematopoietic stem cells using cell surface markers AC133 and CD7. Blood (2000) 95:2813–20.
37. Goussetis E, Theodosaki M, Paterakis G, Tsecoura C, Graphakos S. In vitro identification of a cord blood CD133+CD34-lin+ cell subset that gives rise to myeloid dendritic precursors. Stem Cells (2006) 24:1137–40. doi: 10.1634/stemcells.2005-0283
38. de Wynter EA, Buck D, Hart C, Heywood R, Coutinho LH, Clayton A, et al. CD34+AC133+ cells isolated from cord blood are highly enriched in long-term culture-initiating cells, NOD/SCID-repopulating cells and dendritic cell progenitors. Stem Cells (1998) 16:387–96. doi: 10.1002/stem.160387
39. Matsumoto K, Yasui K, Yamashita N, Horie Y, Yamada T, Tani Y, et al. In vitro proliferation potential of AC133 positive cells in peripheral blood. Stem Cells (2000) 18:196–203. doi: 10.1634/stemcells.18-3-196
40. Pasino M, Lanza T, Marotta F, Scarso L, De Biasio P, Amato S, et al. Flow cytometric and functional characterization of AC133+ cells from human umbilical cord blood. Br J Haematol (2000) 108:793–800. doi: 10.1046/j.1365-2141.2000.01949.x
41. Dichtelmüller H, Stephan W, Prince AM, Gürtler L, Deinhardt F. Inactivation of HIV in plasma derivatives by beta-propiolactone and UV irradiation. Infection (1987) 15:367–9. doi: 10.1007/BF01647745
42. Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol (1986) 59:284–91. doi: 10.1128/JVI.59.2.284-291.1986
43. Doi N, Sakai Y, Adachi A, Nomaguchi M. Generation and characterization of new CCR5-tropic HIV-1rmt clones. J Med Invest (2017) 64:272–9. doi: 10.2152/jmi.64.272
44. Doi N, Fujiwara S, Adachi A, Nomaguchi M. Rhesus M1.3S cells suitable for biological evaluation of macaque-tropic HIV/SIV clones. Front Microbiol (2011) 2:115. doi: 10.3389/fmicb.2011.00115
45. Nomaguchi M, Yokoyama M, Kono K, Nakayama EE, Shioda T, Saito A, et al. Gag-CA Q110D mutation elicits TRIM5-independent enhancement of HIV-1mt replication in macaque cells. Microbes Infect (2013) 15:56–65. doi: 10.1016/j.micinf.2012.10.013
46. Lebkowski JS, Clancy S, Calos MP. Simian virus 40 replication in adenovirus-transformed human cells antagonizes gene expression. Nature (1985) 317:169–71. doi: 10.1038/317169a0
47. Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol (1998) 72:2855–64. doi: 10.1128/JVI.72.4.2855-2864.1998
48. Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J Virol (2009) 83:8289–92. doi: 10.1128/JVI.00709-09
49. Kamada K, Igarashi T, Martin MA, Khamsri B, Hatcho K, Yamashita T, et al. Generation of HIV-1 derivatives that productively infect macaque monkey lymphoid cells. Proc Natl Acad Sci USA (2006) 103:16959–64. doi: 10.1073/pnas.0608289103
50. Willey RL, Smith DH, Lasky LA, Theodore TS, Earl PL, Moss B, et al. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J Virol (1988) 62:139–47. doi: 10.1128/JVI.62.1.139-147.1988
51. Nomaguchi M, Yokoyama M, Kono K, Nakayama EE, Shioda T, Doi N, et al. Generation of rhesus macaque-tropic HIV-1 clones that are resistant to major anti-HIV-1 restriction factors. J Virol (2013) 87:11447–61. doi: 10.1128/JVI.01549-13
52. O’Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol (2000) 74:10074–80. doi: 10.1128/jvi.74.21.10074-10080.2000
53. Xing L, Wang S, Hu Q, Li J, Zeng Y. Comparison of three quantification methods for the TZM-bl pseudovirus assay for screening of anti-HIV-1 agents. J Virol Methods (2016) 233:56–61. doi: 10.1016/j.jviromet.2016.03.008
54. Terahara K, Ishige M, Ikeno S, Mitsuki YY, Okada S, Kobayashi K. Expansion of activated memory CD4+ T cells affects infectivity of CCR5-tropic HIV-1 in humanized NOD/SCID/JAK3null mice. PloS One (2013) 8:e53495. doi: 10.1371/journal.pone.0053495
55. Zou W, Denton PW, Watkins RL, Krisko JF, Nochi T, Foster JL, et al. Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology (2012) 9:44. doi: 10.1186/1742-4690-9-44
56. Watkins RL, Zou W, Denton PW, Krisko JF, Foster JL, Garcia JV. In vivo analysis of highly conserved nef activities in HIV-1 replication and pathogenesis. Retrovirology (2013) 10:125. doi: 10.1186/1742-4690-10-125
57. Watkins RL, Foster JL, Garcia JV. In vivo analysis of Nef’s role in HIV-1 replication, systemic T cell activation and CD4(+) T cell loss. Retrovirology (2015) 12:61. doi: 10.1186/s12977-015-0187-z
58. Miller MD, Warmerdam MT, Gaston I, Greene WC, Feinberg MB. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J Exp Med (1994) 179:101–13. doi: 10.1084/jem.179.1.101
59. Tokunaga K, Ishimoto A, Ikuta K, Adachi A. Growth ability of auxiliary gene mutants of human immunodeficiency virus types 1 and 2 in unstimulated peripheral blood mononuclear cells. Arch Virol (1997) 142:177–81. doi: 10.1007/s007050050068
60. Buffalo CZ, Iwamoto Y, Hurley JH, Ren X. How HIV Nef proteins hijack membrane traffic to promote infection. J Virol (2019) 93:e01322–19. doi: 10.1128/JVI.01322-19
61. Ramirez PW, Sharma S, Singh R, Stoneham CA, Vollbrecht T, Guatelli J. Plasma membrane-associated restriction factors and their counteraction by HIV-1 accessory proteins. Cells (2019) 8:1020. doi: 10.3390/cells8091020
Keywords: HIV-1, humanized mouse, CD133, IBMI, X4-tropic, R5-tropic, Nef
Citation: Koma T, Odaka T, Lee S-I, Doi N, Kondo T, Okuma K, Fujisawa J-I, Adachi A and Nomaguchi M (2023) Humanized mice generated by intra-bone marrow injection of CD133-positive hematopoietic stem cells: application to HIV-1 research. Front. Virol. 3:1192184. doi: 10.3389/fviro.2023.1192184
Received: 23 March 2023; Accepted: 09 June 2023;
Published: 30 June 2023.
Edited by:
Amanda Brown, Johns Hopkins University, United StatesReviewed by:
Hirotaka Ebina, Osaka University, JapanSubodh Kumar Samrat, University of Arizona, United States
Copyright © 2023 Koma, Odaka, Lee, Doi, Kondo, Okuma, Fujisawa, Adachi and Nomaguchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun-Ichi Fujisawa, ZnVqaXNhd2FAaGlyYWthdGEua211LmFjLmpw; Akio Adachi, YWRhY2hpQHRva3VzaGltYS11LmFjLmpw; Masako Nomaguchi, bm9tYWd1Y2hpQHRva3VzaGltYS11LmFjLmpw