1
Department of Chemistry, University of Illinois, Urbana, IL, USA
2
Department of Animal Sciences, University of Illinois, Urbana, IL, USA
Neuropeptides and hormones are signaling molecules that support cellâÂÂcell communication in the central nervous system. Experimentally characterizing neuropeptides requires significant efforts because of the complex and variable processing of prohormone precursor proteins into neuropeptides and hormones. We demonstrate the power and flexibility of the Python language to develop components of an bioinformatic analytical pipeline to identify precursors from genomic data and to predict cleavage as these precursors are en route to the final bioactive peptides. We identified 75 precursors in the rhesus genome, predicted cleavage sites using support vector machines and compared the rhesus predictions to putative assignments based on homology to human sequences. The correct classification rate of cleavage using the support vector machines was over 97% for both human and rhesus data sets. The functionality of Python has been important to develop and maintain NeuroPred (http://neuroproteomics.scs.uiuc.edu/neuropred.html
), a user-centered web application for the neuroscience community that provides cleavage site prediction from a wide range of models, precision and accuracy statistics, post-translational modifications, and the molecular mass of potential peptides. The combined results illustrate the suitability of the Python language to implement an all-inclusive bioinformatics approach to predict neuropeptides that encompasses a large number of interdependent steps, from scanning genomes for precursor genes to identification of potential bioactive neuropeptides.
Neuropeptides are a class of cell–cell peptides that can act as neurotransmitters and hormones and have various paracrine, endocrine, and autocrine effects (Boutrel, 2008
; Heinrichs and Domes, 2008
). Neuropeptides directly influence a diverse set of biological processes from growth and development to learning. For example, oxytocin is known as a mammalian hormone associated with reproduction but also is a neurotransmitter that has been associated with social behavior traits including trust, autism, inhibition of tolerance to additive drugs and impaired learning and memory functions. Furthermore, oxytocin and arginine vasopressin are intermediaries of social behaviors, including attachment, social cognition and stress, anxiety, and aggression (Heinrichs and Domes, 2008
).
Experimental detection of neuropeptides in mammals has been limited to a few species (primarily human, mouse and rat) or the characterization of selected peptide families (such as insulin) across greater numbers of species. This lack of experimental characterization is predominantly because such experimental procedures are resource intense and the presence of neuropeptides varies with species, tissue, developmental stage and even organism state. Genomic sequencing provides the opportunity to discover neuropeptides in other species with limited or no experimental studies on neuropeptides. For example, while the rhesus macaque monkey (Macaca mulatta) is widely used as model organism and its genome has been sequenced (Rhesus Macaque Genome Sequencing and Analysis Consortium, 2007
), only four rhesus prohormone genes have been reported compared to over 90 human prohormone genes. Consequently, accurate bioinformatic identification of neuropeptide genes and characterization of precursors is essential in rhesus neuroscience research.
Several factors make annotating prohormones and their associated peptides difficult. First, neuropeptides result from a complex series of post-translational modifications (PTMs) of precursor proteins. Second, the conserved “bioactive” peptide sequence that interacts with its cognate receptor can be short, only a few amino acids long, with large sections of diverse sequences in the prohormone. Thus, homology to well-studied species is not enough to offer accurate neuropeptide predictions across species.
The typical structure of neuropeptide precursor after translation includes a signal peptide region and a region that contains one or more neuropeptides (Fricker, 2005
; Hook et al., 2008
). After translation, the signal peptide is removed by signal peptidases and the remaining peptide is cleaved by other proteases (notably proprotein or prohormone proteases) that cleave the sequence at basic (Arg or Lys) sites (Fricker, 2005
; Hook, 2006
; Hook et al., 2008
). After cleavage, the N-terminal basic amino acids are typically removed by carboxylases and various additional PTMs such as amidation and glycosylation can occur (Fricker, 2005
; Hook et al., 2008
).
We address these points here with a bioinformatics toolkit to discover and characterize neuropeptides. Essential components of this analytical pipeline are the computational identification of precursor genes in nucleic databases and model-based prediction of cleavage and other PTMs of the precursors. Python is an ideal language to develop this analytical pipeline for the discovery and characterization neuropeptides. The core language has easy to use functions that facilitate complex manipulation of information and integration of results from the multistep analytical pathway. The suitability of Python is further strengthened by third-party modules such as BioPython (http://biopython.org
) for bioinformatics (Bassi, 2007
) and Numerical and Scientific Python (http://www.scipy.org
) for numerical computation (Oliphant, 2007
). The combination of all these features in a single language makes Python an ideal choice for bioinformatic applications (Bassi, 2007
; Kinser, 2008
). In terms of a pipeline, the power and flexibility of Python can be used for the full pipeline or to integrate different components of the pipeline together. We illustrate the use of Python to implement an analytical pipeline that integrates vastly different components necessary to identify rhesus neuropeptides and associated precursors.
An exhaustive survey of neuropeptide precursors in a genome is the first step in the complete characterization of the neuropeptidome of a species. The development of bioinformatics analytical pipeline to discover neuropeptide precursors requires the integration of multiple steps involving multiple tools. Bioinformatic tools including sequence homology search using BLAST (Altschul et al., 1997
) and multiple sequence alignment using T-Coffee (Notredame et al., 2000
) are available as standalone packages or via a web interface. BioPython provides an integrated environment that supports different aspects of bioinformatics including parsing results from bioinformatics tools. For example, Bassi (2007)
illustrates the use of BLAST with BioPython.
In the first step of the prohormone analytical pipeline, rhesus precursors were identified based on precursor information from other mammalian species with extensive neuropeptide research. In particular, a list of human precursors and neuropeptide sequences was collected from the UniProt Knowledgebase database (The UniProt Consortium, 2008
) and literature review (Amare et al., 2006
; Tegge et al., 2008
). The set of human precursors was queried against the database of predicted proteins derived from the rhesus genome (http://www.ncbi.nlm.nih.gov/projects/genome/guide/rhesus_macaque/
) using a standalone version of BLAST (version 2.2.18) using the default parameter settings (e.g., expectation value of 10 and Blosum62 scoring matrix) except for disabling the filter option. Queries were conducted using the complete precursor sequence that included the regions that contain the signal peptide and neuropeptides to maximize the detection of the rhesus precursor. Human precursors were used because of the evolutionary relationships between the rhesus and human species and the completeness of the list in humans. Information from other species (e.g. mouse and rat) can also be used to evaluate the accuracy of the search process. The repetitive process of searching for each human precursor on the rhesus database was implemented by exploiting the ability of BLAST to handle multiple sequences and using Python to parse results. The query input file containing all human precursors was submitted to BLAST and the output was saved in an XML formatted file. An XML format provides structured information in a machine readable format that permits repeated access.
The XML file of BLAST results can be also be parsed directly using standard Python libraries such as the elementtree library to extract the results for each of the human precursors. The script in Listing 1
opens the specified XML file and recursively stores the contents in a Python class that contains the attributes and values specified by the XML docment type definitions used by BLAST. After parsing the BLAST XML file, the script loops across the query sequences and displays the match and the score and e-value of the best match to the query sequence. Using a Python script allows greater control of the output including extracting precursors with the highest scoring BLAST hits, precursors with no hits, all hits that exceed a threshold determined by the user, or all hits. Furthermore, Python provides sufficient flexibility to identify the common scenarios with comparative genome analyses where multiple precursors match the same target or the same precursor matches different targets with similar scores.

Listing 1. Parsing an BLAST XML file in Python.
The complete identification of precursors can require different levels of user input especially related to species divergence. The difficulties imposed by species divergence and available resources can be investigated by evaluating different BLAST specifications (e.g. selection of database, scoring matrices, E-value threshold), different genomic resources (e.g. unassembled sequences) and information from species when this is available. Due to the repetitive nature of these investigations, Python can be used to facilitate the rapid evaluation of the different specifications and combining the information for user assessment.
Although low E-values constitute statistical evidence that supports the detection of homologous sequences between species, false matches and partial matches are possible. The accuracy of the identification of predicted rhesus precursors was accessed by aligning the sequence to corresponding sequences from multiple other species using multiple sequence alignment tools such as T-Coffee. Most multiple sequence alignment tools only perform a single alignment so that it is necessary to perform one alignment for every precursor. Simple Python scripts can be used for the repetitive creation of sequence files including multiple sequences across species for each precursor and subsequent alignment for each precursor. The resulting alignments were then viewed to identify which rhesus precursor predictions are reliable or contain the prediction but are too long (the result of automated predictions and sequencing or assembly errors) or incomplete (due to incomplete coverage of the particular genomic region, sequencing or assembly errors). Based on the final alignments, 67 rhesus neuropeptides precursors were identified solely in the rhesus database of predicted proteins.
Identification of precursors using protein predictions and automated tools is fast and effective. However, this approach misses precursors that are partially predicted or not predicted due to sequencing or assembly issues. In order to identify if a human precursor is present in the genome of the rhesus monkey, the protein sequences of the precursors are queried against the nucleotide sequences from the genome assembly. The result of the BLAST query only provides the locations that sufficiently match the protein sequence and consequently ignore low scoring and intronic regions. The full precursor sequence can be extracted using Wise2 (http://www.ebi.ac.uk/Tools/Wise2/index.html
; Birney et al., 2004
). Wise2 predicts the gene structure by comparing a protein sequence to a genomic DNA sequence and using a gene prediction model that allows for introns and frameshift errors. The genomic sequence required by Wise2 was obtained by using Python to read the genomic DNA sequence of the assembled rhesus genome, identify and extract the relevant chromosomal region and perform the reverse transcription into the complementary strand if necessary. If the extracted region is insufficient to accurately identify the main gene structure components, the genomic region can be expanded and resubmitted to Wise2. An additional advantage of combining the BLAST and Wise2 tools is that the protein sequence, mRNA sequence and the location of the exons are simultaneously available and can be used to confirm the accuracy of the predictions.
The combined strategy of using BLAST and Wise2 directly identified eight additional precursors that were not been previously predicted and provide valuable information for the manual annotation of rhesus precursors. For example, the rhesus CCKN precursor was identified on chromosome 2 but lacked a match to last 28 amino acids of the human CCKN sequence. Examination the genomic sequence showed that a region of 91 unknown bases occurred immediately after the last residue of the mRNA sequence predicted using Wise2. This nucleotide segment most likely codes the missing precursor sequence that corresponded to the last exon of the human precursor gene and was missed in the assembly. The search for the missing region among the rhesus trace archives (a collection of raw sequence traces, http://www.ncbi.nlm.nih.gov/projects/genome/guide/rhesus_macaque/
), uncovered a hit to a contig that contained the missing segment and resulted in the prediction of a complete CCKN precursor. A different scenario was encountered with the NPS precursor because the Wise2 prediction missed the start of the NPS precursor. This failure was most likely due to the structure of the human gene where the first exon only codes for two amino acids. Consequently, the corresponding rhesus exon was identified by a query using the complete human NPS nucleotide and combined with the Wise2 prediction to obtain the complete rhesus NPS precursor.
There were also 17 precursors that could not be recovered solely based on the assembly alone without further examining the trace archives for unassembled or incorrectly assembled contigs. For example, the related crab-eating macaque (M. fascicularis) insulin (INS) precursor has been reported (Wetekam et al., 1982
) and, thus, is expected to be found in the rhesus genome. Queries of the human and M. fascicularis INS sequence on the M. mulatta genome did not permit full recovery of the rhesus INS precursor due to gaps and a stop codon in the genomic assembly. The results from a search of the trace achives indicated that the inclusion of different contig (ti|523766964) would most likely result in the identification of the complete rhesus INS precursor.
The individual precursors undergo a number of additional processing steps before the final bioactive peptides are created. Thus, once the list of precursor protein sequences has been compiled, expected prohormone structural features such as a signal peptide and prohormone cleavage sites are identified for each individual precursor. The signal peptide was predicted using SignalP (Bendtsen et al., 2004
) and the length of the signal peptide was recorded with the sequence. The rhesus precursors lack experimental cleavage information so cleavage sites must be assigned based on homology to other animals or cleavage models. The reliability of the homology-based prediction of cleavage relies on the degree of conservation of the precursor between species available.
Human data were expected to provide the most accurate assignment of cleavage data due to the close evolutionary relationship between the human and rhesus species. Python scripts were developed to assign precursor cleavage information based on homology to human sequences. The human and rhesus sequences of each precursor were first aligned using T-Coffee. The locations of the human cleavage sites were then found in the corresponding aligned rhesus sequence. Finally the rhesus sequence and cleavage data was obtained after removing any gaps that had been entered during the sequence alignment. Assuming that the precursor cleavage assignment based on human information provides a perfect characterization of precursor processing in the rhesus, then the comparison of model-based cleavage predictions and confirmed or homology-based cleavage information will provide the number of true and false positives (cleavage sites) and true and false negatives (non-cleavage sites). These results can be used to construct further indicators of cleavage model performance including correct classification rate (ratio of true versus true and false results), sensitivity (ratio of true positives versus all positives), specificity (ratio of true negatives versus all negatives), positive and negative precision (Southey et al., 2006a
).
Prediction of the cleavage sites within the precursor is essential for identification of the final peptides produced by the prohormones, including the neuropeptides. Previously we have shown that machine learning techniques including logistic regression, artificial neural networks and memory-based reasoning are successful in predicting cleavage sites in neuropeptide precursors in diverse sets of species (Amare et al., 2006
; Hummon et al., 2003
; Southey et al., 2008
; Tegge et al., 2008
). An analytical pipeline to predict cleavage using machine learning involves preparing and processing the sequence and cleavage data, training and testing of prediction models using machine learning techniques to identify the most appropriate model, predict the possible peptides using the most appropriate model and any PTMs present in the predicted peptides.
Python can be used to process the sequence and cleavage data into a generic file that can be used by a single application as well by different applications following the steps outlined by Southey et al. (2008)
. Generally these steps involve: (1) reading the sequence and cleavage data, (2) removing the signal peptide, (3) splitting the remaining sequence into overlapping windows, (4) assigning cleavage status to the window and (5) recoding the amino acids as binary indicators with respect to the actual location within the window. The script in Listing 2
demonstrates how a single neuropeptides sequence with length of signal peptide and cleavage site is processed. First the signal peptide is removed and the resulting sequence is padded to permit windows that may extend past the ends of the sequence. The sequence is then split into overlapping windows and windows with basic amino acids (Lys and Arg) are kept. The amino acids within each window are then recoded with dummy values and cleavage status is assigned. The resulting location within the complete precursor sequence, the window of the sequence, cleavage status of the window and coding of the amino acids is then displayed.

Listing 2. Python script to recode an amino acid sequence into generic format for machine learning applications.
The resulting generic file can be used as input to a stand-alone machine learning package or tool (e.g. R http://www.r-project.org
), or by a tool directly implemented in Python (e.g. the SciKit learn http://www.scipy.org/scipy/scikits/wiki/MachineLearning
), or automatically passed to a stand-alone tool using a Python interface and language bindings. This latter strategy will be illustrated using the Python bindings provided with the LibSVM package (Chang and Lin, 2001
) that implement training and cross-validation of support vector machines in Python. The general use of LibSVM involves the input of data, selection of a support vector machine and associated parameter, training of the support vector machine given the data and parameters and evaluation of trained support vector machine. Following Salzberg (1997)
, the optimal parameters for the support vector machine were identified using cross-validation and a grid search across the parameters of the support vector machine. Preliminary results indicated that the default support vector machine with a radial basis function provided the same performance as other types and had the advantage of only requiring two parameters. The LibSVM also provides k-fold cross-validation where the training data was split into k components of which k – 1 components was used to train a model and the last component was used for testing. The cross-validation approach was repeated such that all data components were used as testing and the overall cleavage miss-classification rate across complete data is obtained.
A Python script was used process generic file previously obtained from the human and rhesus sequence and cleavage data into human and rhesus data sets in the format required by the LibSVM. Part of the script (Listing 3
) loops across the two parameters of a support vector machine with a radial basis function (gamma and C) and within the loop calls the LibSVM cross-validation routine with the parameters of the support vector machine and supplied degree of cross-validation. This script also trains the support vector machine for the supplied parameters on the full training data set and computes the accuracy of this support vector machine on the test and training data sets. This script can be easily extended to evaluate multiple support vector machine specifications including linear and polynomial. In addition to the cross-validation, the script also trained a support vector machine on the full test data set for the supplied parameter values and tested the resulting support vector machine on the full test data and the training data. For data sets where the cross-validation and full data set support vector machine analyses for each combination of parameters becomes prohibitive, the script can be modified such that the support vector machine analysis of the full data set is only executed after the parameter values that provide the lowest miss-classification rate have been identified in a prior cross-validation step.

Listing 3. Python script for training and testing a support vector machine.
The parsing of the results from the Python script that trained and tested the support vector machine models offered insights into the similarities between the human and rhesus cleavage patterns. The rhesus and human cleavage prediction models selected had the highest 5-fold cross-validation accuracy and the fewest prediction errors in the training data. The evaluation of the parameters on the full data sets was also important because support vector machines with similar cross-validation correct classification rates had lower performance on the full and test data sets. For example, in the human support vector machine, the two highest scoring human support vector machines had correct classification rates of 91.0% and 90.6% after cross-validation. However, the highest scoring human support vector machine had correct classification rates of 99.9% and 99.6% in the human full data set and rhesus full data set, respectively. Whereas the second scoring human support vector machine had an approximately 3% lower correct classification rate in human full data set, and rhesus full data set (97.3% and 96.4%, respectively).
The performance of the support vector machine models was compared to the mammalian logistic regression model (Amare et al., 2006
), the human logistic regression and human artificial neural models (Tegge et al., 2008
) and the empirical Known Motif model Southey et al. (2006b)
. On the human data set, the human support vector machine had the highest correct classification rate (99.9%), as expected, followed by the rhesus support vector machine (97.9%), human artificial neural model (92.2%), human logistic regression (90.2%), mammalian logistic regression (82.5%) and finally the Known Motif model (76.6%). The rhesus support vector machine provided perfect classification on the rhesus data set followed by the human support vector machine (99.6%), human artificial neural model (91.3%), human logistic regression (89.6%), mammalian logistic regression (82.4%) and finally the Known Motif model (76.7%). Models trained on human data had better prediction than general mammalian model or empirical known motif model. This result was expected independently of evolutionary relationships because the human cleavage data was used to assign cleavage in the rhesus.
The main reason for the different model performance was the lower number of false positive predictions by the support vector machines relative to the other methodologies. The rhesus support vector machine had slightly lower number of false negative predictions in the human data set than the human artificial neural network. The differences between the different prediction approaches are due to differences in the data sets used to train and test the models and the ability of the methodologies to accommodate linear and non-linear relationships between the input variables and cleavage patterns. Tegge et al. (2008)
used 62 human precursors to train artificial neural network and logistic regression models, Amare et al. (2006)
used 39 mammalian precursors to train logistic models and the human support vector machine model developed in this study were trained on 93 human precursors. The artificial neural network had perfect (100%) classification on the human data set reported in Tegge et al. (2008)
.The lower correct classification rate result by including more human precursors indicating that the human data set used by Tegge et al. (2008)
likely does not contain complete information on cleavage that was used in training the support vector machines.
Across species, the impact of the precursor sequences used to train and test in the model performance can be assessed by comparing the performance of the same model across species. Comparison of the data used to train the support vector machines showed that all rhesus precursors had homologous in the human data set but 20 human precursors were not present on the rhesus data set. Of the 37 sites that received different cleavage classification by the two support vector machines, only 10 sites corresponded to precursors that were present in both species data sets; meanwhile the remaining sites were only present in the human precursor data set. Among the sites with differential cleavage prediction between species, four sites pertained to rhesus sequences that have different amino acids than the human sequence and these amino acids have a strong association with cleavage patterns. For example, the INSL4 precursor in the rhesus includes a window with the amino acid sequence ‘GCGPRFGKR↓MLSYCPMPE’ where ↓ denoted the predicted cleavage site. However, this site was assigned a non-cleavage observed value because the homologous human window, ‘GCGPRFGKHLLSYCPMPE’, has not reported to be cleaved. Similarly, Southey et al. (2006b)
reported a single amino acid difference between human and chimpanzee RFRP precursor that resulted in a false positive prediction in the chimpanzee sequence. These results demonstrate the value of bioinformatic prediction of precursor cleavage, especially in species with limited experimental confirmation. One important use of across species predictions is to eliminate false positive results from experimental consideration. As another use, this same information can also identify potentially species-specific cleavage sites to explain peptides that are unexpected based on homology alone.
The prediction of cleavage sites in a protein sequence requires that the sequence must be processed into a usable format, then the prediction model is applied and finally the actual prediction are returned. Each of these steps requires specialized knowledge ranging from processing the sequence to technical knowledge of applying the models derived from machine learning models. Developing a web application is one approach to remove specialized knowledge because a web form can be provided where the underlying script is responsible to convert the input into required format, apply the prediction models and display resulting predictions.
We developed NeuroPred (http:// neuroproteomics.scs.uiuc.edu/neuropred.html
), a web application in Python, to supports the detection and characterization of the neuropeptidome (Southey et al., 2006a
). The user requires only a sequence basic knowledge of neuropepeptides and there is no requirement for specialized knowledge of areas such as Python or machine learning. Using a simple form, users can enter one or more protein sequences and then select one or more prediction models and different options that control the subsequent processing of the resulting peptides and output. A user can select either simple options where most options have been preselected for the user a more advanced options that provide all possible models and control of the input and output. NeuroPred validates all the inputs, predict cleavage sites for all sequences entered and models selected. Under the default options, NeuroPred will display a cleavage prediction diagram indicating the predicted cleavage locations and optionally the probabilities of cleavage for the sequences entered and model selected (Figure 1
).
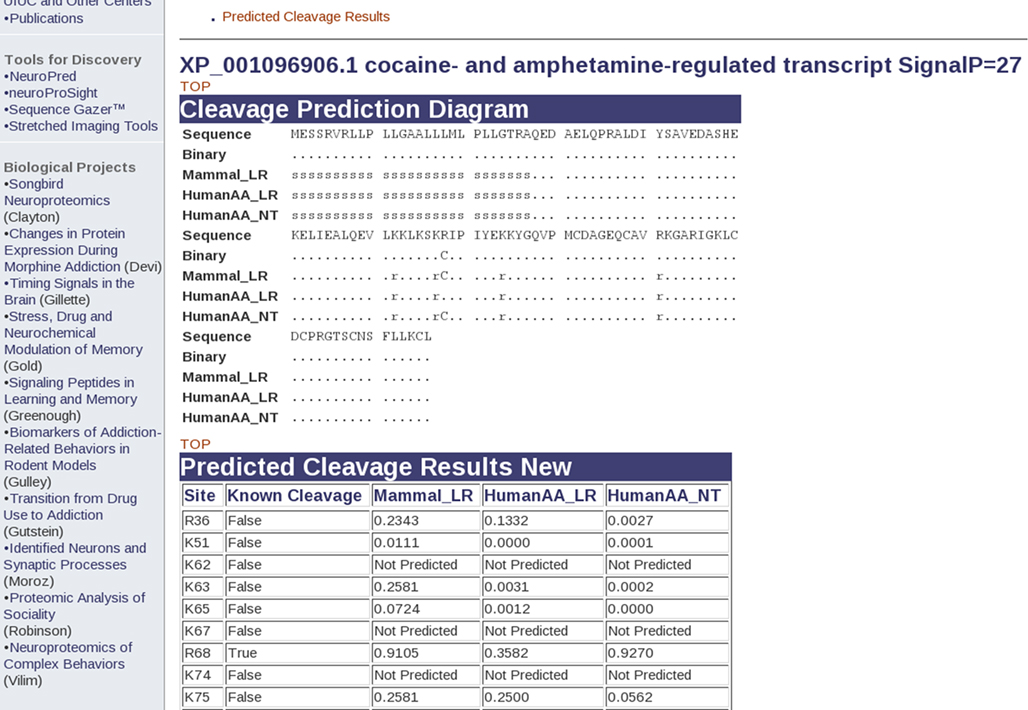
Figure 1. Predicted cleavage sites of the rhesus cocaine- and amphetamine-regulated gene using different models in NeuroPred.
To assist in the experimental studies using mass spectrometry (e.g., Hummon et al., 2005
; Li and Sweedler, 2008
), NeuroPred also computes the predicted mass of peptides including most of the known neuropeptide PTMs. The computation of the mass of the predicted peptides that can be used in high throughput mass spectrometry studies to assist in the identification of peptides. Depending on the options selected, NeuroPred will list the different peptides possible, the source for cleavage for the peptide (such as signal peptidase or prediction from one or more models), PTMs applied to the resulting peptides, predicted mass and full peptide sequence. NeuroPred also joins adjacent peptides to account for false positive cleavages and the presence of intermediate peptides that are eventually cleaved.
NeuroPred provides cleavage predictions using model developed from a vast range of species (including mollusk, insects and mammals) used in neuroscience research. Generally it is expected that the most appropriate model will be trained on the same or closely related species. However, it is expected that there are situations where there is no obvious appropriate model or that there is a requirement for a greater understanding cleavage prediction at different sites. For these types of situations, NeuroPred can compute different model accuracy statistics when cleavage information is uploaded together with sequence. The resulting output enables the comparison of the selected models for individual precursors and for all precursors.
One valuable aspect of using Python was that much of the code developed for the analytical pipeline was reused in NeuroPred and can also be easily packaged into a stand-alone application. For example, the processing of sequence information and application of different cleavage prediction models requires the same code across the different applications. This feature allows the main coding to be focused on integrating components rather than developing a completely new application. Furthermore, additional or more efficient Python code developed for a new application can be reused by previous application. For example, the original prediction equations from different models were implemented using scalar computations. However, faster code was generated by implementing the prediction equations as a series of vector-matrix multiplications in Numerical Python. Improvements in computational speed were beneficial for all applications and particularly for NeuroPred because of the volume of requests handled by this public web service.
The text processing capabilities in Python were important to enable the integration of the NeuroPred application with the visual appearance of the main web site. The main site provides static information that does not change in response to the user. In contrast, the output from NeuroPred is dynamic because the output depends on user interaction. If the html coding recoding is directly used within the script, the script must be changed when the main web site changes. However, the string processing ability of Python permits Python scripts to easily search and replace portions of text. In particular, the template of the main web site or an existing web page in the required format can be directly parsed by Python and the necessary portions replaced such that the web application will provide the same visual appearance as the main web site. Alternatively, Python web frameworks such as Django (http://www.djangoproject.com/
) can be used to develop and maintain extensive web sites.
The Python language is well-suited to implement a bioinformatics approach that encompasses a large number of interdependent steps, from scanning genomes for precursor genes to identification of neuropeptides. We did not encounter any shortcomings with Python that were specific to our application or that hampered our efforts to obtain results. The series of steps encompassed in the analytical pipeline implemented in Python reflect the flexibility of this language to support diverse applications. The versatility of Python across all steps, identification of neuropeptide precursors from genomic sequences, generation and training of cleavage prediction models, and development of a web application to predict cleavage sites, PTMs, and resulting peptides was illustrated.
The components of an neuropeptide analytical pipeline developed using Python supports the examination and annotation of genomes, prediction of cleavage sites, and characterization of resulting peptides, irrespectively of the extent of experimental neuropeptide evidence. The successful application of the discovery aspect of this pipeline led to the identification of 78 rhesus neuropeptide precursors, including 11 precursors that had not been predicted during the automated annotation of the genome. The training and evaluation of models to predict cleavage sites in rhesus precursors resulted in models that had correct classification rate of over 80% based on homologous cleavage assignments from human precursors indicating successful application of the cleavage prediction component of the pipeline. NeuroPred is a direct application of the neuropeptide analytical pipeline to provide an all-inclusive Python web application that allows users to predict precursor cleavage and subsequent PTMs of the resulting peptides. This application supports targeted experimental search for likely predicted peptides and greatly facilitates the laborious search for neuropeptides in mass spectra from high throughput proteomic studies.
The level of user input required to comprehensively identify the precursor complement depends on the available resources and on the divergence of the species under study with respect to other species with known precursor information. In this study we demonstrated how Python routines can aid with many tedious components of genome-wide precursor identification and cleavage prediction such as the processes that must be repeated for each precursor. Our routines help to address the challenges associated with species divergence and in-progress sequencing and assembly processes (e.g. coverage, accuracy) by facilitating the evaluation of different specifications (e.g. databases, scoring matrices, E-value thresholds) and of models from species with different level of divergence.
Results from characterization of the rhesus neuropeptidome using an analytical pipeline and implementation of the pipeline as a public web application that serves the neuroscience community demonstrate the suitability of the Python language for multiplexed and high throughput bioinformatics applications. The object-orientated nature of the Python language enabled considerable reuse of code at the different stages of development. A completely integrated approach can also be achieved by combining the bioinformatics tools in BioPython and the numerical tools in Numerical and Scientific Python.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The support of the National Institute on Drug Abuse Award P30 DA 018310 to the UIUC Neuroproteomics Center on Cell to Cell Signaling is gratefully acknowledged.
Supplementary material can be found online at "http://www.frontiersin.org/neuroinformatics/paper/10.3389/neuro.11/ 007.2008/"
.
Chang, C., and Lin, C. (2001). LIBSVM: a library for support vector machines. Available at: http://www.csie.ntu.edu/ï¾cjlin/libsvm
.