 Bo-Eun Yoon
Bo-Eun Yoon C. Justin Lee
C. Justin Lee- 1Department of Nanobiomedical Science, Dankook University, Chungnam, South Korea
- 2WCI Center for Functional Connectomics, Korea Institute of Science and Technology (KIST), Seoul, South Korea
- 3Center for Neural Science and Center for Functional Connectomics, Korea Institute of Science and Technology (KIST), Seoul, South Korea
Gamma-amino butyric acid (GABA) is the major inhibitory neurotransmitter that is known to be synthesized and released from GABAergic neurons in the brain. However, recent studies have shown that not only neurons but also astrocytes contain a considerable amount of GABA that can be released and activate GABA receptors in neighboring neurons. These exciting new findings for glial GABA raise further interesting questions about the source of GABA, its mechanism of release and regulation and the functional role of glial GABA. In this review, we highlight recent studies that identify the presence and release of GABA in glial cells, we show several proposed potential pathways for accumulation and modulation of glial intracellular and extracellular GABA content, and finally we discuss functional roles for glial GABA in the brain.
Introduction
Recent experimental evidence suggests that glial cells interact closely with neurons and play an active role in the brain. In the central nervous system (CNS), astrocytes, the major type of glia, make direct contacts with neurons via a structure that has been defined as the tripartite synapse, in which the astrocytic process is associated with the pre- and post-synapse areas of neurons (Araque et al., 1999). Indeed, astrocytes sense neuronal activity by expressing various receptors for neurotransmitters (Marcaggi and Attwell, 2004; Schipke and Kettenmann, 2004) and they are also able to release various transmitters that modulate neuronal excitability and synaptic transmission (Volterra and Meldolesi, 2005; Perea and Araque, 2010; Zorec et al., 2012). Of the various transmitters released by glial cells, so-called gliotransmitters, most of the excitatory transmitters, glutamate, ATP and D-serine have received much attention (Volterra and Meldolesi, 2005; Fiacco and McCarthy, 2006; Haydon and Carmignoto, 2006; Oliet and Mothet, 2006). However, classical inhibitory transmitters are also released by glial cells and can modulate neuronal activity. Some studies have suggested that taurine and glycine act as gliotransmitters (Barakat and Bordey, 2002; Hussy, 2002) and some initial reports showed an accumulation of GABA by glial cells (Barres et al., 1990; Gallo et al., 1991; Ochi et al., 1993). Despite these early ideas and recent evidence, the principal CNS inhibitory transmitter, GABA had not been considered as a gliotransmitter (Angulo et al., 2008). However, recent studies in primary cultures (Liu et al., 2000), human astrocytes (Lee et al., 2011b) and acute brain slices of various brain regions including thalamus, olfactory bulb and cerebellum (Barakat and Bordey, 2002; Kozlov et al., 2006; Lee et al., 2010; Jiménez-González et al., 2011) have demonstrated a robust release of GABA from glial cells. Moreover, GABA released by glial cells activates the high affinity GABA receptors to mediate tonic inhibition (Lee et al., 2010; Héja et al., 2012; Wójtowicz et al., 2013). Glial cells have thus begun to make their mark not only as GABAceptive cells, but also as GABAergic cells (Yoon et al., 2012). These exciting new findings raise questions about the gliotransmitter release mechanism by glia and the origin and regulation of glial GABA. Furthermore, alteration of tonic inhibition is found in various pathological states such as absence seizures, after strokes and in Huntington’s disease (Clarkson et al., 2010; Pirttimaki et al., 2013; Wójtowicz et al., 2013). Perhaps these changes in tonic inhibition occur through glial GABA, a possibility that awaits further investigation. It has recently been reported that tonic GABA release from reactive astrocytes is enhanced and memory is impaired in mouse models of Alzheimer’s disease (Jo et al., 2014; Wu et al., 2014). From initial reports showing GABA accumulation by glial cells to the more recent studies that give electrophysiological and pathophysiological evidence of a tonic GABA release that is mediated by glial GABA, one can safely conclude that glial GABA modulates neuronal activity and has an important physiological function. In this review, we present an overview of the cellular localization of GABA, its production and release mechanisms, and the potential roles for glial GABA, that all point towards the new concept of GABA as “a rising gliotransmitter” in the brain.
Cellular Localization of Glial GABA in Various Brain Regions
Initial reports showed that glia contain considerable amounts of GABA under normal physiological conditions. Astrocytes in brainstem were labeled with an anti-GABA antibody at the level of the soma, in the processes surrounding neurons and in astrocyte endfeet in contact with blood vessels (Blomqvist and Broman, 1988). The presence of GABA in glial cells has been studied extensively in rat optic nerve. In cell cultures of rat optic nerve, 1 week after plating, O2A-oligodendrocyte progenitor cells, type 2 astrocytes and differentiated oligodendrocytes were shown to express GABA (Barres et al., 1990). Gamma-amino butyric acid was localized using immunofluorescence with an astrocytic marker, glial fibrillary acidic protein (GFAP), in developing rat optic nerve from embryonic day 20 to postnatal day 28 (Lake, 1992). A transient increase and gradual decrease of GABA in glial cells after postnatal day 20 was reported by immunostaining and high pressure liquid chromatography (HPLC; Ochi et al., 1993). This study showed the developmental time course of astrocytic GABA expression in rat optic nerve. Gamma-amino butyric acid immunostaining was carried out on cultured astrocytes, and on whole optic nerve and GABA immunoreactivity was localized by GFAP staining. Gamma-amino butyric acid staining was most intense in early neonatal optic nerve and became attenuated over 3 weeks of postnatal development. Staining was pronounced in the astrocyte cell bodies and processes but not in the nucleus. There was a paucity of GABA immunoreactivity by postnatal day 20, both in cultured cells and in whole optic nerve. A biochemical assay for optic nerve GABA using HPLC indicated a relatively high concentration of GABA in the neonate, which rapidly attenuated over the first three postnatal weeks (Ochi et al., 1993). Gamma-amino butyric acid immunoreactivity was also observed in glial cells from the adult rat cerebellum (Martínez-Rodríguez et al., 1993). Both GABA- and Glutamic acid decarboxylase (GAD)-immunoreactivities were observed within dendrites and glial cells using different antisera obtained from rabbits immunized with GABA, baclofen and GAD. These results suggested a possible extrasynaptic release of GABA. However, these early findings had been almost forgotten by brain research scientists until recently (Angulo et al., 2008). We ourselves have recently reported strong GABA immunoreactivities in Bergmann glial cells and lamellar astrocytes of the mouse cerebellum using a commercially available anti-GABA antibody in GFAP-GFP transgenic mice (Lee et al., 2010). We have suggested that the amount of glial GABA is variable depending on different brain regions (Figure 1) and that it positively correlates with the degree of tonic inhibition current (Yoon et al., 2011). For example, glial GABA as evidenced by GABA immunoreactivity was high in the cerebellum and correlated well with a significant level of tonic inhibition current, whereas GABA immunoreactivity was very low in the hippocampus CA1 region with a low level of tonic inhibition current (Yoon et al., 2011). These results suggested that some regulatory mechanism exists to control the level of intracellular GABA in astrocytes. More recently, GABA immunoparticles were localized by electron microscopy in synaptic terminals with symmetric synapses as well as in astrocytes from CA1 and CA3 areas, the stratum radiatum and in the dentate gyrus (Le Meur et al., 2012). This would seem to indicate that a large proportion of hippocampal astrocytes contain the inhibitory transmitter GABA. In addition, immunoreactivity for GABA, GAD65, GAD67 and Bestrophin-1 (Best1), a GABA-permeable channel, has been observed in the meninges and the choroid plexus, implying a non-neuronal source for GABA in the developing mouse brain (Tochitani and Kondo, 2013).
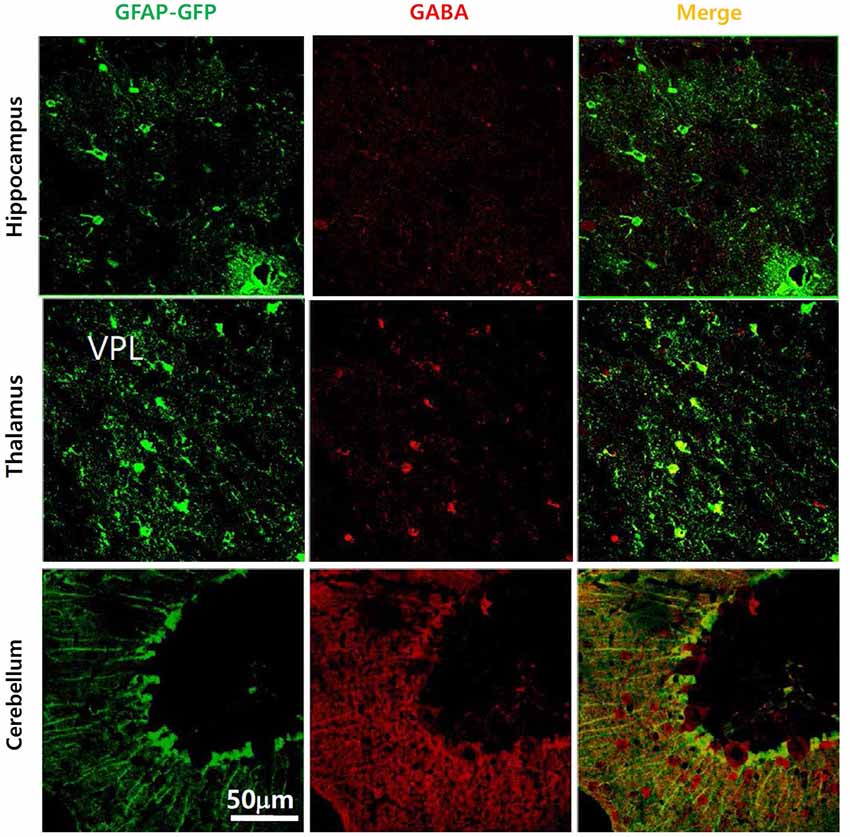
Figure 1. Presence of GABA in glial cells in various brain regions of GFAP-GFP mice. Gamma-amino butyric acid is present in the cerebellum, hippocampus and thalamus, though the GABA-containing portion of the glial cell differs between brain regions. Gamma-amino butyric acid does not co-localize much with GFAP-GFP staining in the hippocampus compared to the cerebellar cortex.
Production of GABA in Glial Cells
With respect to the original source of glial GABA, we wonder whether this GABA is synthesized within cells or taken up from the extracellular space. If a glial cell has its own biosynthetic pathway for GABA, then glial GABA content can be modulated by innate molecular factors and can be independent of neuronal activity. However, if glial GABA comes only from uptake of extracellular GABA via GABA transporters, it should be solely dependent on activity-dependent neuronal GABA release, although this explanation might not explain the varying amount of glial GABA in different brain regions (Yoon et al., 2011). Actually, tonic GABA release from glia in cerebellum is known to be independent of neuronal activity and neuronal GABA release (Rossi et al., 2003). Therefore, it is likely that multiple pathways, rather than one single route, are involved in the biosynthesis and regulation of glial GABA.
Evidence for GAD as a Biosynthetic Enzyme for Glial GABA
Immunoreactivity for the GABA synthesizing enzyme GAD was prominent in neonates but attenuated with development. This was especially the case for GAD67, which was observed in glial cells of neonatal rats but decreased after postnatal day 21. The other isoform, GAD65, was not present in these cells. GABA and GAD are clearly localized in astrocytes of optic nerve and their expression is transient during postnatal development (Ochi et al., 1993). In mature rat cerebellar glia, both GABA and GAD immunoreactivities were found (Martínez-Rodríguez et al., 1993) and more recently, GAD immunoreactivity was reported in astrocyte end feet of the human cerebellar cortex using immunohistochemistry and electron microscopy. In these studies, GAD immunoreactivity was observed in nerve terminals that are in direct contact with the astrocyte perivascular sheath of capillaries and also within astroglial perivascular end feet and endothelial cells (Benagiano et al., 2000). These findings provide further insight into the GABAergic synapse circuitry of the human cerebellar cortex and the detection of “vascular” GAD immunoreactivities suggests that GABAergic mechanisms may regulate cerebellar microvessel function (Benagiano et al., 2000). Glutamic acid decarboxylase was also observed in cultured human astrocytes (Lee et al., 2011b). This work suggested that the intracellular GABA level is about 2.32 mM in untreated cultures of human astrocytes. The authors exported GABA into the culture medium so that an intracellular-extracellular gradient of 3.64-fold was reached. Treatment with inhibitors of the GABA transporters GAT-1, GAT-2, and GAT-3, significantly reduced GABA export in a calcium-independent manner. These findings confirmed the existence of GAD within glial cells but they also emphasized the contribution from GABA transporters in establishing glial GABA content.
Evidence for Monoamine Oxidase B (MAOB) as a Key Biosynthetic Enzyme for Glial GABA
Contrasting reports suggest that GABA is produced by another biosynthetic pathway, which is independent of GAD activity, but that requires putrescine as an initial substrate. This was first suggested in mouse and fish brain (Seiler and Askar, 1971; Seiler et al., 1973). In trout brain, following intraperitoneal and intracerebral injections of [1,4-14C]putrescine.2HCl, GABA was shown to be formed in vivo, via a pathway that does not have glutamic acid as an intermediate. After intracerebral injections of [1-14C] GABA, a half-life of 7 h was obtained for GABA. This slow turnover rate for GABA in trout brain may help to further explain the ineffectiveness of the glutamate decarboxylase inhibitors in lowering the GABA content of fish brain within a few hours (Seiler et al., 1973). Putrescine concentrations can be quantitatively estimated in tissue and have been determined in mouse brain and liver (Seiler and Askar, 1971). This polyamine involved pathway for GABA synthesis was tested in mouse neuroblastoma cells (Kremzner et al., 1975). Polyamine metabolism in cultured mouse neuroblastoma cells was studied with the aim of synthesizing GABA from putrescine and putreanine from spermidine. It was shown that neuroblastoma cells, in the presence of a complete culture medium containing calf serum, readily metabolized [14C] putrescine to GABA. The rate of synthesis is similar to that for the synthesis of spermidine from putrescine. In the absence of serum, the conversion of putrescine to GABA is minimal. In the presence of serum, GABA formation is completely inhibited by the diamine oxidase inhibitor aminoguanidine. Synthesized GABA is not readily metabolized to succinate or homocarnosine. Mouse neuroblastoma cells metabolized [14C] ornithine to putrescine, GABA, and spermidine. Spermidine was metabolized to putrescine, putreanine and spermine (Kremzner et al., 1975). This is the monoamine oxidase pathway of putrescine (Figure 2). Cultured O2A glial progenitor cells of the optic nerve were able to synthesize GABA from putrescine. In these cells, there is no detectable GAD expression, but a strong GABA immunoreactivity was seen and HPLC measurements revealed an increased quantity of GABA in a putrescine-enriched medium (Barres et al., 1990). Glial cell GABA may also be involved in some pathological conditions. A comparison between GABA formation in primary cultured astrocytes from epileptic and normal mice showed that the rate of GABA production from radioactive putrescine was four times higher in epileptic than in normal mice (Laschet et al., 1992). Moreover, after transiently occluding the carotid arteries of adult gerbils, the GFAP immunoreactivity increased in the damaged forebrain tissue and reactive astrocytes were labeled with GABA, but not with GAD antisera. This GABA immunoreactivity persisted in ischemic animals for up to 3 months without GAD activity (Lin et al., 1993). It has recently been reported that glial monoamine oxidase B (MAOB) is the GABA synthesizing enzyme that mediates tonic GABA release. In the cerebellum and striatum of adult mice, general gene-silencing or knockout of MAOB, or treatment with selegiline, eliminated tonic GABA currents recorded from granule neurons and medium spiny neurons. Glial specific rescue of MAOB resulted in a complete rescue of tonic GABA currents. These results identify MAOB as a key synthesizing enzyme of glial GABA, which is released via the Best1 channel to mediate tonic inhibition in the brain (Yoon et al., 2014).
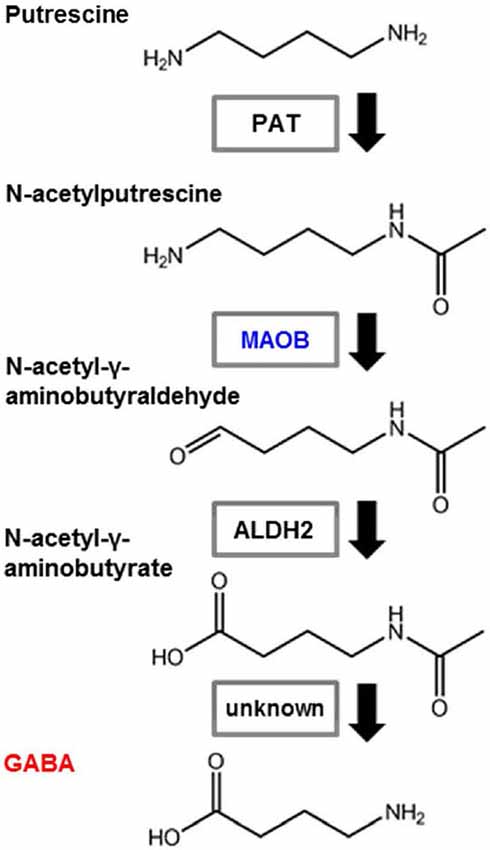
Figure 2. Pathway showing GABA synthesis from putrescine. Astrocytes synthesize GABA from putrescine via monoamine oxidation. PAT: putrescine acetyltransferase; MAOB: monoamine oxidase B; ALDH2: aldehyde dehydrogenase 2.
In summary, there is accumulating evidence for the presence of a GAD or putrescine-derived pathway, suggesting that there are multiple possible routes for the synthesis of glial GABA, as well as the GABA accumulation occurring by uptake through GABA transporters. Until now, most of these findings have been obtained from in vitro experiments and cell culture systems. However, to clarify the GABA production mechanism, in vivo studies are required to confirm the presence of putrescine and to show that MAOB is a key enzyme for GABA production.
Release of GABA from Glial Cells
The synaptic release of GABA and the functional consequences of GABA receptor activation have been extensively studied. The vesicular release of GABA from the axon terminals of inhibitory neurons, which affect nearby postsynaptic targets as well as some extrasynaptic receptors, is a well-established concept of GABAergic signaling. So, questions arising are: Can glial cells release GABA, like neurons? How is glial GABA released? How do glial cells differ in their release of GABA compared to neurons? There is increasing evidence to show that GABA is indeed released from glial cells. Several reports suggest that, unlike neurons, glial GABA can be released via unconventional pathways. For example, GABA may be released from glial cells by the reversal of transporters, or by non-vesicular release mechanisms. We shall review here the recent findings regarding such unconventional mechanisms of glial GABA release and shall discuss the physiological significance of these mechanisms.
Evidence of GABA Release from Glial Cells
Classically, extracellular GABA was thought to be taken up by GABA transporters expressed on glial cells. Consequently, we would expect glial cells to contain considerable amounts of GABA arising from this uptake. However, several studies provide convincing evidence for the active release of GABA from glial cells. Glioma cell lines have been shown to excrete concentrated GABA into the extracellular medium. This is consistent with the theory that, in the brain, the untransformed counterparts of these glial tumor cells may have the capacity to control the extracellular concentration of neuroactive molecules, especially GABA (Schrier and Thompson, 1974). In cultured hippocampal glial cells, Jow et al. (2004) observed that the conditioned medium contained an inhibitory factor that can hyperpolarize and suppress neuronal activity. They investigated this inhibitory factor using biochemistry, electrophysiology, pharmacology, and mass spectrometry. Mass spectrometry analysis of conditioned medium revealed peaks that are identical to those for GABA. A concentration of up to 500 µM GABA was found in conditioned medium from glial cultures, but no GABA was found in conditioned medium from neuronal cultures. According to these findings, hippocampal glial cells make much more GABA than cortical glia (Jow et al., 2004). To overcome the limitations of culture studies, GABA release from glial cells was then detected and characterized in acute brain slices. In the cerebellum, whole-cell patch clamp recordings of Bergmann glia showed an electrogenic GABA efflux that activated neighboring GABAA receptors (Barakat and Bordey, 2002). These authors suggested that the GABA efflux was mediated by a reversal of the GAT-1 expressed in glial cells (Barakat and Bordey, 2002). In acute slices of olfactory bulb, whole-cell recordings from neurons revealed a spontaneous slow GABAA receptor-mediated current (Kozlov et al., 2006). These studies demonstrated that the release of GABA by astrocytes caused a long lasting and synchronous inhibition of mitral and granule cells in the olfactory bulb (Kozlov et al., 2006). This group also observed that astrocytes are capable of releasing glutamate, as well as GABA, leading to a selective activation of granule cell NMDA receptors. Therefore, they suggested that by releasing both excitatory and inhibitory gliotransmitters, astrocytes are capable of a complex modulatory control of the olfactory network through neuron-glia interactions (Kozlov et al., 2006).
Mechanism of GABA Release from Glial Cells
Various candidate pathways have been suggested to account for the mechanism of tonic GABA release from glia. Three possibilities seem to be the most likely: (i) vesicular release; (ii) a reversal of GABA transporters; (iii) non-vesicular channel-mediated release. To date however, there is no clear evidence for vesicular release. The lack of GABA-containing synaptic vesicles makes it unlikely that glial cells utilize a vesicular release mechanism. The remaining two possible mechanisms, reversal of transporters and channel-mediated release are the most often suggested. In cerebellar slices, the GABA transporter has been suggested as a candidate mechanism for GABA release (Barakat and Bordey, 2002). It was shown that with 10 mM GABA in the recording pipette, GABA transporters expressed in Bergmann glia operated in the reverse mode, leading to a release of GABA into the extracellular space, subsequently activating GABAA receptors tonically on the same cell. In contrast, the tonic activation of GABAA receptors expressed in cerebellar granule cells of adult rats has been shown to be an action potential-independent and non-vesicular release (Wall and Usowicz, 1997; Mitchell and Silver, 2003; Rossi et al., 2003). Despite this finding, a reversal of the GABA transporter is also suggested by Richerson and Wu (2003) and Lee et al. (2011a). In particular, GAT-2/3 have been considered as an alternative mechanism of GABA release from astrocytes (Héja et al., 2012; Unichenko et al., 2013). Heja et al. demonstrated that GABA is released from astrocytes by the reverse action of glial GABA transporter and that GABA release can be prevented by blocking glutamate uptake with an inhibitor. They argued that the released GABA contributes to the tonic inhibition of neurons in a network activity-dependent manner. Unichenko et al. (2013) reported a role for GAT-2/3 in developmental stages. However, some of these studies were performed with culture systems or under non-physiological conditions. Moreover, the non-vesicular release and action potential-independent release of GABA were shown to contribute to tonic inhibition and tonic GABAA conductance in other brain areas, including the cerebellum, olfactory bulb and hippocampus (Brickley et al., 1996; Kozlov et al., 2006; Glykys and Mody, 2007). Persistent and slow GABAA receptor-mediated excitatory currents activated by a non-synaptic release of GABA have been reported in rat embryonic and early postnatal hippocampal slices. This tonic and slow current was induced by a Ca2+- and SNARE protein-independent release of GABA, suggesting a non-conventional and non-vesicular release during development (Demarque et al., 2002). These researchers reported the presence of tonic, spontaneous, and evoked currents in embryonic and neonatal CA1 neurons mediated primarily by the activation of GABAA receptors. These currents persist in the presence of calcium channel blockers or botulinum toxin and are observed in Munc18-1-deficient mice in which vesicular release is abolished. Of interest to note, is that this paracrine communication is modulated by glutamate but not by GABA transporters (Demarque et al., 2002). One study performed in a cell line derived from type 2 astrocytes showed that the release of GABA is sensitive to inhibitors of anion channels (Wang et al., 2002). Recently, we have identified and demonstrated a channel-mediated tonic GABA release from glia (Lee et al., 2010), a non-vesicular and non-conventional mechanism for GABA release in cerebellar slices under physiological conditions. We found that GABA permeates directly through the Ca2+-activated anion channel, Best1, to yield GABA release and that tonic inhibition is eliminated by gene silencing of Best1. Using immunohistochemistry, cerebellar Bergmann glial cells and lamellar astrocytes were seen to express both GABA and Best1, and selective expression of Best1 was seen in glial cells. After preventing the general expression of Best1 by Best1 specific shRNA, tonic inhibition was fully rescued by glia specific flanking of the Best1 specific shRNA (Lee et al., 2010).
The precise mechanism underlying tonic GABA release has been difficult to elucidate because this GABA release exhibits several puzzling features that are quite different from those exhibited by the conventional, phasic release of GABA. Our proposed mechanism of channel-mediated release via the Best1 channel can account for each of these properties in cerebellar slices. First, the non-vesicular nature of tonic GABA release is consistent with a channel-mediated mechanism. Second, independence from neuronal activity can be explained by the glial origins of tonic inhibition. Finally, the apparent lack of dependence on external Ca2+ arises from substantial activation of Best1 at resting levels of intracellular Ca2+, leading to constitutive release of GABA at such intracellular Ca2+ levels (Lee et al., 2010). This unprecedented mechanism is consistent with several previous reports that have suggested a non-vesicular and action potential-independent release of GABA (Brickley et al., 1996; Kozlov et al., 2006; Glykys and Mody, 2007) and a well-documented Ca2+- and SNARE protein-independent release of GABA during development (Demarque et al., 2002). A further recent report on GABA release from reactive astrocytes in the hippocampus, supports a role for Best1 channels in the release of GABA from glial cells and has implications not only for physiological states but also for pathological conditions (Jo et al., 2014).
A recently published report by Diaz et al. (2011) raised contradictory evidence against our model of Best1-mediated tonic GABA release. However, after examining the paper in detail, we found that the authors provide only pharmacological evidence using NPPB as a blocker of Best1 channel (Diaz et al., 2011). It is important to note that NPPB is notorious for having many side effects. Authors claimed that NPPB enhanced tonic GABA current within 4–5 min, instead of blocking. We also found that NPPB initially increased tonic GABA current, but if we waited longer time period of about 10 min, we saw a block by NPPB (see Figure Sf9a and S9d of Lee et al. (2010)). Thus, if the authors applied NPPB for longer time, they would have seen the block by NPPB, as we observed previously. Nevertheless, this was precisely why we utilized cell-type specific gene-silencing method by lentiviral shRNA (Lee et al., 2010), which is far more specific to Best1 than NPPB. Diaz et al. (2011) did not use any of genetic approaches. Therefore, their claim as shown in the title of their paper, “Best1 channels are insensitive to ethanol and do not mediate tonic GABAergic currents in cerebellar granule cells,” was an overstatement.
The Role of Glial GABA Release in the Brain
Special Roles of Glial GABA During Development
Depolarization of neurons by astrocytic GABA was first reported in rat embryonic hippocampal neuronal cell cultures (Liu et al., 2000). In this study, neurons exposed to astrocyte-conditioned medium (ACM) showed a highly significant increase in a bicuculline-sensitive persistent GABAA receptor-mediated current, whereas this tonic activation was absent in neurons bathed in normal medium (Liu et al., 2000). These findings implied that astrocyte-released GABA intensifies GABAergic autocrine/paracrine signaling at GABAA receptor/Cl− channels, thus effectively depolarizing differentiating hippocampal neurons near the equilibrium potential of Cl−. The same study also found that ACM could rescue neurite outgrowth in embryonic hippocampal and cortical neurons that had been treated with 3-MPA (to block GAD-derived GABA synthesis), via bicuculline- and nitrendipine-sensitive mechanisms. These results indicated that astrocyte-derived GABA provides a critical depolarizing signal that indirectly stimulates Ca2+ entry through voltage-gated Ca2+ channels, thereby supporting neuritogenesis (Liu et al., 2000). In a separate study, GABA excitation has been shown in rat embryonic and immature hippocampal slices (Demarque et al., 2002). However, during early development, in the marginal zone of mouse neocortex, GABA has been shown to be released via GABA transporters, GAT-2/3, whereas glutamate transporters operate in the uptake mode (Unichenko et al., 2013). This implies that GABA release by glia is essential for development and also that alteration of glial GABA during development can cause neurodevelopmental disorders.
Inhibitory Function of Glial GABA in the Mature and Diseased Brain
Previous reports suggest that glial GABA has a special role in the developing brain, though in the more mature brain, glial GABA appears to have an inhibitory effect. Based on numerous studies, we can infer that glial cells must be the source of an inhibitory transmitter responsible for tonic GABAA receptor-mediated currents. For example, glial cells can release GABA into the extracellular space and tonically activate high-affinity GABAA receptors by volume transmission in cerebellum (Rossi et al., 2003; Lee et al., 2010) maintaining a persistent inhibitory tone in these brain regions. Glial GABA has also been seen to act on GABAB receptors (Benedetti et al., 2011). Tonic inhibitory tones have recently been reported in other brain regions including the cortex (Wlodarczyk et al., 2013) and the hippocampus (Pavlov and Walker, 2013; Song et al., 2013). However, phasic GABA released from presynaptic terminals of neurons activates low-affinity synaptic GABAA receptors. The extracellular tonic GABA is estimated to be around 160 nM (Santhakumar et al., 2006; Lee et al., 2010), whereas that of phasic GABA at the synaptic junctions is around 3 mM (Mozrzymas et al., 2003). The synaptically released GABA is constantly taken up by the high performance GABA transporters that are ready for uptake near the synapses. These GABA transporters serve as a barrier that separates the extrasynaptic space and synaptic junctions. The high affinity extrasynaptic GABAA receptors are non-desensitizing and have a half-maximal effective concentration (EC50) of GABA in the range of 0.3–0.7 µM, whereas synaptic GABAA receptors are strongly desensitizing and have an EC50 of GABA in the range of 6–14 µM. There is a clear forty-fold difference in the affinity of GABAA receptors for GABA. To sum up, different locations, extrasynaptic or synaptic, different extracellular concentrations of GABA, and differing degrees of desensitization and sensitivity of GABAA receptors to GABA, make these two distinct GABAA receptor activation modes, tonic and phasic, function quite differently and independently of each other. Therefore, tonic GABA serves to inhibit target neurons on a slow time scale, whereas phasic GABA serves to inhibit target neurons on a fast time scale in the physiological state.
Indeed, tonic inhibition has been reported to have diverse physiological and pathological roles in the CNS. Blocking tonic inhibition with furosemide was shown to increase the firing frequency of cerebellar granule cells (Hamann et al., 2002). Alcohol, by potentiating extrasynaptic GABAA, was shown to enhance tonic inhibition in the cerebellum and impair motor behavior (Hanchar et al., 2005). Enhanced tonic inhibition in the thalamus was observed in typical genetic and pharmacological absence seizure models (Cope et al., 2009). Reducing excessive GABA-mediated tonic inhibition was shown to promote functional recovery after stroke in the motor cortex (Clarkson et al., 2010). Alterations in tonic GABAA receptor-mediated signaling was not only found in absence seizure models, but also in temporal lobe epilepsy (Pavlov and Walker, 2013). Very recently, an enduring loss of tonic but not phasic GABAA receptor-mediated currents was shown to critically contribute to the prolonged amygdala disinhibition subsequent to chronic stress exposure (Liu et al., 2014).
There are several lines of evidence to show that GABA release from glia is directly associated with pathological conditions. One recent report found an enhanced tonic inhibition in absence epilepsy and observed a dysfunction in the astrocytic GABA transporter, GAT-1 in absence seizure models (Pirttimaki et al., 2013). Reduced tonic inhibition was reported in mouse models of Huntington’s disease, resulting from a loss of astrocytic GABA release (Wójtowicz et al., 2013). Another study reported anti-inflammatory actions of GABA from glia by modulating microglial activity in human astrocyte cultures (Lee et al., 2011b). More recently, two studies from independent research groups showed that reactive astrocytes abnormally release GABA to impair memory in mouse models of Alzheimer’s disease (Jo et al., 2014; Wu et al., 2014). These recent studies propose a novel and crucial role for glial GABA as the major inhibitory gliotransmitter in many pathological conditions.
Concluding Remarks
This review highlights the compelling evidence accumulating for GABA synthesis and release from glia and reveals that this released GABA plays an important role in brain function. Several mechanisms are proposed for glial GABA biosynthesis and release. Further research is needed to substantiate the precise mechanism for each process. However, it is clear that glial GABA is present in various brain regions and has potentially important roles in each region. Finally, we need to develop new genetic animal models, more detailed characterizations of the various pathological conditions associated with glial GABA and perform more detailed investigations of the regulation of glial GABA in order to elucidate the true nature of the in vivo functions of GABA as a gliotransmitter.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported within the framework of the International Cooperation Program managed by the National Research Foundation of Korea (2013K2A2A4003724), the World Class Institute (WCI 2009-003) programs of the National Research Foundation (NRF) funded by the Korean Ministry of Science, Education and Technology (MEST), by the National Agenda Project (NAP) of the Korea Research Council of Fundamental Science and Technology (NAP-09-04), and by a KIST Internal Grant (2E24480).
References
Angulo, M. C., Le Meur, K., Kozlov, A. S., Charpak, S., and Audinat, E. (2008). GABA, a forgotten gliotransmitter. Prog. Neurobiol. 86, 297–303. doi: 10.1016/j.pneurobio.2008.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Araque, A., Parpura, V., Sanzgiri, R. P., and Haydon, P. G. (1999). Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215. doi: 10.1016/s0166-2236(98)01349-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barakat, L., and Bordey, A. (2002). GAT-1 and reversible GABA transport in Bergmann glia in slices. J. Neurophysiol. 88, 1407–1419.
Barres, B. A., Koroshetz, W. J., Swartz, K. J., Chun, L. L., and Corey, D. P. (1990). Ion channel expression by white matter glia: the O-2A glial progenitor cell. Neuron 4, 507–524. doi: 10.1016/0896-6273(90)90109-s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benagiano, V., Virgintino, D., Rizzi, A., Flace, P., Troccoli, V., Bormann, J., et al. (2000). Glutamic acid decarboxylase-positive neuronal cell bodies and terminals in the human cerebellar cortex. Histochem. J. 32, 557–564. doi: 10.1023/A:1004106428844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benedetti, B., Matyash, V., and Kettenmann, H. (2011). Astrocytes control GABAergic inhibition of neurons in the mouse barrel cortex. J. Physiol. 589, 1159–1172. doi: 10.1113/jphysiol.2010.203224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blomqvist, A., and Broman, J. (1988). Light and electron microscopic immunohistochemical demonstration of GABA-immunoreactive astrocytes in the brain stem of the rat. J. Neurocytol. 17, 629–637. doi: 10.1007/bf01260990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brickley, S. G., Cull-Candy, S. G., and Farrant, M. (1996). Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J. Physiol. 497, 753–759.
Clarkson, A. N., Huang, B. S., Macisaac, S. E., Mody, I., and Carmichael, S. T. (2010). Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 468, 305–309. doi: 10.1038/nature09511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cope, D. W., Di Giovanni, G., Fyson, S. J., Orbán, G., Errington, A. C., Lorincz, M. L., et al. (2009). Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat. Med. 15, 1392–1398. doi: 10.1038/nm.2058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demarque, M., Represa, A., Becq, H., Khalilov, I., Ben-Ari, Y., and Aniksztejn, L. (2002). Paracrine intercellular communication by a Ca2+- and SNARE-independent release of GABA and glutamate prior to synapse formation. Neuron 36, 1051–1061. doi: 10.1016/s0896-6273(02)01053-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Diaz, M. R., Wadleigh, A., Hughes, B. A., Woodward, J. J., and Valenzuela, C. F. (2011). Bestrophin1 channels are insensitive to ethanol and do not mediate tonic GABAergic currents in cerebellar granule cells. Front. Neurosci. 5:148. doi: 10.3389/fnins.2011.00148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fiacco, T. A., and McCarthy, K. D. (2006). Astrocyte calcium elevations: properties, propagation and effects on brain signaling. Glia 54, 676–690. doi: 10.1002/glia.20396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gallo, V., Patrizio, M., and Levi, G. (1991). GABA release triggered by the activation of neuron-like non-NMDA receptors in cultured type 2 astrocytes is carrier-mediated. Glia 4, 245–255. doi: 10.1002/glia.440040302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glykys, J., and Mody, I. (2007). The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. 582, 1163–1178. doi: 10.1113/jphysiol.2007.134460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamann, M., Rossi, D. J., and Attwell, D. (2002). Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron 33, 625–633. doi: 10.1016/s0896-6273(02)00593-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanchar, H. J., Dodson, P. D., Olsen, R. W., Otis, T. S., and Wallner, M. (2005). Alcohol-induced motor impairment caused by increased extrasynaptic GABA(A) receptor activity. Nat. Neurosci. 8, 339–345. doi: 10.1038/nn1398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haydon, P. G., and Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009–1031. doi: 10.1152/physrev.00049.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Héja, L., Nyitrai, G., Kékesi, O., Dobolyi, A., Szabó, P., Fiáth, R., et al. (2012). Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 10:26. doi: 10.1186/1741-7007-10-26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hussy, N. (2002). Glial cells in the hypothalamo-neurohypophysial system: key elements of the regulation of neuronal electrical and secretory activity. Prog. Brain Res. 139, 95–112. doi: 10.1016/s0079-6123(02)39010-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiménez-González, C., Pirttimaki, T., Cope, D. W., and Parri, H. R. (2011). Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets delta-subunit-containing GABA(A) receptors. Eur. J. Neurosci. 33, 1471–1482. doi: 10.1111/j.1460-9568.2011.07645.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jo, S., Yarishkin, O., Hwang, Y. J., Chun, Y. E., Park, M., Woo, D. H., et al. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896. doi: 10.1038/nm.3639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jow, F., Chiu, D., Lim, H. K., Novak, T., and Lin, S. (2004). Production of GABA by cultured hippocampal glial cells. Neurochem. Int. 45, 273–283. doi: 10.1016/s0197-0186(03)00292-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kozlov, A. S., Angulo, M. C., Audinat, E., and Charpak, S. (2006). Target cell-specific modulation of neuronal activity by astrocytes. Proc. Natl. Acad. Sci. U S A 103, 10058–10063. doi: 10.1073/pnas.0603741103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kremzner, L. T., Hiller, J. M., and Simon, E. J. (1975). Metabolism of polyamines in mouse neuroblastoma cells in culture: formation of GABA and putreanine. J. Neurochem. 25, 889–894. doi: 10.1111/j.1471-4159.1975.tb04423.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lake, N. (1992). Taurine, GABA and GFAP immunoreactivity in the developing and adult rat optic nerve. Brain Res. 596, 124–132. doi: 10.1016/0006-8993(92)91539-q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laschet, J., Grisar, T., Bureau, M., and Guillaume, D. (1992). Characteristics of putrescine uptake and subsequent GABA formation in primary cultured astrocytes from normal C57BL/6J and epileptic DBA/2J mouse brain cortices. Neuroscience 48, 151–157. doi: 10.1016/0306-4522(92)90345-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, M., McGeer, E. G., and McGeer, P. L. (2011a). Mechanisms of GABA release from human astrocytes. Glia 59, 1600–1611. doi: 10.1002/glia.21202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, M., Schwab, C., and Mcgeer, P. L. (2011b). Astrocytes are GABAergic cells that modulate microglial activity. Glia 59, 152–165. doi: 10.1002/glia.21087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S., Yoon, B. E., Berglund, K., Oh, S. J., Park, H., Shin, H. S., et al. (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796. doi: 10.1126/science.1184334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Le Meur, K., Mendizabal-Zubiaga, J., Grandes, P., and Audinat, E. (2012). GABA release by hippocampal astrocytes. Front. Comput. Neurosci. 6:59. doi: 10.3389/fncom.2012.00059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, R. C., Polsky, K., and Matesic, D. F. (1993). Expression of gamma-aminobutyric acid immunoreactivity in reactive astrocytes after ischemia-induced injury in the adult forebrain. Brain Res. 600, 1–8. doi: 10.1016/0006-8993(93)90394-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Q. Y., Schaffner, A. E., Chang, Y. H., Maric, D., and Barker, J. L. (2000). Persistent activation of GABA(A) receptor/Cl(-) channels by astrocyte-derived GABA in cultured embryonic rat hippocampal neurons. J. Neurophysiol. 84, 1392–1403.
Liu, Z. P., Song, C., Wang, M., He, Y., Xu, X. B., Pan, H. Q., et al. (2014). Chronic stress impairs GABAergic control of amygdala through suppressing the tonic GABAA receptor currents. Mol. Brain 7:32. doi: 10.1186/1756-6606-7-32
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marcaggi, P., and Attwell, D. (2004). Role of glial amino acid transporters in synaptic transmission and brain energetics. Glia 47, 217–225. doi: 10.1002/glia.20027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martínez-Rodríguez, R., Tonda, A., Gragera, R. R., Paz-Doel, R., García-Cordovilla, R., Fernández-Fernández, E., et al. (1993). Synaptic and non-synaptic immunolocalization of GABA and glutamate acid decarboxylase (GAD) in cerebellar cortex of rat. Cell. Mol. Biol. (Noisy-le-grand) 39, 115–123.
Mitchell, S. J., and Silver, R. A. (2003). Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron 38, 433–445. doi: 10.1016/s0896-6273(03)00200-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mozrzymas, J. W., Zarnowska, E. D., Pytel, M., and Mercik, K. (2003). Modulation of GABA(A) receptors by hydrogen ions reveals synaptic GABA transient and a crucial role of the desensitization process. J. Neurosci. 23, 7981–7992.
Ochi, S., Lim, J. Y., Rand, M. N., During, M. J., Sakatani, K., and Kocsis, J. D. (1993). Transient presence of GABA in astrocytes of the developing optic nerve. Glia 9, 188–198. doi: 10.1002/glia.440090304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oliet, S. H., and Mothet, J. P. (2006). Molecular determinants of D-serine-mediated gliotransmission: from release to function. Glia 54, 726–737. doi: 10.1002/glia.20356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pavlov, I., and Walker, M. C. (2013). Tonic GABA(A) receptor-mediated signalling in temporal lobe epilepsy. Neuropharmacology 69, 55–61. doi: 10.1016/j.neuropharm.2012.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perea, G., and Araque, A. (2010). GLIA modulates synaptic transmission. Brain Res. Rev. 63, 93–102. doi: 10.1016/j.brainresrev.2009.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pirttimaki, T., Parri, H. R., and Crunelli, V. (2013). Astrocytic GABA transporter GAT-1 dysfunction in experimental absence seizures. J. Physiol. 591, 823–833. doi: 10.1113/jphysiol.2012.242016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Richerson, G. B., and Wu, Y. (2003). Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J. Neurophysiol. 90, 1363–1374. doi: 10.1152/jn.00317.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rossi, D. J., Hamann, M., and Attwell, D. (2003). Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J. Physiol. 548, 97–110. doi: 10.1111/j.1469-7793.2003.00097.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santhakumar, V., Hanchar, H. J., Wallner, M., Olsen, R. W., and Otis, T. S. (2006). Contributions of the GABAA receptor alpha6 subunit to phasic and tonic inhibition revealed by a naturally occurring polymorphism in the alpha6 gene. J. Neurosci. 26, 3357–3364. doi: 10.1523/jneurosci.4799-05.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schipke, C. G., and Kettenmann, H. (2004). Astrocyte responses to neuronal activity. Glia 47, 226–232. doi: 10.1002/glia.20029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schrier, B. K., and Thompson, E. J. (1974). On the role of glial cells in the mammalian nervous system. Uptake, excretion and metabolism of putative neurotransmitters by cultured glial tumor cells. J. Biol. Chem. 249, 1769–1780.
Seiler, N., al-Therib, M. J., and Kataoka, K. (1973). Formation of GABA from putrescine in the brain of fish (Salmo irideus Gibb.). J. Neurochem. 20, 699–708. doi: 10.1111/j.1471-4159.1973.tb00030.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seiler, N., and Askar, A. (1971). A micro method for the quantitative estimation of putrescine in tissues. J. Chromatog. 62, 121–127. doi: 10.1016/s0021-9673(01)96817-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Song, I., Volynski, K., Brenner, T., Ushkaryov, Y., Walker, M., and Semyanov, A. (2013). Different transporter systems regulate extracellular GABA from vesicular and non-vesicular sources. Front. Cell. Neurosci. 7:23. doi: 10.3389/fncel.2013.00023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tochitani, S., and Kondo, S. (2013). Immunoreactivity for GABA, GAD65, GAD67 and Bestrophin-1 in the meninges and the choroid plexus: implications for non-neuronal sources for GABA in the developing mouse brain. PloS One 8:e56901. doi: 10.1371/journal.pone.0056901
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Unichenko, P., Dvorzhak, A., and Kirischuk, S. (2013). Transporter-mediated replacement of extracellular glutamate for GABA in the developing murine neocortex. Eur. J. Neurosci. 38, 3580–3588. doi: 10.1111/ejn.12380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Volterra, A., and Meldolesi, J. (2005). Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 6, 626–640. doi: 10.1038/nrn1722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wall, M. J., and Usowicz, M. M. (1997). Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur. J. Neurosci. 9, 533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, C. M., Chang, Y. Y., Kuo, J. S., and Sun, S. H. (2002). Activation of P2X(7) receptors induced [(3)H]GABA release from the RBA-2 type-2 astrocyte cell line through a Cl(-)/HCO(3)(-)-dependent mechanism. Glia 37, 8–18. doi: 10.1002/glia.10004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wlodarczyk, A. I., Sylantyev, S., Herd, M. B., Kersanté, F., Lambert, J. J., Rusakov, D. A., et al. (2013). GABA-independent GABAA receptor openings maintain tonic currents. J. Neurosci. 33, 3905–3914. doi: 10.1523/jneurosci.4193-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wójtowicz, A. M., Dvorzhak, A., Semtner, M., and Grantyn, R. (2013). Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT-3. Front. Neural Circuits 7:188. doi: 10.3389/fncir.2013.00188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, Z., Guo, Z., Gearing, M., and Chen, G. (2014). Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzhiemer’s disease model. Nat. Commun. 5:4159. doi: 10.1038/ncomms5159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoon, B. E., Jo, S., Woo, J., Lee, J. H., Kim, T., Kim, D., et al. (2011). The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Mol. Brain 4:42. doi: 10.1186/1756-6606-4-42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoon, B. E., Woo, J., Chun, Y. E., Chun, H., Jo, S., Bae, J. Y., et al. (2014). Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. doi: 10.1113/jphysiol.2014.278754. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoon, B. E., Woo, J., and Lee, C. J. (2012). Astrocytes as GABA-ergic and GABA-ceptive cells. Neurochem. Res. 37, 2474–2479. doi: 10.1007/s11064-012-0808-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zorec, R., Araque, A., Carmignoto, G., Haydon, P. G., Verkhratsky, A., and Parpura, V. (2012). Astroglial excitability and gliotransmission: an appraisal of Ca2+ as a signalling route. ASN Neuro. 4, 103–119. doi: 10.1042/an20110061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: astrocyte, gliotransmitter, glial GABA, tonic inhibition, MAOB
Citation: Yoon B-E and Lee CJ (2014) GABA as a rising gliotransmitter. Front. Neural Circuits 8:141. doi: 10.3389/fncir.2014.00141
Received: 18 August 2014; Accepted: 11 November 2014;
Published online: 17 December 2014.
Edited by:
Alexey Semyanov, University of Nizhny Novgorod, RussiaReviewed by:
Rosemarie Grantyn, Humboldt University Medical School (Charité), GermanyLaszlo Heja, Hungarian Academy of Sciences, Hungary
Copyright © 2014 Yoon and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Justin Lee, Center for Neural Science and Center for Functional Connectomics, Korea Institute of Science and Technology (KIST), 39-1 Hawolgok-dong, Seongbuk-gu, Seoul 136-791, South Korea e-mail:Y2psQGtpc3QucmUua3I=