 Sneha Pallatt
Sneha Pallatt Sibin Nambidi
Sibin Nambidi Subhamay Adhikary1
Subhamay Adhikary1 Antara Banerjee
Antara Banerjee Surajit Pathak
Surajit Pathak Asim K. Duttaroy
Asim K. Duttaroy- 1Medical Biotechnology Lab, Faculty of Allied Health Sciences, Chettinad Academy of Research and Education (CARE), Chettinad Hospital and Research Institute (CHRI), Chennai, India
- 2Department of Nutrition, Institute of Basic Medical Sciences, Faculty of Medicine, University of Oslo, Oslo, Norway
Lynch syndrome (LS) is an autosomal dominant disorder caused by germline mutations in DNA mismatch repair (MMR) genes. These mutations result in frameshift alterations, leading to the accumulation of errors within microsatellites. Individuals with LS have an elevated risk of developing colorectal and distant malignancies, including endometrial cancer (EC), which is one of the most common cancer associated with LS. Despite its significance, the association between EC and LS is often underexplored. Given the slow progression of colorectal cancer (CRC), there is an opportunity for early detection and intervention, which can aid in reducing both incidence and mortality through the identification and management of pre-malignant lesions and early-stage tumors in colorectum/endometrium. Recognizing individuals with a heightened risk of CRC is essential for implementing personalized screening strategies. This review summarizes the original research work on LS to find out the correlation of CRC following an endometrial cancer diagnosis in individuals with MMR gene mutations, may involve refine treatment strategies and moreover this review may help clinicians and researchers to get an up-to date information on LS and its advanced treatment possibilities.
Highlights
• This review comprehensively summarizes the current research findings on LS and possible correlation between CRC development following EC in individuals with MMR gene mutations.
• This review discussed the genetic and molecular pathways, such as MMR gene mutations and microsatellite instability (MSI), that drive the development of both EC and CRC.
• This review finds the key points regarding the role of early detection and surveillance strategies in LS carriers from the original research data available.
1 Overview of Lynch syndrome and associated cancer risks
Lynch syndrome (LS) is a hereditary condition that predisposes individuals to various malignancies, most notably colorectal cancer (CRC) and endometrial cancer (EC) (1). This autosomal dominant disorder is characterized by an increased cancer risk due to defects in DNA mismatch repair (MMR), which compromises genomic stability (2). Microsatellite instability (MSI) is a crucial screening factor for Lynch-associated tumors and underscores the aggressive and rapid progression of these cancers compared to sporadic cases (3, 4). A tumor is classified as microsatellite instability-high (MSI-H) when mutations are detected in two or more of the five microsatellite sequences within the tumor DNA. If only one of these five sequences is altered, the tumor is categorized as microsatellite instability-low (MSI-L). When none of the microsatellite sequences exhibit mutations, the tumor is considered microsatellite stable (MSS) (5). In cases where a tumor is identified as MSI-L, further testing with an extended panel of microsatellite markers is recommended to ensure precise classification (6). In LS, MSI-H tumors are primarily caused by germline mutations, while somatic mutations in the MLH1 and MSH2 genes are observed in only a small percentage of sporadic cases (7). The most common explanation for MSI-H tumors in sporadic cases is the silencing of the MLH1 gene by promoter hyper-methylation, a phenomenon also observed in LS. Additionally, MSI-H tumors are strongly associated with the loss of MLH1 protein expression in sporadic tumors, whereas familial tumors often exhibit a loss of both MLH1 and MSH2 protein expression (8). These genetic alterations create genomic instability, thereby expediting the progression of CRC in patients with LS, frequently advancing from adenoma to carcinoma in an approximate timeframe of 2 years, in stark contrast to the decade-long evolution observed in sporadic cases (9). Beyond LS, additional hereditary syndromes, exemplified by Cowden syndrome, which is marked by mutations in phosphatase and tensin homolog (PTEN), further enhance the risk of developing EC. Lifestyle determinants, such as obesity, physical inactivity, and specific dietary habits, exacerbate the likelihood of both EC and CRC, underscoring the necessity for comprehensive preventive measures (10–12).
A thorough comprehension of the interrelated risks associated with EC and CRC in LS is essential for the enhancement of early detection and therapeutic management. The identification of common genetic mutations and molecular pathways not only augments diagnostic accuracy but also facilitates the development of targeted therapeutic interventions that are efficacious against both forms of cancer. Understanding the genetic and molecular factors underlying this syndrome is crucial for early detection and effective management of affected individuals. This review seeks to elucidate these interconnections, with the objective of informing clinical guidelines and improving prognostic outcomes for individuals afflicted with LS.
2 LS: mechanism and impact
Two major criteria are followed to classify individuals with LS, namely, Amsterdam Criteria II and Revised Bethesda Criteria mutations. The Amsterdam II criteria serve as a guideline for identifying families at high risk for LS, an autosomal dominant disorder that increases susceptibility to cancer. According to these criteria, a family must have at least three members diagnosed with cancers associated with LS, with at least one being a first-degree relative of the other two. Additionally, the disease should affect at least two successive generations, and at least one of the diagnosed individuals may have developed cancer before the age of 50. A confirmed pathological examination is required to verify the presence of tumors, and familial adenomatous polyposis must be ruled out as a possible cause (13). Similarly, Revised Bethesda Criteria is designed to recognize individuals with CRC who may require further evaluation for MSI and serve as a screening tool for LS. These guidelines assist in determining whether a patient’s tumor may be linked to MMR gene mutations, thereby indicating the need for additional genetic testing. One of the key indicators is early-onset CRC, where patients diagnosed before the age of 50 years require additional assessment due to an increased likelihood of hereditary cancer predisposition. Another critical criterion is the presence of synchronous or metachronous LS-associated malignancies, which include cancers of the colorectum, endometrium, stomach, ovaries, small intestine, biliary tract, ureter, or renal pelvis, occurring either concurrently or at different time points, necessitating genetic screening (Figure 1). Additionally, tumors exhibiting MSI-H histopathological features, such as mucinous differentiation, signet-ring cells, Crohn’s-like lymphocytic infiltration, or tumor-infiltrating lymphocytes, particularly when diagnosed before 60 years of age, suggest potential underlying MMR gene mutations and warrant further molecular analysis. Furthermore, a family history of early-onset CRC or LS-associated cancers in a first-degree relative (parent, sibling, or child) diagnosed before 50 years of age serves as another significant criterion for genetic testing. Lastly, the occurrence of CRC or other LS-associated malignancies in at least two first- or second-degree relatives (including grandparents, aunts, uncles, nephews, nieces, or grandchildren) at any age provides further justification for comprehensive genetic evaluation to identify hereditary cancer risks (14, 15). MSI results in changes in the length of microsatellites—short repetitive DNA sequences—and contributes to genomic instability, which drives tumorigenesis by enabling mutations in key oncogenes and tumor-suppressor genes such as TGF-βR2, BAX, and PTEN. MSI-related mutations in TGF-βR2 impair cell proliferation regulation, while alterations in BAX hinder apoptosis, fostering tumor growth (16, 17).
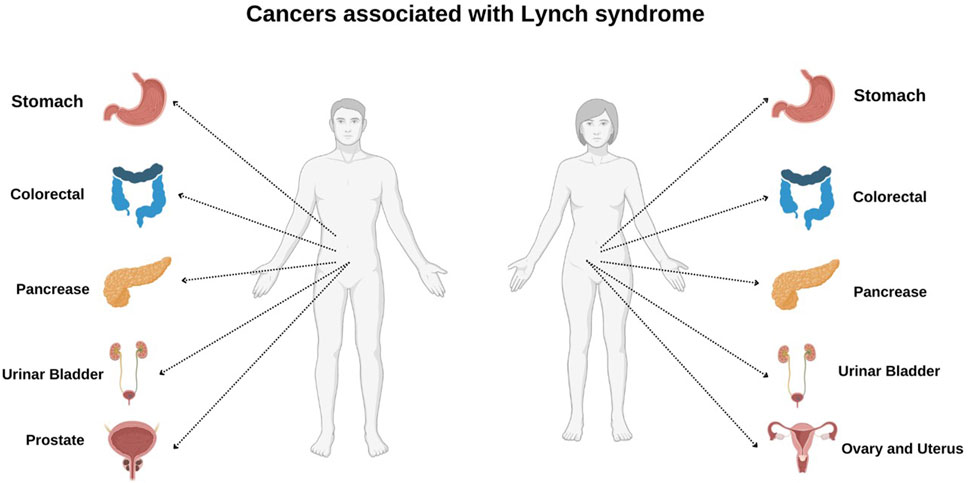
Figure 1. Cancer associated with Lynch syndrome in male and female.
2.1 Cancer spectrum and associated risks based on MMR gene variants
Investigations delineate a significant convergence in the genetic and molecular frameworks that support both EC and CRC. Mutations within mismatch repair genes, including MLH1, MSH2, MSH6, PMS1, and PMS2, play a crucial role in the origin of both malignancies (18). The risk and spectrum of cancers in LS vary depending on which MMR gene harbors the pathogenic variant, with each conferring distinct cancer risks and characteristics.
2.1.1 MutL homolog 1(MLH1) and MutL homolog 2 (MLH2)
Individuals with pathogenic variants in MLH1 and MSH2 have the highest lifetime risk of CRC and EC, estimated between 40% and 80% (19). These individuals are also predisposed to distant colonic malignancies, including gastric, ovarian, urinary tract, hepatobiliary, and small bowel cancers. Among these, stomach cancer risk is particularly high in MLH1 mutation carriers, with MSH2 mutation carriers exhibiting a relatively lower but still significant risk (20). The variation in stomach cancer incidence between MLH1 and MSH2 carriers may be attributed to age-specific hazard ratio (HR) differences, a younger onset for MLH1 carriers, or a higher representation of MLH1 mutations among gastric cancer cases (21). Additionally, there is an increasing evidence for higher incidences of pancreatic cancer in LS carriers, as well as potential associations with breast and prostate cancers, given their frequent presentation with MMR deficiency in Lynch families (22). Moreover, a risk of cervical cancer has been noted, though some cases may be misclassified adenocarcinomas of the lower uterine segment rather than true cervical carcinomas. While the overall cumulative risks of LS-related cancers by age 70 are similar across MLH1, and MSH2 mutation carriers, each mutated gene confers a unique cancer risk profile (23).
2.1.2 MutS homolog 6 (MSH6)
Carriers of pathogenic MSH6 mutations exhibit a distinct cancer risk profile within LS. Recent studies estimate the lifetime CRC risk for MSH6 mutation carriers to range between 10% and 44%, typically presenting at a later age compared to MLH1 or MSH2 mutation carriers. However, the risk of EC is significantly elevated, with lifetime risks between 16% and 49%, often exceeding the risk of CRC (24). Additionally, MSH6 mutations are associated with an increased but variable risk of ovarian cancer (25). Emerging evidence also suggests a heightened susceptibility to breast cancer, indicating a two-fold increased risk among MSH6 and PMS2 carriers compared to the general population. Other malignancies, including urinary tract, stomach, and small intestine cancers, have also been linked to MSH6 mutations, though they occur less frequently (26).
2.1.3 PMS1 homolog 2 (PMS2)
A defective PMS2 gene associated with LS substantially elevates the possibilities of developing specific cancers, particularly CRC and EC in comparison to the general population. However, pathogenic PMS2 variants are associated with the lowest cancer risks among LS-related MMR gene mutations. Studies indicate that the lifetime risk of CRC in individuals with PMS2 mutations ranges between 10% and 20%, significantly lower than that of MLH1, MSH2, and MSH6 mutation carriers (27). Additionally, EC risk in PMS2 carriers is estimated to be between 12% and 15%, also lower than those associated with other MMR genes. The later onset of CRC, typically occurring after age 50, contributes to a less aggressive screening approach. Unlike carriers of MLH1 or MSH2 mutations, who require biennial colonoscopy starting at age 20–25, PMS2 mutation carriers may begin screening at age 35–40, with colonoscopies recommended every 2–3 years instead of annually (28). Recent studies have also suggested that PMS2 carriers may have a lower risk of extra-colonic malignancies, though upper gastrointestinal, ovarian, and urinary tract cancers have been reported at lower frequencies. Due to the reduced overall cancer risk, prophylactic surgeries, such as hysterectomy, are not routinely recommended for PMS2 carriers unless there is a strong family history of EC. PMS2-deficient CRCs tend to exhibit more aggressive behavior and a worse prognosis compared to other MMR-deficient CRCs (29). This distinction is partly attributed to lower levels of intra-tumoral immune infiltration, suggesting that PMS2-deficient CRCs share more biological characteristics with sporadic MMR-proficient CRCs than with other LS-associated CRCs. While it was previously believed that carriers of germline pathogenic PMS2 variants represented a small minority of LS patients, recent studies have challenged this assumption. New investigations indicate that pathogenic PMS2 carriers have the highest population frequency among the four MMR genes, with an estimated prevalence of 1 in 714 individuals (30). Furthermore, studies utilizing IHC staining in CRCs from population-based cohorts have demonstrated that isolated PMS2 loss of expression, indicative of pathogenic PMS2 variants, is observed in 0.5%–1.5% of unselected CRCs. Among MSI CRCs, the fraction of isolated PMS2 loss varies between 1% and 8%, with more than half of these tumors being linked to germline pathogenic PMS2 variants. These findings underscore the importance of refining screening strategies and risk assessment for PMS2-deficient CRCs to improve early detection and patient management (31).
2.1.4 Epithelial cell adhesion molecule (EPCAM)
The EPCAM gene is not an MMR gene, but deletions in EPCAM lead to MSH2 inactivation due to promoter hypermethylation, resulting in a cancer risk profile similar to MSH2 variants (32). Individuals with EPCAM deletions have an increased risk of CRC, with studies reporting a lifetime risk of approximately 75%, comparable to MSH2 mutation carriers. Additionally, the risk of EC in female carriers is estimated to be around 30%, reinforcing the need for targeted surveillance. Unlike other LS-associated mutations, EPCAM deletions do not directly affect DNA mismatch repair function but cause epigenetic silencing of MSH2, leading to a deficiency in MMR and MSI-H (33). This makes individuals with EPCAM deletions susceptible to other LS-associated cancers, including ovarian, gastric, small bowel, and urinary tract malignancies. Colonoscopy screening every 1–2 years starting at age 25 is recommended for EPCAM carriers, along with EC surveillance. However, because EPCAM deletions predominantly affect MSH2 expression, further research is needed to refine cancer risk estimates and optimize screening protocols for affected individuals (34).
The autosomal dominant inheritance of LS results in a 50% probability of passing the condition to offspring, making genetic testing and counseling essential for at-risk families. Early and regular surveillance, such as colonoscopy starting at 20–25 years of age or 2–5 years before the youngest diagnosed family member, significantly reduces cancer-related mortality (35). Prophylactic surgical options, such as colectomy and hysterectomy, are also available for individuals at high risk. Importantly, tumors with MSI-H phenotypes in LS respond well to immune checkpoint inhibitors, particularly anti-PD-1/PD-L1 therapies, offering a targeted treatment approach (36). Advances in molecular diagnostics, including MSI testing and immunohistochemistry for MMR proteins, have greatly improved LS management, enabling timely interventions and personalized treatments to mitigate its impact on affected individuals and their families (37).
3 Endometrial cancer: a central player in LS’s cancer spectrum
EC represents a quintessential neoplasm within LS, frequently manifesting as the chief malignancy preceding the emergence of other tumors associated with LS, including CRC. It is estimated that approximately 40%–60% of female individuals with LS will experience the development of EC during their lifetimes, with the mean age of onset occurring 10–15 years earlier than that observed in sporadic, non-syndromic instances (38). The presence of MSI and germline mutations in MMR genes, particularly in MSH2 and MSH6, is markedly prevalent in Lynch-associated EC, which contributes to genomic instability and tumorigenesis (39). In contrast to sporadic EC, which often relies on estrogen for its progression, Lynch-associated EC is generally non-estrogen-dependent and displays unique molecular subtypes, predominantly categorized as high-grade endometrioid carcinomas. Moreover, Lynch-associated EC is distinguished by a hyper-mutated phenotype, resulting in a high frequency of mutations in genes such as PTEN, KRAS, and PIK3CA (40). Estrogen-dependent EC is linked to factors that elevate lifetime exposure to endogenous or exogenous estrogens. These factors include a higher body mass index (BMI), estrogen replacement therapy, estrogen-secreting tumors, chronic anovulation, tamoxifen therapy, early onset of menstruation, and delayed menopause, all of which contribute to endometrial proliferation stimulated by estrogen (41). In contrast, non-estrogen-dependent EC is not associated with unopposed estrogen exposure and is linked to risk factors such as lower BMI, nulliparity, a history of breast cancer, and being over 55 years old at the time of diagnosis (42).
4 Colorectal cancer: insights from the LS perspective
Although CRC predominantly targets individuals aged 50 and above, those diagnosed with LS experience a considerably elevated risk and are frequently identified at a younger age due to the hereditary predisposition associated with their condition. Approximately 80% of hereditary CRC cases, particularly those associated with LS, arise via the mutation or alternative pathway linked to these MMR gene alterations. This is in contrast to the suppressor or classic pathway, which is responsible for around 80% of sporadic CRC instances, often connected to mutations in genes such as APC, p53, and KRAS (43). CRC associated with LS usually involves activation of the WNT/β-catenin signaling pathway due to secondary mutations in APC or β-catenin (CTNNB1), further advancing tumorigenic processes (9).
In individuals diagnosed with LS, CRC typically initiates as an adenomatous polyp within the intestinal mucosa, with malignant progression occurring at a considerably accelerated rate compared to sporadic cases (44). The typical duration from adenoma to carcinoma in Lynch-associated CRC is roughly 2 years, whereas this timeline extends to approximately 10 years for sporadic cases (27, 28). Unlike sporadic CRC, which often occurs in the distal colon and rectum, LS-associated CRCs predominantly arise in the proximal (right-sided) colon, particularly in the cecum and ascending colon (45). These tumors frequently display mucinous differentiation or signet-ring cell morphology and are poorly differentiated or undifferentiated, highlighting their aggressive nature. A characteristic immune response, marked by peri-tumoral and intra-tumorally lymphoid aggregates, is commonly observed, suggesting active immune surveillance against tumor cells.
Additionally, an increased presence of intraepithelial lymphocytes further reinforces their immunogenic nature, indicating a potential for responsiveness to immunotherapy (46). Some LS-associated CRCs also exhibit serrated glandular architecture or medullary carcinoma-like features, which are relatively uncommon in sporadic cases. A defining aspect of LS-associated CRCs is their rapid progression, transitioning from adenomatous polyps to invasive carcinoma within approximately 2 years, in contrast to the decade-long progression seen in sporadic CRCs (47). The clinical manifestations of CRC in patients possessing LS encompass symptoms including abdominal discomfort, alterations in bowel patterns, weight reduction, nausea, and anemia. Distal tumors are more inclined to induce visible rectal hemorrhage, whereas proximal tumors may lead to occult blood in the feces. In light of the distinctive hereditary risk factors, patients with LS may also exhibit atypical signs of metastasis, such as lymphadenopathy (e.g., Virchow’s node) or hepatomegaly (48).
5 Epidemiological insights and risk factors for EC and CRC
The epidemiology and risk factors for EC and CRC highlight unique and overlapping elements contributing to their development and prevalence. EC primarily impacts women in the postmenopausal stage, with a higher occurrence noted in correlation with advancing age (49). Risk determinants for endometrial carcinoma are closely associated with hormonal dysregulation, notably conditions that lead to extended exposure to estrogen without the counterbalancing effects of progesterone. Obesity, polycystic ovary syndrome (PCOS), nulliparity, and late menopause are significant contributors, as they increase endogenous estrogen levels (50). Estrogen promotes the growth of endometrial cells, raising the risk of hyperplasia (abnormal cell growth) and ultimately leading to EC. Progesterone opposes this effect by balancing estrogen’s action. It induces differentiation in endometrial cells, inhibits proliferation, and facilitates the shedding of the endometrial lining as seen during menstruation (51). When exogenous estrogen is given, such as in hormone replacement therapy (HRT) for postmenopausal women, without the addition of progesterone (unopposed estrogen therapy), the endometrial lining undergoes continuous stimulation without progesterone’s regulatory effects. This prolonged exposure can result in endometrial hyperplasia and markedly heighten the risk of developing EC. Lifestyle factors, including diets high in saturated fats and a lack of physical activity, further amplify this risk (52).
CRC has both genetic and environmental factors playing crucial roles in its epidemiology (53). Lifestyle factors such as diet, physical activity, and smoking are important modifiable risk factors (54). Diets high in red and processed meats, low fiber intake, and excessive alcohol consumption are associated with increased CRC risk. Additionally, chronic conditions such as inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis, elevate the risk of CRC (55). Women diagnosed with LS exhibit a markedly elevated probability of developing EC as their initial malignancy, frequently preceding the occurrence of CRC. This hereditary association emphasizes the critical necessity for systematic screening and vigilant surveillance in individuals possessing a familial predisposition to these malignancies (56,57).
6 LS associated EC and CRC genes
A comprehensive analysis (Li et al., 2022) of data from the TCGA database revealed significant differences in the molecular mechanisms driving the progression of LS to CRC or EC. While LS-CRC progression is closely associated with differential gene expression (DEGs), LS-EC development may rely more on gene methylation processes. For instance, COL11A1, correlated with MSH6 mutations, serves as a key marker for distinguishing MSI-H and microsatellite stable (MSS). CRC, playing a role in extracellular matrix interactions and tumor development (42). From the TCGA database, specific genes were identified that overlap with LS and CRC (SGs-LC) and LS and EC (SGs-LE), comprising 493 and 99 genes, respectively (Li et al., 2022). Enrichment analyses revealed distinct pathways for SGs-LC and SGs-LE, with shared associations in peroxisomal pathways but differing in other functional pathways. For SGs-LC, pathways related to peroxisomal activity and extracellular matrix remodeling may play pivotal roles, as evidenced by genes like CST2 and COL18A1 (58). In contrast, SGs-LE genes like LY6K and MIR27B are implicated in immune response modulation or hormone signaling, both critical in EC. Several genes exhibited notable roles in LS-associated tumor progression. SST, a regulatory peptide, inhibits cellular mitosis and tumor growth in various cancers, including CRC. Similarly, KIF20A and NUF2, implicated in mitotic regulation and tumorigenesis, show significant roles in both CRC and EC (58). Specific survival analyses further underscored unique and overlapping genetic markers influencing patient outcomes in CRC and EC. Genes like COL18A1 and HTR4 modulate the tumor microenvironment and signal transduction in CRC, while CDC45 and WDR31 influence cellular replication processes in EC. SGs-LC, genes such as AADACL2, DHRS7C, KRT24, and LINC00460 exhibit highly significant p-values (59). Both upregulated (e.g., LINC00460) and downregulated (e.g., AADACL2) expressions have been noted, with CST2 being significantly upregulated, suggesting its potential role in CRC tumor progression. Conversely, downregulated genes like NPY2R and KHDRBS2 may contribute to CRC development through their suppression (59). Additional candidates, such as CDH10 and LINC02616, are involved in CRC-specific pathways related to adhesion and cellular communication. For SGs-LE, genes like LINC02691, MIR27B, and LY6K are characterized by less pronounced but still significant differential expression. Notably, IGF2-AS is upregulated, potentially influencing the insulin-like growth factor (IGF) signaling pathway in EC. Meanwhile, genes like ADAMTS9-AS2 and SLC10A4 suggest potential epigenetic or regulatory functions in EC (Figure 2) (59).
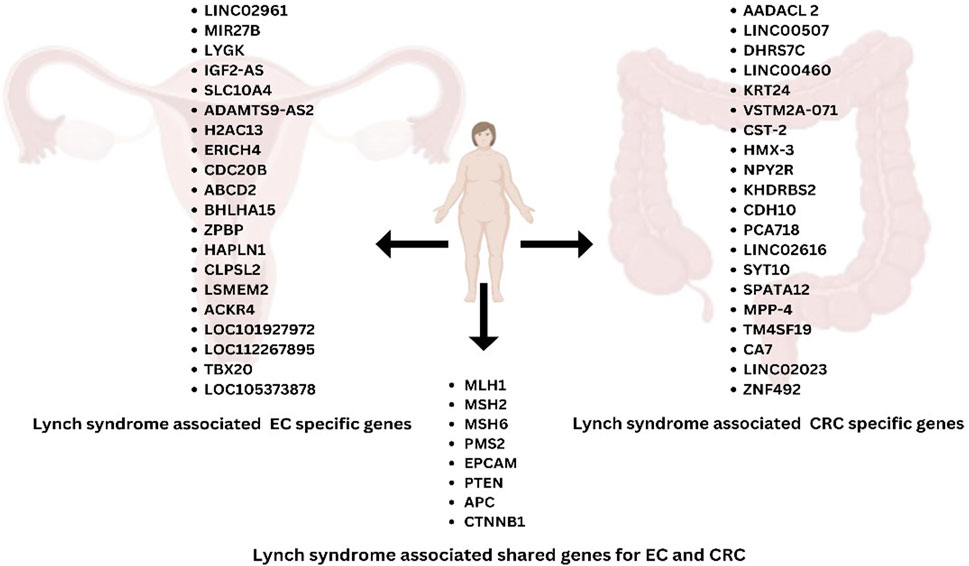
Figure 2. Lynch syndrome-associated genes specific to EC and CRC, highlighting the shared genes between EC and CRC.
7 Molecular alteration and dysregulation pathways in EC: distinction between endometrioid EC and serous EC
Endometrial endometrioid carcinomas (EECs) are marked by frequent genetic mutations and pathway dysregulations that drive their development and progression (60). EECs often exhibit MSI present in about 20% of unselected endometrial tumors and more common in EECs than non-EECs (61). This leads to mutations in various genes involved in tumorigenesis, including Birt-Hogg-Dube (BHD), BAX, insulin-like growth factor type 2 receptor (IGFIIR), Transforming Growth Factor-β Receptor II (TGFβ-RII), and ataxia telangiectasia and Rad3-related (ATR), many of which are part of the DNA damage response (62, 63). The PI3K-PTEN-AKT pathway is also significantly altered in over 80% of EECs, with high-frequency mutations in PIK3R1, PIK3CA, and PTEN, as well as additional alterations like PIK3CA amplification and PTEN promoter methylation. These mutations result in dysregulated cell proliferation, growth, and survival (64).
EECs also feature alterations in the RAS-RAF-MAPK pathway, with KRAS mutations present in 18% of cases, often coexisting with mutations in PTEN, PIK3CA, and PIK3R1 (65). BRAF mutations are rare, occurring in only 1% of EECs. fibroblast growth factor receptor 2 (FGFR2) mutations, found in 12% of EECs, are mostly missense mutations and are mutually exclusive with KRAS mutations but frequently co-occur with PTEN mutations, making FGFR2 a potential therapeutic target (66, 67). The WNT signaling pathway is frequently disrupted through CTNNB1 (β-catenin) mutations in up to 45% of EECs (68). Additionally, ARID1A gene mutations, affecting the BAF250a component of the switch/sucrose nonfermenting (SWI/SNF) chromatin-remodeling complex, are found in approximately 40% of low-grade and 39% of high-grade EECs (Figure 3) (69).
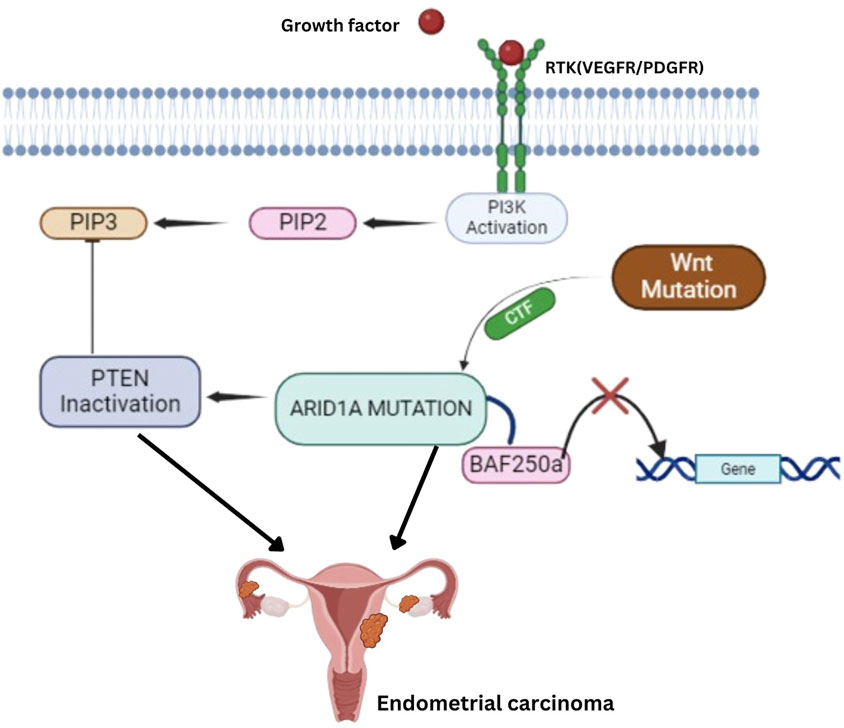
Figure 3. Key Molecular Pathways in Endometrial Carcinoma: ARID1A, PTEN, and Wnt Signaling Mutations, and PI3K Activation lead to Tumorigenesis. Receptor Tyrosine Kinases (RTK) – Growth factors, such as VEGFR/PDGFR, activate RTKs, triggering the PI3K pathway. PI3K Activation–This leads to the conversion of PIP2 to PIP3, which PTEN normally regulates. However, PTEN inactivation disrupts this control, contributing to tumorigenesis. ARID1A Mutation–Mutations in ARID1A disrupt the function of the BAF250a complex, a critical player in chromatin remodeling, further contributing to gene dysregulation and cancer progression. Wnt Pathway Mutation–Mutations in the Wnt signaling pathway also play a role by activating downstream targets that promote cell proliferation and inhibit normal gene regulatory mechanisms.
Serous endometrial carcinomas (ECs) exhibit distinct genetic profiles and clinical behaviors compared to EECs (70). Serous ECs are often characterized by aneuploidy and frequent alterations such as TP53 mutations, overexpression of Cyclin-E and Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2), and p16 dysregulation (71, 72). TP53 mutations are the most common genetic changes in serous ECs, occurring in 53%–90% of tumors, and are often found in early precancerous stages, suggesting a stepwise progression to malignancy (73). These mutations are less common in EECs, with a higher frequency in high-grade cases. The protein phosphatase 2 scaffold subunit Alpha (PPP2R1A) gene, which encodes the scaffolding subunit of the protein phosphatase-2A (PP2A) enzyme, is also frequently mutated in serous ECs (17%–41%) but less so in EECs (5%–7%). These mutations may impair PP2A’s tumor suppressor function, potentially contributing to tumorigenesis (74–76).
The overexpression and amplification of HER-2/ERBB2 are notably more prevalent in serous endometrial carcinomas (ECs) compared to endometrioid endometrial carcinomas (EECs). Research indicates that HER-2/ERBB2 overexpression occurs in 17%–80% of serous EC cases, with gene amplification reported in 17%–42% of these tumors (77, 78). HER-2/ERBB2 status in serous ECs is associated with shorter survival times, suggesting its predictive value (79, 80). Additionally, HER-2/ERBB2-positive serous ECs are more frequently observed in patients with a previous history of breast cancer (81).
7.1 Epigenetic disruption in LS-Associated EC: critical role of aberrant methylation
Aberrant methylation patterns play a critical role in the tumorigenesis of EC, particularly in cases associated with LS. Hypermethylation of tumor suppressor genes and hypomethylation of oncogenes disrupt key cellular pathways, including proliferation, apoptosis, and immune evasion (82). The MLH1 gene is frequently hypermethylated in EC, especially in MSI-H tumors. This methylation silences MLH1 expression, impairing the DNA mismatch repair pathway and allowing the accumulation of genetic mutations. This deficiency in mismatch repair is a hallmark of LS-associated EC, resulting in a high mutational burden and tumor heterogeneity (83). Other tumor suppressor genes commonly affected by hypermethylation include PTEN, RASSF1A, and CDKN2A. Hypermethylation of the PTEN promoter reduces its expression, disrupting the PI3K/AKT pathway, which contributes to uncontrolled cellular proliferation and survival (84). Similarly, hypermethylation of RASSF1A silences its role in regulating cell cycle arrest and apoptosis, thereby enhancing cell proliferation and suppressing apoptotic signaling. Methylation of CDKN2A silences this cyclin-dependent kinase inhibitor, disrupting cell cycle regulation and enabling unchecked cellular growth (84). In contrast, global DNA hypomethylation can activate oncogenes such as C-MYC, which promotes increased proliferation, metabolic reprogramming, and evasion of apoptosis. Additionally, hypomethylation of MEST (Mesoderm-Specific Transcript) leads to its overexpression, enhancing oncogenic signaling and tumor progression (85).
In the context of hormone signaling, hypermethylation of HOXA10 and HOXA11, genes essential for endometrial development, disrupts critical pathways involved in maintaining endometrial homeostasis. These changes alter estrogen receptor (ER) and progesterone receptor (PR) signaling, further contributing to hormone-driven progression of EC (86). Methylation also modulates immune response pathways, as seen with the hypermethylation of SOCS3 (Suppressor of Cytokine Signaling 3), which promotes immune evasion by altering cytokine signalling (87). The clinical implications of these methylation changes in EC are profound. Hypermethylated genes such as MLH1, PTEN, and RASSF1A show promise as diagnostic biomarkers for early detection of EC. Methylation patterns of genes like CDKN2A and MLH1 also serve as prognostic indicators, correlating with tumor stage, grade, and patient outcomes. Notably, MSI-H EC tumors, characterized by MLH1 hypermethylation, often respond favorably to immunotherapy due to their high mutational burden and resultant neoantigen expression (88).
8 Genetic mutation and pathways alteration driving CRC progression
Ahadova et al. (2018) proposed that three distinct pathways explain CRC development in Lynch patients, in contrast to the widely accepted idea that mutations in the Wnt/β-catenin pathway underlie all CRC development in LS. The three signalling pathways frequently affected in LS CRCs are the Wnt/β-catenin the RAF/MEK/ERK and the PI3K/PTEN/AKT pathways, all of which aid in a cell’s road to malignancy when in a deregulated state (89). APC mutations are distributed across the gene and both alleles need to be affected, while CTNNB1 shows gain-of-function mutations usually located in exon 3, an exon that encodes a regulatory domain normally phosphorylated by GSK-3B (90). Additionally, polymorphisms in CCND1, TP53, IGF1, and AURKA influenced age-associated risk for CRC in LS. Reeves et al 2008. confirmed that the IGF1 polymorphism is an important modifier of disease onset in LS. Talseth et al 2008. reported that the CCND1 polymorphism was associated with a significant difference in age of disease onset in patients harboring MSH2 mutations, which was not observed in MLH1 mutation carriers. A shorter CA-repeats is associated with an earlier age at onset of CRC in LS (91–93). The pathway-based approach of Chen et al. 2009. to elucidate genetic risk modifiers influencing age of onset of CRC in patients with LS using CART analysis (classification and regression tree) identified CDKN2A C580T and IGF1 CA-repeat as the initial splits, indicating that the polymorphisms in these genes are the most informative for separating patients into those LS patients who are more likely to develop CRC early versus those who are more likely to develop CRC at a later age. The gene–gene interaction between E2F2 and AURKA as the influence of the AURKA SNP on risk varies depending on the E2F2 genotype (94). A particularly notable finding is that individuals with biallelic mutations in the MUTYH gene face a significantly elevated lifetime risk of developing CRC, with estimates ranging from a 28-fold increase reported by Lubbe et al. (2009) (95). Similarly, to a 93-fold increase was reported and a near-complete penetrance by the age of 60. Moreover, even monoallelic carriers of pathogenic or likely pathogenic MUTYH variants exhibit a moderately increased CRC risk—approximately 1.68-fold. Some monoallelic carriers also harbored mutations in other base excision repair (BER) genes, such as OGG1 and MTH1, underscoring the role that alterations in low-penetrance genes may play in CRC development (96, 97). Statistical analyses from the research findings estimate that approximately 15 or fewer of these mutations are critical drivers of tumor development. Key driver genes in CRC include APC, KRAS, NRAS, BRAF, PIK3CA, and PTEN (98, 99). APC acts as a gatekeeper gene, initiating adenoma formation when mutated. Approximately 40% of CRC harbor KRAS mutations, predominantly at codons 12 and 13, which are critical in the progression of advanced CRC cells (100). NRAS mutations, although less common, occur at codons 12, 13, or 61. BRAF mutations, found in 5%–10% of CRCs, are associated with the CpG island methylator phenotype (CIMP) and an altered adenoma-carcinoma progression pathway (101). The interplay between these mutations and the resulting disruptions in signaling pathways provides valuable insights into the mechanisms of CRC development and progression, paving the way for targeted treatments and better diagnostic tools (102).
9 Mechanism of cancer initiation in EC and progression to CRC
Although LS is primarily driven by mutations in MMR genes (MLH1, MSH2, MSH6, and PMS2), several other genes contribute to EC development in LS patients. These genes regulate crucial cellular processes such as tumor suppression, chromatin remodeling, and cell signaling, which, when disrupted, accelerate tumorigenesis. One of the earliest molecular events in LS-associated EC is the inactivation of PTEN. Loss of PTEN function results in uncontrolled cell proliferation, increased survival, and resistance to apoptosis, hallmark features of cancer progression. Like sporadic EC, PTEN mutations are common in LS-associated cases and contribute to early tumorigenesis (103). Additionally, MSI-induced frameshift mutations in TGFBR2 disrupt TGF-β signaling, which normally functions as a tumor suppressor by regulating cell growth and differentiation. The disruption of this pathway allows for uncontrolled cellular proliferation. The loss of TGF-β signaling leads to unchecked cell proliferation and enhances tumor progression (104). The PI3K/AKT signaling pathway is further affected by mutations in PIK3CA. PIK3CA mutations contribute to sustained activation of the pathway, driving tumor growth and increasing resistance to apoptosis (105). Additionally, ARID1A, a chromatin remodeling gene, is frequently mutated in MSI-H tumors, including LS-associated EC. Loss of ARID1A function disrupts DNA repair mechanisms, leading to genomic instability and increased tumor mutation rates (106). Other oncogenic mutations found in LS-associated EC include KRAS, which affects the RAS/MAPK signaling pathway and promotes uncontrolled cell growth (107, 108). Additionally, overexpression of SOX9, a transcription factor involved in stem cell maintenance and differentiation, has been linked to increased tumorigenicity in MSI-H EC (109).
The Wnt/β-catenin signaling pathway, a critical cell proliferation and differentiation regulator, is frequently altered in LS-associated ECMutations in CTNNB1, which encodes β-catenin, lead to aberrant activation of this pathway, further supporting tumorigenesis (110). Additionally, RNF43, a gene that negatively regulates Wnt signaling, is often mutated in MSI-H ECs, further enhancing tumor growth; the prevalence of truncating mutations at this locus, combined with the rarity of synonymous mutations, strongly indicates that RNF43 mutations have been positively selected during the evolution of EC and CRC (Figure 4) (111). Given the shared genetic basis of LS-associated EC and CRC, it is likely that their molecular pathways exhibit significant similarities. In approximately 50% of LS cases, EC is diagnosed before CRC in instances where the two malignancies are not synchronous, rendering CRC the second primary cancer in these patients. This sequential pattern of cancer development is likely driven by LS’s shared underlying genetic alterations characteristic. As a result, EC may function as a sentinel malignancy, serving as an early indicator of LS in affected individuals and facilitating the identification of at-risk family members through genetic screening and surveillance (38, 112).
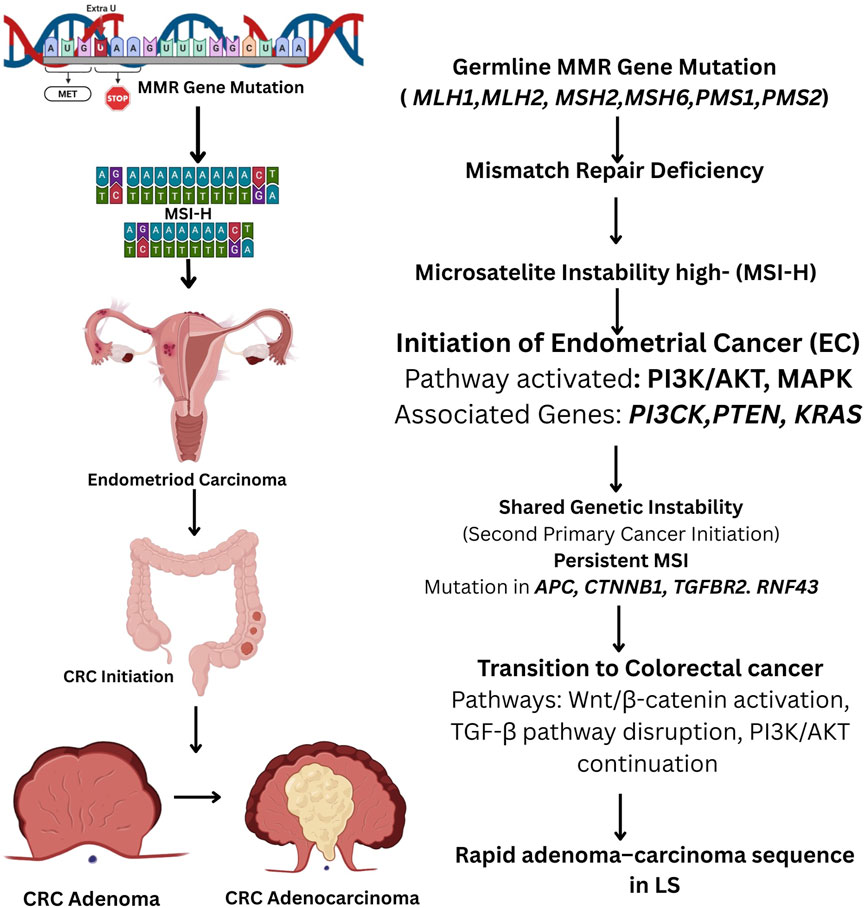
Figure 4. Flowchart illustrating the molecular progression of endometrial cancer (EC) to colorectal cancer (CRC) in Lynch Syndrome (LS). Germline mutations in mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2, EPCAM) lead to MMR deficiency and microsatellite instability (MSI). This instability triggers the initiation of EC via activation of the PI3K/AKT and MAPK pathways, with involvement of genes such as PTEN, PIK3CA, and KRAS. Persistent MSI results in secondary malignancies, including CRC, through mutations in APC, CTNNB1, TGFBR2, and RNF43, promoting Wnt/β-catenin activation, TGF-β pathway disruption, and continued PI3K/AKT signaling. The rapid adenoma–carcinoma sequence in LS accelerates CRC progression.
10 Uncovering the CRC risk in EC: clinical implications
Following the diagnosis of EC as the primary cancer, individuals may face a heightened risk of developing a second primary cancer due to the shared genetic predispositions, environmental exposures, or the impact of treatments for the initial cancer. To address this, there is an immediate necessity for recommendations based on clinical evidence that focus on preventive strategies, including regular screening for secondary cancers among those who have survived from the primary cancer (113). Individuals with LS face up to an 80% lifetime risk of developing CRC (114). Therefore, genetic testing and CRC screening are strongly recommended, if any family member is diagnosed with LS (115, 116).
In 2012, a study (Singh et al 2012) was conducted to assess CRC in women diagnosed with EC. The research comprised a total of 267 women with EC, of whom 2.4% were found to have CRC. Additionally, 13.6% had significant pathological findings, such as adenomatous polyps and tubulovillous histology (117). After that Singh et al. (2013) performed a study that included 3,115 women with EC found that women under 50 years of age had a significantly higher risk of developing CRC of any type, with a hazard ratio (HR) of 4.41 and a 95% confidence interval (CI). The risk was particularly elevated for right-sided CRC, with an HR of 7.48 and a 95% CI. In contrast, no elevated risk of CRC was noted in women aged 51–65 years or older than 65 years. However, women aged 51–65 years with EC had an increased risk of right-sided CRC, with an HR of 2.30 and a 95% CI (116). Another study by Win et al. (2013) reported that women with EC carrying mutations in MMR genes had an elevated risk of developing CRC within the next 20 years. The estimated probability of CRC development was 48%, with a 95% CI. The study also identified a significantly increased risk of CRC, as indicated by a standardized incidence ratio (SIR) of 39.9 (95% CI) compared to the normal population (118). A retrospective cohort study by Liao SC et al. (2021) found that the prevalence of CRC in women with EC was 2.20 times higher compared to controls, with an incidence rate of 1.09 per 1000 person–years. The study also noted that the risk of CRC increased with age, and the hazard ratio for CRC development was highest within 3 years of an EC diagnosis (119). Further (Lai et al., 2021), women diagnosed with EC exhibited significantly elevated SIRs for CRC, irrespective of age. In a sub-site-specific analysis of CRC, EC patients diagnosed before the age of 50 demonstrated higher SIRs for ascending colon. The cumulative incidence of second primary malignancies in EC patients was evaluated over 5, 10, 15, and 20 years of follow-up. Notably, the incidence of CRC showed a progressive increase, rising from 0.7% at 5 years to 3.9% at 20 years. Patients aged ≥50 consistently exhibited a higher incidence than those aged <50, with rates reaching 5.7% and 2.1%, respectively, at 20 years. These findings suggest that EC survivors, particularly those aged ≥50, are at an increased long-term risk of developing CRC (120). This highlights the critical need for ongoing surveillance, risk assessment, and the implementation of targeted preventive strategies in this high-risk population.
11 Prognostic markers of EC and CRC
Predictive biomarkers are crucial in predicting disease progression, independent of treatment. These markers are measurable clinical or biological characteristics that provide insight into a patient’s likely outcome. In EC, blood-based prognostic biomarkers have garnered significant interest among healthcare professionals and patients due to their potential for easy assessment (121). Two protein-based biomarkers have emerged as particularly noteworthy in EC prognosis: Human Epididymis protein 4 (HE4) and cancer antigen 125 (CA125). CA125, in particular, has been the focus of multiple investigations. An increasing amount of evidence indicates a correlation between elevated serum CA125 levels and unfavorable clinicopathological features in EC patients (122). Furthermore, research indicates that higher CA125 concentrations may be associated with poorer outcomes in individuals diagnosed with EC (123). HE4 is a glycoprotein that was initially identified in the epididymis but has been shown to be highly expressed in various cancer types, including EC (Table 1) (124).
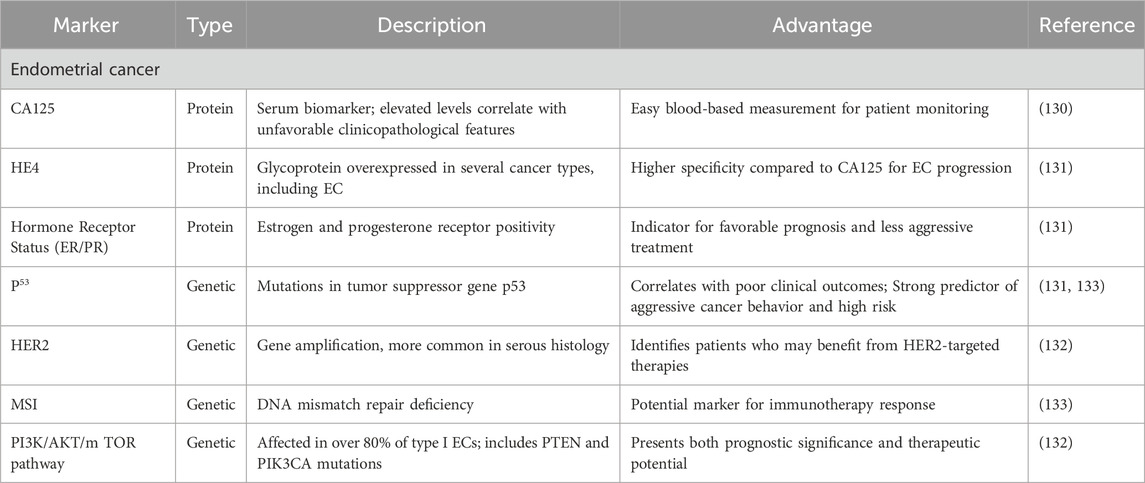
Table 1. Biomarkers in endometrial cancer: types, descriptions, and clinical advantages.
Hormone receptor status, particularly progesterone receptor (PR) and estrogen (ER) positivity, has been recognized as a key prognostic marker linked to a substantial enhancement in disease-free survival. Mutations in the tumor suppressor gene p53 are prominent in type II ECs, with studies reporting mutations in up to 90% of serous carcinomas. p53 mutations correlate with poor clinical outcomes, including an 11-fold elevated risk of death in multivariate analyses adjusting for lymph node metastasis, grade, histology, and FIGO stage (125). HER2 gene amplification, more common in serous histology, has been identified as a distinct prognostic marker associated with reduced overall survival. The PI3K/AKT/mTOR pathway, affected in over 80% of type I ECs, presents both prognostic significance and therapeutic potential, with PTEN and PIK3CA mutations being key components (126). MSI has shown conflicting prognostic implications, with some studies reporting improved 5-year survival rates, while other studies observed no notable variation in relapse or overall survival. These molecular markers and emerging factors, such as microvascular proliferation, are refining our ability to predict EC outcomes and guide personalized treatment strategies (127).
At this point, CRC patient prognosis depends on clinicopathological parameters, with an emphasis on the cancer stage upon diagnosis. The total 5-year rate of survival for stage I is above 90%; it decreases to 70% for the second stage, 58% for stage III, and fewer than 15% for stage IV (128). Prognostic markers are crucial in predicting outcomes and guiding treatment decisions for CRC patients. Carcinoembryonic antigen (CEA), despite its limitations in specificity and accuracy, has shown potential as a distinct prognostic marker for all stages of CRC. A large-scale National Cancer Database data study suggested that serum CEA serves as a reliable prognostic indicator for stage II tumor recurrence (129). Epidermal growth factor receptor (EGFR) expression is observed in 50%–70% of CRC, although its prognostic significance remains inconclusive (130). The homeobox protein CDX2 has emerged as a promising marker of colon cancer cell differentiation and a robust prognostic indicator (Table 2) (131).
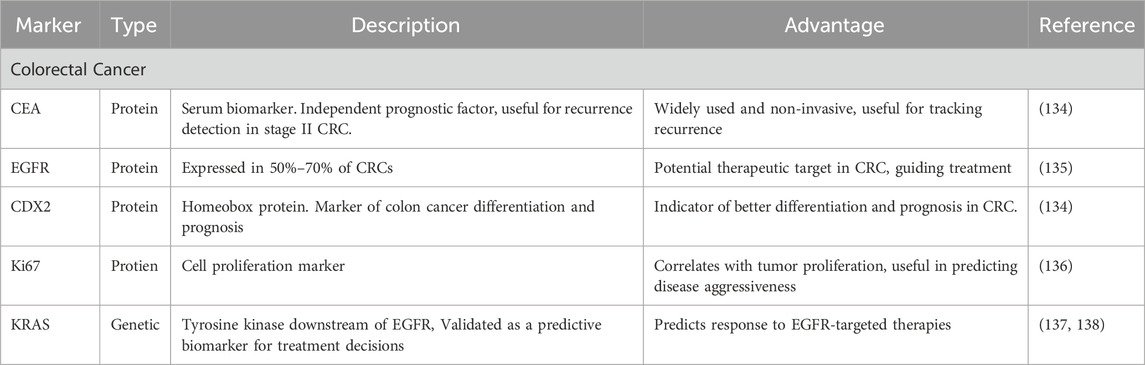
Table 2. Biomarkers in colorectal cancer: types, descriptions, and clinical advantages.
Interestingly, conflicting evidence exists regarding Ki67 expression in CRCs, with some studies suggesting that high expression is associated with good clinical outcomes. At the same time, a meta-analysis showed a strong association between elevated Ki-67 expression and reduced overall survival. disease-free survival (132). KRAS, a tyrosine kinase downstream of the EGFR receptor, stands out as the first validated predictive biomarker in colon cancer. These various prognostic markers collectively enhance our understanding of CRC mechanisms and assist in customizing treatment strategies to achieve better patient outcomes (2, 133).
12 Targeted therapeutics for EC in Lynch syndrome
In the treatment of EC, especially among patients with LS, therapeutic approaches are customized based on the cancer’s progression and the associated risk of relapse. Radiotherapy is often employed in early-stage EC, while chemotherapy is indicated for cases with high-grade histology or advanced chronic conditions (134). As the disease progresses, the risk of recurrence increases significantly. Notably, recurrent vaginal EC tends to respond well to treatments, frequently utilizing radiation therapy as an effective option. Surgery remains the cornerstone of EC treatment, with total hysterectomy (TH) and bilateral salpingo-oophorectomy (BSO) being the standard procedures (135). TH involves the removal of the uterus and cervix, while BSO eliminates the fallopian tubes and ovaries. In patients with LS, oophorectomy is routinely performed during surgery to exclude the presence of ovarian metastases or primary ovarian tumors, given the elevated risk associated with LS (134). Current surgical options include open surgery, laparotomy, and minimally invasive techniques such as laparoscopic surgery (LS) and robot-assisted surgery (RS), which have demonstrated efficacy and reduced recovery times. For advanced stages, particularly Stage III EC, chemotherapy regimens typically include paclitaxel and carboplatin, with alternatives like ifosfamide combined with paclitaxel or cisplatin being explored (136).
Emerging therapeutic pathways in preclinical studies focus on targeting specific molecular mechanisms involved in EC progression, such as cell cycle inhibition, EZH2 inhibition, and modulation of the prorenin pathway. These innovative strategies aim to enhance treatment efficacy and are increasingly integral to personalized medicine approaches. Recent advancements in immunotherapy have notably transformed the treatment landscape for advanced EC (137). The U.S. Food and Drug Administration (FDA) has approved several immune checkpoint inhibitors for this indication, marking significant progress in therapy options for patients, particularly those with dMMR tumors. Notable approvals include durvalumab (Imfinzi) for patients with mismatch repair-deficient tumors, pembrolizumab (Keytruda) for use irrespective of dMMR status, and dostarlimab (Jemperli), which has also been approved for dMMR advanced EC (135). These agents can be employed as first-line therapies or for recurrent cancer after specific prior treatments, significantly expanding the arsenal of options for managing advanced EC and improving patient outcomes, especially for those who previously had limited immunotherapy choices.
13 Targeted therapeutics for CRC in Lynch syndrome
Surgical resection continues to be the foremost intervention for CRC, especially in individuals diagnosed with LS, who exhibit an elevated propensity for the onset of CRC at earlier ages and frequently present with more advanced stages of the disease. In scenarios where the malignancy is classified as non-resectable, a multimodal approach encompassing chemotherapy, radiation therapy, and immunotherapy is generally utilized (139, 140). Radiation therapy constitutes an essential element of CRC management, particularly in the context of rectal cancer, as it employs high-energy X-rays to selectively target and obliterate neoplastic cells through the induction of DNA damage, thereby impeding cellular growth and proliferation (141, 142). This modality proves especially advantageous for rectal tumors that are confined, rendering them more susceptible to radiation intervention. Contemporary chemotherapy protocols for CRC frequently incorporate fluoropyrimidine-based agents, such as 5-fluorouracil (5-FU), in conjunction with combination therapies involving agents such as oxaliplatin (OX), irinotecan (IRI), and capecitabine (143).
Recent innovations in the management of advanced CRC have increasingly centered on targeted therapies, particularly those aimed at inhibiting angiogenesis. Monoclonal antibodies, including bevacizumab, ramucirumab, and aflibercept, have demonstrated a capacity to improve overall survival metrics when administered alongside standard chemotherapy regimens, offering substantial advantages for patients afflicted with advanced disease, including those with LS (144). Oral therapeutic agents such as regorafenib and trifluridine/tipiracil have surfaced as viable alternatives for patients exhibiting refractory CRC, presenting renewed optimism for individuals with constrained treatment options. Although the survival enhancements associated with these novel agents may appear to be modest, they signify considerable progress within the therapeutic domain, enabling patients to potentially experience extended survival and enhanced quality of life (145). Furthermore, research has indicated a plausible role for estrogen/progestin replacement therapy in postmenopausal women, with antecedent findings suggesting a reduced incidence of CRC linked to these therapies, albeit the underlying mechanisms remain elusive. Given the intersection of hormonal influences and cancer risk, further investigation into this association may be warranted, particularly in LS patients who encounter augmented risks for various malignancies, including CRC (77, 146). Overall, the ongoing advancement of therapeutic strategies for CRC, particularly within the framework of LS, underscores the necessity of personalized treatment modalities customized to the distinct genetic and molecular attributes of tumors, thereby enhancing patient outcomes and survival probabilities.
14 Summarizing the landscape of Lynch syndrome-associated cancers
This review underscores the critical link between EC and subsequent CRC in individuals with LS, primarily driven by mutations in MMR genes and the presence of MSI. By consolidating findings from the original research data, it highlights the importance of early identification, genetic screening, and vigilant surveillance in high-risk populations. Both cancers are influenced by common genetic pathways, including the Wnt and PI3K/AKT/mTOR signaling cascades, with critical mutations in genes such as APC, PTEN, and β-catenin. The progression and development of these malignancies are also significantly impacted by lifestyle factors like obesity and hormonal imbalances. Understanding the molecular and genetic commonalities between EC and CRC is crucial for early diagnosis and the formulation of personalized treatment strategies. For patients with LS, the heightened risk of both cancers underscores the need for genetic counselling and regular screenings for these tumors. Advances in diagnostic techniques, including molecular biomarkers and high-throughput omics technologies, have enhanced the detection, treatment, and prognosis of both cancers. Furthermore, emerging therapeutic approaches, especially in the realm of targeted therapy and immunotherapy presents promising opportunities for better patient results. The connection between EC and CRC underscores the need for a comprehensive approach to managing patients, particularly those with genetic predispositions, to mitigate risks and enhance survival rates. This comprehensive review may provide a reference for clinicians and researchers aiming to refine diagnostic and management approaches in LS and explore future advancements in oncology.
15 Future directions in treating EC and CRC in LS
Exploring genetic and molecular interconnections between EC and CRC, especially in LS, is crucial for advancing research and treatment. Identifying shared signaling pathways will facilitate the creation of effective targeted therapies tailored for LS patients. Incorporating these findings into clinical practice may enhance affected individuals’ survival rates and quality of life. Immunotherapy, particularly checkpoint inhibitors, is a promising research area for treating EC and CRC in patients with LS and mismatch repair deficiencies. Exploring immune modulation and combination therapies could lead to innovative strategies that enhance immune responses against these cancers, improving outcomes for advanced-stage patients. Advances in next-generation sequencing (NGS) and high-throughput omics technologies will aid in the discovery of new biomarkers for early diagnosis and prognosis.
Author contributions
SnP: Investigation, Methodology, Visualization, Writing – original draft. SN: Methodology, Writing – original draft. SA: Methodology, Writing – original draft, Investigation. AB: Methodology, Writing – original draft, Supervision, Writing – review and editing. SuP: Conceptualization, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review and editing. AD: Conceptualization, Supervision, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors express their gratitude to Chettinad Academy of Research and Education (CARE) for providing the necessary infrastructure to carry out this work. The authors are also thankful to Department of Biotechnology, Ministry of Science & Technology, Government of India for providing support to Mr. Subhamay Adhikary (Fellowship IDDBT/2021-22/CARE/1592).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Peltomäki, P, Nyström, M, Mecklin, J-P, and Seppälä, TT. Lynch syndrome genetics and clinical implications. Gastroenterology (2023) 164(5):783–99. doi:10.1053/j.gastro.2022.08.058
2. Kuhn, TM, Dhanani, S, and Ahmad, S. An overview of endometrial cancer with novel therapeutic strategies. Curr Oncol (Toronto, Ont.) (2023) 30(9):7904–19. doi:10.3390/curroncol30090574
3. Bhat, GR, Sethi, I, Sadida, HQ, Rah, B, Mir, R, Algehainy, N, et al. Cancer cell plasticity: from cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance. Cancer Metastasis Rev (2024) 43:197–228. doi:10.1007/s10555-024-10172-z
4. Papadopoulou, E, Rigas, G, Fountzilas, E, Boutis, A, Giassas, S, Mitsimponas, N, et al. Microsatellite instability is insufficiently used as a biomarker for Lynch syndrome testing in clinical practice. JCO Precision Oncol (2024) 8(8):e2300332. doi:10.1200/PO.23.00332
5. Li, K, Luo, H, Huang, L, Luo, H, and Zhu, X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int (2020) 20(1):16. doi:10.1186/s12935-019-1091-8
6. Chen, W, and Frankel, WL. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod Pathol (2019) 32(Suppl. 1):1–15. doi:10.1038/s41379-018-0136-1
7. Gupta, D, and Heinen, CD. The mismatch repair-dependent DNA damage response: mechanisms and implications. DNA repair (2019) 78:60–9. doi:10.1016/j.dnarep.2019.03.009
8. Boland, CR, and Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology (2010) 138(6):2073–87.e3. doi:10.1053/j.gastro.2009.12.064
9. Helderman, NC, Bajwa-Ten Broeke, SW, Morreau, H, Suerink, M, Terlouw, D, van der Werf-’ t Lam, AS, et al. The diverse molecular profiles of lynch syndrome-associated colorectal cancers are (highly) dependent on underlying germline mismatch repair mutations. Crit Rev Oncology/Hematology (2021) 163(103338):103338. doi:10.1016/j.critrevonc.2021.103338
10. Friedenreich, CM, Ryder-Burbidge, C, and McNeil, J. Physical activity, obesity and sedentary behavior in cancer etiology: epidemiologic evidence and biologic mechanisms. Mol Oncol (2021) 15(3):790–800. doi:10.1002/1878-0261.12772
11. Felix, AS, and Brinton, LA. Cancer progress and priorities: uterine cancer. Cancer Epidemiol Biomarkers and Prev (2018) 27(9):985–94. doi:10.1158/1055-9965.EPI-18-0264
12. Gambini, D, Ferrero, S, and Kuhn, E. Lynch syndrome: from carcinogenesis to prevention interventions. Cancers (2022) 14(17):4102. doi:10.3390/cancers14174102
13. Vasen, HF, Watson, P, Mecklin, J, and Lynch, H. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology (1999) 116(6):1453–6. doi:10.1016/s0016-5085(99)70510-x
14. Umar, A, Boland, CR, Terdiman, JP, Syngal, S, Chapelle, A, Ruschoff, J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. JNCI J Natl Cancer Inst (2004) 96(4):261–8. doi:10.1093/jnci/djh034
15. Fanale, D, Corsini, LR, Brando, C, Dimino, A, Filorizzo, C, Magrin, L, et al. Impact of different selection approaches for identifying lynch syndrome-related colorectal cancer patients: unity is strength. Front Oncol (2022) 12:827822. doi:10.3389/fonc.2022.827822
16. Velho, S, Fernandes, MS, Leite, M, Figueiredo, C, and Seruca, R. Causes and consequences of microsatellite instability in gastric carcinogenesis. World J Gastroenterol (2014) 20(44):16433–42. doi:10.3748/wjg.v20.i44.16433
17. Woerner, SM, Benner, A, Sutter, C, Schiller, M, Yuan, YP, Keller, G, et al. Pathogenesis of DNA repair-deficient cancers: a statistical meta-analysis of putative Real Common Target genes. Oncogene (2003) 22(15):2226–35. doi:10.1038/sj.onc.1206421
18. Rebuzzi, F, Ulivi, P, and Tedaldi, G. Genetic predisposition to colorectal cancer: how many and which genes to test? Int J Mol Sci (2023) 24(3):2137. doi:10.3390/ijms24032137
19. Bhattacharya, P, and McHugh, T. W. (2024). Lynch Syndrome. PubMed: StatPearls Publishing. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK431096/.
20. Dowty, JG, Win, AK, Buchanan, DD, Lindor, NM, Macrae, FA, Clendenning, M, et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat (2013) 34(3):490–7. doi:10.1002/humu.22262
21. Valle, L, and Monahan, KJ. Genetic predisposition to gastrointestinal polyposis: syndromes, tumour features, genetic testing, and clinical management. The Lancet Gastroenterol and Hepatol (2024) 9(1):68–82. doi:10.1016/S2468-1253(23)00240-6
22. Kastrinos, F. Risk of pancreatic cancer in families with Lynch syndrome. JAMA (2009) 302(16):1790–5. doi:10.1001/jama.2009.1529
23. Nakamura, K, Nakayama, K, Minamoto, T, Ishibashi, T, Ohnishi, K, Yamashita, H, et al. Lynch syndrome-related clear cell carcinoma of the cervix: a case report. Int J Mol Sci (2018) 19(4):979. doi:10.3390/ijms19040979
24. Baglietto, L, Lindor, NM, Dowty, JG, White, DM, Wagner, A, Gomez Garcia, EB, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. JNCI: J Natl Cancer Inst (2010) 102(3):193–201. doi:10.1093/jnci/djp473
25. Belot, A, Grosclaude, P, Bossard, N, Jougla, E, Benhamou, E, Delafosse, P, et al. Cancer incidence and mortality in France over the period 1980-2005. Revue d'epidemiologie et de sante publique (2008) 56(3):159–75. doi:10.1016/j.respe.2008.03.117
26. Roberts, ME, Jackson, SA, Susswein, LR, Zeinomar, N, Ma, X, Marshall, ML, et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med (2018) 20(10):1167–74. doi:10.1038/gim.2017.254
27. Poaty, H, Bouya, LB, Lumaka, A, Mongo-Onkouo, A, and Gassaye, D. PMS2 pathogenic variant in lynch syndrome-associated colorectal cancer with polyps. Glob Med Genet (2023) 10(1 1-5):001–5. doi:10.1055/s-0042-1759888
28. Ten Broeke, SW, van der Klift, HM, Tops, CMJ, Aretz, S, Bernstein, I, Buchanan, DD, et al. Cancer risks for PMS2-associated lynch syndrome. J Clin Oncol : official J Am Soc Clin Oncol (2018) 36(29):2961–8. doi:10.1200/JCO.2018.78.4777
29. Andini, KD, Nielsen, M, Suerink, M, Helderman, NC, Koornstra, JJ, Ahadova, A, et al. PMS2-associated Lynch syndrome: past, present and future. Front Oncol (2023) 13:1127329. doi:10.3389/fonc.2023.1127329
30. Bajwa-Ten Broeke, SW, Ballhausen, A, Ahadova, A, Suerink, M, Bohaumilitzky, L, Seidler, F, et al. The coding microsatellite mutation profile of PMS2-deficient colorectal cancer. Exp Mol Pathol (2021) 122(104668):104668. doi:10.1016/j.yexmp.2021.104668
31. Truninger, K, Menigatti, M, Luz, J, Russell, A, Haider, R, Gebbers, JO, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology (2005) 128(5):1160–71. doi:10.1053/j.gastro.2005.01.056
32. Kastrinos, F, and Stoffel, EM. History, genetics, and strategies for cancer prevention in Lynch syndrome. Clin Gastroenterol Hepatol (2014) 12(5):715–27. doi:10.1016/j.cgh.2013.06.031
33. Kempers, MJE, Kuiper, RP, Ockeloen, CW, Chappuis, PO, Hutter, P, Rahner, N, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. The Lancet Oncol (2011) 12(1):49–55. doi:10.1016/S1470-2045(10)70265-5
34. Edwards, P, and Monahan, KJ. Diagnosis and management of Lynch syndrome. Frontline Gastroenterol (2022) 13:e1 e80–e87. doi:10.1136/flgastro-2022-102123
35. Jasperson, KW, Tuohy, TM, Neklason, DW, and Burt, RW. Hereditary and familial colon cancer. Gastroenterology (2010) 138(6):2044–58. doi:10.1053/j.gastro.2010.01.054
36. Sahin, IH, Akce, M, Alese, O, Shaib, W, Lesinski, GB, El-Rayes, B, et al. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer (2019) 121(10):809–18. doi:10.1038/s41416-019-0599-y
37. Parente, P, Grillo, F, Vanoli, A, Macciomei, MC, Ambrosio, MR, Scibetta, N, et al. The day-to-day practice of MMR and MSI assessment in colorectal adenocarcinoma: what we know and what we still need to explore. Dig Dis (Basel, Switzerland) (2023) 41(5):746–56. doi:10.1159/000531003
38. Wang, Y, Wang, Y, Li, J, Cragun, J, Hatch, K, Chambers, SK, et al. Lynch syndrome related endometrial cancer: clinical significance beyond the endometrium. J Hematol and Oncol (2013) 6(1):22. doi:10.1186/1756-8722-6-22
39. Latham, A, Srinivasan, P, Kemel, Y, Shia, J, Bandlamudi, C, Mandelker, D, et al. Microsatellite instability is associated with the presence of Lynch syndrome pan-cancer. J Clin Oncol (2019) 37(4):286–95. doi:10.1200/jco.18.00283
40. Yang, Y, Wu, SF, and Bao, W. Molecular subtypes of endometrial cancer: implications for adjuvant treatment strategies. Int J Gynecol and Obstet (2024) 164(2):436–59. doi:10.1002/ijgo.14969
41. Makker, V, MacKay, H, Ray-Coquard, I, Levine, DA, Westin, SN, Aoki, D, et al. Endometrial cancer. Nat Rev Dis Primers (2021) 7(1):88. doi:10.1038/s41572-021-00324-8
42. Rodriguez, AC, Blanchard, Z, Maurer, KA, and Gertz, J. Estrogen signaling in endometrial cancer: a key oncogenic pathway with several open questions. Horm Cancer (2019) 10(2–3):51–63. doi:10.1007/s12672-019-0358-9
43. Sung, H, Ferlay, J, Siegel, RL, Laversanne, M, Soerjomataram, I, Jemal, A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clinicians (2021) 71(3):209–49. doi:10.3322/caac.21660
44. Sawicki, T, Ruszkowska, M, Danielewicz, A, Niedźwiedzka, E, Arłukowicz, T, and Przybyłowicz, KE. A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers (2021) 13(9):2025. doi:10.3390/cancers13092025
45. Baran, B, Mert Ozupek, N, Yerli Tetik, N, Acar, E, Bekcioglu, O, and Baskin, Y. Difference between left-sided and right-sided colorectal cancer: a focused review of literature. Gastroenterol Res (2018) 11(4):264–73. doi:10.14740/gr1062w
46. Sung, CO, Seo, JW, Kim, K-M, Do, I-G, Kim, SW, and Park, C-K. Clinical significance of signet-ring cells in colorectal mucinous adenocarcinoma. Mod Pathol (2008) 21(12):1533–41. doi:10.1038/modpathol.2008.170
47. Remo, A, Fassan, M, Vanoli, A, Bonetti, LR, Barresi, V, Tatangelo, F, et al. Morphology and molecular features of rare colorectal carcinoma histotypes. Cancers (2019) 11(7):1036. doi:10.3390/cancers11071036
48. Jasperson, KW, Vu, TM, Schwab, AL, Neklason, DW, Rodriguez-Bigas, MA, Burt, RW, et al. Evaluating Lynch syndrome in very early onset colorectal cancer probands without apparent polyposis. Fam Cancer (2010) 9(2):99–107. doi:10.1007/s10689-009-9290-4
49. Chen, L, Ye, L, and Hu, B. Hereditary colorectal cancer syndromes: molecular genetics and precision medicine. Biomedicines (2022) 10(12):3207. doi:10.3390/biomedicines10123207
50. Barczyński, B, Frąszczak, K, Wnorowski, A, and Kotarski, J. Menopausal status contributes to overall survival in endometrial cancer patients. Cancers (2023) 15(2):451. doi:10.3390/cancers15020451
51. Yu, K, Huang, Z-Y, Xu, X-L, Li, J, Fu, X-W, and Deng, S-L. Estrogen receptor function: impact on the human endometrium. Front Endocrinol (2022) 13:827724. doi:10.3389/fendo.2022.827724
52. Furness, S, Roberts, H, Marjoribanks, J, and Lethaby, A. Hormone therapy in postmenopausal women and risk of endometrial hyperplasia. The Cochrane database Syst Rev (2012) 2012(8):CD000402. doi:10.1002/14651858.CD000402.pub4
53. Valle, L. Genetic predisposition to colorectal cancer: where we stand and future perspectives. World J Gastroenterol (2014) 20(29):9828–49. doi:10.3748/wjg.v20.i29.9828
54. Shetty, C, Rizvi, SMHA, Sharaf, J, Williams, K-AD, Tariq, M, Acharekar, MV, et al. Risk of gynecological cancers in women with polycystic ovary syndrome and the pathophysiology of association. Cureus (2023) 15(4):e37266. doi:10.7759/cureus.37266
55. Ignatov, A, and Ortmann, O. Endocrine risk factors of endometrial cancer: polycystic ovary syndrome, oral contraceptives, infertility, tamoxifen. Cancers (2020) 12(7):1766. doi:10.3390/cancers12071766
56. Kanth, P, Grimmett, J, Champine, M, Burt, R, and Samadder, JN. Hereditary colorectal polyposis and cancer syndromes: a primer on diagnosis and management. Am J Gastroenterol (2017) 112(10):1509–25. doi:10.1038/ajg.2017.212
57. Manski, S, Noverati, N, Policarpo, T, Rubin, E, and Shivashankar, R. Diet and nutrition in inflammatory bowel disease: a review of the literature. Crohn's and Colitis 360 (2024) 6(1):otad077. doi:10.1093/crocol/otad077
58. Li, H, Sun, L, Zhuang, Y, Tian, C, Yan, F, Zhang, Z, et al. Molecular mechanisms and differences in lynch syndrome developing into colorectal cancer and endometrial cancer based on gene expression, methylation, and mutation analysis. Cancer Causes and Control (2022) 33(4):489–501. doi:10.1007/s10552-021-01543-w
59. Xu, W, Wang, B, Cai, Y, Chen, J, Lv, X, Guo, C, et al. ADAMTS9-AS2: a functional long non-coding RNA in tumorigenesis. Curr Pharm Des (2021) 27(23):2722–7. doi:10.2174/1381612827666210325105106
60. Okuda, T, Sekizawa, A, Purwosunu, Y, Nagatsuka, M, Morioka, M, Hayashi, M, et al. Genetics of endometrial cancers. Obstet Gynecol Int (2010) 2010(1):984013. doi:10.1155/2010/984013
61. Duggan, BD, Felix, JC, Muderspach, Ll., Tourgeman, D, Zheng, J, and Shibata, D. Microsatellite instability in sporadic endometrial carcinoma. JNCI J Natl Cancer Inst (1994) 86(16):1216–21. doi:10.1093/jnci/86.16.1216
62. Helderman, NC, Andini, KD, van Leerdam, ME, van Hest, LP, Hoekman, DR, Ahadova, A, et al. MLH1 promotor hypermethylation in colorectal and endometrial carcinomas from patients with Lynch syndrome. The J Mol Diagn (2024) 26(2):106–14. doi:10.1016/j.jmoldx.2023.10.005
63. Shanmugapriya, S, Subramanian, P, and Kanimozhi, S. Geraniol inhibits endometrial carcinoma via downregulating oncogenes and upregulating tumour suppressor genes. Indian J Clin Biochem (2017) 32(2):214–9. doi:10.1007/s12291-016-0601-x
64. Abal, M, Llauradó, M, Doll, A, Monge, M, Colas, E, González, M, et al. Molecular determinants of invasion in endometrial cancer. Clin and Translational Oncol Official Publ Fed Spanish Oncol Societies Natl Cancer Inst Mexico (2007) 9(5):272–7. doi:10.1007/s12094-007-0054-z
65. O'Hara, AJ, and Bell, DW. The genomics and genetics of endometrial cancer. Adv Genomics Genet (2012) 2012(2):33–47. doi:10.2147/AGG.S28953
66. Dixit, G, Gonzalez-Bosquet, J, Skurski, J, Devor, EJ, Dickerson, EB, Nothnick, WB, et al. FGFR2 mutations promote endometrial cancer progression through dual engagement of EGFR and Notch signalling pathways. Clin Translational Med (2023) 13(5):e1223. doi:10.1002/ctm2.1223
67. Gatius, S, Velasco, A, Azueta, A, Santacana, M, Pallares, J, Valls, J, et al. FGFR2 alterations in endometrial carcinoma. Mod Pathol (2011) 24(11):1500–10. doi:10.1038/modpathol.2011.110
68. Parrish, ML, Broaddus, RR, and Gladden, AB. Mechanisms of mutant β-catenin in endometrial cancer progression. Front Oncol (2022) 12:1009345. doi:10.3389/fonc.2022.1009345
69. Bosse, T, ter Haar, NT, Seeber, LM, Diest, PJv, Hes, FJ, Vasen, HFA, et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol (2013) 26(11):1525–35. doi:10.1038/modpathol.2013.96
70. Murali, R, Davidson, B, Fadare, O, Carlson, JA, Crum, CP, Gilks, CB, et al. High-grade endometrial carcinomas: morphologic and immunohistochemical features, diagnostic challenges and recommendations. Int J Gynecol Pathol (2019) 38(Suppl. 1):S40–S63. doi:10.1097/pgp.0000000000000491
71. Lax, SF, Kendall, B, Tashiro, H, Slebos, RJ, and Ellenson, LH. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer (2000) 88(4):814–24. doi:10.1002/(sici)1097-0142(20000215)88:4<814
72. Schultheis, AM, Martelotto, LG, De Filippo, MR, Piscuglio, S, Ng, CKY, Hussein, YR, et al. TP53 mutational spectrum in endometrioid and serous endometrial cancers. Int J Gynecol Pathol (2016) 35(4):289–300. doi:10.1097/pgp.0000000000000243
73. Nagase, S, Suzuki, F, Tokunaga, H, Toyoshima, M, Utsunomiya, H, Niikura, H, et al. Molecular pathogenesis of uterine serous carcinoma. Curr Obstet Gynecol Rep (2014) 3(1):33–9. doi:10.1007/s13669-013-0069-0
74. Shih, I-M, Panuganti, PK, Kuo, K-T, Mao, T-L, Kuhn, E, Jones, S, et al. Somatic mutations of PPP2R1A in ovarian and uterine carcinomas. The Am J Pathol (2011) 178(4):1442–7. doi:10.1016/j.ajpath.2011.01.009
75. Kim, K-R, Choi, J, Hwang, J-E, Baik, Y-A, Shim, JY, Kim, YM, et al. Endocervical-like (Müllerian) mucinous borderline tumours of the ovary are frequently associated with the KRAS mutation. Histopathology (2010) 57(4):587–96. doi:10.1111/j.1365-2559.2010.03673.x
76. Dubé, V, Roy, M, Plante, M, Renaud, M-C, and Têtu, B. Mucinous ovarian tumors of Mullerian-type: an analysis of 17 cases including borderline tumors and intraepithelial, microinvasive, and invasive carcinomas. Int J Gynecol Pathol (2005) 24(2):138–46. doi:10.1097/01.pgp.0000152024.37482.63
77. Vermij, L, Horeweg, N, Leon-Castillo, A, Rutten, TA, Mileshkin, LR, Mackay, HJ, et al. HER2 status in high-risk endometrial cancers (PORTEC-3): relationship with histotype, molecular classification, and clinical outcomes. Cancers (2020) 13(1):44. doi:10.3390/cancers13010044
78. Plotkin, A, Olkhov-Mitsel, E, Huang, W-Y, and Nofech-Mozes, S. Implementation of HER2 testing in endometrial cancer, a summary of real-world initial experience in a large tertiary cancer center. Cancers (2024) 16(11):2100. doi:10.3390/cancers16112100
79. Konecny, GE, Santos, L, Winterhoff, B, Hatmal, M, Keeney, GL, Mariani, A, et al. HER2 gene amplification and EGFR expression in a large cohort of surgically staged patients with nonendometrioid (type II) endometrial cancer. Br J Cancer (2009) 100(1):89–95. doi:10.1038/sj.bjc.6604814
80. Díaz-Montes, TP, Ji, H, Smith Sehdev, AE, Zahurak, ML, Kurman, RJ, Armstrong, DK, et al. Clinical significance of Her-2/neu overexpression in uterine serous carcinoma. Gynecol Oncol (2006) 100(1):139–44. doi:10.1016/j.ygyno.2005.08.017
81. Brodeur, MN, Selenica, P, Ma, W, Moufarrij, S, Dagher, C, Basili, T, et al. ERBB2 mutations define a subgroup of endometrial carcinomas associated with high tumor mutational burden and the microsatellite instability-high (MSI-H) molecular subtype. Mol Oncol (2024) 18(10):2356–68. doi:10.1002/1878-0261.13698
82. Russell, H, Kedzierska, K, Buchanan, DD, Thomas, R, Tham, E, Mints, M, et al. The MLH1 polymorphism rs1800734 and risk of endometrial cancer with microsatellite instability. Clin Epigenetics (2020) 12(1):102. doi:10.1186/s13148-020-00889-3
83. Georgescu, M-M. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes and Cancer (2010) 1(12):1170–7. doi:10.1177/1947601911407325
84. Zhao, R, Choi, BY, Lee, M-H, Bode, AM, and Dong, Z. Implications of genetic and epigenetic alterations of CDKN2A (p16(INK4a)) in cancer. EBioMedicine (2016) 8:30–9. doi:10.1016/j.ebiom.2016.04.017
85. Van Tongelen, A, Loriot, A, and De Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett (2017) 396:130–7. doi:10.1016/j.canlet.2017.03.029
86. Kodaman, PH, and Taylor, HS. Hormonal regulation of implantation. Obstet Gynecol Clin North America (2004) 31(4):745–66. doi:10.1016/j.ogc.2004.08.008
87. Nada, HR, Rashed, LA, Salman, OO, Abdallah, NMA, and Abdelhady, MM. Tissue levels of suppressor of cytokine signaling-3 (SOCS-3) in mycosis fungoides. Arch Dermatol Res (2022) 315(2):165–71. doi:10.1007/s00403-022-02339-x
88. Kaneko, E, Sato, N, Sugawara, T, Noto, A, Takahashi, K, Makino, K, et al. MLH1 promoter hypermethylation predicts poorer prognosis in mismatch repair deficiency endometrial carcinomas. J Gynecol Oncol (2021) 32(6):e79. doi:10.3802/jgo.2021.32.e79
89. Ahadova, A, Gallon, R, Gebert, J, Ballhausen, A, Endris, V, Kirchner, M, et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int J Cancer (2018) 143(1):139–50. doi:10.1002/ijc.31300
90. Johnson, V, Volikos, E, Halford, SE, Eftekhar Sadat, ET, Popat, S, Talbot, I, et al. (2005). Exon 3 beta-catenin mutations are specifically associated with colorectal carcinomas in hereditary non-polyposis colorectal cancer syndrome, Gut, 54, 264–7. doi:10.1136/gut.2004.048132
91. Zahary, MN, Ahmad Aizat, AA, Kaur, G, Yeong Yeh, L, Mazuwin, M, and Ankathil, R. Polymorphisms of cell cycle regulator genes CCND1 G870A and TP53 C215G: association with colorectal cancer susceptibility risk in a Malaysian population. Oncol Lett (2015) 10(5):3216–22. doi:10.3892/ol.2015.3728
92. Reeves, SG, Rich, D, Meldrum, CJ, Colyvas, K, Kurzawski, G, Suchy, J, et al. IGF1 is a modifier of disease risk in hereditary non-polyposis colorectal cancer. Int J Cancer (2008) 123(6):1339–43. doi:10.1002/ijc.23668
93. Talseth, BA, Ashton, KA, Meldrum, C, Suchy, J, Kurzawski, G, Lubinski, J, et al. Aurora-A and Cyclin D1 polymorphisms and the age of onset of colorectal cancer in hereditary nonpolyposis colorectal cancer. Int J Cancer (2008) 122(6):1273–7. doi:10.1002/ijc.23177
94. Chen, J, Etzel, CJ, Amos, CI, Zhang, Q, Viscofsky, N, Lindor, NM, et al. Genetic variants in the cell cycle control pathways contribute to early onset colorectal cancer in Lynch syndrome. Cancer Causes and Control (2009) 20(9):1769–77. doi:10.1007/s10552-009-9416-x
95. Lubbe, SJ, Di Bernardo, MC, Chandler, IP, and Houlston, RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol (2009) 27(24):3975–80. doi:10.1200/JCO.2008.21.6853
96. Castillejo, A, Vargas, G, Castillejo, MI, Navarro, M, Barberá, VM, González, S, et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur J Cancer (Oxford, Engl : 1990) (2014) 50(13):2241–50. doi:10.1016/j.ejca.2014.05.022
97. Magrin, L, Fanale, D, Brando, C, Corsini, LR, Randazzo, U, Di Piazza, M, et al. MUTYH-associated tumor syndrome: the other face of MAP. Oncogene (2022) 41(18):2531–9. doi:10.1038/s41388-022-02304-y
98. Sardo, E, Napolitano, S, Della Corte, CM, Ciardiello, D, Raucci, A, Arrichiello, G, et al. Multi-omic approaches in colorectal cancer beyond genomic data. J Personalized Med (2022) 12(2):128. doi:10.3390/jpm12020128
99. Wu, J-B, Li, X-J, Liu, H, Liu, Y-J, and Liu, X-P. Association of KRAS, NRAS, BRAF and PIK3CA gene mutations with clinicopathological features, prognosis and ring finger protein 215 expression in patients with colorectal cancer. Biomed Rep (2023) 19(6):104. doi:10.3892/br.2023.1686
100. Boutin, AT, Liao, W-T, Wang, M, Hwang, SS, Karpinets, TV, Cheung, H, et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes and Development (2017) 31(4):370–82. doi:10.1101/gad.293449.116
101. Palomba, G, Doneddu, V, Cossu, A, Paliogiannis, P, Manca, A, Casula, M, et al. Prognostic impact of KRAS, NRAS, BRAF, and PIK3CA mutations in primary colorectal carcinomas: a population-based study. J Translational Med (2016) 14(1):292. doi:10.1186/s12967-016-1053-z
102. Malki, A, ElRuz, RA, Gupta, I, Allouch, A, Vranic, S, and Al Moustafa, A-E. Molecular mechanisms of colon cancer progression and metastasis: recent insights and advancements. Int J Mol Sci (2020) 22(1):130. doi:10.3390/ijms22010130
103. Kim, Y-N, Kim, MK, Lee, YJ, Lee, Y, Sohn, JY, Lee, JY, et al. Identification of lynch syndrome in patients with endometrial cancer based on a germline next generation sequencing multigene panel test. Cancers (2022) 14(14):3406. doi:10.3390/cancers14143406
104. Batlle, E, and Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity (2019) 50(4):924–40. doi:10.1016/j.immuni.2019.03.024
105. Rascio, F, Spadaccino, F, Rocchetti, MT, Castellano, G, Stallone, G, Netti, GS, et al. The pathogenic role of PI3K/AKT pathway in cancer onset and drug resistance: an updated review. Cancers (2021) 13(16):3949. doi:10.3390/cancers13163949
106. Xu, S, and Tang, C. The role of ARID1A in tumors: tumor initiation or tumor suppression? Front Oncol (2021) 11(4 Oct):745187. doi:10.3389/fonc.2021.745187
107. Ferreira, A, Pereira, F, Reis, C, Oliveira, MJ, Sousa, MJ, and Preto, A. Crucial role of oncogenic KRAS mutations in apoptosis and autophagy regulation: therapeutic implications. Cells (2022) 11:2183. doi:10.3390/cells11142183
108. Sideris, M, Emin, EI, Abdullah, Z, Hanrahan, J, Stefatou, KM, Sevas, V, et al. The role of KRAS in endometrial cancer: a mini-review. Anticancer Res (2019) 39(2):533–9. doi:10.21873/anticanres.13145
109. Gonzalez, G, Mehra, S, Wang, Y, Akiyama, H, and Behringer, RR. Sox9 overexpression in uterine epithelia induces endometrial gland hyperplasia. Differentiation (2016) 92(4):204–15. doi:10.1016/j.diff.2016.05.006
110. Song, P, Gao, Z, Bao, Y, Chen, L, Huang, Y, Liu, Y, et al. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J Hematol and Oncol (2024) 17(1 46):46. doi:10.1186/s13045-024-01563-4
111. Giannakis, M, Hodis, E, Jasmine Mu, X, Yamauchi, M, Rosenbluh, J, Cibulskis, K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet (2014) 46(12):1264–6. doi:10.1038/ng.3127
112. Watson, P, Vasen, HF, Mecklin, J, Bernstein, I, Aarnio, M, Järvinen, HJ, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer (2008) 123(2):444–9. doi:10.1002/ijc.23508
113. Vasen, HFA, Hendriks, Y, de Jong, AE, van Puijenbroek, M, Tops, C, Bröcker-Vriends, AHJT, et al. Identification of HNPCC by molecular analysis of colorectal and endometrial tumors. Dis Markers (2004) 20(4–5):207–13. doi:10.1155/2004/391039
114. Garg, K, and Soslow, RA. Lynch syndrome (hereditary non-polyposis colorectal cancer) and endometrial carcinoma. J Clin Pathol (2009) 62(8):679–84. doi:10.1136/jcp.2009.064949
115. Blanco, GDV. Familial colorectal cancer screening: when and what to do? World J Gastroenterol (2015) 21(26):7944–53. doi:10.3748/wjg.v21.i26.7944
116. Singh, H, Nugent, Z, Demers, A, Czaykowski, PM, and Mahmud, SM. Risk of colorectal cancer after diagnosis of endometrial cancer: a population-based study. J Clin Oncol (2013) 31(16):2010–5. doi:10.1200/JCO.2012.47.6481
117. Singh, MM, Singh, E, Miller, H, Strum, WB, and Coyle, W. Colorectal cancer screening in women with endometrial cancer: are we following the guidelines? J Gastrointest Cancer (2012) 43(2):190–5. doi:10.1007/s12029-011-9271-3
118. Win, AK, Lindor, NM, Winship, I, Tucker, KM, Buchanan, DD, Young, JP, et al. Risks of colorectal and other cancers after endometrial cancer for women with Lynch syndrome. JNCI: J Natl Cancer Inst (2013) 105(4):274–9. doi:10.1093/jnci/djs525
119. Liao, S-C, Yeh, H-Z, Chang, C-S, Chen, W-C, Muo, C-H, and Sung, F-C. Colorectal cancer risk in women with gynecologic cancers-A population retrospective cohort study. J Clin Med (2021) 10(14):3127. doi:10.3390/jcm10143127
120. Hu, Y, Liu, L, Jiang, Q, Fang, W, Chen, Y, Hong, Y, et al. CRISPR/Cas9: a powerful tool in colorectal cancer research. J Exp and Clin Cancer Res (2023) 42(1):308. doi:10.1186/s13046-023-02901-z
121. Lai, Y-L, Chiang, C-J, Chen, Y-L, You, S-L, Chen, Y-Y, Chiang, Y-C, et al. Increased risk of second primary malignancies among endometrial cancer survivors receiving surgery alone: a population-based analysis. Cancer Med (2021) 10(19):6845–54. doi:10.1002/cam4.3861
122. Min, H, Jo, S-M, and Kim, H-S. Efficient capture and simple quantification of circulating tumor cells using quantum dots and magnetic beads. Small (2015) 11(21):2536–42. doi:10.1002/smll.201403126
123. Njoku, K, Barr, CE, and Crosbie, EJ. Current and emerging prognostic biomarkers in endometrial cancer. Front Oncol (2022) 12:890908. doi:10.3389/fonc.2022.890908
124. Karimi-Zarchi, M, Dehshiri-Zadeh, N, Sekhavat, L, and Nosouhi, F. Correlation of CA-125 serum level and clinico-pathological characteristic of patients with endometriosis. Int J Reprod Biomed (Yazd, Iran) (2016) 14(11):713–8. doi:10.29252/ijrm.14.11.713
125. Behrouzi, R, Barr, CE, and Crosbie, EJ. HE4 as a biomarker for endometrial cancer. Cancers (2021) 13(19):4764. doi:10.3390/cancers13194764
126. Binder, PS, and Mutch, DG. Update on prognostic markers for endometrial cancer. Women’s Health (London, England) (2014) 10(3):277–88. doi:10.2217/whe.14.13
127. Balestra, A, Larsimont, D, and Noël, JC. HER2 amplification in p53-mutated endometrial carcinomas. Cancers (2023) 15(5):1435. doi:10.3390/cancers15051435
128. Alnakli, AAA, Mohamedali, A, Heng, B, Chan, C, Shin, J-S, Solomon, M, et al. Protein prognostic biomarkers in stage II colorectal cancer: implications for post-operative management. BJC Rep (2024) 2(1):13. doi:10.1038/s44276-024-00043-z
129. Walther, A, Johnstone, E, Swanton, C, Midgley, R, Tomlinson, I, and Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer (2009) 9(7):489–99. doi:10.1038/nrc2645
130. Bennedsen, ALB, Cai, L, Hasselager, RP, Özcan, AA, Mohamed, KB, Eriksen, JO, et al. An exploration of immunohistochemistry-based prognostic markers in patients undergoing curative resections for colon cancer. BMC Cancer (2022) 22(1):62. doi:10.1186/s12885-022-09169-0
131. Melling, N, Kowitz, CM, Simon, R, Bokemeyer, C, Terracciano, L, Sauter, G, et al. High Ki67 expression is an independent good prognostic marker in colorectal cancer. J Clin Pathol (2016) 69(3):209–14. doi:10.1136/jclinpath-2015-202985
132. Reddy, S, Vergo, M, and Benson, AB Prognostic and predictive markers in colorectal cancer. Curr Colorectal Cancer Rep (2011) 7(4):267–74. doi:10.1007/s11888-011-0104-3
133. Koncina, E, Haan, S, Rauh, S, and Letellier, E. Prognostic and predictive molecular biomarkers for colorectal cancer: updates and challenges. Cancers (2020) 12(2):319. doi:10.3390/cancers12020319
134. Kalampokas, E, Giannis, G, Kalampokas, T, Papathanasiou, A-A, Mitsopoulou, D, Tsironi, E, et al. Current approaches to the management of patients with endometrial cancer. Cancers (2022) 14(18):4500. doi:10.3390/cancers14184500
135. Corr, B, Cosgrove, C, Spinosa, D, and Guntupalli, S. Endometrial cancer: molecular classification and future treatments. BMJ Med (2022) 1(1):e000152. doi:10.1136/bmjmed-2022-000152
136. Mahdi, H, Chelariu-Raicu, A, and Slomovitz, BM. Immunotherapy in endometrial cancer. Int J Gynecol Cancer (2023) 33(3):351–7. doi:10.1136/ijgc-2022-003675
137. Kumar, A, Gautam, V, Sandhu, A, Rawat, K, Sharma, A, and Saha, L. Current and emerging therapeutic approaches for colorectal cancer: a comprehensive review. World J Gastrointest Surg (2023) 15(4):495–519. doi:10.4240/wjgs.v15.i4.495
138. Krasteva, N, and Georgieva, M. Promising therapeutic strategies for colorectal cancer treatment based on nanomaterials. Pharmaceutics (2022) 14(6):1213. doi:10.3390/pharmaceutics14061213
139. Xie, Y-H, Chen, Y-X, and Fang, J-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduction Targeted Ther (2020) 5(1):22. doi:10.1038/s41392-020-0116-z
140. Bhuin, A, Udayakumar, S, Gopalarethinam, J, Mukherjee, D, Girigoswami, K, Ponraj, C, et al. Photocatalytic degradation of antibiotics and antimicrobial and anticancer activities of two-dimensional ZnO nanosheets. Scientific Rep (2024) 14(1):10406. doi:10.1038/s41598-024-59842-6
141. Golshani, G, and Zhang, Y. Advances in immunotherapy for colorectal cancer: a review. Ther Adv Gastroenterol (2020) 13:1756284820917527. doi:10.1177/1756284820917527
142. Balaji, D, Kalarani, IB, Mohammed, V, and Veerabathiran, R. Potential role of human papillomavirus proteins associated with the development of cancer. Virusdisease (2022) 33(3):322–33. doi:10.1007/s13337-022-00786-8
143. Rennert, G, Rennert, HS, Pinchev, M, Lavie, O, and Gruber, SB. Use of hormone replacement therapy and the risk of colorectal cancer. J Clin Oncol (2009) 27(27):4542–7. doi:10.1200/JCO.2009.22.0764
144. Wang, C, Tran, DA, Fu, MZ, Chen, W, Fu, SW, and Li, X. Estrogen receptor, progesterone receptor, and HER2 receptor markers in endometrial cancer. J Cancer (2020) 11(7):1693–701. doi:10.7150/jca.41943
145. Nakamura, M, Obata, T, Daikoku, T, and Fujiwara, H. The association and significance of p53 in gynecologic cancers: the potential of targeted therapy. Int J Mol Sci (2019) 20(21):5482. doi:10.3390/ijms20215482
146. Bae, JM, Lee, TH, Cho, N-Y, Kim, T-Y, and Kang, GH. Loss of CDX2 expression is associated with poor prognosis in colorectal cancer patients. World J Gastroenterol (2015) 21(5):1457–67. doi:10.3748/wjg.v21.i5.1457
Glossary
LS Lynch Syndrome
EC Endometrial cancer
CRC Colorectal Cancer
MMR Mismatch Repair
MSS Microsatelite Stable
MSI Microsatelite Instability
MSI-H Microsatelite Instability-High
MSI-L Microsatelite Instability-Low
MLH1 MutL homolog 1
MLH2 MutL homolog 2
MSH2 MutS homolog 2
MSH6 MutS homolog 6
PMS1 PMS1 homolog 1
PMS2 PMS1 homolog 2
EPCAM Epithelial Cell Adhesion Molecule
PTEN Phosphatase and Tensin Homolog
TGF-βR2 Transforming growth factor, beta receptor II
BAX BCL-2-associated X protein
HR Hazard Ratio
PD-1 Programmed Death −1
PD-L1 Programmed Death Ligand 1
KRAS Kirsten rat sarcoma viral oncogene homologue
PIK3CA phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
BMI Body Mass Index
APC Adenomatous polyposis coli
CTNNB1 Catenin beta-1
PCOS Polycystic ovary syndrome
HRT Hormone Replacement Therapy
IBD Inflammatory Bowel Disease
TCGA The Cancer Genome Atlas
DEGs Differentially Expressed Genes
LS-CRC Lynch Syndrome Associated Colorectal Cancer
LS-EC Lynch Syndrome Associated Endometrial Cancer
COL11A1 Collagen Type XI Alpha 1 Chain
SG-LC Specific Genes overlaps LS and CRC
SG-LE Specific Genes overlaps LS and EC
CST2 Type 2 cystatin
COL18A1 Collagen Type XVIII Alpha 1 Chain
LY6K Lymphocyte Antigen 6 Family Member K
MIR27B MicroRNA 27b
SST Somatostatin
KIF20A Kinesin-like protein
NUF2 Kinetochore protein Nuf2
HTR4 5-Hydroxytryptamine receptor 4
CDC45 Cell Division Cycle 45
WDR31 WD Repeat Domain 31
AADACL2 Arylacetamide Deacetylase Like 2
DHRS7C Dehydrogenase/Reductase 7C
KRT24 keratin gene
LINC00460 Long Intergenic Non-Protein Coding RNA 460
NPY2R Neuropeptide Y receptor type 2
KHDRBS2 KH RNA Binding Domain Containing, Signal Transduction Associated 2
CDH10 Cadherin 10
LINC02616 Long Intergenic Non-Protein Coding RNA 2616
LINC02691 Long Intergenic Non-Protein Coding RNA 2691
IGF2-AS IGF2 Antisense RNA
IGF Insulin-like growth factor
ADAMTS9-AS2 ADAM Metallopeptidase With Thrombospondin Type 1 Motif 9- Antisense RNA 2
SLC10A4 Solute Carrier Family 10 Member 4
EECs Endometrioid Endometrial Carcinomas
BHD Birt–Hogg–Dubé syndrome
IGFIIR Insulin-like Growth Factor II Receptor
Rad3 A yeast homolog of human ATR
ATR Ataxia Telangiectasia and Rad3-related protein
PIK3R1 Phosphoinositide-3-Kinase Regulatory Subunit 1
BRAF B-Raf Proto-Oncogene, Serine/Threonine Kinase
FGFR2 Fibroblast Growth Factor Receptor 2
ARID1A AT-Rich Interaction Domain 1A
BAF250a BRG1-Associated Factor 250a
SWI/SNF SWItch/Sucrose Non-Fermentable
ERBB2 Erb-B2 Receptor Tyrosine Kinase 2
PPP2R1A Protein Phosphatase 2 Scaffold Subunit A Alpha
PP2A Protein Phosphatase 2A
HER-2 Human Epidermal Growth Factor Receptor 2
RASSF1A Ras Association Domain Family Member 1 isoform A
CDKN2A Cyclin Dependent Kinase Inhibitor 2A
C-MYC MYC Proto-Oncogene, BHLH Transcription Factor
MEST Mesoderm Specific Transcript
HOXA10 Homeobox A10
HOXA11 Homeobox A11
SOCS3 Suppressor of Cytokine Signaling 3
GSK-3B Glycogen Synthase Kinase 3 Beta
CCND1 Cyclin D1
SIR Standardized Incidence Rate
CI Confidence Interval
TP53 Tumor Protein P53
AURKA Aurora Kinase A
CART Cocaine- and Amphetamine-Regulated Transcript
E2F2 E2F Transcription Factor 2 (involved in cell cycle regulation)
SNP Single Nucleotide Polymorphism
MUTYH MutY DNA Glycosylase
BER Base Excision Repair
OGG1 8-Oxoguanine DNA Glycosylase
CIMP CpG Island Methylator Phenotype
HE4 Human Epididymis Protein 4
CA125 Cancer Antigen 125
PR Progesterone Receptor
ER Estrogen Receptor
FIGO International Federation of Gynecology and Obstetrics
CDX2 Caudal Type Homeobox 2
Ki67 Marker of Cellular Proliferation
TH Total Hysterectomy
LS Laproscopic Surgery
RS Robot-assisted Surgery
BSO Bilateral Salpingo-Oophorectomy
RS Recurrence Score
EZH2 Enhancer of Zeste Homolog 2
FDA Food and Drug Administration
dMMR Deficient Mismatch Repair
5-FU 5-Fluorouracil
OX Oxaliplatin
IRI Irinotecan
NGS Next-Generation Sequencing
Keywords: endometrial cancer, colorectal cancer, Lynch syndrome, genetic mutations, risk factors
Citation: Pallatt S, Nambidi S, Adhikary S, Banerjee A, Pathak S and Duttaroy AK (2025) A brief review of Lynch syndrome: understanding the dual cancer risk between endometrial and colorectal cancer. Oncol. Rev. 19:1549416. doi: 10.3389/or.2025.1549416
Received: 21 December 2024; Accepted: 05 May 2025;
Published: 16 May 2025.
Edited by:
Akhil Kapoor, Tata Memorial Hospital, IndiaReviewed by:
Daniele Fanale, Azienda Ospedaliera Universitaria Policlinico Paolo Giaccone, ItalyPoonam Gera, Research and Education in Cancer (ACTREC), India
Copyright © 2025 Pallatt, Nambidi, Adhikary, Banerjee, Pathak and Duttaroy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Surajit Pathak, ZHJzdXJhaml0cGF0aGFrQGNhcmUuZWR1Lmlu; Asim K. Duttaroy, YS5rLmR1dHRhcm95QG1lZGlzaW4udWlvLm5v