 Youshu Zhang
Youshu Zhang Yao Dong1
Yao Dong1 Chuanqiang Dai
Chuanqiang Dai- 1West China Hospital of Sichuan University-Ziyang Hospital, Ziyang Central Hospital, Sichuan, China
- 2Chengdu MAST Medical Technology Co., Ltd, Chengdu, China
Malignant bone tumors, particularly osteosarcoma, pose significant therapeutic challenges due to genomic heterogeneity, chemoresistance, and stagnant survival rates. The PI3K/AKT/mTOR pathway emerges as a central driver of tumor progression, metastasis, and therapeutic resistance. Everolimus (EVR), a rapamycin-derived mTORC1 inhibitor, demonstrates multifaceted antitumor effects in osteosarcoma by suppressing protein synthesis, metabolic reprogramming, angiogenesis, and osteoclastogenesis. Preclinical studies highlight EVR’s synergistic potential with targeted agents (e.g., sorafenib, zoledronic acid), chemotherapy (e.g., doxorubicin), and proteasome inhibitors (e.g., bortezomib), achieving >50% tumor volume reduction and metastasis suppression in xenograft models through dual mTORC1/2 blockade, stress-apoptosis activation, and microenvironment remodeling. Clinically, phase II trials report a 45% 6-month progression-free survival (PFS) rate for EVR-sorafenib combinations in refractory osteosarcoma, albeit with manageable toxicity. Precision oncology approaches, such as EVR combined with tumor-treating fields (TTFields) and immune checkpoint inhibitors, further reveal its role in DNA repair-deficient subtypes and TME modulation. However, challenges persist, including mTORC2-mediated resistance, limited intratumoral bioavailability (<20% plasma levels), and biomarker scarcity. Future strategies emphasize bone-targeted nanoparticle delivery systems, dual-target inhibitors (e.g., RapaLink-1), and dynamic multi-omics predictive models to optimize EVR’s precision. By integrating organoid platforms, AI-driven drug screening, and international trials, EVR is poised to evolve from a broad-spectrum agent into a molecularly guided therapeutic hub, bridging “anti-tumor, bone-protective, and immune-regulatory” mechanisms. This paradigm shift promises to transform osteosarcoma management from empirical combinations to biomarker-driven precision therapy, ultimately improving survival and quality of life for patients.
1 Background
Malignant bone tumors, predominantly comprising osteosarcoma, chondrosarcoma, and Ewing sarcoma, represent the most common primary bone malignancies in children and adolescents (1). Data from the German Childhood Cancer Registry (1987–2011) indicate an age-standardized incidence rate (ASR) of 5.5 cases per million population annually among children under 15 years, with osteosarcoma and Ewing sarcoma accounting for the majority of subtypes (2). According to the Chinese Cancer Registry Annual Report (2000–2015), the crude incidence and mortality rates of malignant bone tumors in China were 1.77/100,000 and 1.31/100,000, respectively, with a significantly higher disease burden observed in males and rural populations (urban areas showed an annual decline of 2.2%, while rural areas exhibited a downward trend post-2007). The nationwide decline in incidence and mortality may correlate with early screening initiatives and standardized chemotherapy protocols (3). However, despite these epidemiological improvements, clinical therapeutic advancements remain stagnant. Approximately 10%–15% of newly diagnosed osteosarcoma patients present with metastases (primarily pulmonary) at initial diagnosis. While the 5-year survival rate for localized osteosarcoma is approximately 60%, it plummets to 20% for metastatic or recurrent cases, with no significant survival improvements observed over the past 3 decades (4). Despite its low global incidence (ASR <1.5 per million), malignant bone tumors impose multidimensional burdens on adolescents, including developmental impairment, limb dysfunction, and psychological distress, underscoring the urgent need for effective therapeutic strategies.
Therapeutic paradigms for osteosarcoma have evolved substantially. Surgically, limb-salvage procedures (now utilized in 90% of cases) have replaced traditional amputations, combined with neoadjuvant chemotherapy (10–12 weeks preoperatively) to evaluate prognosis via histologic necrosis rates (≥90% indicating favorable response (5)). Patients achieving favorable responses exhibit a 5-year survival rate of 75%, compared to 45% in poor responders (6, 7). The MAP regimen (methotrexate, doxorubicin, and cisplatin), established in 1982, elevated postoperative 2-year progression-free survival from 17% to 66% and remains the standard regimen for adolescents (neoadjuvant + adjuvant chemotherapy) (4). Nevertheless, three critical scientific challenges persist: (1) High genomic heterogeneity impedes molecular subtyping and precise prognostic stratification; (2) Incomplete understanding of tumor microenvironment (TME) regulatory networks limits targeted/immunotherapy development; and (3) Intrinsic chemoresistance mechanisms (e.g., aberrant DNA damage repair, drug efflux pump overexpression) lead to poor MAP responses in subsets of patients. Recent genomic advances have identified dysregulated signaling cascades—including PI3K/AKT/mTOR, JAK/STAT3, PD-1/PD-L1, and Wnt/β-catenin—as drivers of tumorigenesis, progression, metastasis, and therapeutic resistance, revealing actionable targets such as mTOR inhibition to address these challenges (8). The interplay between these biological traits and clinical hurdles has resulted in diminishing returns from conventional therapies. Targeted therapies, however, offer dual solutions: (1) Genomically informed inhibitors may overcome chemoresistance and enable precision intervention; and (2) Targeting TME regulators (e.g., angiogenesis, immunosuppressive pathways) could synergize with immunotherapies to disrupt TME barriers, establishing novel combinatorial paradigms (9).
Everolimus (EVR), an oral mTOR inhibitor, has been widely adopted in oncology with established indications across multiple tumor types (10–15). In renal cell carcinoma, EVR demonstrated significant PFS benefit in VEGF-refractory disease (RECORD-1 trial), leading to its approval as a standard second-line option (16). In hormone receptor–positive, HER2-negative advanced breast cancer, EVR combined with exemestane improved median PFS from 2.8 to 6.9 months (BOLERO-2 trial, local assessment; 10.6 vs 4.1 months by central review) (17). Similarly, in advanced pancreatic and non-pancreatic neuroendocrine tumors, EVR prolonged median PFS by approximately 6–7 months versus placebo (RADIANT-3 and RADIANT-4 trials) (18, 19), underscoring its broad antitumor activity through mTOR pathway inhibition. These clinical successes in diverse malignancies provide a rationale for investigating EVR in osteosarcoma, particularly in refractory settings where therapeutic options remain limited.
As mTOR-selective inhibitors gain traction in bone oncology, EVR has emerged as a research focus due to its robust bone matrix penetration and favorable safety profile. In addressing osteosarcoma (OS) chemoresistance, EVR demonstrates synergistic efficacy by blocking compensatory pathways: A landmark 2015 study by Grignani et al. (Candiolo Cancer Institute, Italy) revealed that sorafenib monotherapy induces resistance via mTORC2 activation, while EVR combination therapy suppresses this compensatory mechanism, achieving a 6-month PFS rate of 45% in advanced OS patients—a significant improvement over historical controls (20, 21). This synergy extends to other agents (e.g., proteasome inhibitor bortezomib, CDK4/6 inhibitor palbociclib, and osteoclast inhibitor zoledronic acid) (22–24), offering multimodal strategies for multidrug-resistant cases. Notably, EVR uniquely combines antitumor and osteoprotective effects: Preclinical studies confirm that mTOR signaling promotes osteoclast survival via RANKL/OPG axis regulation, while EVR induces osteoclast apoptosis (62% increase, p < 0.01) and improves bone microarchitecture (25–27). A 2017 ovariectomy-induced bone loss model (Dresden University of Technology, Germany) demonstrated EVR’s dual capacity to inhibit tumor progression and restore bone metabolic homeostasis, highlighting its remodeling of the tumor-bone microenvironment (25, 28). Despite these advances, fragmented understanding of EVR’s mechanistic network persists. This review systematically elucidates EVR’s molecular mechanisms in OS, integrating multidimensional preclinical and translational evidence to unravel its “dual-effect” logic, evaluate its potential as a cornerstone of precision bone oncology, and propose biomarker-driven therapeutic frameworks to advance targeted therapy paradigms (Figure 1).
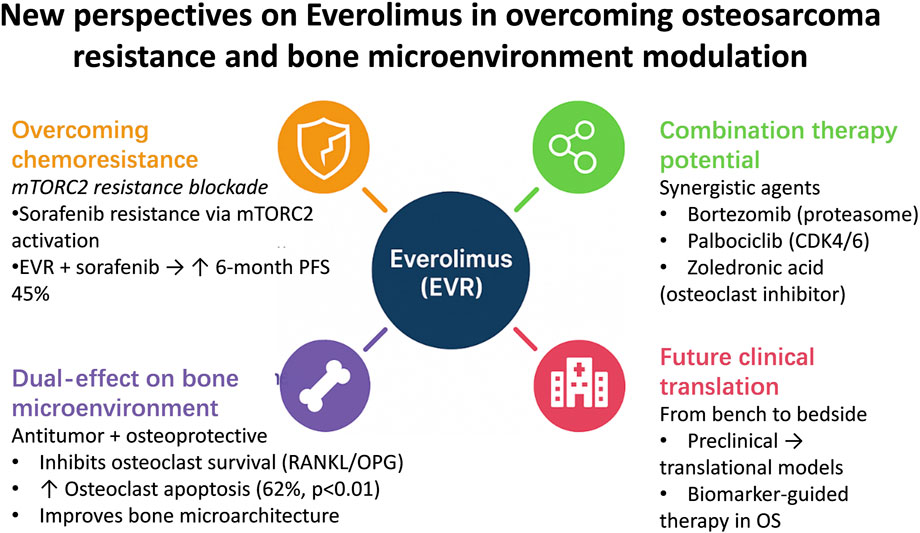
Figure 1. Schematic illustration of novel therapeutic perspectives of everolimus in osteosarcoma. The diagram summarizes key mechanisms including mTORC2 inhibition to overcome sorafenib resistance, synergistic effects with targeted and bone-modifying agents, and dual antitumor–osteoprotective actions through RANKL/OPG modulation.
2 Everolimus-driven modulation of the mTOR signaling axis in osteosarcoma inhibition
2.1 Structure and functional dynamics of PI3K/AKT/mTOR pathway
The canonical PI3K/AKT/mTOR signaling pathway serves as a central hub in cellular signal transduction, governing diverse biological processes such as metabolism, proliferation, and angiogenesis (29–31), with pivotal roles in malignant transformation and progression (32). The elucidation of this pathway began in the mid-1980s, marked by seminal discoveries over nearly 5 decades. A breakthrough emerged from Lewis Cantley’s team during investigations into viral oncogenesis, where they identified a phosphatidylinositol kinase (PI3K) activity linked to cellular transformation, laying the foundation for subsequent signal transduction research. In 1988, Cantley et al. (33) published a landmark study in Nature detailing the catalytic mechanism of class I PI3K: this heterodimeric enzyme, comprising a regulatory subunit (p85α) and a catalytic subunit (p110α), phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3) at the plasma membrane (34–36). PIP3 acts as a secondary messenger, recruiting phosphoinositide-dependent kinase 1 (PDK1), AKT, and serum/glucocorticoid-regulated kinase (SGK) to the membrane, thereby initiating downstream signaling cascades (37–39).
During the 1990s, subsequent studies unraveled key downstream regulatory mechanisms. AKT, a central effector kinase, requires dual phosphorylation at Thr308 (by PDK1) and Ser473 (by mTORC2) for full activation (40–43). Activated AKT phosphorylates downstream targets such as tuberous sclerosis complex 2 (TSC2) and forkhead box O (FOXO) proteins, directly or indirectly modulating cell growth and survival (44, 45). mTOR, a highly conserved serine/threonine kinase and critical AKT substrate, orchestrates cellular processes via two distinct complexes: mTORC1 and mTORC2 (46, 47). mTORC1 activation is regulated by TSC phosphorylation: AKT inhibits the TSC1-TSC2 complex (composed of hamartin [TSC1] and tuberin [TSC2]) via TSC2 phosphorylation, relieving its GTPase-activating protein (GAP) suppression on Ras homolog enriched in brain (Rheb), thereby activating mTORC1 (48, 49). Active mTORC1 drives oncogenic progression by phosphorylating ribosomal S6 kinase 1 (S6K1/p70S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1), enhancing protein synthesis, ribosome biogenesis, and cell cycle progression (50–52). Additionally, mTORC1 suppresses autophagy, promotes lipid synthesis, and upregulates hypoxia-inducible factor 1α (HIF-1α) to stimulate angiogenesis (53).
Further mechanistic insights revealed PTEN (phosphatase and tensin homolog), a lipid phosphatase, as a critical tumor suppressor that counteracts PI3K signaling by dephosphorylating PIP3 to PIP2 (54). By the early 2000s, mTOR’s dual roles through mTORC1 and mTORC2 in regulating protein synthesis, cell growth, and survival were firmly established (55). The advent of CRISPR-Cas9 gene editing (widely adopted post-2013) enabled functional validation of PI3Kα (encoded by PIK3CA) as a driver in oncogenesis, solidifying its status as a therapeutic target (56). Landmark studies published in Nature (circa 2015) resolved the full atomic structure of PI3K, enabling structure-guided design of allosteric inhibitors like alpelisib and accelerating their clinical translation (56). Collectively, these discoveries have constructed a robust framework bridging molecular insights to therapeutic innovation, positioning the PI3K/AKT/mTOR pathway as a cornerstone for targeted therapy in malignancies, including osteosarcoma (Figure 2).
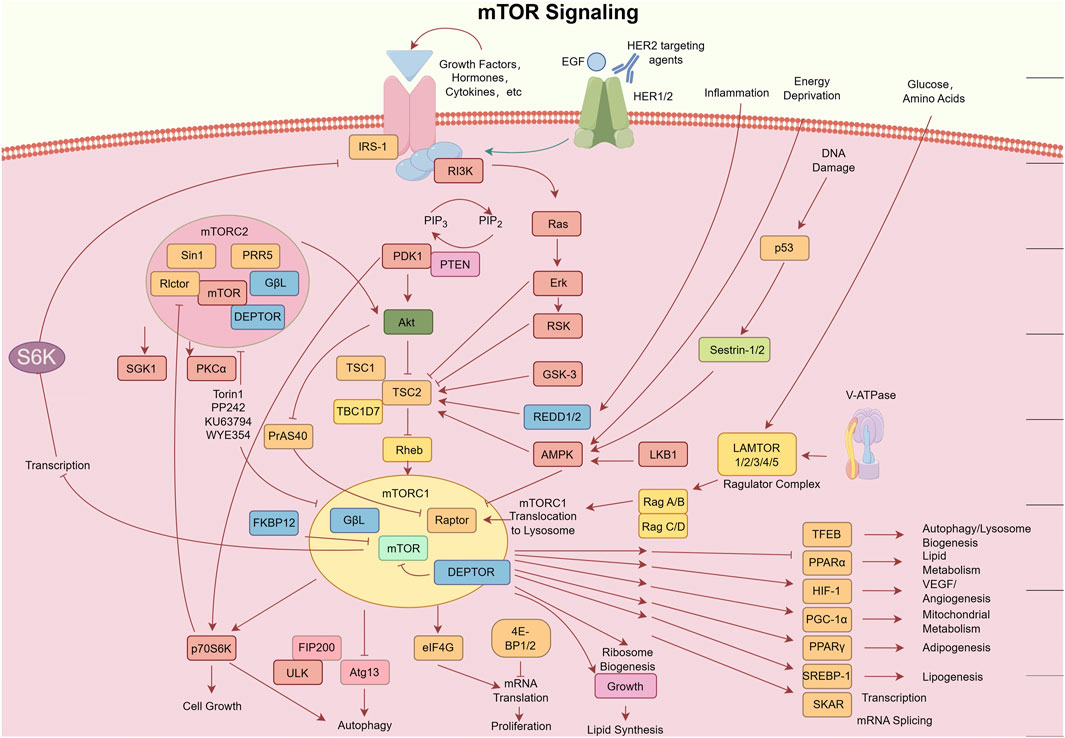
Figure 2. Schematic diagram of the mTOR signaling pathway. Key components include: Deptor (DEP domain-containing mTOR-interacting protein); EGF (epidermal growth factor); eIF4E (eukaryotic translation initiation factor 4E); 4E-BP (eukaryotic initiation factor 4E-binding protein); FKBP (FK506-binding protein); HER (human epidermal growth factor receptor); IGF(R) [insulin-like growth factor (receptor)]; IRS1 (insulin receptor substrate 1); LKB1 (liver kinase B1); AMPK (AMP-activated protein kinase); mLST8 (mammalian lethal with SEC13 protein 8); mTOR (mechanistic target of rapamycin); mTORC (mTOR complex); PDK1 (3-phosphoinositide-dependent protein kinase 1); PI3K (phosphatidylinositol 3-kinase); PRAS (proline-rich AKT1 substrate 1); Proctor (protein observed with Rictor); PTEN (phosphatase and tensin homolog); Rag and Rheb (small GTPases); Raptor (regulatory-associated protein of mTOR); Rictor (rapamycin-insensitive companion of mTOR); S6K (ribosomal protein S6 kinase); SIN1 (stress-activated protein kinase-interacting protein 1). Provided by Figdraw with authorized copyright (ID: ORPAP460e0).
2.2 Dysregulation of the PI3K/AKT/mTOR pathway in osteosarcoma progression
Aberrant activation of the PI3K/AKT/mTOR signaling pathway is a central driver of malignant progression in osteosarcoma (OS). Clinical evidence reveals characteristic expression patterns of key pathway components in OS patients. A study analyzing 130 paired tumor/normal bone tissue samples demonstrated significant downregulation of miR-99a in OS tissues, which inversely correlated with mTOR mRNA overexpression. Low miR-99a or high mTOR expression was associated with advanced Enneking stage (III), increased metastasis, postoperative recurrence, and chemoresistance, underscoring the miR-99a/mTOR axis as a critical regulator of OS aggressiveness (57). Similarly, STEAP2 overexpression in OS cell lines and clinical specimens has been linked to malignant phenotypes and poor prognosis. Mechanistically, STEAP2 activates PI3K/AKT/mTOR signaling to induce epithelial-mesenchymal transition (EMT), thereby enhancing OS cell invasion and migration (58). Immunohistochemical quantification further confirmed elevated phosphorylation levels of downstream mTOR effectors (p-mTOR, p-4E-BP1, and p-S6K1) in OS tissues compared to normal bone (p < 0.001), corroborating sustained pathway activation at the protein level (59).
Molecular investigations highlight multifaceted contributions of PI3K/AKT/mTOR components to OS pathogenesis. Activation of mTOR effectors (e.g., p70S6K and 4EBP1) directly promotes tumor proliferation and survival (57), while additional regulators amplify oncogenic signaling.
• KIF21B upregulation modulates OS cell proliferation and apoptosis via PI3K/AKT signaling (60);
• NRSN2 co-activates PI3K/AKT/mTOR and Wnt/β-catenin pathways to drive OS growth (61);
• SLC35A2 regulates mitochondrial autophagy through PI3K/AKT/mTOR to influence OS cell migration and invasion (62).
Genomic analyses reveal PIK3CA mutations in ∼80% of OS cases, predominantly clustering in kinase (exon 20) and helical (exon 9) domains. These mutations enhance catalytic activity of the class I PI3K p110α subunit, activating AKT via lipid and protein kinase functions (63, 64). Hyperactivated AKT phosphorylates multiple substrates to regulate proliferation, differentiation, and apoptosis, with its overexpression often correlating with PI3K/HER4 co-amplification—a hallmark of OS metastasis and poor prognosis. Conversely, PTEN loss or downregulation removes inhibitory control over PI3K/AKT signaling, exacerbating malignant phenotypes (65, 66). Additionally, AKT-mediated VEGF-A activation fosters tumor angiogenesis, providing microenvironmental support for OS invasion and metastasis (67).
Targeting this pathway has yielded promising preclinical and clinical advances. PI3K inhibition (e.g., LY294002) reduces p-AKT levels and induces OS apoptosis (68), while modulating non-coding RNAs (e.g., lncRNA MSC-AS1 silencing or miR-191-5p activation) restores chemosensitivity (69). As the pathway’s central integrator, mTOR activation drives cell cycle progression (e.g., G1/S transition) and upregulates pro-proliferative factors (cyclin D1/B1), making it a prime therapeutic target. Preclinical studies demonstrate that rapamycin analogs (e.g., temsirolimus) suppress OS xenograft growth and synergize with cisplatin. Dual-target inhibitors (e.g., NVP-BEZ235 [PI3K/mTOR] and CC-115 [mTOR/DNA-PK]) show superior efficacy in overcoming resistance. Translational evidence supports clinical potential.
• Ridaforolimus monotherapy prolongs PFS in metastatic sarcoma;
• Everolimus combined with sorafenib improves response rates in high-grade OS.
Mechanistically, everolimus binds FKBP12 to selectively inhibit mTORC1, reducing phosphorylation of p70S6K and 4E-BP1. This blockade suppresses cell cycle progression, protein synthesis, and VEGF-A-mediated angiogenesis, providing a molecular rationale for precision therapy in OS (20–22, 70). In summary, PI3K/AKT/mTOR dysregulation in OS constitutes a systemic network driving proliferation, invasion, metastasis, and chemoresistance. Current evidence solidifies mTOR-targeted therapy as a cornerstone of OS precision medicine. Future advances in multi-target inhibitors, combination regimens, and biomarker-driven strategies hold promise for developing low-toxicity, high-efficacy therapies for OS patients.
2.3 Antitumor molecular mechanisms of everolimus
Preclinical evidence shows that EVR inhibits proliferation in diverse human cancer cell lines (e.g., breast, gastric, renal) at subnanomolar concentrations, mainly via multidimensional modulation of the PI3K/AKT/mTOR network. By binding FKBP-12 and selectively inhibiting mTORC1, EVR suppresses S6K1 phosphorylation and releases 4E-BP1–mediated inhibition of oncogenic mRNA translation (Cyclin D1, c-MYC, HIF-1α (71)), leading to G1-phase arrest, activation of mitochondrial apoptosis pathways, and autophagy induction. In parallel, EVR reprograms cancer cell metabolism by downregulating HK2 and LDHA through mTORC1–HIF-1α suppression, thereby inhibiting glycolysis, and blocking SREBP-driven lipid synthesis, which disrupts membrane signaling platforms.
Although EVR is widely known for its immunosuppressive role in transplantation via mTORC1 inhibition in T lymphocytes, emerging evidence suggests it may also modulate the immune microenvironment in OS. Preclinical studies indicate EVR reduces M2 polarization of tumor-associated macrophages, promoting a shift toward the M1 phenotype (72). It enhances dendritic cell antigen presentation (upregulating CD80/CD86), limits expansion of regulatory T cells, and downregulates immunosuppressive cytokines such as IL-10 and TGF-β (EVR has been reported to inhibit IL-10 expression in immune cells (73)). While combination strategies with immune checkpoint inhibitors have shown feasibility in other cancers, OS-specific studies remain limited and often lack immune cell profiling, leaving this a promising but underexplored direction.
In addition, EVR exerts dual anti-angiogenic effects—directly inhibiting endothelial cell proliferation and indirectly suppressing HIF-1α–driven VEGF expression—while prolonged exposure may activate compensatory PI3K/AKT signaling via IRS-1 stabilization. Such resistance mechanisms can be mitigated through combination regimens, for example, with PI3K or CDK4/6 inhibitors, to enhance therapeutic durability.
3 Advances in everolimus therapy for osteosarcoma
Through systematic searches of authoritative databases (PubMed, Web of Science, Embase, Scopus), we identified 14 studies meeting predefined criteria (spanning research centers in six countries) to comprehensively evaluate EVR’s preclinical and clinical progress in OS. These studies encompass.
• In vitro models: SaOS-2 and U2OS human OS cell lines;
• Genetically engineered models: Patient-derived orthotopic xenografts (PDOX) and murine xenografts;
• Clinical trials: Phase I/II trials testing EVR monotherapy or combinations (e.g., sorafenib, doxorubicin, tumor-treating fields).
Table 1 summarizes critical study parameters. Using evidence-based methodology, we integrate EVR’s molecular mechanisms in OS to construct a translational framework bridging foundational research and clinical practice.

Table 1. Research progress of everolimus (EVR) in osteosarcoma therapy.
3.1 Preclinical research advancements
Recent years have witnessed multidimensional breakthroughs in elucidating the mechanisms of everolimus (EVR) in combinatorial osteosarcoma (OS) therapy. Preclinical investigations have evolved from single-pathway inhibition to cross-network molecular regulation, accumulating robust laboratory evidence. In the realm of metabolic reprogramming and resistance reversal, Moriceau et al. (22) demonstrated that zoledronic acid (ZOL) synergizes with EVR via dual mechanisms: by suppressing the mevalonate pathway, ZOL reduces Ras protein isoprenylation, thereby inhibiting Ras/ERK pro-proliferative signaling while enhancing EVR’s suppression of PI3K/mTOR. In SaOS-2 and U2OS cell models, this combination increased proliferation inhibition by 30% and significantly reduced GTP-Ras activity. Notably, in C57BL/6 osteoblastic and NOD/SCID osteolytic murine models, the EVR + ZOL regimen achieved 60% and 50% tumor volume reduction, respectively, without added toxicity. This study pioneered the link between OS metabolic vulnerability (mevalonate pathway dependency) and targeted therapy resistance, establishing a mechanistic paradigm for subsequent combinatorial strategies.
Building on metabolic insights, Pignochino et al. (21) systematically dissected the synergy between sorafenib and EVR. Using seven OS cell lines (including MNNG-HOS and U-2OS) and NOD/SCID xenografts, they revealed that sorafenib monotherapy inhibits mTORC1 (reduced p-S6) but paradoxically activates mTORC2 (elevated p-mTOR Ser2481), explaining limited clinical responses. EVR combination therapy counteracted this resistance via ROS-mediated AMPK activation (suppressing mTORC1) and conformational interference (blocking mTORC2), achieving complete mTOR pathway suppression. In vivo, this dual inhibition reduced tumor volume by 70%, lung metastases by 50%, and microvessel density by 60%. Higuchi et al. (2019) (75) validated this in doxorubicin (DOX)-resistant patient-derived orthotopic xenograft (PDOX) models (nu/nu mice): EVR + sorafenib emerged as the sole regimen inducing significant tumor regression (p < 0.001 vs control and DOX groups), with histopathology revealing extensive necrosis and stromal remodeling. Sorafenib or EVR monotherapy showed moderate efficacy (p < 0.001 vs control), while DOX was ineffective (p = 0.2 vs control). Crucially, neither study observed cumulative toxicity, underscoring the regimen’s superior therapeutic window.
Advances in cell cycle modulation and apoptosis induction further highlight EVR’s combinatorial potential. Oshiro et al. (24) validated the synergy of palbociclib (CDK4/6 inhibitor) with EVR in DOX-resistant PDOX models. In completely DOX-refractory tumors, combination therapy significantly reduced tumor volume (p = 0.018 vs control and DOX groups), with histology showing coagulative necrosis. EVR monotherapy partially suppressed proliferation (p = 0.04 vs control), while palbociclib and DOX failed. Mechanistically, dual blockade of G1/S transition (via CDK4/6 inhibition) and mTOR-mediated metabolic support synergistically disrupted tumor proliferation. Similarly, Garofalo et al. (74) demonstrated that NT157 (IRS-1/2 inhibitor) degrades IRS-1 to block IGF-1R/AKT/mTOR signaling, synergizing with EVR (combination index <1). In MG-63 and U-2OS cells, NT157-induced G2/M arrest created a time-dependent therapeutic window for EVR’s cytostatic effects, offering a novel non-apoptotic strategy for sequential chemotherapy.
Dual inhibition of proteasome and mTOR pathways exhibits unique therapeutic advantages. Nakamura et al. (23) confirmed that EVR combined with bortezomib activates JNK/p38/ERK MAPK apoptotic pathways while suppressing AKT and c-MYC in HT1080 fibrosarcoma and LM8 OS models, yielding significant synergy in vitro (p < 0.05). In vivo, this combination reduced primary tumor volume (p < 0.05) and lung metastases (p < 0.01). Asanuma et al. (76) mechanistically validated this synergy: in MG-63 cells, bortezomib upregulated JNK/p38 phosphorylation to amplify stress signaling, while EVR inhibited mTORC1 survival pathways, elevating caspase-3/8/9 and PARP cleavage. In 143B xenografts, dose-dependent antitumor efficacy was achieved without overt toxicity, highlighting clinical translatability.
Tumor microenvironment (TME) remodeling has emerged as a novel therapeutic frontier. Sapio et al. (77) reported that AdipoRon (synthetic adiponectin receptor agonist) differentially regulates ERK/mTOR signaling in Saos-2 and U2OS cells: monotherapy activates pro-survival ERK1/2 while inhibiting mTORC1/p70S6K. EVR combination abolished ERK-driven survival, inducing a 2.3-fold increase in G0/G1 arrest in U2OS cells. Oshiro et al. (78) validated “vessel normalization” in lung-metastatic PDOX models: EVR + pazopanib reduced vasculature length by 58% (p < 0.05) and hypoxic regions via mTORC1-VEGF suppression and VEGFR blockade. This dual targeting of tumor-autonomous secretion and TME crosstalk provides a mechanistic foundation for controlling metastatic spread in advanced OS.
3.2 Clinical research progress
Building on preclinical validation of EVR’s multifaceted antitumor effects, such as metabolic modulation, cell cycle arrest, and microenvironment remodeling, its therapeutic potential has now advanced into clinical trials for osteosarcoma.
In adults, Grignani et al. (20) conducted a nonrandomized phase II trial evaluating sorafenib plus EVR as salvage therapy for unresectable or recurrent high-grade OS refractory to standard treatments. Among 38 patients, the regimen achieved a 6-month PFS rate of 45% (95% CI 28–61), narrowly missing the prespecified 50% target but demonstrating modest clinical activity. Dose adjustments or interruptions were required in 66% of cases due to grade 3–4 adverse events—most commonly lymphopenia (16%), hypophosphatemia (16%), and hand–foot syndrome (13%)—though no treatment-related deaths occurred. While mTOR pathway-mediated resistance limited efficacy, the findings underscored the regimen’s potential in refractory OS and highlighted the need for dosing refinement or incorporation of additional targeted agents.
In pediatric populations, an earlier pilot study (n = 14) (79) in heavily pretreated refractory OS showed predominantly stable disease, with median PFS and OS of 4.4 and 6 months, respectively, and a manageable toxicity profile characterized mainly by low-grade skin and hematologic events. Building on these findings, a more recent pediatric trial (n = 12) (80) reported partial responses in 33% of patients and 6-/12-month PFS rates of 54% and 36%, further supporting feasibility in younger patients and underscoring the importance of age-specific optimization. Beyond systemic therapy alone, Anderson et al. (81) pioneered a multimodal regimen integrating α-particle therapy (radium-223) with systemic treatments—including one case combining EVR and a PD-1 inhibitor—and radiotherapy, revealing synergistic potential for bone metastasis control via tumor microenvironment modulation and immune activation.
Precision oncology further illustrates EVR’s versatility. Yi et al. (82) described a case of glioblastoma with secondary OS in which exome sequencing-guided therapy—combining EVR, tumor-treating fields (TTFields), pembrolizumab, and temozolomide—achieved exceptional benefit through dual mechanisms: (1) targeting PTEN loss in glioblastoma to suppress mTORC1-driven proliferation and enhance temozolomide chemosensitivity; and (2) exploiting MSH3/ERCC4 mutation-associated DNA repair defects in secondary OS by disrupting mTOR signaling. The regimen synergistically remodeled the tumor microenvironment (e.g., reduced regulatory T-cell infiltration) and amplified antitumor efficacy through complementary actions, although EVR’s precise contribution and dose-response effects warrant further investigation.
4 Challenges and future perspectives
The clinical use of EVR in osteosarcoma is hindered by evolving drug resistance and limited intratumoral delivery. Although preclinical evidence demonstrates broad antitumor activity through mTORC1 inhibition, metabolic reprogramming, and antiangiogenesis, its monotherapy efficacy is often reduced by compensatory mTORC2 activation, IGF-1R/ERK bypass signaling, and poor bone penetration (<20% of plasma levels). The lack of predictive biomarkers limits patient stratification, while its dual immunomodulatory effects—such as Treg suppression versus potential T-cell inhibition—complicate combinations with immune checkpoint inhibitors. Future advances in bone-targeted nanoparticle delivery, biomarker-guided patient selection, dual mTORC1/2 blockade, and fine-tuned immune modulation may enable EVR to evolve from a broad-spectrum inhibitor into a precision therapeutic platform for osteosarcoma.
5 Conclusion
Everolimus, a pivotal mTOR inhibitor, integrates antitumor, osteoprotective, and immunomodulatory actions in osteosarcoma, targeting oncogenic signaling, metabolic reprogramming, angiogenesis, and the bone–immune interface. Clinical evidence supports its synergy with chemotherapy, targeted agents, and TTFields, achieving promising disease control in refractory cases. Overcoming resistance, optimizing delivery, and refining patient selection through molecular profiling remain key priorities. Advances in smart drug platforms, dual mTORC1/2 blockade, and biomarker-driven strategies—validated by multicenter trials—may establish EVR as a precision-based therapeutic cornerstone in OS management.
Author contributions
YoZ: Supervision, Visualization, Writing – original draft, Data curation, Validation, Conceptualization, Methodology, Investigation, Writing – review and editing. YDo: Writing – review and editing, Writing – original draft. YaZ: Writing – review and editing, Writing – original draft. GL: Writing – review and editing, Writing – original draft. GY: Methodology, Validation, Visualization, Software, Writing – review and editing. DZ: Writing – original draft, Writing – review and editing. CD: Software, Writing – original draft, Writing – review and editing, Validation, Methodology, Supervision, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Supported by: the 2024 Medical Quality (Evidence-Based) Management Research Project of the National Institute of Hospital Administration, National Health Commission: “Research on the Analysis of Influencing Factors and Improvement Strategies for Quality Control Indicators in Oncology Under the National Evaluation Background” (Project No. YLZLXZ24G044).
Conflict of interest
Author GY was employed by Chengdu MAST Medical Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jackson, TM, Bittman, M, and Granowetter, L. Pediatric malignant bone tumors: a review and update on current challenges, and emerging drug targets. Curr Probl Pediatr Adolesc Health Care (2016) 46(7):213–28. doi:10.1016/j.cppeds.2016.04.002
2. Kaatsch, P, Strothotte, J, Becker, C, Bielack, S, Dirksen, U, and Blettner, M. Pediatric bone tumors in Germany from 1987 to 2011: incidence rates, time trends and survival. Acta Oncologica (2016) 55(9-10):1145–51. doi:10.1080/0284186x.2016.1195509
3. Xi, Y, Qiao, L, Na, B, Liu, H, Zhang, S, Zheng, R, et al. Primary malignant bone tumors incidence, mortality, and trends in China from 2000 to 2015. Chin Med J (2023) 136(17):2037–43. doi:10.1097/cm9.0000000000002547
4. Meltzer, PS, and Helman, LJ. New Horizons in the treatment of osteosarcoma. N Engl J Med (2021) 385(22):2066–76. doi:10.1056/nejmra2103423
5. Rosen, G, Marcove, RC, Caparros, B, Nirenberg, A, Kosloff, C, and Huvos, AG. Primary osteogenic sarcoma: the rationale for preoperative chemotherapy and delayed surgery. Cancer (1979) 43(6):2163–77. doi:10.1002/1097-0142(197906)43:6<2163::aid-cncr2820430602>3.0.co;2-s
6. Ferrari, S, Mercuri, M, Bacci, G, Bielack, SS, and Jürgens, H. Comment on “prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols”. J Clin Oncol (2002) 20(12):2910–1. doi:10.1200/jco.2002.20.12.2910
7. Bacci, G, Bertoni, F, Longhi, A, Ferrari, S, Forni, C, Biagini, R, et al. Neoadjuvant chemotherapy for high-grade central osteosarcoma of the extremity. Histologic response to preoperative chemotherapy correlates with histologic subtype of the tumor. Cancer (2003) 97(12):3068–75. doi:10.1002/cncr.11456
8. Ji, Z, Shen, J, Lan, Y, Yi, Q, and Liu, H. Targeting signaling pathways in osteosarcoma: mechanisms and clinical studies. MedComm (2020) (2023) 4(4):e308. doi:10.1002/mco2.308
9. Khan, M, Patel, R, Youssef, M, Banerjee, R, Pardiwala, A, and Belen, C. A systemic review of primary malignant long bone tumors in children and adolescents. Acta chirurgiae orthopaedicae et traumatologiae Cechoslovaca (2024) 91(2):77–87. doi:10.55095/achot2024/010
10. Wedel, S, Hudak, L, Seibel, JM, Makarević, J, Juengel, E, Tsaur, I, et al. Impact of combined HDAC and mTOR inhibition on adhesion, migration and invasion of prostate cancer cells. Clin Exp Metastasis (2011) 28(5):479–91. doi:10.1007/s10585-011-9386-8
11. Boffa, DJ, Luan, F, Thomas, D, Yang, H, Sharma, VK, Lagman, M, et al. Rapamycin inhibits the growth and metastatic progression of non-small cell lung cancer. Clin Cancer Res (2004) 10(Pt 1):293–300. doi:10.1158/1078-0432.ccr-0629-3
12. Nishikawa, T, Takaoka, M, Ohara, T, Tomono, Y, Hao, H, Bao, X, et al. Antiproliferative effect of a novel mTOR inhibitor temsirolimus contributes to the prolonged survival of orthotopic esophageal cancer-bearing mice. Cancer Biol and Ther (2013) 14(3):230–6. doi:10.4161/cbt.23294
13. Meissner, MA, McCormick, BZ, Karam, JA, and Wood, CG. Adjuvant therapy for advanced renal cell carcinoma. Expert Rev Anticancer Ther (2018) 18(7):663–71. doi:10.1080/14737140.2018.1469980
14. Hasskarl, J. Everolimus. Recent Results Cancer Res (2018) 211:101–23. doi:10.1007/978-3-319-91442-8_8
15. Houghton, PJ. Everolimus. Clin Cancer Res (2010) 16(5):1368–72. doi:10.1158/1078-0432.ccr-09-1314
16. Motzer, RJ, Escudier, B, Oudard, S, Hutson, TE, Porta, C, Bracarda, S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. The Lancet (2008) 372(9637):449–56. doi:10.1016/s0140-6736(08)61039-9
17. Baselga, J, Campone, M, Piccart, M, Burris, hA, Rugo, HS, Sahmoud, T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med (2012) 366:520–9. doi:10.1056/nejmoa1109653
18. Yao, JC, Shah, MH, Ito, T, Bohas, CL, Wolin, EM, Van Cutsem, E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. New Engl J Med (2011) 364(6):514–23. doi:10.1056/nejmoa1009290
19. Yao, JC, Fazio, N, Singh, S, Buzzoni, R, Carnaghi, C, Wolin, E, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. The Lancet (2016) 387(10022):968–77. doi:10.1016/s0140-6736(15)00817-x
20. Grignani, G, Palmerini, E, Ferraresi, V, D'Ambrosio, L, Bertulli, R, Asaftei, SD, et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol (2015) 16(1):98–107. doi:10.1016/s1470-2045(14)71136-2
21. Pignochino, Y, Dell'Aglio, C, Basiricò, M, Capozzi, F, Soster, M, Marchiò, S, et al. The combination of sorafenib and everolimus abrogates mTORC1 and mTORC2 upregulation in osteosarcoma preclinical models. Clin Cancer Res (2013) 19(8):2117–31. doi:10.1158/1078-0432.CCR-12-2293
22. Moriceau, G, Ory, B, Mitrofan, L, Riganti, C, Blanchard, F, Brion, R, et al. Zoledronic acid potentiates mTOR inhibition and abolishes the resistance of osteosarcoma cells to RAD001 (everolimus): pivotal role of the prenylation process. Cancer Res (2010) 70(24):10329–39. doi:10.1158/0008-5472.can-10-0578
23. Nakamura, K, Asanuma, K, Okamoto, T, Iino, T, Hagi, T, Nakamura, T, et al. Combination of everolimus and bortezomib inhibits the growth and metastasis of bone and soft tissue sarcomas via JNK/p38/ERK MAPK and AKT pathways. Cancers (Basel) (2023) 15(9):2468. doi:10.3390/cancers15092468
24. Oshiro, H, Tome, Y, Miyake, K, Higuchi, T, Sugisawa, N, Kanaya, F, et al. Combination of CDK4/6 and mTOR inhibitors suppressed doxorubicin-resistant osteosarcoma in a patient-derived orthotopic xenograft mouse model: a translatable strategy for recalcitrant disease. Anticancer Res (2021) 41(7):3287–92. doi:10.21873/anticanres.15115
25. Browne, AJ, Kubasch, ML, Göbel, A, Hadji, P, Chen, D, Rauner, M, et al. Concurrent antitumor and bone-protective effects of everolimus in osteotropic breast cancer. Breast Cancer Res (2017) 19(1):92. doi:10.1186/s13058-017-0885-7
26. Gnant, M, Baselga, J, Rugo, HS, Noguchi, S, Burris, HA, Piccart, M, et al. Effect of everolimus on bone marker levels and progressive disease in bone in BOLERO-2. JNCI: J Natl Cancer Inst (2013) 105(9):654–63. doi:10.1093/jnci/djt026
27. Hadji, P, Coleman, R, and Gnant, M. Bone effects of Mammalian target of rapamycin (mTOR) inhibition with everolimus. Crit Rev Oncology/Hematology (2013) 87(2):101–11. doi:10.1016/j.critrevonc.2013.05.015
28. Glantschnig, H, Fisher, JE, Wesolowski, G, Rodan, GA, and Reszka, AA. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ (2003) 10(10):1165–77. doi:10.1038/sj.cdd.4401285
29. Song, S, Zhang, G, Chen, X, Zheng, J, Liu, X, Wang, Y, et al. HIF-1α increases the osteogenic capacity of ADSCs by coupling angiogenesis and osteogenesis via the HIF-1α/VEGF/AKT/mTOR signaling pathway. J Nanobiotechnology (2023) 21(1):257. doi:10.1186/s12951-023-02020-z
30. Tewari, D, Patni, P, Bishayee, A, Sah, AN, and Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: a novel therapeutic strategy. Semin Cancer Biol (2022) 80:1–17. doi:10.1016/j.semcancer.2019.12.008
31. Li, J, Jiang, M, Yu, Z, Xiong, C, Pan, J, Cai, Z, et al. Artemisinin relieves osteoarthritis by activating mitochondrial autophagy through reducing TNFSF11 expression and inhibiting PI3K/AKT/mTOR signaling in cartilage. Cell Mol Biol Lett (2022) 27(1):62. doi:10.1186/s11658-022-00365-1
32. Derwich, A, Sykutera, M, Bromińska, B, Rubiś, B, Ruchała, M, and Sawicka-Gutaj, N. The role of activation of PI3K/AKT/mTOR and RAF/MEK/ERK pathways in aggressive pituitary adenomas-new potential therapeutic Approach-A systematic review. Int J Mol Sci (2023) 24(13):10952. doi:10.3390/ijms241310952
33. Whitman, M, Downes, CP, Keeler, M, Keller, T, and Cantley, L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature (1988) 332(6165):644–6. doi:10.1038/332644a0
34. Wang, JF, Wang, YP, Xie, J, Zhao, ZZ, Gupta, S, Guo, Y, et al. Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood (2021) 138(9):806–10. doi:10.1182/blood.2020009417
35. Shen, Q, Han, Y, Wu, K, He, Y, Jiang, X, Liu, P, et al. MrgprF acts as a tumor suppressor in cutaneous melanoma by restraining PI3K/Akt signaling. Signal Transduction Targeted Ther (2022) 7(1):147. doi:10.1038/s41392-022-00945-9
36. Yamasaki, E, Ali, S, Sanchez Solano, A, Thakore, P, Smith, M, Wang, X, et al. Faulty TRPM4 channels underlie age-dependent cerebral vascular dysfunction in gould syndrome. Proc Natl Acad Sci U S A (2023) 120(5):e2217327120. doi:10.1073/pnas.2217327120
37. Jiang, Q, Zhang, X, Dai, X, Han, S, Wu, X, Wang, L, et al. S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions. Nat Commun (2022) 13(1):1548. doi:10.1038/s41467-022-28910-8
38. Levina, A, Fleming, KD, Burke, JE, and Leonard, TA. Activation of the essential kinase PDK1 by phosphoinositide-driven trans-autophosphorylation. Nat Commun (2022) 13(1):1874. doi:10.1038/s41467-022-29368-4
39. Leroux, AE, and Biondi, RM. The choreography of protein kinase PDK1 and its diverse substrate dance partners. Biochem J (2023) 480(19):1503–32. doi:10.1042/bcj20220396
40. Yu, L, Wei, J, and Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol (2022) 85:69–94. doi:10.1016/j.semcancer.2021.06.019
41. Huang, L, Zhang, XO, Rozen, EJ, Sun, X, Sallis, B, Verdejo-Torres, O, et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat Commun (2022) 13(1):3955. doi:10.1038/s41467-022-31645-1
42. Xiong, Y, Ju, L, Yuan, L, Chen, L, Wang, G, Xu, H, et al. KNSTRN promotes tumorigenesis and gemcitabine resistance by activating AKT in bladder cancer. Oncogene (2021) 40(9):1595–608. doi:10.1038/s41388-020-01634-z
43. Xia, X, Li, X, Li, F, Wu, X, Zhang, M, Zhou, H, et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol Cancer (2019) 18(1):131. doi:10.1186/s12943-019-1056-5
44. Goldbraikh, D, Neufeld, D, Eid-Mutlak, Y, Lasry, I, Gilda, JE, Parnis, A, et al. USP1 deubiquitinates Akt to inhibit PI3K-Akt-FoxO signaling in muscle during prolonged starvation. EMBO Rep (2020) 21(4):e48791. doi:10.15252/embr.201948791
45. García Coronado, PL, Franco Molina, MA, Zárate Triviño, DG, Hernández Martínez, SP, Castro Valenzuela, BE, Zapata Benavides, P, et al. Exosomes isolated from IMMUNEPOTENT CRP, a hemoderivative, to accelerate diabetic wound healing. Front Bioeng Biotechnol (2024) 12:1356028. doi:10.3389/fbioe.2024.1356028
46. Simcox, J, and Lamming, DW. The central moTOR of metabolism. Develop Cell (2022) 57(6):691–706. doi:10.1016/j.devcel.2022.02.024
47. Szwed, A, Kim, E, and Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev (2021) 101(3):1371–426. doi:10.1152/physrev.00026.2020
48. Prentzell, MT, Rehbein, U, Cadena Sandoval, M, De Meulemeester, AS, Baumeister, R, Brohée, L, et al. G3BPs tether the TSC complex to lysosomes and suppress mTORC1 signaling. Cell (2021) 184(3):655–74.e27. doi:10.1016/j.cell.2020.12.024
49. Fitzian, K, Brückner, A, Brohée, L, Zech, R, Antoni, C, Kiontke, S, et al. TSC1 binding to lysosomal PIPs is required for TSC complex translocation and mTORC1 regulation. Mol Cell (2021) 81(13):2705–21.e8. doi:10.1016/j.molcel.2021.04.019
50. Hosios, AM, Wilkinson, ME, McNamara, MC, Kalafut, KC, Torrence, ME, Asara, JM, et al. mTORC1 regulates a lysosome-dependent adaptive shift in intracellular lipid species. Nat Metab (2022) 4(12):1792–811. doi:10.1038/s42255-022-00706-6
51. Hua, H, Kong, Q, Zhang, H, Wang, J, Luo, T, and Jiang, Y. Targeting mTOR for cancer therapy. J Hematol Oncol (2019) 12(1):71. doi:10.1186/s13045-019-0754-1
52. Wang, W, Tan, J, Liu, X, Guo, W, Li, M, Liu, X, et al. Cytoplasmic endonuclease G promotes nonalcoholic fatty liver disease via mTORC2-AKT-ACLY and endoplasmic reticulum stress. Nat Commun (2023) 14(1):6201. doi:10.1038/s41467-023-41757-x
53. Duan, S, Wang, C, Xu, X, Zhang, X, Su, G, Li, Y, et al. Peripheral serum exosomes isolated from patients with acute myocardial infarction promote endothelial cell angiogenesis via the miR-126-3p/TSC1/mTORC1/HIF-1α pathway. Int J Nanomedicine (2022) 17:1577–92. doi:10.2147/ijn.s338937
54. Steck, PA, Pershouse, MA, Jasser, SA, Yung, WK, Lin, H, Ligon, AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet (1997) 15(4):356–62. doi:10.1038/ng0497-356
55. Sarbassov, DD, Ali, SM, and Sabatini, DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol (2005) 17(6):596–603. doi:10.1016/j.ceb.2005.09.009
56. André, F, Ciruelos, E, Rubovszky, G, Campone, M, Loibl, S, Rugo, HS, et al. Alpelisib for PIK3CA-Mutated, hormone receptor-positive advanced breast cancer. N Engl J Med (2019) 380(20):1929–40. doi:10.1056/nejmoa1813904
57. Zhao, J, Chen, F, Zhou, Q, Pan, W, Wang, X, Xu, J, et al. Aberrant expression of microRNA-99a and its target gene mTOR associated with malignant progression and poor prognosis in patients with osteosarcoma. OncoTargets Ther (2016) 9:1589–97. doi:10.2147/ott.s102421
58. Zhang, D, Liu, H, Wang, W, Xu, G, Yin, C, and Wang, S. STEAP2 promotes osteosarcoma progression by inducing epithelial-mesenchymal transition via the PI3K/AKT/mTOR signaling pathway and is regulated by EFEMP2. Cancer Biol and Ther (2022) 23(1):1–16. doi:10.1080/15384047.2022.2136465
59. Ma, BL, Shan, MH, Sun, G, Ren, GH, Dong, C, Yao, X, et al. Immunohistochemical analysis of phosphorylated mammalian target of rapamycin and its downstream signaling components in invasive breast cancer. Mol Med Rep (2015) 12(4):5246–54. doi:10.3892/mmr.2015.4037
60. Ni, S, Li, J, Qiu, S, Xie, Y, Gong, K, and Duan, Y. KIF21B expression in osteosarcoma and its regulatory effect on osteosarcoma cell proliferation and apoptosis through the PI3K/AKT pathway. Front Oncol (2021) 10:606765. doi:10.3389/fonc.2020.606765
61. Keremu, A, Maimaiti, X, Aimaiti, A, Yushan, M, Alike, Y, Yilihamu, Y, et al. NRSN2 promotes osteosarcoma cell proliferation and growth through PI3K/Akt/MTOR and Wnt/β-catenin signaling. Am J Cancer Res (2017) 7(3):565–73.
62. Luo, X, Zhang, J, Guo, C, Jiang, N, Zhang, F, Jiao, Q, et al. Solute carrier family 35 member A2 regulates mitophagy through the PI3K/AKT/mTOR axis, promoting the proliferation, migration, and invasion of osteosarcoma cells. Gene (2024) 898:148110. doi:10.1016/j.gene.2023.148110
63. Samuels, Y, Wang, Z, Bardelli, A, Silliman, N, Ptak, J, Szabo, S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science (2004) 304(5670):554. doi:10.1126/science.1096502
64. Ligresti, G, Militello, L, Steelman, LS, Cavallaro, A, Basile, F, Nicoletti, F, et al. PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle (2009) 8(9):1352–8. doi:10.4161/cc.8.9.8255
65. Li, X, Huang, Q, Wang, S, Huang, Z, Yu, F, and Lin, J. HER4 promotes the growth and metastasis of osteosarcoma via the PI3K/AKT pathway. Acta Biochim Biophys Sinica (2020) 52(4):345–62. doi:10.1093/abbs/gmaa004
66. Yu, C, Zhang, B, Li, Y-L, and Yu, X-R. SIX1 reduces the expression of PTEN via activating PI3K/AKT signal to promote cell proliferation and tumorigenesis in osteosarcoma. Biomed and Pharmacother (2018) 105:10–7. doi:10.1016/j.biopha.2018.04.028
67. Jiang, B-H, and Liu, L-Z. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets (2008) 8(1):19–26. doi:10.2174/156800908783497122
68. Long, XH, Zhong, ZH, Peng, AF, Zhu, LB, Wang, H, Zhang, GM, et al. LY294002 suppresses the malignant phenotype and sensitizes osteosarcoma cells to pirarubicin chemotherapy. Mol Med Rep (2014) 10(6):2967–72. doi:10.3892/mmr.2014.2617
69. Zhang, L, Zhao, G, Ji, S, Yuan, Q, and Zhou, H. Downregulated long non-coding RNA MSC-AS1 inhibits osteosarcoma progression and increases sensitivity to cisplatin by binding to MicroRNA-142. Med Sci Monit (2020) 26:e921594. doi:10.12659/msm.921594
70. Chen, Y, Liu, R, Wang, W, Wang, C, Zhang, N, Shao, X, et al. Advances in targeted therapy for osteosarcoma based on molecular classification. Pharmacol Res (2021) 169:105684. doi:10.1016/j.phrs.2021.105684
71. Huang, H, Yan, J, Xu, X, Feng, Y, Liu, H, Liu, J, et al. Everolimus inhibits hepatoblastoma by inducing autophagy-dependent ferroptosis. Drug Development Res (2024) 85(1):e22140. doi:10.1002/ddr.22140
72. Mercalli, A, Calavita, I, Dugnani, E, Citro, A, Cantarelli, E, Nano, R, et al. Rapamycin unbalances the polarization of human macrophages to M 1. Immunology (2013) 140(2):179–90. doi:10.1111/imm.12126
73. Iwasaki, K, Kitahata, N, Miwa, Y, Uchida, K, Matsuoka, Y, Horimi, K, et al. Suppressive effect of everolimus on IL-2, IL-10, IL-21, and IFNγ levels: implications for the successful minimization of calcineurin inhibitor use in transplantation. Ther Drug Monit (2019) 41(3):371–5. doi:10.1097/ftd.0000000000000630
74. Garofalo, C, Capristo, M, Mancarella, C, Reunevi, H, Picci, P, and Scotlandi, K. Preclinical effectiveness of selective inhibitor of IRS-1/2 NT157 in osteosarcoma cell lines. Front Endocrinol (Lausanne) (2015) 6:74. doi:10.3389/fendo.2015.00074
75. Higuchi, T, Sugisawa, N, Miyake, K, Oshiro, H, Yamamoto, N, Hayashi, K, et al. Combination treatment with sorafenib and everolimus regresses a doxorubicin-resistant osteosarcoma in a PDOX mouse model. Anticancer Res (2019) 39(9):4781–6. doi:10.21873/anticanres.13662
76. Asanuma, K, Nakamura, T, Nakamura, K, Hagi, T, Okamoto, T, Kita, K, et al. Compound library screening for synergistic drug combinations: mTOR inhibitor and proteasome inhibitor effective against osteosarcoma cells. Anticancer Res (2022) 42(9):4319–28. doi:10.21873/anticanres.15932
77. Sapio, L, Nigro, E, Ragone, A, Salzillo, A, Illiano, M, Spina, A, et al. AdipoRon affects cell cycle progression and inhibits proliferation in human osteosarcoma cells. J Oncol (2020) 2020:1–12. doi:10.1155/2020/7262479
78. Oshiro, H, Tome, Y, Miyake, K, Higuchi, T, Sugisawa, N, Kanaya, F, et al. An mTOR and VEGFR inhibitor combination arrests a doxorubicin resistant lung metastatic osteosarcoma in a PDOX mouse model. Sci Rep (2021) 11(1):8583. doi:10.1038/s41598-021-87553-9
79. Fedenko, AA, Senzhapova, E, Aliev, M, Dzampaev, A, and Bokhyan, B. Everolimus/sorafenib combination in the treatment of refractory pediatric osteosarcomas: single center experience. J Clin Oncol (2016) 34:e22501. doi:10.1200/jco.2016.34.15_suppl.e22501
80. Asamedinov, N, Abdikarimov, K, Rakhimberdiyevich, DR, Sultonov, B, Nurjabov, A, and Kamalovich, N. 125P possibility of using the combination everolimus/sorafenib combination of medications in the treatment of children and adolescents with refractory osteosarcoma and ewing sarcoma. ESMO Open (2025) 10:104438. doi:10.1016/j.esmoop.2025.104438
81. Anderson, PM, Scott, J, Parsai, S, Zahler, S, Worley, S, Shrikanthan, S, et al. 223-Radium for metastatic osteosarcoma: combination therapy with other agents and external beam radiotherapy. ESMO Open (2020) 5(2):e000635. doi:10.1136/esmoopen-2019-000635
82. Yi, GZ, Zhu, TC, Que, TS, Li, ZY, and Huang, GL. Individualized combination therapies based on whole-exome sequencing displayed significant clinical benefits in a glioblastoma patient with secondary osteosarcoma: case report and genetic characterization. BMC Neurol (2022) 22(1):390. doi:10.1186/s12883-022-02920-x
Keywords: mTOR inhibitor, osteosarcoma, metabolic reprogramming, tumor microenvironment, precision oncology
Citation: Zhang Y, Dong Y, Zhang Y, Liang G, Yu G, Zhang D and Dai C (2025) The role of everolimus in malignant bone tumor therapy: Molecular mechanisms, preclinical evidence, and advances in clinical applications. Oncol. Rev. 19:1630239. doi: 10.3389/or.2025.1630239
Received: 17 May 2025; Accepted: 18 August 2025;
Published: 04 September 2025.
Edited by:
Mauro Cives, University of Bari Aldo Moro, ItalyReviewed by:
Valeria Simone, Ospedale Vito Fazzi, ItalyAndrea Giglio, University of Bari Aldo Moro, Italy
Copyright © 2025 Zhang, Dong, Zhang, Liang, Yu, Zhang and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuanqiang Dai, ZGFpcXExNTlzY0AxNjMuY29t