 Amol Tatode1
Amol Tatode1 Anis Ahmad Chaudhary2
Anis Ahmad Chaudhary2 Mohammad Qutub1*
Mohammad Qutub1* Rashmi Trivedi3*
Rashmi Trivedi3* Milind Umekar1
Milind Umekar1 Mohamed A. M. Ali2
Mohamed A. M. Ali2 Tanvi Premchandani1
Tanvi Premchandani1- 1Division of Onco-Therapeutics, Department of Pharmaceutics, Smt. Kishoritai Bhoyar College of Pharmacy, Kamptee, Nagpur, Maharashtra, India
- 2Department of Biology, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh, Saudi Arabia
- 3Department of Quality Assurance, Smt. Kishoritai Bhoyar College of Pharmacy, Kamptee, Nagpur, Maharashtra, India
Colorectal cancer (CRC) progresses through defined stages, from localized carcinoma in situ (Stage 0) to metastatic disease (Stage IV), with treatment strategies evolving from surgery in early stages to systemic therapies in advanced stages. Advances in biomarkers and genomic profiling have enabled personalized approaches, enhancing precision medicine. Nitric oxide (NO) plays a multifaceted role in CRC, acting as both a promoter and an inhibitor of cancer progression depending on its concentration, timing, and cellular context. At low concentrations, NO promotes angiogenesis, enabling tumor growth and metastasis. Conversely, high concentrations can exert anti-tumor effects, including the induction of cell death. Notably, its role in ferroptosis is biphasic: while high, exogenously delivered concentrations of NO can induce this iron-dependent cell death, lower, endogenously regulated levels can be protective by terminating lipid peroxidation. NO influences CRC by modulating the tumor microenvironment, mechanostress responses during metastasis, and signaling through extracellular vesicles (EVs), thereby aiding immune evasion. It also reprograms CRC cell metabolism, enhancing glucose utilization and mitochondrial activity to support growth in hypoxic conditions. The three nitric oxide synthases (NOS)—inducible NOS (iNOS), endothelial NOS (eNOS), and neuronal NOS (nNOS)—interact with hydrogen sulfide (H2S) to regulate oxidative stress and tumor growth. Targeting NO-related processes, such as ferroptosis, metabolic adaptations, and immune modulation, offers promising therapeutic advances to improve CRC treatment outcomes. This review highlights the dual role of NO in CRC, with particular focus on its novel mechanisms in ferroptosis, metabolism, immune modulation, and tumor–microenvironment interactions.
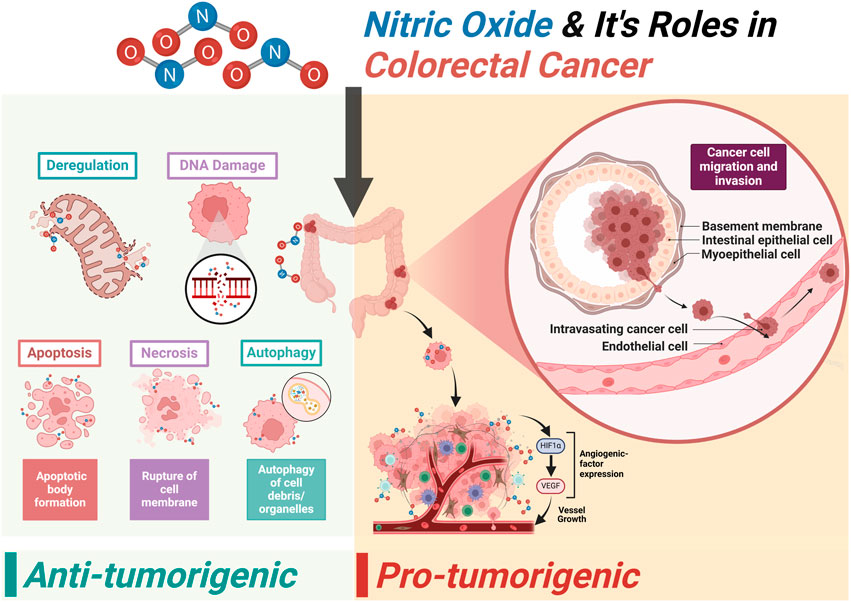
Highlights
• Early screening reduces CRC mortality with accessible healthcare.
• Molecular oncology reveals CRC’s genetic and environmental complexities.
• NO modulates metastasis via mechanical force responses.
• Immune-cell-derived NO inhibits T-cell activity, enabling evasion.
• NO promotes angiogenesis, supporting tumor growth and survival.
• NO impacts gene expression, affecting apoptosis and DNA repair.
• NO–ROS interactions enhance CRC survival and chemoresistance.
1 Introduction
Colorectal cancer (CRC) is one of the most prevalent cancers globally (1). It originates in the colon or rectum, primarily from adenomatous polyps that may develop into malignancies over time (2, 3). Epidemiological data from 2023 reveal a shift in age-related trends, with CRC incidences increasing among younger populations (4, 5). Despite significant advancements in treatment, disparities in healthcare access continue to pose challenges in the management of CRC (6, 7). CRC progresses through well-defined stages, each reflecting the depth of tumor invasion, lymph node involvement, and distant metastasis (8, 9). The earliest stage, Stage 0 represents carcinoma in situ, with abnormal cells confined to the inner lining of the colon or rectum and minimal risk if treated early (10). In Stage I, the cancer invades the muscular layer but remains localized, without lymph node or distant spread (11). Stage II involves tumor extension through the colon wall, sometimes reaching adjacent organs, but lymph nodes remain unaffected, often requiring surgery and adjuvant therapies (12, 13). Stage III marks lymph node involvement, with systemic interventions such as chemotherapy becoming essential (14, 15). Stage IV is metastatic, with cancer spreading to distant organs such as the liver or lungs, necessitating complex therapies and palliative care (16, 17). Accurate staging is critical for personalized treatment planning (18, 19).
CRC remains a leading cause of cancer-related mortality worldwide, emphasizing the urgent need for effective treatment strategies (20–22). Advances in molecular oncology have revealed that a combination of genetic, environmental, and biochemical factors influences CRC progression. Among these, nitric oxide (NO) has gained attention due to its complex role in CRC development (23, 24). NO, a reactive molecule produced by nitric oxide synthase (NOS) enzymes, can either promote or inhibit cancer progression depending on its concentration, cellular location, and biochemical context (25, 26). Crucially, the biological effects of NO in CRC are not uniform; they are determined by a delicate balance of its concentration, cellular localization, tumor stage, and the surrounding redox environment. This review will dissect this dualism: at low, physiological concentrations, NO often exhibits pro-tumorigenic properties by promoting angiogenesis, supporting immune evasion, and preventing certain forms of cell death. Conversely, high, supraphysiological concentrations, often achieved through therapeutic delivery, typically exert anti-tumorigenic effects by inducing DNA damage, apoptosis, and ferroptosis. A central theme of this review is to delineate the context-dependent mechanisms that determine the functional role of NO. This dual nature of NO makes it both a challenging and promising target in CRC research, with potential applications across disease stages, from early tumor initiation to immune evasion and metastasis (27, 28). At low concentrations, NO can support tumor growth by facilitating angiogenesis, the process by which tumors form new blood vessels to sustain their nutrient and oxygen demands. This is particularly important for CRC as rapidly expanding tumors require an enhanced blood supply (29–31). NO contributes to this process by promoting the production of vascular endothelial growth factor (VEGF) and activating signaling pathways such as cGMP, both of which play roles in vascular development (32–34). In certain contexts, NO induces ferroptosis, although it can also inhibit this process depending on concentration and cellular state. In support of its ferroptosis-inducing role, recent evidence suggests that NO-releasing drugs, such as NCX4040, can directly trigger ferroptotic cell death in CRC cells. Specifically, NCX4040 treatment led to increased reactive oxygen species (ROS), lipid peroxidation, and activation of ferroptosis-related genes such as CHAC1, GPX4, and NOX4. These effects were reversed by ferrostatin-1, a specific inhibitor of ferroptosis, confirming the ferroptosis-dependent nature of NO-induced cell death in CRC models (35). Additionally, the development of self-catalyzing nitric oxide nanocomplexes has revealed another mechanism through which NO can promote ferroptosis (36). Moreover, combinations of NO with traditional ferroptosis inducers such as erastin have demonstrated synergistic effects, further supporting the pro-ferroptotic potential of NO-based therapies in CRC (35). Targeting this vulnerability could be valuable in CRC treatment by exploiting cancer cells’ sensitivity to oxidative damage. Thus, NO’s dual nature, with its tumor-promoting effects at low levels and cytotoxic effects at higher levels, presents both challenges and opportunities for CRC therapy (37–39). The therapeutic potential of targeting NO pathways in CRC has led to a variety of novel approaches (40). NOS inhibitors have shown promise in preclinical models by reducing CRC cell proliferation, angiogenesis, and immune suppression (41–43). Advances in drug delivery, such as NO-releasing nanoparticles, offer enhanced specificity by delivering therapeutic levels of NO directly to tumor sites, thus maximizing anti-tumor efficacy while minimizing off-target effects. These approaches underscore NO modulation as a potential strategy for addressing CRC’s therapeutic challenges (44–49).
This review examines NO’s role in CRC progression, highlighting its effects on molecular pathways, immune modulation, and metabolism. It explores NO’s influence on critical signaling pathways (Wnt/β-catenin, PI3K/AKT/mTOR, and NF-κB), immune modulation through pro-tumorigenic immune cell polarization, and metabolic reprogramming to sustain tumor growth and therapy resistance. The review also evaluates therapeutic strategies targeting NO, such as nitric oxide synthase inhibitors and NO delivery systems, while addressing challenges such as site-specific modulation and off-target effects.
2 Nitric oxide signaling networks in colorectal cancer
In CRC, NO signaling has been implicated in multiple aspects of tumorigenesis, including tumor initiation, progression, metastasis, immune evasion, and therapeutic resistance (50, 51). The molecular and cellular mechanisms through which NO exerts its effects on CRC involve complex interactions among signaling pathways, redox biology, immune responses, and metabolic regulation (Figure 1; (52, 53)).
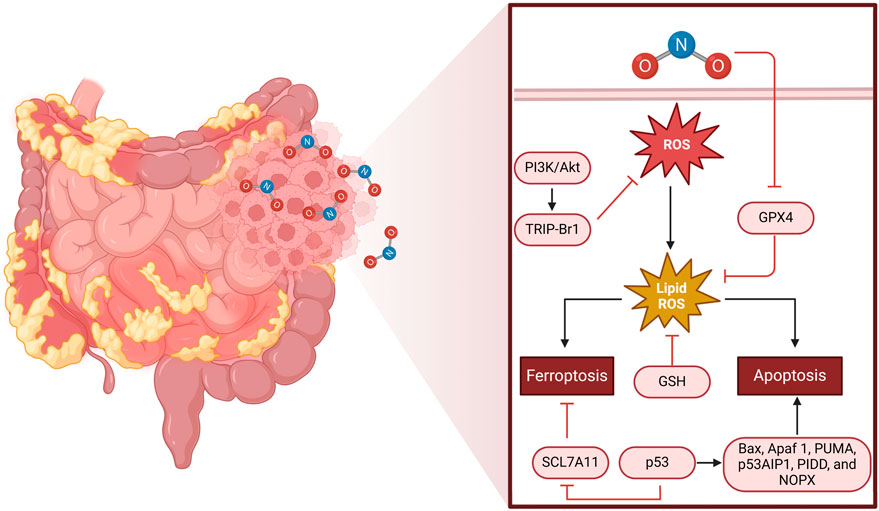
Figure 1. Mechanism of ROS-induced cell death in colorectal cancer. The left half of the figure depicts a portion of the human intestine, with emphasis on CRC tissue. The magnified area shows the molecular mechanisms of oxidative stress-induced cell death. NO triggers the production of ROS, which in turn causes lipid peroxidation (lipid ROS). This process can trigger ferroptosis through SCL7A11 inhibition and glutathione (GSH) loss or apoptosis through p53 activation and downstream targets (Bax, Apaf1, PUMA, p53AIP1, PIDD, and NOX). The PI3K/AKT pathway and TRIP-Br1 regulate ROS production, whereas GPX4 is an antioxidant protective mechanism against lipid peroxidation. The equilibrium among these pathways dictates CRC cell fate, either supporting survival or triggering programmed cell death.
2.1 NO synthase isoforms and their roles in CRC
Each isoform plays a unique role in the pathophysiology of CRC, as mentioned in Table 1. Neuronal NOS (nNOS/NOS1) has emerging evidence suggesting a role in neuronal-like signaling within the tumor microenvironment (54). Expression of nNOS has been observed in certain CRC subtypes, where it may influence tumor cell migration and invasion (55, 56). Wang et al. (57) demonstrated that mtNOS1 suppresses cisplatin-induced mitochondrial superoxide accumulation and apoptosis in colon cancer cells by enhancing SIRT3 activity, which stabilizes SOD2—a key antioxidant enzyme. The mtNOS1–SIRT3–SOD2 axis underscores a mechanism through which cancer cells evade oxidative stress-driven death. Notably, geldanamycin, an Hsp90 inhibitor, blocked NOS1 mitochondrial translocation, reversing its anti-apoptotic effects and restoring chemosensitivity. These findings position mtNOS1 as an actionable target in overcoming therapy resistance. Expanding on this, Qiu et al. (58) revealed a novel link between hypercholesterolemia and NOS1-driven CRC progression. Their work showed that oxidized LDL (oxLDL) activates oxidant stress and hypoxia signaling, transcriptionally upregulating NOS1 in CRC cells. This pathway, mediated by the LOX-1 receptor, connects elevated cholesterol levels to enhanced NO production, fostering a tumor-permissive microenvironment. Crucially, pharmacological inhibition of NOS1 with Nω-propyl-L-arginine selectively curbed tumor growth in hypercholesterolemic models, suggesting a therapeutic strategy with reduced off-target toxicity. Together, these studies illuminate NOS1 as a dual regulator of chemoresistance and cholesterol-mediated tumorigenesis. The convergence of mitochondrial redox regulation (via SIRT3–SOD2) and hypercholesterolemia-induced hypoxia signaling on NOS1 highlights its centrality in CRC pathophysiology. Targeting NOS1 either through Hsp90 inhibitors to block its mitochondrial localization or via specific inhibitors such as Nω-propyl-L-arginine may offer dual benefits: mitigating therapy resistance and addressing metabolic risk factors in CRC (57, 58). Inducible NOS (iNOS/NOS2) is upregulated in response to pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interferon-gamma (IFN-γ). It is highly expressed in the inflammatory microenvironment of CRC and is associated with increased NO production, which promotes chronic inflammation, angiogenesis, and immune evasion (40, 59). NO generated by iNOS can induce DNA damage through the formation of peroxynitrite (ONOO−), a reactive nitrogen species that can cause mutagenesis and genomic instability, thus driving tumor progression (60).
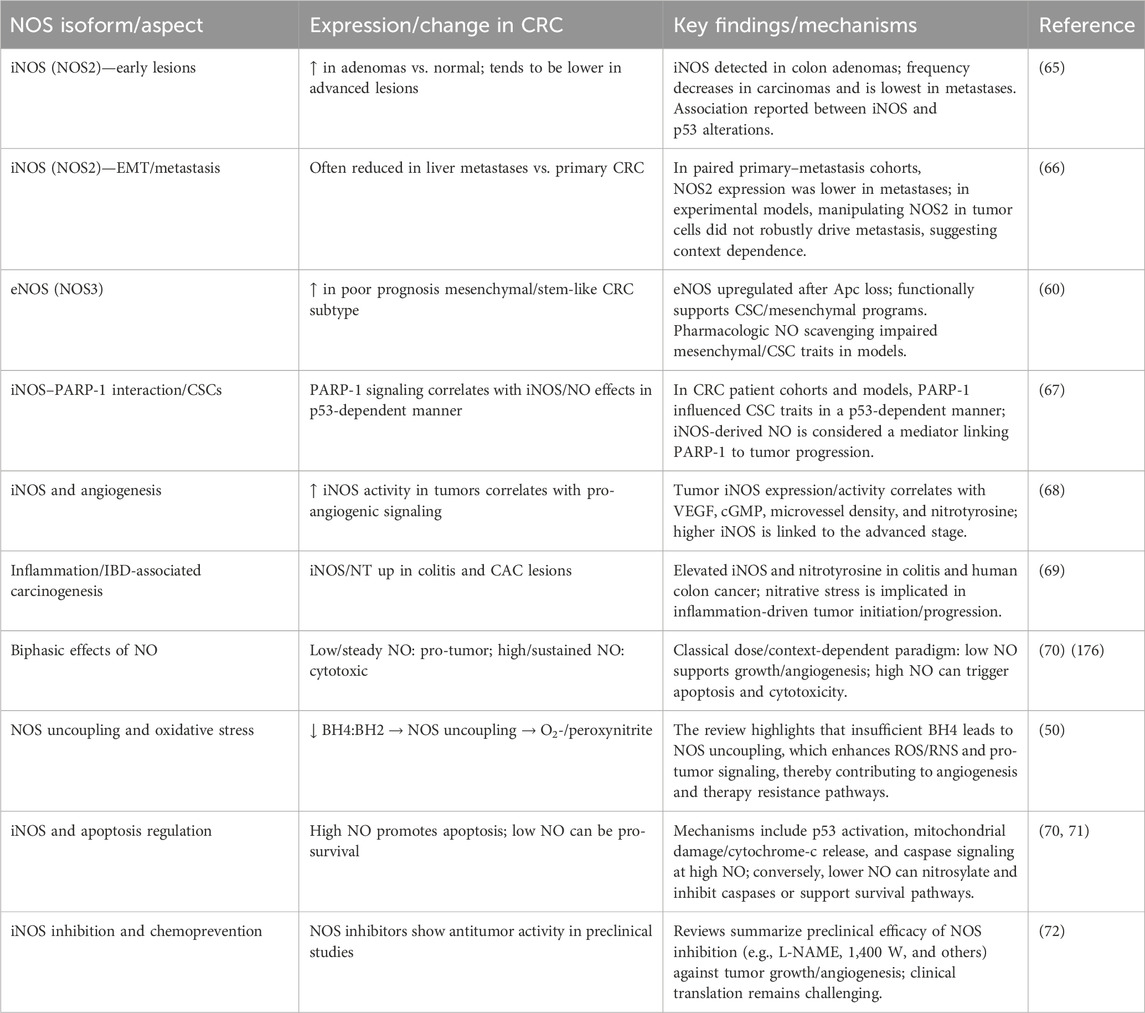
Table 1. Roles of NOS isoforms in CRC: expression patterns, mechanisms, and therapeutic implications.
Endothelial NOS (eNOS/NOS3) is primarily expressed in endothelial cells, where it regulates vascular tone and angiogenesis (61). In CRC, eNOS expression is often elevated in tumor-associated endothelial cells, contributing to the formation of an abnormal vasculature that supports tumor growth. However, eNOS can also have tumor-suppressive effects, particularly through the generation of low levels of NO, which can promote apoptosis and enhance immune surveillance (62). Garza Treviño et al. (63) and Lu et al. (64) indicated that eNOS is upregulated in human mesenchymal CRC tumors and may represent an active stem-cell regulatory point in cancer and a possible target for therapy against aggressive human tumors (63, 64).
2.2 NO and immune modulation in CRC
Nitric oxide functions as a double-edged mediator in CRC. Depending on its source, concentration, and the surrounding environment, it can either promote or inhibit tumor growth. Its main function involves interacting with key immune cells such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and cancer-killing cytotoxic T lymphocytes (CTLs).
A major strategy to combat CRC involves targeting arginine metabolism to reactivate the immune system. For example, engineered microparticles can be used to inhibit the enzyme arginase while activating NOS. This cleverly shifts tumor-friendly M2-like TAMs into pro-inflammatory, cancer-fighting M1 types. This switch enhances endogenous NO production, which synergizes with photodynamic therapy to create a hostile tumor environment, leading to significant regression in resistant CRC models (73). Similarly, low doses of the PARP inhibitor olaparib can reduce the immunosuppressive activity of MDSCs by downregulating arginase-1 (ARG1) and iNOS, thereby restoring the activity of CTLs. When combined with anti-PD-1 therapy, this approach completely eradicated microsatellite instability-high (MSI-high) tumors and significantly reduced the size of microsatellite stable (MSS) tumors (74). These findings underscore the therapeutic potential of modulating arginine metabolism to enhance the efficacy of immune checkpoint therapies.
The tumor microenvironment (TME) often functions as a barrier that restricts immune cells infiltration. However, these barriers can be overcome. Nanoemulsions delivering melittin and NO donors can reverse the activation of cancer-associated fibroblasts (CAFs), reduce the dense collagen that shields the tumor, and normalize blood vessels, thereby facilitating CTL infiltration. When combined with anti-CTLA-4 therapy, this remodeling approach effectively suppressed tumor growth in CRC models rich in CAFs (75). Another approach involves using all-trans retinoic acid (AtRA) to suppress signaling pathways such as TLR4/NF-κB, which reduces the expression of iNOS and TNF-α in colitis-associated CRC (76). These strategies highlight the importance of targeting the tumor’s supportive structures to overcome immune exclusion.
Natural phytochemicals also show great promise. Phloretin, for example, has powerful anti-inflammatory effects, reducing NO and ROS levels in co-cultures of cancer and immune cells. It works by inhibiting NF-κB, which helps protect the gut lining and reduces inflammatory signals (77). Extracts from organisms such as Micractinium sp. can also suppress key inflammatory enzymes such as COX-2 and iNOS in macrophages while halting the cancer cell cycle (78). Combining different therapies is also incredibly effective. Angiotensin II receptor blockers (ARBs) can disrupt MDSC function, which boosts anti-PD-L1 activity and substantially increases the number of tumor-specific CD8+ T-cells (79). In another study, the combination of cyclophosphamide with Toll-like receptor agonists (TLRAs) eliminated MDSCs and activated tumoricidal myeloid cells, leading to complete tumor regression in an NO-dependent manner (80). Even in clinical settings, combining cetuximab with chemotherapy reduces plasma iNOS and NO levels in patients with metastatic CRC, which correlates with better T-cell responses (81).
Although these preclinical studies are promising, translating them into clinical practice requires caution. It is important to account for different tumor subtypes and determine the appropriate dosing to balance the beneficial and detrimental effects of NO as excessive NO can promote DNA damage and tumor growth. Future research needs to focus on finding biomarkers, such as ARG1/iNOS expression or TAM polarization status, to predict which patients are most likely to benefit. The ultimate goal is to develop personalized combinations of NO modulators and checkpoint inhibitors. A deep and nuanced understanding of the context-dependent roles of NO is essential to fully unlock its therapeutic potential and advance treatment for CRC.
2.3 NO-mediated ferroptosis in CRC
The relationship between NO and ferroptosis in CRC represents a critical and complex paradox. Initially regarded as a straightforward pro-ferroptotic agent due to its role in generating reactive nitrogen species (RNS), emerging evidence reveals a potent, context-dependent anti-ferroptotic function. Understanding this duality is essential as endogenous NO within the tumor microenvironment may protect cancer cells from ferroptotic death, while exogenous, high-dose NO delivery represents a promising therapeutic strategy to forcibly induce it.
At high or bolus concentrations, typically achieved with therapeutic NO-donors, NO strongly promotes ferroptosis through several interconnected mechanisms. The primary pathway involves its rapid reaction with superoxide radicals (O2⋅−) to form peroxynitrite (ONOO−), a highly potent and damaging oxidant. This potent oxidant induces ferroptosis by depleting glutathione (GSH), the cell’s principal soluble antioxidant, thereby compromising the primary defense against lipid peroxides. This effect is further compounded by the inactivation of glutathione peroxidase 4 (GPX4) as ONOO− can modify the selenocysteine residue within its active site, thereby disabling the only enzyme capable of directly reducing complex lipid hydroperoxides in biological membranes. The resulting accumulation of lipid ROS, coupled with the ability of ONOO− to directly initiate lipid peroxidation, creates a vicious cycle of membrane damage. Furthermore, NO can modulate iron homeostasis by promoting the release of iron from ferritin, which increases the labile iron pool (Fe2+) available to catalyze the Fenton reaction and amplify lipid peroxidation. This pro-ferroptotic role of NO provides the rationale for using NO-releasing drugs to induce ferroptotic cell death in CRC (Tables 2, 3).
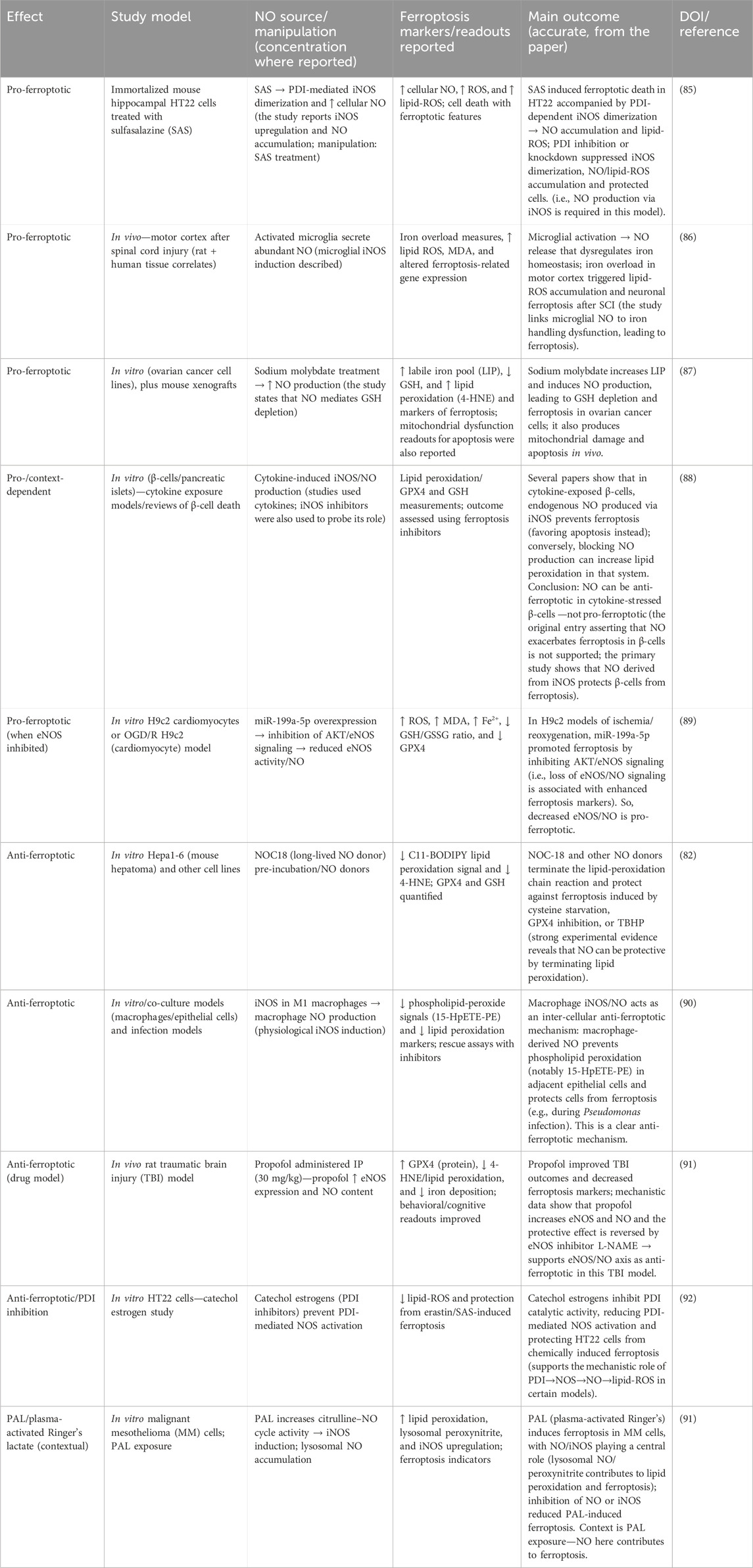
Table 2. Nitric oxide-mediated modulation of ferroptosis: pro- and anti-ferroptotic mechanisms across various study models.
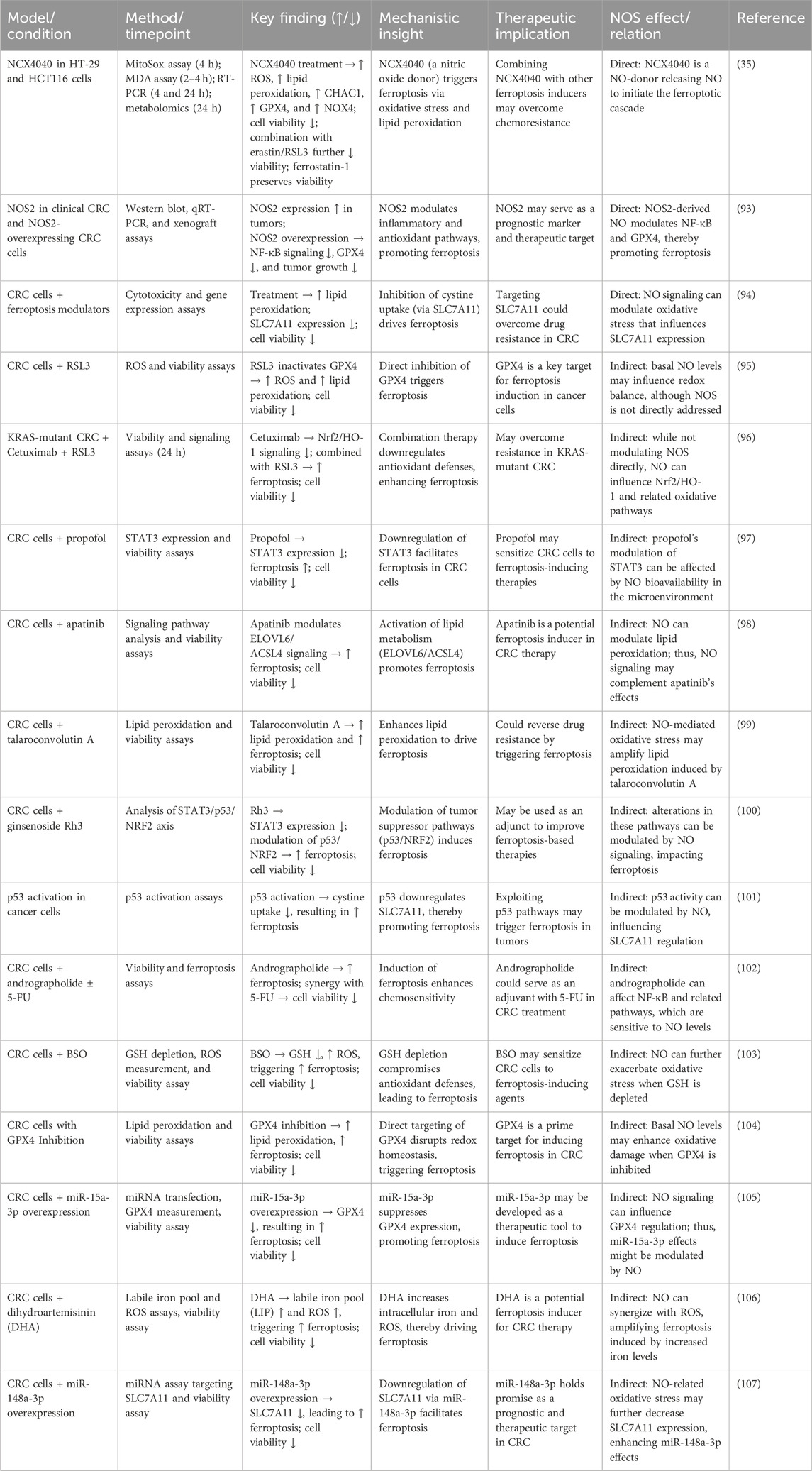
Table 3. Ferroptosis in colorectal cancer and the role of nitric oxide.
Conversely, at lower, physiologically relevant concentrations, often produced endogenously by NOS enzymes, NO can act as a powerful inhibitor of ferroptosis. This protective function is primarily attributed to its intrinsic properties as a radical species. As demonstrated compellingly by Homma et al. (82), NO can directly react with and terminate lipid peroxyl radicals (LOO⋅), effectively acting as a chain-breaking antioxidant that inhibits the propagation of lipid peroxidation and protects cells from ferroptosis induced by GPX4 inhibition or cysteine starvation. Beyond this direct chemical defense, NO also functions as a critical signaling molecule, modulating protein function through regulatory S-nitrosylation to actively suppress the ferroptotic cascade. For instance, NOS1-mediated S-nitrosylation of PTEN leads to its degradation and subsequent activation of the pro-survival AKT/mTOR pathway, which can suppress autophagy-dependent ferroptosis (82, 83).
Moreover, this ferroptosis-suppressive effect appears to be conserved in vivo. NOS2 knockout mice exhibit enhanced inflammation and lipid peroxidation under stress, supporting the idea that endogenous NO production has cytoprotective, anti-ferroptotic effects in mammalian tissues (84). Additionally, the microbiome’s role in modulating NO–ferroptosis crosstalk via microbial metabolites influencing iron availability or ROS levels remains largely unexplored, although recent studies have begun to elucidate this topic. For example, gut microbiota metabolites such as short-chain fatty acids (SCFAs) and tryptophan derivatives indirectly affect ferroptosis by regulating oxidative stress and iron metabolism. SCFAs can upregulate NLRP6 expression and promote RIG-I/MAVS-mediated mitophagy, thereby inhibiting ferroptosis. Tryptophan metabolites such as kynurenine (KYN) can scavenge ROS and activate the Nrf2-dependent pathway to regulate cellular ferroptosis. Although not extensively studied, these findings suggest that the gut microbiota may influence NO–ferroptosis crosstalk through metabolic pathways, warranting further investigation.
The switch between NO’s pro- and anti-ferroptotic roles is determined by a delicate interplay of concentration and the local redox environment. In a high-oxidative-stress environment rich in superoxide, NO is rapidly converted to pro-ferroptotic ONOO −. In contrast, when superoxide levels are lower, NO can persist and exert its anti-ferroptotic, radical-scavenging effects. Therefore, the low, sustained levels of NO often found in the CRC tumor microenvironment may confer a survival advantage by protecting cancer cells against ferroptotic stress. This presents a therapeutic challenge as endogenous NO could contribute to resistance against ferroptosis-inducing chemotherapies. However, it also highlights a therapeutic opportunity: overwhelming this protective system with high, localized doses delivered via NO-releasing nanocarriers can trigger ferroptotic cell death in CRC cells (Table 3). This biphasic nature positions the NO signaling pathway as a sophisticated, druggable node in the regulation of CRC cell death.
The seemingly contradictory roles of NO in ferroptosis, as detailed in Tables 2, 3, can be reconciled by considering several key factors. The balance between pro- and anti-ferroptotic outcomes is largely dictated by the net effect of NO on the cellular redox state, specifically the ROS/GSH balance, and its targeted S-nitrosylation of key regulatory proteins. At high concentrations, NO can react with superoxide to form peroxynitrite (ONOO−), a potent oxidant that depletes GSH and initiates lipid peroxidation, thereby driving ferroptosis. Conversely, at lower, controlled concentrations, NO can act as a radical-trapping antioxidant, directly terminating lipid peroxidation chain reactions and preventing the accumulation of toxic lipid peroxides. Furthermore, S-nitrosylation of specific targets can either promote (e.g., inhibiting GPX4) or inhibit (e.g., activating survival pathways) ferroptosis. Therefore, the ultimate effect of NO is not absolute but is instead a product of its concentration, the local redox environment, and the specific molecular machinery of the target cell.
3 Interaction between NO and H2S in CRC
The interplay between NO and hydrogen sulfide (H2S) constitutes a dynamic regulatory axis in CRC, with profound implications for redox balance, therapeutic resistance, and immune evasion. Mechanistically, their crosstalk is bidirectional and often antagonistic. These gasotransmitters engage in reciprocal post-translational modifications of NO via S-nitrosylation and H2S via S-sulfhydration to modulate critical signaling pathways. For instance, NO suppresses cystathionine β-synthase (CBS), the primary H2S-producing enzyme, thereby reducing endogenous H2S levels and impairing tumor survival (108). Conversely, H2S inhibits endothelial nitric oxide synthase (eNOS), curtailing NO bioavailability and altering vascular dynamics. This antagonistic relationship extends to mitochondrial function H2S scavenges ONOO−, a cytotoxic reactive nitrogen species, an effect that can be mitigated by H2S, thereby creating a complex regulatory loop that governs mitochondrial dysfunction and redox balance (109). These interactions underscore the delicate redox equilibrium that governs CRC progression.
In multidrug-resistant CRC, the NO–H2S axis emerges as a therapeutic vulnerability. Co-treatment with NO and H2S donors downregulates P-glycoprotein (MDR1), reducing chemotherapeutic efflux and resensitizing tumors to conventional agents. This synergy highlights their potential to circumvent resistance mechanisms rooted in drug transporter overexpression. Furthermore, the TME dictates context-specific effects. It has been experimentally demonstrated that NO, particularly when produced endogenously under hypoxic conditions, can stabilize hypoxia-inducible factor-1α (HIF-1α). In colon carcinoma HCT116 cells, Chowdhury et al. (110) showed that endogenously generated NO and ROS contribute to HIF-1α accumulation by inhibiting prolyl hydroxylase domain proteins (PHDs) via S-nitrosation of PHD2, thus preventing HIF-1α degradation during hypoxia (110). Further supporting this, Lee et al. (111) demonstrated that in Caco-2 epithelial cells, nitric oxide produced during exposure to Clostridium difficile toxin facilitated HIF-1α stabilization through iNOS-dependent S-nitrosylation. Inhibition of iNOS reduced HIF-1α accumulation and worsened epithelial damage, emphasizing NO’s protective role via this pathway (111–113).
These platforms exploit the pro-death effects of gasotransmitter fluxes, achieving localized redox disruption without compromising healthy tissues. Immunologically, the NO–H2S axis modulates dendritic cell activation and suppresses MDSC expansion, thereby restoring antitumor immunity. This immunomodulatory role, coupled with their ability to induce ferroptosis, provides a rationale for combining gasotransmitter-targeted therapies with immune checkpoint inhibitors. Future investigations should unravel the epigenetic implications of NO–H2S crosstalk, particularly their influence on histone acetylation and DNA methylation patterns that drive CRC aggressiveness. Additionally, spatial mapping of gasotransmitter fluxes within the TME, coupled with single-cell analyses, could identify niche-specific vulnerabilities. The development of dual NO/H2S modulators, capable of fine-tuning their synergistic or antagonistic effects, represents a frontier in precision oncology.
A significant gap in the current understanding is the influence of the gut microbiome on this crosstalk as microbial metabolites could modulate the local availability of both gasotransmitters and their precursors, thereby influencing CRC progression. Furthermore, developing dual NO/H2S modulators capable of fine-tuning their synergistic or antagonistic effects represents a promising frontier in precision oncology.
3.1 Molecular crosstalk between NO and H2S
NO plays a multifaceted role in CRC, influencing tumor progression, angiogenesis, and therapeutic responses through diverse molecular mechanisms. Recent studies have demonstrated that uncoupled NOS activity contributes to CRC progression by generating reactive oxygen/nitrogen species (ROS/RNS). Alam et al. (50) reported that sepiapterin, a tetrahydrobiopterin precursor, recoupled NOS in CRC cell lines (HCT116 and HT29), restoring the tetrahydrobiopterin: dihydrobiopterin ratio, which is significantly lower in tumors than in normal tissues. This intervention reduced proliferation and induced apoptosis via AKT/GSK-3β-mediated β-catenin downregulation. In murine models, oral sepiapterin decreased metabolic uptake of fluorodeoxyglucose and significantly increased apoptosis in azoxymethane/dextran sodium sulfate-induced CRC tumors. Extracellular vesicles (EVs) derived from CRC cells further amplify NO-mediated pathways. Ikeda et al. identified CAT1-positive EVs in CRC patients, which enhanced arginine transport and NO synthesis in endothelial cells, promoting angiogenesis. Plasma EV-CAT1 levels were significantly elevated in CRC patients, correlating with increased vascular endothelial cell growth and tubule formation (115).
Immunomodulatory strategies targeting NO pathways have shown therapeutic promise. (116) engineered exosomes (exoASO-STAT6) to silence STAT6 in TAMs, inducing M1 polarization through NOS2 upregulation. This approach generated a substantial amount of NO in triple-negative breast and CRC cells, leading to significant tumor growth inhibition and a high rate of complete remission in syngeneic CRC models. Ferroptosis induction via NO donors has also been explored. (35) demonstrated that NCX4040, a non-steroidal NO donor, generated ROS in CRC cells (HT29 and HCT116) without glutathione depletion. Co-treatment with ferroptosis inducers (erastin or RSL3) synergistically enhanced cell death, while ferrostatin-1, a ferroptosis inhibitor, markedly reduced cytotoxicity. Lipid peroxidation increased dose-dependently, and metabolomic profiling revealed upregulated expression of CHAC1, GPX4, and NOX4, key regulators of ferroptosis. Real-time NO detection methodologies have further advanced therapeutic targeting. Daw et al. (117) developed an oxyhemoglobin-based assay quantifying NO production in cytokine-stimulated CRC cells. IFN-γ, IL1-β, and TNF-α induced NOS2 expression, producing a quantifiable amount of NO. The NOS2 inhibitor 1,400 W exhibited a potent IC50 value in 4T1 cells, with rapid inhibition that persisted for an extended period.
H2S, another gaseous signaling molecule, has been implicated in CRC vascular function, although research remains limited. Hassan et al. investigated the vasodilatory effects of NO and H2S in human mesenteric arteries obtained from CRC patients. Sodium nitroprusside (SNP)-induced NO-mediated relaxation was significantly reduced by tetraethylammonium (TEA), a K+ channel blocker, compared to controls. H2S-induced vasorelaxation involved KATP channels, although its specific role in CRC progression remains underexplored. The study highlighted that H2S and NO interactions may regulate vascular tone in CRC, with potential implications for tumor microenvironment modulation (118).
NO exerts profound effects on CRC through angiogenesis, immunomodulation, and ferroptosis, with therapeutic strategies targeting NOS coupling, EV-mediated pathways, and ferroptosis induction showing significant promise. H2S, while less studied, appears to modulate vascular function in CRC, warranting further investigation. Translating these preclinical findings into clinical applications requires validation of exact molecular mechanisms and dose-response relationships, particularly for H2S, to develop targeted therapies for CRC.
4 Novel therapeutic approaches of NO signaling in CRC
The therapeutic potential of targeting NO signaling and its associated molecular pathways has attracted significant attention in CRC research in recent years (119, 120). Several studies have explored various aspects of NO signaling in CRC, including its impact on ferroptosis, mitochondrial dysfunction (Figure 2), angiogenesis, immune regulation, and metabolic reprogramming, as discussed in Table 3.
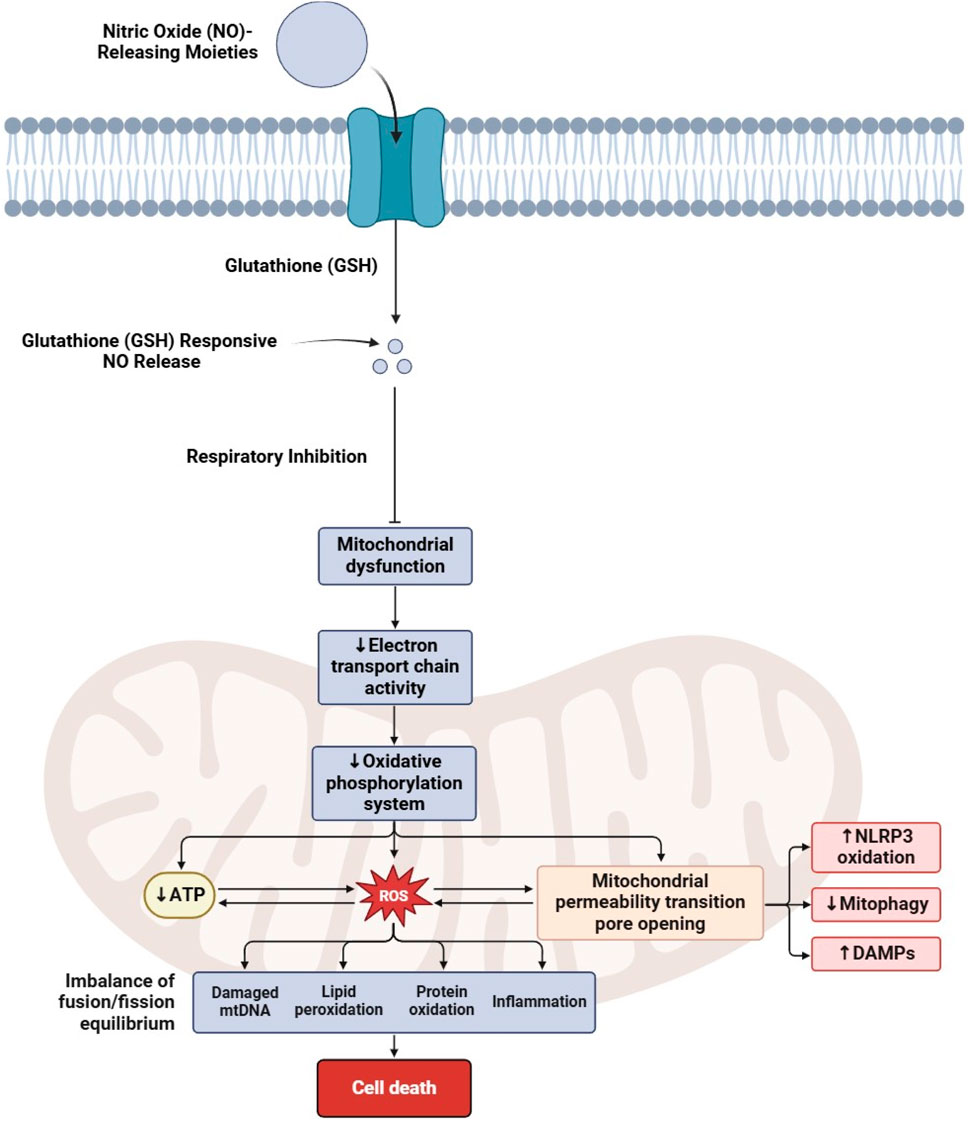
Figure 2. Mechanism of nitric oxide-induced mitochondrial dysfunction leading to cell death.
4.1 Enhancing ferroptosis in CRC via NO modulation
The interplay between NO signaling and ferroptosis in CRC has garnered significant attention, focusing on exploiting redox imbalances to induce iron-dependent cell death. Ferroptosis, characterized by lipid peroxide accumulation and GSH depletion, is modulated by NO through direct enzyme inhibition, iron metabolism regulation, and potentiation of immunogenic cell death (ICD). Various NO donors induce ferroptosis in CRC cells. Compounds such as NCX4040 deplete intracellular GSH and elevate lipid peroxidation markers, and their effects are significantly enhanced when combined with ferroptosis inducers such as erastin or RSL3 (35). Other agents, including coumarin–furoxan hybrids and phenylsulfonyl furoxan derivatives, suppress the expression of solute carrier family 7 member 11 (SLC7A11) and covalently inhibit glutathione peroxidase 4 (GPX4), sensitizing cells to lipid peroxidation and helping reverse multidrug resistance ((121); 177). Sensitivity to ferroptosis is also regulated by other key proteins. Acyl-CoA synthase long-chain family member 4 (ACSL4) expression correlates with susceptibility, while NO-driven upregulation of heme oxygenase-1 (HMOX1) increases labile iron pools, amplifying lipid peroxidation (122). Lipidomic profiling confirms that NO donors lead to a marked increase in peroxidized phosphatidylethanolamines (PEs), a hallmark of ferroptosis (123).
GPX4, the primary enzyme for detoxifying lipid hydroperoxides, is a central target in NO-mediated ferroptosis. NO can directly inhibit GPX4 activity through mechanisms such as S-nitrosylation (124). Consequently, NO donors synergize with GPX4 inhibitors such as RSL3 to elevate markers of lipid peroxidation, while the ferroptosis inhibitor ferrostatin-1 can restore GPX4 activity and reduce lipid ROS (179, 125). The role of NO is context-dependent; while it can be protective in some cell types under specific conditions, higher NO concentrations in CRC consistently deplete GSH and inhibit GPX4, promoting a pro-ferroptotic outcome (82). Furthermore, NO produced by iNOS in M1 macrophages can exacerbate lipid peroxidation and ferroptosis in co-cultured CRC cells (123).
Advanced strategies using nanoplatforms and combinatorial therapies have shown promise. Nanocarriers such as AZOSH or IS@ATF enable triggered or targeted NO release, leading to GSH depletion, ONOO− generation, and significant tumor weight reduction in murine models (36, 126, 127). Integrating NO donors with other treatments enhances therapeutic efficacy. Combination with immunotherapy (e.g., anti-PD-1) promotes immunogenic ferroptosis by increasing the infiltration of CD8+ T-cells, while combination with chemotherapy (e.g., 5-fluorouracil) increases chemosensitization in CRC cells (35, 36). Plasma-activated Ringer’s lactate, which generates NO and RNS, also induces ferroptosis effectively (128). Despite these promising results, challenges remain, including off-target vascular toxicity and the risk of systemic iron overload (36, 127). Future studies must prioritize tumor-specific delivery systems and the use of biomarkers such as ACSL4 and GPX4 for patient stratification to translate these findings into clinical practice.
4.2 NO’s role in mitochondrial dysfunction and oxidative stress
The activity of mitochondrial nitric oxide synthase (mtNOS) is a key driver of mitochondrial dysfunction, oxidative stress, and progression in colorectal cancer (CRC). Research shows that mitochondrial oxidative damage is a critical feature of CRC, with markers such as TBARS and protein carbonyls being significantly elevated in tumor tissues. The function of the electron transport chain (ETC) is also compromised, as shown by the reduced activity of key mitochondrial enzymes. Notably, mtNOS activity significantly increases in advanced-stage CRC, which correlates directly with markers of oxidative damage. This suggests that NO and hydrogen peroxide produced by mtNOS act as diffusible “toxohormones,” promoting oxidative stress in nearby non-tumor tissues and thereby facilitating tumor progression. This problem is further exacerbated by an imbalance in the cell’s antioxidant defenses as the activity of Cu, Zn-superoxide dismutase (SOD) is markedly reduced in advanced tumors. These findings directly implicate mtNOS hyperactivity in driving mitochondrial problems in CRC (129).
This destructive interplay between NO, ROS, and mitochondria is not unique to CRC; it is a common mechanism in many other cancers. For instance, in liver cancer, the drug sorafenib was found to increase intracellular NO and superoxide, leading to the formation of the highly reactive molecule peroxynitrite (ONOO−). This caused a drastic reduction in oxygen consumption and severe mitochondrial damage, a process that mirrors mtNOS-driven disruption observed in CRC (130). Similarly, in melanoma, the overexpression of a protein called UT-B was shown to increase NO levels, leading to mitochondrial depolarization, a spike in ROS, and reduced cell viability. This effect could be reversed with an antioxidant, directly linking NO overproduction to the observed oxidative stress (131).
NO overproduction in the tumor the microenvironment also plays a crucial role in promoting cancer progression. The loss of a protein called caveolin-1 (Cav-1) in stromal fibroblasts can lead to NO overproduction and a metabolic shift toward aerobic glycolysis. This phenomenon, known as the “Reverse Warburg Effect,” causes these fibroblasts to secrete lactate, which is subsequently used by cancer cells to enhance their mitochondrial activity and support proliferation. This shows how NO and ROS signaling from stromal cells creates a pro-tumor environment, reinforcing the “toxohormone” concept, where signals from one cell type promote cancer growth in another (129, 132). Similar metabolic shifts driven by NO and ROS have also been observed in lung cancer cells (133).
Interestingly, scientists are now turning this destructive mechanism into a therapeutic strategy. Mitochondria-targeted nanoplatforms that are designed to generate both NO and ROS have shown great promise. In hepatocellular carcinoma models, this approach produced high levels of ONOO− inside mitochondria, causing irreversible damage to the ETC, inhibiting ATP production, and triggering cancer cell death, which significantly reduced tumor volume (134). Similar results have been observed using photodynamic therapy in lung cancer and with combination drug treatments in breast cancer, where increased NO and ROS lead to mitochondrial stress, autophagy, and apoptosis (121, 135, 136). In summary, mtNOS upregulation in CRC is a central cause of mitochondrial damage, driving oxidative stress and weakening the cell’s defenses. This mechanism is conserved across many cancers and even extends to the tumor microenvironment. Targeting this interplay between NO and mitochondrial function is, therefore, a highly promising therapeutic avenue for CRC and other cancers.
4.3 NO-releasing agents in overcoming tumor hypoxia
Tumor hypoxia, a hallmark of solid malignancies such as CRC, drives therapeutic resistance, immunosuppression, and metastasis. NO, a gaseous signaling molecule, has emerged as a promising agent for modulating hypoxic microenvironments (Table 4). This discussion synthesizes findings from preclinical studies on NO-releasing strategies, emphasizing their applicability to CRC. Tu et al. (137) demonstrated that a micellar NO donor (TPGS-NO) enhanced radiotherapy efficacy in hypoxic tumors by improving oxygenation and reducing HIF-1α expression. TPGS-NO prolonged NO release in tumors, leading to increased angiogenesis and apoptosis (Figure 3) while inhibiting DNA repair post-radiation. Although tested in a non-CRC model, this mechanism is highly relevant to CRC, where hypoxia-driven resistance limits radiotherapy outcomes (137). Similarly, Dou et al. (138) developed radiation-activated nanoagents (NSC@SiO2-SNO NPs) that release NO upon X-ray irradiation. These nanoparticles reduced hypoxia and improved tumor oxygenation, monitored via BOLD/DWI imaging. This approach significantly inhibited tumor growth in vivo, suggesting potential for CRC applications where hypoxia compromises radiation efficacy (138). Zhang et al. (139) utilized ultrasound-stimulated microbubbles (USMBs) to enhance tumor perfusion and NO release in the MC38 murine colon cancer model. At a mechanical index (MI) of 0.3–0.5, USMBs increased tissue oxygen partial pressure (pO2) and reduced HIF-1α and lactate levels. Repeated treatments sustained hypoxia alleviation without resistance, highlighting a translatable strategy for CRC (139).
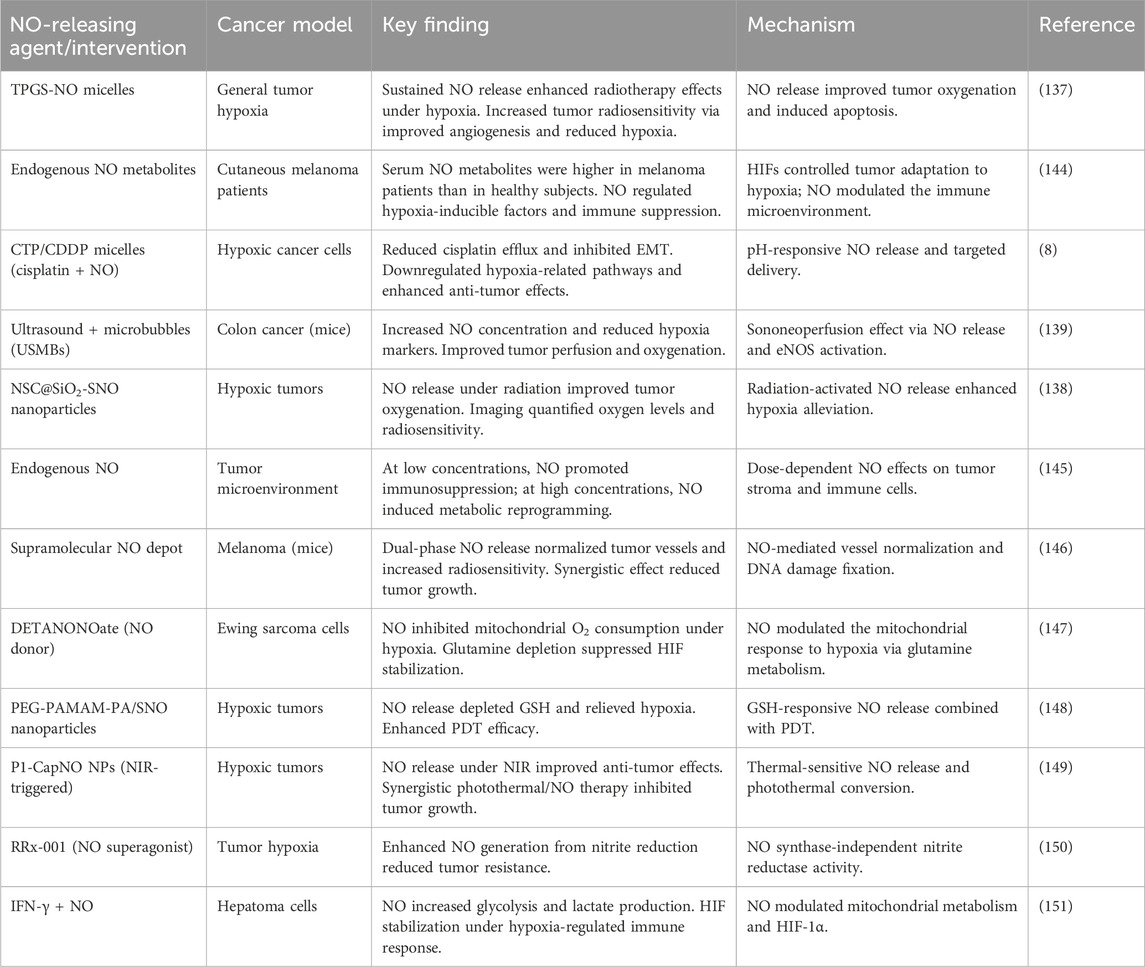
Table 4. Comprehensive analysis of nitric oxide-releasing agents in overcoming tumor hypoxia.
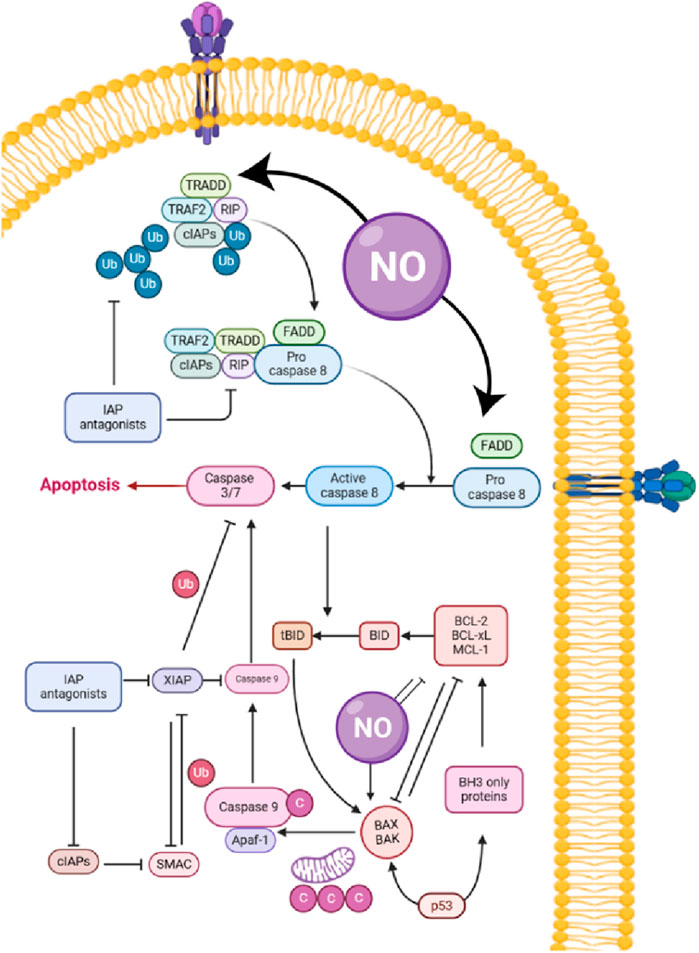
Figure 3. Role of nitric oxide (NO) in apoptosis pathways: mechanisms and interactions with cellular proteins.
Zhao et al. (140) combined ultrasound-targeted microbubble destruction (UTMD) with NO-generating nanodroplets (L-Arg@PTX). UTMD-triggered NO release reversed hypoxia, reduced cisplatin efflux, and enhanced cytotoxic T-lymphocyte infiltration. This dual approach improved chemoimmunotherapy outcomes, underscoring NO’s role in overcoming CRC immunosuppression (140). Chen et al. (141) designed chitosan-coated micelles (CTP/CDDP) co-delivering cisplatin and NO. In hypoxic cancer cells, NO downregulated HIF-1α, GSH, and multidrug resistance-associated protein 2 (MRP2), reversing cisplatin resistance. Although these findings were observed in non-CRC models, they are highly relevant to CRC, where hypoxia-driven chemoresistance remains a significant challenge (141).
Hypoxia stabilizes HIF-1α, promoting CRC progression. NO disrupts this pathway by inhibiting HIF-1α accumulation, as shown by Graham et al. (142), where NO/cGMP signaling blocked hypoxia-induced immune escape mechanisms. In CRC, this could enhance NK cell-mediated lysis by preserving surface MICA expression (142). This has direct implications for immunotherapy as hypoxia is known to increase PD-L1 expression via HIF-1α, enabling immune escape. As demonstrated by Barsoum et al. (143), NO donors such as nitroglycerin can attenuate this PD-L1 upregulation, thereby restoring T-cell cytotoxicity. This mechanism provides a strong rationale for combining NO-based therapies with checkpoint inhibitors such as anti-PD-1/PD-L1 to overcome hypoxia-induced immune resistance in CRC (143).
Although preclinical data are promising, clinical translation requires addressing NO’s biphasic effects; low doses alleviate hypoxia, whereas high doses may promote metastasis. Targeted delivery systems, such as CRC-specific nanoparticles or ultrasound-responsive agents, could mitigate off-target effects. Furthermore, there is a clear need for studies in CRC-specific models to optimize dosing and delivery schedules to ensure clinical viability. Additionally, combining NO donors with immunotherapy or hypoxia-activated prodrugs may amplify efficacy in CRC. NO-releasing agents represent a multifaceted strategy to combat CRC hypoxia. By enhancing perfusion, downregulating HIF-1α, reversing chemoresistance, and modulating immunity, NO synergizes with radiotherapy, chemotherapy, and immunotherapy. Further studies in CRC-specific models are warranted to optimize dosing and delivery, ensuring clinical viability.
4.4 Oxicam analogs and modulation of the NO pathway
Oxicam analogs exhibit novel capabilities in modulating the NO pathway offering unprecedented therapeutic potential. Unlike traditional anti-inflammatory agents, these compounds uniquely influence NO-related mechanisms, making them a promising frontier in treating inflammation and cancer (152). Krzystek-Korpacka et al. (153) and Dowling et. al. (154) highlighted the metabolic reprogramming in CRC, which includes the overexpression of enzymes such as ARG1, PRMTs, and DDAHs, in addition to NOS2. In the context of CRC, the NO pathway metabolites are found to be altered, with enzymes such as ARG1, PRMT1, and PRMT5 being overexpressed in both tumor and tumor-adjacent tissues. Notably, DDAH2 is overexpressed solely in tumor-adjacent tissue. The expression of ARG1 in tumors has been observed to increase with tumor grade and reflects lymph node involvement, indicating a potential role in disease progression (153, 154).
The modulation of this pathway by oxicam analog presents a promising therapeutic role (154, 155). Classic and novel oxicam analogs have been assessed for their impact on enzyme expression and intracellular metabolite concentration in CRC cell lines such as Caco-2, HCT116, and HT-29 (156). Novel oxicam analogs, particularly those with an arylpiperazine moiety at the thiazine ring, have shown greater efficacy in downregulating DDAHs and PRMTs and upregulating ARG2 compared to traditional oxicams such as piroxicam and meloxicam (157, 158). Oxicam derivatives significantly impact macrophage-associated chemokine expression, which is crucial in colorectal cancer pathophysiology. Their work suggested that these derivatives not only modulate the NO pathway but also exhibit dual COX-1/COX-2 inhibition, amplifying their anti-inflammatory properties (157). Similarly, Szczuka et al. (159) explored the interplay between oxicam compounds and heat shock proteins (HSPA1 and HSP90AA1), demonstrating potential therapeutic targeting in colorectal polyps and other malignancies (159). Moreover, (180) demonstrated the synergistic cytotoxic effects of oxicam derivatives with simvastatin, showing apoptosis induction in drug-resistant colon cancer cells (160).
In experimental models, Abdul Wanees El-Awdan et al. (161) tested combinations of meloxicam with octreotide, observing improved anti-inflammatory outcomes mediated through NO-dependent pathways (161). Research into stable lipoxin analogs underscores the connection between NO modulation and broader anti-inflammatory mechanisms, offering insights into drug development strategies (162). These studies collectively underscore the importance of oxicam analogs in NO pathway modulation, providing robust frameworks for clinical application in inflammatory disorders and oncology. These findings further suggest that metabolic reprogramming in CRC is not limited to tumor tissue and can be affected by novel oxicam analogs. This may provide a potential strategy for chemoprevention and therapy (Table 5).
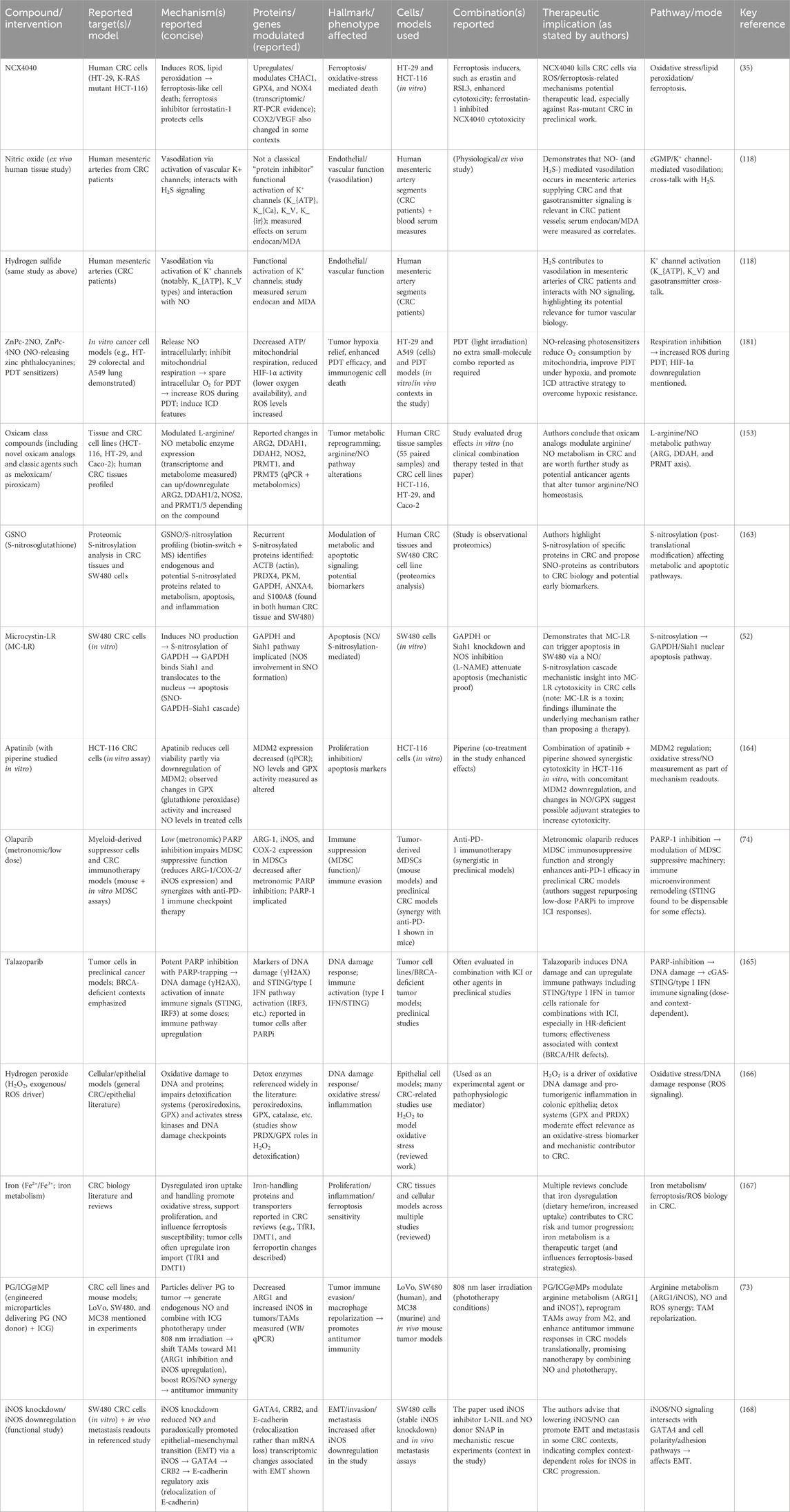
Table 5. Recent advances in therapeutic compounds targeting nitric oxide and related pathways in colorectal cancer (CRC).
5 Therapeutic implications and future perspectives in NO-Targeted colorectal cancer treatments
Emerging therapeutics target NO pathways to address these complexities. EV-CAT1, which inhibits arginine transport via the CAT1 transporter, not only suppresses NO-related angiogenesis but also enhances diagnostic accuracy when combined with carcinoembryonic antigen (CEA). Similarly, compounds such as NAD(P)H stimulate NO synthesis, identifying aggressive angiogenic phenotypes through cGMP-PKG signaling and providing avenues for therapeutic intervention. Furthermore, the interplay of NO and H2S in vascular modulation, particularly through agents such as SNP and Na2S, offers potential in regulating endothelial dysfunction and oxidative stress. These compounds activate K+ channels (e.g., KATP and KV), revealing a novel strategy to manage CRC progression. Future directions highlight integrating genetic insights and tumor microenvironment dynamics into precision therapies. Combining NO pathway modulators with innovative diagnostic tools, such as EV-CAT1, could significantly improve treatment efficacy. The synergistic targeting of NO and gasotransmitter pathways, such as H2S, represents an exciting frontier in developing robust anti-cancer strategies for CRC.
Therapeutic strategies targeting NO have also shown promise in preclinical models and present potential for CRC treatment (Table 6). NCX4040, targeting CHAC1 and GPX4, induces ferroptosis in Ras-mutated CRC cells, suggesting future applications in oxidative stress-focused therapies. ZnPc-2NO and ZnPc-4NO inhibit mitochondrial respiration, reducing oxygen consumption and offering strategies against hypoxic tumors via ICD. Modulating ARG1 and iNOS polarizes TAMs from M2 to M1 phenotypes, enhancing immune responses and positioning ARG1 as a key therapeutic target for macrophage-directed interventions (181).
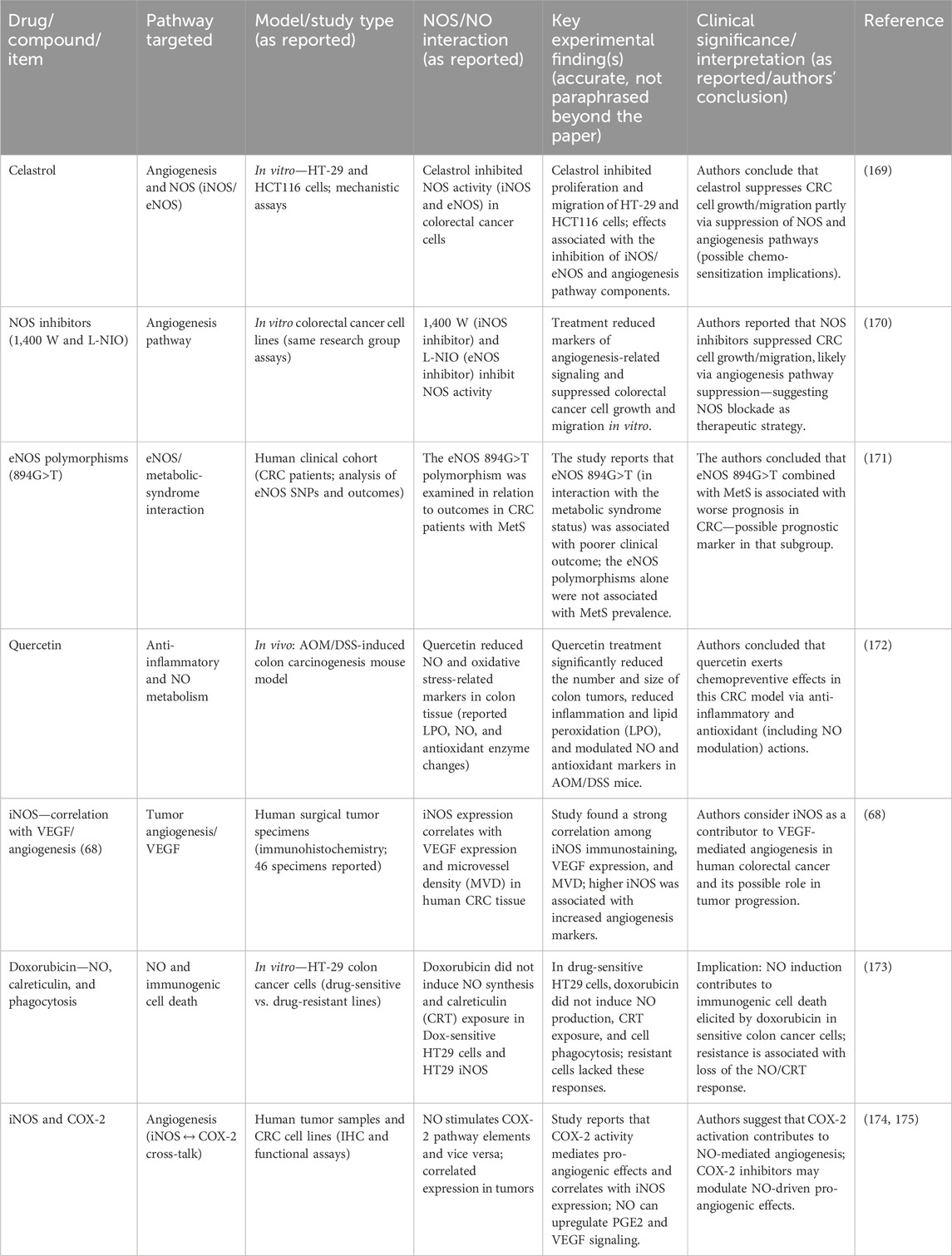
Table 6. Experimental and clinical findings on NOS-targeting agents in colorectal cancer research.
Olaparib’s capacity to suppress MDSCs and enhance T-cell function, particularly in combination with anti-PD-1 therapy, underscores its potential role in immunotherapy for MSS tumors. NOS inhibitors, such as 1,400 W and L-NIO, combined with 5-fluorouracil, enhance anti-CRC effects, paving the way for dual-pathway treatments. Celastrol’s and atorvastatin’s anti-tumor activities suggest potential as chemopreventive agents targeting NO signaling.
NO-releasing nanotechnologies, delivering localized therapeutic doses, minimize systemic toxicity and address CRC’s adaptive resistance mechanisms. These advancements align with future CRC treatments focusing on precise NO modulation and combination regimens to overcome resistance and optimize outcomes.
6 Conclusion
NO has emerged as a critical mediator in the complex biology of CRC, exhibiting a dual role as a tumor promoter and a tumor suppressor. This duality presents significant challenges and opportunities for therapeutic intervention. NO exerts diverse effects on CRC pathophysiology, including promoting ferroptosis, modulating the tumor immune microenvironment, driving metabolic reprogramming, and enhancing metastatic capacity. The complexity of NO signaling is shaped by its concentration, intracellular localization, and interactions with other molecular pathways, such as H2S, underscoring the importance of these contextual factors. Current research efforts focused on the precise modulation of NO or selective targeting of NO-related pathways represent a promising frontier for enhancing CRC treatment efficacy. These approaches hold the potential to overcome resistance mechanisms and enhance the effectiveness of existing therapies. Developing targeted therapies that leverage NO’s complex roles will require a more comprehensive understanding of the delicate balance between its tumor-promoting and tumor-suppressing activities. Achieving this understanding is essential for creating interventions that can strategically influence NO’s effects in CRC, potentially leading to more refined and effective treatment strategies.
Author contributions
AT: Conceptualization, Investigation, Writing – original draft, Writing – review and editing. AC: Writing – original draft, Writing – review and editing. MQ: Conceptualization, Data curation, Formal analysis, Writing – original draft, Writing – review and editing. RT: Writing – original draft, Writing – review and editing. MU: Writing – original draft, Writing – review and editing. MA: Writing – original draft, Writing – review and editing. TP: Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-DDRSP2501.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Marcellinaro, R, Spoletini, D, Grieco, M, Avella, P, Cappuccio, M, Troiano, R, et al. Colorectal Cancer: Current Updates and Future Perspectives. J Clin Med (2023) 13(1):40. doi:10.3390/jcm13010040
2. Mohammed, AM, Al-sarray, AAM, and Rudha, AR. Clinical study of colorectal cancer in al najaf al ashraf. Obstet and Gynaecol Forum (2024) 3:2423–34.
3. Zheng, S, Yan, J, Wang, J, Wang, X, Kang, YE, Koo, BS, et al. Unveiling the Effects of Cruciferous Vegetable Intake on Different Cancers: A Systematic Review and Dose–Response Meta-analysis. Nutr Rev (2024) 83:842–58. doi:10.1093/nutrit/nuae131
4. Stephens, KR, Donica, WRF, Egger, ME, Philips, P, Scoggins, CR, McMasters, KM, et al. Observed Changes in the Distribution of Colon Cancer Metastasis: A National Cancer Database Review and Institutional Experience. Ann Surg Oncol (2024) 32:418–23. doi:10.1245/s10434-024-16330-5
5. Wang, X-Y, Hao, Y, Wang, Z-J, Xu, X-L, and Yang, J-H. Clinicopathological differences between patients with schistosomal appendicitis and non schistosomal appendicitis: A retrospectively study of past ten years. World J Clin Cases (2025) 13(2):96557. doi:10.12998/wjcc.v13.i2.96557
6. Escaron, A, Lara, R, Ramirez, K, Jacobo, A, Schneider, J, Rivelli, J, et al. Abstract A141: Exploring patient barriers and facilitators to colorectal cancer screening during the SARS-CoV-2 pandemic. Cancer Epidemiol Biomarkers and Prev (2024) 33(9_Suppl. ment):A141. doi:10.1158/1538-7755.DISP24-A141
7. Kpossou, AR, Vignon, RK, Hadjete, J, Sokpon, CNM, Gnangnon, FHR, Séidou, F, et al. Colorectal cancers in cotonou from 2013 to 2023: epidemiological, diagnostic, therapeutic and prognostic aspects. West Afr J Med (2024) 41(11 Suppl. 1):S3–S4.
8. Chen, K, Collins, G, Wang, H, and Toh, JWT. Pathological Features and Prognostication in Colorectal Cancer. Curr Oncol (2021) 28(6):5356–83. doi:10.3390/curroncol28060447
9. Ma, Y, Ma, D, Xu, X, Li, J, and Guan, Z. Progress of MRI in predicting the circumferential resection margin of rectal cancer: A narrative review. Asian J Surg (2024) 47(5):2122–31. doi:10.1016/j.asjsur.2024.01.131
10. Duan, B, Zhao, Y, Bai, J, Wang, J, Duan, X, Luo, X, et al. Colorectal Cancer: An Overview. In: Gastrointestinal cancers. Brisbane, AU: Exon Publications (2022). p. 1–12. doi:10.36255/exon-publications-gastrointestinal-cancers-colorectal-cancer
11. Wu, Q, Chen, P, Shu, C, Chen, L, Jin, Z, Huang, J, et al. Survival outcomes of stage I colorectal cancer: development and validation of the ACEPLY model using two prospective cohorts. BMC Med (2023) 21(1):3. doi:10.1186/s12916-022-02693-7
12. Baxter, NN, Kennedy, EB, Bergsland, E, Berlin, J, George, TJ, Gill, S, et al. Adjuvant Therapy for Stage II Colon Cancer: ASCO Guideline Update. J Clin Oncol (2022) 40(8):892–910. doi:10.1200/JCO.21.02538
13. Xiong, Z-Z, Xie, M-H, Li, X-Z, Jin, L-Y, Zhang, F-X, Yin, S, et al. Risk factors for postoperative recurrence in patients with stage II colorectal cancer. BMC Cancer (2023) 23(1):658. doi:10.1186/s12885-023-11093-w
14. Pricolo, VE, Steingrimsson, J, McDuffie, TJ, McHale, JM, McMillen, B, and Shparber, M. Tumor Deposits in Stage III Colon Cancer. Am J Clin Oncol (2020) 43(2):133–8. doi:10.1097/COC.0000000000000645
15. Stachtea, X, Loughrey, MB, Salvucci, M, Lindner, AU, Cho, S, McDonough, E, et al. Stratification of chemotherapy-treated stage III colorectal cancer patients using multiplexed imaging and single-cell analysis of T-cell populations. Mod Pathol (2022) 35(4):564–76. doi:10.1038/s41379-021-00953-0
16. Cervantes, A, Adam, R, Roselló, S, Arnold, D, Normanno, N, Taïeb, J, et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol (2023) 34(1):10–32. doi:10.1016/j.annonc.2022.10.003
17. Hernandez Dominguez, O, Yilmaz, S, and Steele, SR. Stage IV Colorectal Cancer Management and Treatment. J Clin Med (2023) 12(5):2072. doi:10.3390/jcm12052072
18. Koulis, C, Yap, R, Engel, R, Jardé, T, Wilkins, S, Solon, G, et al. Personalized Medicine—Current and Emerging Predictive and Prognostic Biomarkers in Colorectal Cancer. Cancers (2020) 12(4):812. doi:10.3390/cancers12040812
19. Lin, H-H, Wei, N-C, Chou, T-Y, Lin, C-C, Lan, Y-T, Chang, S-C, et al. Building personalized treatment plans for early-stage colorectal cancer patients. Oncotarget (2017) 8(8):13805–17. doi:10.18632/oncotarget.14638
20. Chen, Y, Shao, Z, and Wu, S. Research progress on the tsRNA biogenesis, function, and application in lung cancer. Non-Coding RNA Res (2025) 10:63–9. doi:10.1016/j.ncrna.2024.09.004
21. Xu, Z, Li, W, Dong, X, Chen, Y, Zhang, D, Wang, J, et al. Precision medicine in colorectal cancer: Leveraging multi-omics, spatial omics, and artificial intelligence. Clinica Chim Acta (2024) 559:119686. doi:10.1016/j.cca.2024.119686
22. Yang, G, Cao, Y, Yang, X, Cui, T, Tan, NZV, Lim, YK, et al. Advancements in nanomedicine: Precision delivery strategies for male pelvic malignancies – Spotlight on prostate and colorectal cancer. Exp Mol Pathol (2024) 137:104904. doi:10.1016/j.yexmp.2024.104904
23. Gao, D, Asghar, S, Hu, R, Chen, S, Niu, R, Liu, J, et al. Recent advances in diverse nanosystems for nitric oxide delivery in cancer therapy. Acta Pharmaceutica Sinica B (2023) 13(4):1498–521. doi:10.1016/j.apsb.2022.11.016
24. Vinay, DS, Ryan, EP, Pawelec, G, Talib, WH, Stagg, J, Elkord, E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol (2015) 35:S185–S198. doi:10.1016/j.semcancer.2015.03.004
25. Khan, FH, Dervan, E, Bhattacharyya, DD, McAuliffe, JD, Miranda, KM, and Glynn, SA. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? Int J Mol Sci (2020) 21(24):9393. doi:10.3390/ijms21249393
26. Somasundaram, V, Basudhar, D, Bharadwaj, G, No, JH, Ridnour, LA, Cheng, RYS, et al. Molecular Mechanisms of Nitric Oxide in Cancer Progression, Signal Transduction, and Metabolism. Antioxid and Redox Signaling (2019) 30(8):1124–43. doi:10.1089/ars.2018.7527
27. Vannini, F, Kashfi, K, and Nath, N. The dual role of iNOS in cancer. Redox Biol (2015) 6:334–43. doi:10.1016/j.redox.2015.08.009
28. Zhang, S, Xiao, X, Yi, Y, Wang, X, Zhu, L, Shen, Y, et al. Tumor initiation and early tumorigenesis: molecular mechanisms and interventional targets. Signal Transduction Targeted Ther (2024) 9(1):149. doi:10.1038/s41392-024-01848-7
29. Cheng, X, Zhao, F, Ke, B, Chen, D, and Liu, F. Harnessing Ferroptosis to Overcome Drug Resistance in Colorectal Cancer: Promising Therapeutic Approaches. Cancers (2023) 15(21):5209. doi:10.3390/cancers15215209
30. Song, Y-Q, Yan, X-D, Wang, Y, Wang, Z-Z, Mao, X-L, Ye, L-P, et al. Role of ferroptosis in colorectal cancer. World J Gastrointest Oncol (2023) 15(2):225–39. doi:10.4251/wjgo.v15.i2.225
31. Yang, L, Zhang, Y, Zhang, Y, and Fan, Z. Mechanism and application of ferroptosis in colorectal cancer. Biomed and Pharmacother (2023) 158:114102. doi:10.1016/j.biopha.2022.114102
32. Andrabi, SM, Sharma, NS, Karan, A, Shahriar, SMS, Cordon, B, Ma, B, et al. Nitric Oxide: Physiological Functions, Delivery, and Biomedical Applications. Adv Sci (2023) 10(30):2303259. doi:10.1002/advs.202303259
33. He, H, Venema, VJ, Gu, X, Venema, RC, Marrero, MB, and Caldwell, RB. Vascular Endothelial Growth Factor Signals Endothelial Cell Production of Nitric Oxide and Prostacyclin through Flk-1/KDR Activation of c-Src. J Biol Chem (1999) 274(35):25130–5. doi:10.1074/jbc.274.35.25130
34. Papapetropoulos, A, García-Cardeña, G, Madri, JA, and Sessa, WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest (1997) 100(12):3131–9. doi:10.1172/JCI119868
35. Sinha, BK, Bortner, CD, Jarmusch, AK, Tokar, EJ, Murphy, C, Wu, X, et al. Ferroptosis-Mediated Cell Death Induced by NCX4040, The Non-Steroidal Nitric Oxide Donor, in Human Colorectal Cancer Cells: Implications in Therapy. Cells (2023) 12(12):1626. doi:10.3390/cells12121626
36. Zhu, L, Leng, D, Guo, Z, Zhao, Y, Leung, KT, Dai, Y, et al. Self-catalyzed nitric oxide nanocomplexes induce ferroptosis for cancer immunotherapy. J Controlled Release (2025) 377:524–39. doi:10.1016/J.JCONREL.2024.11.048
37. Bian, W, Li, H, Chen, Y, Yu, Y, Lei, G, Yang, X, et al. Ferroptosis mechanisms and its novel potential therapeutic targets for DLBCL. Biomed and Pharmacother (2024) 173:116386. doi:10.1016/j.biopha.2024.116386
38. de Souza, I, Ramalho, MCC, Guedes, CB, Osawa, IYA, Monteiro, LKS, Gomes, LR, et al. Ferroptosis Modulation: Potential Therapeutic Target for Glioblastoma Treatment. Int J Mol Sci (2022) 23(13):6879. doi:10.3390/ijms23136879
39. Sun, S, Shen, J, Jiang, J, Wang, F, and Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduction Targeted Ther (2023) 8(1):372. doi:10.1038/s41392-023-01606-1
40. Wang, H, Wang, L, Xie, Z, Zhou, S, Li, Y, Zhou, Y, et al. Nitric Oxide (NO) and NO Synthases (NOS)-Based Targeted Therapy for Colon Cancer. Cancers (2020) 12(7):1881. doi:10.3390/cancers12071881
41. Merhi, M, Ahmad, F, Taib, N, Inchakalody, V, Uddin, S, Shablak, A, et al. The complex network of transcription factors, immune checkpoint inhibitors and stemness features in colorectal cancer: A recent update. Semin Cancer Biol (2023) 89:1–17. doi:10.1016/j.semcancer.2023.01.001
42. Xie, Y-H, Chen, Y-X, and Fang, J-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduction Targeted Ther (2020) 5(1):22. doi:10.1038/s41392-020-0116-z
43. Yang, Z, Zhang, X, Bai, X, Xi, X, Liu, W, and Zhong, W. Anti-angiogenesis in colorectal cancer therapy. Cancer Sci (2024) 115(3):734–51. doi:10.1111/cas.16063
44. Alhujaily, M. Glyoxalase System in Breast and Ovarian Cancers: Role of MEK/ERK/SMAD1 Pathway. Biomolecules (2024) 14(5):584. doi:10.3390/biom14050584
45. Wang, L, Tian, Y, Lai, K, Liu, Y, Liu, Y, Mou, J, et al. An Ultrasound-Triggered Nanoplatform for Synergistic Sonodynamic-Nitric Oxide Therapy. ACS Biomater Sci and Eng (2023) 9(2):797–808. doi:10.1021/acsbiomaterials.2c01431
46. Wang, S, Guo, S, Guo, J, Du, Q, Wu, C, Wu, Y, et al. Cell death pathways: molecular mechanisms and therapeutic targets for cancer. MedComm (2024) 5(9):e693. doi:10.1002/mco2.693
47. Wang, W, Niu, Y, Zhang, N, Wan, Y, Xiao, Y, Zhao, L, et al. Cascade-Catalyzed Nanogel for Amplifying Starvation Therapy by Nitric Oxide-Mediated Hypoxia Alleviation. ACS Appl Mater and Inter (2024) 16(14):17313–22. doi:10.1021/acsami.4c01866
48. Xu, W, Liu, LZ, Loizidou, M, Ahmed, M, and Charles, IG. The role of nitric oxide in cancer. Cell Res (2002) 12(5–6):311–20. doi:10.1038/sj.cr.7290133
49. Xu, S, Xie, X, He, P, Zhu, S, Li, X, Chen, Q, et al. Nitric Oxide-Producing Multiple Functional Nanoparticle Remodeling Tumor Microenvironment for Synergistic Photodynamic Immunotherapy against Hypoxic Tumor. ACS Nano. (2025). doi:10.1021/ACSNANO.4C16329/SUPPL_FILE/NN4C16329_SI_001.PDF
50. Alam, A, Smith, SC, Gobalakrishnan, S, McGinn, M, Yakovlev, VA, and Rabender, CS. Uncoupled nitric oxide synthase activity promotes colorectal cancer progression. Front Oncol (2023) 13:1165326. doi:10.3389/fonc.2023.1165326
51. Li, J, Chen, D, and Shen, M. Tumor Microenvironment Shapes Colorectal Cancer Progression, Metastasis, and Treatment Responses. Front Med (2022) 9:869010. doi:10.3389/fmed.2022.869010
52. Li, K, Huang, M, Xu, P, Wang, M, Ye, S, Wang, Q, et al. Microcystins-LR induced apoptosis via S-nitrosylation of GAPDH in colorectal cancer cells. Ecotoxicology and environmental safety (2020) 190.
53. You, M, Xie, Z, Zhang, N, Zhang, Y, Xiao, D, Liu, S, et al. Signaling pathways in cancer metabolism: mechanisms and therapeutic targets. Signal Transduction Targeted Ther (2023) 8(1):196. doi:10.1038/s41392-023-01442-3
54. Takahashi, R, Ijichi, H, and Fujishiro, M. The Role of Neural Signaling in the Pancreatic Cancer Microenvironment. Cancers (2022) 14(17):4269. doi:10.3390/cancers14174269
55. Hamaoka, R, Yaginuma, Y, Takahashi, T, Fujii, J, Koizumi, M, Seo, HG, et al. Different expression patterns of nitric oxide synthase isozymes in various gynecological cancers. J Cancer Res Clin Oncol (1999) 125(6):321. doi:10.1007/s004320050281
56. Korde Choudhari, S, Chaudhary, M, Bagde, S, Gadbail, AR, and Joshi, V. Nitric oxide and cancer: a review. World J Surg Oncol (2013) 11(1):118. doi:10.1186/1477-7819-11-118
57. Wang, Q, Ye, S, Chen, X, Xu, P, Li, K, Zeng, S, et al. Mitochondrial NOS1 suppresses apoptosis in colon cancer cells through increasing SIRT3 activity. Biochem Biophysical Res Commun (2019) 515(4):517–23. doi:10.1016/j.bbrc.2019.05.114
58. Qiu, W, Zhao, L, Liu, H, Xu, P, and Qian, C. Hypoxia-induced NOS1 as a therapeutic target in hypercholesterolemia-related colorectal cancer. Cancer and Metab (2024) 12(1):14. doi:10.1186/s40170-024-00338-2
59. Bhat, NR, Zhang, P, and Bhat, AN. Cytokine Induction of Inducible Nitric Oxide Synthase in an Oligodendrocyte Cell Line. J Neurochem (1999) 72(2):472–8. doi:10.1046/j.1471-4159.1999.0720472.x
60. Peñarando, J, López-Sánchez, LM, Mena, R, Guil-Luna, S, Conde, F, Hernández, V, et al. A role for endothelial nitric oxide synthase in intestinal stem cell proliferation and mesenchymal colorectal cancer. BMC Biol (2018) 16(1):1–14. doi:10.1186/S12915-017-0472-5/FIGURES/6
61. Bosma, EK, Darwesh, S, Habani, YI, Cammeraat, M, Serrano Martinez, P, van Breest Smallenburg, ME, et al. Differential roles of eNOS in late effects of VEGF-A on hyperpermeability in different types of endothelial cells. Scientific Rep (2023) 13(1):21436. doi:10.1038/s41598-023-46893-4
62. Tran, N, Garcia, T, Aniqa, M, Ali, S, Ally, A, and Nauli, SM. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: in Physiology and in Disease States. Am J Biomed Sci and Res (2022) 15(2):153–77. doi:10.34297/ajbsr.2022.15.002087
63. Garza Treviño, EN, Quiroz Reyes, AG, Rojas Murillo, JA, de la Garza Kalife, DA, Delgado Gonzalez, P, Islas, JF, et al. Cell Therapy as Target Therapy against Colon Cancer Stem Cells. Int J Mol Sci (2023) 24(9):8163. doi:10.3390/ijms24098163
64. Lu, J, Kornmann, M, and Traub, B. Role of Epithelial to Mesenchymal Transition in Colorectal Cancer. Int J Mol Sci (2023) 24(19):14815. doi:10.3390/ijms241914815
65. Ambs, S, Merriam, WG, Ogunfusika, MO, Bennett, WP, Ishibe, N, Hussain, SP, et al. p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat Med (1998) 4(12):1371–6. doi:10.1038/3957
66. Du, J, Filipović, MR, Wagner, BA, and Buettner, GR. Ascorbate mediates the non-enzymatic reduction of nitrite to nitric oxide. Adv Redox Res (2023) 9:100079. doi:10.1016/J.ARRES.2023.100079
67. del Moral-Martinez, M, Sánchez-Uceta, P, Clemente-Gonzalez, R, Moreno-SanJuan, S, Puentes-Pardo, JD, Khaldy, H, et al. iNOS-Produced Nitric Oxide from Cancer Cells as an Intermediate of Stemness Regulation by PARP-1 in Colorectal Cancer. Biomolecules (2025) 15(1):125. doi:10.3390/biom15010125
68. Cianchi, F, Cortesini, C, Fantappiè, O, Messerini, L, Schiavone, N, Vannacci, A, et al. Inducible nitric oxide synthase expression in human colorectal cancer: Correlation with tumor angiogenesis. The Am J Pathol (2003) 162(3):793–801. doi:10.1016/S0002-9440(10)63876-X
69. Gochman, E, Mahajna, J, Shenzer, P, Dahan, A, Blatt, A, Elyakim, R, et al. The expression of iNOS and nitrotyrosine in colitis and colon cancer in humans. Acta Histochem (2012) 114(8):827–35. doi:10.1016/J.ACTHIS.2012.02.004
70. Myśliwiec, A, Bartusik-Aebisher, D, and Aebisher, D. The Role of Nitric Oxide in Cancer Treatment: Ally or Foe? Molecules (2025) 30(13):2802. doi:10.3390/MOLECULES30132802
71. Brüne, B. Nitric oxide: NO apoptosis or turning it ON? Cell Death and Differ (2003) 10(8):864–9. doi:10.1038/sj.cdd.4401261
72. Reddy, TP, Glynn, SA, Billiar, TR, Wink, DA, and Chang, JC. Targeting Nitric Oxide: Say NO to Metastasis. Clin Cancer Res official J Am Assoc Cancer Res (2023) 29(10):1855–68. doi:10.1158/1078-0432.CCR-22-2791
73. Wang, J, Deng, S, Cheng, D, Gu, J, Qin, L, Mao, F, et al. Engineered microparticles modulate arginine metabolism to repolarize tumor-associated macrophages for refractory colorectal cancer treatment. J Translational Med (2024) 22(1):908. doi:10.1186/S12967-024-05652-3
74. Ghonim, MA, Ibba, SV, Tarhuni, AF, Errami, Y, Luu, HH, Dean, MJ, et al. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J ImmunoTherapy Cancer (2021) 9(1):e001643. doi:10.1136/JITC-2020-001643
75. Shen, W, Li, Y, Yang, Z, Li, W, Cao, Y, Liu, Y, et al. Tumor microenvironment reprogramming combined with immunogenic enhancement by nanoemulsions potentiates immunotherapy. J Nanobiotechnology (2024) 22(1):154. doi:10.1186/s12951-024-02401-y
76. Rafa, H, Benkhelifa, S, Aityounes, S, Saoula, H, Belhadef, S, Belkhelfa, M, et al. All-Trans Retinoic Acid Modulates TLR4/NF-κB Signaling Pathway Targeting TNF-α and Nitric Oxide Synthase 2 Expression in Colonic Mucosa during Ulcerative Colitis and Colitis Associated Cancer. Mediators Inflamm (2017) 2017(1):1–16. doi:10.1155/2017/7353252
77. Kapoor, S, and Padwad, YS. Phloretin suppresses intestinal inflammation and maintained epithelial tight junction integrity by modulating cytokines secretion in in vitro model of gut inflammation. Cell Immunol (2023) 391-392:104754–392. doi:10.1016/j.cellimm.2023.104754
78. Suh, SS, Hong, JM, Kim, EJ, Jung, SW, Kim, SM, Kim, JE, et al. Anti-inflammation and anti-cancer activity of ethanol extract of antarctic freshwater microalga, Micractinium sp. Int J Med Sci (2018) 15(9):929–36. doi:10.7150/IJMS.26410
79. Nakamura, K, Yaguchi, T, Ohmura, G, Kobayashi, A, Kawamura, N, Iwata, T, et al. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci (2018) 109(1):54–64. doi:10.1111/cas.13423
80. Manrique, SZ, Dominguez, AL, Mirza, N, Spencer, CD, Bradley, JM, Finke, JH, et al. Definitive activation of endogenous antitumor immunity by repetitive cycles of cyclophosphamide with interspersed Tolllike receptor agonists. Oncotarget (2016) 7(28):42919–42. doi:10.18632/ONCOTARGET.10190
81. Benkhelifa, S, Rafa, H, Belhadef, S, Ait-kaci, H, Medjeber, O, Belkhelfa, M, et al. Aberrant up-regulation of iNOS/NO system is correlated with an increased abundance of Foxp3+ cells and reduced effector/memory cell markers expression during colorectal cancer: immunomodulatory effects of cetuximab combined with chemotherapy. Inflammopharmacology (2019) 27(4):685–700. doi:10.1007/S10787-019-00566-9
82. Homma, T, Kobayashi, S, Conrad, M, Konno, H, Yokoyama, C, and Fujii, J. Nitric oxide protects against ferroptosis by aborting the lipid peroxidation chain reaction. Nitric Oxide (2021) 115:34–43. doi:10.1016/j.niox.2021.07.003
83. Zhu, L, Zhang, C, and Liu, Q. PTEN S-nitrosylation by NOS1 inhibits autophagy in NPC cells. Cell Death and Dis (2019) 10(4):306–3. doi:10.1038/s41419-019-1542-0
84. Kelleher, ZT, Potts, EN, Brahmajothi, MV, Foster, MW, Auten, RL, Michael Foster, W, et al. NOS2 regulation of LPS-induced airway inflammation via S-nitrosylation of NF-κB p65. Am J Physiol - Lung Cell Mol Physiol (2011) 301(3):327–33. doi:10.1152/AJPLUNG.00463.2010/ASSET/IMAGES/LARGE/ZH50091159280005.JPEG
{kind=link}
85. Wu, Y, and Zhu, BT. Role of protein disulfide isomerase in mediating sulfasalazine-induced ferroptosis in HT22 cells: The PDI−NOS–NO−ROS/lipid-ROS cascade. Arch Biochem Biophys (2025) 768:110366. doi:10.1016/J.ABB.2025.110366
86. Feng, Z, Min, L, Chen, H, Deng, W, Tan, M, Liu, H, et al. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol (2021) 43:101984. doi:10.1016/J.REDOX.2021.101984
87. Mao, G, Xin, D, Wang, Q, and Lai, D. Sodium molybdate inhibits the growth of ovarian cancer cells via inducing both ferroptosis and apoptosis. Free Radic Biol Med (2022) 182:79–92. doi:10.1016/j.freeradbiomed.2022.02.023
88. Krümmel, B, Plötz, T, Jörns, A, Lenzen, S, and Mehmeti, I. The central role of glutathione peroxidase 4 in the regulation of ferroptosis and its implications for pro-inflammatory cytokine-mediated beta-cell death. Biochim Biophys Acta - Mol Basis Dis (2021) 1867(6):166114. doi:10.1016/j.bbadis.2021.166114
89. Zhang, GY, Gao, Y, Guo, XY, Wang, GH, and Guo, CX. MiR-199a-5p promotes ferroptosis-induced cardiomyocyte death responding to oxygen–glucose deprivation/reperfusion injury via inhibiting Akt/eNOS signaling pathway. The Kaohsiung J Med Sci (2022) 38(11):1093–102. doi:10.1002/KJM2.12605
90. Dar, HH, Anthonymuthu, TS, Ponomareva, LA, Souryavong, AB, Shurin, GV, Kapralov, AO, et al. A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO⋅ sabotage of theft-ferroptosis. Redox Biol (2021) 45:102045. doi:10.1016/j.redox.2021.102045
91. Zheng, ZL, Wang, XP, Hu, YF, Li, WG, Zhou, Q, Xu, F, et al. Propofol Suppresses Ferroptosis via Modulating eNOS/NO Signaling Pathway to Improve Traumatic Brain Injury. Brain Behav (2024) 14(12):e70187. doi:10.1002/BRB3.70187
92. Huang, X, Hou, MJ, and Zhu, BT. Protection of HT22 neuronal cells against chemically-induced ferroptosis by catechol estrogens: protein disulfide isomerase as a mechanistic target. Scientific Rep (2024) 14(1):23988. doi:10.1038/s41598-024-74742-5
93. Li, H, Feng, X, Hu, Y, Wang, J, Huang, C, and Yao, X. Development of a prognostic model based on ferroptosis-related genes for colorectal cancer patients and exploration of the biological functions of NOS2 in vivo and in vitro. Front Oncol (2023) 13:1133946. doi:10.3389/fonc.2023.1133946
94. Guo, Z, Zhuang, H, and Shi, X. Therapeutic efficacy of ferroptosis in the treatment of colorectal cancer (Review). Oncol Lett (2024) 28(6):563. doi:10.3892/ol.2024.14697
95. Sui, X, Zhang, R, Liu, S, Duan, T, Zhai, L, Zhang, M, et al. RSL3 drives ferroptosis through GPX4 inactivation and ros production in colorectal cancer. Front Pharmacol (2018) 9(NOV):425764. doi:10.3389/FPHAR.2018.01371/BIBTEX
96. Yang, J, Mo, J, Dai, J, Ye, C, Cen, W, Zheng, X, et al. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death and Dis (2021) 12(11):1079–11. doi:10.1038/s41419-021-04367-3
97. Zhao, X, and Chen, F. Propofol induces the ferroptosis of colorectal cancer cells by downregulating STAT3 expression. Oncol Lett (2021) 22(5):767. doi:10.3892/ol.2021.13028
98. Tian, X, Li, S, and Ge, G. Apatinib Promotes Ferroptosis in Colorectal Cancer Cells by Targeting ELOVL6/ACSL4 Signaling. Cancer Management Res (2021) 13:1333–42. doi:10.2147/CMAR.S274631
99. Xia, Y, Liu, S, Li, C, Ai, Z, Shen, W, Ren, W, et al. Discovery of a novel ferroptosis inducer-talaroconvolutin A—killing colorectal cancer cells in vitro and in vivo. Cell Death and Dis (2020) 11(11):988. doi:10.1038/s41419-020-03194-2
100. Wu, Y, Pi, D, Zhou, S, Yi, Z, Dong, Y, Wang, W, et al. Ginsenoside Rh3 induces pyroptosis and ferroptosis through the Stat3/p53/NRF2 axis in colorectal cancer cells. Acta Biochim Biophys Sinica (2023) 55(4):587–600. doi:10.3724/abbs.2023068
101. Jiang, L, Kon, N, Li, T, Wang, S-J, Su, T, Hibshoosh, H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi:10.1038/nature14344
102. Zhao, Y, Wang, C, and Goel, A. Andrographis overcomes 5-fluorouracil-associated chemoresistance through inhibition of DKK1 in colorectal cancer. Carcinogenesis (2021) 42(6):814–25. doi:10.1093/carcin/bgab027
103. Cao, JY, and Dixon, SJ. Mechanisms of ferroptosis. Cell Mol Life Sci (2016) 73(11–12):2195–209. doi:10.1007/s00018-016-2194-1
104. Zhang, Y, and Xie, J. Targeting ferroptosis regulators by natural products in colorectal cancer. Front Pharmacol (2024) 15:1374722. doi:10.3389/fphar.2024.1374722
105. Liu, L, Yao, H, Zhou, X, Chen, J, Chen, G, Shi, X, et al. MiR-15a-3p regulates ferroptosis via targeting glutathione peroxidase GPX4 in colorectal cancer. Mol Carcinogenesis (2022) 61(3):301–10. doi:10.1002/mc.23367
106. Du, J, Wang, T, Li, Y, Zhou, Y, Wang, X, Yu, X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med (2019) 131:356–69. doi:10.1016/j.freeradbiomed.2018.12.011
107. Martino, E, Balestrieri, A, Aragona, F, Bifulco, G, Mele, L, Campanile, G, et al. MiR-148a-3p Promotes Colorectal Cancer Cell Ferroptosis by Targeting SLC7A11. Cancers (2023) 15(17):4342. doi:10.3390/cancers15174342
108. Jiang, Z, Liu, Y, Zhang, C-H, Chu, T, Yang, Y-L, Zhu, Y-W, et al. Emerging roles of hydrogen sulfide in colorectal cancer. Chemico-Biological Interactions (2024) 403:111226. doi:10.1016/j.cbi.2024.111226
109. Bogdan, C, Röllinghoff, M, and Diefenbach, A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol (2000) 12(1):64–76. doi:10.1016/S0952-7915(99)00052-7
110. Chowdhury, R, Godoy, LC, Thiantanawat, A, Trudel, LJ, Deen, WM, and Wogan, GN. Nitric oxide produced endogenously is responsible for hypoxia-induced HIF-1α stabilization in colon carcinoma cells. Chem Res Toxicol (2012) 25(10):2194–202. doi:10.1021/tx300274a
111. Lee, JY, Hirota, SA, Glover, LE, Armstrong, GD, Beck, PL, and Macdonald, JA. Effects of Nitric Oxide and Reactive Oxygen Species on HIF-1a Stabilization Following Clostridium DifficileToxin Exposure of the Caco-2 Epithelial Cell Line. Cell Physiol Biochem (2013) 32(2):417–30. doi:10.1159/000354448
112. Lee, VY, McClintock, DS, Santore, MT, Budinger, GRS, and Chandel, NS. Hypoxia sensitizes cells to nitric oxide-induced apoptosis. J Biol Chem (2002) 277(18):16067–74. doi:10.1074/jbc.M111177200
113. Lee, M, Wang, C, Jin, SW, Labrecque, MP, Beischlag, TV, Brockman, MA, et al. Expression of human inducible nitric oxide synthase in response to cytokines is regulated by hypoxia-inducible factor-1. Free Radic Biol Med (2019) 130:278–87. doi:10.1016/J.FREERADBIOMED.2018.10.441
115. Ikeda, A, Nagayama, S, Sumazaki, M, Konishi, M, Fujii, R, Saichi, N, et al. Colorectal Cancer–Derived CAT1-Positive Extracellular Vesicles Alter Nitric Oxide Metabolism in Endothelial Cells and Promote Angiogenesis. Mol Cancer Res (2021) 19(5):834–46. doi:10.1158/1541-7786.MCR-20-0827
116. Kamerkar, S, Leng, C, Burenkova, O, Jang, SC, McCoy, C, Zhang, K, et al. Exosome-mediated genetic reprogramming of tumor-associated macrophages by exoASO-STAT6 leads to potent monotherapy antitumor activity. Sci Adv (2022) 8(7):eabj7002. doi:10.1126/SCIADV.ABJ7002
117. Daw, J, Chung, S, Chen, CY, Heimark, RL, and Montfort, WR. Real-time nitric oxide detection in cytokine stimulated cancer cells and macrophages. Nitric Oxide (2025) 156:42–9. doi:10.1016/J.NIOX.2025.02.004
118. Hassan, AY, Maulood, IM, and Salihi, A. The vasodilatory mechanism of nitric oxide and hydrogen sulfide in the human mesenteric artery in patients with colorectal cancer. Exp Ther Med (2021) 21(3):214. doi:10.3892/ETM.2021.9646
119. Cao, M, Wang, Y, Lu, G, Qi, H, Li, P, Dai, X, et al. Classical Angiogenic Signaling Pathways and Novel Anti-Angiogenic Strategies for Colorectal Cancer. Curr Issues Mol Biol (2022) 44(10):4447–71. doi:10.3390/cimb44100305
120. Chen, C, Jiang, X, and Zhao, Z. Inhibition or promotion, the potential role of arginine metabolism in immunotherapy for colorectal cancer. All Life (2023) 16(1):2163306. doi:10.1080/26895293.2022.2163306
121. Li, M, Cao, F, Wang, W, Ma, Y, Yu, Z, Wang, K, et al. Coumarin-Furoxan Hybrid Suppressed the Proliferation and Metastasis of Triple-Negative Breast Cancer by Activating Mitochondrial Stress and Cell Apoptosis. ACS Pharmacol and translational Sci (2024) 7(5):1278–90. doi:10.1021/acsptsci.3c00329
122. Ding, K, Liu, C, Li, L, Yang, M, Jiang, N, Luo, S, et al. Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chin Med J (2023) 136(21):2521–37. doi:10.1097/CM9.0000000000002533
123. Kapralov, AA, Yang, Q, Dar, HH, Tyurina, YY, Anthonymuthu, TS, Kim, R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16(3):278–90. doi:10.1038/S41589-019-0462-8
124. Fujii, J, and Osaki, T. Involvement of Nitric Oxide in Protecting against Radical Species and Autoregulation of M1-Polarized Macrophages through Metabolic Remodeling. Molecules (2023) 28(2):814. doi:10.3390/MOLECULES28020814
125. Negi, M, Kaushik, N, Lamichhane, P, Patel, P, Jaiswal, A, Choi, EH, et al. Nitric oxide water-driven immunogenic cell death: Unfolding mitochondrial dysfunction’s role in sensitizing lung adenocarcinoma to ferroptosis and autophagic cell death. Free Radic Biol Med (2024) 222:1–15. doi:10.1016/J.FREERADBIOMED.2024.05.033
126. Wang, M, Zhang, Z, Li, Q, Liu, R, Li, J, and Wang, X. Multifunctional nanoplatform with near-infrared triggered nitric-oxide release for enhanced tumor ferroptosis. J Nanobiotechnology (2024) 22(1):656. doi:10.1186/s12951-024-02942-2
127. Yang, Z, Yang, C, Yang, D, Zhang, Y, Yang, Q, Qu, F, et al. l-Arginine-Modified CoWO4/FeWO4 S-Scheme Heterojunction Enhances Ferroptosis against Solid Tumor. Adv Healthc Mater (2023) 12(18):2203092. doi:10.1002/ADHM.202203092
128. Jiang, L, Zheng, H, Lyu, Q, Hayashi, S, Sato, K, Sekido, Y, et al. Lysosomal nitric oxide determines transition from autophagy to ferroptosis after exposure to plasma-activated Ringer’s lactate. Redox Biol (2021) 43:101989. doi:10.1016/J.REDOX.2021.101989
129. Sanchez-Pino, MJ, Moreno, P, and Navarro, A. Mitocondrial dysfunction in human colorectal cancer progression. Front Biosci (2007) 12(4):1190–9. doi:10.2741/2137
130. Cruz-Ojeda, Pd. l., Navarro-Villarán, E, Fuertes-Agudo, M, Mata, A, López-Lluch, G, Navas, P, et al. Peroxynitrite is involved in the mitochondrial dysfunction induced by Sorafenib in liver cancer cells. Free Radic Biol Med (2025) 229:251–63. doi:10.1016/J.FREERADBIOMED.2024.12.053
131. Li, J, Sun, Y, Yan, R, Wu, X, Zou, H, and Meng, Y. Urea transporter B downregulates polyamines levels in melanoma B16 cells via p53 activation. Biochim Biophys Acta - Mol Cell Res (2022) 1869(5):119236. doi:10.1016/J.BBAMCR.2022.119236
132. Martinez-Outschoorn, UE, Balliet, RM, Rivadeneira, DB, Chiavarina, B, Pavlides, S, Wang, C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution. Cell Cycle (2010) 9(16):3276–96. doi:10.4161/CC.9.16.12553
133. Concato-Lopes, VM, Silva, TF, Detoni, MB, Cruz, EMS, Gonçalves, MD, da Silva Bortoleti, BT, et al. 3,3’,5,5’-Tetramethoxybiphenyl-4,4’diol triggers oxidative stress, metabolic changes, and apoptosis-like process by reducing the PI3K/AKT/NF-κB pathway in the NCI-H460 lung cancer cell line. Biomed and Pharmacother (2024) 170:115979. doi:10.1016/J.BIOPHA.2023.115979
134. Dai, W, Deng, Y, Chen, X, Huang, Y, Hu, H, Jin, Q, et al. A mitochondria-targeted supramolecular nanoplatform for peroxynitrite-potentiated oxidative therapy of orthotopic hepatoma. Biomaterials (2022) 290:121854. doi:10.1016/J.BIOMATERIALS.2022.121854
135. Zheng, L, Li, Z, Wang, R, Wang, J, Liu, B, Wang, Y, et al. A novel photosensitizer DTPP-mediated photodynamic therapy induces oxidative stress and apoptosis through mitochondrial pathways in LA795 cells. Photodiagnosis Photodynamic Ther (2024) 45:103894. doi:10.1016/J.PDPDT.2023.103894
136. Guizzardi, S, Picotto, G, Rodriguez, V, Welsh, J, Narvaez, C, Bohl, L, et al. Combined treatment of menadione and calcitriol increases the antiproliferative effect by promoting oxidative/nitrosative stress, mitochondrial dysfunction, and autophagy in breast cancer mcf-7 cells. Can J Physiol Pharmacol (2020) 98(8):548–56. doi:10.1139/CJPP-2019-0585
137. Tu, J, Tu, K, Xu, H, Wang, L, Yuan, X, Qin, X, et al. Improving tumor hypoxia and radiotherapy resistance via in situ nitric oxide release strategy. Eur J Pharmaceutics Biopharmaceutics (2020) 150:96–107. doi:10.1016/J.EJPB.2020.03.003
138. Dou, Y, Zhao, F, Li, X, and Guo, Y. Monitoring Nitric Oxide-Induced Hypoxic Tumor Radiosensitization by Radiation-Activated Nanoagents under BOLD/DWI Imaging. ACS Biomater Sci and Eng (2021) 7(11):5242–54. doi:10.1021/acsbiomaterials.1c00543
139. Zhang, Y, Zhang, J, Luo, T, Cai, Z, Yang, G, Li, H, et al. Sononeoperfusion effect by ultrasound and microbubble promotes nitric oxide release to alleviate hypoxia in a mouse MC38 tumor model. Ultrason Sonochem (2023) 100:106619. doi:10.1016/j.ultsonch.2023.106619
140. Zhao, Y, Shi, D, Guo, L, Shang, M, Sun, X, Meng, D, et al. Ultrasound targeted microbubble destruction-triggered nitric oxide release via nanoscale ultrasound contrast agent for sensitizing chemoimmunotherapy. J Nanobiotechnology (2023) 21(1):35. doi:10.1186/S12951-023-01776-8
141. Chen, Y, Fang, L, Zhou, W, Chang, J, Zhang, X, He, C, et al. Nitric oxide-releasing micelles with intelligent targeting for enhanced anti-tumor effect of cisplatin in hypoxia. J Nanobiotechnology (2021) 19(1):1–20. doi:10.1186/S12951-021-00989-Z/FIGURES/10
142. Graham, C, Barsoum, I, Kim, J, Black, M, and Siemens, RD. Mechanisms Of Hypoxia-Induced Immune Escape In Cancer And Their Regulation By Nitric Oxide. Redox Biol (2015) 5:417. doi:10.1016/j.redox.2015.09.022
143. Barsoum, IB, Hamilton, TK, Li, X, Cotechini, T, Miles, EA, Siemens, DR, et al. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res (2011) 71(24):7433–41. doi:10.1158/0008-5472.CAN-11-2104
144. Ene, CD, and Nicolae, I. Hypoxia-Nitric Oxide Axis and the Associated Damage Molecular Pattern in Cutaneous Melanoma. J Personalized Med (2022) 12(10):1646. doi:10.3390/JPM12101646
145. Vedenko, A, Panara, K, Goldstein, G, Ramasamy, R, and Arora, H. Tumor Microenvironment and Nitric Oxide: Concepts and Mechanisms. Adv Exp Med Biol (2020) 1277:143–58. doi:10.1007/978-3-030-50224-9_10
146. Yang, C, Mu, G, Zhang, Y, Gao, Y, Zhang, W, Liu, J, et al. Supramolecular Nitric Oxide Depot for Hypoxic Tumor Vessel Normalization and Radiosensitization. Adv Mater (2022) 34(37):2202625. doi:10.1002/adma.202202625
147. Xing, D, Benavides, GA, Johnson, MS, Tian, R, Barnes, S, and Darley-Usmar, VM. Glutamine-dependent effects of nitric oxide on cancer cells subjected to hypoxia-reoxygenation. Nitric Oxide (2023) 130:22–35. doi:10.1016/J.NIOX.2022.11.003
148. Yao, W, Wang, K, Guo, Y, Wei, R, Luo, S, Tang, W, et al. Nitric oxide nano-prodrug platform with synchronous glutathione depletion and hypoxia relief for enhanced photodynamic cancer therapy. Biomater Adv (2022) 133:112616. doi:10.1016/J.MSEC.2021.112616
149. Zhang, S, Li, M, Wang, J, Zhou, Y, Dai, P, Zhao, M, et al. NIR-Triggered On-Demand Nitric Oxide Release for Enhanced Synergistic Phototherapy of Hypoxic Tumor. Bioconjug Chem (2023) 34(7):1327–35. doi:10.1021/acs.bioconjchem.3c00250
150. Fens, MH, Cabrales, P, Scicinski, J, Larkin, SK, Suh, JH, Kuypers, FA, et al. Targeting tumor hypoxia with the epigenetic anticancer agent, RRx-001: a superagonist of nitric oxide generation. Med Oncol (2016) 33(8):1–11. doi:10.1007/S12032-016-0798-9/METRICS
151. Chattopadhyay, A, Jagdish, S, Karhale, AK, Ramteke, NS, Zaib, A, and Nandi, D. IFN-γ lowers tumor growth by increasing glycolysis and lactate production in a nitric oxide-dependent manner: implications for cancer immunotherapy. Front Immunol (2023) 14:1282653. doi:10.3389/fimmu.2023.1282653
152. Du, T, and Han, J. Arginine Metabolism and Its Potential in Treatment of Colorectal Cancer. Front Cell Developmental Biol (2021) 9:658861. doi:10.3389/fcell.2021.658861
153. Krzystek-Korpacka, M, Szczęśniak-Sięga, B, Szczuka, I, Fortuna, P, Zawadzki, M, Kubiak, A, et al. L-Arginine/Nitric Oxide Pathway Is Altered in Colorectal Cancer and Can Be Modulated by Novel Derivatives from Oxicam Class of Non-Steroidal Anti-Inflammatory Drugs. Cancers (2020) 12(9):2594. doi:10.3390/CANCERS12092594
154. Dowling, JK, Afzal, R, Gearing, LJ, Cervantes-Silva, MP, Annett, S, Davis, GM, et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat Commun (2021) 12(1):1460. doi:10.1038/S41467-021-21617-2
155. Vanella, L, Di Giacomo, C, Acquaviva, R, Santangelo, R, Cardile, V, Barbagallo, I, et al. The DDAH/NOS pathway in human prostatic cancer cell lines: Antiangiogenic effect of L-NAME. Int J Oncol (2011) 39:1303–10. doi:10.3892/ijo.2011.1107
156. Szczęśniak-Sięga, BM, Gębczak, K, Gębarowski, T, and Maniewska, J. Synthesis, COX-1/2 inhibition and antioxidant activities of new oxicam analogues designed as potential chemopreventive agents. Acta Biochim Pol (2018) 65(2):199–207. doi:10.18388/abp.2018_2614
157. Lewandowska, P, Szczuka, I, Bednarz-Misa, I, Szczęśniak-Sięga, BM, Neubauer, K, Mierzchała-Pasierb, M, et al. Modulating Properties of Piroxicam, Meloxicam and Oxicam Analogues against Macrophage-Associated Chemokines in Colorectal Cancer. Molecules (Basel, Switzerland) (2021) 26(23):7375. doi:10.3390/MOLECULES26237375
158. Szczęśniak-Sięga, BM, Mogilski, S, Wiglusz, RJ, Janczak, J, Maniewska, J, Malinka, W, et al. Synthesis and pharmacological evaluation of novel arylpiperazine oxicams derivatives as potent analgesics without ulcerogenicity. Bioorg and Med Chem (2019) 27(8):1619–28. doi:10.1016/j.bmc.2019.03.007
159. Szczuka, I, Wierzbicki, J, Serek, P, Szczęśniak-Sięga, BM, and Krzystek-Korpacka, M. Heat Shock Proteins HSPA1 and HSP90AA1 Are Upregulated in Colorectal Polyps and Can Be Targeted in Cancer Cells by Anti-Inflammatory Oxicams with Arylpiperazine Pharmacophore and Benzoyl Moiety Substitutions at Thiazine Ring. Biomolecules (2021) 11(11):1588. doi:10.3390/biom11111588
160. Środa-Pomianek, K, Michalak, K, Palko-Łabuz, A, Uryga, A, Szczęśniak-Sięga, B, and Wesołowska, O. Simvastatin Strongly Augments Proapoptotic, Anti-inflammatory and Cytotoxic Activity of Oxicam Derivatives in Doxorubicin-resistant Colon Cancer Cells. Anticancer Res (2019) 39(2):727–34. doi:10.21873/anticanres.13169
161. Abdul Wanees El-Awdan, S, Al-Shafeey, N, Salam, OA, Ibrahim El-Iraqy, W, and Abdul Bakky Kenawy, S. Modulation of the pharmacological properties of meloxicam by octreotide in rats. J Saudi Chem Soc (2015) 19(2):123–32. doi:10.1016/j.jscs.2012.01.002
162. Gilroy, DW. New insights into the anti-inflammatory actions of aspirin-induction of nitric oxide through the generation of epi-lipoxins. Memórias do Instituto Oswaldo Cruz (2005) 100(Suppl. 1):49–54. doi:10.1590/S0074-02762005000900009
163. Liang, F, Wang, S, Guo, Y, Mu, Y, Shang, FJ, and Wang, M. Proteome profiling of endogenous and potential S-nitrosylation in colorectal cancer. Front Endocrinol (2023) 14:1153719. doi:10.3389/FENDO.2023.1153719
164. Mohammadian, M, Rostamzadeh Khameneh, Z, Emamgholizadeh Minaei, S, Ebrahimifar, M, and Esgandari, K. Regulatory Effects of Apatinib in Combination with Piperine on MDM- 2 Gene Expression, Glutathione Peroxidase Activity and Nitric Oxide Level as Mechanisms of Cytotoxicity in Colorectal Cancer Cells. Adv Pharm Bull (2022) 12(2):404–9. doi:10.34172/APB.2022.040
165. Staniszewska, AD, Armenia, J, King, M, Michaloglou, C, Reddy, A, Singh, M, et al. PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors. OncoImmunology (2022) 11(1):2083755. doi:10.1080/2162402X.2022.2083755
166. Lamaudière, MTF, Arasaradnam, R, Weedall, GD, and Morozov, IY. The Colorectal Cancer Microbiota Alter Their Transcriptome To Adapt to the Acidity, Reactive Oxygen Species, and Metabolite Availability of Gut Microenvironments. MSphere (2023) 8(2):e0062722–22. doi:10.1128/MSPHERE.00627-22
167. Xue, X, Ramakrishnan, SK, Weisz, K, Triner, D, Xie, L, Attili, D, et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab (2016) 24(3):447–61. doi:10.1016/J.CMET.2016.07.015
168. Du, Q, Liu, S, Dong, K, Cui, X, Luo, J, and Geller, DA. Downregulation of iNOS/NO Promotes Epithelial-Mesenchymal Transition and Metastasis in Colorectal Cancer. Mol Cancer Res (2023) 21(2):102–14. doi:10.1158/1541-7786.MCR-22-0509
169. Gao, Y, Zhou, S, Pang, L, Yang, J, Li, HJ, Huo, X, et al. Celastrol suppresses nitric oxide synthases and the angiogenesis pathway in colorectal cancer. Free Radic Res (2019) 53(3):324–34. doi:10.1080/10715762.2019.1575512
170. Gao, Y, Zhou, S, Xu, Y, Sheng, S, Qian, SY, and Huo, X. Nitric oxide synthase inhibitors 1400W and L-NIO inhibit angiogenesis pathway of colorectal cancer. Nitric Oxide (2019) 83:33–9. doi:10.1016/J.NIOX.2018.12.008
171. Ko, EJ, Kim, EJ, Cho, HJ, Oh, J, Park, HS, Ryu, CS, et al. Prognostic significance of three endothelial nitric oxide synthase (eNOS) polymorphisms and metabolic syndrome (MetS) in patients with colorectal cancer. Genes and Genomics (2022) 44(6):659–70. doi:10.1007/S13258-022-01246-9
172. Lin, R, Piao, M, Song, Y, and Liu, C. Quercetin Suppresses AOM/DSS-Induced Colon Carcinogenesis through Its Anti-Inflammation Effects in Mice. J Immunol Res (2020) 2020:1–10. doi:10.1155/2020/9242601
173. Kopecka, J, Campia, I, Brusa, D, Doublier, S, Matera, L, Ghigo, D, et al. Nitric oxide and P-glycoprotein modulate the phagocytosis of colon cancer cells. J Cell Mol Med (2011) 15(7):1492–504. doi:10.1111/J.1582-4934.2010.01137.X
174. Cianchi, F, Cortesini, C, Fantappiè, O, Messerini, L, Sardi, I, Lasagna, N, et al. Cyclooxygenase-2 activation mediates the proangiogenic effect of nitric oxide in colorectal cancer. Clin Cancer Res : An Official J Am Assoc Cancer Res (2004) 10(8):2694–704. doi:10.1158/1078-0432.CCR-03-0192
175. Coutinho, LL, Femino, EL, Gonzalez, AL, Moffat, RL, Heinz, WF, Cheng, RYS, et al. NOS2 and COX-2 Co-Expression Promotes Cancer Progression: A Potential Target for Developing Agents to Prevent or Treat Highly Aggressive Breast Cancer. Int J Mol Sci (2024) 25(11):6103. doi:10.3390/IJMS25116103
176. Ridnour, L. A., Thomas, D. D., Donzelli, S., Espey, M. G., Roberts, D. D., Wink, D. A., et al. (2006). The biphasic nature of nitric oxide responses in tumor biolog. Antioxid Redox Signal 8 (7-8), 1329–37. doi:10.1089/ars.2006.8.1329
177. Gao, C., Li, X., Liu, T., Wang, W., Wu, J., et al. (2025). An overview of phenylsulfonylfuroxan-based nitric oxide donors for cancer treatment. Bioorg Chem. 154, 108020. doi:10.1016/j.bioorg.2024.108020
178. Tian, X., Li, S., and Ge, G. (2021). Apatinib Promotes Ferroptosis in Colorectal Cancer Cells by Targeting ELOVL6/ACSL4 Signaling. Cancer Manag Res.13, 1333–1342. doi:10.2147/CMAR.S274631
180. Środa-pomianek, K, Michalak, K, Palko-łabuz, A, Uryga, A, Szczęśniak-sięga, B, Wesołowska, O, et al. Simvastatin Strongly Augments Proapoptotic, Anti-inflammatory and Cytotoxic Activity of Oxicam Derivatives in Doxorubicin-resistant Colon Cancer Cells. Anticancer Research (2019) 39(2):727–734.
181. Xu, F, Wang, M, Dotse, E, Chow, KT, and Lo, PC. Inducing immunogenic cancer cell death through oxygen-economized photodynamic therapy with nitric oxide-releasing photosensitizers. Angewandte Chemie (2024) 63(37).
Glossary
APC Adenomatous polyposis coli
ALOX15 Arachidonate 15-lipoxygenase
ADMA Asymmetric dimethyl arginine
CHAC1 Cation transport regulator homolog 1
CTCs Circulating tumor cells
CBS Cystathionine β-synthase
CSE Cystathionine γ-lyase
COX-2 Cyclooxygenase-2
DDAH1 Dimethylarginine dimethylaminohydrolase 1
eNOS Endothelial nitric oxide synthase
EMT Epithelial–mesenchymal transition
ERK Extracellular signal-regulated kinases
EVs Extracellular vesicles
GAPDH Glyceraldehyde-3-phosphate dehydrogenase
GLS2 Glutaminase 2
GPX4 Glutathione peroxidase 4
CAT1 High-affinity cationic amino acid transporter 1
H2S Hydrogen sulfide
iNOS Inducible nitric oxide synthase
IFN-γ Interferon-gamma
IL-6 Interleukin-6
Substrate for nitric oxide synthesis L-arginine
MAPK Mitogen-activated protein kinases
MDSCs Myeloid-derived suppressor cells
NOX4 NADPH oxidase 4
nNOS Neuronal nitric oxide synthase
NO Nitric oxide
NOS Nitric oxide synthase
PPP Pentose phosphate pathway
ONOO− Peroxynitrite
PDE2A Phosphodiesterase 2A
PDE5A Phosphodiesterase 5A
PRMT1 Protein arginine methyltransferase 1
PRMT5 Protein arginine methyltransferase 5
PKG Protein kinase G
RNS Reactive nitrogen species
ROS Reactive oxygen species
SNO S-nitrosylation
sGC Soluble guanylate cyclase
TMEM180 Transmembrane protein 180
TAMs Tumor-associated macrophages
TNF-α Tumor necrosis factor-alpha
VEGF Vascular endothelial growth factor
Keywords: ferroptosis, angiogenesis, metabolic reprogramming, extracellular vesicles, oxidative stress, tumor microenvironment
Citation: Tatode A, Chaudhary AA, Qutub M, Trivedi R, Umekar M, Ali MAM and Premchandani T (2025) Dissecting the opposing regulatory functions of endogenous nitric oxide production in colorectal cancer initiation, adaptive immune response alterations, and ferroptosis execution. Oncol. Rev. 19:1671235. doi: 10.3389/or.2025.1671235
Received: 22 July 2025; Accepted: 11 September 2025;
Published: 05 November 2025.
Edited by:
Priyanka Sharma, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Lei Yin, Shanghai Jiaotong University School of Medicine, ChinaSitaramaraju Adduri, University of Texas at Tyler, United States
Copyright © 2025 Tatode, Chaudhary, Qutub, Trivedi, Umekar, Ali and Premchandani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Qutub, cXV0dWJtYWxpa3FtQGdtYWlsLmNvbQ==; Rashmi Trivedi, cmFzaG1pdHJpdmVkaW1pc2hyYUBnbWFpbC5jb20=