 Cătălin Vasile Munteanu1,2
Cătălin Vasile Munteanu1,2 Diana Luisa Lighezan3,4*
Diana Luisa Lighezan3,4* Alexandru Capcelea1,5
Alexandru Capcelea1,5 Adela Chiriță-Emandi2,6,7
Adela Chiriță-Emandi2,6,7 Adrian Pavel Trifa6,8,9
Adrian Pavel Trifa6,8,9- 1Doctoral School, Victor Babeş University of Medicine and Pharmacy, Timişoara, Romania
- 2Regional Center of Medical Genetics Timiş, Louis Țurcanu Clinical Emergency Hospital for Children, Timişoara, Romania
- 3Department of Hematology, Victor Babes University of Medicine and Pharmacy, Timişoara, Romania
- 4Multidisciplinary Research Center for Malignant Hematological Diseases, Victor Babes University of Medicine and Pharmacy, Timişoara, Romania
- 5Department of Medical Oncology, OncoHelp Oncology Center, Timişoara, Romania
- 6Department of Microscopic Morphology, Genetics Discipline, Victor Babeş University of Medicine and Pharmacy, Timişoara, Romania
- 7Center for Genomic Medicine, Victor Babeş University of Medicine and Pharmacy, Timişoara, Romania
- 8Center of Expertise on Rare Pulmonary Diseases, Victor Babeş Clinical Hospital of Infectious Diseases and Pneumophysiology, Timişoara, Romania
- 9Breast Cancer Center, The Oncology Institute “Prof. Dr. Ion Chiricuta”, Cluj-Napoca, Romania
Constitutional mismatch repair deficiency (CMMRD) is a rare pediatric cancer predisposition syndrome primarily characterised by central nervous system (CNS), gastro-intestinal (GI) tumours and hematological malignancies, along with NF1-like cutaneous features. The PMS2-related subtype (PMS2-CMMRD) is the most common molecular form of CMMRD, exhibiting variable severity and both early and late-onset clinical presentations. Although pathogenic and likely pathogenic PMS2 heterozygous variants are relatively frequent in healthy population, CMMRD incidence is generally rare in humans and genotype-phenotype correlations are still limited. To better characterise PMS2-CMMRD group, we collected clinical cases described in literature, using three alternative methods (VarChat, VarSome and LitVar2), starting from 102 pathogenic/likely pathogenic PMS2 variants (<50 bp) reported in ClinVar by clinical and research laboratories. PMS2-CMMRD cases were split into two distinct groups based on tumour onset age: early (diagnosis under 10 years) and later-onset (diagnosis after 10 years). Significant differences in tumour distribution were observed, with CNS tumours being most prevalent in the early-onset group, while GI tumours were more common in the later-onset group. Six PMS2 variants were associated with either early or later-onset CMMRD. Future validation through larger prospective cohort studies is necessary to confirm our findings and better understand the natural history of PMS2-CMMRD to inform clinical decision-making in PMS2-Lynch syndrome (PMS2-LS).
Introduction
Constitutional mismatch repair deficiency (CMMRD) syndrome (OMIM #276300, #619096, #619097, #619101) is a rare autosomal recessive cancer predisposition syndrome manifesting in childhood, associated with biallelic germline variants in mismatch repair (MMR) genes, MLH1, MSH2, MSH6 and PMS2. Affected individuals typically develop early-onset malignancies, with central nervous system, hematological and gastro-intestinal tumours being the most prevalent neoplasias in this group (1–3). As clinical phenotype in CMMRD overlaps with other rare genetic diseases, such as neurofibromatosis type 1 (NF1) and Legius syndrome (4–6), timely diagnosis plays an essential role for appropriate clinical care and genetic counselling.
Among reported CMMRD cases, those associated with biallelic PMS2 variants are the most prevalent in literature, compared to presentations involving other Lynch syndrome-associated MMR genes (7–9). In contrast, heterozygous PMS2 variants are typically associated with lower penetrance and later-onset disease, with PMS2-associated Lynch syndrome (PMS2-LS) considered the mildest and the most frequently underdiagnosed form of LS documented to date (8,10–12). However, genotype–phenotype correlations in both PMS2-CMMRD and PMS2-LS remain poorly defined. Despite the presumed high prevalence of pathogenic PMS2 variants in the general population, clinical data on disease progression in relation to specific genotypes remain scarce. For other similar recessive cancer predisposition syndromes, including Fanconi anemia (FA), emerging genotypic data are demonstrating the role of specific variants in disease development (13–15). These insights not only impact the clinical management of biallelic carriers, but also provide valuable data regarding heterozygous carriers of low penetrance variants associated with milder cancer predisposition phenotypes, contributing to more accurate risk assessment and enabling personalized follow-up strategies distinct from conventional gene-based approaches (15–17).
In this context, we aimed to systematically investigate PMS2-related CMMRD cases documented in scientific literature to date and reported in ClinVar, the most widely used clinical genomic database worldwide. The primary source of data was represented, in the vast majority of instances, by case reports and case series from which both clinical and molecular information were extracted. In our endeavour, we primarily focused on detailed genotype and phenotype characterisation of the cases under analysis, as well as on discovering potential genotype-phenotype correlations relevant for clinical practice.
Methods
Variant selection
All PMS2 variants submitted by clinical and research laboratories to ClinVar were analysed, with ClinVar serving as the genomic database for this study (last accessed 1 May 2025). Only short variants (<50 bp) classified as pathogenic (class 5) and likely pathogenic (class 4) were included. Clinical significance for all variants were established according to the ACMG/AMP 2015 guidelines (18) by independent clinical and research laboratories or expert panels (19), with all variants meeting the ClinVar one-star criteria at least. Seven variants with conflicting interpretations (uncertain significance versus pathogenic/likely pathogenic) were not considered. Variants associated with constitutional mismatch repair deficiency (CMMRD) were selected based on the presence of one of the following terms in ClinVar records: “CMMRD,” “constitutional,” “homozygous” and “compound heterozygous.”
Clinical cases discovery
After variant selection, 102 PMS2 variants were further evaluated for supporting publications in the scientific literature. PMS2 variants were annotated following the Human Genome Variation Society (HGVS) nomenclature guidelines (https://hgvs-nomenclature.org/stable/), using the MANE Select transcript (NM_000535.7; ENST00000265849.12) as reference, where c.1 denotes the first coding nucleotide. The literature review was conducted in a semi-automated manner based on the HGVS nomenclature of each variant, using three alternative tools: VarSome (https://varsome.com/) (20), LitVar2 (https://www.ncbi.nlm.nih.gov/research/litvar2/) (21) and VarChat (https://varchat.engenome.com/) (22). Moreover, citations supporting the germline classification of variants in ClinVar, as provided by other submitting laboratories, were manually reviewed. For variants with no publications identified using mentioned resources, an additional manual literature review in PubMed was performed by two independent researchers to ensure a comprehensive analysis (Figure 1).
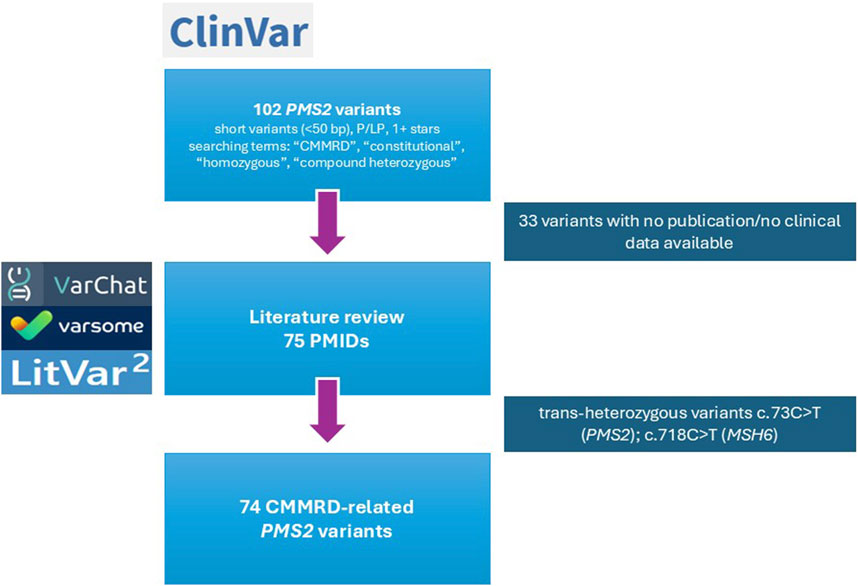 T (PMS2) and c.718C>T (MSH6). The flow is guided with arrows connecting each stage. Logos for VarChat, VarSome, and LitVar2 are on the left." id="F1" loading="lazy">
T (PMS2) and c.718C>T (MSH6). The flow is guided with arrows connecting each stage. Logos for VarChat, VarSome, and LitVar2 are on the left." id="F1" loading="lazy">
Figure 1. Flowchart of CMMRD-related PMS2 variant selection. The process involved initial variant retrieval from ClinVar, literature review using VarChat, VarSome and LitVar2 and final manual curation. Excluded variants (n = 33) were subjected to an additional manual review.
Variants with no literature evidence supporting an association with CMMRD were excluded, resulting in an initial list of 69 PMS2 variants. Following literature review, the number of short variants (<50 bp) increased to 74 and 8 exonic copy number variants (CNVs, >50 bp) were found in trans with the original variants (Figure 1).
Based on 75 PubMed indexed articles (2, 6, 10), (23–40), (41–60), (61–80), (81–94), we build a clinical database with 133 entries comprising patients and/or families with CMMRD. Two cases were excluded: 1) one case with the genotype c.[746_753del]; [1738A>T] and clinical presentation not suggestive of CMMRD (colorectal cancer at age 69) (31), 2) one case with trans-heterozygosity for c.73C>T (PMS2) and c.718C>T (MSH6), presenting with CMMRD features (glioblastoma at age 8 and café-au-lait macules) (27). Five cases with only one pathogenic PMS2 variant reported but with clinical features suggestive of CMMRD were included, under the assumption that the second variant might have gone undetected due to technical limitations. A small number of CMMRD cases incidentally identified during the literature review, for which the genotypes were not reported in ClinVar, were not further assessed.
We constructed an internal review database comprising 133 entries of individuals and families with constitutional mismatch repair deficiency (CMMRD). In the vast majority of cases, each entry represents an individual. However, for three entries, the data reflect families rather than single individuals, due to insufficient clinical details in the original publications to distinguish separate cases. When publications provided enough clinical data and genotype inference was possible, typically for individuals identified through cascade testing or those with very suggestive phenotypes, they were included separately, even if not specifically mentioned in the original papers.
Control variants
The 74 variants to study were compared with a control group consisting of 733 PMS2 short variants (<50 bp) concordantly classified as pathogenic (class 5) or likely pathogenic (class 4) in all ClinVar submissions but not associated with CMMRD. Control variants were identified by excluding any variants retrieved using the CMMRD-related keywords described in the variant selection section. Variants that were initially considered for the study group but subsequently excluded were not included in the control group.
Variant annotation and statistical analysis
Both study variants and control variants were annotated using GeneBe (https://genebe.net/) (95) and Ensembl Variant Effect Predictor (VEP) (https://ensembl.org/Homo_sapiens/Tools/VEP/). The reference human genome used was GRCh38. Variant nomenclature followed the Human Genetic Variation Society guidelines (https://hgvs-nomenclature.org/stable/). The MANE Select transcript (NM_000535.7, ENST00000265849.12) represented the reference sequence, with position c.1 being the first coding nucleotide. Splicing impact was predicted in silico using three complementary tools, SpliceAI (https://spliceailookup.broadinstitute.org/) (96), SpliceAI-visual (https://mobidetails.chu-montpellier.fr/) (97) and SPiCEv2.1 (98). Statistical analysis was performed using IBM SPSS Statistics 27. Statistical significance was defined for p-values <0.05.
Results
Targeted gene testing–the major approach for establishing definitive molecular diagnostic in CMMRD
For the majority of cases, 73/133 (53.2%), the first-tier molecular testing available was targeted PMS2 gene testing, typically guided by initial immunohistochemistry (IHC) results. This included DNA sequence analysis (based on long-range PCR, Sanger sequencing and MLPA), RNA sequencing (based on RT-PCR and Sanger sequencing) and combined testing (both DNA and RNA). In 14/133 cases (10.2%), NGS panels were the preferred diagnostic tool. Exome sequencing (both standard and enhanced versions) was used in 10/133 cases (7.3%), while genome sequencing was employed in 3/133 cases (2.1%). In 37/133 cases (27.0%), the preferred testing approach could not be definitively determined.
Brain tumours followed by gastro-intestinal tumours represent the most common sequence in the natural history of PMS2-CMMRD
In 129/133 (97%) of cases, individuals with CMMRD developed at least one tumour (Figure 2), with central nervous system tumours being the most common neoplasia in the natural history (p < 0.001, χ2). In 67/133 (50.3%) of cases, a second tumour occurred, most commonly in the gastro-intestinal tract. Only 29/133 (21.8%) of cases developed a third tumour during the disease course, with hematological (10/29, 34%) and gastro-intestinal (9/29, 31%) tumours being the most common.
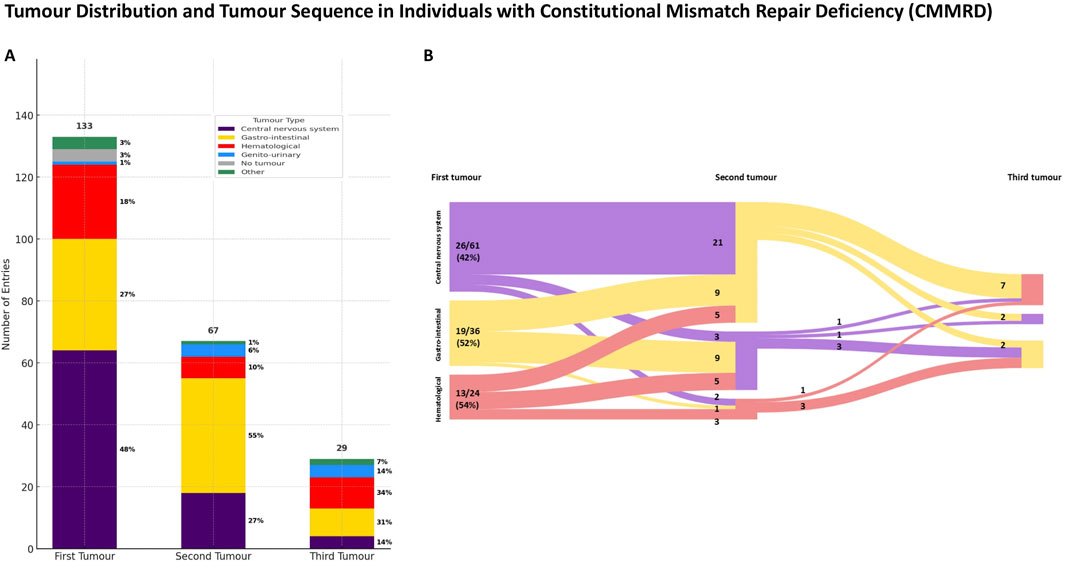
Figure 2. Tumour distribution (A) and tumour sequence (B) in individuals with PMS2-reated constitutional mismatch repair deficiency (PMS2-CMMRD). (A) Note that central nervous system tumours were the most prevalent as an initial presentation, followed by gastro-intestinal malignancies that occurred more frequently subsequently during the disease course. (B) Diagram B displays only reported cases with two or more tumours, with both absolute and relative numbers shown. Percentages in diagram B represent the proportion of cases presenting at least two neoplasms. For simplicity, cases with other tumour types were excluded.
Brain tumours are the most frequent first neoplasia in early-onset PMS2-CMMRD, while gastro-intestinal tumours predominate in later-onset cases
First neoplasia developed before age 10 in 55/129 (42.6%) of cases, whereas 66/129 (51.1%) had a later onset (Figure 3). Notably, gastro-intestinal tumours were rare as first presentations in early-onset (<10 years) CMMRD (p < 0.001, χ2), but represented 50% of initial tumours in later-onset cases (≥10 years). For 8/133 (6%) of cases, the age of onset could not be determined and the remaining 4/133 (3%) were tumour-free at the moment of reporting. While observed in both groups, the relative reduction in the number of brain tumours (p = 0.007, χ2) and the increase in the proportion of gastro-intestinal tumours (p < 0.001, χ2) from the first to the second neoplasia were significant only in the early-onset group. Even though haematological tumours were not the most common first presentation, they occurred earlier at a median age of 6 years, whereas CNS tumours were diagnosed at a median age of 7.5 years; nevertheless, the difference was not statistically significant (p = 0.41, Mann–Whitney U test).
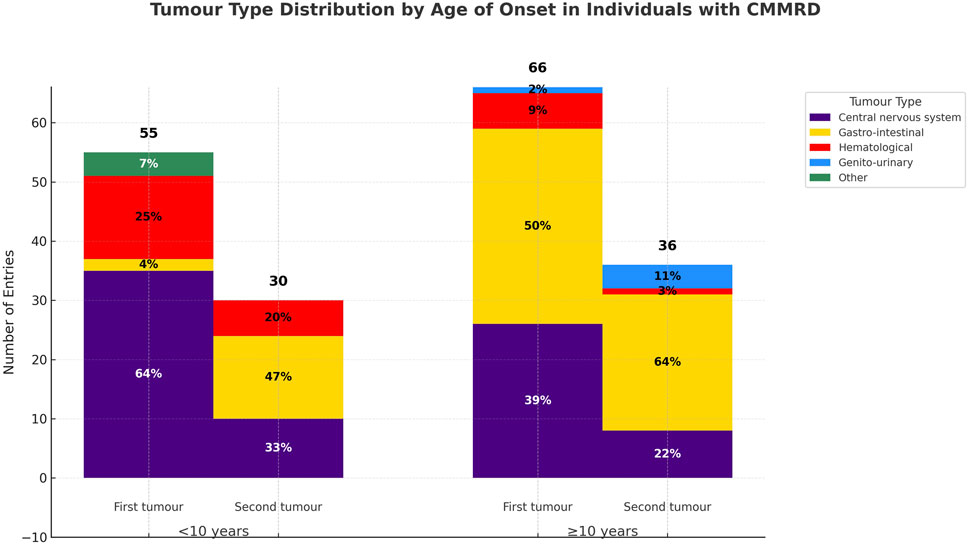
Figure 3. Tumour distribution in early and later-onset PMS2-CMMRD cases. Central nervous system tumours were the most common initial malignancy in early-onset cases, while gastro-intestinal tumours predominated as a first presentation in later-onset cases. Gastro-interstinal tumours were the major presentation in the evolution of both groups.
Rare tumoural and other non-tumoural phenotypes in PMS2-associated constitutional mismatch repair deficiency
Apart from early-onset central nervous tumours, gastro-intestinal and hematological neoplasms, the CMMRD cases included in this study presented with other clinical features, mainly dermatological, and less commonly reported immunological (3/133, 2.25%) and neuropsychiatric (5/133, 3.75%) manifestations. Rare neoplasms were also reported in several cases (Table 1).
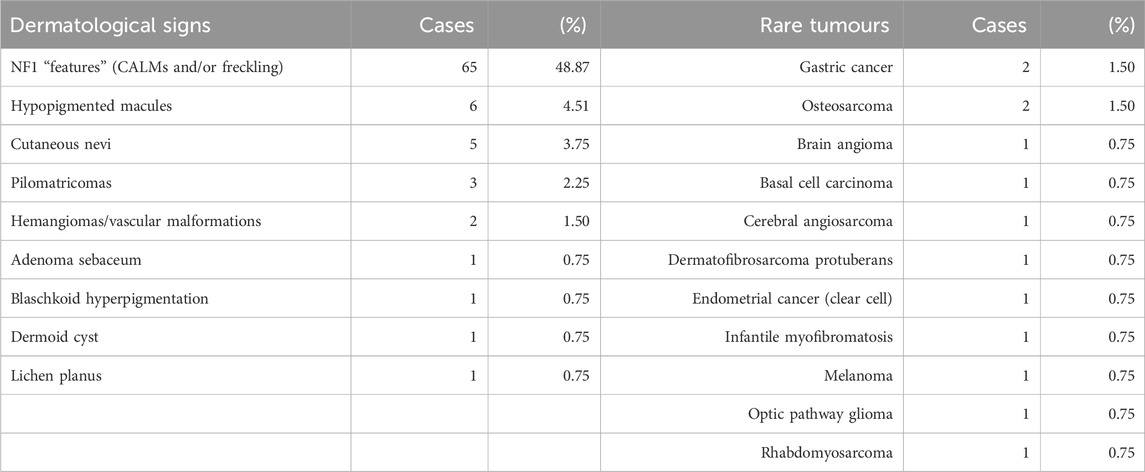
Table 1. Summary of cutaneous manifestations and rare tumours in CMMRD cases. NF1-like phenotype was the most frequently reported clinical non-tumoural presentation in the CMMRD group. Both absolute and relative counts are provided for each clinical feature.
Genotypic characteristics of the study group
We identified 74 short PMS2 variants associated with CMMRD cases that were reported in ClinVar (Figure 4). Among them, 28/74 (37.83%) were frameshift variants, 18/74 (24.32%) stopgain variants (including both nonsense and frameshifts variants creating stop codons at the same genomic site), 13/74 (17.56%) missense, 11/74 (14.86%) splicing (excluding missense and frameshifts located at canonical splice sites), 3/74 (4.05%) startloss and 1/74 (1.35%) synonymous. Homozygosity was noted in 79/133 (59.39%) of entries and 33/74 (44.59%) of variants, while 54/133 (40.60%) of entries and 48/74 (64.86%) of variants were compound heterozygous. The large proportion of homozygous cases coincided with a large number of homozygous variants reported in consanguineous families (16 out of 36 homozygous variants, 44.44%) and founder/recurrent variants (10 out of 36 homozygous variants, 27.77%). In 15/133 (11.12%) of entries, compound heterozygosity involved exonic copy number variants (deletions or duplications). Genotypes weighted by their frequency in our database are illustrated in Figure 4.
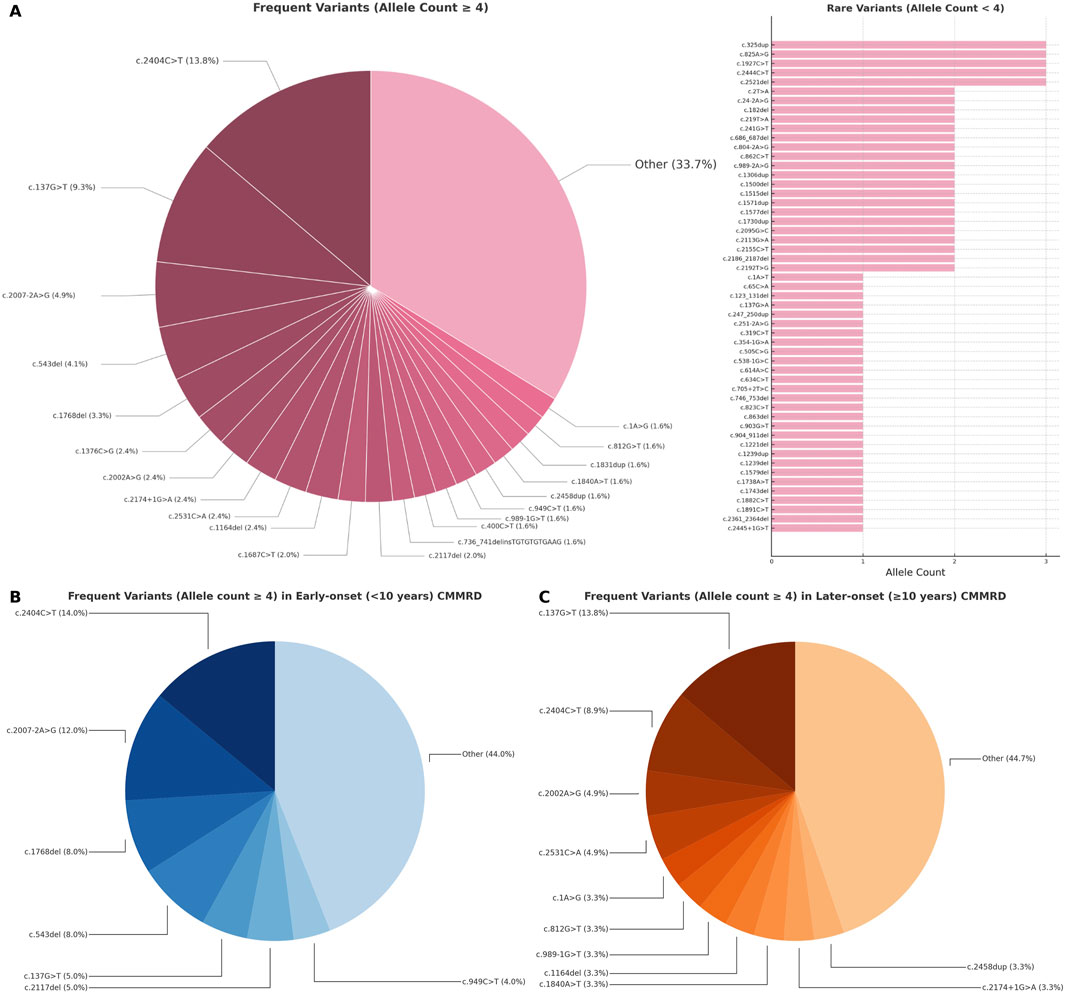
Figure 4. Distribution of short PMS2 variants (<50 bp) in CMMRD cases: (A) all reported cases, (B) early-onset and (C) later-onset presentations. Variants are shown proportionally, according to allelic frequency in the review database.
Later-onset PMS2-CMMRD cases are enriched in splicing variants with mild predicted impact
Of the variants reported in cases with onset after 10 years of age, 12/48 (25%) had a predicted splicing impact, whereas in cases with onset before 10 years, 7/39 (17.94%) involved splicing variants. In the later-onset group (≥10 years), in-frame exon skipping and leaky splicing events were more frequently predicted (Table 2). Non-spliceogenic truncating variants were a more frequent occurrence in the early-onset group. However, in the later-onset cases, most variants were clustered in exon 11 of the gene.
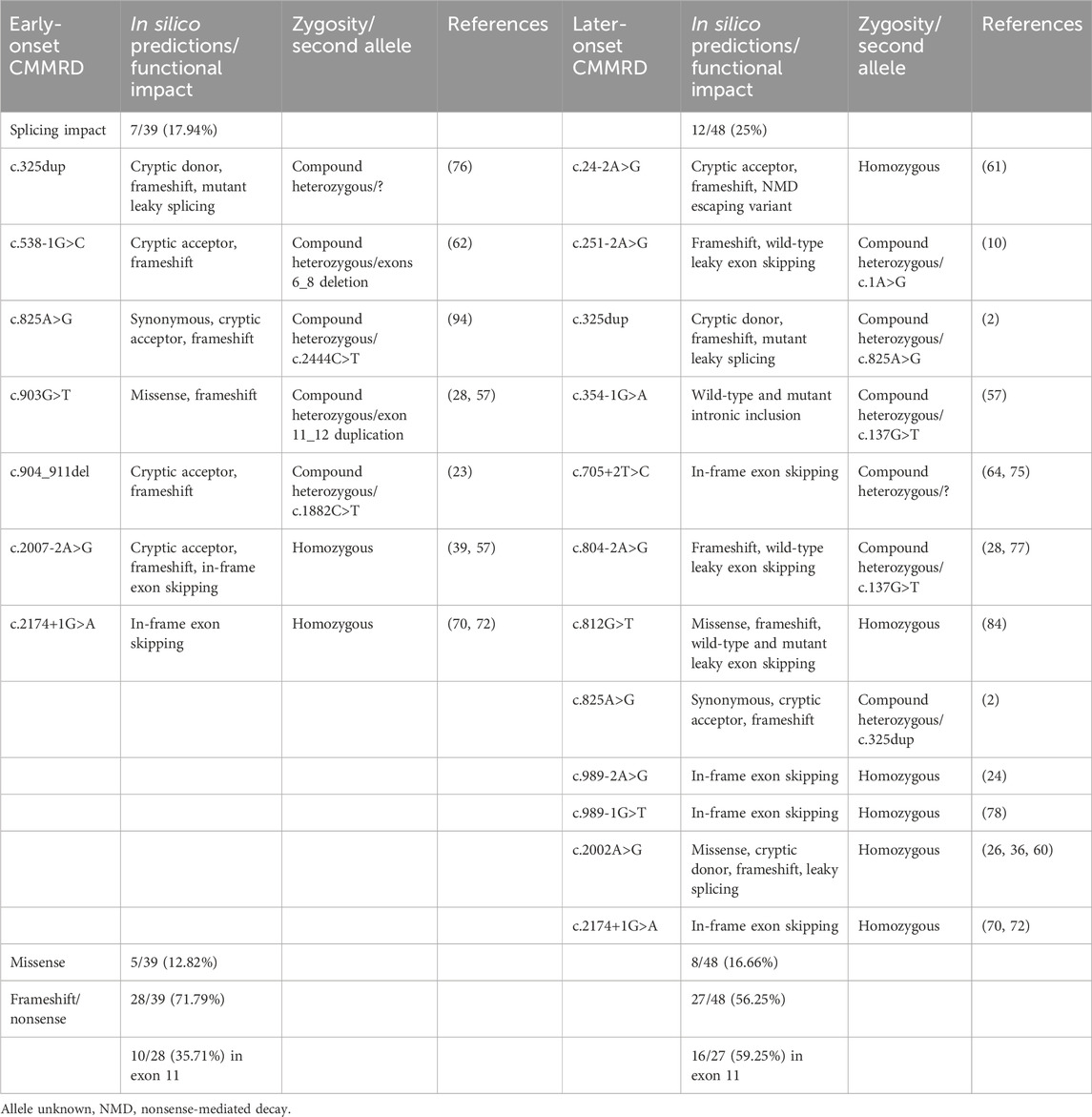
Table 2. Comparison between predicted spliceogenic effects of PMS2 variants identified in early-onset (<10 years) and later-onset (≥10 years) CMMRD groups. In silico predictions were generated using SpliceAI, SpliceAI-visual, SPiCE and the Ensembl Variant Effect Predictor (VEP).
PMS2 variants associated with early-onset (<10 years) and later-onset (≥10 years) CMMRD
To minimize the impact of shared polygenic background and non-genetic environmental exposures that may additionally contribute to phenotypic similarity among family members, we filtered out PMS2 variants reported exclusively within a single family. Due to the limited number of case reports in the review database, variants observed in at least three unrelated individuals were considered sufficiently significant for further analysis. Moreover, inclusion was restricted to variants with an allele count greater than four, thereby excluding those documented solely in two homozygous individuals. Variants occurring exclusively in individuals with CMMRD presenting with early (<10 years) or later (≥10 years) onset were reported in Table 3. Two PMS2 variants were exclusively identified in early-onset CMMRD cases (c.2007-2A>G and c.2117del), while four variants were uniquely observed in later-onset cases (c.1A>G, c.2002A>G, c.2458dup and c.2531C>A).
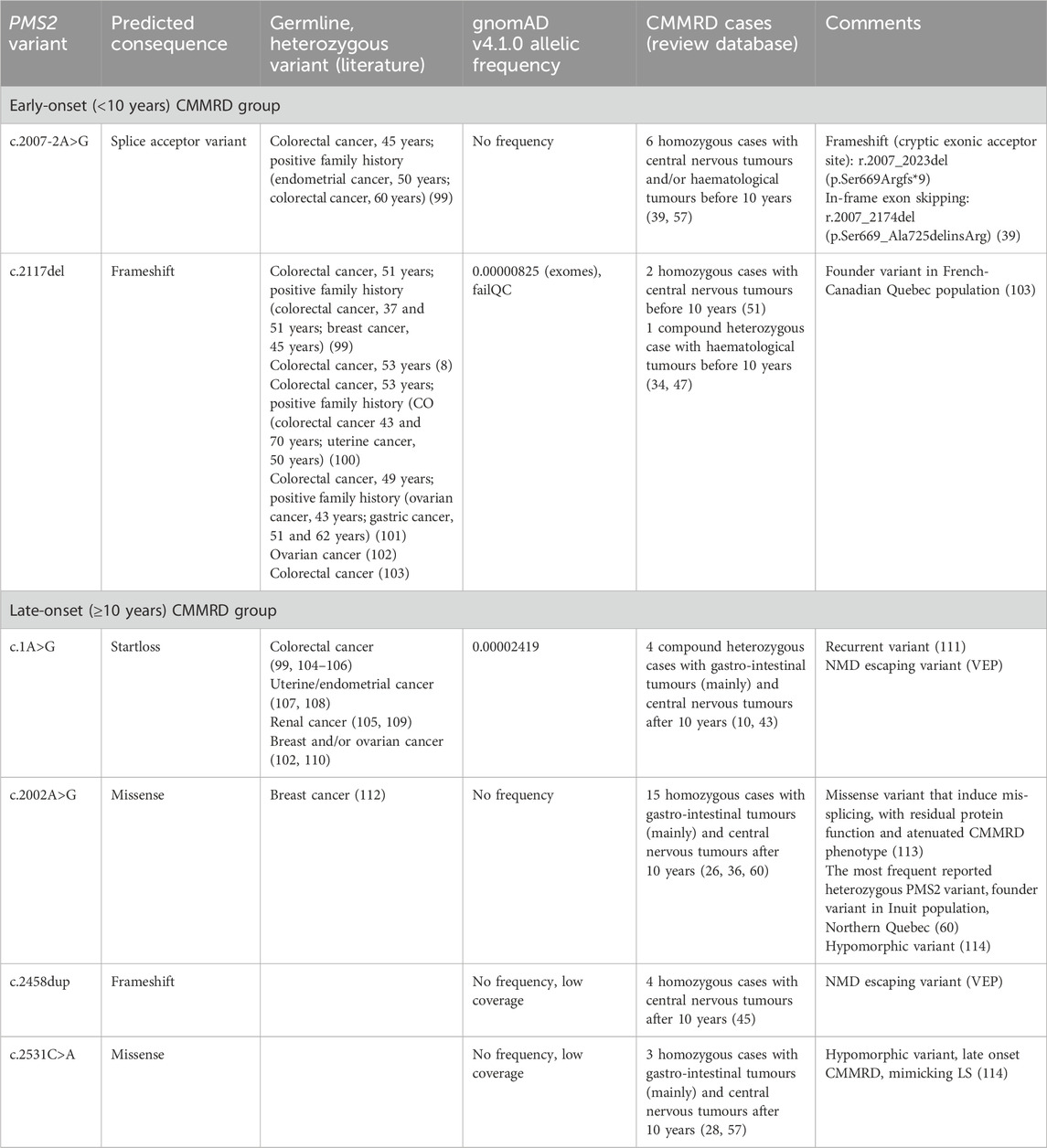
Table 3. PMS2 variants exclusively reported in early (<10 years) and later-onset (≥10 years) CMMRD groups.
CMMRD-related PMS2 variants are more frequent than controls in gnomAD v4
Out of 74 variants identified in CMMRD cases, the paralogous specific variant (PSV) c.2186_2187del was, as expected, very frequent in the gnomAD v4 dataset (115). The remaining 73 CMMRD-associated variants were more frequent in the general (presumably healthy) population compared to the control group of 733 PMS2 variants (p < 0.001, Mann–Whitney U test). The most prevalent CMMRD variants in gnomAD v4 are illustrated in Figure 5.
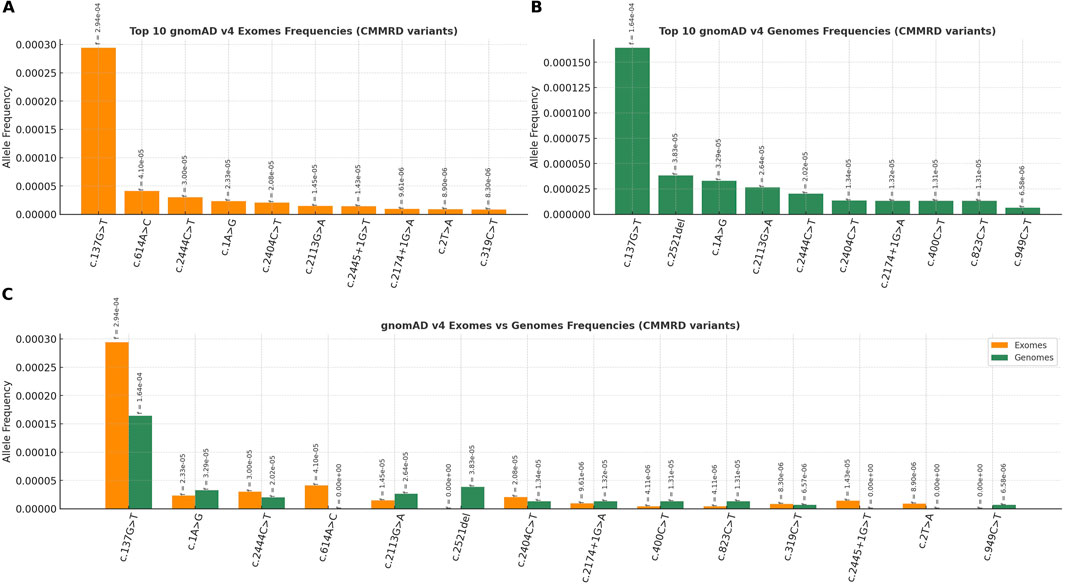
Figure 5. The most frequent PMS2-CMMRD variants in healthy population (gnomAD v4), ranked by allelic frequency: (A) gnomAD exomes, (B) gnomAD genomes and (C) gnomAD exomes and genomes (combined).
The combined VarChat–VarSome strategy is the most effective method for variant detection in literature
All 102 CMMRD-related PMS2 variants included in ClinVar were investigated using three complementary approaches (VarChat, VarSome and LitVar2) to find supporting literature. A combined search employing both VarChat and VarSome identified PubMed indexed publications for 67/68 (98.5%) confirmed CMMRD-related PMS2 variants, outperforming each individual tool (Figure 6). None of the tools were able to find relevant literature for the PMS2, c.2458dup CMMRD variant, making it the only variant missed by the combined approach.
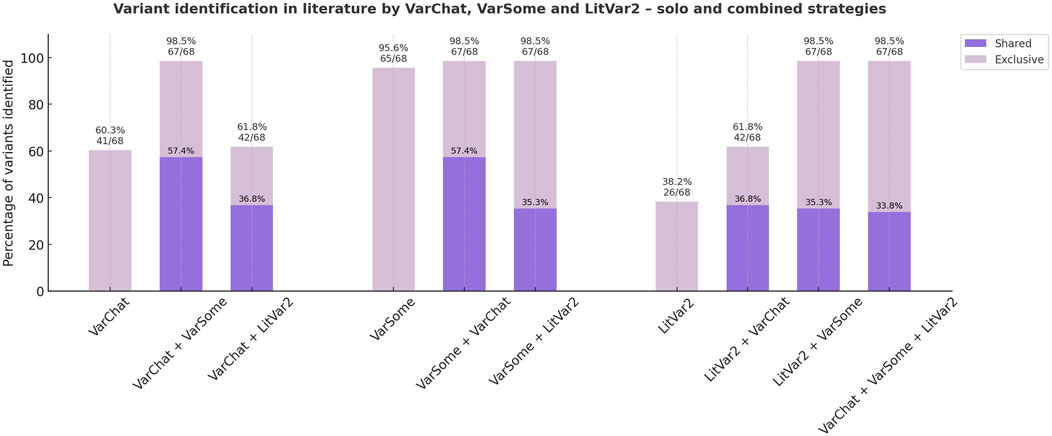
Figure 6. Literature discovery strategies for previously reported PMS2-CMMRD short variants (ClinVar). Both individual and combined approaches to literature review are illustrated. Variants identified by at least two individual tools are highlighted in dark purple, while those detected by only one tool are shown in light purple.
Of the 102 variants analysed, 13 (12.74%) were located in the same canonical splice sites as other PMS2 splicing variants previously reported in the literature but lacked direct evidence supporting their role in CMMRD. Similarly, 5/102 variants (4.90%) without supporting literature affected the start codon. Two frameshift and nonsense variants, c.320_321insT and c.543T>G, had the same predicted protein consequence as other confirmed CMMRD variants. One false positive, c.1376C>A, was erroneously reported by testing laboratories in a CMMRD case (Variation ID: 135936, Accession: SCV005474595.1), where the actual variant was c.1376C>G (93). Another variant, c.1731_1732delinsAGT, was misreported (Variation ID: 9244, Accession: SCV001173401.5), possibly due to a nomenclature-related issue regarding c.1730dup variant, which was described as c.1730_1731insA in the original publication (85).
Discussion
Our study provides a comprehensive review of reported cases of constitutional mismatch repair deficiency (CMMRD) associated with PMS2 pathogenic and likely pathogenic variants submitted to ClinVar by diagnostic and research laboratories. CMMRD is a rare autosomal recessive cancer predisposition syndrome, characterised by early onset of malignancies during childhood, most commonly involving central nervous system, gastro-intestinal tract and hematopoietic system (1–3). By analysing 133 cases and families with PMS2-associated CMMRD, we confirmed this characteristic pattern of tumor predisposition observed in the syndrome. Brain tumors occurred earliest in the course of disease, most frequently followed by gastro-intestinal malignancies. However, in line with the literature, we also identified a subset of cases with later-onset disease, often associated with previously recognized hypomorphic PMS2 variants, as well as new candidate variants predicted to have a mild impact on protein function (60,114). Splicing variants predicted to cause in-frame exon skipping or affecting exons with constitutive leaky splicing were enriched in cases of late-onset neoplasias. Several of these variants involved exons 4, 6, 8 and 10, which our group previously identified as being prone to exon skipping (116). These cases commonly presented with gastro-intestinal tumors as the first malignancy. Moreover, a significant proportion of the analysed cases presented with cutaneous findings, mainly café-au-lait macules and freckles, which are known features shared with other rare genetic disorders such as neurofibromatosis type 1 and Legius syndrome. In this context, our study reinforces the importance of early recognition of these clinical features and timely molecular diagnosis in distinguishing CMMRD from phenocopies, thereby ensuring precise clinical management and appropriate genetic counselling (4–6).
Two truncating PMS2 variants, c.2007-2A>G and c.2117del, were exclusively associated with a highly penetrant phenotype in our cohort, with brain tumors as the main clinical presentation occurring before the age of 10. However, these findings provide only limited information regarding the penetrance of these variants in heterozygous state. PMS2 is generally considered a low to moderate penetrance cancer predisposition gene, typically associated with milder forms of Lynch syndrome (8,10–12). Moreover, many pathogenic PMS2 variants are expected not to manifest any cancer phenotype over the course of individual’s lifetime (8). In LS families related to both c.2007-2A>G and c.2117del PMS2 variants, a high penetrance of cancer phenotype across several generations could be observed, with neoplasia mostly occurring after the age of 40 (99–101). For c.2007-2A>G, calculated constitutional microsatellite instability (cMSI) score in blood of homozygous individuals with CMMRD showed a significantly increased value compared to other PMS2 variants, indicating a more severe phenotype (117). Whether c.2007-2A>G and c.2117del truly confer a higher penetrance compared to other PMS2 variants remains unclear and represents an important topic for future research.
Moreover, the PMS2 variants under analysis were significantly more frequent in the healthy population, which might be indicative of a role for lower-penetrance variants in the pathogenesis of CMMRD. Consistent with this observation, an enrichment of variants with milder predicted functional impact was noted among cases where the first neoplasia occurred after the age of 10. Further studies on larger datasets are needed to determine whether lower-penetrance PMS2 variants must be coinherited with another pathogenic allele to cause CMMRD, similar to the genetic model recently described in Fanconi anemia (FA) (15), in which one BRCA1 or BRCA2 variant retains partial protein function to ensure embryonic viability. Additionally, it remains unclear whether variants exclusively associated with early and later-onset CMMRD significantly differ in penetrance, an important distinction to be made, with potential clinical implications for the management of individuals with PMS2-LS.
Limitations
This systematic review was performed retrospectively, with all recognized limitations for this specific type of study, including potential selection and reporting biases. The current study did not include CMMRD cases for which causative variants were not reported in ClinVar. However, this is not expected to significantly alter the main observations regarding disease phenotype. The literature review relied predominantly on semi-automated tools, which could have influenced the detection or inclusion of some relevant cases. Nonetheless, this approach enabled the development of a combined literature review method, that allows a faster and more scalable data extraction. Moreover, our findings underscore that manual literature review remains essential following the initial screening process to ensure the accuracy and reliability of the information. In the reviewed cases, molecular diagnosis was primarily established through targeted PMS2 gene testing, as a direct consequence to the known issue of gene–pseudogene interference in clinical practice (69,72,118–120). This strategy, however, does not allow for identification of other genetic modifiers that may modulate the phenotype. Tools based on low-pass genome sequencing, such as LOGIC, aim to address this gap (121,122). Although the MMRDness score shows correlations with age at onset and severity of the phenotype, this method falls short in detecting other genomic variants that may account for phenotypic variability among cases. Future research involving larger datasets and prospective cohort studies, using whole genome sequencing as the testing method would be valuable for further expanding and validating genotype–phenotype correlations.
Conclusion
In summary, PMS2-associated constitutional mismatch repair deficiency (PMS2-CMMRD) represents a heterogenous clinical and genetic entity. Our study highlights emerging genotype–phenotype correlations in PMS2-associated CMMRD, which may contribute to refining prognosis and guiding clinical care in both CMMRD and LS. Notably, we identified several candidate PMS2 variants that represent promising targets for future penetrance studies. Larger datasets and prospective cohort studies are warranted to further validate these observations.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
CM: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review and editing, Formal Analysis, Methodology, Project administration, Resources, Software, Validation, Visualization. DL: Methodology, Validation, Writing – original draft, Writing – review and editing. AC: Conceptualization, Data curation, Formal Analysis, Investigation, Writing – original draft, Writing – review and editing. AC-E: Conceptualization, Formal Analysis, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review and editing. AT: Conceptualization, Methodology, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing.
Funding
The authors declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
AcknowledgementsWe would like to acknowledge Victor Babes University Of Medicine And Pharmacy Timisoara for their support in covering the costs of publication for this research paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BRCA1, Breast Cancer 1, Early Onset; BRCA2, Breast Cancer 2, Early Onset; CMMRD, Constitutional Mismatch Repair Deficiency; CNS, Central Nervous System; cMSI, Constitutional Microsatellite Instability; DNA, Deoxyribonucleic Acid; GI, Gastrointestinal; IHC, Immunohistochemistry; LS, Lynch Syndrome; MLH1, MutL Homolog 1, Mismatch Repair Protein; MLPA, Multiplex Ligation-dependent Probe Amplification; MMR, Mismatch Repair (inferred from context, commonly paired with MMRDness); MSH2, MutS Homolog 2, Mismatch Repair Protein; MSH6, MutS Homolog 6, Mismatch Repair Protein; NGS, Next-Generation Sequencing; NF1, Neurofibromin 1; PCR, Polymerase Chain Reaction; PMS2, PMS1 Homolog 2, Mismatch Repair Protein; RNA, Ribonucleic Acid; RT-PCR, Reverse Transcription Polymerase Chain Reaction; VEP, Variant Effect Predictor.
References
1. Ripperger, T, and Schlegelberger, B. Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet (2016) 59(3):133–42. doi:10.1016/j.ejmg.2015.12.014
2. Johannesma, P, van der Klift, H, van Grieken, N, Troost, D, te Riele, H, Jacobs, M, et al. Childhood brain tumours due to germline bi-allelic mismatch repair gene mutations. Clin Genet (2011) 80(3):243–55. doi:10.1111/j.1399-0004.2011.01635.x
3. Aronson, M, Gallinger, S, Cohen, Z, Cohen, S, Dvir, R, Elhasid, R, et al. Gastrointestinal findings in the largest series of patients with hereditary biallelic mismatch repair deficiency syndrome: report from the international consortium. Am J Gastroenterol (2016) 111(2):275–84. doi:10.1038/ajg.2015.392
4. Wimmer, K, Rosenbaum, T, and Messiaen, L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet (2017) 91(4):507–19. doi:10.1111/cge.12904
5. Suerink, M, Ripperger, T, Messiaen, L, Menko, FH, Bourdeaut, F, Colas, C, et al. Constitutional mismatch repair deficiency as a differential diagnosis of neurofibromatosis type 1: consensus guidelines for testing a child without malignancy. J Med Genet (2019) 56(2):53–62. doi:10.1136/jmedgenet-2018-105664
6. Perez-Valencia, JA, Gallon, R, Chen, Y, Koch, J, Keller, M, Oberhuber, K, et al. Constitutional mismatch repair deficiency is the diagnosis in 0.41% of pathogenic NF1/SPRED1 variant negative children suspected of sporadic neurofibromatosis type 1. Genet Med (2020) 22(12):2081–8. doi:10.1038/s41436-020-0925-z
7. Wimmer, K, and Etzler, J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet (2008) 124(2):105–22. doi:10.1007/s00439-008-0542-4
8. Goodenberger, ML, Thomas, BC, Riegert-Johnson, D, Boland, CR, Plon, SE, Clendenning, M, et al. PMS2 monoallelic mutation carriers: the known unknown. Genet Med (2016) 18(1):13–9. doi:10.1038/gim.2015.27
9. Wimmer, K, Kratz, CP, Vasen, HFA, Caron, O, Colas, C, Entz-Werle, N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘Care for CMMRD’ (C4CMMRD). J Med Genet (2014) 51(6):355–65. doi:10.1136/jmedgenet-2014-102284
10. Senter, L, Clendenning, M, Sotamaa, K, Hampel, H, Green, J, Potter, JD, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology (2008) 135(2):419–28.e1. doi:10.1053/j.gastro.2008.04.026
11. Dominguez-Valentin, M, Sampson, JR, Seppälä, TT, ten Broeke, SW, Plazzer, JP, Nakken, S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the prospective Lynch syndrome database. Genet Med (2020) 22(1):15–25. doi:10.1038/s41436-019-0596-9
12. ten Broeke, SW, Suerink, M, and Nielsen, M. Response to roberts 2018: is breast cancer truly caused by MSH6 and PMS2 variants or is it simply due to a high prevalence of these variants in the population? Genet Med (2019) 21(1):256–7. doi:10.1038/s41436-018-0029-1
13. Sawyer, SL, Tian, L, Kähkönen, M, Schwartzentruber, J, Kircher, M, Majewski, J, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov (2015) 5(2):135–42. doi:10.1158/2159-8290.CD-14-1156
14. Howlett, NG, Taniguchi, T, Olson, S, Cox, B, Waisfisz, Q, De Die-Smulders, C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science (2002) 297(5581):606–9. doi:10.1126/science.1073834
15. Pal, T, Mundt, E, Richardson, ME, Chao, E, Pesaran, T, Slavin, TP, et al. Reduced penetrance BRCA1 and BRCA2 pathogenic variants in clinical germline genetic testing. npj Precision Oncol (2024) 8(1):247–9. doi:10.1038/s41698-024-00741-4
16. Aronson, M, Colas, C, Shuen, A, Hampel, H, Foulkes, WD, Baris Feldman, H, et al. Diagnostic criteria for constitutional mismatch repair deficiency (CMMRD): recommendations from the international consensus working group. J Med Genet (2022) 59(4):318–27. doi:10.1136/jmedgenet-2020-107627
17. Colas, C, Guerrini-Rousseau, L, Suerink, M, Gallon, R, Kratz, CP, Ayuso, É, et al. ERN GENTURIS guidelines on constitutional mismatch repair deficiency diagnosis, genetic counselling, surveillance, quality of life, and clinical management. Eur J Hum Genet (2024) 32(12):1526–41. doi:10.1038/s41431-024-01708-6
18. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi:10.1038/gim.2015.30
19. Thompson, BA, Spurdle, AB, Plazzer, JP, Greenblatt, MS, Akagi, K, Al-Mulla, F, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet (2014) 46(2):107–15. doi:10.1038/ng.2854
20. Kopanos, C, Tsiolkas, V, Kouris, A, Chapple, CE, Albarca Aguilera, M, Meyer, R, et al. VarSome: the human genomic variant search engine. Bioinformatics (2019) 35(11):1978–80. doi:10.1093/bioinformatics/bty897
21. Allot, A, Wei, CH, Phan, L, Hefferon, T, Landrum, M, Rehm, HL, et al. Tracking genetic variants in the biomedical literature using LitVar 2.0. Nat Genet (2023) 55(6):901–3. doi:10.1038/s41588-023-01414-x
22. De Paoli, F, Berardelli, S, Limongelli, I, Rizzo, E, and Zucca, S. VarChat: the generative AI assistant for the interpretation of human genomic variations. Bioinformatics (2024) 40(4):btae183. doi:10.1093/bioinformatics/btae183
23. Kroeze, E, Weijers, DD, Hagleitner, MM, De Groot-Kruseman, HA, Jongmans, MCJ, Kuiper, RP, et al. High prevalence of constitutional mismatch repair deficiency in a pediatric T-cell lymphoblastic lymphoma cohort. HemaSphere (2022) 6(1):E668. doi:10.1097/HS9.0000000000000668
24. Fiala, EM, Jayakumaran, G, Mauguen, A, Kennedy, JA, Bouvier, N, Kemel, Y, et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat Cancer (2021) 2(3):357–65. doi:10.1038/s43018-021-00172-1
25. Tan, S, Wu, X, Wang, A, and Ying, L. Diagnostic challenges in a CMMRD patient with a novel mutation in the PMS2 gene: a case report. BMC Med Genomics (2021) 14(1):184. doi:10.1186/s12920-021-01031-9
26. Rittberg, R, Harlos, C, Rothenmund, H, Das, A, Tabori, U, Sinha, N, et al. Immune checkpoint inhibition as primary adjuvant therapy for an IDH1-mutant anaplastic astrocytoma in a patient with CMMRD: a case report—usage of immune checkpoint inhibition in CMMRD. Curr Oncol (2021) 28(1):757–66. doi:10.3390/curroncol28010074
27. Chhabda, S, Sudhakar, S, Mankad, K, Jorgensen, M, Carceller, F, Jacques, TS, et al. Constitutional mismatch repair deficiency (CMMRD) presenting with high-grade glioma, multiple developmental venous anomalies and malformations of cortical development—a multidisciplinary/multicentre approach and neuroimaging clues to clinching the diagnosis. Child’s Nerv Syst (2021) 37(7):2375–9. doi:10.1007/s00381-020-04986-9
28. Guerrini-Rousseau, L, Varlet, P, Colas, C, Andreiuolo, F, Bourdeaut, F, Dahan, K, et al. Constitutional mismatch repair deficiency-associated brain tumors: report from the european C4CMMRD consortium. Neuro-Oncology Adv (2019) 1(1):vdz033. doi:10.1093/noajnl/vdz033
29. Siraj, AK, Masoodi, T, Bu, R, Parvathareddy, SK, Siraj, S, Alassiri, A, et al. The study of Lynch syndrome in a special population reveals a strong founder effect and an unusual mutational mechanism in familial adenomatous polyposis. Gut (2020) 69(11):2048–9. doi:10.1136/gutjnl-2019-320511
30. Saǧ, E, Erkut, M, Saygin, İ, Çebi, AH, Bahadir, A, Erduran, E, et al. Constitutional Mismatch Repair Gene Defect Syndrome Presenting with Adenomatous Polyposis and Cafe au Lait Spots: a Case Report. J Pediatr Hematology/Oncology (2020) 42(7):e689–91. doi:10.1097/MPH.0000000000001614
31. Okkels, H, Lagerstedt-Robinsson, K, Wikman, FP, Hansen, TVO, Lolas, I, Lindberg, LJ, et al. Detection of PMS2 mutations by screening hereditary nonpolyposis Colon cancer families from Denmark and Sweden. Genet Test Mol Biomarkers (2019) 23(9):688–95. doi:10.1089/gtmb.2018.0316
32. Pavelka, Z, Zitterbart, K, Nosková, H, Bajčiová, V, Slabý, O, and Štěrba, J. Effective immunotherapy of glioblastoma in an adolescent with constitutional mismatch repair-deficiency syndrome. Klin Onkol. (2019) 32(1):70–4. doi:10.14735/amko201970
33. Gallon, R, Mühlegger, B, Wenzel, SS, Sheth, H, Hayes, C, Aretz, S, et al. A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum Mutat (2019) 40(5):649–55. doi:10.1002/humu.23721
34. Henn, J, Spier, I, Adam, RS, Holzapfel, S, Uhlhaas, S, Kayser, K, et al. Diagnostic yield and clinical utility of a comprehensive gene panel for hereditary tumor syndromes. Hered Cancer Clin Pract (2019) 17(1):5. doi:10.1186/s13053-018-0102-4
35. D’Arcy, BM, Blount, J, and Prakash, A. Biochemical and structural characterization of two variants of uncertain significance in the PMS2 gene. Hum Mutat (2019) 40(4):458–71. doi:10.1002/humu.23708
36. Shuen, AY, Lanni, S, Panigrahi, GB, Edwards, M, Yu, L, Campbell, BB, et al. Functional repair assay for the diagnosis of constitutional mismatch repair deficiency from non-neoplastic tissue. J Clin Oncol (2019) 37(6):461–70. doi:10.1200/JCO.18.00474
37. Baig, SM, Fatima, A, Tariq, M, Khan, TN, Ali, Z, Faheem, M, et al. Hereditary brain tumor with a homozygous germline mutation in PMS2: pedigree analysis and prenatal screening in a family with constitutional mismatch repair deficiency (CMMRD) syndrome. Fam Cancer (2019) 18(2):261–5. doi:10.1007/s10689-018-0112-4
38. Hildreth, A, Valasek, MA, Thung, I, Savides, T, Sivagnanam, M, Ramamoorthy, S, et al. Biallelic mismatch repair deficiency in an adolescent female. Case Rep Genet (2018) 2018:1–5. doi:10.1155/2018/8657823
39. Stefanovic, E, Kilcawley, KN, Roces, C, Rea, MC, O’Sullivan, M, Sheehan, JJ, et al. Evaluation of the potential of Lactobacillus paracasei adjuncts for flavor compounds development and diversification in short-aged cheddar cheese. Front Microbiol (2018) 9:1506. doi:10.3389/fmicb.2018.01506
40. Leenders, EKSM, Westdorp, H, Brüggemann, RJ, Loeffen, J, Kratz, C, Burn, J, et al. Cancer prevention by aspirin in children with constitutional mismatch repair deficiency (CMMRD). Eur J Hum Genet (2018) 26(10):1417–23. doi:10.1038/s41431-018-0197-0
41. Gröbner, SN, Worst, BC, Weischenfeldt, J, Buchhalter, I, Kleinheinz, K, Rudneva, VA, et al. The landscape of genomic alterations across childhood cancers. Nature (2018) 555(7696):321–7. doi:10.1038/nature25480
42. Marks, LJ, Oberg, JA, Pendrick, D, Sireci, AN, Glasser, C, Coval, C, et al. Precision medicine in children and young adults with hematologic malignancies and blood disorders: the Columbia university experience. Front Pediatr (2017) 5:265. doi:10.3389/fped.2017.00265
43. Mardis, ER, Potter, SL, Schieffer, KM, Varga, EA, Mathew, MT, Costello, HM, et al. Germline susceptibility from broad genomic profiling of pediatric brain cancers. Neuro-Oncology Adv (2024) 6(1):vdae099. doi:10.1093/noajnl/vdae099
44. Andrianova, MA, Chetan, GK, Sibin, MK, Mckee, T, Merkler, D, Narasinga, RKVL, et al. Germline PMS2 and somatic POLE exonuclease mutations cause hypermutability of the leading DNA strand in biallelic mismatch repair deficiency syndrome brain tumours. The J Pathol (2017) 243(3):331–41. doi:10.1002/path.4957
45. Suerink, M, Potjer, TP, Versluijs, AB, ten Broeke, SW, Tops, CM, Wimmer, K, et al. Constitutional mismatch repair deficiency in a healthy child: on the spot diagnosis? Clin Genet (2018) 93(1):134–7. doi:10.1111/cge.13053
46. Ramchander, NC, Ryan, NAJ, Crosbie, EJ, and Evans, DG. Homozygous germ-line mutation of the PMS2 mismatch repair gene: a unique case report of constitutional mismatch repair deficiency (CMMRD). BMC Med Genet (2017) 18(1):40. doi:10.1186/s12881-017-0391-x
47. Adam, R, Spier, I, Zhao, B, Kloth, M, Marquez, J, Hinrichsen, I, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. The Am J Hum Genet (2016) 99(2):337–51. doi:10.1016/j.ajhg.2016.06.015
48. van der Klift, HM, Mensenkamp, AR, Drost, M, Bik, EC, Vos, YJ, Gille, HJJP, et al. Comprehensive mutation analysis of PMS2 in a large cohort of probands suspected of Lynch syndrome or constitutional mismatch repair deficiency syndrome. Hum Mutat (2016) 37(11):1162–79. doi:10.1002/humu.23052
49. Ortiz, MV, Kobos, R, Walsh, M, Slotkin, EK, Roberts, S, Berger, MF, et al. Integrating genomics into clinical pediatric oncology using the molecular tumor board at the memorial sloan kettering cancer center. Pediatr Blood Cancer (2016) 63(8):1368–74. doi:10.1002/pbc.26002
50. Mork, ME, Borras, E, Taggart, MW, Cuddy, A, Bannon, SA, You, YN, et al. Identification of a novel PMS2 alteration c.505C>G (R169G) in trans with a PMS2 pathogenic mutation in a patient with constitutional mismatch repair deficiency. Fam Cancer (2016) 15(4):587–91. doi:10.1007/s10689-016-9902-8
51. Bouffet, E, Larouche, V, Campbell, BB, Merico, D, De Borja, R, Aronson, M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol (2016) 34(19):2206–11. doi:10.1200/JCO.2016.66.6552
52. Shirts, BH, Casadei, S, Jacobson, AL, Lee, MK, Gulsuner, S, Bennett, RL, et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med (2016) 18(10):974–81. doi:10.1038/gim.2015.212
53. Baris, HN, Barnes-Kedar, I, Toledano, H, Halpern, M, Hershkovitz, D, Lossos, A, et al. Constitutional mismatch repair deficiency in Israel: high proportion of founder mutations in MMR genes and consanguinity. Pediatr Blood Cancer (2016) 63(3):418–27. doi:10.1002/pbc.25818
54. Palova, H, Das, A, Pokorna, P, Bajciova, V, Pavelka, Z, Jezova, M, et al. Precision immuno-oncology approach for four malignant tumors in siblings with constitutional mismatch repair deficiency syndrome. npj Precision Oncol (2024) 8(1):110. doi:10.1038/s41698-024-00597-8
55. Urganci, N, Genc, DB, Kose, G, Onal, Z, and Vidin, OO. Colorectal cancer due to constitutional mismatch repair deficiency mimicking neurofibromatosis I. Pediatrics (2015) 136(4):e1047–50. doi:10.1542/peds.2015-1426
56. Li, J, Dai, H, Feng, Y, Tang, J, Chen, S, Tian, X, et al. A comprehensive strategy for accurate mutation detection of the highly homologous PMS2. The J Mol Diagn (2015) 17(5):545–53. doi:10.1016/j.jmoldx.2015.04.001
57. Lavoine, N, Colas, C, Muleris, M, Bodo, S, Duval, A, Entz-Werle, N, et al. Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet (2015) 52(11):770–8. doi:10.1136/jmedgenet-2015-103299
58. Bodo, S, Colas, C, Buhard, O, Collura, A, Tinat, J, Lavoine, N, et al. Diagnosis of constitutional mismatch repair-deficiency syndrome based on microsatellite instability and lymphocyte tolerance to methylating agents. Gastroenterology (2015) 149(4):1017–29.e3. doi:10.1053/j.gastro.2015.06.013
59. Daou, B, Zanello, M, Varlet, P, Brugieres, L, Jabbour, P, Caron, O, et al. An unusual case of constitutional mismatch repair deficiency syndrome with Anaplastic ganglioglioma, colonic adenocarcinoma, osteosarcoma, acute myeloid leukemia, and signs of neurofibromatosis type 1: case report. Neurosurgery (2015) 77(1):E145–52. doi:10.1227/NEU.0000000000000754
60. Li, L, Hamel, N, Baker, K, McGuffin, MJ, Couillard, M, Gologan, A, et al. A homozygous PMS2 founder mutation with an attenuated constitutional mismatch repair deficiency phenotype. J Med Genet (2015) 52(5):348–52. doi:10.1136/jmedgenet-2014-102934
61. Ten Broeke, SW, Brohet, RM, Tops, CM, van der Klift, HM, Velthuizen, ME, Bernstein, I, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol (2015) 33(4):319–25. doi:10.1200/JCO.2014.57.8088
62. Bakry, D, Aronson, M, Durno, C, Rimawi, H, Farah, R, Alharbi, QK, et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer (2014) 50(5):987–96. doi:10.1016/j.ejca.2013.12.005
63. Walter, AW, Ennis, S, Best, H, Vaughn, CP, Swensen, JJ, Openshaw, A, et al. Constitutional mismatch repair deficiency presenting in childhood as three simultaneous malignancies. Pediatr Blood Cancer (2013) 60(11):E135–E136. doi:10.1002/pbc.24613
64. Chmara, M, Wernstedt, A, Wasag, B, Peeters, H, Renard, M, Beert, E, et al. Multiple pilomatricomas with somatic CTNNB1 mutations in children with constitutive mismatch repair deficiency. Genes, Chromosomes and Cancer (2013) 52(7):656–64. doi:10.1002/gcc.22061
65. Onishi, S, Yamasaki, F, Kuraoka, K, Taguchi, A, Takayasu, T, Akagi, K, et al. Diagnostic and therapeutic challenges of glioblastoma as an initial malignancy of constitutional mismatch repair deficiency (CMMRD): two case reports and a literature review. BMC Med Genomics (2023) 16(1):6. doi:10.1186/s12920-022-01403-9
66. Yeung, JT, Pollack, IF, Shah, S, Jaffe, R, Nikiforova, M, and Jakacki, RI. Optic pathway glioma as part of a constitutional mismatch-repair deficiency syndrome in a patient meeting the criteria for neurofibromatosis type 1. Pediatr Blood and Cancer (2013) 60(1):137–9. doi:10.1002/pbc.24254
67. Vasovcak, P, Krepelova, A, Menigatti, M, Puchmajerova, A, Skapa, P, Augustinakova, A, et al. Unique mutational profile associated with a loss of TDG expression in the rectal cancer of a patient with a constitutional PMS2 deficiency. DNA Repair (Amst) (2012) 11(7):616–23. doi:10.1016/j.dnarep.2012.04.004
68. Wernstedt, A, Valtorta, E, Armelao, F, Togni, R, Girlando, S, Baudis, M, et al. Improved multiplex ligation-dependent probe amplification analysis identifies a deleterious PMS2 allele generated by recombination with crossover between PMS2 and PMS2CL. Genes, Chromosomes and Cancer (2012) 51(9):819–31. doi:10.1002/gcc.21966
69. Vaughn, CP, Hart, KJ, Samowitz, WS, and Swensen, JJ. Avoidance of pseudogene interference in the detection of 3’ deletions in PMS2. Hum Mutat (2011) 32(9):1063–71. doi:10.1002/humu.21540
70. Herkert, JC, Niessen, RC, Olderode-Berends, MJW, Veenstra-Knol, HE, Vos, YJ, van der Klift, HM, et al. Paediatric intestinal cancer and polyposis due to bi-allelic PMS2 mutations: case series, review and follow-up guidelines. Eur J Cancer (2011) 47(7):965–82. doi:10.1016/j.ejca.2011.01.013
71. Leenen, C, Geurts-Giele, W, Dubbink, H, Reddingius, R, van den Ouweland, A, Tops, C, et al. Pitfalls in molecular analysis for mismatch repair deficiency in a family with biallelic PMS2 germline mutations. Clin Genet (2011) 80(6):558–65. doi:10.1111/j.1399-0004.2010.01608.x
72. Vaughn, CP, Robles, J, Swensen, JJ, Miller, CE, Lyon, E, Mao, R, et al. Clinical analysis of PMS2: mutation detection and avoidance of pseudogenes. Hum Mutat (2010) 31(5):588–93. doi:10.1002/humu.21230
73. Kratz, CP, Holter, S, Etzler, J, Lauten, M, Pollett, A, Niemeyer, CM, et al. Rhabdomyosarcoma in patients with constitutional mismatch-repair-deficiency syndrome. J Med Genet (2009) 46(6):418–20. doi:10.1136/jmg.2008.064212
74. Roy, S, Raskin, L, Raymond, VM, Thibodeau, SN, Mody, RJ, and Gruber, SB. Pediatric duodenal cancer and biallelic mismatch repair gene mutations. Pediatr Blood and Cancer (2009) 53(1):116–20. doi:10.1002/pbc.21957
75. Wachter-Giner, T, Bieber, I, Warmuth-Metz, M, Bröcker, E, and Hamm, H. Multiple pilomatricomas and gliomatosis cerebri - a new association? Pediatr Dermatol (2009) 26(1):75–8. doi:10.1111/j.1525-1470.2008.00827.x
76. Douglas, SPM, Lahtinen, AK, Koski, JR, Leimi, L, Keränen, MAI, Koskenvuo, M, et al. Enrichment of cancer-predisposing germline variants in adult and pediatric patients with acute lymphoblastic leukemia. Sci Rep (2022) 12(1):10670. doi:10.1038/s41598-022-14364-x
77. Giunti, L, Cetica, V, Ricci, U, Giglio, S, Sardi, I, Paglierani, M, et al. Type A microsatellite instability in pediatric gliomas as an indicator of Turcot syndrome. Eur J Hum Genet (2009) 17(7):919–27. doi:10.1038/ejhg.2008.271
78. Sjursen, W, Bjørnevoll, I, Engebretsen, LF, Fjelland, K, Halvorsen, T, and Myrvold, HE. A homozygote splice site PMS2 mutation as cause of Turcot syndrome gives rise to two different abnormal transcripts. Fam Cancer (2009) 8(3):179–86. doi:10.1007/s10689-008-9225-5
79. Péron, S, Metin, A, Gardès, P, Alyanakian, MA, Sheridan, E, Kratz, CP, et al. Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. The J Exp Med (2008) 205(11):2465–72. doi:10.1084/jem.20080789
80. Jackson, CC, Holter, S, Pollett, A, Clendenning, M, Chou, S, Senter, L, et al. Café-au-lait macules and pediatric malignancy caused by biallelic mutations in the DNA mismatch repair (MMR) gene PMS2. Pediatr Blood and Cancer (2008) 50(6):1268–70. doi:10.1002/pbc.21514
81. Etzler, J, Peyrl, A, Zatkova, A, Schildhaus, HU, Ficek, A, Merkelbach-Bruse, S, et al. RNA-based mutation analysis identifies an unusual MSH6 splicing defect and circumvents PMS2 pseudogene interference. Hum Mutat (2008) 29(2):299–305. doi:10.1002/humu.20657
82. Kratz, CP, Niemeyer, CM, Jüttner, E, Kartal, M, Weninger, A, Schmitt-Graeff, A, et al. Childhood T-cell non-hodgkin’s lymphoma, colorectal carcinoma and brain tumor in association with café-au-lait spots caused by a novel homozygous PMS2 mutation. Leukemia (2008) 22(5):1078–80. doi:10.1038/sj.leu.2405008
83. Gururangan, S, Frankel, W, Broaddus, R, Clendenning, M, Senter, L, McDonald, M, et al. Multifocal anaplastic astrocytoma in a patient with hereditary colorectal cancer, transcobalamin II deficiency, agenesis of the corpus callosum, mental retardation, and inherited PMS2 mutation. Neuro-Oncology (2008) 10(1):93–7. doi:10.1215/15228517-2007-037
84. Krüger, S, Kinzel, M, Walldorf, C, Gottschling, S, Bier, A, Tinschert, S, et al. Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet (2008) 16(1):62–72. doi:10.1038/sj.ejhg.5201923
85. Auclair, J, Leroux, D, Desseigne, F, Lasset, C, Saurin, JC, Joly, MO, et al. Novel biallelic mutations in MSH6 and PMS2 genes: gene conversion as a likely cause of PMS2 gene inactivation. Hum Mutat (2007) 28(11):1084–90. doi:10.1002/humu.20569
86. De Vos, M, Hayward, BE, Charlton, R, Taylor, GR, Glaser, AW, Picton, S, et al. PMS2 mutations in childhood cancer. JNCI: J Natl Cancer Inst (2006) 98(5):358–61. doi:10.1093/jnci/djj073
87. Arslan Ates, E, Turkyilmaz, A, Alavanda, C, Yildirim, O, and Guney, AI. Multigene panel testing in Turkish hereditary cancer syndrome patients. Medeniyet Med J (2022) 37(2):150–8. doi:10.4274/MMJ.galenos.2022.22556
88. Agostini, M, Tibiletti, MG, Lucci-Cordisco, E, Chiaravalli, A, Morreau, H, Furlan, D, et al. Two PMS2 mutations in a Turcot syndrome family with small bowel cancers. The Am J Gastroenterol (2005) 100(8):1886–91. doi:10.1111/j.1572-0241.2005.50441.x
89. De Vos, M, Hayward, BE, Picton, S, Sheridan, E, and Bonthron, DT. Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. The Am J Hum Genet (2004) 74(5):954–64. doi:10.1086/420796
90. De Rosa, M, Fasano, C, Panariello, L, Scarano, MI, Belli, G, Iannelli, A, et al. Evidence for a recessive inheritance of Turcot’s syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene (2000) 19(13):1719–23. doi:10.1038/sj.onc.1203447
91. Miyaki, M, Nishio, J, Konishi, M, Kikuchi-Yanoshita, R, Tanaka, K, Muraoka, M, et al. Drastic genetic instability of tumors and normal tissues in Turcot syndrome. Oncogene (1997) 15(23):2877–81. doi:10.1038/sj.onc.1201668
92. Hamilton, SR, Liu, B, Parsons, RE, Papadopoulos, N, Jen, J, Powell, SM, et al. The molecular basis of turcot’s syndrome. N Engl J Med (1995) 332(13):839–47. doi:10.1056/NEJM199503303321302
93. AlAli, MN, Zikry, AH, AlShammari, SA, Zayed, MA, Alswayyed, M, and AlObeed, OA. A constitutional mismatch repair deficiency syndrome presented with an advanced rectal cancer in a juvenile female: a case report and literature review. Cureus (2022) 14(4):e24615. doi:10.7759/cureus.24615
94. Mishra, AK, Achari, RB, Zameer, L, Achari, G, Gehani, A, Roy, P, et al. Germline biallelic mismatch repair deficiency in childhood glioblastoma and implications for clinical management. Neurol India (2022) 70(2):772–4. doi:10.4103/0028-3886.344608
95. Stawiński, P, and Płoski, R. Genebe.net: implementation and validation of an automatic ACMG variant pathogenicity criteria assignment. Clin Genet (2024) 106(2):119–26. doi:10.1111/cge.14516
96. Jaganathan, K, Kyriazopoulou Panagiotopoulou, S, McRae, JF, Darbandi, SF, Knowles, D, Li, YI, et al. Predicting splicing from primary sequence with deep learning. Cell (2019) 176(3):535–48.e24. doi:10.1016/j.cell.2018.12.015
97. de Sainte Agathe, JM, Filser, M, Isidor, B, Besnard, T, Gueguen, P, Perrin, A, et al. SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation. Hum Genomics (2023) 17(1):7. doi:10.1186/s40246-023-00451-1
98. Leman, R, Gaildrat, P, Gac, GL, Ka, C, Fichou, Y, Audrezet, MP, et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in Silico/in vitro studies: an international collaborative effort. Nucleic Acids Res (2018) 46(15):11656–7. doi:10.1093/nar/gky979
99. Wang, Q, Leclerc, J, Bougeard, G, Olschwang, S, Vasseur, S, Cassinari, K, et al. Characterisation of heterozygous PMS2 variants in French patients with Lynch syndrome. J Med Genet (2020) 57(7):487–99. doi:10.1136/jmedgenet-2019-106256
100. Yurgelun, MB, Kulke, MH, Fuchs, CS, Allen, BA, Uno, H, Hornick, JL, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol (2017) 35(10):1086–95. doi:10.1200/JCO.2016.71.0012
101. Blount, J, and Prakash, A. The changing landscape of Lynch syndrome due to PMS2 mutations. Clin Genet (2018) 94(1):61–9. doi:10.1111/cge.13205
102. Susswein, LR, Marshall, ML, Nusbaum, R, Vogel Postula, KJ, Weissman, SM, Yackowski, L, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med (2016) 18(8):823–32. doi:10.1038/gim.2015.166
103. Chong, AL, Mejia-Garcia, A, Behl, S, El Haffaf, Z, Chénier, S, Maranda, B, et al. PMS2 c.2117del (p.Lys706Serfs*19) is the Most frequent cancer-associated founder pathogenic variant in the French-Canadian population of Quebec, Canada. Clin Genet (2025) 108:747–51. doi:10.1111/cge.14784
104. Singh, AK, Talseth-Palmer, B, Xavier, A, Scott, RJ, Drabløs, F, and Sjursen, W. Detection of germline variants with pathogenic potential in 48 patients with familial colorectal cancer by using whole exome sequencing. BMC Med Genomics (2023) 16(1):126. doi:10.1186/s12920-023-01562-3
105. Talseth-Palmer, BA, McPhillips, M, Groombridge, C, Spigelman, A, and Scott, RJ. MSH6 and PMS2 mutation positive Australian Lynch syndrome families: novel mutations, cancer risk and age of diagnosis of colorectal cancer. Hered Cancer Clin Pract (2010) 8(1):5. doi:10.1186/1897-4287-8-5
106. Brennan, B, Hemmings, CT, Clark, I, Yip, D, Fadia, M, and Taupin, DR. Universal molecular screening does not effectively detect Lynch syndrome in clinical practice. Therap Adv Gastroenterol (2017) 10(4):361–71. doi:10.1177/1756283X17690990
107. Sjursen, W, McPhillips, M, Scott, RJ, and Talseth-Palmer, BA. Lynch syndrome mutation spectrum in New South Wales, Australia, including 55 novel mutations. Mol Genet and Genomic Med (2016) 4(2):223–31. doi:10.1002/mgg3.198
108. Tian, W, Bi, R, Ren, Y, He, H, Shi, S, Shan, B, et al. Screening for hereditary cancers in patients with endometrial cancer reveals a high frequency of germline mutations in cancer predisposition genes. Int J Cancer (2019) 145(5):1290–8. doi:10.1002/ijc.32389
109. Hartman, TR, Demidova, EV, Lesh, RW, Hoang, L, Richardson, M, Forman, A, et al. Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer. Sci Rep (2020) 10(1):13518. doi:10.1038/s41598-020-70449-5
110. Desmond, A, Kurian, AW, Gabree, M, Mills, MA, Anderson, MJ, Kobayashi, Y, et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol (2015) 1(7):943–51. doi:10.1001/jamaoncol.2015.2690
111. Tomsic, J, Senter, L, Liyanarachchi, S, Clendenning, M, Vaughn, CP, Jenkins, MA, et al. Recurrent and founder mutations in the PMS2 gene. Clin Genet (2013) 83(3):238–43. doi:10.1111/j.1399-0004.2012.01898.x
112. Hu, L, Sun, J, Li, Z, Qu, Z, Liu, Y, Wan, Q, et al. Clinical relevance of pathogenic germline variants in mismatch repair genes in Chinese breast cancer patients. npj Breast Cancer (2022) 8(1):52–10. doi:10.1038/s41523-022-00417-x
113. Guerrini-Rousseau, L, Gallon, R, Pineda, M, Brugières, L, Baert-Desurmont, S, Corsini, C, et al. Report of the sixth meeting of the European Consortium ‘Care for CMMRD’ (C4CMMRD), Paris, France, November 16th 2022. Fam Cancer (2024) 23(4):447–57. doi:10.1007/s10689-024-00403-1
114. Gallon, R, Brekelmans, C, Martin, M, Bours, V, Schamschula, E, Amberger, A, et al. Constitutional mismatch repair deficiency mimicking Lynch syndrome is associated with hypomorphic mismatch repair gene variants. npj Precision Oncol (2024) 8(1):119. doi:10.1038/s41698-024-00603-z
115. Segura, AVC, da Silva, SIO, Santiago, KM, Brianese, RC, Carraro, DM, and Torrezan, GT. Misclassification of a frequent variant from PMS2CL pseudogene as a PMS2 loss of function variant in Brazilian patients. Fam Cancer (2024) 23(4):653–7. doi:10.1007/s10689-024-00411-1
116. Munteanu, CV, Marian, C, Chiriță-Emandi, A, Puiu, M, and Trifa, AP. In silico splicing analysis of the PMS2 gene: exploring alternative molecular mechanisms in PMS2-associated Lynch syndrome. BMC Genomic Data (2024) 25(1):1–14. doi:10.1186/s12863-024-01281-3
117. Gallon, R, Phelps, R, Hayes, C, Brugieres, L, Guerrini-Rousseau, L, Colas, C, et al. Constitutional microsatellite instability, genotype, and phenotype correlations in constitutional mismatch repair deficiency. Gastroenterology (2023) 164(4):579–92.e8. doi:10.1053/j.gastro.2022.12.017
118. Gould, GM, Grauman, PV, Theilmann, MR, Spurka, L, Wang, IE, Melroy, LM, et al. Detecting clinically actionable variants in the 3′ exons of PMS2 via a reflex workflow based on equivalent hybrid capture of the gene and its pseudogene. BMC Med Genet (2018) 19(1):176–13. doi:10.1186/s12881-018-0691-9
119. Vaughn, CP, Baker, CL, Samowitz, WS, and Swensen, JJ. The frequency of previously undetectable deletions involving 3’ exons of the PMS2 gene. Genes, Chromosomes and Cancer (2013) 52(1):107–12. doi:10.1002/gcc.22011
120. Hayward, BE, De Vos, M, Valleley, EMA, Charlton, RS, Taylor, GR, Sheridan, E, et al. Extensive gene conversion at the PMS2 DNA mismatch repair locus. Hum Mutat (2007) 28(5):424–30. doi:10.1002/humu.20457
121. Chung, J, Negm, L, Bianchi, V, Stengs, L, Das, A, Liu, ZA, et al. Genomic microsatellite signatures identify germline mismatch repair deficiency and risk of cancer onset. J Clin Oncol (2023) 41(4):766–77. doi:10.1200/JCO.21.02873
Keywords: constitutional mismatch repair deficiency, PMS2, genotype, Lynch, VarChat
Citation: Munteanu CV, Lighezan DL, Capcelea A, Chiriță-Emandi A and Trifa AP (2025) Genotype-phenotype correlations in PMS2-associated constitutional mismatch repair deficiency: a systematic literature review. Oncol. Rev. 19:1679576. doi: 10.3389/or.2025.1679576
Received: 04 August 2025; Accepted: 31 October 2025;
Published: 17 November 2025.
Edited by:
Claudia Maletzki, University Hospital Rostock, GermanyReviewed by:
Joseph Lam, University of British Columbia, CanadaYurong Song, National Cancer Institute at Frederick (NIH), United States
Copyright © 2025 Munteanu, Lighezan, Capcelea, Chiriță-Emandi and Trifa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Luisa Lighezan, ZGlhbmFsaWdoZXphbkBnbWFpbC5jb20=