 Fawaz Awad1,2*
Fawaz Awad1,2* Razan Abukhaizaran3Shahira Al Jabi3Mustafa Nabilsi2
Razan Abukhaizaran3Shahira Al Jabi3Mustafa Nabilsi2 Laith A. Ayasa2Majd A. AbuAlrob2,†Haneen Owienah4
Laith A. Ayasa2Majd A. AbuAlrob2,†Haneen Owienah4 Hanin Kassem1
Hanin Kassem1 Moien Kanaan1,5
Moien Kanaan1,5
- 1Department of Molecular Genetics Laboratory, Istishari Arab Hospital, Ramallah, Palestine
- 2Molecular Genetics and Microbiology Research Laboratory, Faculty of Medicine, Al-Quds University, Jerusalem, Palestine
- 3Razan Centre for Infertility, Ramallah, Palestine
- 4Department of Radiology, Istishari Arab Hospital, Ramallah, Palestine
- 5Molecular Genetic Lab, Bethlehem University, Bethlehem, Palestine
Aims: Multiple morphological abnormalities of the sperm flagella (MMAF) represents a rare and severe form of male infertility, characterized by defects in sperm flagella. Mutations in genes essential for flagellar function, such as CFAP43, have been implicated in MMAF. Flagella and motile cilia share a conserved axonemal structure essential for their motile function and the asthenospermia-related infertility of MMAF overlaps with primary ciliary dyskinesia (PCD) symptoms, characterized by chronic airway disease and infertility due to ciliary and flagellar dysfunction. This study investigates the genetic basis of MMAF in two siblings, who also exhibited respiratory symptoms.
Methods: Clinical assessment and semen analysis were conducted for two brothers presenting with infertility and chronic respiratory symptoms. Whole-exome sequencing (WES) was performed to identify potential genetic defects.
Results: Both siblings exhibited classic MMAF features, including asthenospermia with various flagellar abnormalities, in addition to chronic respiratory symptoms including sinusitis and wet cough. WES identified a novel homozygous missense genetic variation in CFAP43 [c.421T>A p.(Trp141Arg)].
Conclusion: Our findings provide additional evidence of the genetic contribution of CFAP43 in MMAF and suggest an expanded phenotypic spectrum of CFAP43-associated conditions to encompass chronic respiratory symptoms attributed to airway ciliary dysfunction. Further research is needed to uncover the underlying mechanisms linking CFAP43 mutations to these phenotypes.
Introduction
Male infertility is a rapidly growing concern, contributing to around 50% of infertility cases. While the revolutionary technique of intracytoplasmic sperm injection (ICSI) has been a treatment option for many cases of male infertility, the aetiology of male infertility remains elusive in nearly 50% of cases (1). A shift towards identifying the underlying causes of infertility and exploring advanced personalized management have gained momentum. This endeavour has been aided by advanced genetic testing, which has uncovered several previously unknown genetic conditions contributing to infertility.
Multiple morphological abnormalities of the sperm flagella (MMAF) is a rare yet severe cause of asthenospermia-related infertility, where sperm flagella show a wide range of malformations, including short, coiled, absent, and irregular flagella forms leading to impairment in sperm motility and morphology, two critical factors in successful fertilization and normal reproductive function. These abnormalities are primarily a consequence of alterations in the ultrastructure of the flagellar axoneme, which can include defects in the highly organized microtubules or proteins involved in their assembly, as well as the surrounding dense fibers (2).
The axoneme, typically organized in a ″9 + 2″ microtubule arrangement, consists of nine microtubule doublets surrounding a central pair of microtubules. This structure is crucial for the motility of both sperm flagella and motile cilia. The microtubule doublets serve as a structural scaffold, supporting the movement, while the central pair complex stabilizes and coordinates movement through radial spokes. These spokes help transmit signals for synchronized motion. The dynein arms, motor proteins on the outer doublets, generate the force needed for axonemal bending by moving along adjacent microtubules (2).
The use of high-throughput sequencing techniques such as whole exome sequencing (WES) has significantly advanced the genetic diagnosis of MMAF. About 43 MMAF-associated genes have so far been identified, with mutations in the DNAH1, CFAP43, and CFAP44 genes responsible for approximately one-third of MMAF cases (3). However, the molecular mechanisms linking these defects to the MMAF phenotype are still largely poorly understood. Additionally, the exact relationship of these defects with patients' phenotypes is not well established.
Cilia and flagella have similar axonemal structure sharing many essential constitutional proteins. This can be clinically manifested in PCD, in which patients have manifestations of ciliary dysfunction, including chronic airway disease and, in most cases, infertility due to flagellar dysfunction. Nevertheless, it is noteworthy that cilia and flagella may have differences in the molecular associated function indicated by the fact that not all PCD patients experience infertility, and that most individuals with MMAF do not present respiratory symptoms (4).
Recently, biallelic loss-of-function mutations in the CFAP43 gene (cilia- and flagella-associated protein 43, MIM#: 617592), which encodes a protein necessary for normal sperm flagella axonemal organization, were reported as a causative factor of MMAF in patients with infertility (5, 6). Since then, about 29 CFAP43 MMAF-causing genetic variations (pathogenic or likely pathogenic) have been described in the literature (7). However, none of the described patients showed symptoms related to defects with airway cilia function. It is worth noting that a nonsense variation in the CFAP43 gene has been linked to adult-onset normal-pressure hydrocephalus, suggesting that this gene may also be involved in the functioning of cilia in the ventricles of the brain (8).
In this study, we aimed to elucidate the molecular basis of MMAF-induced male infertility in a Palestinian family with two affected siblings who exhibited chronic respiratory symptoms indicative of ciliary dysfunction of the airways. We conducted WES in the two siblings and identified a novel missense variation in the CFAP43 gene.
Method
Patients
Two male brothers diagnosed with MMAF referred from Razan Centre for Infertility (Palestine) were enrolled in this study. Both patients sought infertility treatment from the years 2005 to 2007. They were enrolled in this study for genetic diagnosis and evaluation of respiratory symptoms. A standardized clinical information form was filled out for each patient.
Semen analysis
Semen samples were collected via masturbation following 2–7 days of sexual abstinence. The samples were examined after liquefaction for 30 min at 37°C. Semen analyses were repeated twice for each individual six weeks apart. Semen analysis, including morphological assessment of sperm, was performed per the 4th edition of the WHO manual.
Genetic investigations
Molecular diagnosis was performed at the molecular genetic laboratory of the Istishari Arab Hospital (Ramallah, Palestine). Genomic DNA was extracted from 5 ml peripheral blood samples collected from both patients and an apparently healthy unrelated control. WES was performed using the TruSeq Capture Exome Kit (Illumina), targeting 45 Mb of exonic content to obtain at least 20× coverage depth for >98% of the targeted bases. Sequencing was conducted on the NextSeq500 platform, and data were aligned to the human genome reference (hg19) using the BWA aligner and ANNOVAR was used for variant annotation. Copy-number variants (CNVs) were called using an in-house bioinformatics CNV detection pipeline.
A stepwise filtering approach was applied to prioritize variants relevant to the phenotype (Supplementary Figure 1). Variants with a read depth <5 were excluded, and common variants were filtered out based on a minor allele frequency (MAF) threshold of >1% in gnomAD (v2.1.1) and >5% in our in-house exome/genome database. The analysis focused on coding exons and flanking +/−10 intronic nucleotides. Variants predicted to have a high or moderate impact on protein function including missense, nonsense, frameshift, and splice site variations were included. Synonymous and intronic variants beyond 10 nucleotides from exon-intron boundaries were excluded unless they were known to affect splicing or previously linked to disease. Remaining variants were evaluated for pathogenicity using prediction tools such as PolyPhen-2, SIFT, AlphaMissense, and CADD. Following this filtration process, a final list of candidate variants was generated (Supplementary Table 1). The CFAP43 variant identified through WES was subsequently confirmed by Sanger sequencing using the following primers for PCR amplification and sequencing: F: 5′-ACTTGGACTTGATAAGCACAGC-3′; R: 5′-GCCTTGGTATAGCATATGACAGT-3′.
Results
Clinical presentation of MMAF patients
The two patients (Patient 1 and Patient 2) are siblings born to a consanguineous union (first-degree cousins) from a village in the West Bank (Palestine) who both consulted for primary male infertility. Semen analysis of both patients revealed normal sperm count. In contrast, an almost complete asthenospermia with the absence of progressive sperm motility was observed in both patients. In addition, very few spermatozoa with normal morphology were present (1% and 2% respectively) (Table 1). An initial diagnosis of MMAF was established based on the following criteria: Infertility in addition to the presence of spermatozoa with 5 morphological abnormalities: short, absent, coiled, bent, and irregular flagella. None of the siblings reported exposure to gonadotoxic factors.
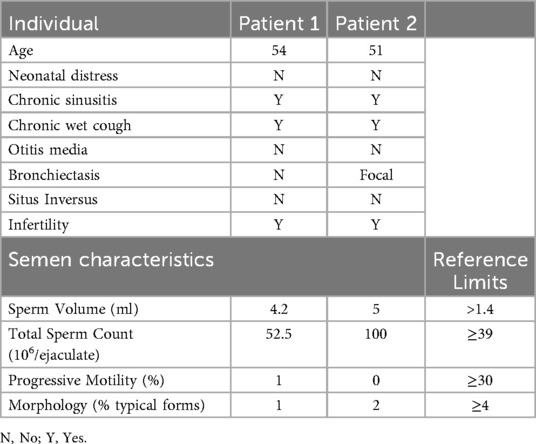
Table 1. Clinical and semen characteristics of the individuals with the identified CFAP43 genetic variation.
Both patients are married and have a son each via assisted reproductive technology. Patient 1 (Figure 1A, IV2), who is now 54 years old, has a history of 8 ICSI trials, and one of them was successful. Patient 2 (Figure 1A, IV3), who is now 51 years old, also has a history of 4 ICSI trials, and one of them was successful. As per the genealogical tree shown in Figure 1A, they have 3 brothers and 5 sisters, all of them had naturally conceived infants.
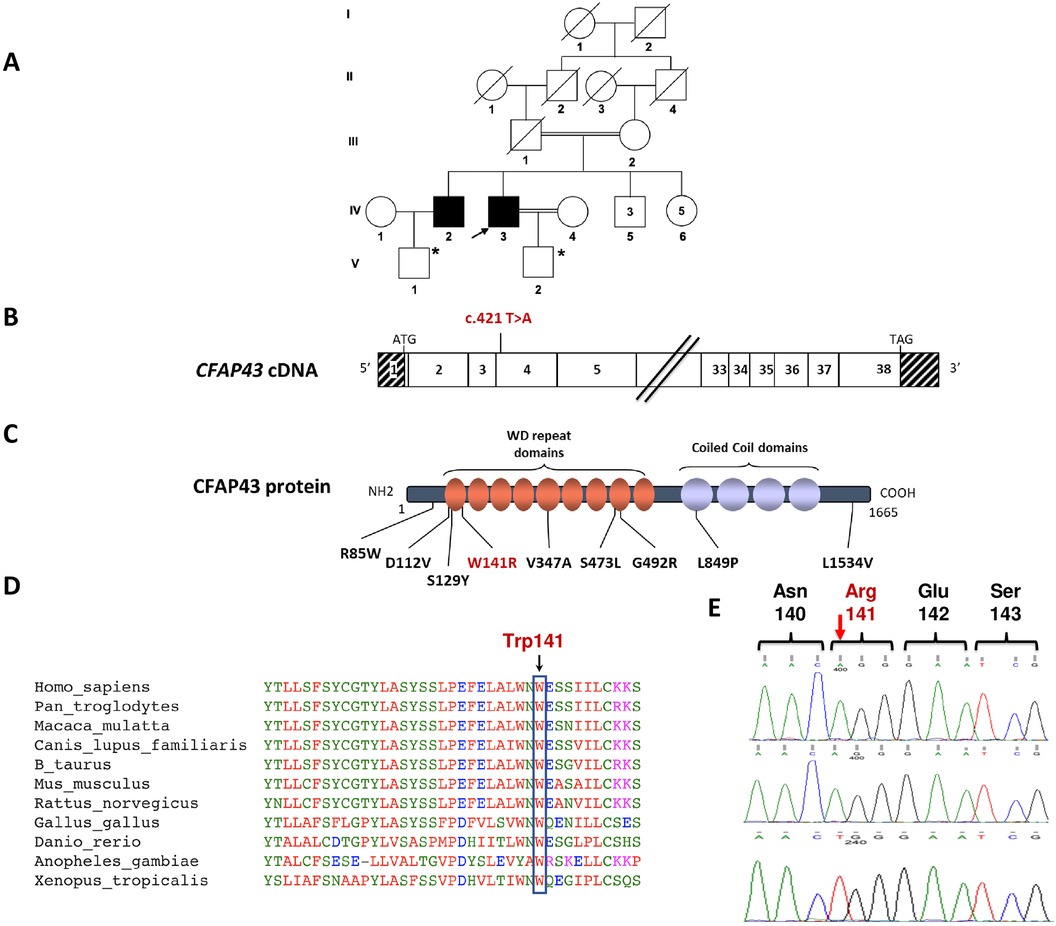
Figure 1. Identification of the homozygous c.421T>A; p.(Trp141Arg) missense variation in the CFAP43 gene. (A) Genealogical tree of the studied family. The black arrow shows the Proband. *Products of assisted reproductive technology through ICSI. (B) Exonic organization of the human CFAP43 cDNA in which the genetic variation described in this study is shown. The exons of CFAP43 are indicated by empty or hashed boxes depicting translated or untranslated sequences, respectively. (C) Domain-organization model of the CFAP43 protein as described by Uniprot server with a summary of previously reported MMAF-associated missense variations (in black font) and the novel variation identified in this study (in red font). (D) A partial protein sequence alignment of CFAP43 among various species shows the evolutionary conservation of the amino acid substitution identified in this study. Sequence alignment shows that Trp141 is a highly conserved residue. (E) Sanger sequencing confirmed the presence of the genetic variation (c.421T>A) in CFAP43, which was identified by WES in the two brother patients (upper and middle electropherograms). The sequence of the identified CFAP43 variation locus is also shown in a healthy control, showing the wild-type (WT) genotype (lower electropherogram).
Importantly, both patients reported that they also suffer from respiratory symptoms characterized by chronic sinusitis and chronic wet cough since infancy (Table 1) and therefore, they were referred to a pulmonology clinic for further evaluation and follow-up. As shown in Figure 2, a chest computerised tomography (CT) scan of patient 2 revealed a localized area of bronchiectasis in the medial segment of the right middle lobe of the lung. Moreover, cystic bronchiectasis accompanied by mucus plugging was also observed, particularly in the lower lobes. There were no signs of laterality defects in the CT scan, which was expected based on the patient's medical history. Patient 1 refused to be evaluated for the respiratory symptoms.
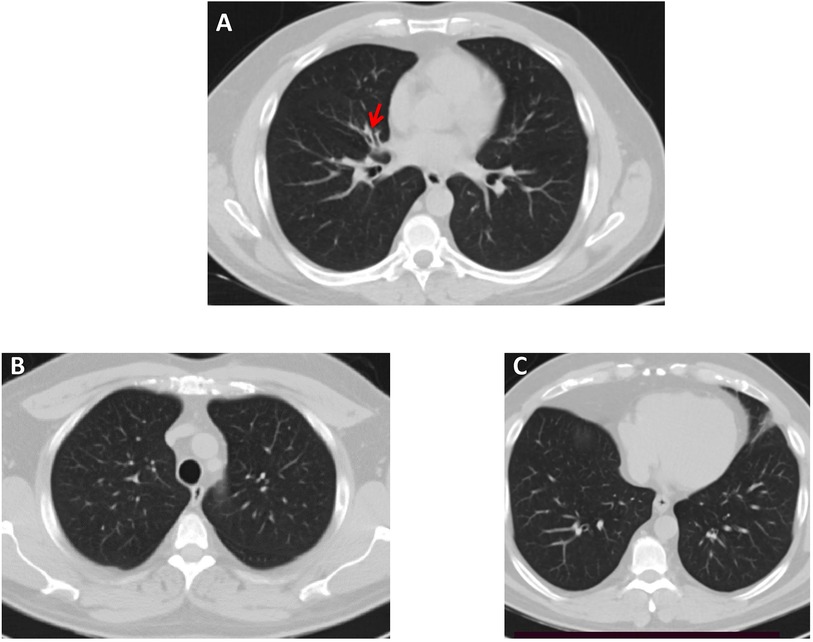
Figure 2. High-resolution chest CT scan—lung window, of Patient 2. (A) A small area of bronchiectasis in the medial segment of the right middle lobe, indicated by a red arrow. (B,C) Both lung fields in the upper and lower lobes appear normal.
Identification of a missense variation in the CFAP43 gene
To identify the underlying genetic cause of this condition, we performed WES for both affected siblings. WES analysis led to the identification of a homozygous c.421T>A p.(Trp141Arg) variant in exon 4 of the CFAP43 gene in both patients (Figure 1B). The allele frequency of this variant was extremely low in gnomAD and was absent from 1,000 healthy ancestry-matched control samples. Additionally, the identified sequence variation is predicted to be deleterious according to SIFT, PolyPhen-2, AlphaMissense (score = 0.987) and CADD (Phred score = 27.4) (Supplementary Table 1). This variant leads to amino acid substitution at codon 141 involving the amino acid tryptophan that is conserved throughout evolution and located within the WD repeat domains of the CFAP43 protein (Figures 1C,D). No variants have been previously described as disease-causing in the literature or reported in clinical variant databases at the same nucleotide (c.421T) or at adjacent nucleotides affecting the same codon that would result in an identical or different amino acid substitution. The identified CFAP43 variation was further verified by Sanger sequencing in both patients (Figure 1E). Unfortunately, family-based segregation analysis could not be performed due to the unavailability of DNA samples from the parents and healthy siblings of the patients.
To rule out other potential genetic contributors to the phenotype, we reviewed all variants identified in known MMAF and PCD genes, irrespective of their predicted pathogenicity (Supplementary Table 2). Each variant underwent assessment and was classified as benign or likely benign, or found in a monoallelic state (one heterozygous variant) in genes with a recessive mode of inheritance, thus ruling out their involvement in the patients' phenotype. These findings collectively suggest that the c.421T>A p.Trp141Arg missense variation in CFAP43 is causative for the MMAF phenotype and associated PCD-like symptoms in this family.
Discussion
In this study, we investigated two brothers diagnosed with MMAF associated with chronic respiratory manifestations, both carrying a homozygous variation [c.421T>A p.(Trp141Arg)] in the CFAP43 gene. Our findings provide the first evidence of a CFAP43 genetic variation causing asthenospermia-related infertility in Palestine. Additionally, they highlight the potential expansion of the CFAP43 gene's phenotypic spectrum to include respiratory manifestations.
Although direct evaluation of the functional and structural impact of the p.(Trp141Arg) missense variation on patients' spermatozoa was not feasible due to the lack of fresh biological samples, we postulate its causality based on similarities observed in the semen analysis of our patients compared to previously reported patients with CFAP43 mutations (6). Furthermore, several lines of molecular evidence support the potential pathogenicity of the identified CFAP43 variation. Firstly, it resides in a critical hotspot within the WD repeat domains, where most reported pathogenic missense CFAP43 variations are found (Figure 1C) (7). Secondly, the evolutionary conservation of the Trp141 residue underscores its significance in the biological role of the CFAP43 protein. Additionally, the identified p.(Trp141Arg) variation in CFAP43 protein is predicted to be deleterious using mutation-prediction bioinformatic tools such as SIFT, Polyphen-2 and CADD.
While most previously identified CFAP43 variations are loss-of-function truncating mutations (frameshift, splicing, or stop mutations), our findings reveal a novel missense variation p.(Trp141Arg), that needs to be assessed by functional studies. Interestingly, previous reports have indicated that missense variants in CFAP43 are associated with milder symptoms manifested by mild asthenospermia (5). However, this was not observed in the studied family, as both siblings suffered from almost complete asthenospermia in addition to the chronic respiratory symptoms.
Several studies have reported genetic variations in CFAP43 as a causative factor for MMAF in infertility patients, and this was further confirmed by functional studies and various knockout mouse models (5, 6). Moreover, Urbanska et al. proposed that CFAP43 also plays a role in ciliary beating. Their study on Tetrahymena thermophila described a novel ciliary complex composed of two conserved WD-repeat proteins, Fap43 (the ortholog of human CFAP43) and Fap44, located near the two-headed inner dynein arm, IDA I1. Loss of these proteins altered ciliary waveform, beat stroke, and reduced swimming speed (9). Additionally, Rachev et al. demonstrated that Cfap43 modulates ciliary beating in mouse and Xenopus, and that loss of Cfap43 leads to mucociliary clearance defects and hydrocephalus in addition to known male infertility (10).
While CFAP43 protein is implicated in both ciliary and flagellar structure and function, reported cases with CFAP43 genetic variations have not typically exhibited clinical manifestations of ciliary dysfunction in the airways. In our study, both patients exhibited symptoms consistent with respiratory ciliary dysfunction, including chronic sinusitis and chronic wet cough since early infancy. Additionally, chest CT scan revealed bronchiectasis, providing further evidence of ciliary impairment. Notably, previous reports have documented a similar clinical presentation, wherein variations in genes initially associated with MMAF, such as SPEF2 and CFAP47, were identified as the underlying cause for both ciliary and flagellar phenotypes (11, 12). These reports indicate that defects in some MMAF genes may cause a phenotypic spectrum ranging from MMAF without respiratory manifestations to infertile patients with PCD-like symptoms.
In conclusion, this study identified a novel variation in the CFAP43 gene in two siblings presenting with MMAF and chronic respiratory symptoms. While our findings provide preliminary evidence suggesting a potential role of CFAP43 mutations in both reproductive and respiratory phenotypes, further functional studies are needed to validate the direct effect of the identified variant on protein function and confirm its pathogenicity. Additionally, further research is warranted to identify other disease-causing genetic variations that may contribute to the MMAF phenotype with impaired motile cilia.
Data availability statement
De-identified summary data supporting the conclusions of this study are provided as Supplementary Table 2.
Ethics statement
The studies involving humans were approved by the local ethics committee of the Istishari Arab Hospital (Ramallah, Palestine). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
FA: Methodology, Writing – original draft, Formal analysis, Supervision, Writing – review & editing, Software, Visualization, Conceptualization, Validation, Funding acquisition, Project administration. RA: Visualization, Formal analysis, Conceptualization, Writing – review & editing. SA: Writing – review & editing, Methodology, Formal analysis. MN: Methodology, Formal analysis, Writing – original draft. LA: Formal analysis, Writing – original draft, Methodology. MA: Writing – original draft, Formal analysis, Methodology. HO: Methodology, Writing – review & editing, Formal analysis. HK: Writing – review & editing, Methodology, Formal analysis. MK: Validation, Methodology, Writing – review & editing, Writing – original draft, Visualization, Formal analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The study was funded by a grant from Razan Centre for Infertility, Ramallah, State of Palestine.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frph.2025.1609938/full#supplementary-material
Supplementary Figure 1 | Exome data analysis filtering approach. Illustration of the stepwise variant filtration approach used in the exome analysis of two affected siblings (Patient 1 and Patient 2). MAF, minor allele frequency; MS, missense; FS, frameshift; NFS, non-frameshift; SS, splice site; NS, nonsense.
Supplementary Table 1 | Details of the final list of homozygous candidate variants shared by the two siblings after applying the whole exome sequencing filtering strategy. AA, amino acid; gnomAD, genome aggregation database; OMIM, online Mendelian inheritance in man; N/A, not available.
Supplementary Table 2 | Comprehensive list of all variants identified in known MMAF and PCD genes in the two affected siblings, regardless of predicted pathogenicity.
References
1. Esteves SC, Zini A, Coward RM, Evenson DP, Gosálvez J, Lewis SEM, et al. Sperm DNA fragmentation testing: summary evidence and clinical practice recommendations. Andrologia. (2021) 53:e13874. doi: 10.1111/and.13874
2. Ben Khelifa M, Coutton C, Zouari R, Karaouzène T, Rendu J, Bidart M, et al. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. (2014) 94:95–104. doi: 10.1016/j.ajhg.2013.11.017
3. Wang J, Wang W, Shen L, Zheng A, Meng Q, Li H, et al. Clinical detection, diagnosis and treatment of morphological abnormalities of sperm flagella: a review of literature. Front Genet. (2022) 13:1034951. doi: 10.3389/fgene.2022.1034951
4. Jiao S-Y, Yang Y-H, Chen S-R. Molecular genetics of infertility: loss-of-function mutations in humans and corresponding knockout/mutated mice. Hum Reprod Update. (2021) 27:154–89. doi: 10.1093/humupd/dmaa034
5. Coutton C, Vargas AS, Amiri-Yekta A, Kherraf Z-E, Mustapha SFB, Tanno PL, et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in trypanosoma and human. Nat Commun. (2018) 9:686. doi: 10.1038/s41467-017-02792-7
6. Tang S, Wang X, Li W, Yang X, Li Z, Liu W, et al. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. (2017) 100:854–64. doi: 10.1016/j.ajhg.2017.04.012
7. Ma J, Long S-H, Yu H-B, Xiang Y-Z, Tang X-R, Li J-X, et al. Patients with MMAF induced by novel biallelic CFAP43 mutations have good fertility outcomes after intracytoplasmic sperm injection. Asian J Androl. (2023) 25:564–71. doi: 10.4103/aja2022118
8. Morimoto Y, Yoshida S, Kinoshita A, Satoh C, Mishima H, Yamaguchi N, et al. Nonsense mutation in CFAP43 causes normal-pressure hydrocephalus with ciliary abnormalities. Neurology. (2019) 92:e2364–74. doi: 10.1212/WNL.0000000000007505
9. Urbanska P, Joachimiak E, Bazan R, Fu G, Poprzeczko M, Fabczak H, et al. Ciliary proteins Fap43 and Fap44 interact with each other and are essential for proper cilia and flagella beating. Cell Mol Life Sci CMLS. (2018) 75:4479–93. doi: 10.1007/s00018-018-2819-7
10. Rachev E, Schuster-Gossler K, Fuhl F, Ott T, Tveriakhina L, Beckers A, et al. CFAP43 modulates ciliary beating in mouse and xenopus. Dev Biol. (2020) 459:109–25. doi: 10.1016/j.ydbio.2019.12.010
11. Ge H, Zhou W, He M, Zheng H, Zhao X, Zhang T, et al. Mutations in CFAP47, a previously reported MMAF causative gene, also contribute to the respiratory defects in patients with PCD. Mol Genet Genomic Med. (2024) 12:e2278. doi: 10.1002/mgg3.2278
Keywords: CFAP43, male infertility, asthenoazoospermia, multiple morphological abnormalities of the flagella, primary ciliary diskynesia
Citation: Awad F, Abukhaizaran R, Al Jabi S, Nabilsi M, Ayasa LA, AbuAlrob MA, Owienah H, Kassem H and Kanaan M (2025) Expanding the phenotypic spectrum associated with CFAP43 mutations: a case report of familial male infertility with respiratory manifestations. Front. Reprod. Health 7:1609938. doi: 10.3389/frph.2025.1609938
Received: 7 May 2025; Accepted: 22 October 2025;
Published: 18 November 2025.
Edited by:
Poonam Mehta, University of Massachusetts Medical School, United StatesReviewed by:
Santasree Banerjee, Jilin University, ChinaGuoliang Jiang, Central South University, China
Ao Ma, University of Science and Technology of China, China
Copyright: © 2025 Awad, Abukhaizaran, Al Jabi, Nabilsi, Ayasa, AbuAlrob, Owienah, Kassem and Kanaan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fawaz Awad, ZmF3YXouYXdhZEBpYWgucHM=
†Present Address: Majd A. AbuAlrob, Neurology department of Hamad Medical Corporation, Doha, Qatar