 Leidy Erandy Hernández-Magaña1†
Leidy Erandy Hernández-Magaña1† Alfredo Mosqueda-Gracida1†Víctor Javier Cruz-Holguín2†Macario Martínez-Castillo3
Alfredo Mosqueda-Gracida1†Víctor Javier Cruz-Holguín2†Macario Martínez-Castillo3 Ezequiel M. Fuentes-Pananá4Tomasz Rozmyslowicz5
Ezequiel M. Fuentes-Pananá4Tomasz Rozmyslowicz5 Moisés León-Juárez2
Moisés León-Juárez2 Haruki Arévalo-Romero1*
Haruki Arévalo-Romero1*- 1Laboratorio de Inmunología y Microbiología Molecular, División Académica Multidisciplinaria de Jalpa de Méndez, Departamento de Genómica, Universidad Juárez Autónoma de Tabasco, Jalpa de Méndez, Mexico
- 2Laboratorio de Virología Perinatal, Departamento de Inmunobioquímica, Instituto Nacional de Perinatología Isidro Espinosa de los Reyes, Ciudad de México, Mexico
- 3Sección de Estudios de Posgrado e Investigación, Escuela Superior de Medicina, Instituto Politécnico Nacional, Ciudad de México, Mexico
- 4Unidad de Investigación en Virología y Cáncer, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico
- 5Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Human Adenoviruses are a diverse family of viruses that can infect a variety of tissues causing acute or persistent infection. Viruses induce numerous cellular alterations as they hijack cellular functions to promote viral progeny. Recent research has shed light on the functions of viral proteins in orchestrating viral production, revealing that many of these functions overlap with oncogenesis or metabolic disruption. Studies of the Adenovirus family (Adenoviridae) have identified oncogenic members, such as Adenovirus (Ad-)2, 5, 9, and 12, and also Ad-36, which is most extensively studied for its ability to induce metabolic alterations. Specifically, Adenoviruses encode a gene product known as early region 4 open reading frame 1 (E4orf1), which has emerged as an oncoprotein and regulator of metabolism depending on the lineage of the infected host cell. This article aims to provide insight into the functions of the viral protein E4orf1 and the overlapping similarities between the oncogenic process and cell metabolism.
Introduction
Cancer and obesity are two leading causes of morbidity and mortality worldwide, and recent research suggest an interconnection between the two (1). The study of tumor viruses has been instrumental in advancing our understanding of molecular oncology, leading to the discovery of oncogenes and tumor suppressor genes, and the elucidation of the cellular processes that they regulate (2). In fact, the International Agency for Research on Cancer has reported that approximately 10% of cancer cases worldwide are attributable to chronic viral infection (3).
Although direct viral-induced cancer is rare, viruses can promote oncogenic transformation through chronic infections and the resulting cellular alterations. As intracellular parasites, viruses depend on the reprogramming of cellular metabolism to facilitate their replication; this reprogramming also occurs in oncogenesis and obesity. The degree of oncogenic or metabolic alteration varies depending on the viral family and may involve direct alteration to cellular proteins or signaling pathways.
The Adenovirus (AdV) family is an example of a family of viruses that induces both oncogenic and metabolic alterations, through direct alteration and signaling pathway misregulation. Although initially thought to transform only rodent cell lines, several AdV members have been found to transform certain human primary cells (4). We focus in this article on AdVs and their ability to induce oncogenesis and metabolic alterations.
Studies mapping the oncogenic activity of AdVs have revealed that the E1 transcriptional unit is responsible for cell transformation. This unit encodes for the oncogenic proteins E1A and E1B, which must be co-expressed to achieve full transformation (5). Analysis of the E1 region has contributed enormously to the development of molecular biology, as it has shown the initial mechanisms of oncogenic transformation through the deregulation of DNA synthesis and proliferation. Through direct or indirect interactions, E1A activities lead to induction of S phase, inactivation of differentiation genes, and activation of proliferation in the host cell. The classic example of direct interaction is the deregulation of the pRb protein, whereas an example of indirect interaction is through histone acetylation of scaffolding proteins (6, 7).
Another essential mechanism for understanding cell cycle control is the inhibition of p53 by E1B to avoid apoptosis. The molecular mechanism involves suppressing p53 transcriptional activity by sequestering the protein in perinuclear bodies, inducing its degradation, or through posttranslational modifications (8, 9). The study of AdVs has also contributed greatly to the understanding of RNA splicing, a process involving a series of reactions catalyzed by a complex of small nuclear ribonucleoproteins (spliceosome) to generate a mature transcript. In the 1980s, in contrast to what was known regarding transcription in prokaryotic cells, little was known about transcription in eukaryotic cells. However, in 1977, two independent groups described the new messenger RNA (mRNA) biosynthesis mechanism using hybridization and electron microscopy assays. To date, almost 1.2 million transcripts have been described using differential RNA sequencing (dRNA-seq), making AdVs the most transcriptionally complex viruses studied in terms of splicing sites (10–12).
As previously discussed, it was initially thought that only E1 region genes were involved in oncogenic transformation. However, it was soon discovered that genes encoded in the E4 region also play a role in altering cellular mechanisms. The transformation potential is attributed to three gene products, namely E4orf1, E4orf3, and E4orf6. Although the last two have overlapping functions with E1A and E1B, and support the oncogenic process in cell models, the E4orf1 of Ad-9 operates independently of E1A and E1B functions. In fact, E4orf1 was first identified as a new oncoprotein because its sole expression was sufficient to induce anchorage-independent growth in rat cells (13).
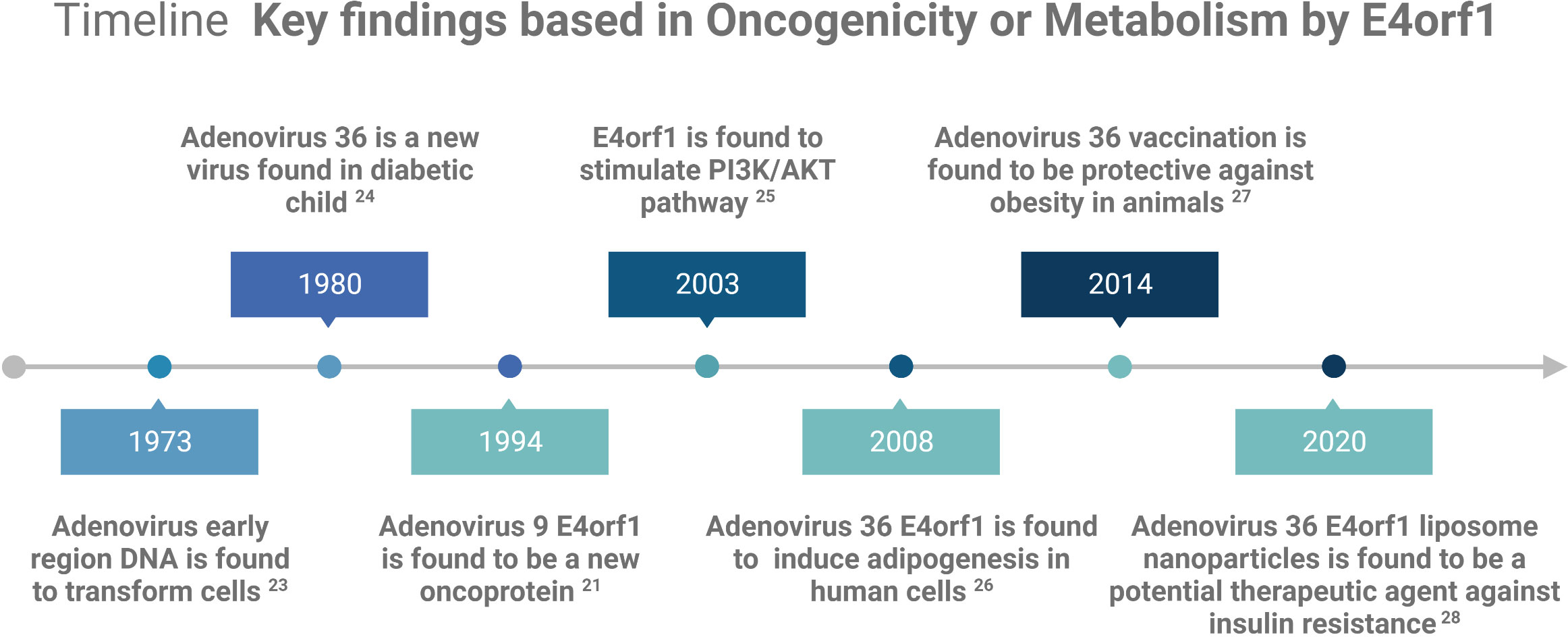
E4orf1 in human AdVs reprogram cellular signaling pathways in infected cells to shape an optimal microenvironment for viral replication. Although not all AdV family members possess the oncogenic capacity of E4orf1, many can deregulate crucial cellular metabolic processes that overlap with oncogenic processes. Consequently, E4orf1 has been investigated as both an oncogenic model and a potential therapeutic agent for obesity and diabetes (see Figure 1 Timeline). This mini review focuses on the roles of E4orf1 in targeting cellular proteins and its involvement in oncogenic and metabolic pathways.
The E4 region organization
The transcription of the AdV genome, on infection, depends on the sequential activation of early and late transcriptional units. There are five early units identified as E1a, E1b, E2a/b, E3, and E4, while examples of later units include IX, IV2a, and L1–L5 genes (21). Initially, the E4 unit was reported to contain eight open reading frames (orfs), encoding proteins with sizes from 11 to 34 kDa (22). However, it is now more widely accepted that E4 encodes for seven different proteins that collectively sculpt the intracellular milieu, optimal to yield viral progeny (21, 23).
Many AdVs share the same E4 unit organization, which is transcribed in the leftward direction and situated at the right end of the genome. The expression of the E4 products is regulated both transcriptionally and posttranscriptionally. The E4 promoter spans approximately 200 bp upstream from the transcription start site and contains binding sites for the viral transcription factor E1A, as well as cellular factors ATF-2 and E4F1 (24, 25).
E4 expression is also importantly regulated by alternative splicing. Initially, it was demonstrated that there are at least 10 splicing sites on E4, with some transcripts generating two or more polypeptides (26). Subsequently, the 10 splice sites were characterized either as a combination of four donor and six acceptor sites or as eight donor and two acceptor sites, encoding a total of seven unique proteins (23, 27). These seven unique products are E4orf1, E4orf2, E4orf3, E4orf4, E4orf6, E4orf6/E4orf7, and E4orf3/E4orf4, based on their orf arrangement. We focus on E4orf1 because it possesses oncogenic potential in several AdVs and has a unique ability, in Ad-36, to deregulate cellular metabolism.
Biochemical features of the E4orf1 protein
E4orf1 is a protein expressed in various AdVs, including Ad-2, Ad-3, Ad-5, Ad-9, Ad-12, and Ad-36. The E4orf1 proteins from these viruses vary from 125 to 128 amino acids in length and have sizes ranging from 14 to 17 kDa. They share 50%–65% identity and possess several potential glycosylation sites, which may contribute to the diversity of their sizes (14, 22). E4orf1 is primarily localized in cytoplasm, where it modulates cellular signaling pathways. However, one study reported E4orf1 forming nuclear complexes and participating in gene transcription (28).
Three E4orf1 regions, termed RI, RII, and RIII, are conserved across several AdVs and have been deemed significant through mutational analysis (Figure 2) (29). The function of RI is unclear, but it appears to form a functional domain with RII. This RI–RII domain seems crucial for the transforming activity of certain E4orf1 protein members (30). RII exhibits some degree of homology with eukaryotic dUTPases, but no enzymatic activity has been demonstrated for E4orf1 so far (30). dUTPases can complex with transcription factors and inhibit their function and it is possible that E4orf1 evolved to mimic this function, as bioinformatic analysis revealed that E4orf1 shares a very similar three-dimensional (3D) structure to dUTPases (29, 30).
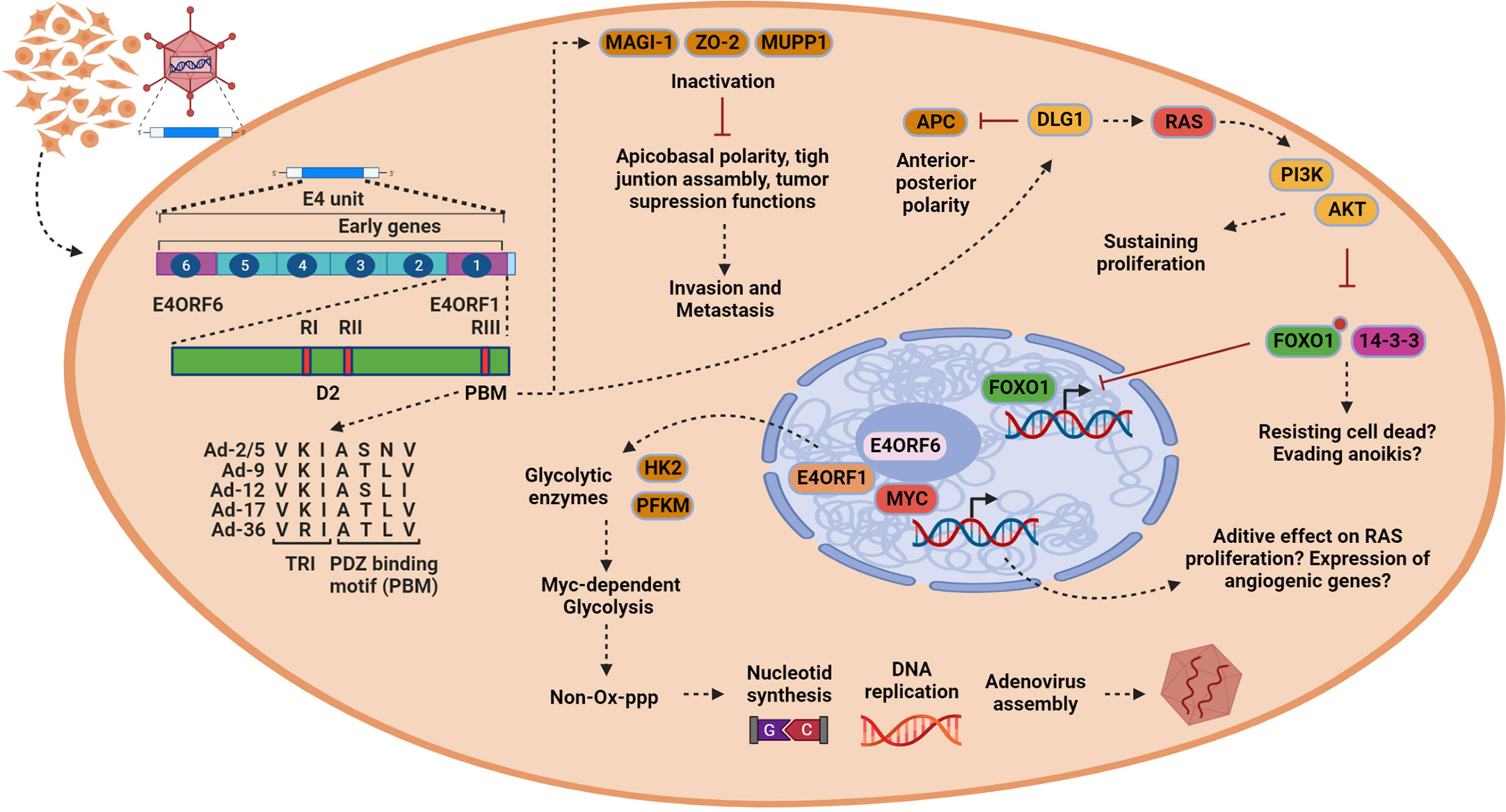
Figure 2 In epithelial cells, E4orf1 induces processes associated with oncogenesis and tumor progression. Once the viral genome is released (left), the transcription of the early genes (only E4 unit is indicated) begins. E4orf1 (green bar) consists of two protein domains. The first of them is designated domain 2 (D2), which is formed by the region RI and RII described initially by mutational analysis. This region shows homology with eukaryotic dUTPases that beyond their enzymatic activity can interact with transcription factors to inhibit their function. E4orf1 through this domain interacts with E4orf6 to form a complex with Myc transcription factor, but, in contrary to dUTPases, the transcriptional function of MYC is activated. The second domain is the PDZ domain-binding motif (PBM) initially called region RIII, which allows E4orf1 to interact with several cellular proteins. The trimerization element (TRI) and PBM sequence alignment is conserved among various members of the Adenoviruses (AdVs) (Ad-2/5, Ad-9, Ad-12, Ad-17, Ad-36) and is shown by an arrow from PBM in the figure (bottom left). E4orf1 dysregulates Dlg1 through the PBM and TRI element, activating a sustained proliferation through the RAS/AKT signaling pathway (upper right). Moreover, it is possible that through PI3K/AKT E4orf1 may promote cell survival by inactivating the FoxO1 transcription factor, by inactivating the pro-apoptotic protein Bad, or by inhibiting pro-caspase 9, all of which promote escape from anoikis (bottom right). However, E4orf1 dysregulates the proteins MUPP1, MAGI-1, and ZO-2. These proteins participate in modulating cell polarization and have tumor suppression activities, which can lead to cell invasiveness (top in the middle). Finally, E4orf1 forms a complex with E4orf6 through D2 domain to interact with MYC transcription factor, inducing enzymes [hexokinase 2 (HK2) and phosphofructokinase 1 (PFKM)] responsible for glycolysis (bottom in the middle). This MYC-dependent glycolysis (MDG) contributes to generating intermediary products used for nucleotide synthesis via the non-oxidative pentose phosphate pathway (PPP), resulting in efficient viral assembly. It is possible that MYC activation also has an effect on cell proliferation or the expression of angiogenic genes, but this must be verified. This figure was created with BioRender.com.
E4orf1 interacts with several cellular proteins through RIII, which features a C-terminal bona fide PDZ domain-binding motif (PBM) with the consensus sequence S/T–X/V/I (X denotes any amino acid) (31, 32). The PDZ domain of RIII is present in several AdV members, such as Ad-2, Ad-3, Ad-5, Ad-9, Ad-12, Ad-17, and Ad-36 (Figure 2). Unlike other PDZ proteins where only 10–15 C-terminal amino acid residues retain the binding activity, mutational analysis indicates that E4orf1 requires an intact 3D structure to function, as seen in Ad-9 (21, 32). No phosphorylation or other posttranslational modifications have been reported for E4orf1; however, most of the associated cellular proteins are phosphorylated. E4orf1 binds to key cellular proteins involved in signal transduction, cell proliferation, and tight junction assembly, and modulates the PI3K-AKT signaling pathway and glycolytic metabolism (17). Most cellular targets of E4orf1 are located in the cytoplasm, with MYC being the only exception. MYC is also modulated in the nucleus by a ternary complex formed by E4orf1–E4orf6–MYC, which activates their transcriptional activity (Figure 2) (28).
E4orf1 in oncogenesis
Viruses alter host cells to support the metabolic demands of their viral progeny. In persistent infections, viruses also alter the replicative and survival capacity of host cells, with resulting consequences for oncogenesis. The first cellular protein identified as a partner of E4orf1 was hDlg/SAP97 (Dlg1). Dlg1 is a protein rich in protein–protein interaction domains, containing three PDZ, one SH3, and one GuK domain. In drosophila, mutations in Dlg result in epithelial tumors exhibiting loss of apicobasal polarity, leading to the initial classification of Dlg1 as a tumor suppressor protein. Dlg1 has been associated with the development of cancer because it controls two very critical signaling pathways for cell proliferation: the Wnt pathway and the PI3K pathway. By interacting with APC (Adenomatous polyposis coli) tumor suppressor gene, it negatively regulates cell cycle progression toward S phase, while its interaction with PTEN regulates the PI3K pathway (33, 34).
It is important to note that the second PDZ domain, which is the target of E4orf1 interaction, also regulates the interaction with APC. Therefore, it has been proposed that E4orf1 promotes oncogenic transformation through this deregulation (32, 35). Moreover, E4orf1 forms a complex with Dlg1 using their trimerization and PMB domains, which is then utilized to recruit PI3K to the plasma membrane. This process subsequently activates AKT kinase, ultimately enhancing viral replication and inducing cell transformation (36, 37). The ability to deregulate the PI3K pathway is conserved among various AdV members, such as Ad-3, Ad-5, Ad-9, Ad-12, and Ad-36, all of which can activate the pathway in a Dlg1-dependent manner. However, Ad-36 is not classified as oncogenic despite inducing substrate-independent growth in soft agar assays. These findings show that cells have acquired the ability to avoid apoptosis and maintain constant proliferation as a consequence of providing a favorable environment for viral progeny assembly (38).
A crucial point in regulating the PI3K signaling pathway is the involvement of the phosphatase and tensin homolog (PTEN), which is often mutated in many cancers. This mutation promotes uncontrolled cell growth by prolonging PI3K signaling (39). In addition to recruiting PI3K through its interaction with Dlg1, E4orf1 might also recruit PTEN. If this were to occur, E4orf1 would ensure a state of constant proliferation without the negative feedback of PTEN. However, it has not been reported that the E4orf1/Dlg1 complex can deregulate the function of PTEN, although it is known that Dlg1 interacts with PTEN (40).
Last, further evidence that points towards Dlg1 deregulation playing a role in cancer is that Dlg1 is a target of other oncogenic viruses. For example, the Tax protein of human T-cell leukemia virus type 1 and E6 proteins from high-risk human papillomavirus (HPV) types, such as HPV-16 and HPV-18, can interact with and disrupt the function of Dlg1 (32).
A second target protein, MAGI-1, was identified following the discovery of E4orf1-mediated dysregulation of Dlg1. MAGI-1 has been implicated in regulating various processes, such as cell–cell adhesion, migration, proliferation, signaling, and survival. Like Dlg1, MAGI-1 acts as a tumor suppressor in different types of cancer, including liver, colorectal, cervical, brain, and gastric, because it also regulates crucial signaling pathways, such as PI3K/AKT and Wnt/β-catenin. Three isoforms of MAGI-1 (a, b, and c) are generated through alternate splicing, and they are widely expressed in different tissues. Interestingly, E4orf1 can interact with isoforms b and c via PDZ domains 1 and 3 to sequester the proteins at particular points in the cell cytoplasm, thereby inactivating their cellular functions (Figure 2) (41).
This observation aligns with the oncogenic capacity of E4orf1, as MAGI-1 also interacts with PTEN, which allows the virus to maintain a constant state of proliferation in the host cell, as mentioned above. The deregulation of PDZ proteins is a common strategy used by other pathogenic viruses, with several examples of MAGI-1 activity control being well documented. For instance, the Tax1 protein disrupts MAGI-1’s function, the E6 protein sends it to degradation, and the NS1 protein of influenza seemingly regulates the antiviral response mediated by interferon through its interaction with MAGI-1 (42–44).
Another tight junction protein targeted by E4orf1 is the multi-PDZ domain protein 1 (MUPP1), which was cloned from human fetal brain cDNA library through interaction with the 5-HT2C receptor. The human gene encodes a 2054 amino acid protein with 13 PDZ domains. MUPP1 is expressed in various tissues and was initially proposed as a novel scaffold protein involved in cell signaling because of its similar structural organization with InaD from Drosophila melanogaster, which regulates the rhodopsin signaling pathway (45). E4orf1 sequesters MUPP1 into the cytoplasm and inactivates its function through PDZ domains 7 and 10, while the 18E6 oncoprotein inactivates its function by sending it to degradation (46).
The most likely role of this deregulation is to overcome the blockage of cell cycle progression, as some evidence suggests MUPP1 acts as a negative regulator of proliferation. E4orf1, by sequestering the protein in the cytoplasm, would circumvent this negative control. Supporting this proposal, a study using a lung cancer model showed that MUPP1 functions as a tumor suppressor by inhibiting cell proliferation through regulation of the Hippo-YAP signaling pathway (47).
However, through the same PDZ domain, deregulated by E4orf1 (PDZ10), MUPP1 seems to regulate the activity of the proto-oncogene c-Kit. In vitro interaction assays have shown that the constitutively active form of c-Kit does not interact with MUPP1, while the wild type does. This suggests that MUPP1 may regulate the receptor activity prior to activation with its ligand (48). This is important because c-Kit induces signaling pathways that regulate proliferation, differentiation, or metabolism, which are involved in the development of various types of tumors (49).
In addition, other evidence supporting MUPP1 as a negative regulator of proliferation has shown that the TAPP1 protein was identified as an adapter protein to recruit MUPP1 to the plasma membrane in response to signals where there are high levels of PtdIns(4,5)P2 (50). This again coincides with the PI3K signaling pathway, as after generating PtdIns(3,4,5)P3 for proliferative AKT signaling it is necessary to return to the basal state and generate PtdIns(4,5)P2. This homeostasis is carried out by phosphatases such as PTEN, and it has been reported that MUPP1 interacts with PTEN through its PDZ domain. Therefore, it is conceivable that this interaction increases the concentration of phosphatase in the plasma membrane and regulates proliferative signaling.
Finally, another cell adhesion molecule deregulated by E4orf1 is ZO-2. This protein is encoded by the TJP2 gene and can be present in both tight and adherent junctions. ZO-2 is a scaffolding protein that has three PDZ domains in its N-terminal domain, allowing it to regulate the formation of tight junctions. The protein can be found in the cytoplasm, tight junctions, or in the cell nucleus. In fact, it has been reported that in the nucleus it can act as a transcription suppressor and block proliferation by interacting with the Myc transcription factor, preventing the latter from activating one of its target genes, CCND1 (51). E4orf1 is capable of sequestering ZO-2 in the cytoplasm, and the objective of this seems to be overcoming the cell cycle entry block induced by ZO-2 (52).
However, it has been mentioned that E4orf1 activates the AKT pathway, which has an inhibitory effect on GSK3B kinase by phosphorylating it at the Ser9 residue. GSK3B inactivation releases the β-catenin protein, which can enter the nucleus and activate proliferation through Myc expression. It is important to mention that ZO-2 prevents GSK3B phosphorylation by AKT, which in turn blocks the proliferation (53). Again, E4orf1, by sequestering ZO-2, ensures a constant proliferative state by allowing β-catenin to act in the nucleus. Therefore, ZO-2 is considered a tumor suppressor, which coincides with reports indicating that its expression is silenced in various types of cancer (54).
In normal epithelium, cells that lose contact with the extracellular matrix (EM) undergo apoptosis. This type of death is called anoikis, and its purpose is to prevent cells from proliferating in inappropriate places (55). Physiologically, cells are protected from anoikis by signal transduction through integrins and growth factor receptors, which generally lead to activation of the PI3K/AKT pathway. Activation of this pathway promotes cell survival by inactivating the pro-apoptotic protein Bad, by inhibiting pro-caspase 9 or by inducing the nuclear exclusion of FoxO transcription factor (56–59). As mentioned previously, PI3K/AKT is the main pathway altered by E4orf1, and so it is plausible that anoikis is also deregulated in virus-infected cells. This proposal aligns with the fact that one of the most notable properties of cells expressing E4orf1 is anchorage-independent growth, a test that precisely indicates that the cells are evading anoikis in addition to acquiring the ability to constantly proliferate. Furthermore, in 3D cultures, it was shown that apoptosis is important for the formation of the lumen in glandular epithelia. Cells in contact with the EM undergo apicobasal polarization, while cells in the lumen (non-polarized and not in contact with the matrix) die by apoptosis. Finally, the proliferation of the polarized cells is suppressed to give rise to the glandular acinus (60). Therefore, tumor cells dysregulate polarization, apoptosis, and proliferation at the beginning of carcinogenesis. Returning to the proteins altered by E4orf1, several (Dlg1, MUPP1, MAGI-1, and ZO-2) participate in modulating both polarization and proliferation, explaining the oncogenic potential of the viral protein. However, we speculate that evasion of anoikis also contributes to the oncogenic potential of the viral protein, as the main pathway altered by E4orf1 (PI3K/AKT) protects against this type of cell death and there is a report of the involvement of Dlg1 in conferring resistance to anoikis (61). Overall, E4orf1 induces loss of control of cell proliferation and apoptosis, two important events at the beginning of the oncogenic process, and also induces loss of polarity that is involved in the invasiveness of tumor cells. These three tumor-acquired capabilities are considered important hallmarks of cancer (39).
E4orf1 in metabolism dysregulation
Among the members of the AdV family, Ad-36 possesses the highest correlation with obesity in both animal models and humans. Since its identification in 1980, the first report of the adipogenic potential of Ad-36 was demonstrated by infecting chickens and mice. In this study, the animals developed an increase in visceral adipose tissue and viral DNA was isolated from adipose tissue, suggesting that the principal target cells are adipocytes. Other studies indicate, however, that the virus can spread to other organs in infected animals (62, 63). In another study using monkey models, a significant association was reported between antibodies against Ad-36 and increased body weight, compared with uninfected animals, indicating the adipogenic potential of this virus (64). Moreover, in a chicken model, it was shown that Ad-36 can be transmitted horizontally and induce obesity, suggesting that transmission in humans can take place by a similar mechanism (65). Finally, various meta-analyses have demonstrated an association with the risk of developing obesity [mean odds ratio (OR) = 2.2] in infected adults, and the analysis in different countries reveals a 30% prevalence of infection among obese individuals. Therefore, these studies in animal models and humans highlight the significant role of Ad-36 infection in altering adiposity and contributing to the development of obesity (63).
Using the 3T3-L1 pre-adipocyte cell model, it was demonstrated that Ad-36 infection has an adipogenic effect. On day 2 post infection, the virus begins its replication but is abortive; however, the mRNA expression peak of early genes (including E4orf1) coincides with the differentiation of adipocytes. The pharmacological inhibition of E4orf1 expression decreases lipid accumulation and the expression of the C/EBPβ gene, which is important for lipogenesis, suggesting the participation of the viral protein in the induction of adipogenesis (66). Subsequently, using in vitro and ex vivo models, it was reported that the adipogenic effect of Ad-36 is partly due to the modulation of leptin expression and glucose metabolism. In both 3T3-L1 cells and in primary rat adipocytes or in their fatty tissue, it was observed that infection by Ad-36 reduces leptin expression, with the concomitant increase in glucose uptake. In turn, a progressive increase in lipid accumulation was observed due to the expression of genes involved in their synthesis, such as FAS and ACC1, revealing part of the mechanism behind the positive effect on adipogenesis (67).
To evaluate if the adipogenic effect was present in humans, experiments were conducted using primary human adipose-derived stem/stromal cells (hASC). In this model, it was observed that Ad-36 increases the expression of PPARγ and C/EBPβ, which are master regulators of adipocyte differentiation. Concurrently, an increase in fatty acid-binding protein (AP2), which is a marker of adipocyte differentiation, was observed. This was also present in LPL, which is a key enzyme for the uptake of triglyceride-delivered fatty acids to adipose tissue. Using hASC extracted from subjects infected with Ad-36, a greater accumulation of lipids was observed, demonstrating that the virus effectively induces the commitment and differentiation toward adipocytes (68).
However, using a microarray assay and gene set enrichment analysis with a human mesenchymal stem cell model, it was confirmed that Ad-36 induces the expression of at least 35 genes involved in adipocyte differentiation, such as LPL, C/EBP, ATF3, and FABP4. Interestingly, the importance of the PPARγ signaling pathway was highlighted again, as genes such as ADIPOQ, APOC3, and PLTP were upregulated and are involving in enhancing glucose utilization and fatty acid combustion (69). Finally, in the search for the specific viral gene responsible for adipogenesis, it was found that Ad-36 does so through E4orf1. In both 3T3-L1 cells and hASCs infected with Ad-36, an induction of adipogenesis accompanied by cell proliferation was observed through signals mediated by the phosphorylation of AKTser473 and P38thr180. Through the expression of the recombinant protein in the 3T3-L1 cells, the adipogenic effect was confirmed, because if the expression of the viral protein is inhibited by using siRNA technology, then the adipogenic effect is reversed, showing that E4orf1 is the viral adipogenic regulator (18). In addition, the recombinant protein induced the redistribution of FoxO1 from the nucleus to the cytoplasm in an AKT-dependent manner, which in adipocytes is important to allow their differentiation (70, 71). Overall, these studies have shown that E4orf1 can induce hypertrophy and hyperplasia in adipocytes by increasing their capacity to store glucose and induce proliferative signaling pathways (Figure 3).
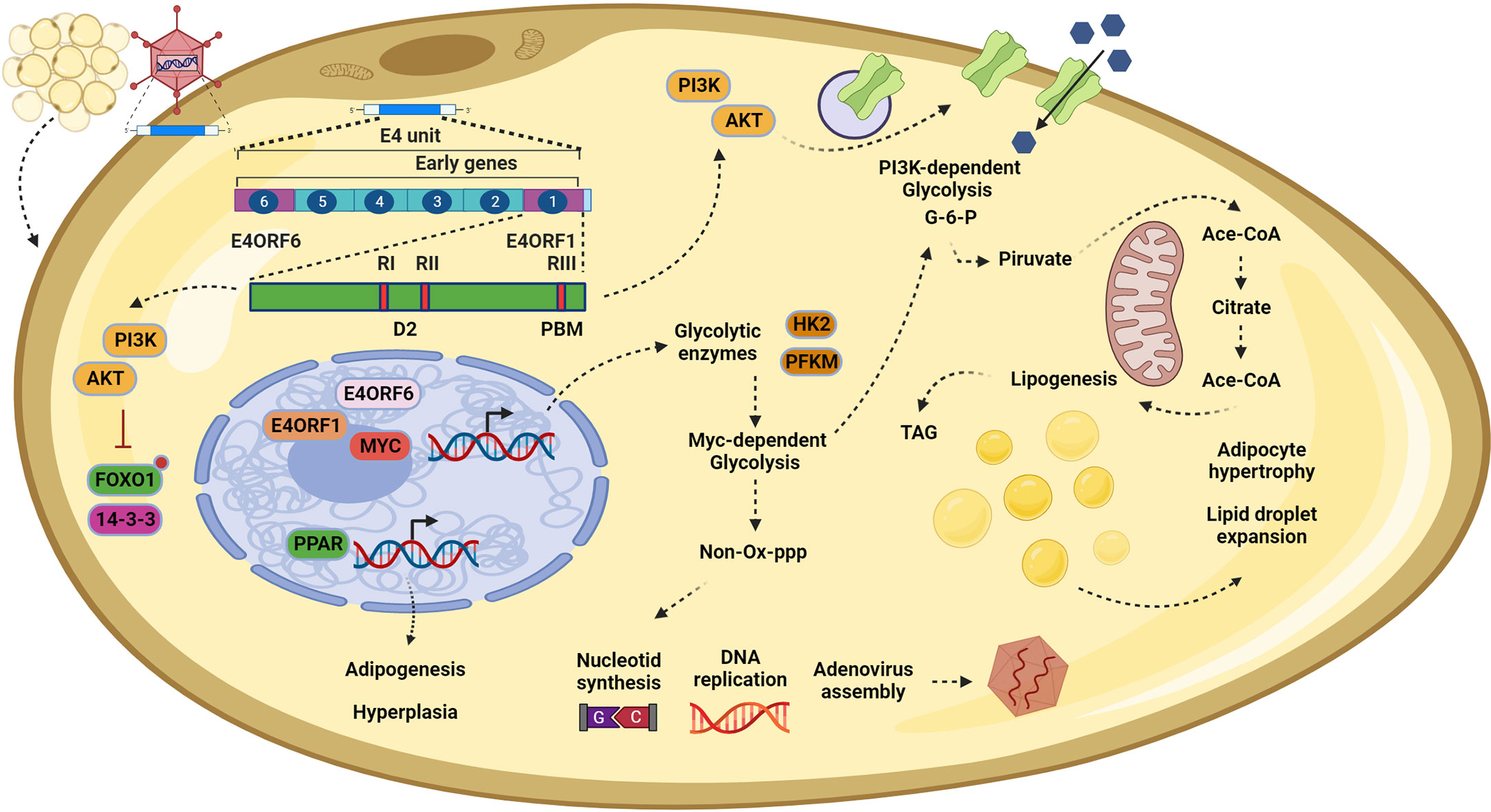
Figure 3 In adipocytes, E4orf1 promotes cell hypertrophy and hyperplasia. Infection by Adenovirus (Ad)-36 in adipocytes also disrupts the PI3K/AKT signaling pathway. Once E4orf1 is expressed (upper left) it also uses its PDZ domain-binding motif (PBM) to trigger the expression of glucose transporters on the cell membrane, inducing a PI3K-dependent glycolysis (top in the middle). The excess of glucose is converted to glucose-6-phosphate (G-6-P), which enters the mitochondria to synthetize acetyl-CoA and finally triacylglycerols (TAG) that are stored in lipid droplets (in the center). This process promotes lipogenesis in the infected cells and causes lipid droplet expansion, culminating in cellular hypertrophy. Moreover, it is possible that the mechanism described in epithelial cells (MDG) also participates in promoting the hypertrophy mentioned before by generating more G-6-P and creating positive feedback (top in the middle). However, in adipocytes, the MDG may also generate nucleotides via the pentose phosphate pathway (PPP) to sustain viral replication (bottom in the middle). Finally, E4orf1, through modulating the PI3K/AKT pathway, regulates the forkhead transcription factor FoxO1, inducing their nuclear exclusion (bottom left). Once FoxO1 is excluded, the brake for the adipoyte differentiation is liberated, allowing the expression of the master regulator for the adipogenesis program PPARγ, which in turn upregulates genes involved in commitment to adipocyte cell fate and progress to fat tissue hyperplasia. This figure was created with BioRender.com.
Molecularly, the metabolic deregulation of Ad-36 presents a conserved alteration of the PI3K/AKT pathway, as mentioned before. E4orf1 upregulates the expression of glucose transporter 1 (GLUT1) and glucose transporter 4 (GLUT4) in adipocytes through the Ras-PI3K pathway. The increase in glucose uptake is dependent on E4orf1–Dlg1 interaction and H-Ras isoform activation (72). In this context of metabolic modulation, the only commonality among the proteins deregulated by E4orf1 is Dlg1. Recently, this protein has emerged as an important mediator of vesicular transport in adipocytes. Through interaction with Sec8 (a protein of the exocyst complex), Dlg1 modulates the anchoring of GLUT4 to the plasma membrane for glucose uptake (73, 74). This deregulation of vesicle trafficking by the viral protein makes sense, as it seems to ensure efficient viral production by promoting constant proliferation through the PI3K/AKT pathway on one hand, and creating positive feedback through glucose uptake on the other (Figure 3). This strategy is also used by other viruses. For example, the E5 oncoprotein of HPV deregulates the endosomal route to recycling EGF receptors to create positive feedback on EGF stimulation (75).
The essence of the oncogenic process lies in chronic and sustained cell proliferation, often accompanied by adjustments in cell metabolism to support the demands of cell growth and division. Certain viruses converge in perturbing the cell metabolism particularly by increasing glycolysis even in the presence of oxygen, a phenomena called the Warburg effect in cancer (39). In cancer cells, such reprograming is self-defeating because ATP production by glycolysis is very low. However, this problem is overcome by upregulating the GLUT1 to increase glucose import into the cell.
It is worth noting that E4orf1 derived from Ad-36 mirror this metabolic switch in adipose tissue and muscle, and, in fact, utilizes the same mechanism of glucose transporters and oncogenes as cancer cells, such as RAS and MYC (28).
Similar to cancer cells, the benefit for the virus of this metabolic shift is related to the versatility of the glycolysis intermediates, which contribute to the production of nucleotides and amino acids to facilitate viral assembly. E4orf1 derived from Ad-36, by forming a trimer with E4orf6 and MYC, promotes the expression of glycolytic enzymes hexokinase2 and phosphofructokinase 1 to induce PI3K-independent glycolysis (28). This mechanism, in turn, provides intermediaries that enter the pentose phosphate pathway to ultimately provide nucleotides for adenoviral DNA replication (Figure 3). It is important to highlight that we believe that PI3K-dependent glycolysis and MYC-dependent glycolysis are not mutually exclusive. Instead, they both work together in the oncogenic process and the metabolic shift, having an additive effect on cell proliferation or virus replication.
Another interesting correlation is the reliance on glycolysis under hypoxic conditions. It is well documented that the tumor microenvironment operates under fluctuating oxygen gradients, favoring the existence of cell populations that are completely dependent on glucose metabolism, while others do not. This metabolic condition is also shared in adipose tissue of obese individuals, which, in turn, leads to mitochondrial dysfunction and, consequently, low ATP production. This cellular microenvironment is ideal for some Ad members, such as Ad-36, to evolve and modulate key cellular processes through E4orf1 in favor of viral production. In the context of the oncogenesis, when the virus reaches this cellular microenvironment as a consequence of driving the construction of new virions, some hallmarks of cancer emerge.
Conclusions
Adenoviruses can infect different cells and modulate their physiology to ensure their persistence and viral production. The E4 region encodes the oncoprotein E4orf1, which works to induce optimal viral replication. The E4orf1-induced cell metabolic changes could have the side effect of causing cell transformation. We describe significant changes induced by E4orf1 to modulate signaling pathways and checkpoints of cellular processes, promoting cell cycle entry, avoiding apoptosis, and causing the loss of polarity in epithelial cells; these alterations are considered the hallmarks of cancer. Moreover, E4orf1 promotes increased glycolytic flux in epithelial cells, which, in turn, serves to produce nucleotides for optimal replication. In adipocytes, E4orf1 conserves the disruption of checkpoints and signaling pathways to also induce cell cycle entry, producing hyperplasia and promoting greater glucose uptake, causing hypertrophy. These events are associated with the possible role of E4orf1 in obesity development. Understanding these mechanisms has been essential for describing fine-tuned molecular events in the oncogenic process and has positioned E4orf1 as a potential therapeutic agent for clinical intervention in diabetes.
Author contributions
HA-R and ML-J designed the concept. EF-P, TR, and HA-R completed the final editing of the manuscript. All authors contributed to writing of the manuscript. VC-H, LH-M, and AM-G prepared the figures. All authors have read and approved the manuscript for publication. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grant CB2017-2018 A1-S-10352 to HA-R from Consejo Nacional de Ciencia y Tecnología (CONACYT, México). LH-M and VC-H received a master and predoctoral fellowship, respectively, from CONACYT.
Acknowledgments
MM-C, EF-P, ML-J, and HA-R acknowledge their membership of the National System of Researchers (SNI) CONACYT, México.
Conflict of interest
The authors EF-P and ML-J declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cheng E, Kirley J, Cespedes Feliciano EM, Caan BJ. Adiposity and cancer survival: a systematic review and meta-analysis. Cancer Causes Control (2022) 33(10):1219–46. doi: 10.1007/s10552-022-01613-7
2. Morales-Sánchez A, Fuentes-Pananá EM. Human viruses and cancer. Viruses (2014) 6(10):4047–79. doi: 10.3390/v6104047
3. de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol (2012) 13(6):607–15. doi: 10.1016/S1470-2045(12)70137-7
4. Ip WH, Dobner T. Cell transformation by the adenovirus oncogenes E1 and E4. FEBS Lett (2020) 594(12):1848–60. doi: 10.1002/1873-3468.13717
5. Van den Elsen P, Houweling A, van der Eb A. Expression of region E1b of human Adenoviruses in the absence of region E1a is not sufficient for complete transformation. Virology (1983) 128(2):377–90. doi: 10.1016/0042-6822(83)90264-7
6. Reichel R, Kovesdi I, Nevins JR. Developmental control of a promoter-specific factor that is also regulated by the E1A gene product. Cell (1987) 48(3):501–6. doi: 10.1016/0092-8674(87)90200-5
7. Deleu L, Shellard S, Alevizopoulos K, Amati B, Land H. Recruitment of TRRAP required for oncogenic transformation by E1A. Oncogene (2001) 20(57):8270–5. doi: 10.1038/sj.onc.1205159
8. Liu Y, Colosimo AL, Yang XJ, Liao D. Adenovirus E1B 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol Cell Biol (2000) 20(15):5540–53. doi: 10.1128/MCB.20.15.5540-5553.2000
9. Steegenga WT, Riteco N, Jochemsen AG, Fallaux FJ, Bos JL. The large E1B protein together with the E4orf6 protein target p53 for active degradation in Adenovirus infected cells. Oncogene (1998) 16(3):349–57. doi: 10.1038/sj.onc.1201540
10. Berget SM, Moore C, Sharp PA. Spliced segments at the 5' terminus of Adenovirus 2 late mRNA. Proc Natl Acad Sci USA (1977) 74(8):3171–5. doi: 10.1073/pnas.74.8.3171
11. Chow LT, Gelinas RE, Broker TR, Roberts RJ. An amazing sequence arrangement at the 5' ends of Adenovirus 2 messenger RNA. Cell (1977) 12(1):1–8. doi: 10.1016/0092-8674(77)90180-5
12. Donovan-Banfield I, Turnell AS, Hiscox JA, Leppard KN, Matthews DA. Deep splicing plasticity of the human Adenovirus type 5 transcriptome drives virus evolution. Commun Biol (2020) 3(1):124. doi: 10.1038/s42003-020-0849-9
13. Thomas DL, Shin S, Jiang BH, Vogel H, Ross MA, Kaplitt M, et al. Early region 1 transforming functions are dispensable for mammary tumorigenesis by human Adenovirus type 9. J Virol (1999) 73(4):3071–9. doi: 10.1128/JVI.73.4.3071-3079.1999
14. Javier RT. Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J Virol (1994) 68(6):3917–24. doi: 10.1128/jvi.68.6.3917-3924.1994
15. Graham FL, van der Eb AJ. Transformation of rat cells by DNA of human Adenovirus 5. Virology (1973) 54(2):536–9. doi: 10.1016/0042-6822(73)90163-3
16. Wigand R, Gelderblom H, Wadell G. New human Adenovirus (candidate Adenovirus 36), a novel member of subgroup d. Arch Virol (1980) 64(3):225–33. doi: 10.1007/BF01322702
17. Frese KK, Lee SS, Thomas DL, Latorre IJ, Weiss RS, Glaunsinger BA, et al. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the Adenovirus E4-ORF1 oncoprotein. Oncogene (2003) 22(5):710–21. doi: 10.1038/sj.onc.1206151
18. Rogers PM, Fusinski KA, Rathod MA, Loiler SA, Pasarica M, Shaw MK, et al. Human Adenovirus ad-36 induces adipogenesis via its E4 orf-1 gene. Int J Obes (Lond) (2008) 32(3):397–406. doi: 10.1038/sj.ijo.0803748
19. Na HN, Nam JH. Proof-of-concept for a virus-induced obesity vaccine; vaccination against the obesity agent Adenovirus 36. Int J Obes (Lond) (2014) 38(11):1470–4. doi: 10.1038/ijo.2014.41
20. Feizy Z, Peddibhotla S, Khan S, Hegde V, Wang S, Dhurandhar NV. Nanoparticle-mediated in vitro delivery of E4orf1 to preadipocytes is a clinically relevant delivery system to improve glucose uptake. Int J Obes (2020) 44(7):1607–16. doi: 10.1038/s41366-020-0526-6
21. Täuber B, Dobner T. Adenovirus early E4 genes in viral oncogenesis. Oncogene (2001) 20(54):7847–54. doi: 10.1038/sj.onc.1204914
22. Hérissé J, Rigolet M, de Dinechin SD, Galibert F. Nucleotide sequence of Adenovirus 2 DNA fragment encoding for the carboxylic region of the fiber protein and the entire E4 region. Nucleic Acids Res (1981) 9(16):4023–42. doi: 10.1093/nar/9.16.4023
23. Virtanen A, Gilardi P, Näslund A, LeMoullec JM, Pettersson U, Perricaudet M. mRNAs from human Adenovirus 2 early region 4. J Virol (1984) 51(3):822–31. doi: 10.1128/jvi.51.3.822-831.1984
24. Gilardi P, Perricaudet M. The E4 promoter of Adenovirus type 2 contains an E1A dependent cis-acting element. Nucleic Acids Res (1986) 14(22):9035–49. doi: 10.1093/nar/14.22.9035
25. Watanabe H, Imai T, Sharp PA, Handa H. Identification of two transcription factors that bind to specific elements in the promoter of the Adenovirus early-region 4. Mol Cell Biol (1988) 8(3):1290–300. doi: 10.1128/mcb.8.3.1290-1300.1988
26. Freyer GA, Katoh Y, Roberts RJ. Characterization of the major mRNAs from Adenovirus 2 early region 4 by cDNA cloning and sequencing. Nucleic Acids Res (1984) 12(8):3503–19. doi: 10.1093/nar/12.8.3503
27. Tigges MA, Raskas HJ. Splice junctions in Adenovirus 2 early region 4 mRNAs: multiple splice sites produce 18 to 24 RNAs. J Virol (1984) 50(1):106–17. doi: 10.1128/jvi.50.1.106-117.1984
28. Graham NA, Braas D, Nehil M, Komisopoulou E, Kurdistani SK, et al. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab (2014) 19(4):694–701. doi: 10.1016/j.cmet.2014.03.009
29. Weiss RS, Gold MO, Vogel H, Javier RT. Mutant Adenovirus type 9 E4 ORF1 genes define three protein regions required for transformation of CREF cells. J Virol (1997) 71(6):4385–94. doi: 10.1128/jvi.71.6.4385-4394.1997
30. Weiss RS, Lee SS, Prasad BV, Javier RT. Human Adenovirus early region 4 open reading frame 1 genes encode growth-transforming proteins that may be distantly related to dUTP pyrophosphatase enzymes. J Virol (1997) 71(3):1857–70. doi: 10.1128/jvi.71.3.1857-1870.1997
31. Weiss RS, Javier RT. A carboxy-terminal region required by the Adenovirus type 9 E4 ORF1 oncoprotein for transformation mediates direct binding to cellular polypeptides. J Virol (1997) 71(10):7873–80. doi: 10.1128/jvi.71.10.7873-7880.1997
32. Lee SS, Weiss RS, Javier RT. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA (1997) 94(13):6670–5. doi: 10.1073/pnas.94.13.6670
33. Ishidate T, Matsumine A, Toyoshima K, Akiyama T. The APC-hDLG complex negatively regulates cell cycle progression from the G0/G1 to s phase. Oncogene (2000) 19(3):365–72. doi: 10.1038/sj.onc.1203309
34. Humbert PO, Grzeschik NA, Brumby AM, Galea R, Elsum I, Richardson HE. Control of tumourigenesis by the Scribble/Dlg/Lgl polarity module. Oncogene (2008) 27(55):6888–907. doi: 10.1038/onc.2008.341
35. Matsumine A, Ogai A, Senda T, Okumura N, Satoh K, Baeg GH, et al. Binding of APC to the human homolog of the drosophila discs large tumor suppressor protein. Science (1996) 272(5264):1020–3. doi: 10.1126/science.272.5264.1020
36. Kong K, Kumar M, Taruishi M, Javier RT. The human Adenovirus E4-ORF1 protein subverts discs large 1 to mediate membrane recruitment and dysregulation of phosphatidylinositol 3-kinase. PloS Pathog (2014) 10(5):e1004102. doi: 10.1371/journal.ppat.1004102
37. Javier RT. Cell polarity proteins: common targets for tumorigenic human viruses. Oncogene (2008) 27(55):7031–46. doi: 10.1038/onc.2008.352
38. Kumar M, Kong K, Javier RT. Hijacking Dlg1 for oncogenic phosphatidylinositol 3-kinase activation in human epithelial cells is a conserved mechanism of human Adenovirus E4-ORF1 proteins. J Virol (2014) 88(24):14268–77. doi: 10.1128/JVI.02324-14
39. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
40. Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J (2004) 382(Pt 1):1–11. doi: 10.1042/BJ20040825
41. Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. Interactions of the PDZ-protein MAGI-1 with Adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene (2000) 19(46):5270–80. doi: 10.1038/sj.onc.1203906
42. Makokha GN, Takahashi M, Higuchi M, Saito S, Tanaka Y, Fujii M. Human T-cell leukemia virus type 1 tax protein interacts with and mislocalizes the PDZ domain protein MAGI-1. Cancer Sci (2013) 104(3):313–20. doi: 10.1111/cas.12087
43. Kumar M, Liu H, Rice AP. Regulation of interferon-β by MAGI-1 and its interaction with influenza a virus NS1 protein with ESEV PBM. PloS One (2012) 7(7):e41251. doi: 10.1371/journal.pone.0041251
44. Massimi P, Shai A, Lambert P, Banks L. HPV E6 degradation of p53 and PDZ containing substrates in an E6AP null background. Oncogene (2008) 27(12):1800–4. doi: 10.1038/sj.onc.1210810
45. Ullmer C, Schmuck K, Figge A, Lübbert H. Cloning and characterization of MUPP1, a novel PDZ domain protein. FEBS Lett (1998) 424(1-2):63–8. doi: 10.1016/S0014-5793(98)00141-0
46. Lee SS, Glaunsinger B, Mantovani F, Banks L, Javier RT. Multi-PDZ domain protein MUPP1 is a cellular target for both Adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J Virol (2000) 74(20):9680–93. doi: 10.1128/JVI.74.20.9680-9693.2000
47. Liu W, Huang Y, Wang D, Han F, Chen H, Chen J, et al. MPDZ as a novel epigenetic silenced tumor suppressor inhibits growth and progression of lung cancer through the hippo-YAP pathway. Oncogene (2021) 40(26):4468–85. doi: 10.1038/s41388-021-01857-8
48. Mancini A, Koch A, Stefan M, Niemann H, Tamura T. The direct association of the multiple PDZ domain containing proteins (MUPP-1) with the human c-kit c-terminus is regulated by tyrosine kinase activity. FEBS Lett (2000) 482(1-2):54–8. doi: 10.1016/S0014-5793(00)02036-6
49. Roskoski R Jr. Signaling by kit protein-tyrosine kinase–the stem cell factor receptor. Biochem Biophys Res Commun (2005) 337(1):1–13. doi: 10.1016/j.bbrc.2005.08.055
50. Kimber WA, Trinkle- Mulcahy L, Cheung PC, Deak M, Marsden LJ, Kieloch A, et al. Evidence that the tandem-pleckstrin-homology-domain-containing protein TAPP1 interacts with Ptd(3,4)P2 and the multi-PDZ-domain-containing protein MUPP1 in vivo. Biochem J (2002) 361(Pt 3):525–36. doi: 10.1042/bj3610525
51. Huerta M, Muñoz R, Tapia R, Soto-Reyes E, Ramírez L, Recillas-Targa F, et al. Cyclin D1 is transcriptionally down-regulated by ZO-2 via an e box and the transcription factor c-myc. Mol Biol Cell (2007) 18(12):4826–36. doi: 10.1091/mbc.e07-02-0109
52. Glaunsinger BA, Weiss RS, Lee SS, Javier R. Link of the unique oncogenic properties of Adenovirus type 9 E4-ORF1 to a select interaction with the candidate tumor suppressor protein ZO-2. EMBO J (2001) 20(20):5578–86. doi: 10.1093/emboj/20.20.5578
53. Tapia R, Huerta M, Islas S, Avila-Flores A, Lopez-Bayghen E, Weiske J, et al. Zona occludens-2 inhibits cyclin D1 expression and cell proliferation and exhibits changes in localization along the cell cycle. Mol Biol Cell (2009) 20(3):1102–17. doi: 10.1091/mbc.e08-03-0277
54. González-Mariscal L, Gallego-Gutiérrez H, González-González L, Hernández-Guzmán C. ZO-2 is a master regulator of gene expression, cell proliferation, cytoarchitecture, and cell size. Int J Mol Sci (2019) 20(17):4128. doi: 10.3390/ijms20174128
55. Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol (1994) 124(4):619–26. doi: 10.1083/jcb.124.4.619
56. Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science (1998) 282(5392):1318–21. doi: 10.1126/science.282.5392.1318
57. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell (1997) 91(2):231–41. doi: 10.1016/S0092-8674(00)80405-5
58. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell (1999) 96(6):857–68. doi: 10.1016/S0092-8674(00)80595-4
59. Jackson JG, Kreisberg JI, Koterba AP, Yee D, Brattain MG. Phosphorylation and nuclear exclusion of the forkhead transcription factor FKHR after epidermal growth factor treatment in human breast cancer cells. Oncogene (2000) 19(40):4574–81. doi: 10.1038/sj.onc.1203825
60. Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell (2002) 111(1):29–40. doi: 10.1016/S0092-8674(02)01001-2
61. Massimi P, Zori P, Roberts S, Banks L. Differential regulation of cell-cell contact, invasion and anoikis by hScrib and hDlg in keratinocytes. PloS One (2012) 7(7):e40279. doi: 10.1371/journal.pone.0040279
62. Dhurandhar NV, Israel BA, Kolesar JM, Mayhew GF, Cook ME, Atkinson RL. Increased adiposity in animals due to a human virus. Int J Obes Relat Metab Disord (2000) 24(8):989–96. doi: 10.1038/sj.ijo.0801319
63. Kim J, Na H, Kim JA, Nam JH. What we know and what we need to know about Adenovirus 36-induced obesity. Int J Obes (Lond) (2020) 44(6):1197–209. doi: 10.1038/s41366-020-0536-4
64. Dhurandhar NV, Whigham LD, Abbott DH, Schultz-Darken NJ, Israel BA, Bradley SM, et al. Human Adenovirus ad-36 promotes weight gain in male rhesus and marmoset monkeys. J Nutr (2002) 132(10):3155–60. doi: 10.1093/jn/131.10.3155
65. Dhurandhar NV, Israel BA, Kolesar JM, Mayhew G, Cook ME, Atkinson RL. Transmissibility of Adenovirus-induced adiposity in a chicken model. Int J Obes Relat Metab Disord (2001) 25(7):990–6. doi: 10.1038/sj.ijo.0801668
66. Rathod M, Vangipuram SD, Krishnan B, Heydari AR, Holland TC, Dhurandhar NV. Viral mRNA expression but not DNA replication is required for lipogenic effect of human Adenovirus ad-36 in preadipocytes. Int J Obes (Lond) (2007) 31(1):78–86. doi: 10.1038/sj.ijo.0803358
67. Vangipuram SD, Yu M, Tian J, Stanhope KL, Pasarica M, Havel PJ, et al. Adipogenic human Adenovirus-36 reduces leptin expression and secretion and increases glucose uptake by fat cells. Int J Obes (Lond) (2007) 31(1):87–96. doi: 10.1038/sj.ijo.0803366
68. Pasarica M, Mashtalir N, McAllister EJ, Kilroy GE, Koska J, Permana P, et al. Adipogenic human Adenovirus ad-36 induces commitment, differentiation, and lipid accumulation in human adipose-derived stem cells. Stem Cells (2008) 26(4):969–78. doi: 10.1634/stemcells.2007-0868
69. Na HN, Kim H, Nam JH. Novel genes and cellular pathways related to infection with Adenovirus-36 as an obesity agent in human mesenchymal stem cells. Int J Obes (Lond) (2012) 36(2):195–200. doi: 10.1038/ijo.2011.89
70. Chaudhary N, Gonzalez E, Chang SH, Geng F, Rafii S, Altorki NK, et al. Adenovirus protein E4-ORF1 activation of PI3 kinase reveals differential regulation of downstream effector pathways in adipocytes. Cell Rep (2016) 17(12):3305–18. doi: 10.1016/j.celrep.2016.11.082
71. Nakae J, Kitamura T, Kitamura Y, Biggs WH 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell (2003) 4(1):119–29. doi: 10.1016/S1534-5807(02)00401-X
72. Dhurandhar EJ, Dubuisson O, Mashtalir N, Krishnapuram R, Hegde V, Dhurandhar NV. E4orf1: a novel ligand that improves glucose disposal in cell culture. PloS One (2011) 6(8):e23394. doi: 10.1371/journal.pone.0023394
73. Inoue M, Chiang SH, Chang L, Chen XW, Saltiel AR. Compartmentalization of the exocyst complex in lipid rafts controls Glut4 vesicle tethering. Mol Biol Cell (2006) 17(5):2303–11. doi: 10.1091/mbc.e06-01-0030
74. Walch L. Emerging role of the scaffolding protein Dlg1 in vesicle trafficking. Traffic (2013) 14(9):964–73. doi: 10.1111/tra.12089
Keywords: Adenovirus-36, adipocytes, oncogenesis, E4orf1, metabolism
Citation: Hernández-Magaña LE, Mosqueda-Gracida A, Cruz-Holguín VJ, Martínez-Castillo M, Fuentes-Pananá EM, Rozmyslowicz T, León-Juárez M and Arévalo-Romero H (2023) E4orf1 as a key modulator in oncogenesis and of metabolism in Adenovirus infection. Front. Virol. 3:1195717. doi: 10.3389/fviro.2023.1195717
Received: 28 March 2023; Accepted: 15 May 2023;
Published: 14 June 2023.
Edited by:
Benjamin M. Liu, George Washington University, United StatesReviewed by:
Timothy Keiffer, Louisiana State University Health Shreveport, United StatesCopyright © 2023 Hernández-Magaña, Mosqueda-Gracida, Cruz-Holguín, Martínez-Castillo, Fuentes-Pananá, Rozmyslowicz, León-Juárez and Arévalo-Romero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haruki Arévalo-Romero, YXJldmFsb18yMjAzQGhvdG1haWwuY29t
†These authors have contributed equally to this work and share first authorship