 Zachary Lozier
Zachary Lozier Lilyahna Hill2
Lilyahna Hill2 W. Allen Miller
W. Allen Miller- 1Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA, United States
- 2Plant Pathology, Entomology and Microbiology Department, Iowa State University, Ames, IA, United States
Tombusviridae is a large family of single-stranded, positive-sense RNA plant viruses with uncapped, non-polyadenylated genomes encoding 4–7 open reading frames (ORFs). Previously, we discovered, by high-throughput sequencing of maize and teosinte RNA, a novel genome of a virus we call Maize-associated tombusvirus (MaTV). Here we determined the precise termini of the MaTV genome by using 5’ and 3’ rapid amplification of cDNA ends (RACE). In GenBank, we discovered eleven other nearly complete viral genomes with MaTV-like genome organizations and related RNA-dependent RNA polymerase (RdRp) sequences. These genomes came from diverse plant, fungal, invertebrate and vertebrate organisms, and some have been found in multiple organisms across the globe. The available 5’ untranslated regions (UTRs) of these genomes are remarkably long: at least 438 to 727 nucleotides (nt), in contrast to those of other tombusvirids, which are <150 nt. Moreover these UTRs contain 6 to 12 AUG triplets that are unlikely to be start codons, because - with the possible exception of MaTV - there are no large or conserved ORFs in the 5’ UTRs. Such features suggest an internal ribosome entry site (IRES), but the only conserved features we found were that the 50 nt upstream of and adjacent to the ORF1 start codon are cytosine-rich and guanosine-poor. ORF2 (RdRp gene) appears to be translated by in-frame ribosomal readthrough of the ORF1 stop codon. In all twelve genomes we identified RNA structures known in other tombusvirids to facilitate this readthrough. ORF4 overlaps with ORF3 (coat protein gene) and may initiate with a non-AUG start codon. ORF5 is predicted to be translated by readthrough of the ORF3 stop codon. The proteins encoded by ORFs 4 and 5 diverge highly from each other and from those of the similarly organized luteo- and poleroviruses. We also found no obvious 3’ cap-independent translation elements, which are present in other tombusvirids. The twelve genomes diverge sufficiently from other tombusvirids to warrant classification in a new genus. Because they contain two leaky stop codons and a potential leaky start codon, we propose to name this genus Rimosavirus (rimosa = leaky in Latin).
1 Introduction
Metagenomics has revolutionized virus discovery. Searching for viruses by sequencing total RNA from environmental samples (metagenomics), such as soil (1), seawater (2), or organisms has resulted in an exponential increase in known viruses, or viral genomes in the past decade (3, 4). The viruses associated with these newly discovered genomes are mostly uncharacterized, but the obvious viral nature of the genomes indicate that those viruses exist (5). Because virus particles can be highly abundant and stable, viruses isolated from an organism may not actually infect that organism, but may be just “hitching a ride”. For example, plant viruses have been identified in human intestinal microbiome (6), bat guano (7), and in aphid vectors that transmit them (8) but which are not infected by them. Hence, viruses known only by sequence and the organism from which they are isolated are labeled as “associated” with that organism.
In large scale metagenomics projects, thousands of viral genome sequences have been automatically assembled, annotated, and deposited in GenBank, in some cases with very little direct scrutiny by humans (9, 10). Because of the numerous noncanonical translation mechanisms used by many RNA viruses, these autoannotated genomes are often mis-annotated (11). Also, if the viral genomes are not of interest to the sequencers, or if the sequencers simply lack the time to study these viral genomes, they may remain essentially undiscovered in GenBank. Here we describe several genomes of viruses in the Tombusviridae family (tombusvirids) that appear to fall in this category.
The Tombusviridae family contains over 100 virus species (tombusvirids) in eighteen genera officially named by the International Committee on Virus Taxonomy (ICTV). This large and diverse family includes many economically costly pathogens, such as maize chlorotic mottle virus, which has devastated maize production in mixed infection with a potyvirus in East Africa (12), and the barley yellow dwarf viruses which comprise the most ubiquitous viruses of wheat, barley and oat, worldwide (13, 14). In addition, tombusvirids have proved to be excellent models. For example, the first X-ray crystal structure of an icosahedral virus was that of tomato bushy stunt virus (for which the family is named) (15). Also the roles of host proteins and subcellular structures in every stage of the virus life cycle are better understood for TBSV than almost any other RNA virus (16–18). Tombusvirids contain a positive strand RNA genome of 4–6 kb which lacks a 5’ cap and a poly(A) tail, as the genome terminates in CCC (19, 20). Dianthovirus is the only tombusvirid genus that has a bipartite genome (21).
The tombusvirid genome encodes 4–7 open reading frames (ORFs). ORF1 encodes a replication protein that lines membrane-bound replication vesicles (18). ORF2 encodes the RNA-dependent RNA polymerase (RdRp) and is translated as a long C-terminal extension of ORF1 via ribosomal readthrough of – or frameshift around – the ORF1 stop codon (22–25). Downstream ORFs encode movement proteins, suppressors of RNA silencing, coat protein and vector transmission components (19, 26–28). They are translated – often via various leaky start and stop codons (23, 29–31) – from subgenomic mRNAs that are 5’-truncated versions of genomic RNA (32–34). To allow translation of the uncapped genomic and subgenomic mRNAs, a 3’ cap-independent translation element (3’ CITE) is present in the 5’ end of the 3’ UTR (35, 36). The 3’ CITE is followed at the 3’ end by RNA structures and sequences required for RNA replication (20), and in some cases structures that regulate readthrough or frameshifting at the ORF1 stop codon via long-distance base pairing (37, 38).
After discovery by deep sequencing of the genome of the novel tombusvirid maize-associated tombusvirus (MaTV) in maize and teosinte leaves (39), we searched GenBank for related sequences. Here we describe eleven other viral genomes, all found by metagenomic sequencing, that are related to the MaTV. Based on (i) genome organization, (ii) sequences of RNA-dependent RNA polymerase (RdRp) and coat protein (CP), and (iii) conserved RNA secondary structures, all of these viruses clearly belong in the Tombusviridae family. However their sequences and genome features, such as extremely long 5’ UTR, place them in a clade sufficiently distinct to merit classification in a new genus.
2 Materials and methods
2.1 Rapid amplification of cDNA ends
Sequences of oligonucleotides used for RACE are listed in Supplementary Table S1.
2.1.1 Source of RNA
The RNA used for RACE was from the same total RNA extract from maize leaf that was used for sequencing the MaTV genome (39). The leaf was collected from an unhealthy, possibly diseased maize plant near Irapuato, Mexico in October 2017. Total RNA was extracted using a Zymo Direct-zol RNA Miniprep Plus kit (Zymo Research, Irvine, CA, USA), depleted for ribosomal RNA using an Illumina Ribo-Zero rRNA Removal Kit (Plant Leaf) (Illumina, San Diego, CA, USA), concentrated with a Zymo RNA Clean & Concentrator-5 kit, and stored at −80°C. Details are described in Lappe et al. (39).
2.1.2 First-strand cDNA synthesis for 5’ RACE
Using Millipore Sigma’s 5’/3’ RACE Kit, 2nd Generation, MaTV cDNA was synthesized using random primers on 1 µg of the above total maize leaf RNA. Following the kit’s instructions, the reaction mixture was incubated at 55°C for 60 minutes and for another 5 minutes at 85°C. Immediately after first-strand cDNA synthesis, the cDNA products were purified using Sigma’s High Pure PCR Product Purification Kit, following the specific instructions for cDNA purification outlined in the 5’ RACE protocol as opposed to the protocol that comes with the purification kit.
2.1.3 PCR on the cDNA sample for 5’ RACE
PCR was carried out following the purification step after first-strand cDNA synthesis, using Sigma-Aldrich’s Expand High Fidelity PCR System. Using the Expand High Fidelity Buffer with 15 mM MgCl2, thermal cycler PCR conditions were set according to Sigma’s RACE protocol with an altered annealing temperature to accommodate both the MaTV-specific primer and the oligo dT anchor primer in the RACE kit. Three rounds of PCR were conducted on the samples using a different nested primer each round (Nested primers 1, 2, and 3), without purifying the PCR product or diluting the PCR product before the next round was started. The final PCR product was subjected to gel electrophoresis and gel extraction followed by ethanol precipitation and resuspension in 50 µl of nuclease-free water. The resulting DNA was sequenced at the Iowa State University DNA Facility using MaTV-specific 5’ RACE sequencing primer, 5R1 (Supplementary Table S1).
2.1.4 Ligation of an artificial poly(A) tail to MaTV RNA for 3’ RACE
The Millipore Sigma 3’ RACE kit utilizes a poly(A) tail on the RNA sample. Because viruses in the family Tombusviridae, which includes MaTV, do not have a poly(A) tail, an oligonucleotide [3’poly(A)] containing a poly(A) tract was ligated onto the 3’ end of the total RNA sample using NEB’s T4 RNA Ligase 1 kit. This sequence is complementary to the oligo d(T) anchor primer from the RACE kit and was designed with both a 5’ and 3’ phosphate to prevent the oligo from ligating to itself and ligating to RNA in the total RNA sample that already possesses a poly(A) tail. Using 1 µl of the total RNA sample including Thermo Fisher’s RNaseOut RNA inhibitor, the reaction mixture was incubated at 25°C for 2 hours and the reaction was stopped by column cleanup using the NEB Monarch RNA Cleanup Kit.
2.1.5 First-strand cDNA synthesis for 3’ RACE
Following Millipore Sigma’s 5’/3’ RACE Kit, 2nd Generation, 5ul of the newly polyadenylated total RNA sample containing the ligated oligo(dT) anchor primer was used at a concentration of 100 ng/µl. After incubation at 55°C for 60 min immediately followed by incubation at 85°C for 5 min, the cDNA was ready for PCR. No purification step was necessary after cDNA synthesis.
2.1.6 PCR on the cDNA sample for 3’ RACE
PCR was carried out immediately following first-strand cDNA synthesis using Sigma-Aldrich’s Expand High Fidelity PCR System. Using the Expand High Fidelity Buffer with 15 mM MgCl2, thermal cycler conditions were set according to Sigma’s RACE protocol altered to accommodate both the MaTV-specific primer, 3F1 (Supplementary Table S1), and the oligo dT anchor primer in the RACE kit. The PCR product was ethanol precipitated and resuspended in 50 µl of nuclease-free water.
2.1.7 Gel electrophoresis and gel extraction
16 µl of purified PCR product at a concentration of 100 ng/µl was run on a 2% agarose gel for 30 minutes at 150V to isolate the MaTV cDNA produced from the total RNA sample. A visible band at ~400 bp (estimated product size at the 3’ end of MaTV genome) was gel extracted using Qiagen’s QIAquick PCR & Gel Cleanup Kit. After measuring the concentration of cDNA in the eluted sample after gel extraction (~5 ng/µl), an additional round of PCR was conducted to increase the cDNA concentration (~300 ng/µl).
2.1.8 3’ end sequencing of MaTV
Following PCR and purification of the MaTV cDNA, the samples were sent to Iowa State University’s DNA Sequencing Facility for dideoxy sequencing using MaTV-specific primer 3F2 (Supplementary Table S1) that was nested 3’ of the MaTV-specific primer used for PCR.
2.2 Multiple sequence alignment and phylogeny prediction
The RNA-dependent RNA polymerase (RdRp), coat protein (CP), and read-through domain (RTD) amino acid sequences of select viruses from families Tombusviridae and Solemoviridae were aligned with Muscle v3.8.31 using default parameters in SnapGene. RNA alignments were imaged using Jalview (40, 41). For phylogenetic tree construction, multiple sequence alignments were passed to FastTree 2.1.11 with the flags “-lg -gamma” to predict phylogenies where branch split reliability values are estimated with the Shimodaira-Hasegawa test (42). The resultant trees were drawn with FigTree v1.4.4 and re-rooted using the nearest relative outside of the taxonomic group of the compared sequences (43): Providence virus (NC_014126.1) for the RdRp tree and Ourmia melon virus (NC_011070.1) for the coat protein tree. Accession numbers are the GenBank accession numbers from which the respective amino acid sequences were taken.
2.3 RNA structures
RNA secondary structures were predicted using MFOLD (44), Vienna package (45), Scanfold2 (46) under default parameters, in iterations with multiple sequence alignments and inspection with the human eye. Secondary structures were drawn using RNAcanvas (47).
3 Results
3.1 Identification of viruses similar to MaTV: a new genus
Previously, we discovered and assembled genomic sequences of MaTV from maize (GenBank no. OK0180181) and teosinte (OK0180182). These genomes are 99.5% identical and none of the base differences affect lengths of open reading frames (ORFs) (39). Thus they are isolates of the same virus; for all subsequent comparisons in this paper, we use the maize isolate. To identify other viruses related to MaTV, we performed a BLAST search of GenBank seeking matches with the RNA-dependent RNA polymerase (RdRp) gene (ORF2) of MaTV. We use the RdRp because a major component of RNA virus classification is based on sequence similarities of the RdRps, as it is the key replication protein encoded by all RNA viruses (48, 49). We also searched for genes with similarity to MaTV ORF4 which seemed to be unique to this genus, when compared to known tombusvirids.
The BLAST searches revealed nearly complete genomes of eleven other viruses with substantial sequence similarity in the RdRp ORF and similar genome organizations to that of MaTV (Figure 1). This includes apple virus E (AVE), which we reported previously as being similar to MaTV (39). Like the MaTV genome, all eleven of these genomes were discovered in metagenomics sequencing projects using Illumina sequencing (9, 10, 50–52). As discussed below, all of the genomes were misannotated because ORFs were assumed to begin with an AUG codon. Yet, by comparison with other members of the Tombusviridae, there appear to be two ORFs (ORFs 2 and 5) that are translated via in-frame readthrough of the stop codon of the preceding ORF. Thus, these ORFs begin immediately after the stop codon rather than at the first AUG codon of the ORF. Because these genomes were found in large, exploratory metagenomics sequencing projects, we do not know what hosts they infect or if any symptoms were associated with these viruses, but they have been found associated with a remarkable variety of organisms including plants, fungi, invertebrates and vertebrates (Table 1). Some virus species have been found more than once, in organisms of diverse kingdoms across the globe (Table 1).
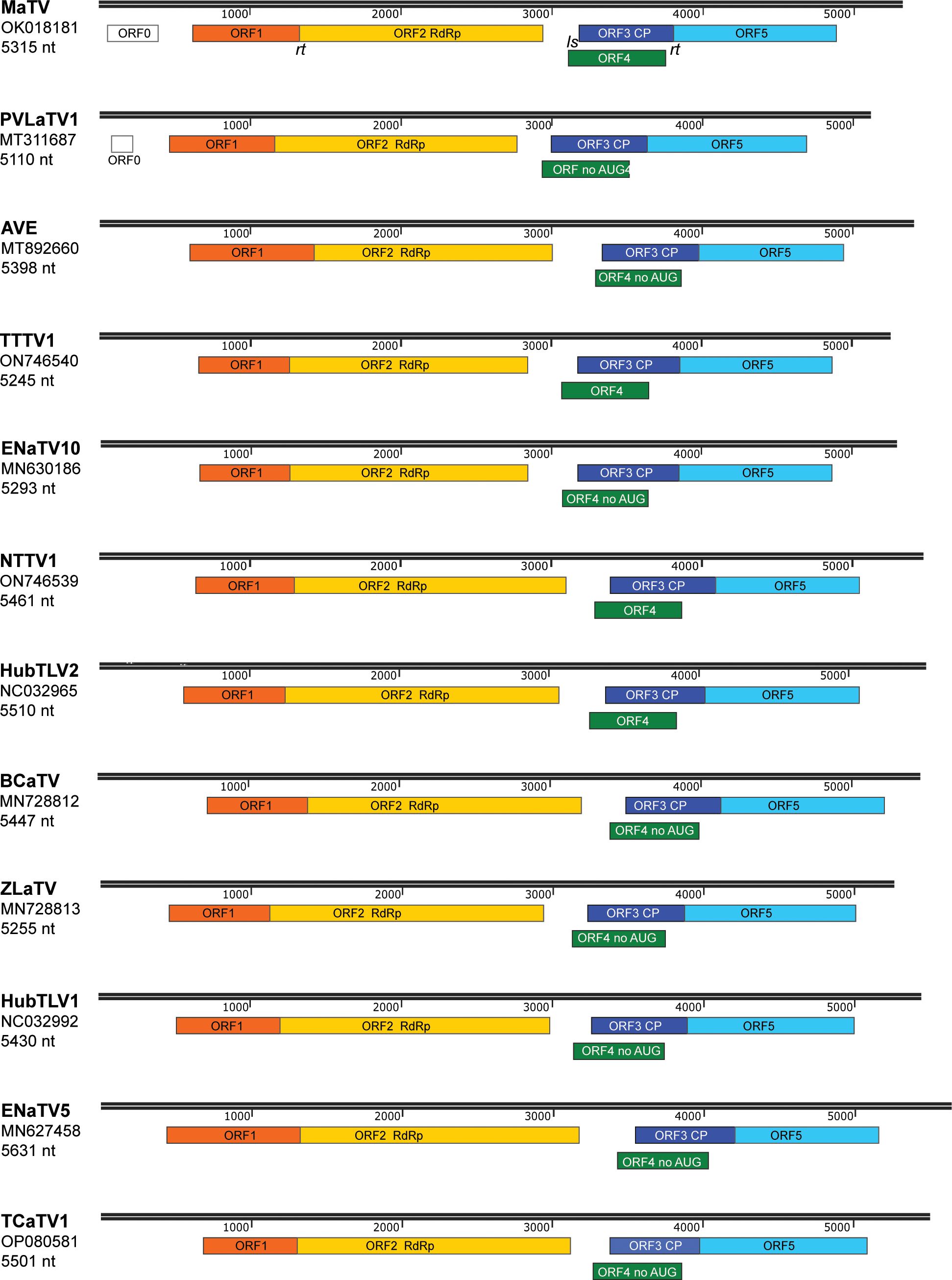
Figure 1. Maps of genomes in proposed new genus, Rimosavirus. Virus acronym, GenBank accession numbers used throughout this manuscript, and length of genome sequence for that accession number are shown at left. Colored boxes indicate ORFs, with functions of the protein products of ORF2 (RdRp) and ORF3 (CP) indicated. Scale bar (in nt) is indicated in black above each genome map. ORFs 2 and 5 are translated by in-frame readthrough (rt) of the ORF1 and ORF3 stop codons, respectively. ORFs 4 of the indicated viruses (no AUG) lack an in-frame AUG start codon upstream of ORF3 AUG start codon (see text), which is predicted to be translated from a subgenomic mRNA via leaky scanning (ls).
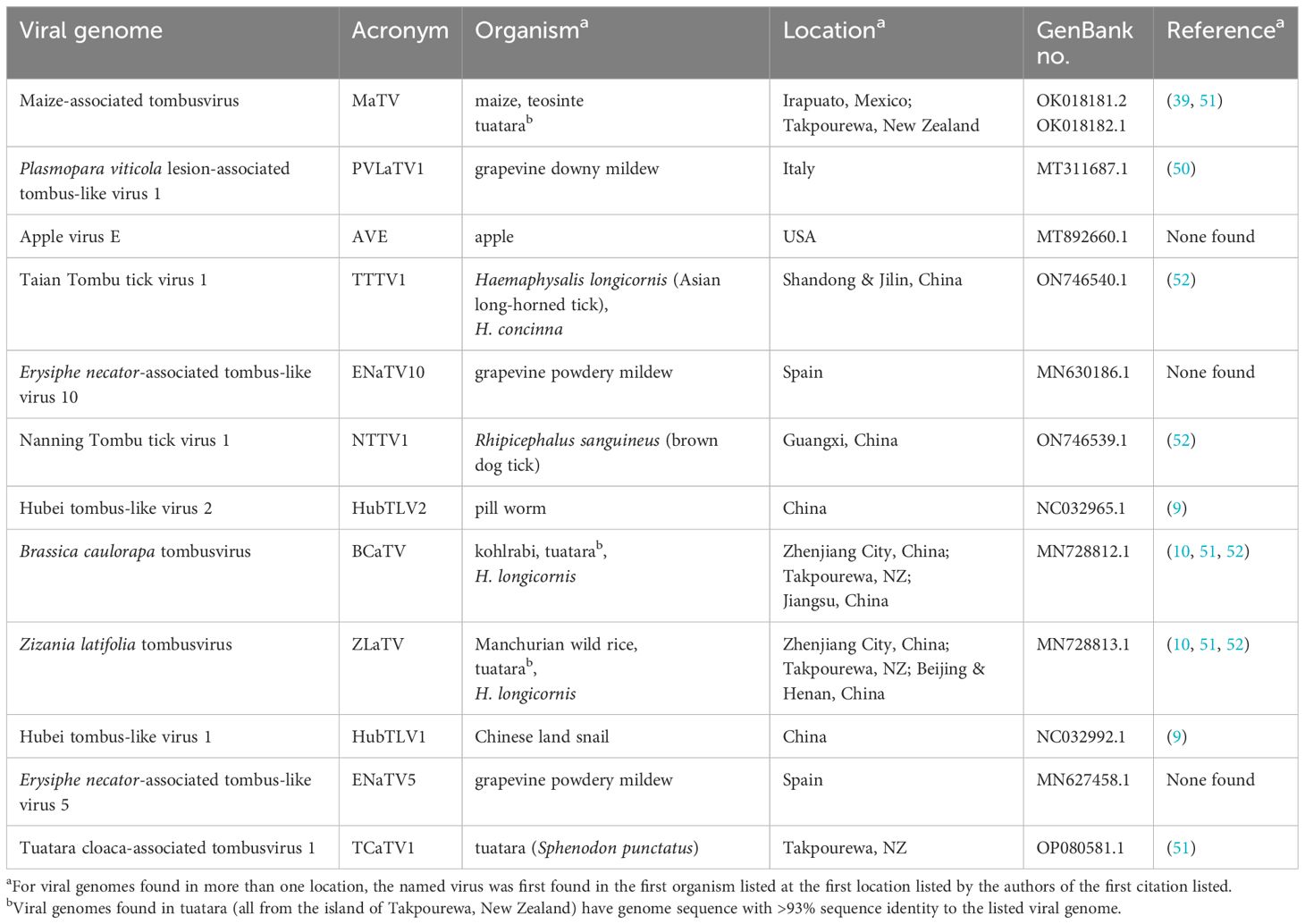
Table 1. Genomes and sources of viruses in this study.
In addition to high sequence similarity in the RdRp ORF, these genomes share the following features. (i) They contain an extremely long putative 5’ untranslated region (UTR) ranging from at least 438 nt to 727 nt, although most are almost certainly longer than listed because it is unlikely that most have been sequenced to the 5’ end (below); (ii) The presumed first translated ORF (ORF1, MW 23–34 kDa, Supplementary Table S2) is followed immediately by ORF2, which encodes the RdRp (59–69 kDa) in an arrangement that suggests translation of ORF2 via ribosomal readthrough of the ORF1 stop codon. This arrangement is present in 15 of the 18 Tombusviridae genera, the exceptions being Luteovirus, Dianthovirus and Umbravirus genera, members of which employ a -1 ribosomal frameshift for translation of the RdRp. (iii) ORF2 is followed by a noncoding region followed by ORF3, which encodes the coat protein (CP, 22–26 kDa), and an overlapping ORF4 (~17–20 kDa) (Figure 1; Supplementary Table S2). The stop codon of ORF3 is followed immediately in the same reading frame by ORF5, suggesting translation of ORF5 via readthrough of the ORF3 stop codon to generate a 34–41 kDa C-terminal extension to the CP (Figure 1; Supplementary Table S2). This arrangement of ORFs 3, 4, and 5 resembles that of viruses in the genera Luteovirus (Tombusviridae) and Polerovirus (Solemoviridae) (53), with the exception that ORF4 likely initiates upstream of ORF3 in these new viruses. In all luteo- polero- and enamoviruses (L/P/E viruses), ORF5 has been shown to be translated via readthrough of the ORF3 stop codon (31, 54, 55), and the protein product of ORF5 (the readthrough domain – RTD) is required for aphid transmission (26, 56–58), and participates in virus cell to cell movement in the luteo- and poleroviruses (59–61). In the new tombusvirids reported here, ORF5 is followed by a 3’ UTR with minimum lengths of 229 to 458 nt, but it is unlikely that the 3’ UTR sequences are complete all the way to the 3’ end, except for those of MaTV and possibly Erysiphe necator-associated tombus-like virus 10 (ENaTV10) (discussed below).
Whole genome comparisons revealed that Taian tombu tick virus 1 (TTTV1) and ENaTV10 are 95% identical, thus they are strains of the same virus even though one was obtained from an invertebrate parasite of mammals and the other from a plant pathogenic fungus. MaTV and Plasmopara viticola lesion-associated tombus-like virus 1 (PVLaTV1) genomes also show close similarity at 68.5% nucleotide sequence identity (73.4% identity in the RdRp ORF), but are clearly different species based on ICTV demarcation criteria for tombusvirids, which is <85% amino acid (aa) sequence identity in the CP, as the CPs show 71.2% aa identity (76.6% nt identity).
We propose that these twelve viruses comprise a new genus in the Tombusviridae family. This is based on the distance of the clade in the phylogenetic tree of RdRp sequences from the nearest relative, oat necrotic dwarf virus (genus Avenavirus) (Figure 2), and their distinctive genome organizations, which differ in the same ways from those of other tombusvirids. Because all twelve of these viruses have two probable leaky stop codons (at the ends of ORFs 1 and 3), and may initiate translation of the ORF3 via ribosomal leaky scanning, we propose to call this new genus Rimosavirus (rimosa, Latin for leaky). While this name is only provisional, for convenience throughout this manuscript, we refer to the twelve viruses on which this paper focuses (Table 1) as rimosaviruses.
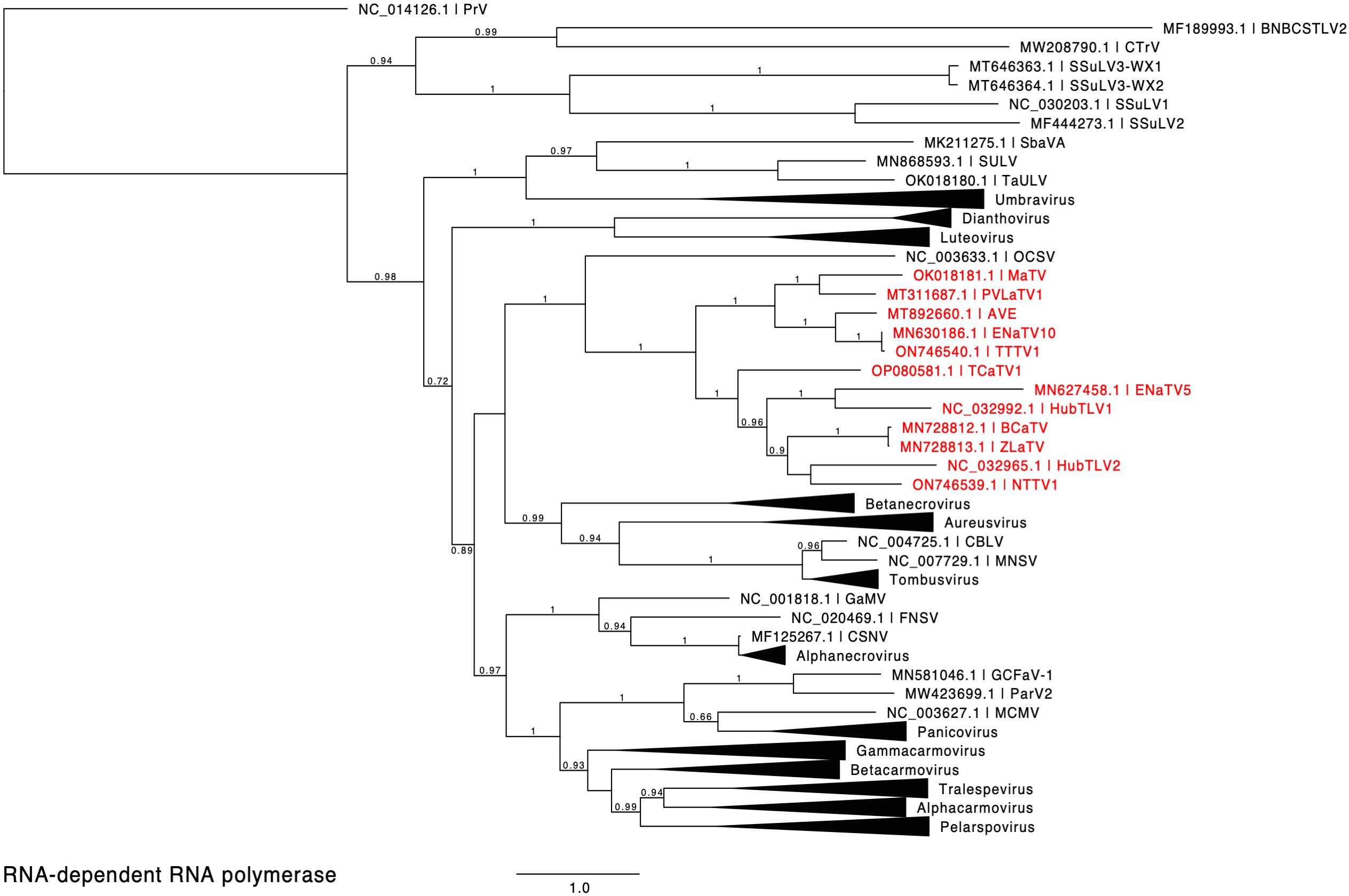
Figure 2. Phylogenetic tree predicting the relationship of viruses based on the amino acid sequences of full-length RdRps (ORF1-ORF2 fusion products). Red entries indicate those sequences belonging to the proposed new Rimosavirus genus. Branch support values are shown for splits > 0.5 and are calculated from 1,000 resamples of the Shimodaira-Hasegawa test (SH-like local supports). Branch lengths indicate arbitrary units of evolutionary distances. Providence virus (PrV) was used as outgroup because it is the nearest relative outside of the Tombusviridae (43). For individual viruses (single member genus or unassigned to genus), GenBank accession numbers and virus acronyms are shown.
3.2 5’ and 3’ untranslated regions
As mentioned above, it is not clear if any of the rimosaviral genomes were sequenced completely to the 5’ and 3’ ends. To determine the terminal sequences of MaTV, we performed 5’ and 3’ RACE (rapid amplification of cDNA ends) (62) on the same preparation of total maize RNA from which the nearly complete MaTV genome sequence was obtained. RACE revealed 19 additional nt at the 5’ end (GAAAAUAUUUAGGGUACUA) and 62 nt at the 3’ end (UUCCAAACUGCUCAGUAAUGAGAACUUCAAUUACAGUACAGCUAGACAGAUCUGUAAUGCCC) that were not found in the Illumina sequence assembly (accession no. OK018181.1) (39). That sequence has been updated to include the above RACE results (accession no. OK018181.2). The complete genome length is 5315 nt with 622 nt upstream of ORF1, which we call the 5’ UTR (below), and a 434 nt 3’ UTR. Assuming the same length of sequence is present at the ends of the 99% identical teosinte isolate, the GenBank sequence (OK018182.1) lacks 20 nt and 50 nt at the 5’ and 3’ ends, respectively, but we have not performed 5’ or 3’ RACE on that sample.
3.2.1 5’ untranslated region
The 5’ end of the MaTV genome starts with GAAAAUAU, which is highly similar to that of apple virus E (AVE), GAAAAUCU. These sequences resemble the 5’ ends of other tombusvirids which start with a purine (usually a G) followed by a purine-rich, C-poor tract. Thus, the GenBank sequence of AVE (accession no. MT892660) appears to have a complete 5’ end. The GenBank sequence of PLVaTV1 (MT311687) appears to be missing 18 nt from its 5’ terminus, based on alignment with its close relative MaTV (Supplementary Figure S1). The 5’ ends of the other nine rimosavirus sequences found in GenBank do not begin with a similar sequence, and alignments (Supplementary Figure S1) suggest numerous bases are missing from the 5’ ends.
The 5’ termini of characterized tombusvirid genomes form a stem-loop of modest stability (63, 64). We predicted the secondary structures of the 5’-terminal 40 nt of MaTV and AVE genomes and indeed found such stem-loops (Figure 3). For comparison, the known terminal structures of tombusvirids barley yellow dwarf virus (BYDV, Luteovirus) and saguaro cactus virus (SCV, Carmovirus) are shown. This further supports that the MaTV and AVE 5’ ends are complete.
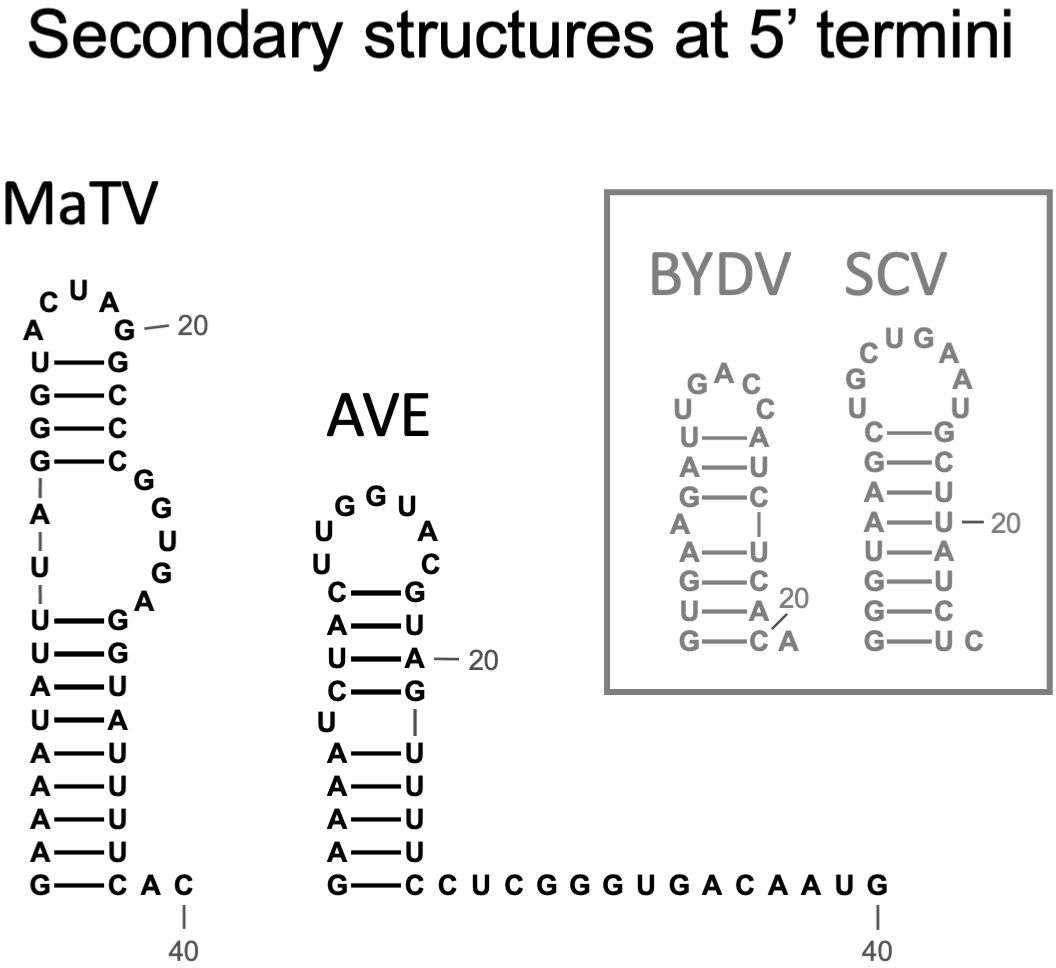
Figure 3. Predicted secondary structures of the 5’ termini of MaTV and AVE, the only two rimosaviruses for which the 5’ end is known (MaTV) or predicted (AVE). For comparison, secondary structures determined by chemical probing of tombusvirids barley yellow dwarf virus [BYDV, Luteovirus (63)], and saguaro cactus virus [SGV, Carmovirus (64)] are shown.
We predict that the sequence upstream of ORF1 is untranslated in rimosaviruses based on the following observations. (i) There are no conserved ORFs upstream of ORF1 with the following possible exception. MaTV contains a predicted AUG-initiating ORF of 336 nt (112 codons), that we call ORF0, starting at nt 85, but it is absent in the other rimosaviruses, save for a truncated version of ORF0 (177 nt, 58 codons) in PVLaTV1 (Figure 1; Supplementary Figure S2B). (ii) There are 6 to 12 AUG triplets scattered in different positions among the rimosavirus genomes upstream of the ORF1 start codon (Table 2; Supplementary Figure S1), including upstream and downstream of ORF0 in MaTV and PVLaTV1, but they result in short, non-conserved ORFs. (iii) Finally, ORF1 is the 5’-proximal ORF in all other tombusvirids.
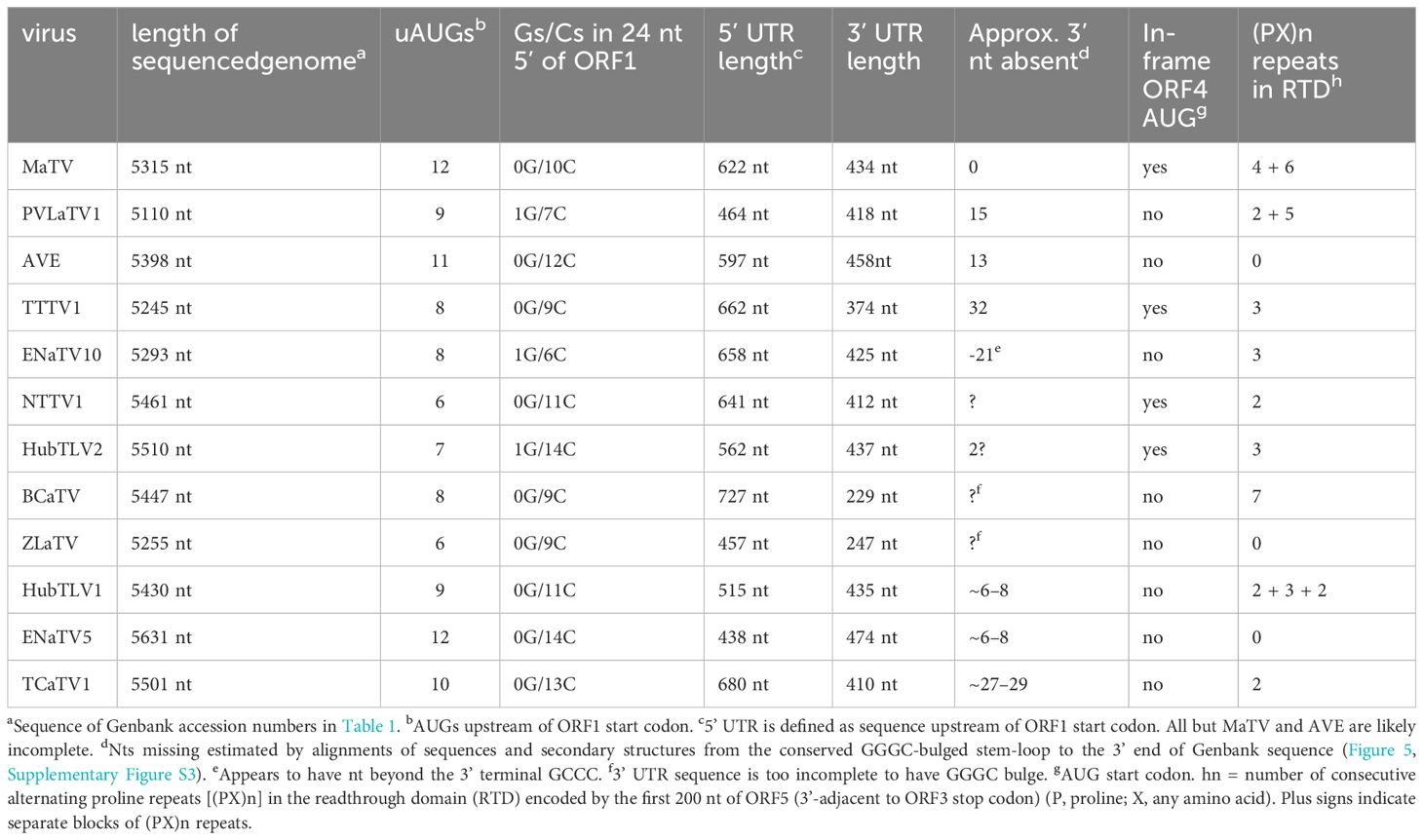
Table 2. Properties of rimosavirus genomes.
Such a 5’ UTR consisting of many hundreds of bases (Table 2) and containing numerous nontranslated AUG triplets suggests presence of an internal ribosome entry site (IRES) or possibly a long ribosomal shunt system. IRESes in the genomes of picornaviruses, hepaciviruses and many other animal RNA viruses are highly structured RNAs that co-opt various host proteins to recruit the ribosome shortly upstream of the first actual start codon for highly efficient cap-independent translation (65–67). In shunting, scanning ribosomes jump across a long structured stem-loop, then rejoin the mRNA to scan to the start codon (68). However, we detect little sequence conservation among the 5’ UTRs of the twelve rimosaviruses, nor were we able to predict any conserved secondary structure, as would be expected for an IRES or a shunting mechanism. One conserved feature is that the sequence upstream of and near the ORF1 AUG is G-poor and C-rich (Figure 4). Specifically, in all twelve viruses, the 24 nt immediately upstream of ORF1 start codon have at most one guanosine base (Table 2). The C-richness usually extends 50 or more bases upstream of the ORF1 AUG (Figure 4). Certain IRESes have cytosine- or pyrimidine-tracts at about this position relative to the start codon (69), and the region just upstream of well-translated start codons in plants is enriched in C-rich tracts (70). In summary, the 5’ UTR is enigmatic, as it contains numerous AUG codons and small nonconserved ORFs, that seem to rule out a conventional scanning mechanism for initiation of translation of ORF1, but it appears to lack conserved secondary structures or sequences (except potential C-rich motif), as are known in IRESes or shunting structures.
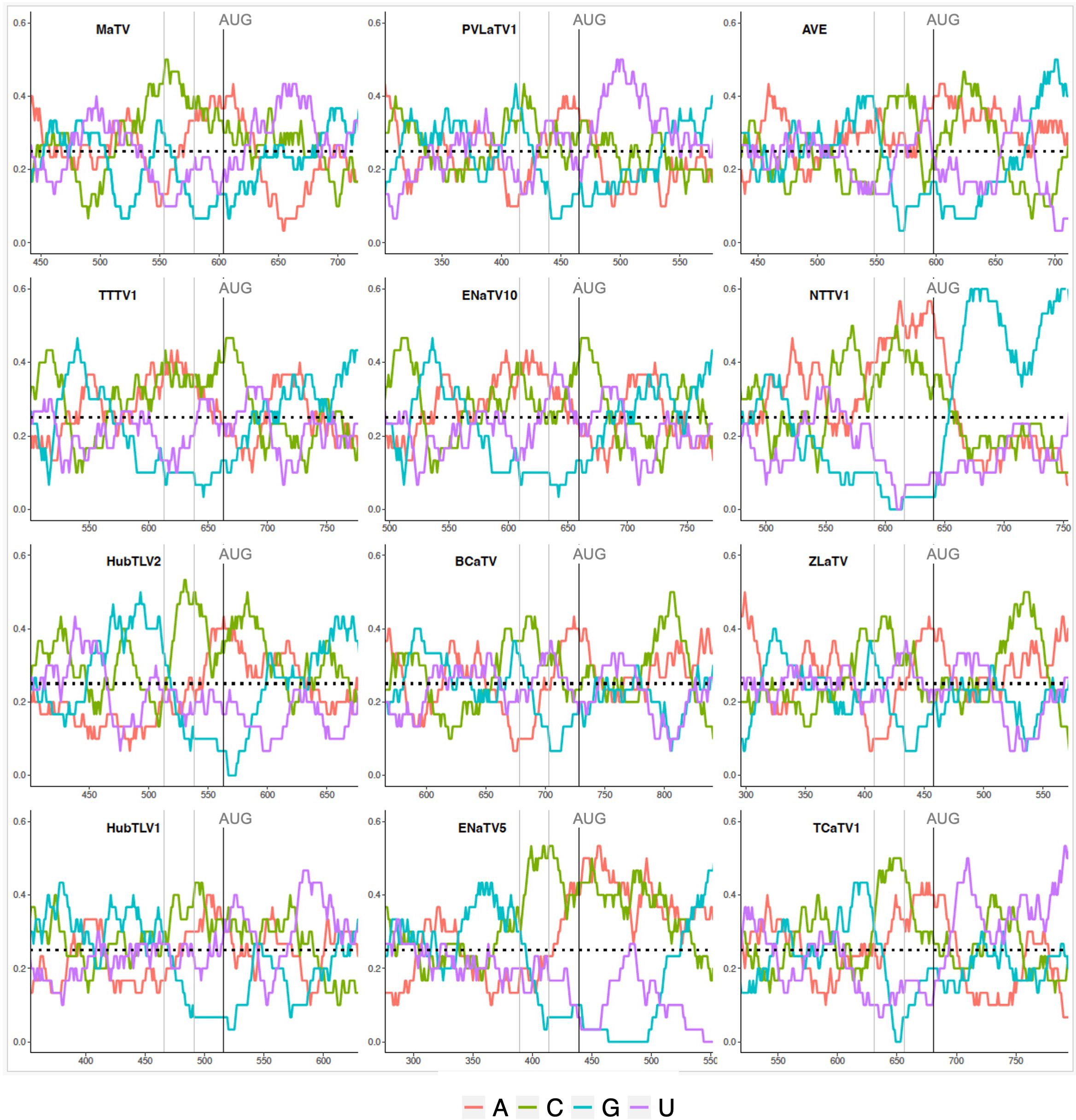
Figure 4. Base compositions flanking the ORF1 start codons (AUG). Plots depicting relative nucleotide frequencies calculated using a sliding window approach plus or minus one-hundred fifty or one-hundred nucleotides relative to the ORF1 start codon, respectively. The X-axis shows genomic position, the Y-axis represents the frequency of each base in a 50 nucleotide window as depicted by the colors in the legend.
3.2.2 3’ untranslated region
The 3’ terminus of MaTV is CCC, the trinucleotide present at the 3’ end of virtually all tombusvirid genomes for which the 3’ sequence has been confirmed. The eleven posted rimosavirus sequences other than that of MaTV do not end in CCC, so we predict they are incomplete. To estimate roughly how many bases are missing from the 3’ ends, we took advantage of known structures near the 3’ end. Tombusvirids have a distinct conserved secondary structure at the 3’ end in which the final four bases, usually GCCC, form a pseudoknot by base pairing to a GGGC bulge in a nearby upstream stem-loop (20, 71–74). Indeed, we predict these structures in MaTV RNA, with the bulged stem-loop terminating 61 nt upstream of the 3’ end of the genome (Figure 5A).
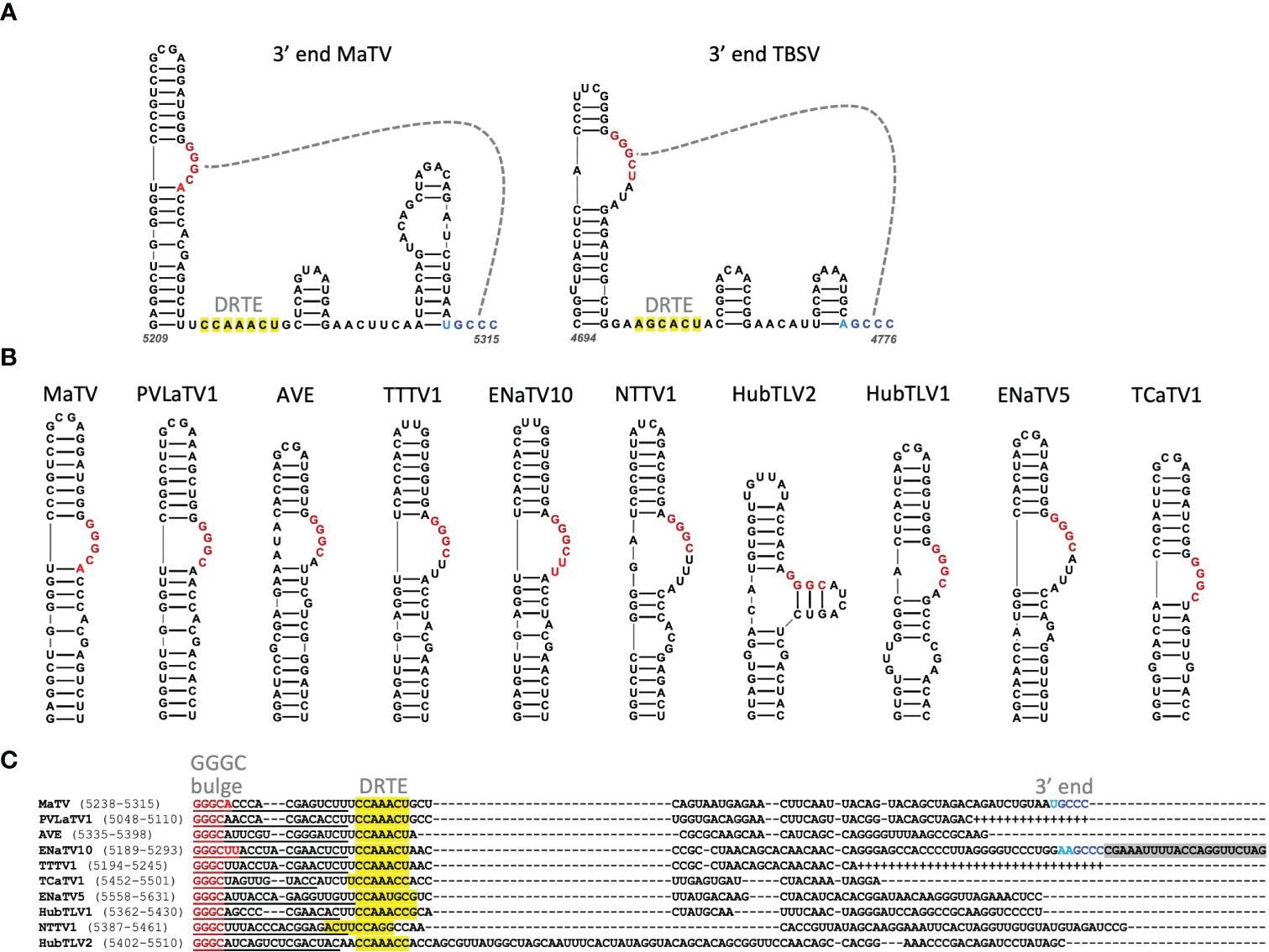
Figure 5. (A). Predicted secondary structure using Mfold and Scanfold of the 3’-terminal 107 nt of the MaTV genome, and the known secondary structure of the 3’ end of the TBSV (Tombusvirus) genome (23, 75). Gray dashed line indicates pseudoknot base pairing. Dark red and blue bases indicate conserved GGGC: GCCC base pairing, while lighter red and blue indicate additional base complementarity. Yellow highlighting indicates bases in the putative distal readthrough element (DRTE) predicted to pair to the bulged stem-loop adjacent to ORF1 stop codon (proximal readthrough element, PRTE, Figure 9). (B). GGGC-bulge-containing stem-loops upstream of 3’ terminus of genome as found in other tombusvirids. BCaTV and ZLaTV are not shown because their GenBank sequences presumably terminate upstream of the GGGC bulge. (C). Alignments of 3’ ends (MUSCLE) of available 3’ terminal sequences starting at the GGGC bulge (dark red). Known (MaTV) and predicted (ENaTV10) 3’ terminal GCCC are in blue (see also Supplementary Figure S3). Additional potential base pairing between GGGC bulge region and 3’ end are shown in lighter shades of red and blue, respectively. Plus symbols in place of dashes indicate missing bases based on closely related sequence above the sequence. Predicted distal readthrough element (DRTE) capable of base pairing to a bulge in the proximal readthrough element (PRTE, Figure 10) is highlighted in yellow. Predicted nonviral sequencing adapter-derived sequence is highlighted in gray (ENaTV10). Numbers in parentheses indicate base positions in the available genome sequence.
To estimate how many bases are missing from the 3’ ends of the other rimosaviruses, we searched for the 3’-proximal GGGC tract in a bulged stem-loop in each genome. All but two viral genomes gave a discrete stem-loop with the GGGC bulge (Figure 5B). (Although weak intra-bulge base pairs were predicted in HubTLV2). The two exceptions, BCaTV and ZLaTV, have much shorter 3’ UTRs in the GenBank sequences (229 nt and 247 nt, respectively) than the others, which range from 374 to 474 nt (Table 2); thus the posted BCaTV and ZLaTV 3’ UTR sequences are both probably incomplete to the extent that they lack the entire GGGC bulged stem-loop. Based on the number of bases downstream of this bulged stem-loop in the other rimosavirus sequences, we roughly estimated the number of bases that would be missing from each 3’ end if the number of bases from the bulged stem-loop to the 3’ end is the same as for MaTV (Table 2). However, the HubTLV2 and ENaTV10 3’UTRs extend beyond the predicted position for the terminal GCCC sequence and do not terminate in GCCC (Figure 5C). An additional stem-loop adjacent to the terminal GCCC is much larger than that of MaTV, giving HubTLV2 and ENaTV10 longer 3’UTRs (Supplementary Figure S3). ENaTV10 has an additional 21 nt 3’ of a GCCC tract. We speculate that adapter bases used for sequencing were not trimmed from what is the true 3’ end of the viral genome (GCCC). Interestingly, the predicted terminal six bases, AAGCCC can form a six-base pseudoknot with the upstream GGGCUU bulge, instead of the usual four base pairs (Supplementary Figure S3).
Tombusvirid RNAs are uncapped (76), so they carry a cap-independent translation element (CITE) usually in the 5’ end of the 3’ untranslated region (36). In this region of the rimosavirus genomes we did not identify any secondary structures conserved across all twelve viruses. We predict structures conserved between closely related viruses MaTV and PVLaTV1, and between ENaTV10 and TTTV1 (Supplementary Figure S4). However, these two pairs of structures differ from each other and do not resemble known CITEs. That said, CITEs can be difficult to identify, given the variety of structures possible, varying from a bulged stem-loop with some conserved motifs to more complex branched structures (77–80). Thus, we cannot rule out the presence of a 3’ CITE in the 3’ UTRs of rimosaviruses, given that the 3’ UTRs are certainly long enough to contain a 3’ CITE.
3.3 Rimosavirus ORFs and encoded proteins
3.3.1 ORF0
As mentioned above, MaTV and PLaTLV1 encode what we call ORF0, although that of PLaTLV1 is only 60% as long as that of MaTV (Supplementary Figure S2A). This includes a 37 codon block of high sequence similarity (28/37 amino acids are identical; Supplementary Figure S2B). Close inspection reveals three codons that vary in the wobble position, conserving would-be amino acid sequence (Supplementary Figure S2C). This may indicate selection for amino acid and thus that ORF0 encodes functional protein. However, numerous other differences alter amino acid sequences, and there are indels outside of the conserved coding region that completely alter amino acid sequence (including introduction of the stop codon that truncates the PLaTLV1 ORF0), while the nucleotide sequence remains highly conserved. These observations, combined with the absence of ORF0 in the other rimosaviruses leads us to think that ORF0 is unlikely to encode functional protein.
3.3.2 ORFs 1–2
ORF1 is predicted to be translated as the protein P1 and also fused with ORF2, via occasional stop codon readthrough, to produce P1-P2 protein. As expected, P2 contains the RdRp active site with the highly conserved D(X)3ϕD, SG(X)3T(X)3N(X)25GDD motifs (81). In addition to forming a clade distinct from those of other tombusvirids (Figure 2), the RdRps also fall into two subclades with MaTV, PVLaTV1, ENaTV10, TTTV and AVE in one group. The other seven rimosaviruses contain a 67 to 81 amino acid insertion between the positions of amino acids 85 and 86 in the RdRps of MaTV, PVLaTV1, ENaTV10, TTTV and AVE (Supplementary Figure S5).
3.3.3 ORF3
ORF3 encodes the coat protein, as evidenced by homology with those of other tombusvirids. The CPs of the twelve rimosaviruses fall into a distinct clade like the RdRps, but some relationships within the proposed genus differ from those of the RdRps (Figure 6). For example, the CPs of TTTV1/ENaTV10 are in a distant branch from MaTV and PLVaTV1, but the RdRps of these four viruses fall in the same subclade (Figure 2). While the RdRps of BCaTV and ZLaTV are closely related, their CPs are not. Interestingly the CPs of other tombusvirids – with the exception of luteoviruses – are more closely related to those of viruses in genus Sobemovirus of the Solemoviridae family (e.g. rice yellow mottle virus, RYMV), than they are to those of the rimosaviruses (Figure 6).
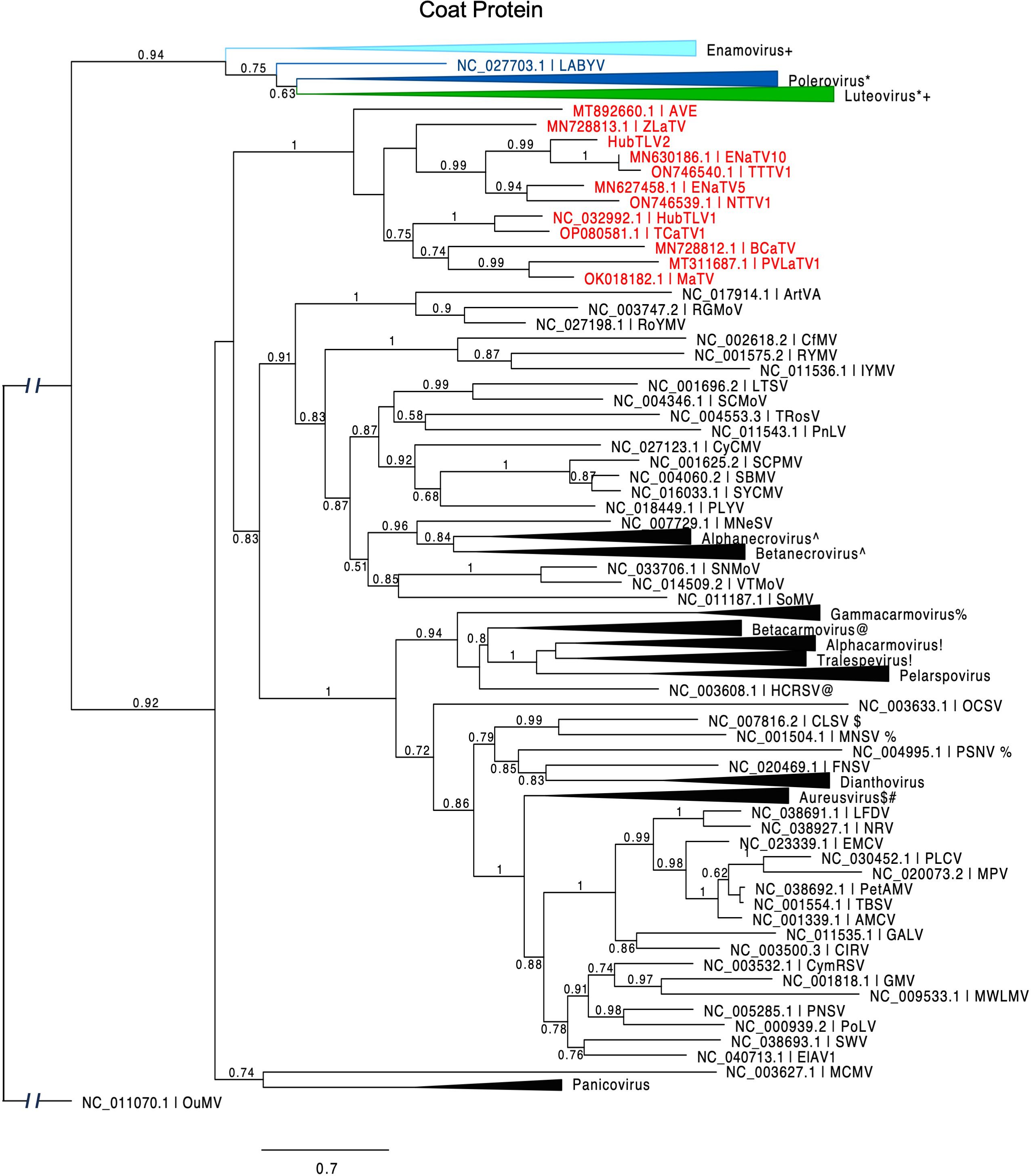
Figure 6. Phylogenetic tree predicting the relationship of viral coat proteins (ORF3) in Tombusviridae and Solemoviridae. Red entries indicate those members of the proposed new Rimosavirus genus. Each collapsed tree consists primarily of sequences belonging to the indicated genus. Modifier symbols (+, *, ^, %, etc.) indicate genera where one or more CP sequences from one genus are grouped with the other genus containing same modifier symbol. Branch support values are shown for splits > 0.5 and are calculated from 1,000 resamples of the Shimodaira-Hasegawa test (SH-like local supports). Branch lengths indicate arbitrary units of evolutionary distances. Spell-outs of viral acronym can be found in GenBank via the indicated accession number. Because solemovirids (Pisuviricota) and tombusvirids (Kitrinoviricota) belong to separate phyla, the outgroup CP sequence (Ourmia melon virus, OuMV) was chosen from a third phylum, Lenarviricota.
3.3.4 ORF4
Poleroviruses and most luteoviruses (but not enamoviruses) encode a movement protein (MP) in ORF4, that overlaps with ORF3, initiating a few nucleotides downstream of the ORF3 start codon but in a different frame and terminating shortly before the ORF3 stop codon (82). In these genera, ORF4 is translated from the same subgenomic mRNA (sgRNA1) as ORF3 (83, 84), via leaky scanning, in which some scanning ribosomes skip the ORF3 AUG and instead initiate on the ORF4 AUG codon (29, 85). In contrast, in four of the rimosaviruses, what we call ORF4 appears to initiate with an AUG codon upstream of the ORF3 start codon. In the other eight, there is a similar ORF overlapping with most of ORF3, but it lacks an in-frame methionine start codon and appears to be disrupted by frameshift mutations upstream of the ORF3 overlap (Figure 7). Upon aligning the RNA sequence at the beginning of MaTV ORF4 with the closely related PVLaTV1 sequence, we see an AUG in PVLaTV1 sequence that aligns with the second AUG of MaTV ORF4 as does the remaining sequence until position 48 at which a U insertion occurs in PVLaTV1 RNA (Figure 7B). This places the upstream portion of what would encode PVLaTV1 ORF4 out of frame with the rest of the ORF. A U insertion at a similar position also disrupts ORF4 in ENaTV10 ORF4 relative to the 95% identical TTTV1 genome, which has an intact AUG-initiated ORF4 (Figure 7B). Similar frameshift mutations near the 5’ end of ORF4 may explain why the ORFs 4 of AVE, BCaTV, ZLaTV, HubTLV1, ENaTV5 and TCaTV1 also appear to lack an in-frame AUG start codon (Supplementary Figure S6).
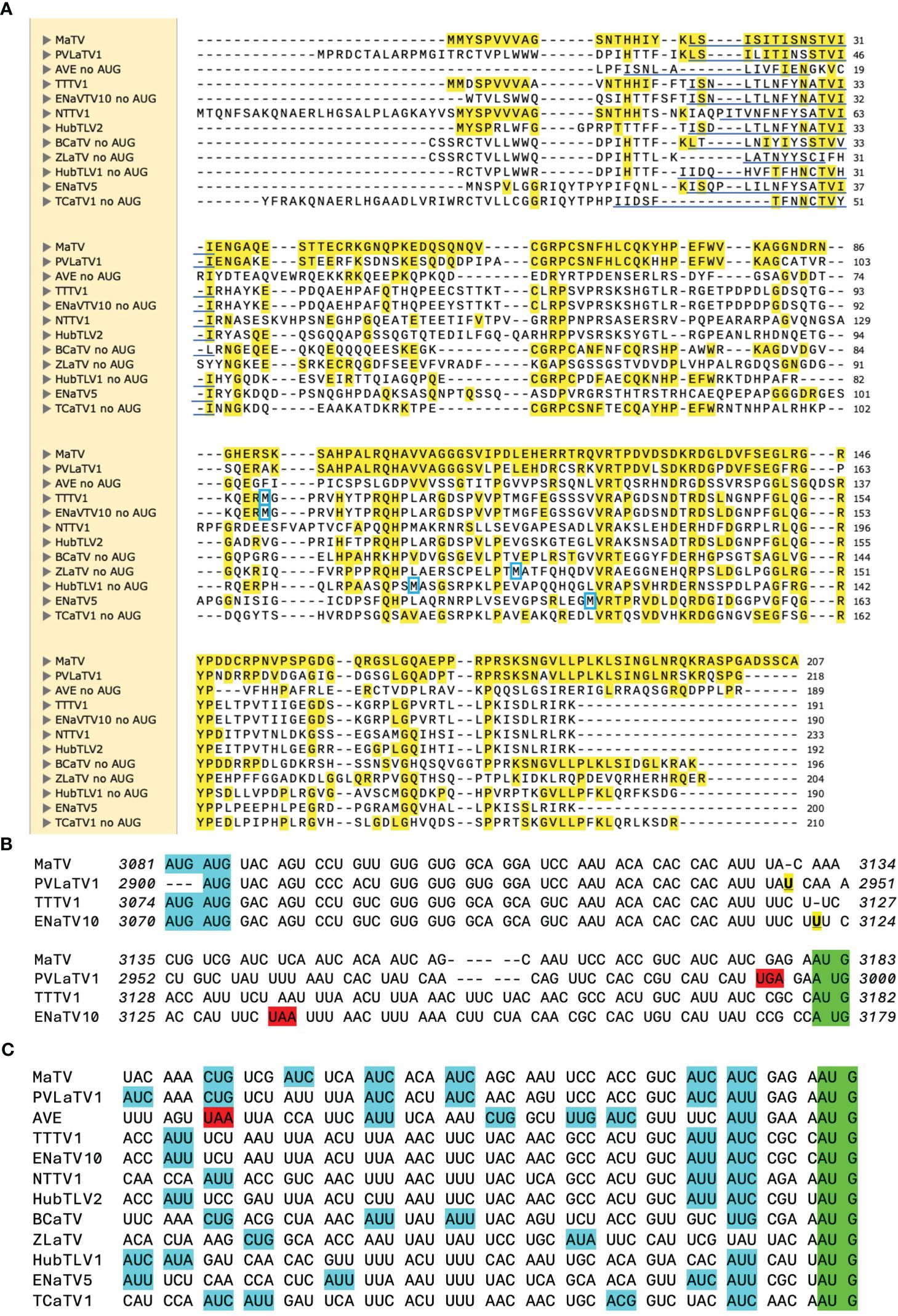
Figure 7. ORF4 comparisons. (A). Alignment (MUSCLE) of predicted P4 proteins, starting from an in-frame methionine or immediately downstream of an in-frame stop codon. However, actual predicted N-terminus of P4 is predicted to be from translation initiation at a non-AUG codon in the underlined region [per ungapped nt alignment, (C)]. First met translated from an AUG located downstream (in the ORF4 frame) of the ORF3 AUG is indicate by blue box. MaTV, PVLaTV1, AVE, HubTLV2, BCaTV and TCaTV1 have no such AUGs (i.e. no internal mets). Yellow highlighting: aa identical to that of MaTV P4. (B). Alignment of the regions of ORF4 from their potential AUG start codons (blue) to the ORF3 start codons in a different frame (green), with spacing to show codons starting from the AUGs. The U insertions (bold, underlined, yellow) in PVLaTV1 relative to MaTV, and in ENaTV10 relative to TTTV1, disrupt the reading frames, leading to stop codons (red). For PVLaTV1 and ENaTV10, the potential initiator AUGs are in a different frame than the amino termi in panel (A). Italic numbering indicates genomic positions of first and last nucleotide on each line. See Supplementary Figure S6 for alignments of this portion of all 12 rimosavirus genomes without codon spacing. (C) Possible non-AUG start codons (blue) (86–88) for ORF4 are shown in the 52 nt upstream of the ORF3 start codon (green). Stop codon is highlighted in red.
Other possibilities were considered. If the 5’ end of the sgRNA1 is downstream of the predicted ORF4 AUG start codon but upstream of the ORF3 start codon, then we would expect that ORF4 would initiate at the next AUG downstream of the ORF3 start codon, as in the luteo- and poleroviruses (29). However, AVE, BCaTV1, TCaTV1 and PVLaTV1 have no AUG codons anywhere in ORF4, and ORFs 4 of MaTV and HubTLV2 have no AUG codons downstream of the ORF3 start codon (methionine residues, Figure 7A). The AUG codons in ORFs 4 of the other rimosaviruses are in the middle or C-terminus of the ORF and not in conserved locations (Figure 7A). Thus, initiation by leaky scanning downstream of the ORF3 start codon is highly unlikely. Instead, the most plausible explanation is that ORF4 initiates with a non-AUG codon. The non-AUG codons ACG, AUC, AUU, AUA, UUG, CUG, GUG have been observed to serve as start codons, albeit much less efficiently than AUG (86–88). To allow translation of ORF4 in all twelve rimosaviruses, a non-AUG start codon would have to be located downstream of the homologous positions to the frame-disrupting insertions in ENaTV10 and PVLaTV1 and an in-frame stop codon in AVE (Figure 7C), and upstream of the ORF3 AUG start codon, as initiation of translation at a non-AUG is not likely to take place downstream of the highly efficient AUG. Alignment of this portion of the rimosavirus genome revealed numerous non-AUG codons, known to be capable of initiation (Figure 7C). Thus, we propose that ORF4 is translated by initiation at one of these non-AUG start codons. This arrangement allows leaky scanning initiation at the AUG of ORF3, resembling the arrangement of ORF3a in luteo- and poleroviruses, which also initiates with a non-AUG codon and overlaps with ORF3 (89).
Regardless of the precise start site, it is clear that ORF4 is present in all 12 rimosaviruses (Figure 1). Starting from the potential non-AUG start codon closest to the CP AUG (Figure 7C), we translated ORFs 4 in silico and aligned the protein sequences with those of polero- and luteoviruses. The phylogenetic tree generated from this alignment revealed the rimosavirus P4 proteins diverge highly from those of the polero/luteoviruses and from each other (Figure 8). One rimosavirus, AVE, clusters with a luteovirus clade, but it is so divergent and bootstrap value so low (<0.5) that this branch assignment is not certain.
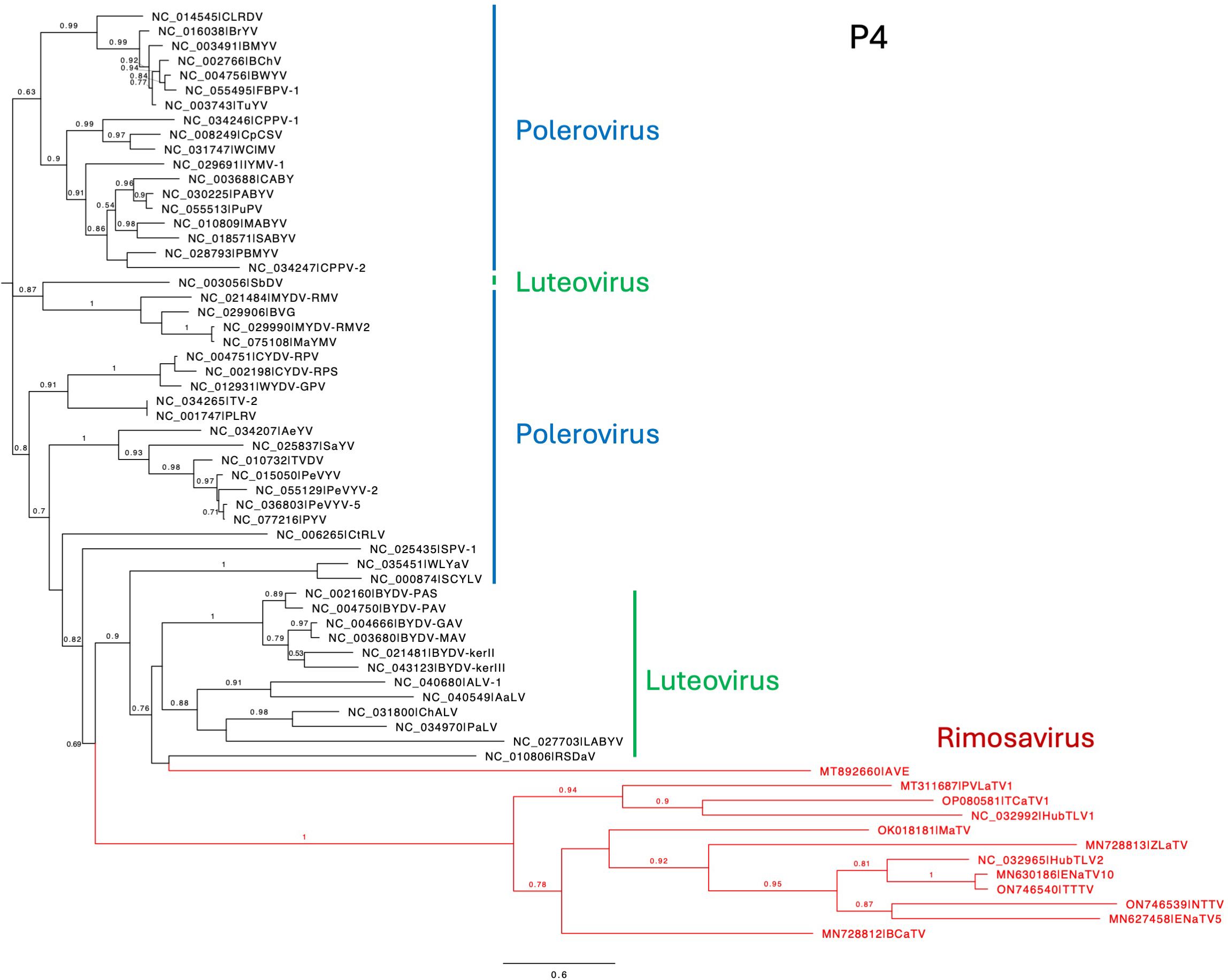
Figure 8. Phylogenetic tree predicting the relationships of P4 encoded by ORF4. The N-terminus of P4 was chosen as using the non-AUG initiator nearest to the ORF3 AUG as start codon. (See Figure 7C.) Rimosavirus sequences are shown in red. Branch support values are shown for splits > 0.5 and are calculated from 1,000 resamples of the Shimodaira-Hasegawa test (SH-like local supports). Branch lengths indicate arbitrary units of evolutionary distances. Full virus names can be looked up via the indicated GenBank accession number.
3.3.5 ORF5
The arrangement of rimosavirus ORFs 3 and 5 resembles that in the L/P/E genera because ORF5 appears to be translated by readthrough of the ORF3 stop codon, creating a large C-terminal readthrough domain (RTD) extension to the CP. The L/P/E RTDs all are more closely related to each other than to any of those from proposed genus Rimosavirus, despite the fact that luteoviruses (Tombusviridae), and polero- and enamoviruses (both Solemoviridae) belong to different families. Interestingly, polerovirus RTDs (dark blue in Figure 9) fall into two major subclades. One clade also includes a luteovirus (green) RTD (bean leafroll virus, BLRV), while the other polerovirus clade includes enamovirus RTDs (light blue). The RTDs of the rimosaviruses (red, Figure 9) all diverge highly from those of L/P/E viruses and from each other. Although some bootstrap values are low, RTDs of the branches representing HubTLV2/ENaTV10/TTTV1, MaTV/PLVaTV1, AVE, and ZLaTV all are more divergent from each other than all of the divergence among all the L/P/E viruses.

Figure 9. Phylogenetic tree predicting the relationships of RTDs (ORF5) based on the amino acid sequences. Genus is color coded in red for Rimosavirus, green for Luteovirus, dark blue for Polerovirus and light blue for Enamovirus. Branch support values are shown for splits > 0.5 and are calculated from 1,000 resamples of the Shimodaira-Hasegawa test (SH-like local supports). Branch lengths indicate arbitrary units of evolutionary distances. Full virus names can be looked up via the indicated GenBank accession number.
Features of ORF5 and the RTD it encodes reflect these extreme differences from the L/P/E RTDs. For example, ORF5 of L/P/E virus RNAs contains about eight to sixteen direct repeats of the sequence CCXXXX (X = any base) shortly downstream of the ORF3 stop codon. This encodes an alternating proline repeat (PX)n, which is thought to serve as a spacer between the CP and functional domains of the RTD protein (58, 59, 90). However, most rimosaviruses have few, if any CCXXXX or PX repeats in the RNA and encoded protein, respectively (Table 2).
3.4 Readthrough elements
Readthrough of stop codons in viral RNAs is usually facilitated by RNA structures located immediately 3’ of the stop codon (23, 31, 54, 55, 91, 92). The ~100 nt tract adjacent to the ORF1 stop codons of all twelve rimosaviruses are well-conserved in sequence and secondary structure (Figure 10). This UAG-proximal structure consists of a stem-loop with four to five helices separated by bulged regions, including a distal bulge with a run of 3 C’s, and a more proximal bulge with the consensus RGUUUGG (red, Figure 10). We predict this conserved sequence base pairs with downstream sequences to form a pseudoknotted structure that facilitates readthrough, as shown for other tombusvirids (17, 23). Indeed, in the 3’ UTR, just downstream of the GGGC bulged stem-loop is a conserved CCAAAYY sequence in a region predicted to be single stranded (Figure 4C, Supplementary Figure S3). This is the exact position of the distal readthrough element (DRTE) which base pairs to a bulge in the stop codon-proximal readthrough element (PRTE) to facilitate readthrough in carnation Italian ringspot virus (CIRV, genus Tombusvirus) (23). A DRTE is also at or near this position in the genomes of at least six other tombusvirid genera, all base pairing to the PRTE with different sequences (23).
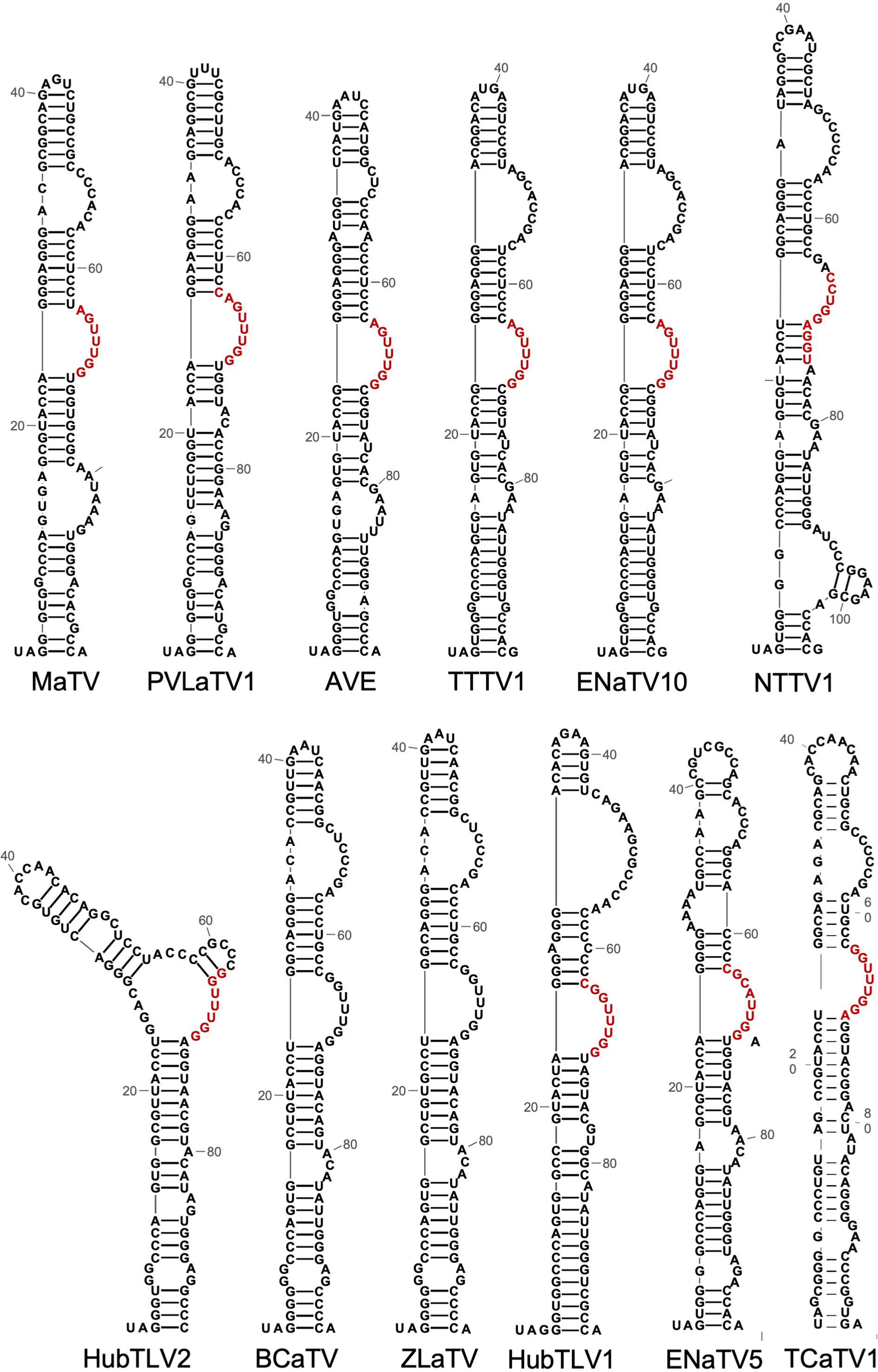
Figure 10. Predicted secondary structures (RNA Alifold, Mfold) of sequences beginning with the putatively leaky ORF1 stop codon (UAG in all rimosaviruses). Based on studies of other tombusvirids, these structures comprise the proximal readthrough element (PRTE). Bases in red are predicted to base pair to the distal readthrough element (DRTE, highlighted in yellow in Figure 5). Note the co-variations in which the sequences vary but maintain at least seven consecutive base pairs in all 10 viruses where the base pairing can be predicted. This long-distance base pairing cannot be predicted for BCaTV and ZLaTV PRTEs because the available genome sequences lack the region containing the PRTE.
For readthrough of the predicted leaky ORF3 stop codon, we look to research on RNA sequences and structures that control readthrough of the homologous stop codon in L/P/E viruses. Mutagenesis of the (CCXXXX)8-16 repeat sequence prevented efficient readthrough for BYDV (54) and PLRV (55). Andy White’s lab then did a more comprehensive analysis of the Pea enation mosaic virus 1 (PEMV1) (Enamovirus) readthrough structure which revealed four separate long-distance base pairings that coaxially stack in the readthrough-facilitating structure (31). That publication also revealed a different arrangement of long-distance base pairings needed for PLRV readthrough. Finally, they showed that local base pairing can compete with the long-distance base pairing to perhaps comprise a switch to regulate readthrough efficiency (31). We predict diverse stem-loop structures adjacent to the ORF5 stop codon, with all except MaTV, PVLaTV1 and AVE having a G-C-rich helix that contains a G-C base pair 7 nt downstream of the stop codon (boxed helices, Supplementary Figure S7). Thus, the RTD for rimosaviruses is novel, not only in its high amino acid sequence variation in its encoded RTD, but also for lack of obvious conserved sequence (e.g. CCXXXX repeats) or obvious L/P/E-like secondary structure to facilitate the CP ORF stop codon readthrough.
4 Discussion
4.1 Remarkable distribution and diversity of sources of rimosaviruses
Rimosavirus sequences were collected from diverse organisms around the globe in large environmental metagenomics sequencing projects with no reports of actual hosts in which they replicate or disease symptoms they cause. Because of their worldwide distribution, it is perhaps surprising that the viruses associated with these genomes have not been discovered previously. It is remarkable that the genome of the virus we call MaTV because its genome was found first in maize and its ancestor teosinte in Mexico, was also found in the cloaca of a tuatara on the tiny uninhabited island of Takpourewa (also known as Stephens Island) in New Zealand (Table 1), a wildlife refuge on which no crops are cultivated (93). Moreover, BCaTV and ZLaTV sequences, first described in kohlrabi and Manchurian wild rice, respectively, in China (10) were also found in the Asian long-horned tick in China (52) and in the cloaca of a tuatara in New Zealand (51).
These wide distributions suggest these viruses may have wide host ranges, and perhaps rather cryptic symptomatology. However, the host range is likely limited to plants, based on their clear membership in the Tombusviridae family, despite the fact that several were isolated from various invertebrates, a reptile, and from plant pathogenic fungi (Table 1). The rimosaviruses associated with Erisyphe necator (powdery mildew of grapevine) and Plasmopara viticola (downy mildew of grapevine) may have infected plant material contaminating the mildew preparation for sequencing, or the mildew may be a vector of the virus. The tombusvirid cucumber necrosis virus is transmitted by zoospores of the soil fungus Olpidium bornovanus (94). Recently, cucumber mosaic virus (not a tombusvirid), which has a wide plant host range, was shown to infect and replicate in the plant pathogenic fungus Rhizoctonia solani (95). Thus, we cannot rule out that these apparent plant viruses may infect the mildews with which they associate.
The animal-associated rimosavirus genomes may have been acquired from plant material in the diet of Chinese land snails (HubTLV1) or pill worms (HubTLV2). For the carnivorous tuatara, the rimosavirus found in its cloaca could be derived from a herbivorous insect in its diet. Plant viruses, including tombusvirids, have been found in other carnivores such as dragonflies (7) and bats (7), and were assumed to have been obtained this way. Ticks are blood feeders, but even in that case, plant viral sequences, including those of tombusvirids have been identified in the human blood virome, albeit at very low abundance (96). This wide diversity of associations by rimosaviruses is a testament to the high abundance and particle stability of tombusvirids. Clearly, additional experiments are necessary to determine the actual hosts in which these rimosaviruses replicate.
4.2 Gene organization and protein function
As mentioned above, the rimosavirus genome encodes P1 and P2 almost certainly via a readthrough mechanism, which is standard for most tombusvirids. In TBSV, and probably all tombusvirids, P1 binds viral RNA and lines the membrane-bound replication vesicles, while P1-P2 fusion has the RNA-dependent RNA polymerase activity to replicate the genome and transcribe subgenomic mRNA (16). ORF3 clearly encodes the CP to form the T=3 icosahedral virion, based on sequence similarity to other tombusvirids.
We expect P4 plays a role in virus movement and other functions, based on the fact that ORF4, which also overlaps with ORF3 in polero- and luteoviruses, encodes a movement protein (MP) in these viruses (26, 97, 98). It can also boost virus infection by suppressing (i) antiviral RNA silencing (99), (ii) host catalase activity (100), and (iii) thiamine synthesis (101). ORF4 differs (i) markedly in sequence compared to that of the luteo/poleroviruses, (ii) in that we predict its translation initiates upstream instead of downstream of the ORF3 start codon, and (iii) at a non-AUG start codon. Thirdly, we detected no ORF3a, which encodes another MP in the ORF4-encoding viruses (89, 102).
While unlikely, it cannot be ruled out that ORF4 may not be translated in the rimosaviruses that lack an in-frame AUG start codon for ORF4. Recently some luteoviruses were discovered that lack ORFs 4 and 3a (103, 104). None of the polero-like enamoviruses encode ORFs 3a or 4 (89). For some enamoviruses, the movement functions are provided by a co-infecting umbravirus (105, 106), but for other enamoviruses and the luteoviruses lacking ORFs 3a and 4, no co-infecting partner is known. Thus these and many rimosaviruses may have found other ways to move within the host plant, perhaps by commandeering a host phloem protein as has been observed recently for certain umbravirus-like viruses in the Tombusviridae (107). However, given the presence of ORF4 overlapping with most of ORF3 in all twelve rimosavirus genomes, we favor our hypothesis that ORF4 is translated via initiation at a non-AUG shortly upstream of the ORF3 AUG start codon.
Based on its position and homology to ORF5 in L/P/E viruses, ORF5 is highly likely to be translated by readthrough of the ORF3 stop codon, and thus encodes the RTD. In the L/P/E/s, the RTD is essential for the persistent, circulative, nonreplicative transmission by aphids (26, 58, 60, 108, 109). Thus, we speculate that the rimosaviruses may be also transmitted by aphids, but given the extreme sequence differences of many of the rimosavirus RTDs from those of L/P/Es, we wonder if some rimosaviruses may be transmitted by other insect species or possibly non-insect vectors. For example, the unrelated beet necrotic yellow vein virus (Benyviridae) encodes an RTD extension on the CP that facilitates fungal transmission of the virus (110). In the poleroviruses, the C-terminal half of the RTD also has been shown to play a role in virus movement in the phloem (111–113). Thus, it appears that ORFs 3a, 4, and C-terminus of ORF5 may act together to ensure efficient, phloem-limited virus movement in the infected plant (102). Other functions are possible. Recently, a CP-RTD protein of an ilarvirus was shown to have silencing suppressor activity (114). The role(s), if any, the rimosavirus RTD plays in vector transmission, virus movement, or silencing suppression is one of the many interesting questions about this cryptic genus that remains to be answered.
Finally, based on the phylogenetic tree showing that all L/P/E RTDs fall into one clade that is less diverse than the branches that include only rimosaviruses, we propose that rimosavirus RTDs have a very ancient origin, and/or have been undergoing more rapid selection and evolution than the L/P/E RTDs. The polero- and enamoviruses have a replication apparatus so different from the rimosaviruses and luteoviruses that they fall into a different phylum (Pisuviricota) from the Tombusviridae (Kitrinoviricota) (53). Thus, we speculate that a sobemo-like ancestor of the polero- and enamoviruses may have acquired its RTD by recombination in mixed infection with an ancestral rimosa-like or luteo-like virus.
4.3 Noncanonical translation
4.3.1 Potential IRES in the 5’ UTR
Perhaps the most unusual feature of rimosaviruses is the long tract at the 5’ end upstream of the ORF1 initiation codon. MaTV encodes a significant size ORF (ORF0) in this region, and PVLaTV1 encodes a truncated version of this ORF, however (i) there are AUGs upstream of ORF0, (ii) no ORF of substantial size is present or conserved upstream of ORF1 in the other rimosaviruses, and (iii) there are numerous AUGs scattered at different positions upstream of ORF1 in all rimosavirus genomes (Supplementary Figure S1). Thus, we speculate that the 5’UTR may have IRES or ribosome shunting activity, which would be novel, because genomes in the other tombusvirid genera contain a short 5’ UTR (maximum 142 nt in BYDV), relying on the 3’ CITE that facilitates translation by ribosome scanning from the 5’ end. The rimosavirus 5’ UTR has potential to contain an IRES or shunting structure, but we found no significant, conserved secondary structures in the 5’ UTRs. A conserved G-poor tract immediately upstream of the ORF1 start codon is a feature in some translation enhancers such as the tobacco mosaic virus 5’ leader (115), and we also found the tract upstream of and including the G-poor tract is C-rich, which is a feature in some IRESes (116). Although the 3’ UTR is long enough to encode a 3’ CITE (36), we found no secondary structures in the 3’ UTR that resembled known CITEs. Additional computational approaches and of course lab experiments are necessary to determine how translation initiates on rimosavirus genomes.
4.3.2 Leaky start and stop codons
One translation feature that is not mysterious is the secondary structure that controls readthrough of the ORF1 stop codon to allow translation of the RdRp encoded in ORF2. All twelve viruses have obvious bulged stem-loops adjacent to the ORF1 stop codon that can base pair to a 3’ distal readthrough element (DRTE) as has been shown to facilitate readthrough in many tombusviruses (23). Also, the stem-loop near the 3’ end, with the GGGC bulge capable of pseudoknot base pairing to the extreme 3’ terminal bases GCCC resembles the replication structure present in all other studied tombusvirid genomes (20, 71).
By analogy with luteo- and poleroviruses (29, 117), we speculate that ORFs 4, 3 and 5 are translated from sgRNA1, initiating upstream of the ORF3 start codon, as the CP of all tombusvirids is translated from a sgRNA. If our hypothesis of non-AUG translation initiation of ORF4 is correct, then the 5’ end of sgRNA1 must be downstream of any AUG triplets located upstream of the ORF3 AUG, because if the 5’ end of sgRNA1 included those AUGs, scanning ribosomes would initiate at those, rather than the predicted non-AUG start codon and subsequent AUG initiator of ORF3. Bearing this in mind, we sought conserved sequences and secondary structures that may be required for generating the 5’ end of sgRNA1, because such structures have been shown to be required for sgRNA1 synthesis in many tombusvirids (34, 118). Indeed, a conserved sequence and stem-loop is predicted in this region of rimosavirus genomes (underlined bases in Supplementary Figure S6). Co-variations in the few base differences among these sequences support the existence of the stem-loop, implying that base pairing is required, even if the sequences that comprise those base pairs vary (compare underlined regions in PVLaTV1 and AVE in Supplementary Figure S6).
In conclusion, phylogenetic comparisons reveal that the twelve genome sequences found in GenBank described here clearly belong to viruses in a new genus in the Tombusviridae family. The biological properties of the viruses associated with these genomes, such as host range, symptomatology, vector specificity, remain to be elucidated. It is clear that the diverse catalog of noncanonical translation mechanisms that are a hallmark of tombusvirid gene expression is enriched by this puzzling new collection of viral genomes.
Author’s note
A paper published online after this manuscript was accepted identifies genomes of more probable rimosaviruses: Kim, J., Jeon, E. J., Jun, M., Lee, D.-S., Lee, S.-J., and Lim, S. (2014). Complete genome sequences of two tombusviruslike viruses identified in Echinacea purpurea seeds. Virus Genes doi: 10.1007/s11262-024-02092-5.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OK018181.2.
Author contributions
ZL: Formal analysis, Visualization, Writing – original draft, Writing – review & editing. LH: Investigation, Methodology, Writing – review & editing. ES: Formal analysis, Visualization, Writing – review & editing. WM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research is supported by the Iowa State University Plant Sciences Institute Scholars Program. This paper is also a product of the Iowa Agriculture and Home Economics Experiment Station, Ames, Iowa, project No. IOW4308 supported by USDA/NIFA and State of Iowa funds.
Acknowledgments
The authors thank Megan Harrison, Seema Raychaudhuri, and Abigail Maue for assistance in preparing this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2024.1422934/full#supplementary-material
Supplementary Figure 1 | 5’ UTR alignments using default parameters for MUSCLE version 3.8.1551 in SnapGene. Sequences end at the start codon for ORF1. Bases with ≥50% sequence identity at each position are shaded in yellow, with intensity of shading proportional to number of sequences in which the base is conserved at each position. All AUG triplets are highlighted in green.
Supplementary Figure 2 | Alignment of ORF0 and P0 sequences of MaTV and PLaTLV1. (A) Alignments of complete ORFs. Note PLaTLV1 ORF0 is 60% as long as MaTV ORF0. Identical bases are highlighted in blue. (B) Alignment of P0 encoded by ORF0. (C) Codons encoding the region of highly conserved potential amino acids. Base differences in wobble position that do not change amino acid are in green. Base differences that change the encoded amino acid are in red. In all three panels, gold box outlines the nts (A, C) that encode the conserved block of 37 codons, or the 37 amino acid sequence itself (B). Note that most of the identical bases in the first row of the alignment in (A) are in different reading frames, hence no aa sequence identity is encoded until bases 70/52 for MaTV and PLaTLV1 ORFs 0, respectively.
Supplementary Figure 3 | Alignment of amino acid sequences of RdRp domains encoded by ORF2. Alignment was made using MUSCLE version 3.8.1551 in SnapGene using default parameters.
Supplementary Figure 4 | Predicted (MFOLD) secondary structures of the 3’ termini of GenBank sequences of the indicated viral genomes. HubTLV2 sequence is likely incomplete, as it does not terminate in GCCC. Predicted 3’ terminus of ENaTV10 genome is indicated. Bases we speculate are adapter-derived are in gray. In this portion of the genome, TTTV1 differs from ENaTV10 at only one base position (indicated) and the TTTV1 GenBank sequence terminates before the actual predicted 3’ end, which is the same as that predicted for ENaTV10. Dashed line indicates pseudoknot base pairing conserved in all tombusvirids (dark red and dark blue). Extended potential base pairing beyond conserved 4 base pairs is indicated in lighter shades of red and blue. Downstream translational readthrough element (DTRE), predicted to base pair to a bulge in the stem-loop that facilitates readthrough of the ORF1 stop codon are highlighted in yellow.
Supplementary Figure 5 | Predicted secondary structures of regions in the 5’ end of the 3’ UTRs of the indicated viruses. Sequences begin with the ORF5 stop codon.
Supplementary Figure 6 | Alignment of nucleotide sequences of the AUGs that might be predicted to start ORF4, except for frame changes caused by indels before ORF3 in most cases, through the start codon of ORF3, which overlaps ORF4 in a different frame. Shade of yellow highlighting increases with increased sequence identity at each position. The two underlined tracts are predicted (MFOLD) to base pair to each other to form a stem-loop that may contribute to generating the 5’ end of sgRNA1.
Supplementary Figure 7 | Predicted base pairing 3’ proximal to the CP ORF stop codon (UAG in red). 203 nt were used in each prediction. Base numbering starts with the first base of the stop codon. Only the UAG-proximal helices are shown. Dashed line indicates long structured region not shown. Box indicates G-C-rich base-paired region starting with the G-C base pair 7 nt downstream of the stop codon. TTTV1 and ENAV10 sequences are identical with the exception of the blue U in ENaTV10 vs A in TTTV1 at position UAG +3.
References
1. Nicolas AM, Sieradzki ET, Pett-Ridge J, Banfield JF, Taga ME, Firestone MK, et al. A subset of viruses thrives following microbial resuscitation during rewetting of a seasonally dry California grassland soil. Nat Commun. (2023) 14:5835. doi: 10.1038/s41467-023-40835-4
2. Culley AI, Lang AS, Suttle CA. Metagenomic analysis of coastal RNA virus communities. Science. (2006) 312:1795–8. doi: 10.1126/science.1127404
3. Roossinck MJ. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. (2017) 239:82–6. doi: 10.1016/j.virusres.2016.11.019
4. Harvey E, Holmes EC. Diversity and evolution of the animal virome. Nat Rev Microbiol. (2022) 20:321–34. doi: 10.1038/s41579-021-00665-x
5. Simmonds P, Adams MJ, Benkő M, Breitbart M, Brister JR, Carstens EB, et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat Rev Microbiol. (2017) 15:161–8. doi: 10.1038/nrmicro.2016.177
6. Tiamani K, Luo S, Schulz S, Xue J, Costa R, Khan mirzaei M, et al. The role of virome in the gastrointestinal tract and beyond. FEMS Microbiol Rev. (2022) 46:1–12. doi: 10.1093/femsre/fuac027
7. Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, et al. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. (2010) 84:6955–65. doi: 10.1128/JVI.00501-10
8. Feng Y, Krueger EN, Liu S, Dorman K, Bonning BC, Miller WA. Discovery of known and novel viral genomes in soybean aphid by deep sequencing. Phytobiomes. (2017) 1:36–45. doi: 10.1094/PBIOMES-11-16-0013-R
9. Shi M, Lin X-D, Tian J-H, Chen L-J, Chen X, Li C-X, et al. Redefining the invertebrate RNA virosphere. Nature. (2016) 540:539–43. doi: 10.1038/nature20167
10. Yang S, Mao Q, Wang Y, He J, Yang J, Chen X, et al. Expanding known viral diversity in plants: virome of 161 species alongside an ancient canal. Environ Microbiome. (2022) 17:58. doi: 10.1186/s40793-022-00453-x
11. Cobbin JC, Charon J, Harvey E, Holmes EC, Mahar JE. Current challenges to virus discovery by meta-transcriptomics. Curr Opin Virol. (2021) 51:48–55. doi: 10.1016/j.coviro.2021.09.007
12. Redinbaugh MG, Stewart LR. Maize lethal necrosis: an emerging, synergistic viral disease. Annu Rev Virol. (2018) 5:301–22. doi: 10.1146/annurev-virology-092917-043413
13. Trebicki P, Nancarrow N, Cole E, Bosque-Perez NA, Constable FE, Freeman AJ, et al. Virus disease in wheat predicted to increase with a changing climate. Glob Chang Biol. (2015) 21:3511–9. doi: 10.1111/gcb.12941
14. Peters JS, Aguirre BA, Dipaola A, Power AG. Ecology of yellow dwarf viruses in crops and grasslands: interactions in the context of climate change. Annu Rev Phytopathol. (2022) 60:283–305. doi: 10.1146/annurev-phyto-020620-101848
15. Winkler FK, Schutt CE, Harrison SC, Bricogne G. Tomato bushy stunt virus at 5.5-Å resolution. Nature. (1977) 265:509–13. doi: 10.1038/265509a0
16. Nagy PD. Tombusvirus-host Interactions: Co-opted evolutionarily conserved host factors take center court. Annu Rev Virol. (2016) 3:491–515. doi: 10.1146/annurev-virology-110615-042312
17. Chkuaseli T, White KA. Intragenomic long-distance RNA-RNA interactions in plus-strand RNA plant viruses. Front Microbiol. (2018) 9:529. doi: 10.3389/fmicb.2018.00529
18. Nagy PD, Feng Z. Tombusviruses orchestrate the host endomembrane system to create elaborate membranous replication organelles. Curr Opin Virol. (2021) 48:30–41. doi: 10.1016/j.coviro.2021.03.007
19. White KA, Nagy PD. Advances in the molecular biology of tombusviruses: gene expression, genome replication, and recombination. Prog Nucleic Acid Res Mol Biol. (2004) 78:187–226. doi: 10.1016/S0079-6603(04)78005-8
20. Simon AE. 3’UTRs of carmoviruses. Virus Res. (2015) 206:27–36. doi: 10.1016/j.virusres.2015.01.023
21. Okuno T, Hiruki C. Molecular biology and epidemiology of dianthoviruses. Adv Virus Res. (2013) 87:37–74. doi: 10.1016/B978-0-12-407698-3.00002-8
22. Barry JK, Miller WA. A -1 ribosomal frameshift element that requires base pairing across four kilobases suggests a mechanism of regulating ribosome and replicase traffic on a viral RNA. Proc Natl Acad Sci U.S.A. (2002) 99:11133–8. doi: 10.1073/pnas.162223099
23. Cimino PA, Nicholson BL, Wu B, Xu W, White KA. Multifaceted regulation of translational readthrough by RNA replication elements in a tombusvirus. PloS Pathog. (2011) 7:e1002423. doi: 10.1371/journal.ppat.1002423
24. Tajima Y, Iwakawa HO, Kaido M, Mise K, Okuno T. A long-distance RNA-RNA interaction plays an important role in programmed -1 ribosomal frameshifting in the translation of p88 replicase protein of Red clover necrotic mosaic virus. Virology. (2011) 417:169–78. doi: 10.1016/j.virol.2011.05.012
25. Kuhlmann MM, Chattopadhyay M, Stupina VA, Gao F, Simon AE. An RNA element that facilitates programmed ribosomal readthrough in turnip crinkle virus adopts multiple conformations. J Virol. (2016) 90:8575–91. doi: 10.1128/JVI.01129-16
26. Chay CA, Gunasinge UB, Dineshkumar SP, Miller WA, Gray SM. Aphid transmission and systemic plant infection determinants of barley yellow dwarf luteovirus-PAV are contained in the coat protein readthrough domain and 17-kDa protein, respectively. Virology. (1996) 219:57–65. doi: 10.1006/viro.1996.0222
27. Kong Q, Oh JW, Carpenter CD, Simon AE. The coat protein of turnip crinkle virus is involved in subviral RNA- mediated symptom modulation and accumulation. Virology. (1997) 238:478–85. doi: 10.1006/viro.1997.8853
28. Lakatos L, Szittya G, Silhavy D, Burgyan J. Molecular mechanism of RNA silencing suppression mediated by p19 protein of tombusviruses. EMBO J. (2004) 23:876–84. doi: 10.1038/sj.emboj.7600096
29. Dinesh-Kumar SP, Miller WA. Control of start codon choice on a plant viral RNA encoding overlapping genes. Plant Cell. (1993) 5:679–92. doi: 10.1105/tpc.5.6.679
30. Johnston JC, Rochon DM. Both codon context and leader length contribute to efficient expression of two overlapping open reading frames of a cucumber necrosis virus bifunctional subgenomic mRNA. Virology. (1996) 221:232–9. doi: 10.1006/viro.1996.0370
31. Chkuaseli T, White KA. Complex and simple translational readthrough signals in pea enation mosaic virus 1 and potato leafroll virus, respectively. PloS Pathog. (2022) 18:e1010888. doi: 10.1371/journal.ppat.1010888
32. Wang J, Simon AE. Analysis of the two subgenomic RNA promoters for turnip crinkle virus in vivo and in vitro. Virology. (1997) 232:174–86. doi: 10.1006/viro.1997.8550
33. Koev G, Miller WA. A positive strand RNA virus with three very different subgenomic RNA promoters. J Virol. (2000) 74:5988–96. doi: 10.1128/JVI.74.13.5988-5996.2000
34. Jiwan SD, White KA. Subgenomic mRNA transcription in Tombusviridae. RNA Biol. (2011) 8:287–94. doi: 10.4161/rna.8.2.15195
35. Fabian MR, White KA. 5’-3’ RNA-RNA interaction facilitates cap- and poly(A) tail-independent translation of tomato bushy stunt virus mRNA: a potential common mechanism for Tombusviridae. J Biol Chem. (2004) 279:28862–72. doi: 10.1074/jbc.M401272200
36. Simon AE, Miller WA. 3’ cap-independent translation enhancers of plant viruses. Annu Rev Microbiol. (2013) 67:21–42. doi: 10.1146/annurev-micro-092412-155609
37. Miller WA, White KA. Long distance RNA-RNA interactions in plant virus gene expression and replication. Ann Rev Phytopathol. (2006) 44:447–67. doi: 10.1146/annurev.phyto.44.070505.143353
38. Nicholson BL, White KA. Functional long-range RNA-RNA interactions in positive-strand RNA viruses. Nat Rev Microbiol. (2014) 12:493–504. doi: 10.1038/nrmicro3288
39. Lappe RR, Elmore MG, Lozier ZR, Jander G, Miller WA, Whitham SA. Metagenomic identification of novel viruses of maize and teosinte in North America. BMC Genomics. (2022) 23:767. doi: 10.1186/s12864-022-09001-w
40. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. (2009) 25:1189–91. doi: 10.1093/bioinformatics/btp033
41. Procter JB, Carstairs GM, Soares B, Mourão K, Ofoegbu TC, Barton D, et al. Alignment of Biological Sequences with Jalview. In: Katoh K, editor. Multiple Sequence Alignment: Methods and Protocols. Springer US, New York, NY (2021). p. 203–24.
42. Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. (1999) 16:1114–4. doi: 10.1093/oxfordjournals.molbev.a026201
43. DeSalle R, Narechania A, Tessler M. Multiple outgroups can cause random rooting in phylogenomics. Mol Phylogenet Evol. (2023) 184:107806. doi: 10.1016/j.ympev.2023.107806
44. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. (2003) 31:3406–15. doi: 10.1093/nar/gkg595
45. Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, et al. ViennaRNA package 2.0. Algo Mol Biol. (2011) 6:26. doi: 10.1186/1748-7188-6-26
46. Andrews RJ, Roche J, Moss WN. ScanFold: an approach for genome-wide discovery of local RNA structural elements-applications to Zika virus and HIV. PeerJ. (2018) 6:e6136. doi: 10.7717/peerj.6136
47. Johnson PZ, Simon AE. RNAcanvas: interactive drawing and exploration of nucleic acid structures. Nucleic Acids Res. (2023) 51:W501–8. doi: 10.1093/nar/gkad302
48. Dolja VV, Krupovic M, Koonin EV. Deep roots and splendid boughs of the global plant virome. Annu Rev Phytopathol. (2020) 58:23–53. doi: 10.1146/annurev-phyto-030320-041346
49. Simmonds P, Adriaenssens EM, Zerbini FM, Abrescia NGA, Aiewsakun P, Alfenas-Zerbini P, et al. Four principles to establish a universal virus taxonomy. PloS Biol. (2023) 21:e3001922. doi: 10.1371/journal.pbio.3001922
50. Chiapello M, Rodríguez-Romero J, Ayllón MA, Turina M. Analysis of the virome associated to grapevine downy mildew lesions reveals new mycovirus lineages. Virus Evol. (2020) 6:veaa058. doi: 10.1093/ve/veaa058
51. Waller SJ, Lamar S, Perry BJ, Grimwood RM, Holmes EC, Geoghegan JL. Cloacal virome of an ancient host lineage – The tuatara (Sphenodon punctatus) – Reveals abundant and diverse diet-related viruses. Virology. (2022) 575:43–53. doi: 10.1016/j.virol.2022.08.012
52. Ni X-B, Cui X-M, Liu J-Y, Ye R-Z, Wu Y-Q, Jiang J-F, et al. Metavirome of 31 tick species provides a compendium of 1,801 RNA virus genomes. Nat Microbiol. (2023) 8:162–73. doi: 10.1038/s41564-022-01275-w
53. Miller WA, Lozier Z. Yellow dwarf viruses of cereals: taxonomy and molecular mechanisms. Annu Rev Phytopathol. (2022) 60:121–41. doi: 10.1146/annurev-phyto-121421-125135
54. Brown CM, Dinesh-Kumar SP, Miller WA. Local and distant sequences are required for efficient read-through of the barley yellow dwarf virus-PAV coat protein gene stop codon. J Virol. (1996) 70:5884–92. doi: 10.1128/jvi.70.9.5884-5892.1996
55. Xu Y, Ju HJ, Deblasio S, Carino EJ, Johnson R, Maccoss MJ, et al. A stem-loop structure in potato leafroll virus open reading frame 5 (ORF5) is essential for readthrough translation of the coat protein ORF stop codon 700 bases upstream. J Virol. (2018) 92:e01544-17. doi: 10.1128/JVI.01544-17
56. Brault V, Van Den Heuvel JFJM, Verbeek M, Ziegler-Graff V, Reutenauer A, Herrbach E, et al. Aphid transmission of beet western yellows luteovirus requires the minor capsid read-through protein P74. EMBO J. (1995) 14:650–9. doi: 10.1002/embj.1995.14.issue-4
57. Brault V, Perigon S, Reinbold C, Erdinger M, Scheidecker D, Herrbach E, et al. The polerovirus minor capsid protein determines vector specificity and intestinal tropism in the aphid. J Virol. (2005) 79:9685–93. doi: 10.1128/JVI.79.15.9685-9693.2005
58. Schiltz CJ, Wilson JR, Hosford CJ, Adams MC, Preising SE, Deblasio SL, et al. Polerovirus N-terminal readthrough domain structures reveal molecular strategies for mitigating virus transmission by aphids. Nat Commun. (2022) 13:6368. doi: 10.1038/s41467-022-33979-2
59. Mutterer JD, Stussi-Garaud C, Michler P, Richards KE, Jonard G, Ziegler-Graff V. Role of the beet western yellows virus readthrough protein in virus movement in Nicotiana clevelandii. J Gen Virol. (1999) 80:2771–8. doi: 10.1099/0022-1317-80-10-2771
60. Brault V, Mutterer J, Scheidecker D, Simonis MT, Herrbach E, Richards K, et al. Effects of point mutations in the readthrough domain of the beet western yellows virus minor capsid protein on virus accumulation in planta and on transmission by aphids. J Virol. (2000) 74:1140–8. doi: 10.1128/JVI.74.3.1140-1148.2000
61. Peter KA, Liang D, Palukaitis P, Gray SM. Small deletions in the potato leafroll virus readthrough protein affect particle morphology, aphid transmission, virus movement and accumulation. J Gen Virol. (2008) 89:2037–45. doi: 10.1099/vir.0.83625-0
62. Yeku O, Frohman MA. Rapid amplification of cDNA ends (RACE). Methods Mol Biol. (2011) 703:107–22. doi: 10.1007/978-1-59745-248-9_8
63. Guo L, Allen E, Miller WA. Base-pairing between untranslated regions facilitates translation of uncapped, nonpolyadenylated viral RNA. Mol Cell. (2001) 7:1103–9. doi: 10.1016/S1097-2765(01)00252-0
64. Chattopadhyay M, Shi K, Yuan X, Simon AE. Long-distance kissing loop interactions between a 3’ proximal Y-shaped structure and apical loops of 5’ hairpins enhance translation of Saguaro cactus virus. Virology. (2011) 417:113–25. doi: 10.1016/j.virol.2011.05.007
65. Filbin ME, Kieft JS. Toward a structural understanding of IRES RNA function. Curr Opin Struct Biol. (2009) 19:267–76. doi: 10.1016/j.sbi.2009.03.005
66. Fraser CS, Hershey JW, Doudna JA. The pathway of hepatitis C virus mRNA recruitment to the human ribosome. Nat Struct Mol Biol. (2009) 16:397–404. doi: 10.1038/nsmb.1572
67. Yamamoto H, Unbehaun A, Spahn CMT. Ribosomal chamber music: toward an understanding of IRES mechanisms. Trends Biochem Sci. (2017) 42:655–68. doi: 10.1016/j.tibs.2017.06.002
68. Ryabova LA, Pooggin MM, Hohn T. Viral strategies of translation initiation: ribosomal shunt and reinitiation. Prog Nucleic Acid Res Mol Biol. (2002) 72:1–39. doi: 10.1016/S0079-6603(02)72066-7
69. Jaramillo-Mesa H, Gannon M, Holshbach E, Zhang J, Roberts R, Buettner M, et al. The Triticum Mosaic Virus Internal Ribosome Entry Site Relies on a Picornavirus-Like YX-AUG Motif To Designate the Preferred Translation Initiation Site and To Likely Target the 18S rRNA. J Virol. (2019) 93:e01705-18. doi: 10.1128/JVI.01705-18
70. Wu T-Y, Li Y-R, Chang K-J, Fang J-C, Urano D, Liu M-J. Modeling alternative translation initiation sites in plants reveals evolutionarily conserved cis-regulatory codes in eukaryotes. Genome Res. (2024) 34:272–85. doi: 10.1101/gr.278100.123
71. Koev G, Liu S, Beckett R, Miller WA. The 3’-terminal structure required for replication of barley yellow dwarf virus RNA contains an embedded 3’ end. Virology. (2002) 292:114–26. doi: 10.1006/viro.2001.1268
72. Pogany J, Fabian MR, White KA, Nagy PD. A replication silencer element in a plus-strand RNA virus. EMBO J. (2003) 22:5602–11. doi: 10.1093/emboj/cdg523
73. Na H, Fabian MR, White KA. Conformational organization of the 3’ untranslated region in the tomato bushy stunt virus genome. RNA. (2006) 12:2199–210. doi: 10.1261/rna.238606
74. Mccormack JC, Yuan X, Yingling YG, Kasprzak W, Zamora RE, Shapiro BA, et al. Structural domains within the 3’ untranslated region of Turnip crinkle virus. J Virol. (2008) 82:8706–20. doi: 10.1128/JVI.00416-08
75. Na H, White KA. Structure and prevalence of replication silencer-3’ terminus RNA interactions in Tombusviridae. Virology. (2006) 345:305–16. doi: 10.1016/j.virol.2005.09.008
76. Allen E, Wang S, Miller WA. Barley yellow dwarf virus RNA requires a cap-independent translation sequence because it lacks a 5’ cap. Virology. (1999) 253:139–44. doi: 10.1006/viro.1998.9507
77. Mizumoto H, Tatsuta M, Kaido M, Mise K, Okuno T. Cap-independent translational enhancement by the 3’ untranslated region of red clover necrotic mosaic virus RNA1. J Virol. (2003) 77:12113–21. doi: 10.1128/JVI.77.22.12113-12121.2003
78. Fabian MR, White KA. Analysis of a 3’-translation enhancer in a tombusvirus: a dynamic model for RNA-RNA interactions of mRNA termini. RNA. (2006) 12:1304–14. doi: 10.1261/rna.69506
79. Wang Z, Parisien M, Scheets K, Miller WA. The cap-binding translation initiation factor, eIF4E, binds a pseudoknot in a viral cap-independent translation element. Structure. (2011) 19:868–80. doi: 10.1016/j.str.2011.03.013
80. Nicholson BL, Zaslaver O, Mayberry LK, Browning KS, White KA. Tombusvirus Y-shaped translational enhancer forms a complex with eIF4F and can be functionally replaced by heterologous translational enhancers. J Virol. (2013) 87:1872–83. doi: 10.1128/JVI.02711-12
81. Charon J, Buchmann JP, Sadiq S, Holmes EC. RdRp-scan: A bioinformatic resource to identify and annotate divergent RNA viruses in metagenomic sequence data. Virus Evol. (2022) 8:1–15. doi: 10.1093/ve/veac082
82. Domier LL. Family Luteoviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy: Ninth Report of the International Committee on the Taxonomy of Viruses, vol. 1045-1053 . Elsevier Academic Press, Amsterdam (2012).
83. Koev G, Mohan BR, Miller WA. Primary and secondary structural elements required for synthesis of barley yellow dwarf virus subgenomic RNA1. J Virol. (1999) 73:2876–85. doi: 10.1128/JVI.73.4.2876-2885.1999
84. Juszczuk M, Paczkowska E, Sadowy E, Zagorski W, Hulanicka DM. Effect of genomic and subgenomic leader sequences of potato leafroll virus on gene expression. FEBS Lett. (2000) 484:33–6. doi: 10.1016/S0014-5793(00)02122-0
85. Miras M, Miller WA, Truniger V, Aranda MA. Non-canonical translation in plant RNA viruses. Front Plant Sci. (2017) 8:494. doi: 10.3389/fpls.2017.00494
86. Gordon K, Futterer J, Hohn T. Efficient initiation of translation at non-AUG triplets in plant cells. Plant J. (1992) 2:809–13. doi: 10.1111/j.1365-313X.1992.tb00150.x
87. Diaz De Arce AJ, Noderer WL, Wang CL. Complete motif analysis of sequence requirements for translation initiation at non-AUG start codons. Nucleic Acids Res. (2018) 46:985–94. doi: 10.1093/nar/gkx1114
88. Fang JC, Liu MJ. Translation initiation at AUG and non-AUG triplets in plants. Plant Sci. (2023) 335:111822. doi: 10.1016/j.plantsci.2023.111822
89. Smirnova E, Firth AE, Miller WA, Scheidecker D, Brault V, Reinbold C, et al. Discovery of a small non-AUG-initiated ORF in poleroviruses and luteoviruses that is required for long-distance movement. PloS Pathog. (2015) 11:e1004868. doi: 10.1371/journal.ppat.1004868
90. Bonning BC, Pal N, Liu S, Wang Z, Sivakumar S, Dixon PM, et al. Toxin delivery by the coat protein of an aphid-vectored plant virus provides plant resistance to aphids. Nat Biotechnol. (2014) 32:102–5. doi: 10.1038/nbt.2753
91. Newburn LR, Nicholson BL, Yosefi M, Cimino PA, White KA. Translational readthrough in Tobacco necrosis virus-D. Virology. (2014) 450-451:258–65. doi: 10.1016/j.virol.2013.12.006
92. Newburn LR, White KA. Atypical RNA Elements Modulate Translational Readthrough in Tobacco necrosis virus-D. J Virol. (2017) 91:e02443-16. doi: 10.1128/JVI.02443-16
93. East KT, East MR, Daugherty CH. Ecological restoration and habitat relationships of reptiles on Stephens Island, New Zealand. New Z J Zool. (1995) 22:249–61. doi: 10.1080/03014223.1995.9518040
94. Rochon D, Kakani K, Robbins M, Reade R. Molecular aspects of plant virus transmission by olpidium and plasmodiophorid vectors. Annu Rev Phytopathol. (2004) 42:211–41. doi: 10.1146/annurev.phyto.42.040803.140317
95. Andika IB, Wei S, Cao C, Salaipeth L, Kondo H, Sun L. Phytopathogenic fungus hosts a plant virus: A naturally occurring cross-kingdom viral infection. Proc Natl Acad Sci U.S.A. (2017) 114:12267–72. doi: 10.1073/pnas.1714916114
96. Cebriá-Mendoza M, Bracho MA, Arbona C, Larrea L, Díaz W, Sanjuán R, et al. Exploring the diversity of the human blood virome. Viruses. (2021) 13:2322. doi: 10.3390/v13112322
97. Schmitz J, Stussi-Garaud C, Tacke E, Prufer D, Rohde W, Rohfritsch O. In situ localization of the putative movement protein (pr17) from potato leafroll luteovirus (PLRV) in infected and transgenic potato plants. Virology. (1997) 235:311–22. doi: 10.1006/viro.1997.8679
98. Link K, Vogel F, Sonnewald U. PD trafficking of potato leaf roll virus movement protein in arabidopsis depends on site-specific protein phosphorylation. Front Plant Sci. (2011) 2:18. doi: 10.3389/fpls.2011.00018
99. Fusaro AF, Barton DA, Nakasugi K, Jackson C, Kalischuk ML, Kawchuk LM, et al. The luteovirus P4 movement protein is a suppressor of systemic RNA silencing. Viruses. (2017) 9:294. doi: 10.3390/v9100294
100. Tian S, Song Q, Zhou W, Wang J, Wang Y, An W, et al. A viral movement protein targets host catalases for 26S proteasome-mediated degradation to facilitate viral infection and aphid transmission in wheat. Mol Plant. (2024) 17:614–30. doi: 10.1016/j.molp.2024.03.004
101. Han X, Yang X, Chen S, Wang H, Liu X, Wang D, et al. Barley yellow dwarf virus-GAV 17K protein disrupts thiamine biosynthesis to facilitate viral infection in plants. Plant J. (2024) 119:432–44. doi: 10.1111/tpj.16772
102. Deblasio SL, Xu Y, Johnson RS, Rebelo AR, Maccoss MJ, Gray SM, et al. The interaction dynamics of two potato leafroll virus movement proteins affects their localization to the outer membranes of mitochondria and plastids. Viruses. (2018) 10:585. doi: 10.3390/v10110585
103. Khalili M, Candresse T, Koloniuk I, Safarova D, Brans Y, Faure C, et al. The expanding menagerie of prunus-infecting luteoviruses. Phytopathology. (2023) 113:345–54. doi: 10.1094/PHYTO-06-22-0203-R
104. Stainton D, Villamor DEV, Sierra Mejia A, Srivastava A, Mollov D, Martin RR, et al. Genomic analyses of a widespread blueberry virus in the United States. Virus Res. (2023) 333:199143. doi: 10.1016/j.virusres.2023.199143
105. Ryabov EV, Fraser G, Mayo MA, Barker H, Taliansky M. Umbravirus gene expression helps potato leafroll virus to invade mesophyll tissues and to be transmitted mechanically between plants. Virology. (2001) 286:363–72. doi: 10.1006/viro.2001.0982
106. Ryabov EV, Robinson DJ, Taliansky M. Umbravirus-encoded proteins both stabilize heterologous viral RNA and mediate its systemic movement in some plant species. Virology. (2001) 288:391–400. doi: 10.1006/viro.2001.1078
107. Ying X, Bera S, Liu J, Toscano-Morales R, Jang C, Yang S, et al. Umbravirus-like RNA viruses are capable of independent systemic plant infection in the absence of encoded movement proteins. PloS Biol. (2024) 22:e3002600. doi: 10.1371/journal.pbio.3002600
108. Gunasinghe UB, Banerjee N, Gray SM. Regions of the barley yellow dwarf virus readthrough protein that are required for aphid transmission. Phytopathology. (1997) 87:S36–7.
109. Gray S, Gildow FE. Luteovirus-aphid interactions. Annu Rev Phytopathol. (2003) 41:539–66. doi: 10.1146/annurev.phyto.41.012203.105815
110. Tamada T, Schmitt C, Saito M, Guilley H, Richards K, Jonard G. High resolution analysis of the readthrough domain of beet necrotic yellow vein virus readthrough protein: a KTER motif is important for efficient transmission of the virus by Polymyxa betae. J Gen Virol. (1996) 77:1359–67. doi: 10.1099/0022-1317-77-7-1359
111. Peter KA, Gildow F, Palukaitis P, Gray SM. The C terminus of the polerovirus p5 readthrough domain limits virus infection to the phloem. J Virol. (2009) 83:5419–29. doi: 10.1128/JVI.02312-08
112. Boissinot S, Erdinger M, Monsion B, Ziegler-Graff V, Brault V. Both structural and non-structural forms of the readthrough protein of cucurbit aphid-borne yellows virus are essential for efficient systemic infection of plants. PloS One. (2014) 9:e93448. doi: 10.1371/journal.pone.0093448
113. Xu Y, Da Silva WL, Qian Y, Gray SM. An aromatic amino acid and associated helix in the C-terminus of the potato leafroll virus minor capsid protein regulate systemic infection and symptom expression. PloS Pathog. (2018) 14:e1007451. doi: 10.1371/journal.ppat.1007451
114. Lukhovitskaya N, Brown K, Hua L, Pate AE, Carr JP, Firth AE. A novel ilarvirus protein CP-RT is expressed via stop codon readthrough and suppresses RDR6-dependent RNA silencing. PloS Pathog. (2024) 20:e1012034. doi: 10.1371/journal.ppat.1012034
115. Sleat DE, Gallie DR, Jefferson RA, Bevan MW, Turner PC, Wilson T. Characterisation of the 5’-leader sequence of tobacco mosaic virus RNA as a general enhancer of translation in vitro. Gene. (1987) 60:217–25. doi: 10.1016/0378-1119(87)90230-7
116. Pilipenko EV, Gmyl AP, Maslova SV, Svitkin YV, Sinyakov AN, Agol VI. Prokaryotic-like cis elements in the cap-independent internal initiation of translation on picornavirus RNA. Cell. (1992) 68:119–31. doi: 10.1016/0092-8674(92)90211-T
117. Tacke E, Prufer D, Salamini F, Rohde W. Characterization of a potato leafroll luteovirus subgenomic RNA: differential expression by internal translation initiation and UAG suppression. J Gen Virol. (1990) 71:2265–72. doi: 10.1099/0022-1317-71-10-2265
Keywords: metagenomics, stop codon readthrough, leaky scanning, internal ribosome entry site, untranslated regions, luteovirus
Citation: Lozier Z, Hill L, Semmann E and Miller WA (2024) A proposed new Tombusviridae genus featuring extremely long 5’ untranslated regions and a luteo/polerovirus-like gene block. Front. Virol. 4:1422934. doi: 10.3389/fviro.2024.1422934
Received: 24 April 2024; Accepted: 17 July 2024;
Published: 09 August 2024.
Edited by:
Lev G. Nemchinov, United States Department of Agriculture (USDA), United StatesReviewed by:
Alexander Karasev, University of Idaho, United StatesJingyuan Liu, United States Department of Agriculture, United States
Copyright © 2024 Lozier, Hill, Semmann and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. Allen Miller, d2FtaWxsZXJAaWFzdGF0ZS5lZHU=