 Amukelani Marivate
Amukelani Marivate Shani Bekker
Shani Bekker Muhammad Qasim Fish
Muhammad Qasim Fish Salerwe Mosebi
Salerwe Mosebi- 1Biomedical Group, Advanced Materials Division, Mintek, Randburg, South Africa
- 2Department of Life and Consumer Sciences, College of Agricultural and Environmental Sciences, University of South Africa, Roodepoort, South Africa
Millions of people have died and a worldwide economic catastrophe has been brought on by the coronavirus disease 2019 (COVID-19) pandemic. Infections caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may presently be treated with less than 10 antiviral drugs such as Remdesivir. The need for medical intervention due to sickness has led to unprecedented research efforts to study the biology of coronaviruses. Additionally, there is a strong likelihood that coronaviruses will cause pandemics in the future. All viruses cannot replicate optimally due to host restriction factors. Given that they are genetically more stable than viral targets and may be shared by similar viruses, these antiviral host factors provide appealing targets for antiviral treatment. The identification of antiviral host factors that are a component of human innate immunity and that prevent the completion of the SARS-CoV-2 life cycle has been made possible by the deployment of several “omics” technologies. In this review, we provide an overview of the antiviral host factors that limit the replication of SARS-CoV-2 in this, which were mostly discovered using functional genetic and interactome screening. Important cellular mechanisms for the SARS-CoV-2 life cycle are covered. Finally, we highlight host restriction factors that could be targeted by clinically approved molecules and the induction of these factors as potential antiviral therapies for COVID-19.
1 Introduction
Multiple zoonotic cross-species transmission episodes in humans led to the emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in Wuhan, China, in November 2019, causing an outbreak of severe pneumonia. The disease caused by this virus was termed COVID-19. The World Health Organization (WHO) then proclaimed the COVID-19 pandemic. It is believed that this coronavirus originated in horseshoe bats and most likely spread to humans via an unidentified intermediate host. Due to its high transmissibility and the prevalence of asymptomatic carriers, SARS-CoV-2 spread quickly over the world, taking lives and impeding social and economic activities even as vaccine campaigns are still in progress (1–7). Over 770 million COVID-19 confirmed cases have been documented between November 2019 and September 2023 (8).The four genera of coronaviruses are betacoronavirus, gammacoronavirus, deltacoronavirus, and alphacoronavirus. Coronaviruses are positive-strand RNA viruses that fall within the subfamily Orthocoronavirinae of the family Coronaviridae (International Committee on Taxonomy of Viruses). Animals such as domestic animals and cattle can have intestinal and respiratory diseases due to coronaviruses (9). Four of the seven identified human coronaviruses (HCoVs) cause mild respiratory infections (HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1). Notably, new pathogenic coronaviruses can enter the human population by zoonotic transmission, as demonstrated by the introduction of two extremely virulent betacoronaviruses in 2002 (the severe acute respiratory syndrome coronavirus, or SARS-CoV) and 2012 (the Middle East respiratory syndrome coronavirus, or MERS-CoV) (10).
The first line of defence for the body against infections, including SARS-CoV-2, is the innate immune system. The development of adaptive immunity is accelerated and coordinated by innate immune responses, which also help detect and eliminate infected cells and restrict viral entrance, translation, replication, and assembly. Pathogen-associated molecular patterns (PAMPs) elicit inflammatory reactions and programmed cell death through cell surface, endosomal, and cytosolic pattern recognition receptors (PRRs), which in turn restrict viral infection and facilitate clearance (11). On the other hand, serious illness and systemic inflammation can result from overstimulating the immune system.
Adaptive immune responses are primarily triggered by viral particle contacts with antigen-presenting cells or B cell receptors after SARS-CoV-2 infection. These encounters result in further biological interactions that protect the host against the virus. The development of immunological memory occurs after the virus has been warded off (Figure 1) (12). It has been demonstrated that, despite the undetectable humoral component, the SARS-CoV cellular immunity endures up to 17 years after the infection. Analysing interferon-gamma levels after stimulating heparinised blood with virus-specific peptides has shown similar results for the SARS-CoV-2 T cell memory in a shorter amount of time. T cells are also essential for a humoral immune response because they provide the foundation of a cellular immunological response (Figure 1). They supply the signals for the humoral response’s maturity, competence, and memory as well as for the activation of B cells. As the initial component of the defence mechanism, mucosal immunity has been demonstrated to be significantly influenced by B cell production of IgA (12).
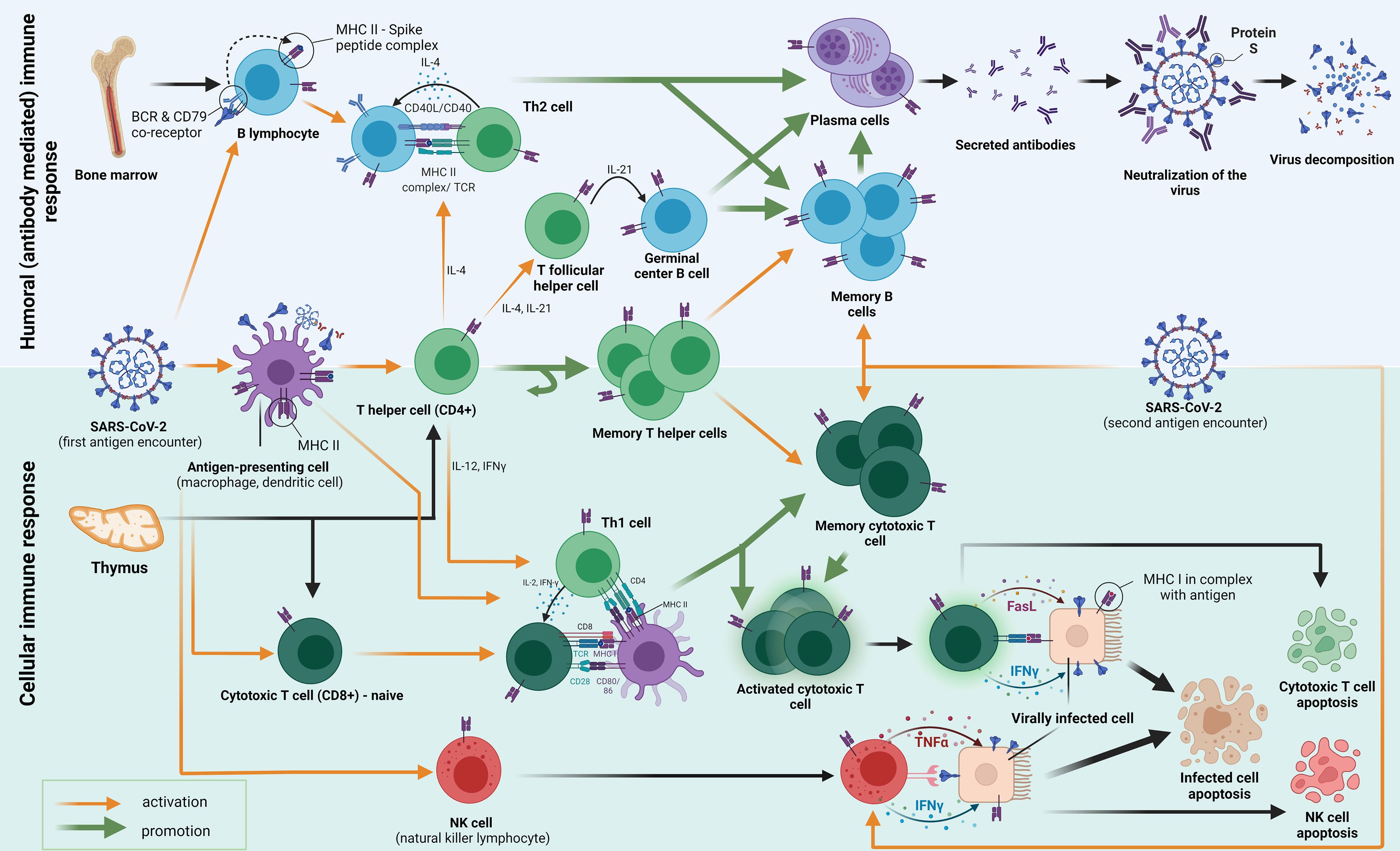
Figure 1. An adaptive immune response to the first and second exposure to the SARS-CoV-2 antigen (S protein) is depicted in the diagram. Pattern recognition receptors (PRRs) to pathogen-associated molecular patterns (PAMPs) are the initial mechanism by which innate immunity functions. Strong antiviral type-I-interferon responses are elicited by antiviral innate membrane (TLR7, TLR8, and TLR9) or cytosolic related receptors (RIG-I-like receptors). From “Adaptive immune responses and immunity to SARS-CoV-2” by Primorac et al. (12), Frontiers in immunology, 13, p. 848582 (https://doi.org/10.3389/fimmu.2022.848582). CC BY-NC.
In addition to several attempts to create vaccines and antiviral treatments, the pandemic has sparked several basic research projects aimed at improving our understanding of coronavirus biology. As opposed to vaccination countermeasures, there are still fewer possibilities for antiviral treatments. The FDA has approved the use of remdesivir, a viral polymerase inhibitor, for patients who are hospitalised. Additionally, molnupiravir and paxlovid, which are combinations of nirmatrelvir and ritonavir, as well as baricitinib, a Janus kinase (JAK) inhibitor, have been approved for use in conjunction with remdesivir. Other approved treatments include tocilizumab, an anti-interleukin (IL)-6 receptor monoclonal antibody, and sotrovimab and monoclonal antibody cocktails, which are all neutralising monoclonal antibodies that bind the viral spike protein. Among them, systemic steroid treatment has been combined with immunomodulatory tactics such as baricitinib and tocilizumab to suppress SARS-CoV-2-induced inflammation (13). Despite these approvals and EUAs, many of these medications have narrow therapeutic indications, and most serious illness cases globally go untreated for lack of appropriate therapy.
Numerous research groups have examined the host factor restriction of coronaviruses, including SARS-CoV-2, since cellular host restriction factors are promising targets for antiviral drugs (14). Preventing future viral infections through proactive measures instead of reacting to emerging viruses as they emerge is the most important lesson learned from the COVID-19 pandemic. Prevention steps involve anticipating and accurately predicting future pandemics and reducing negative impacts by developing treatment options such as vaccinations and, broad-spectrum antiviral medications.
As new information on the immuno-modulatory effects of the virus and viral proteins is discovered, so is our understanding of the host immune response to SARS-CoV-2. This review will discuss the human cellular restriction mechanisms that try to prevent the SARS-CoV-2 life cycle from progressing to various stages. The present state of research emphasises the importance of understanding the host immune response since it can direct new treatment plans and influence public health initiatives.
The enveloped single-stranded positive-sense RNA genome of SARS-CoV-2, codes for four structural proteins i.e. the envelope protein (E), membrane protein (M), nucleocapsid protein (N), and spike protein (S) (2, 4, 5). The E protein is a tiny integrated membrane protein that is essential for the pathogenicity of viruses because it shapes the viral envelope (15). The M protein is involved in the assembly of viruses and the generation of their particles. It is present in the virion as a dimer that causes the membrane to curve and binds to the N protein (15). The N protein participates in the binding, replication and assembly of the viral RNA genome and also regulates the host cell response during viral infection (15). The S protein, which is made up of the two subunits S1 and S2, is the most noticeable surface glycoprotein. Through interactions with receptors on nearby cells, the S protein mediates the formation of syncytia during viral infection, i.e. the fusion of neighbouring cells via receptor interactions. The N- and C-terminal regions of S1 are structurally folded into two separate domains that each include the N- and C-terminal domains (NTD and CTD), respectively. The receptor binding domains (RBDs) are found in the CTD. The S2 subunits play a critical role in controlling membrane fusion and facilitating viral RNA genome transport into host cells (16).
The initial step of the SARS-CoV-2 life cycle, which determines the virus host range and cellular tropism, is recognised as the binding of the S glycoprotein to cell surface receptors (Figure 2) (18, 19). Whether SARS-CoV-2 enters the host cell through the plasma membrane or endocytosis is primarily determined by the proteolytic and hydrolytic cleavage of the S protein, which is mediated by cellular proteases (20). After membrane fusion, viral RNA is released into the host cytoplasm, where it is subsequently used by the host cell’s internal machinery to create viral replication and transcription complexes. Before they mature and spread, the new virus particles are assembled in the endoplasmic reticulum (Figure 2).
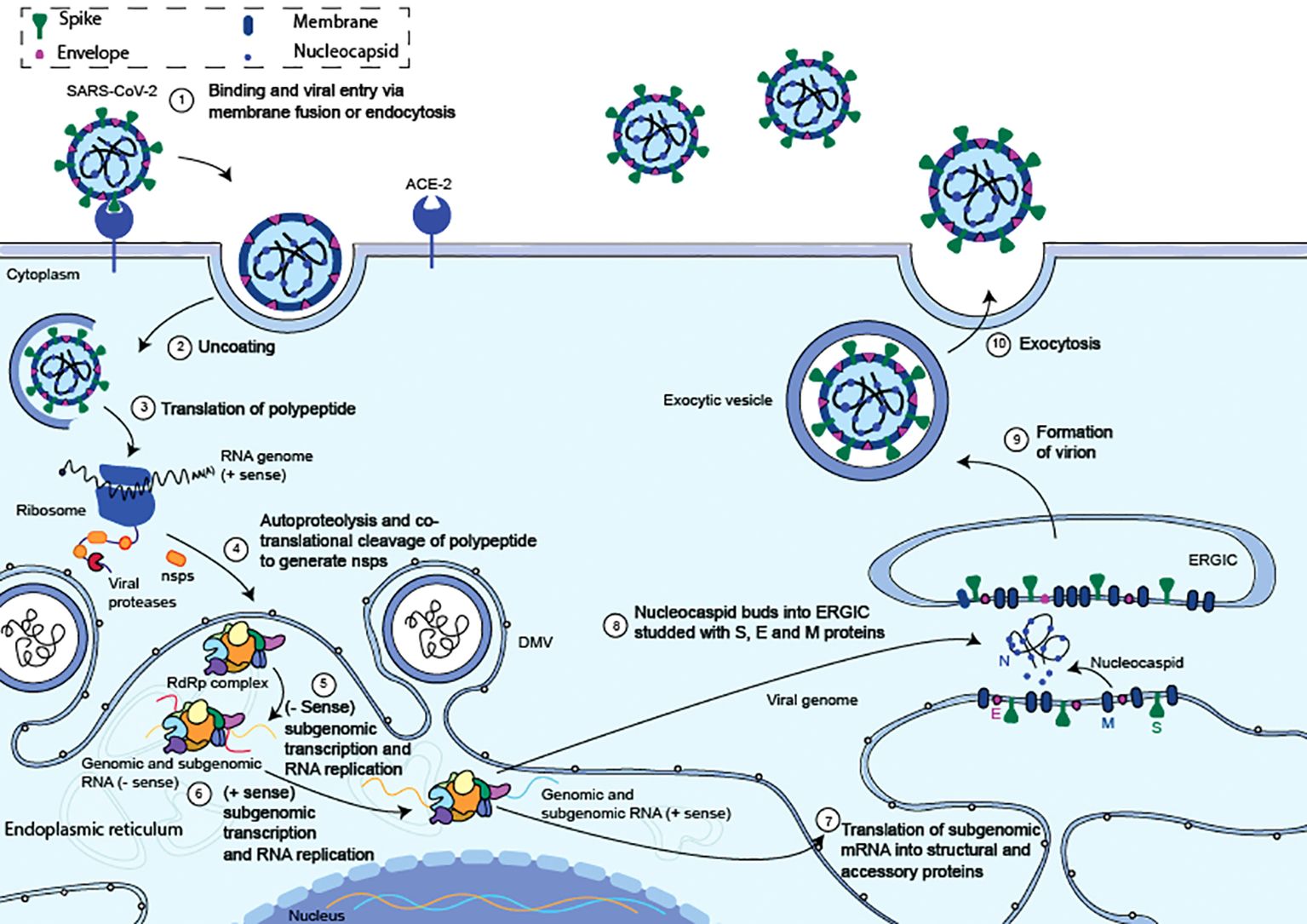
Figure 2. Summary of the entrance and replication cycles for the SARS-CoV-2 virus. (1) The S protein S1 domain binds to the ACE2 and triggers the start of a SARS-CoV-2 entrance. (2) The viral membrane and cellular membranes fuse resulting in the release of the viral RNA into the cytosol. (3) Viral RNA translation begins as it is released into the cytosol. (4) ORF1ab is translated to produce polyproteins pp1a and pp1ab. Within the endoplasmic reticulum, the replication transcription complex (RTC) and the RNA-dependent RNA polymerase (RdRp) is formed. (5) The RTC creates complementary negative-strand genomes and negative-strand subgenomic RNAs within the ER, inside unique double-membrane vesicles (DMV). (6) The progeny genomes and subgenomic mRNAs are created using the negative strands as a template. (7) The subgenomic mRNAs are translated into structural and auxiliary proteins that enclose the viral genome. The S, E, and M structural proteins are translated at the ER membrane while the N protein is translated in the cytoplasm. (8) The N-coated viral genome will bud into the S, E, and M protein-containing ERGIC complex. (9) A vesicle attached to the cellular membrane will contain the budded virion. (10) After the vesicle and cellular membrane have fused, the fully formed virus is exocytised. Adapted from “The molecular virology of coronaviruses”, by Hartenian et al. (17), Journal of Biological chemistry, 295(37), p. 12910 (https://doi.org/10.1074/jbc.REV120.013930). CC BY-NC.
There is growing evidence that host restriction factors provide a defence against SARS-CoV-2 replication. This review aims to shed light on the role of these antiviral components in SARS-CoV-2 infection, as well as provide a broader perspective for future research and prediction of emerging viral infections.
2 Innate immunity and proinflammatory response
The early identification and containment of infections, as well as the subsequent induction of the adaptive immune response, depend on the innate immune system, a conserved defensive mechanism. Effective innate immunity activation requires pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), to identify pathogen-associated molecular patterns (PAMPs) (21). PRRs recruit adaptor proteins in response to PAMP activation, which starts intricate signalling cascades involving many kinases (Figure 3). In the end, this results in the activation of critical transcription factors, proteins that use their recognition of certain DNA sequences to regulate transcription and chromatin, creating a sophisticated mechanism that directs the expression of the genome (23). Transcription factors such as activation protein 1 (AP-1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and interferon regulatory factor 3 (IRF3) are activated.
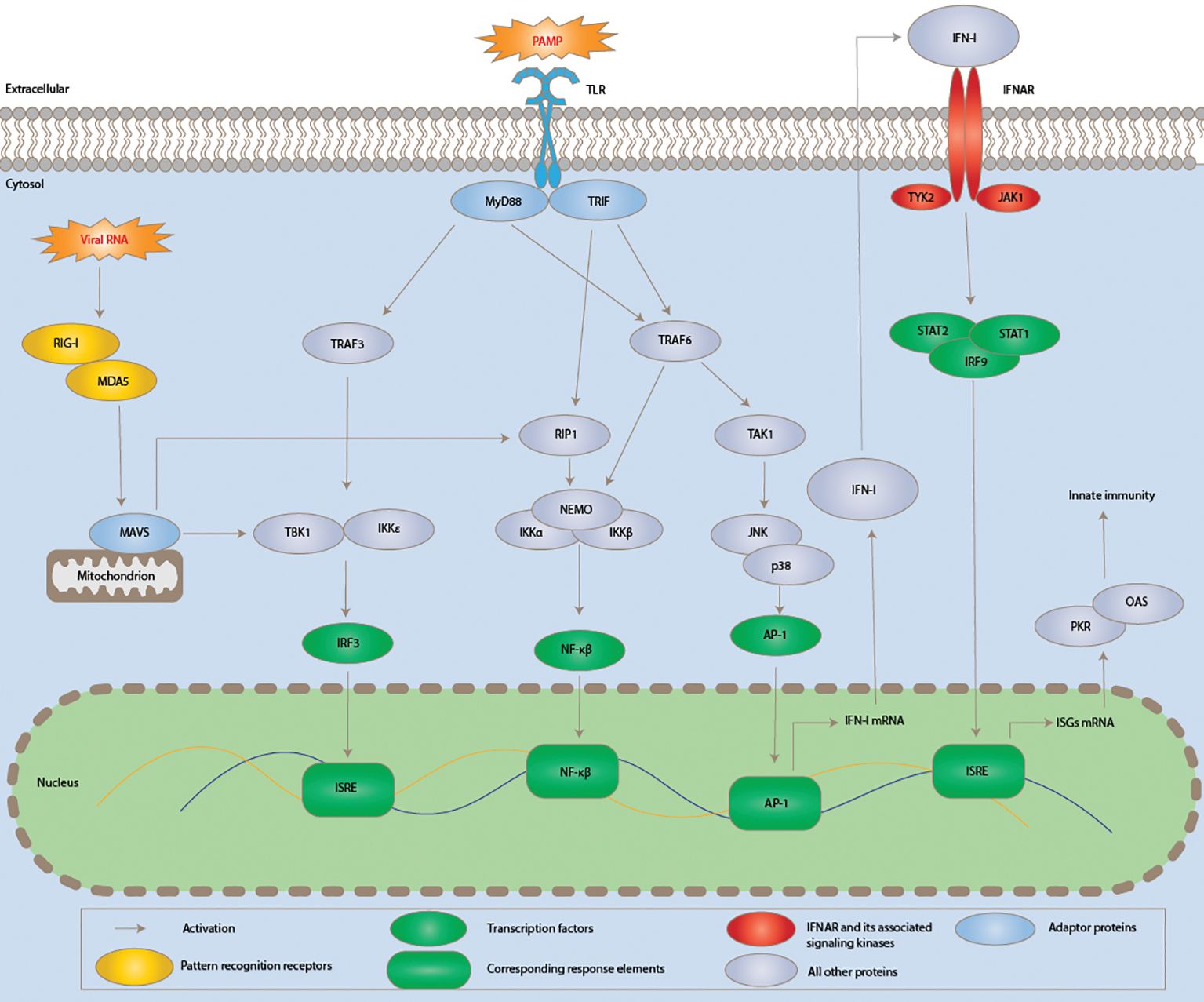
Figure 3. Schematic diagram showing the induction of innate immunity and signalling pathways of type I interferon during SARS-CoV-2 infection. Viral components modulating the pathway are typed in red. Acronyms are AP-1, activator protein 1; IFNAR, IFN-α/β receptor; IKKα, IκB kinase α. JAK1, Janus kinase 1; TRIF, TIR domain–containing adaptor inducing interferon-beta; JNK, c-Jun N-terminal kinase; TAK1, TGF-β-activated kinase; 1IRF3, interferon regulatory factor 3; ISRE, interferon-stimulated response element; mitogen-associated antiviral signalling protein, or MAVS; PAMP, pathogen-associated molecular pattern; MyD88, myeloid differentiation primary response 88; TBK1, TANK-binding kinase 1; NEMO, NF-κB essential modulator; RIG-I, retinoic acid-inducible gene I; PKR, protein kinase RNA-activated; MDA5, melanoma differentiation-associated protein 5; RIP1, receptor-interacting serine/threonine kinase 1:TRAF3, TNF receptor-associated factor 3; MDA5, melanoma differentiation-associated protein 5 and TYK2, tyrosine kinase 2. Adapted from “Human Coronavirus: Host-Pathogen Interaction” by Fung and Liu (22), Annual Review of Microbiology, 73, p. 529 (https://doi.org/10.1146/annurev-micro-020518-115759). CC BY-NC.
Together, these elements promote the synthesis of type I interferons (IFN-I), which are then released and attach to the IFN-α/β receptor (IFNAR) to affect nearby cells (Figure 3) (21). Many interferon-stimulated genes (ISGs) are induced in response to interferon-I, and these ISGs counteract viral replication through a variety of ways, therefore mediating the antiviral action of IFN-I. In the meantime, the inflammatory response is also triggered by the induction of cytokines and chemokines, which can occasionally result in significant tissue damage and other immunopathies linked to SARS-CoV-2 infection (24).
The humoral innate immune response encompasses various components that play a role in combating viral infections, including but not limited to:
● Cytokines: These soluble extracellular glycoproteins play a critical role as intercellular regulators and mobilisers of cells involved in both innate and adaptive inflammatory host defences, angiogenesis, cell growth, differentiation, and death, as well as development and repair processes meant to restore homeostasis (25).
● IFNs: These set of signalling proteins are produced and secreted by host cells in reaction to many viruses. Interferons are released when a virus invades a cell, alerting neighbouring cells to strengthen their defences against the pathogen (26). These proteins are a part of the broader family of chemicals called cytokines, which help cells communicate with one another and trigger the body’s defence mechanisms in response to infections (26).
● ISGs: Genes known as ISGs have the ability to express in response to interferons. Interferons induce protein-signalling pathways inside of cells when they attach to receptors on the cell surface. As a result of this interaction, several genes involved in the innate immune system response are expressed (27).
● Selectins: Selectins play a role in the body’s immune response and inflammation processes by facilitating the binding and adhesion of cells to each other. These glycoproteins are found on the surface of certain cells such as platelets (P-selectin), leukocytes (L-selectin), and endothelial (E-selectin) cells. Selectins are responsible for trafficking of their target cells via sialylated and fucosylated glycoprotein ligand binding (28).
● TLRs: The innate immune system depends heavily on this class of transmembrane proteins. These receptors are often expressed by sentinel cells such as dendritic and macrophage cells. Their primary responsibility is to find structurally conserved chemicals from microorganisms. When these pathogens penetrate physical barriers like the skin or the mucosa of the digestive system, TLRs recognise them and initiate inflammatory responses and the development of antigen-specific adaptive immunity (29).
A vital initial line of defence against viral infections is provided by antiviral restriction factors, which are varied cellular proteins in both structure and function. While many cell types produce these components at low levels during normal conditions, effective control and elimination of invasive viruses often require their activation in response to viral replication and exposure (30). These antiviral restriction factors are known to be effectively produced by type I interferons (IFNs). There is mounting evidence that other IFN subtypes, together with specific cytokines like IL-27 and other cellular activators, can also boost the synthesis of restriction factors, which in turn induces the cell to enter an antiviral state (30).
Agents that efficiently produce these restriction factors, boost their activity, or impart resistance to viral antagonists without causing significant side effects or widespread inflammation may open up new therapeutic or preventive avenues.
3 Binding and entry restriction
The S protein forms part of the outer surface of the SARS-CoV-2 virion by assembling into trimers that form large spikes that protrude from the outer surface giving the virion a crown-like appearance. The S proteins are responsible for host cell entry via receptor binding and virus fusion with target cells. Within the trimer, each S protein monomer is a type I fusion protein that mediates receptor binding via its extracellular S1 subunit and facilitates fusion with the host cell membrane via the transmembrane domain of its S2 subunit (31). Cleavage of the S protein into two subunits is required for binding and entry. Many viral surface glycoproteins are cleaved into two subunits either during virion biosynthesis or when they reach their next target cell. Although unique from SARS-CoV and similar to most known coronaviruses,SARS-CoV-2 S protein is cleaved into two subunits via a furin proteolytic cleavage site prior to exit from its producer cell. Cleavage is essential for effective viral entry, dissemination, and pathogenesis (31).
Many coronaviruses employ their S proteins to bind to and activate a range of proviral host receptors, including sialic acid moieties, dipeptidyl peptidase 4, aminopeptidase-N, and angiotensin-converting enzyme 2 (ACE2). The wide range of receptors that these infections exploit may help to explain in part why there is such a high risk of zoonotic transmission of coronaviruses to humans. The human body has several antiviral factors that play crucial roles in defending the body against viral infection by preventing receptor binding and entry into host cells. SARS-CoV-2 encodes three main groups of proteins: structural proteins, non-structural proteins (nsps), and accessory proteins (5). It has been revealed that a number of these proteins obstruct IFN responses at various stages, including detecting viral RNA, inhibiting IFN transcription/translation, and focussing on ISG effector activities. Numerous proteins interfere with IFN signalling through several mechanisms, and some of them have identical targets, which gives them redundancy in their actions and underscores how crucial it is to modify IFN responses for SARS-CoV-2 to successfully replicate. (5). Interferons, cellular receptors, restriction factors, and cytokines are examples of antiviral factors that work in concert to provide a robust defence against viral infections.
The interplay between these mechanisms contributes to the complexity and effectiveness of the human immune response to viral entry and infection. The human restriction factors involved in the restriction of SARS-CoV-2 entry include lymphocyte antigen 6 complex locus E (LY6E), cholesterol-25-hydroxylase (CH25H), human defensin 5 (HD5), p-selectin glycoprotein ligand-1 (PSGL-1), tripartite motif protein 28 (TRIM28), phospholipid scramblase 1 (PLSCR1) and serine incorporator 5 (SERINC5). These factors trigger an intense immune response upon SARS-CoV-2 infection and inhibit virus entry in a variety of ways (Figure 4). Below we highlight the antiviral factors and function of the innate immune sensors in limiting SARS-CoV-2 binding and entry.
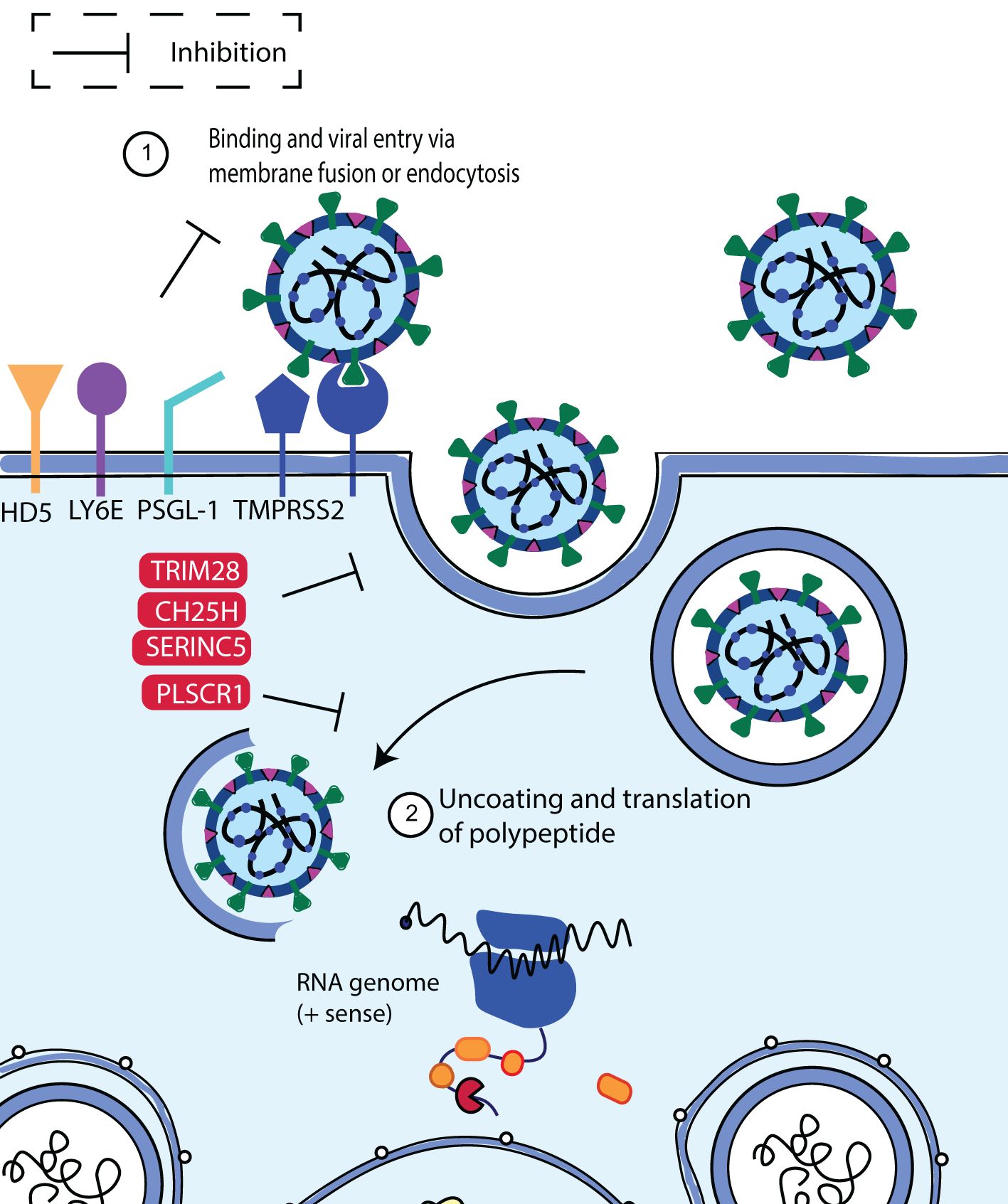
Figure 4. Schematic diagram showing SARS-CoV-2 binding and entry inhibition. Entry of SARS-CoV-2 into the host cell is facilitated by viral S protein binding to the ACE2 cellular receptor promoting endocytosis. Upon entry, SARS-CoV-2 viral particles escape the endosome to initiate viral replication. It was discovered that LY6E, CH25H, HD5, PSGL-1, PLSCR1 and SERINC5 inhibited S-mediated cell fusion and virion entry. Adapted from “The molecular virology of coronaviruses”, by Hartenian et al. (17), Journal of Biological chemistry, 295(37), p. 12910 (https://doi.org/10.1074/jbc.REV120.013930). CC BY-NC.
3.1 LY6E
IFNs play a role in innate antiviral defence within cells by triggering the activation of numerous ISGs. LY6E is an example of such an ISG that modifies viral infection, particularly viral entrance (32). Functioning as a cell surface protein, LY6E is anchored to the surface via a glycosylphosphatidylinositol (GPI) anchor. Notably, the conserved functional location L36 plays a critical role, as evidenced by studies involving the mutation of the GPI anchor and the L36A mutation, which did not impact the restriction of human CoV entry (33–35). LY6E effectively prevents the spike protein-mediated development of syncytia, proving that it inhibits virus entrance by fusing the viral and cellular membranes. This discovery aligns with the outcomes of a quantitative fusion test (34). Surprisingly, LY6E inhibited viral infection irrespective of the presence of cell surface and endosomal proteases, indicating that spike protein activation was not necessary for viral fusion limitation (35). The evidence shows that LY6E is a broad-spectrum coronavirus antiviral factor that blocks coronavirus spike protein-mediated membrane fusion to impede the spread of coronavirus, including SARS-CoV-2 (35).
3.2 CH25H
CH25H, one of the antiviral ISGs, encoded an ER-associated enzyme that facilitated the conversion of cholesterol to oxysterol 25-hydroxycholesterol (25HC) (36, 37). Previous research has demonstrated that 25HC prevents enveloped DNA and RNA viruses from entering host cells (37, 38). The generation of 25HC by CH25H and the activation of acyl-CoA: cholesterol acyltransferase (ACAT), which leads to the reduction of accessible cholesterol from the plasma membrane, are the two main factors that inhibit SARS_CoV-2 virus-cell fusion (39). Overexpression of CH25H consistently impeded SARS-CoV-2 entrance. According to Wang et al. (39), 25HC prevented the SARS-CoV-2 virus-cell fusion by activating the ER-localised ACAT, which mobilised the plasma membrane’s available cholesterol and did not promote spike-induced syncytia. Because 25HC therapy reduced the expression of anthrolysin O domain 4 (ALOD4), which might trap accessible cholesterol at the plasma membrane, it is possible that 25HC reduced plasma membrane cholesterol to prevent fusion between SARS-CoV-2 virus-infected cells (39–41). Both SARS-CoV and MERS-CoV pseudoviruses have had similar impacts (39). As a naturally occurring substance with no documented toxicity at therapeutic quantities, 25HC offers a prospective treatment option for COVID-19 and other emerging viral diseases (39).
3.3 HD5
Paneth cells of the small intestine are the only cells that make and express HD5, one of the defensins that can bind lipids and glycosylated proteins (42–44). It has been found that the binding of HD5 to the ligand-binding domain (LBD) of ACE2 interferes with SARS-CoV-2 entry by obstructing S1 binding (45). The idea was that the glycosylated residue of HD5 might connect to the S1 protein and stop viral invasion (46, 47). In the presence of HD5, SARS-CoV-2 S1 recruitment and adhesion to host cells was dramatically decreased, indicating that HD5 prevented SARS-CoV-2 entry (45).
3.4 PSGL-1
An integral membrane protein PSGL-1 binds to P-, E-, and L-selectins and encourages immune cells to first bind and group together before moving on to migrate into inflamed tissue (48, 49). PSGL-1 acted as a functional receptor for the penetration and reproduction of enterovirus A71 (EV-A71), but it has also been shown to prevent viral infection as an IFN-induced HIV inhibitory factor (50, 51).
Virus-like particle (VLP) incorporation of PSGL-1 on SARS-CoV and SARS-CoV-2 VLPs led to considerably lower infectivity than SARS-CoV-2 in the virus-producing cells, indicating that PSGL-1 effectively reduced SARS-CoV-2 infectivity (52). A virion attachment assay revealed that PSGL-1 reduced viral infectivity by obstructing its structural attachment to target cells (52). In PSGL-1 expressed cells, the release of SARS-CoV and SARS-CoV-2 pseudovirions was modestly inhibited, showing that PSGL-1 expression had little impact on viral release (52, 53). In general, PSGL-1 has been found to decrease SARS-CoV or SARS-CoV-2 infectivity by hindering the S proteins’ integration and preventing viral attachment, principally by interfering with particle binding to the host cell (52). It is interesting to note that PSGL-1 levels were downregulated by viral accessory proteins to counteract impacts during HIV-1 infection (51, 53). More research is required to determine whether the viral evasion of PSGL-1 occurs in coronavirus infection.
3.5 TRIM28
It has been discovered that type II pneumocytes co-express TRIM28, an antiviral restriction protein, and the SARS-CoV-2 receptor. Furthermore, it is known that TRIM28 supports endogenous retrovirus latency, antiviral limitation, and the immune system. Wang et al. (54), identified TRIM28 as a novel regulator of ACE2 expression and SARS-CoV-2 cell entry. Their findings demonstrate that TRIM28 knockdown facilitates pseudotyped SARS-CoV-2 infection of both A549 cells and primary pulmonary alveolar epithelial cells (PAEpiCs). Furthermore, they discovered that NK cells’ reduction of TRIM28 and elevation of ACE2 expression in lung epithelial cells in co-culture were only partially reversed by interleukin-2 depletion and granzyme B blocking in the co-culture medium. Additionally, TRIM28 knockdown boosted IFN-induced ACE2 expression in A549 and PAEpiCs through activation of IFN-receptor 2 (IFNGR2). Dexamethasone partially inhibited the elevated ACE2 brought on by the co-culture of NK cells and TRIM28 knockdown in A549 cells (55).
3.6 PLSCR1
In simultaneous genome-wide clustered regularly interspaced short palindromic repeats (CRISPR)–Cas9 screenings of human hepatocytes and lung epithelia before and after activation with IFNγ, PLSCR1 was found to be a powerful SARS-CoV-2 restriction factor (56). Using CRISPR-Cas9 engineering to eliminate the signal transducer and activator of transcription-1 (STAT1), which is necessary for IFNγ-induced gene expression, the strong anti-SARS-CoV-2 action of IFNγ was verified (56). Beyond restricting SARS-CoV-2 USA-WA1/2020, IFNγ-induced PLSCR1 also showed efficacy against the Omicron BA.1 and Delta B.1.617.2 lineages. It was functionally conserved in mice and bats, and its strong activity extended to other highly pathogenic coronaviruses. Furthermore, it hindered the absorption of SARS-CoV-2 by both the TMPRSS2-dependent and endocytic approaches (56).
Bipartite nano-reporter assays and whole-cell 4Pi single-molecule switching nanoscopy revealed that PLSCR1 selectively targeted SARS-CoV-2-containing vesicles to prevent S-mediated fusion and viral escape (56). The β-barrel domain of PLSCR1’s C-terminus was necessary for fusogenic blocking, but the lipid scramblase activity was not. This mechanistic investigation identifies an anti-coronavirus protein that obstructs at a late entrance phase prior before the RNA release into the host-cell cytoplasm and reveals that PLSCR1 mutations associated with COVID-19 have been detected in some susceptible patients (57, 58).
3.7 SERINC5
A variety of viruses, notably SARS-CoV-2, are effectively inhibited by SERINC5, a cellular multi-pass transmembrane protein involved in sphingolipid and phosphatidylserine biosynthesis (59). Pneumocytes and lung tissue are the natural targets of SARS-CoV-2 for all members of the SERINC family, except for SERINC4 (60). Additionally, it was demonstrated that SERINC5 is integrated inside viral particles and that it has the most impact on SARS-CoV-2 entrance utilizing SARS-CoV-2 S pseudoviruses, VLPs, and infectious SARS-CoV-2 (59, 61, 62).
After SARS-CoV-2 attaches to the hACE2 receptor on the cell surface, target cell proteases activate the S protein, which causes the viral genome to be released within the cell and the viral-host membranes to fuse. As is the case with retroviruses, it was demonstrated here that SERINC5 blocks SARS-CoV-2 entrance by interrupting the virus-cell membrane fusion process (63). Therefore, to carry out its antiviral action against two different virus families, SERINC5 employs a conserved mechanism. Entry of SARS-CoV-2 is reliant on the S protein. S proteins from various variants of SARS-CoV-2 exhibit polymorphism and exhibit varying levels of entrance effectiveness based on the cell line (64). SERINC5 prevented SARS-CoV-2 entrance for S variants derived from the Alpha (B.1.1.7), Beta (B.1.351), Gamma (P1),and Delta (B.1.617) respectively (59). Therefore, these results imply that vulnerability to SERINC5 is never lost, even in the face of variation in S protein across the many SARS-CoV-2 variants.
Upon activation of the immune response, IFNs are released that lead to the translation and expression of ISGs such as those mentioned in this section that form part of the host restriction factors restricting SARS-CoV-2 entry. These host restriction factors all interact with the SARS-CoV-2 S protein during viral entry and play different roles in impeding the S proteins role in facilitating the virus’ entry into the host cell. The viral binding and entry host restrictions and their proposed mechanisms are summarised in Table 1.
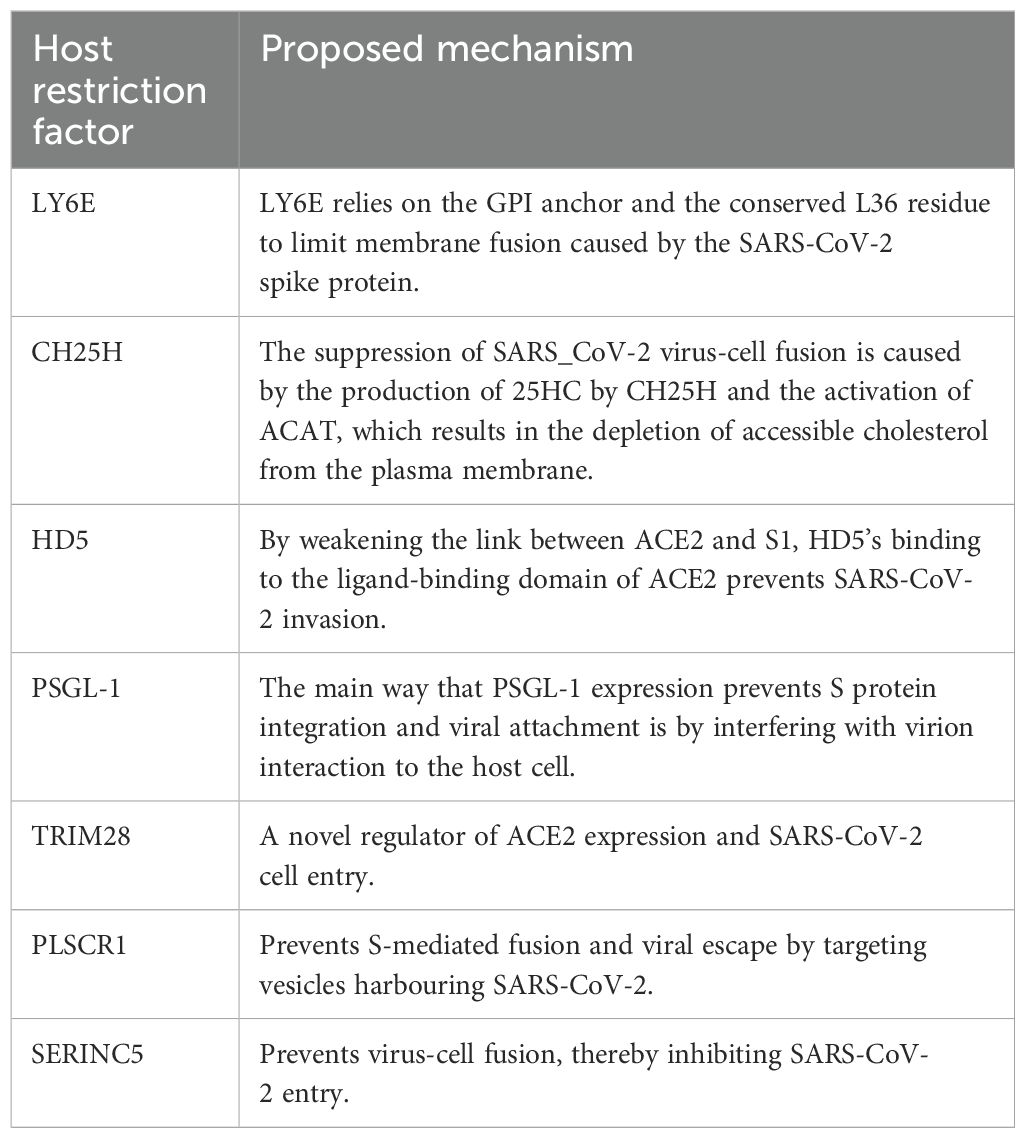
Table 1. Viral binding and entry host restriction factors and their proposed mechanism.
4 Translation and RNA replication restriction
Non-structural proteins (nsps) are produced when SARS-CoV-2 is released into the cytosol via the transcription and translation of the replicase gene from the viral genomic RNA. The replication of viral genomic, sub-genomic RNA and, protein synthesis are made possible by the replicase-transcriptase complex (RTC), which is assembled in the ER by these nsps (Figure 5) (66). A viral infection triggers the activation of cellular stress responses in a cell. Stress granules arise in the cytoplasm as part of this reaction. RNA-binding proteins and mRNAs make up stress granules. The human antiviral factors involved in the restriction of SARS-CoV-2 RNA translation and replication include death-associated protein 6 (DAXX), RNA-binding motif protein 24 (RBM24), spermatogenesis associated serine-rich 2-like protein (SPAT2SL), La related protein 1 (LARP1), interferon-induced proteins with tetratricopeptide repeats (IFIT) family and DnaJ heat shock protein family member C6 (DNAJC6).
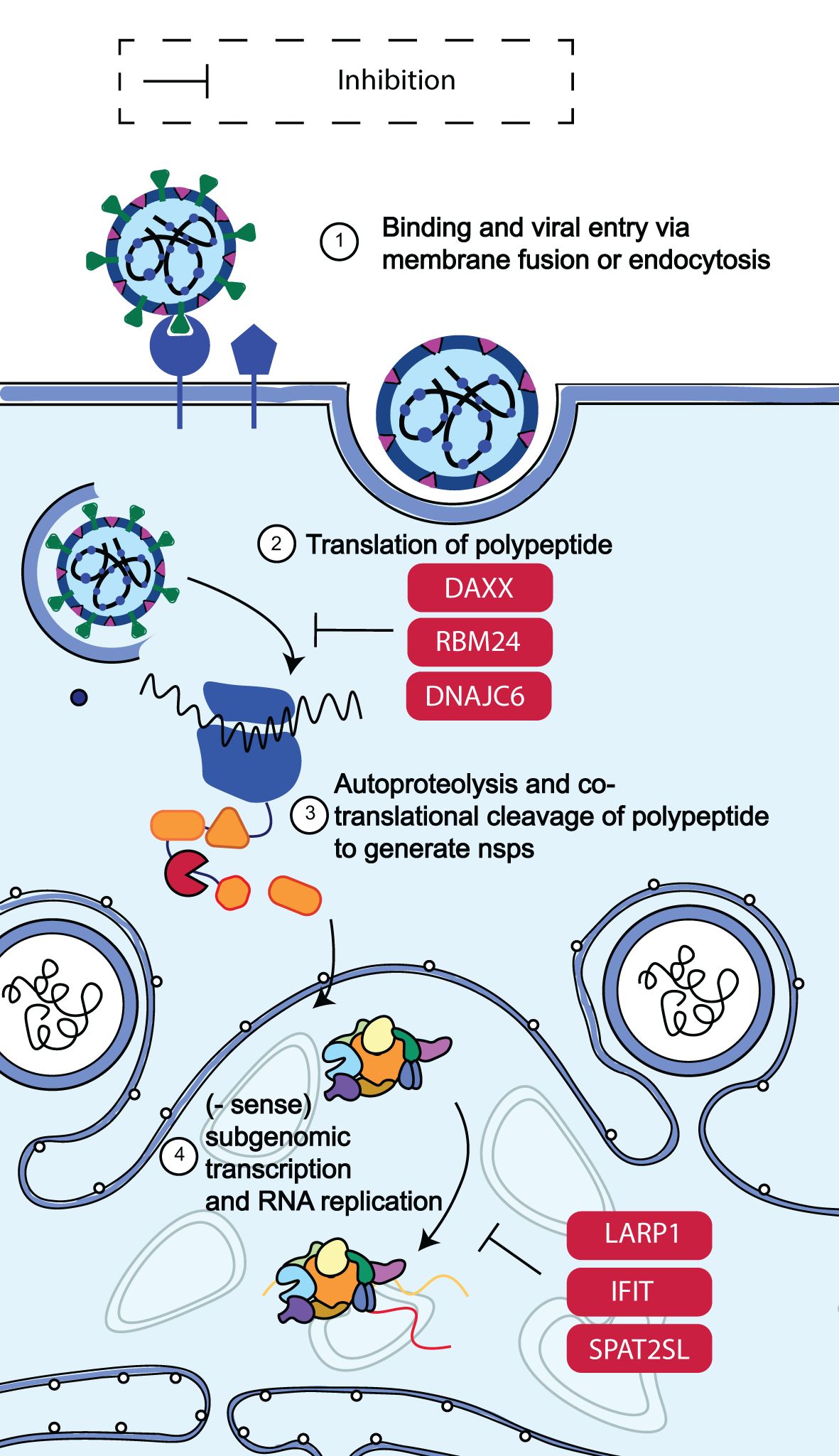
Figure 5. Schematic diagram of SARS-CoV-2 RNA translation and replication restriction. Following the release of SARS-CoV-2 into the cytosol, transcription and translation of the replicase gene begin, leading to the production of nsps from the viral genomic RNA. These nsps control the construction of the replicase-transcriptase complex, which facilitates viral RNA replication and protein synthesis. It was discovered that DAXX, RBM24, DNAJC6, SPAT2SL, LARP1, the IFIT family of proteins, and LARP1 all reduced SARS-CoV-2 RNA levels by more than 50% (65). Adapted from “The molecular virology of coronaviruses”, by Hartenian et al. (17), Journal of Biological chemistry, 295(37), p. 12910 (https://doi.org/10.1074/jbc.REV120.013930). CC BY-NC.
4.1 DAXX
It has been discovered that DAXX, a scaffold protein found in promyelocytic leukaemia (PML) nuclear bodies and known to block DNA virus and retrovirus replication, is a strong inhibitor of SARS-CoV-2 and SARS-CoV replication in human cells (67). SARS-CoV-2 replication can be inhibited by basal expression of DAXX, and infection can be further limited by DAXX overexpression. An early, post-entry stage of the SARS-CoV-2 life cycle is restricted by DAXX (67). The D/E domain of DAXX, which is also required for its protein-folding action, is what allows it to bind SARS-CoV-2 and is not reliant on the SUMOylation route. DAXX caused a significant decrease in the amounts of viral transcripts at 8 hours after infection and in the levels of S protein at 16 hours after infection, despite having no effect on S-mediated viral entry (67). This implies that DAXX inhibits a replication cycle post-entry stage, such as viral transcription (67).
The SARS-CoV-2 infection causes DAXX to relocalise to cytoplasmic locations and encourages its breakdown. The proteasome and viral papain-like protease (PLpro) mediate this process mechanistically (67). The abovementioned findings show that SARS-CoV-2 has evolved a defence mechanism against DAXX’s inhibition of it.
4.2 RBM24
Research has shown that the RNA binding protein RBM24 interacts with the SARS-CoV-2 RNA genome through its RNA recognition submotifs (RNPs) (68). The results showed that in the 5′- untranslated region (UTR) of SARS-CoV-2, RBM24 identifies and binds to the GUGUG element at stem-loop 4 (SL4) (68). Polyprotein translation and SARS-CoV-2 replication are inhibited by the association between RBM24 and 5′-UTR, which hinders 80S ribosome assembly (68).
Remarkably, one or more G(U/C/A)GUG sequences in the 5′-UTR, which is also targeted by RBM24, are found in other RNA viruses, such as West Nile virus, SARS-CoV, Ebolavirus, MERS-CoV, rhinovirus, Zika virus, hepatitis C virus, Japanese encephalitis virus, yellow fever virus and HIV-1 and also leads to their replication restriction based on the same mode of action as what occurs with SARS-CoV-2 (68, 69).
4.3 SPAT2SL
Stress granules impede translation and trap viral RNAs to stop the spread of viruses (70). In research performed by May et al. (71), it was shown that the ISG SPAT2SL, which was identified to prevent the replication of the SARS-CoV-2 RNA and is involved in the creation of stress granules, was associated with viral N protein (65, 72). The SPAT2SL gene encodes a protein that reduces viral RNA levels through direct binding. Following stress stimuli, SPAT2SL is recruited into cytoplasmic stress granules, where viral RNA can be isolated. These findings indicate that the interaction between N protein and SPAT2SL may be crucial for the mechanism through which SARS-CoV-2 is thought to counteract stress granules to avoid immunological responses (73).
Eight gene ontology (GO) keywords were discovered to be highly enriched in the SPATS2L interactome (p-values ranged from 0.01 to 0.05). The majority of these GO keywords have to do with various facets of ribosomal and chromosomal biology (for example, GO:0051276, chromosome structure, p-value = 0.019; GO:0042273, ribosomal biogenesis, p-value = 0.021) (74), indicating that SPAT2SL plays a crucial during restriction of viral RNA translation and replication.
4.4 LARP1
SARS-CoV-2 replication in cells was suppressed by phosphoinositide 3 kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) inhibition (75). Translation of the truncated osteopontin (TOP) mRNA is limited by the La-related protein 1 (LARP1), a new target of mTORC1 (76). As a result, LARP1 could have a significant adverse effect on SARS-CoV-2 replication. The SARS-CoV-2 5’-leader sequence has a terminal oligopyrimidine-like (TOP-like) sequence motif, and the enhanced UV-crosslinking and immunoprecipitation (eCLIP) experiment on host RNAs demonstrated that LARP1 linked to this pattern, demonstrating a direct interaction of LARP1 with subgenomic mRNAs (77).
Compared to wild-type cells, LARP1 deletion cells produced infectious virus at levels that were about five times higher, whereas LARP1 overexpression reduced the generation of infectious virus and intracellular viral RNA (77).
4.5 IFIT FAMILY
IFIT proteins, which are generated following IFN-I or IRF3-dependent signalling, assist in the defence against certain viruses by interacting with components of the eukaryotic initiation factor 3 (eIF3) translation initiation complex and inhibiting protein translation (78). Five members of the IFIT family (IFIT1, IFIT1B, IFIT2, IFIT3, and IFIT5) work to stop the reproduction of active viral RNA by identifying and sequestering single-stranded RNA that is 5’-ppp or 2’O-unmethylated (79). Three members of this family, IFIT1, IFIT3, and IFIT5, were shown to suppress SARS-CoV-2 replication in a research investigation, indicating that this family is crucial for the restriction of SARS-CoV-2 (65).
4.6 DNAJC6
ISG DNAJC6, a member of the HSP40 family, was shown to have an impact on the SARS-CoV-2 replicative stage (Figure 5) HSP40 family members are known to be essential for protein transport, folding, and structural disassembly. They have been found to bind the 3’-UTR of the mouse hepatitis virus (MHV) and SARS-CoV (80, 81). DNAJC6, which binds to the 3′ UTR of SARS-CoV-2 viral RNA, sequesters viral RNA into stress granule ribonucleoprotein (RNP) complexes and inhibits mRNA translation (82).
The abovementioned host restriction factors interact with viral RNA leading to the impediment of their translation and replication. Furthermore, some of these factors have similar modes of action which are the suppression virus translation by inducing the formation of stress granules and decreasing viral RNA levels in infected cells. The viral RNA translation and replication host restriction factors and their proposed mechanisms are summarised in Table 2.
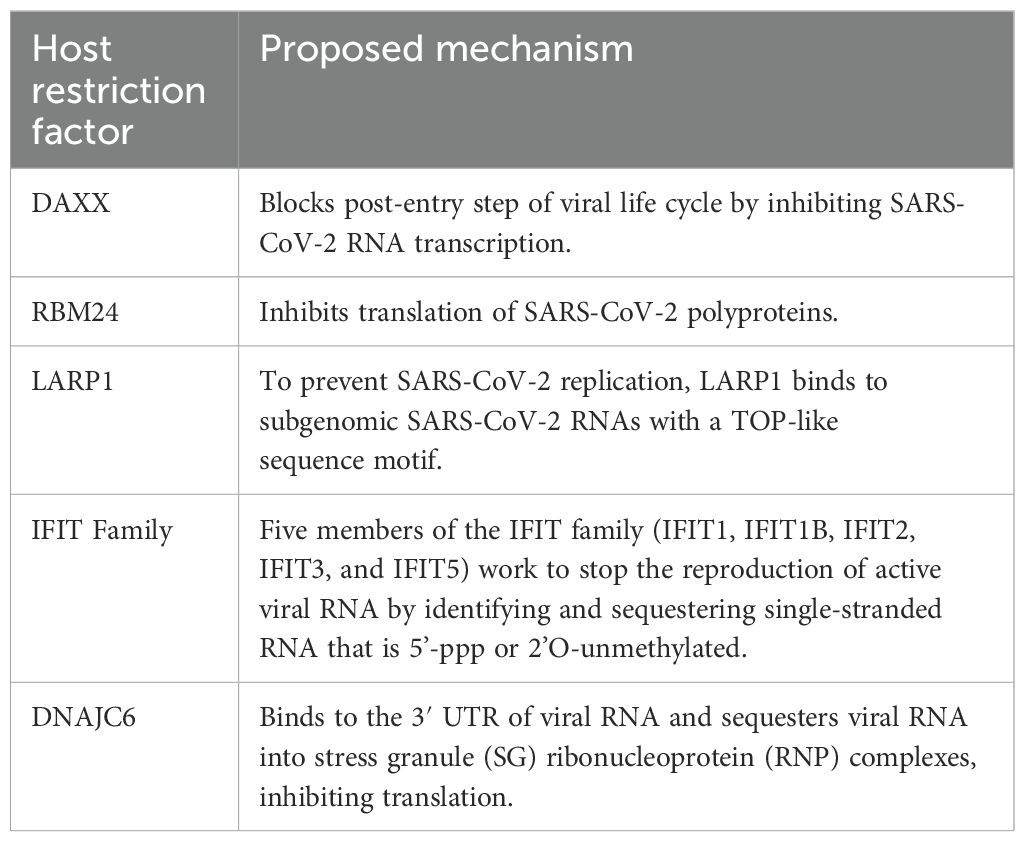
Table 2. Viral RNA translation and replication host restriction factors.
5 Assembly and budding restriction
Two subgenomic mRNAs in the ER membrane also create the structural proteins S, E, M, and N. Following their insertion into the ER, S, E, and M are trafficked down the secretory pathway to begin viral assembly in the ER-Golgi intermediate compartment (ERGIC). In particular, virions are produced from the ERGIC by the collaboration of M, S, and E with viral genomes that the N protein has encapsulated. Viruses are transported in vesicles through the trans-Golgi network, and they are later released by exocytosis (Figure 6). Notably, it was discovered that the majority of ISGs in the study conducted by Martin-Sancho et al. (65) (16/35, 55%) limited late viral replication phases. These late-stage ISGs were then clustered based on their known impact on (ER)-associated protein degradation (ERAD) and vesicle trafficking pathways (65).
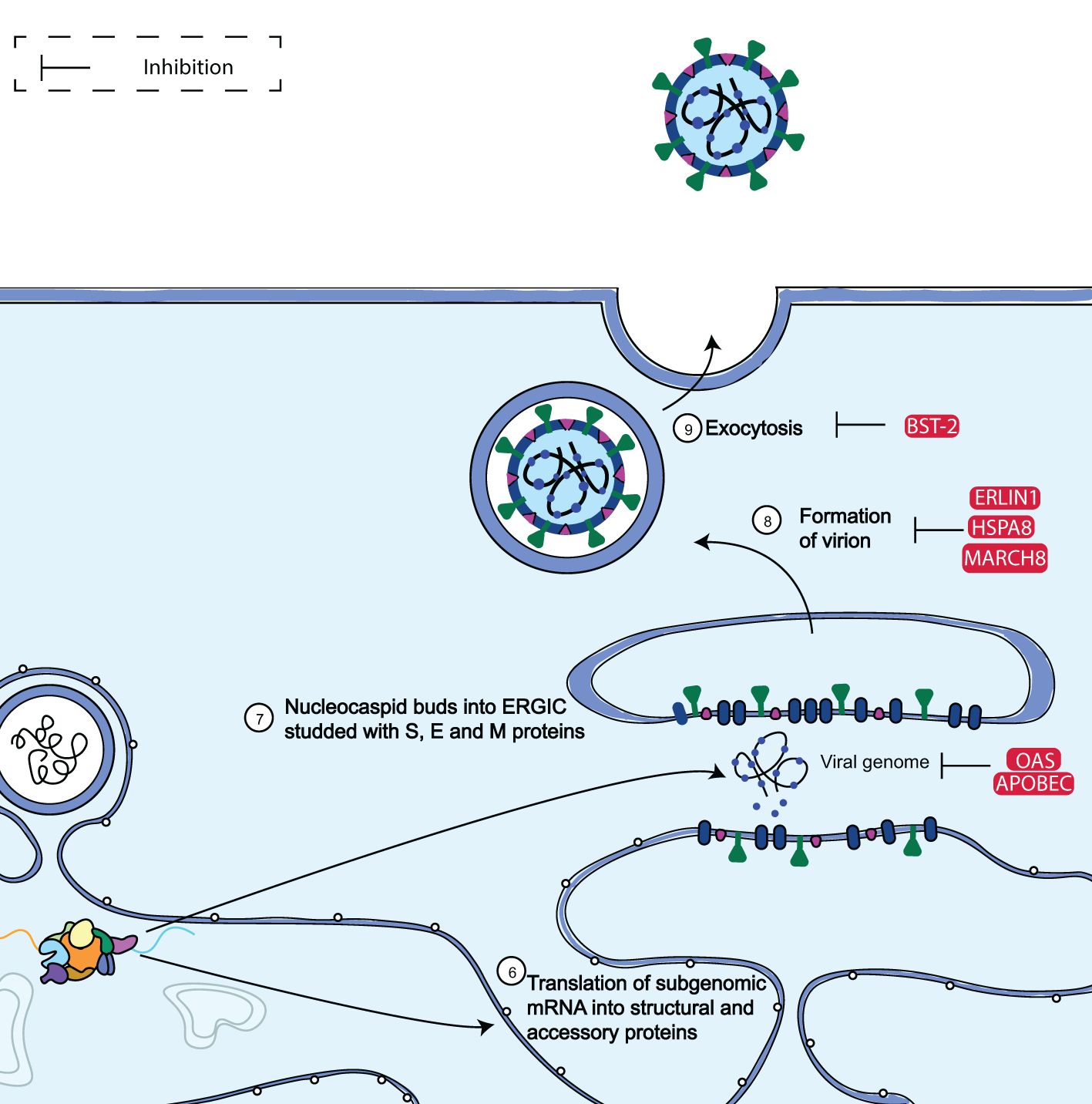
Figure 6. Diagram showing the assembly and budding limitation of SARS-CoV-2. SARS-CoV-2 subgenomic mRNAs are translated into accessory proteins and structural proteins S, M, E, and N at the ER membrane. The bulk of the ISGs examined by Martin-Sancho et al. (65) such as MARCH8, OAS, ERLIN1, and BST-2, were discovered to inhibit the late phases of viral replication. Adapted from “The molecular virology of coronaviruses”, by Hartenian et al. (17), Journal of Biological chemistry, 295(37), p. 12910 (https://doi.org/10.1074/jbc.REV120.013930). CC BY-NC.
The human antiviral factors involved in the restriction of SARS-CoV-2 assembly and budding include apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) family of proteins, oligoadenylate synthetase 1 (OAS1), membrane-associated RING-CH 8 protein (MARCH8) heat shock 70 kDa protein 8 (HSPA8), endoplasmic reticulum raft-associated protein 1 (ERLIN-1) and bone stromal antigen 2 (BST-2).
5.1 APOBEC
The APOBEC family comprises restriction factors that are active against a wide variety of viruses, including as herpesviruses, retroviruses, hepatitis C virus (HCV), and foot and mouth disease virus. (83, 84). The three main members of this family—APOBEC1, APOBEC3A, and APOBEC3G—are capable of single-stranded RNA deamination (85). Viral replication is aberrated by executing fatal editing through the action of cytidine deaminase in a setting that is reliant on both sequence and structure (86). Exoribonuclease confers a certain degree of “immunity” to CoVs against APOBEC editing-induced mutagenesis. Nonetheless, APOBEC-mediated C-to-U transition signals were found in global SARS-CoV-2 consensus sequences that were deposited in the Global Initiative on Sharing All Influenza Data (GISAID). Up to 46% of nucleotide alterations in SARS-CoV-2 are caused by this mutation, which is the most common mutation (87–89). The asymmetric abundance in C-to-U transitions in particular dinucleotide contexts (TC, AC, or CC) and the high ratio of nonsynonymous to synonymous mutations support the frequency of this mutation. This indicates non-neutral evolution driven by additional mutational mechanisms beyond random changes observed in genetic drift (84, 90–93).
5.2 OAS1
Members of the OAS family are widely active ISGs crucial for innate antiviral defence against a variety of viruses (94). RNaseL, an OAS1-3 downstream effector, restricts viral spread by destroying cellular and viral RNA when activated. Recently, against SARS-CoV-2, RNaseL activity has been explicitly linked to host protection against several coronaviruses (95). MERS-CoV inhibits the OAS/RNaseL pathway by destroying the 2’-5’ A species produced by OAS proteins, which prevents RNaseL activation (96). Both A549-SunTag ACE2 and A549-STAT1-SunTag ACE2 cells exhibited OAS1 as a SARS-CoV-2 restriction factor in the screening procedures performed by Danziger et al. (97). The tests further proved that OAS1’s effects on SARS-CoV-2 depend on its catalytic activity (97).
According to this finding, SARS-CoV-2 may not directly inhibit the production of 2’-5’A like MERS-CoV and is hence still vulnerable to RNaseL effector activities (95). A rising collection of genetic, epidemiological, and clinical evidence supports OAS1’s significant contribution to host defence against SARS-CoV and SARS-CoV-2. In the SARS-CoV outbreak, OAS1 genetic variations were connected to infection and high levels of morbidity (98). More recently, single nucleotide polymorphisms (SNPs) at OAS loci have been linked to COVID-19 mortality by genome-wide association studies (GWAS) (99).
Clinical investigations have demonstrated that higher plasma OAS1 levels are linked with decreased COVID-19 hospitalization and mortality, and that an OAS1 isoform with Neanderthal ancestry amplifies these benefits (100). OAS1 was discovered as a SARS-CoV-2 antiviral gene by two additional research groups (101, 102). Wickenhagen et al. (102) and Soveg et al. (101) emphasised the role of OAS1 in the pathophysiology of SARS-CoV-2 by describing a connection between a prenylated isoform of OAS1 (p46) and COVID-19 outcomes. Prenylated OAS1 is shown in both investigations to be directed to the endomembranous sites of SARS-CoV-2 replication.
5.3 MARCH8
Numerous investigations have shown that MARCH8 can break down the SARS-CoV-2 S glycoprotein through a variety of methods (103). In addition to recognising the S protein’s cytoplasmic lysine residues, which lead to ubiquitination and lysosomal destruction, MARCH8 has can trap the S protein on the cell surface into an intracellular lysosomal associated membrane protein 1 (LAMP-1) compartment for degradation (104). Furthermore, because MARCH8 and the highly glycosylated form of M protein colocalise at the Golgi complex, MARCH8 degrades the highly glycosylated form of SARS-CoV-2 M protein (105). Here, we discovered that overexpressing MARCH8 decreased SARS-CoV-2 replication and that endogenous MARCH8 deletion restored IRAV-induced N protein degradation. These findings suggest that MARCH8 is in charge of blocking SARS-CoV-2 replication which is mediated by early growth response protein 1 (EGR1) and IFN-regulated antiviral protein (IRAV).
In a lysosome-dependent mechanism, the cargo receptor nuclear dot protein 52 kDa (NDP52) engaged with N protein to stimulate its degradation via the E3 ubiquitin ligase MARCH8. This was facilitated by the promotion of IRAV production by EGR1 (106). K48-linked poly-ubiquitination of N protein at lysine residue 143 was catalysed by MARCH8, and IRAV-mediated degradation of N protein was prevented by endogenous MARCH8 deletion. Furthermore, SARS-CoV-2 replication may be inhibited by MARCH8 overexpression (106).
5.4 HSPA8
It was discovered that an enhanced route implicated in SARS-CoV-2 was trans-Golgi vesicle budding (65). Heat shock protein HSPA8, a protein in this network, has been linked to late-stage viral replication inhibition (65). Vesicle uncoating is reported to be facilitated by HSPA8. HSPA8 is essential for human cellular functions, especially for its control of autophagy (107).
Ghosh et al. (108) used GO enrichment analysis to determine the relevance of the functions that HSPA8 plays in biological processes. In this study, it was discovered that open reading frame 10 (ORF10) and E protein both target HSPA8, with the E protein having an enrichment value of GO:0016071: mRNA metabolic process with FDR- corrected p-value 4.13E-123 and ORF10 targeting HSPA8 with an enrichment value of GO:0016071: mRNA metabolic process and an FDR-corrected p-value of 6.46E-132.
5.5 ERLIN1
It has been shown that Usp25 plays a critical function in regulating cholesterol biosynthesis flux to stimulate TLR3-dependent immune activation and prevent viral infection. Usp25 was once recognised as a deubiquitinating enzyme (DUB) that was triggered by viral infection (109). Its function has since been defined in the initiation of a metabolic inflammatory pathway that plays a crucial role in host defence. It has been demonstrated that intracellular lipids are used by viruses as a resource for replication, assembly, and secretion (110–113). Thus, lowering lipid production during infection helps to reduce the resources that viruses may access.
These studies show that this pathway is controlled by the Usp25-Erlin1/2 complex in order to maintain proper immunological responses to infection. The mevalonate pathway and IFN-I signalling pathway were postulated to be parts of a metabolic inflammatory pathway. Endoplasmic reticulum (ER)-associated protein degradation (ERAD) may be brought on by the build-up of viral proteins during virion formation at the ER-Golgi interface. As a result, it was discovered that the ERAD regulator ERLIN1 significantly reduced the late stages of SARS-CoV-2 replication (65, 114).
5.6 BST-2
As a powerful inhibitor of SARS-CoV-2 replication, BST-2; also known as CD317 or tetherin, was discovered by Wrapp et al. (47). BST-2 is found in the plasma membrane and in endosomes and is transported via the ER and Golgi. It has been demonstrated to prevent the release of various enveloped viruses, including HIV-1, human coronavirus 229E, and SARS-CoV-1, by anchoring their virions to the cell surface or internal membranes. These viruses bud either at the plasma membrane or at the ERGIC (115–118).
It was shown that at 24 and 48 hours after infection, ACE2/TMPRSS2-expressing 293T and Huh7 cells demonstrated SARS-CoV-2 replication restriction by BST-2 (65). Martin-Sancho et al. (65) then carried out loss-of-function research using HeLa cells, which naturally express BST-2 (115, 116, 118). Researchers discovered that over time, cells depleted of BST-2 generated much more infectious particles (65). Since the reduction of BST-2 in Calu-3 cells led to a considerable rise in SARS-CoV-2 titres at 24 and 38 hours after infection, they further confirmed this finding using human lung epithelial cells (65). Overall, these findings provide compelling evidence that BST-2 is involved in limiting SARS-CoV-2 replication.
The viral assembly and budding host restriction factors and their proposed mechanisms are summarised in Table 3. SARS-CoV-2 has developed countermeasures to the antiviral activity of host restriction factors, facilitating its replication. These evasion strategies are discussed next.

Table 3. Viral assembly and budding host restriction factors.
6 Viral innate immune evasion strategies
Inducing an inflammatory response that restricts viral replication is one of the innate immune system’s main functions during a viral infection. In order to get over this host defence, coronaviruses have developed a number of evasion techniques. By lowering IFN levels, SARS-CoV-2 can selectively avoid antiviral innate immune responses; individuals with mild to moderate COVID-19 have low blood levels of type I and III IFNs (119). Moreover, SARS-CoV-2 infection inhibits the generation of type I and III IFNs at the post-transcriptional stage by blocking the release of mRNA from transcription sites and/or inducing the nucleus to degrade transcripts (120).
SARS-CoV-2 also encodes many proteins that disrupt signalling, effector activity, RLR sensing pathways, and IFN induction. For example, the MDA5 caspase activation and recruitment domain (CARD) requires ISG15 conjugation, also known as ISGylation, in order for it to be activated following RNA virus infection. SARS-CoV-2 PLpro de-ISGylates MDA5 to inhibit MDA5 activation (121). SARS-CoV-2 ORF9b, N, and M proteins can also inhibit the production of pro-inflammatory cytokines and IFN-β by interfering with the RIG-I and MDA5 pathways (122–126).
The TLR3 pathway can also be blocked by ORF9b (124). Compared to its SARS-CoV orthologue, ORF3b inhibits the induction of type I IFN more effectively (127). IFN-β expression and ISG activation are inhibited by SARS-CoV-2 ORF6 and ORF8 (123). NSP1 and NSP14 from SARS-CoV-2, as well as perhaps other viral proteins, can obstruct translation and stop the production of IFN signalling pathway components (128–130). It appears that the SARS-CoV-2 N protein inhibits the aggregation of viral RNA with MAVS to avoid the stimulation of the IFN pathway (131).
Encoding antagonists of certain ISGs is another viral tactic; this has been shown for BST-2, which otherwise prevents the release of formed virus particles, such as HIV-1 (115, 116). Known viral BST-2 antagonists include the Ebola virus glycoprotein (132), KSHV K5 (133, 134), and the lentiviral accessory proteins Vpu and Nef (135–138). There is growing evidence that ORF7a is a BST-2 antagonist for SARS-CoV and SARS CoV-2, capable of directly binding to BST-2 and changing its glycosylation pattern (65, 118).
According to a similar research, the SARS-CoV S protein functions as a BST-2 antagonist (139). This finding was validated, and it was extended to the S protein of SARS-CoV-2 (140). While the BST-2 cell surface levels in several cell lines were actively reduced by the SARS CoV and SARS-CoV-2 S proteins, ORF7a showed little to no such activity, which is consistent with the two viral proteins’ complementary but distinct ways of BST-2 antagonistic interactions (140). Furthermore, albeit to varying degrees, S and ORF7a both altered overall BST-2 levels. SARS-CoV-2 S and ORF7a of the virus only slightly decreased total cellular BST-2, while SARS-CoV-2 S (which was also intrinsic to SARS-CoV-2 ORF7a) potently decreased total BST-2 protein levels (140).
More understanding of SARS-CoV-2 immune evasion tactics and the PRRs, IFN, and cytokine-production pathways that might oppose them may lead to the identification of additional mechanistic targets for treatment development.
7 Host-directed therapies
Given the rise in drug resistance among pathogens, there is an urgent need to refocus attention on host-directed therapies for the effective treatment of infectious illnesses. This becomes particularly significant when it comes to viral infections, because the pathogen encodes few genes and is mainly dependent on the host for persistence and proliferation. As a result, future research on antiviral medications should focus more on finding molecules of host pathways that are regularly hacked by several viruses. These host cellular proteins are an attractive target for the broad synthesis of anticipatory antivirals.
Many disease outbreaks have impacted the world over the last few decades, including SARS-CoV (2003), H1N1 (2009), MERS-CoV (2012), Ebola (2014), and SARS-CoV-2 (2019). Improving our understanding of the processes behind host-pathogen interactions and infectious disease states is essential if we are to prevent imminent damage. With these findings in hand, proactive steps to stop such epidemics in the future can be taken. The ability to systematically assess and anticipate the host features that contribute to the disease cycle would be a more effective tool for controlling outbreaks. Finding a subgroup of viruses with the capacity to spread quickly must thus be a key goal for research.
The emergence of COVID-19 has prompted the investigation of several therapeutic options. Over 7,000 clinical studies for COVID-19 have been registered, an astounding number by any standard (141). Vaccine approaches, widely discussed elsewhere (142), and therapeutic approaches might be classified as immunomodulatory or antiviral. FDA approval of remdesivir and EUA for the antivirals molnupiravir, paxlovid (a combination of nirmatrelvir and ritonavir), sotrovimab, and monoclonal antibody cocktails came from clinical trials of these medicines. Furthermore, evidence suggested that CRISPR/Cas was also usable for diagnosis and treatment of SARS-CoV-2 infection. -Next, we discuss host-directed therapies directed against SARS-CoV-2.
7.1 Genetic approaches to harness restriction factors for therapy and prevention
Restriction factors can target conserved viral components or produce an unfavourable cellular environment for replication, hence impeding the development of resistance in viral infections. Furthermore, antiviral effectors may “overpower” viral antagonists or evasion mechanisms at higher expression levels in genetic drift, despite the fact that well-adapted pathogens, like SARS-CoV-2 variants, are typically largely resistant to restriction factors at expression levels naturally achieved in infected individuals (30).
Therefore, it is highly desirable to induce endogenous restriction factors under control without triggering systemic immunological activation. For instance, it was possible to specifically induce the expression of APOBEC3G and 3B in HEK293T and CEM-SS cells, which do not ordinarily exhibit these restriction factors (143), using an engineered Cas9-based transcriptional activation system developed by Konermann et al. (144).
In this method, a single guide RNA (sgRNA) recruits a DNA-cleavage-incompetent Cas9 nuclease, coupled to the powerful VP64 transactivation domain, to certain promoter regions. The two terminal loops on the sgRNA are especially involved in interactions with the MS2 bacteriophage coat protein. This protein is produced as a fusion that comprises transcription activation domains that are derived from heat shock factor 1 and the NF-κB subunit p65. Expression of particular target genes was potently stimulated upon binding of this complex, known as synergistic activation mediator (SAM), to its target site upstream of the transcriptional start site. Significantly, proviral APOBEC signature alterations were caused in target cells by the activation of APOBEC3G and APOBEC3B (143), providing proof of concept evidence that increased expression of restriction factors have suppressive effects on viral replication in vitro.
Although the removal of viral dependence factors is an intriguing strategy, it is only feasible if these cellular proteins do not perform important physiological roles. Using restriction factor modification to make them resistant to viral antagonists or active against SARS-CoV-2 offers another exciting possibility for the use of genome editing techniques in clinical development. For instance, N protein alterations make SARS-CoV-2 resistant to human TRIM5α’s premature uncoating, but they offer no defence against other species’ TRIM5α orthologues (145). Therefore, methods to prevent SARS-CoV-2 replication might include modifying endogenous human TRIM5α or expressing heterologous TRIM5α. Feline immunodeficiency virus (FIV), a lentivirus that is vulnerable to species-specific restriction factors similar to SARS-CoV-2, finds a natural host in cats, providing proof of concept (146). The introduction of TRIMCyp into the cat germline completely protected its lymphocytes from FIV infection (146). Preventing viral antagonists from inactivating restriction factors would be another tactic. Indeed, it has been demonstrated that human APOBEC3G is spared from destruction by the HIV-1 auxiliary protein Vif due to a single amino acid alteration (D128K) (147). Therefore, human restriction factors that are unlikely to have negative consequences might entirely restore their action against human diseases like SARS-CoV-2 by changing just a few, or even a single, amino acid.
7.1.1 Potential challenges for genomics-based antiviral therapy
There have been several reported effective uses of CRISPR/Cas9 technology thus far for anti-viral modification of significant viral illnesses. However, there are many obstacles to overcome before it can be implemented as human antiviral therapy. Prioritising the reduction of any off-target action is necessary to guarantee safety in an in vivo application. A disastrous consequence for a host cell might arise from the incorrect digestion of a crucial host gene that bears a strong resemblance to the 20 bp seeding plus protospacer adjacent motif (PAM) sequence of the CRISPR/Cas9 target site. A different version of Cas9 called Cas9nickase, which has a reduced off-target property due to its induction of a single strand DNA cleavage instead of a double strand one, is highly recommended for human use in order to tightly control this potential off-target activity of the CRISPR/Cas9 system (148, 149). For the clinical implementation of a CRISPR/Cas9-based antiviral approach, three CRISPR/Cas systems with high gene-editing efficiency and little off-target cleavage should be a far safer option (150–152).
The second a major obstacle to the successful clinical translation of this new technology is the delivery of CRISPR/Cas9 components into each virus-infected cell. This is because residual virus-infected cells that have not been corrected may act as a new viral reservoir, dispersing a new virion to previously corrected normal cells.
Third, another issue that must be resolved before the CRISPR/Cas9 system is implemented in clinical settings is the emergence of viral resistance to it via the creation of a viral escape mutant. It is imperative to carefully choose the most conserved and important viral genome regions while designing gRNA in order to prevent this problem. The most exacting and ideal gRNA selection is necessary for the effective CRISPR/Cas9-mediated control of a viral infection brought on by a population of viruses with a wide range of sequence variations (quasi-species). Furthermore, it has been demonstrated that a number of single gRNA-based monoplex strategies become ineffective against viruses as a result of the selection of a mutant virus strain with a changed target site that the CRISPR/Cas9 system can no longer cleave (153–157).
Fourth, a more thorough validation of the safety of the CRISPR/Cas9-based host-targeting antiviral strategy is required. One common example of an indirect use of the CRISPR/Cas9 system is the suggestion of eliminating host components necessary for a certain stage of a viral life cycle. The host-targeting technique aided by CRISPR/Cas9 appears to benefit from far lower viral resistance as compared to directly altering a virus’s DNA. It is not simple to choose a host dependence factor, though, as they are both necessary for a virus and optional for a host. To forecast the potential adverse effects of this antiviral medication, a thorough investigation of the human immunological response to the CRISPR/Cas system must also be carried out. Practically speaking, the majority of infected individuals should benefit from this CRISPR/Cas9-based antiviral medication if its cost is kept within a reasonable range (158).
8 Conclusion and future perspectives
The fast global appearance and dissemination of SARS-CoV-2 over the past few years has significantly altered virus research. This dynamic has seen the monthly production of more than 10,000 new SARS-CoV-2 papers (159). In order to provide a thorough overview of the host immune response to SARS-CoV-2 at this time, this review includes some of the most pertinent and cited immune response information. This collection of data reflects the context and current level of knowledge in a dynamic and constantly changing field. The development of targeted therapies, such as antivirals and immunomodulatory medicines, which can lessen the severity of infection and restrict viral propagation, can benefit from an understanding of the interactions between the virus and the host immune system. Additionally, by assisting in the identification of possible treatment targets and the development of efficient preventive measures, this knowledge can aid with pandemic preparedness.
As new data are provided globally, the subjects addressed here need to be continually re-evaluated. Through such contemplation, pharmacological target selection may be improved, and the molecular underpinnings of disease can be clarified and understood. Understanding the molecular mechanism by which SARS-CoV-2 inhibits IFN production and signalling for drug targets and pathogenesis can point to additional potential treatments, such as IFN mimicry, small molecules, solubilizable ACE2 in place of monoclonal antibodies (160), prophylactic RIG-I agonists (161), or nanobody-fusions targeting particular ORFs. Clinical trials might further investigate a variety of already available and authorised IFN receptor agonists (ropeginterferon alfa-2b, peginterferon alfa-2b, etc.). The management of COVID-19, SARS-CoV-2, and upcoming pandemics may be done in the most efficient way thanks to these points of constant assessment.
An interesting, if complicated, field of study is the continuous molecular arms race between viruses and their hosts. It is astounding that certain restriction factors may remain to suppress modern viral infections even after existing for many millions of years (67, 97, 162). Since restriction factors are generally widely active and viral infections often cannot simply acquire resistance through point mutations, they typically have certain benefits over other antiviral medicines. However, certain well adapted viral infections have developed specialised antagonists and are therefore little impacted by the constraints present in their native hosts. However, if restriction factors are resistant to viral antagonists or expressed at abnormally high levels, then even well-adapted viruses are often successfully suppressed. Therefore, effective restriction factor induction or genetic alteration constitute viable approaches to improve viral replication control. According to recent research, millions of years of coevolution between the virus and the host may have fuelled the development of cytokines that may be utilised to raise certain subsets of restriction factors to levels high enough to prevent viral reproduction without inducing deleterious inflammation (30). We however still don’t fully understand how the many inducers and effectors of this first-line antiviral defence interact with one another. Infectious viral infections continue to grow and pose a serious threat to public health despite massive scientific efforts and notable advancements. As a result, a deeper comprehension of the framework including effectors and modulators of the antiviral immune response is imperative and might result in the generation of novel therapeutic or preventative strategies.
Author contributions
AM: Conceptualization, Writing – original draft, Writing – review & editing. SB: Supervision, Writing – review & editing. MF: Supervision, Writing – review & editing. SM: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by Mintek Science Vote, Project number ADR-22305, ADR-22401, ADR-22404 and ADR-22501.
Acknowledgments
We would like to acknowledge Mintek and the University of South Africa for the provision of resources during the course of writing. All the figures were created and edited with Adobe Illustrator CC.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. (2018) 34:4121–3. doi: 10.1093/bioinformatics/bty407
2. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. (2020) 26:450–2. doi: 10.1038/s41591-020-0820-9
3. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
4. Rambaut A, Lipkin W, Holmes E, Kristian G. The proximal origin of SARS-CoV-2. Nature. (2020) 26:450–2. doi: 10.1038/s41591-020-0820-9
5. Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9.
6. Maison DP, Ching LL, Cleveland SB, Tseng AC, Nakano E, Shikuma CM, et al. Dynamic SARS-CoV-2 emergence algorithm for rationally-designed logical next-generation vaccines. Commun Biol. (2022) 5:1081. doi: 10.1038/s42003-022-04030-3
7. Worobey M, Levy JI, Malpica Serrano L, Crits-Christoph A, Pekar JE, Goldstein SA, et al. The Huanan Seafood Wholesale Market in Wuhan was the early epicenter of the COVID-19 pandemic. Science. (2022) 377:951–9. doi: 10.1126/science.abp8715
8. WHO. WHO coronavirus (COVID-19) dashboard (2023). Available online at: https://covid19.who.int/ (Accessed July 1, 2024).
9. Baggen J, Vanstreels E, Jansen S, Daelemans D. Cellular host factors for SARS-CoV-2 infection. Nat Microbiol. (2021) 6:1219–32. doi: 10.1038/s41564-021-00958-0
10. Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
11. Kanneganti T-D. Intracellular innate immune receptors: Life inside the cell. Immunol Rev. (2020) 297:5. doi: 10.1111/imr.12912
12. Primorac D, Vrdoljak K, Brlek P, Pavelić E, Molnar V, Matišić V, et al. Adaptive immune responses and immunity to SARS-CoV-2. Front Immunol. (2022) 13:848582. doi: 10.3389/fimmu.2022.848582
13. FDA. Coronavirus disease 2019 (COVID-19) EUA information. Silver Springs, Maryland: Emergency Use Authorization (2024). Available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorizationcovid19euas.
14. Chemudupati M, Kenney AD, Bonifati S, Zani A, McMichael TM, Wu L, et al. From APOBEC to ZAP: Diverse mechanisms used by cellular restriction factors to inhibit virus infections. Biochim Et Biophys Acta (BBA)-Molecular Cell Res. (2019) 1866:382–94. doi: 10.1016/j.bbamcr.2018.09.012
15. Liang Y, Wang M-L, Chien C-S, Yarmishyn AA, Yang Y-P, Lai W-Y, et al. Highlight of immune pathogenic response and hematopathologic effect in SARS-CoV, MERS-CoV, and SARS-Cov-2 infection. Front Immunol. (2020) 11:1022. doi: 10.3389/fimmu.2020.01022
16. Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. (2020) 181:894–904. e899. doi: 10.1016/j.cell.2020.03.045
17. Hartenian E, Nandakumar D, Lari A, Ly M, Tucker JM, Glaunsinger BA. The molecular virology of coronaviruses. J Biol Chem. (2020) 295:12910–34. doi: 10.1074/jbc.rev120.013930
18. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. (2020) 5:562–9. doi: 10.1038/s41564-020-0688-y
19. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci. (2020) 117:11727–34. doi: 10.1073/pnas.2003138117
20. Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. (2020) 11:1620. doi: 10.1038/s41467-020-15562-9
21. Lim YX, Ng YL, Tam JP, Liu DX. Human coronaviruses: a review of virus–host interactions. Diseases. (2016) 4:26. doi: 10.3390/diseases4030026
22. Fung TS, Liu DX. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol. (2019) 73:529–57. doi: 10.1146/annurev-micro-020518-115759
23. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. (2018) 172:650–65.
24. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. (2021) 22:829–38. doi: 10.1038/s41590-021-00937-x
25. Oppenheim JJ. Cytokines: past, present, and future. Int J Hematol. (2001) 74:3–8. doi: 10.1007/BF02982543
26. Taylor MW. Interferons. In: Viruses and man: A history of interactions. Springer International Publishing, Cham (2014). p. 101–19.
27. Schoggins JW. Interferon-stimulated genes: what do they all do? Annu Rev Virol. (2019) 6:567–84. doi: 10.1146/annurev-virology-092818-015756
28. Ley K. The role of selectins in inflammation and disease. Trends Mol Med. (2003) 9:263–8. doi: 10.1016/s1471-4914(03)00071-6
29. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. (2003) 21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126
30. Hotter D, Kirchhoff F. Interferons and beyond: Induction of antiretroviral restriction factors. J leukocyte Biol. (2018) 103:465–77. doi: 10.1002/jlb.3mr0717-307r
31. Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, et al. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol. (2020) 27:763–7. doi: 10.1038/s41594-020-0468-7
32. Yu J, Liu S-L. Emerging role of LY6E in virus–host interactions. Viruses. (2019) 11:1020. doi: 10.3390/v11111020
33. Mar KB, Rinkenberger NR, Boys IN, Eitson JL, McDougal MB, Richardson RB, et al. LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat Commun. (2018) 9:3603. doi: 10.1038/s41467-018-06000-y
34. Pfaender S, Mar KB, Michailidis E, Kratzel A, Boys IN, V’kovski P, et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat Microbiol. (2020) 5:1330–9. doi: 10.1038/s41564-020-0769-y
35. Zhao X, Zheng S, Chen D, Zheng M, Li X, Li G, et al. LY6E restricts entry of human coronaviruses, including currently pandemic SARS-CoV-2. J Virol. (2020) 94. doi: 10.1128/jvi.00562-00520
36. Lund EG, Kerr TA, Sakai J, Li W-P, Russell DW. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem. (1998) 273:34316–27. doi: 10.1074/jbc.273.51.34316
37. Liu S-Y, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity. (2013) 38:92–105. doi: 10.1016/j.immuni.2012.11.005
38. Li C, Deng Y-Q, Wang S, Ma F, Aliyari R, Huang X-Y, et al. 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity. (2017) 46:446–56. doi: 10.1016/j.immuni.2017.02.012
39. Wang S, Li W, Hui H, Tiwari SK, Zhang Q, Croker BA, et al. Cholesterol 25-Hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J. (2020) 39:e106057. doi: 10.15252/embj.2020106057
40. Johnson KA, Endapally S, Vazquez DC, Infante RE, Radhakrishnan A. Ostreolysin A and anthrolysin O use different mechanisms to control movement of cholesterol from the plasma membrane to the endoplasmic reticulum. J Biol Chem. (2019) 294:17289–300. doi: 10.1074/jbc.ra119.010393
41. Abrams ME, Johnson KA, Perelman SS, Zhang L-S, Endapally S, Mar KB, et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat Microbiol. (2020) 5:929–42. doi: 10.1038/s41564-020-0701-5
42. Jones DE, Bevins CL. Paneth cells of the human small intestine express an antimicrobial peptide gene. J Biol Chem. (1992) 267:23216–25. doi: 10.1016/s0021-9258(18)50079-x
43. Ericksen B, Wu Z, Lu W, Lehrer RI. Antibacterial activity and specificity of the six human α-defensins. Antimicrobial Agents Chem. (2005) 49:269–75. doi: 10.1128/aac.49.1.269-275.2005
44. Wang C, Shen M, Gohain N, Tolbert WD, Chen F, Zhang N, et al. Design of a potent antibiotic peptide based on the active region of human defensin 5. J Medicinal Chem. (2015) 58:3083–93. doi: 10.1021/jm501824a
45. Wang C, Wang S, Li D, Wei D-Q, Zhao J, Wang J. Human intestinal defensin 5 inhibits SARS-CoV-2 invasion by cloaking ACE2. Gastroenterology. (2020) 159:1145–1147. e1144. doi: 10.1053/j.gastro.2020.05.015
46. Niv Y. Defensin 5 for prevention of SARS-CoV-2 invasion and Covid-19 disease. Med Hypotheses. (2020) 143:110244. doi: 10.1016/j.mehy.2020.110244
47. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 367:1260–3. doi: 10.1126/science.abb2507
48. Sako D, Chang X-J, Barone KM, Vachino G, White HM, Shaw G, et al. Expression cloning of a functional glycoprotein ligand for P-selectin. Cell. (1993) 75:1179–86. doi: 10.1016/0092-8674(93)90327-m
49. Somers WS, Tang J, Shaw GD, Camphausen RT. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P-and E-selectin bound to SLeX and PSGL-1. Cell. (2000) 103:467–79.
50. Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med. (2009) 15:794–7. doi: 10.1038/nm.1961
51. Liu Y, Fu Y, Wang Q, Li M, Zhou Z, Dabbagh D, et al. Proteomic profiling of HIV-1 infection of human CD4+ T cells identifies PSGL-1 as an HIV restriction factor. Nat Microbiol. (2019) 4:813–25. doi: 10.1038/s41564-019-0372-2
52. He S, Hetrick B, Dabbagh D, Akhrymuk IV, Kehn-Hall K, Freed EO, et al. PSGL-1 blocks SARS-CoV-2 S protein-mediated virus attachment and infection of target cells. bioRxiv. (2020).
53. Fu Y, He S, Waheed AA, Dabbagh D, Zhou Z, Trinité B, et al. PSGL-1 restricts HIV-1 infectivity by blocking virus particle attachment to target cells. Proc Natl Acad Sci. (2020) 117:9537–45. doi: 10.1073/pnas.1916054117
54. Wang Y, Fan Y, Huang Y, Du T, Liu Z, Huang D, et al. TRIM28 regulates SARS-CoV-2 cell entry by targeting ACE2. Cell Signalling. (2021) 85:110064. doi: 10.1016/j.cellsig.2021.110064
55. Raheem Juhi Al-Kaabi N, Khameneh SC, Montazeri M, Mardasi M, Amroabadi JM, Sakhaee F, et al. On the relationship between tripartite motif-containing 22 single-nucleotide polymorphisms and COVID-19 infection severity. Hum Genomics. (2022) 16:33. doi: 10.1186/s40246-022-00394-z
56. Xu D, Jiang W, Wu L, Gaudet RG, Park E-S, Su M, et al. PLSCR1 is a cell-autonomous defence factor against SARS-CoV-2 infection. Nature. (2023) 619:819–27. doi: 10.1038/s41586-023-06322-y
57. Kousathanas A, Pairo-Castineira E, Rawlik K, Stuckey A, Odhams CA, Walker S, et al. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature. (2022) 607:97–103.
58. Pairo-Castineira E, Rawlik K, Bretherick AD, Qi T, Wu Y, Nassiri I, et al. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature. (2023) 617:764–8.
59. Timilsina U, Umthong S, Ivey EB, Waxman B, Stavrou S. SARS-CoV-2 ORF7a potently inhibits the antiviral effect of the host factor SERINC5. Nat Commun. (2022) 13:2935. doi: 10.1038/s41467-022-30609-9
60. Schaefer I-M, Padera RF, Solomon IH, Kanjilal S, Hammer MM, Hornick JL, et al. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Modern Pathol. (2020) 33:2104–14. doi: 10.1038/s41379-020-0595-z
61. Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature. (2015) 526:212–7. doi: 10.1038/nature15399
62. Usami Y, Wu Y, Göttlinger HG. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature. (2015) 526:218–23. doi: 10.1038/nature15400
63. Sood C, Marin M, Chande A, Pizzato M, Melikyan GB. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J Biol Chem. (2017) 292:6014–26. doi: 10.1074/jbc.m117.777714
64. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–280. e278. doi: 10.1016/j.cell.2020.02.052
65. Martin-Sancho L, Lewinski MK, Pache L, Stoneham CA, Yin X, Becker ME, et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol Cell. (2021) 81:2656–2668. e2658.
66. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses: Methods Protoc. (2015), 1–23.
67. Mac Kain A, Maarifi G, Aicher S-M, Arhel N, Baidaliuk A, Munier S, et al. Identification of DAXX as a restriction factor of SARS-CoV-2 through a CRISPR/Cas9 screen. Nat Commun. (2022) 13:2442. doi: 10.1038/s41467-022-30134-9
68. Yao Y, Sun H, Chen Y, Tian L, Huang D, Liu C, et al. RBM24 inhibits the translation of SARS-CoV-2 polyproteins by targeting the 5′-untranslated region. Antiviral Res. (2023) 209:105478. doi: 10.1016/j.antiviral.2022.105478
69. Cao H, Zhao K, Yao Y, Guo J, Gao X, Yang Q, et al. RNA binding protein 24 regulates the translation and replication of hepatitis C virus. Protein Cell. (2018) 9:930–44. doi: 10.1007/s13238-018-0507-x
70. Zhao J, Feng D, Zhao Y, Huang M, Zhang X, Zhang G. Role of stress granules in suppressing viral replication by the infectious bronchitis virus endoribonuclease. J Virol. (2022) 96:e0068622. doi: 10.1128/jvi.00686-22
71. May DG, Martin-Sancho L, Anschau V, Liu S, Chrisopulos RJ, Scott KL, et al. A BioID-derived proximity interactome for SARS-CoV-2 proteins. Viruses. (2022) 14:611. doi: 10.3390/v14030611
72. Miller CL. Stress granules and virus replication. Future Virol. (2011) 6:1329–38. doi: 10.2217/fvl.11.108
73. Cascarina SM, Ross ED. A proposed role for the SARS-CoV-2 nucleocapsid protein in the formation and regulation of biomolecular condensates. FASEB J. (2020) 34:9832–42. doi: 10.1096/fj.202001351
74. Palau N, Julià A, Ferrándiz C, Puig L, Fonseca E, Fernández E, et al. Genome-wide transcriptional analysis of T cell activation reveals differential gene expression associated with psoriasis. BMC Genomics. (2013) 14:825. doi: 10.1186/1471-2164-14-825
75. Klann K, Bojkova D, Tascher G, Ciesek S, Münch C, Cinatl J. Growth factor receptor signaling inhibition prevents SARS-CoV-2 replication. Mol Cell. (2020) 80:164–174. e164. doi: 10.1016/j.molcel.2020.08.006
76. Fonseca BD, Zakaria C, Jia J-J, Graber TE, Svitkin Y, Tahmasebi S, et al. La-related protein 1 (LARP1) represses terminal oligopyrimidine (TOP) mRNA translation downstream of mTOR complex 1 (mTORC1). J Biol Chem. (2015) 290:15996–6020. doi: 10.1074/jbc.m114.621730
77. Schmidt N, Lareau CA, Keshishian H, Ganskih S, Schneider C, Hennig T, et al. The SARS-CoV-2 RNA–protein interactome in infected human cells. Nat Microbiol. (2021) 6:339–53. doi: 10.1038/s41564-020-00846-z
78. Shi G, Kenney AD, Kudryashova E, Zani A, Zhang L, Lai KK, et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. (2021) 40:e106501. doi: 10.15252/embj.2020106501
79. Metz P, Reuter A, Bender S, Bartenschlager R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J Hepatol. (2013) 59:1331–41. doi: 10.1016/j.jhep.2013.07.033
80. Nanda S, Johnson R, Liu Q, Leibowitz J. Mitochondrial HSP70, HSP40, and HSP60 bind to the 3′ untranslated region of the Murine hepatitis virus genome. Arch Virol. (2003) 149:93–111. doi: 10.1007/s00705-003-0196-4
81. Rosenzweig R, Nillegoda NB, Mayer MP, Bukau B. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. (2019) 20:665–80. doi: 10.1038/s41580-019-0133-3
82. Daly JL, Simonetti B, Klein K, Chen K-E, Williamson MK, Antón-Plágaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. (2020) 370:861–5.
83. Cheng A, Moraes S, Shaban N, Fanunza E, Bierle C, Southern P, et al. APOBECs and herpesviruses. Viruses. (2021) 13:390.
84. Simmonds P, Ansari MA. Extensive C-> U transition biases in the genomes of a wide range of mammalian RNA viruses; potential associations with transcriptional mutations, damage-or host-mediated editing of viral RNA. PloS Pathog. (2021) 17:e1009596. doi: 10.1371/journal.ppat.1009596
85. Nakata Y, Ode H, Kubota M, Kasahara T, Matsuoka K, Sugimoto A, et al. Cellular APOBEC3A deaminase drives mutations in the SARS-CoV-2 genome. Nucleic Acids Res. (2023) 51:783–95. doi: 10.1093/nar/gkac1238
86. McDaniel YZ, Wang D, Love RP, Adolph MB, Mohammadzadeh N, Chelico L, et al. Deamination hotspots among APOBEC3 family members are defined by both target site sequence context and ssDNA secondary structure. Nucleic Acids Res. (2020) 48:1353–71. doi: 10.1093/nar/gkz1164
87. Smith EC, Blanc H, Vignuzzi M, Denison MR. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PloS Pathog. (2013) 9:e1003565. doi: 10.1371/journal.ppat.1003565
88. Azgari C, Kilinc Z, Turhan B, Circi D, Adebali O. The mutation profile of SARS-CoV-2 is primarily shaped by the host antiviral defense. Viruses. (2021) 13:394. doi: 10.3390/v13030394
89. Rice AM, Castillo Morales A, Ho AT, Mordstein C, Mühlhausen S, Watson S, et al. Evidence for strong mutation bias toward, and selection against, U content in SARS-CoV-2: implications for vaccine design. Mol Biol Evol. (2021) 38:67–83. doi: 10.1093/molbev/msaa188
90. Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med. (2013) 5:1–13. doi: 10.1186/gm508
91. van Dorp L, Richard D, Tan CC, Shaw LP, Acman M, Balloux F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat Commun. (2020) 11:5986. doi: 10.1038/s41467-020-19818-2
92. Ratcliff J, Simmonds P. Potential APOBEC-mediated RNA editing of the genomes of SARS-CoV-2 and other coronaviruses and its impact on their longer term evolution. Virology. (2021) 556:62–72. doi: 10.1016/j.virol.2020.12.018
93. Yi K, Kim SY, Bleazard T, Kim T, Youk J, Ju YS. Mutational spectrum of SARS-CoV-2 during the global pandemic. Exp Mol Med. (2021) 53:1229–37. doi: 10.1038/s12276-021-00658-z
94. Hornung V, Hartmann R, Ablasser A, Hopfner K-P. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol. (2014) 14:521–8. doi: 10.1038/nri3719
95. Li Y, Renner DM, Comar CE, Whelan JN, Reyes HM, Cardenas-Diaz FL, et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc Natl Acad Sci. (2021) 118:e2022643118. doi: 10.1073/pnas.2022643118
96. Thornbrough JM, Jha BK, Yount B, Goldstein SA, Li Y, Elliott R, et al. Middle East respiratory syndrome coronavirus NS4b protein inhibits host RNase L activation. MBio. (2016) 7. doi: 10.1128/mbio.00258-00216
97. Danziger O, Patel RS, DeGrace EJ, Rosen MR, Rosenberg BR. Inducible CRISPR activation screen for interferon-stimulated genes identifies OAS1 as a SARS-CoV-2 restriction factor. PloS Pathog. (2022) 18:e1010464. doi: 10.1371/journal.ppat.1010464
98. Hamano E, Hijikata M, Itoyama S, Quy T, Phi NC, Long HT, et al. Polymorphisms of interferon-inducible genes OAS-1 and MxA associated with SARS in the Vietnamese population. Biochem Biophys Res Commun. (2005) 329:1234–9. doi: 10.1016/j.bbrc.2005.02.101
99. Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell Host Microbe. (2020) 28:455–464. e452. doi: 10.1016/j.chom.2020.07.005
100. Hoagland DA, Møller R, Uhl SA, Oishi K, Frere J, Golynker I, et al. Leveraging the antiviral type I interferon system as a first line of defense against SARS-CoV-2 pathogenicity. Immunity. (2021) 54:557–570. e555. doi: 10.1016/j.immuni.2021.01.017
101. Soveg FW, Schwerk J, Gokhale NS, Cerosaletti K, Smith JR, Pairo-Castineira E, et al. Endomembrane targeting of human OAS1 p46 augments antiviral activity. Elife. (2021) 10:e71047. doi: 10.7554/elife.71047
102. Wickenhagen A, Sugrue E, Lytras S, Kuchi S, Noerenberg M, Turnbull ML, et al. A prenylated dsRNA sensor protects against severe COVID-19. Science. (2021) 374:eabj3624.
103. Yu C, Wang G, Liu Q, Zhai J, Xue M, Li Q, et al. Host antiviral factors hijack furin to block SARS-CoV-2, ebola virus, and HIV-1 glycoproteins cleavage. Emerging Microbes infections. (2023) 12:2164742. doi: 10.1080/22221751.2022.2164742
104. Lun CM, Waheed AA, Majadly A, Powell N, Freed EO. Mechanism of viral glycoprotein targeting by membrane-associated RING-CH proteins. MBio. (2021) 12. doi: 10.1128/mbio.00219-00221
105. Umthong S, Lynch B, Timilsina U, Waxman B, Ivey EB, Stavrou S. Elucidating the antiviral mechanism of different MARCH factors. MBio. (2021) 12. doi: 10.1128/mbio.03264-03220
106. Zhao Y, Sui L, Wu P, Li L, Liu L, Ma B, et al. EGR1 functions as a new host restriction factor for SARS-CoV-2 to inhibit virus replication through the E3 ubiquitin ligase MARCH8. J Virol. (2023) 97:e01028–01023. doi: 10.1128/jvi.01028-23
107. Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. (2013) 9:1937–54.
108. Ghosh N, Saha I, Sharma N. Interactome of human and SARS-CoV-2 proteins to identify human hub proteins associated with comorbidities. Comput Biol Med. (2021) 138:104889. doi: 10.1016/j.compbiomed.2021.104889
109. Jahan AS, Biquand E, Munoz-Moreno R, Le Quang A, Mok CK-P, Wong HH, et al. OTUB1 is a key regulator of RIG-I-dependent immune signaling and is targeted for proteasomal degradation by influenza A NS1. Cell Rep. (2020) 30:1570–1584. e1576. doi: 10.1016/j.celrep.2020.01.015
110. Zhang J, Lan Y, Li MY, Lamers MM, Fusade-Boyer M, Klemm E, et al. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe. (2018) 23:819–831. e815. doi: 10.1016/j.chom.2018.05.005
112. Teo QW, van Leur SW, Sanyal S. Escaping the Lion’s Den: redirecting autophagy for unconventional release and spread of viruses. FEBS J. (2021) 288:3913–27. doi: 10.1111/febs.15590
113. van Leur SW, Heunis T, Munnur D, Sanyal S. Pathogenesis and virulence of flavivirus infections. Virulence. (2021) 12:2814–38. doi: 10.1080/21505594.2021.1996059
114. Pearce MM, Wormer DB, Wilkens S, Wojcikiewicz RJ. An endoplasmic reticulum (ER) membrane complex composed of SPFH1 and SPFH2 mediates the ER-associated degradation of inositol 1, 4, 5-trisphosphate receptors. J Biol Chem. (2009) 284:10433–45.
115. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. (2008) 451:425–30. doi: 10.1038/nature06553
116. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. (2008) 3:245–52. doi: 10.1016/j.chom.2008.03.001
117. Wang S-M, Huang K-J, Wang C-T. BST2/CD317 counteracts human coronavirus 229E productive infection by tethering virions at the cell surface. Virology. (2014) 449:287–96. doi: 10.1016/j.virol.2013.11.030
118. Taylor JK, Coleman CM, Postel S, Sisk JM, Bernbaum JG, Venkataraman T, et al. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J Virol. (2015) 89:11820–33. doi: 10.1128/jvi.02274-15
119. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. (2020) 181:1036–1045. e1039. doi: 10.1016/j.cell.2020.04.026
120. Burke JM, St Clair LA, Perera R, Parker R. SARS-CoV-2 infection triggers widespread host mRNA decay leading to an mRNA export block. Rna. (2021) 27:1318–29. doi: 10.1261/rna.078923.121
121. Liu G, Lee J-H, Parker ZM, Acharya D, Chiang JJ, van Gent M, et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat Microbiol. (2021) 6:467–78. doi: 10.1038/s41564-021-00884-1
122. Chen K, Xiao F, Hu D, Ge W, Tian M, Wang W, et al. SARS-CoV-2 nucleocapsid protein interacts with RIG-I and represses RIG-mediated IFN-β production. Viruses. (2020) 13:47. doi: 10.3390/v13010047
123. Li J-Y, Liao C-H, Wang Q, Tan Y-J, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus research 286. (2020) 198074.
124. Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. J Med Virol. (2021) 93:5376–89. doi: 10.1002/jmv.27050
125. Sui L, Zhao Y, Wang W, Wu P, Wang Z, Yu Y, et al. SARS-CoV-2 membrane protein inhibits type I interferon production through ubiquitin-mediated degradation of TBK1. Front Immunol. (2021) 12:662989. doi: 10.3389/fimmu.2021.662989
126. Wu J, Shi Y, Pan X, Wu S, Hou R, Zhang Y, et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. (2021) 34. doi: 10.1016/j.celrep.2021.108761
127. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. (2020) 32. doi: 10.1016/j.celrep.2020.108185
128. Ho B-C, Yu S-L, Chen JJW, Chang S-Y, Yan B-S, Hong Q-S, et al. Enterovirus-Induced miR-141 Contributes to Shutoff of Host Protein Translation by Targeting the Translation Initiation Factor eIF4E. Cell Host Microbe. (2011) 9:58–69. doi: 10.1016/j.chom.2010.12.001
129. Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. (2020) 369:1249–55. doi: 10.1126/science.abc8665
130. Xia H, Cao Z, Xie X, Zhang X, Chen J, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. (2020) 33:108234. doi: 10.1016/j.celrep.2020.108234
131. Wang S, Dai T, Qin Z, Pan T, Chu F, Lou L, et al. Targeting liquid–liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat Cell Biol. (2021) 23:718–32. doi: 10.1038/s41556-021-00710-0
132. Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci. (2009) 106:2886–91. doi: 10.1073/pnas.0811014106
133. Mansouri M, Viswanathan K, Douglas JL, Hines J, Gustin J, Moses AV, et al. Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J Virol. (2009) 83:9672–81. doi: 10.1128/jvi.00597-09
134. Pardieu C, Vigan R, Wilson SJ, Calvi A, Zang T, Bieniasz P, et al. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PloS Pathog. (2010) 6:e1000843. doi: 10.1371/journal.ppat.1000843
135. Douglas JL, Viswanathan K, McCarroll MN, Gustin JK, Früh K, Moses AV. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a βTrCP-dependent mechanism. J Virol. (2009) 83:7931–47. doi: 10.1128/jvi.00242-09
136. Sauter D, Schindler M, Specht A, Landford WN, Münch J, Kim K-A, et al. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe. (2009) 6:409–21. doi: 10.1016/j.chom.2009.10.004
137. Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, et al. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe. (2009) 6:54–67. doi: 10.1016/j.chom.2009.05.008
138. Mangeat B, Cavagliotti L, Lehmann M, Gers-Huber G, Kaur I, Thomas Y, et al. Influenza virus partially counteracts restriction imposed by tetherin/BST-2. J Biol Chem. (2012) 287:22015–29. doi: 10.1074/jbc.m111.319996
139. Wang SM, Huang KJ, Wang CT. Severe acute respiratory syndrome coronavirus spike protein counteracts BST2-mediated restriction of virus-like particle release. J Med Virol. (2019) 91:1743–50. doi: 10.1002/jmv.25518
140. Hagelauer E, Lotke R, Kmiec D, Hu D, Hohner M, Stopper S, et al. Tetherin restricts SARS-CoV-2 replication despite antagonistic effects of Spike and ORF7a. J Med Virol. (2023) 91:1743–1750.
141. Akinbolade S, Coughlan D, Fairbairn R, McConkey G, Powell H, Ogunbayo D, et al. Combination therapies for COVID-19: An overview of the clinical trials landscape. Br J Clin Pharmacol. (2022) 88:1590–7. doi: 10.1111/bcp.15089
142. WHO. Solidarity” clinical trial for COVID-19 treatments. (2020) Geneva, Switzerland: World health organization.
143. Bogerd HP, Kornepati AV, Marshall JB, Kennedy EM, Cullen BR. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc Natl Acad Sci. (2015) 112:E7249–56. doi: 10.1073/pnas.1516305112
144. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. (2015) 517:583–8. doi: 10.1038/nature14136
145. Sakuma R, Noser JA, Ohmine S, Ikeda Y. Rhesus monkey TRIM5α restricts HIV-1 production through rapid degradation of viral Gag polyproteins. Nat Med. (2007) 13:631–5. doi: 10.1038/nm1562
146. Wongsrikeao P, Saenz D, Rinkoski T, Otoi T, Poeschla E. Antiviral restriction factor transgenesis in the domestic cat. Nat Methods. (2011) 8:853–9. doi: 10.1038/nmeth.1703
147. Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, et al. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci. (2004) 101:5652–7. doi: 10.1073/pnas.0400830101
148. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. (2013) 31:833–8. doi: 10.1038/nbt.2675
149. Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell. (2013) 154:1380–9. doi: 10.1016/j.cell.2013.08.021
150. Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-cas system. Cell. (2015) 163:759–71. doi: 10.1016/j.cell.2015.09.038
151. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. (2016) 529:490–5. doi: 10.1038/nature16526
152. Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. (2016) 351:84–8. doi: 10.1126/science.aad5227
153. Ueda S, Ebina H, Kanemura Y, Misawa N, Koyanagi Y. Anti-HIV-1 potency of the CRISPR/Cas9 system insufficient to fully inhibit viral replication. Microbiol Immunol. (2016) 60:483–96. doi: 10.1111/1348-0421.12395
154. Wang G, Zhao N, Berkhout B, Das AT. CRISPR-cas9 can inhibit HIV-1 replication but NHEJ repair facilitates virus escape. Mol Ther. (2016) 24:522–6. doi: 10.1038/mt.2016.24
155. Yoder KE, Bundschuh R. Host double strand break repair generates HIV-1 strains resistant to CRISPR/cas9. Sci Rep. (2016) 6:29530. doi: 10.1038/srep29530
156. Tang Y-D, Liu J-T, Wang T-Y, Sun M-X, Tian Z-J, Cai X-H. CRISPR/Cas9-mediated multiple single guide RNAs potently abrogate pseudorabies virus replication. Arch Virol. (2017) 162:3881–6. doi: 10.1007/s00705-017-3553-4
157. Hübner A, Petersen B, Keil GM, Niemann H, Mettenleiter TC, Fuchs W. Efficient inhibition of African swine fever virus replication by CRISPR/Cas9 targeting of the viral p30 gene (CP204L). Sci Rep. (2018) 8:1449. doi: 10.1038/s41598-018-19626-1
158. Lee C. CRISPR/Cas9-based antiviral strategy: current status and the potential challenge. Molecules. (2019) 24:1349. doi: 10.3390/molecules24071349
159. Chen Q, Allot A, Lu Z. LitCovid: an open database of COVID-19 literature. Nucleic Acids Res. (2021) 49:D1534–40. doi: 10.1093/nar/gkaa952
160. Krishnamurthy S, Lockey RF, Kolliputi N. Soluble ACE2 as a potential therapy for COVID-19. Am Physiol Soc. (2021).
161. Marx S, Kümmerer BM, Grützner C, Kato H, Schlee M, Renn M, et al. RIG-I-induced innate antiviral immunity protects mice from lethal SARS-CoV-2 infection. Mol Therapy-Nucleic Acids. (2022) 27:1225–34. doi: 10.1016/j.omtn.2022.02.008
Keywords: SARS-CoV-2, host factors, host immune response, COVID-19, host directed therapy
Citation: Marivate A, Bekker S, Fish MQ and Mosebi S (2024) Human cellular restriction factors that target SARS-CoV-2 replication. Front. Virol. 4:1462283. doi: 10.3389/fviro.2024.1462283
Received: 09 July 2024; Accepted: 03 October 2024;
Published: 28 October 2024.
Edited by:
Stefania Varchetta, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, Milan, ItalyReviewed by:
Siyu Huang, The University of Iowa, United StatesLing Ding, University of Pittsburgh Medical Center, United States
Copyright © 2024 Marivate, Bekker, Fish and Mosebi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salerwe Mosebi, bW9zZWJzQHVuaXNhLmFjLnph