 Rachid Tahzima1,2,3,4*
Rachid Tahzima1,2,3,4* Justine Charon5
Justine Charon5 Adrian Diaz3Kris De Jonghe2
Adrian Diaz3Kris De Jonghe2 Sebastien Massart1
Sebastien Massart1 Thierry Michon5
Thierry Michon5 Wim Vranken3,4,6,7
Wim Vranken3,4,6,7- 1Laboratory of Plant Pathology, TERRA, Gembloux Agro-BioTech, University of Liège (ULg), Gembloux, Belgium
- 2Department of Plant Sciences, Flanders Research Institute for Agriculture, Fisheries and Food (ILVO), Ghent, Belgium
- 3Interuniversity Institute of Bioinformatics in Brussels (ULB/VUB), Brussels, Belgium
- 4Artificial Intelligence Lab, Vrije Universiteit Brussel (VUB), Brussels, Belgium
- 5Fruit Biology and Pathology Unit, National Research Institute for Agriculture, Food and Environment (INRAE)/University of Bordeaux, Bordeaux, France
- 6Chemistry Department, Vrije Universiteit Brussel (VUB), Brussels, Belgium
- 7Structural Biology Brussels, Vrije Universiteit Brussel (VUB), Brussels, Belgium
The functions of RNA-dependent RNA polymerases (RdRps) in RNA viruses are demonstrably modulated by native substrates of dynamic and interconvertible conformational ensembles. Many of these are populated by essential flexible or intrinsically disordered regions (IDRs) that lack a stable three-dimensional (3D) structure and that make up nearly 16% of the conserved RdRp domains across Riboviria lineages. Typical structural models of RdRps are conversely generally agnostic of multiple conformations and their fluctuations, whether derived from protein structure predictors or from experimentally resolved structures from crystal states or dynamic conformer sets. In this review, we highlight how biophysics-inspired prediction tools combined with advanced deep learning algorithms, such as AlphaFold2 (AF2), can help efficiently infer the conformational heterogeneity and dynamics of RdRps. We discuss the use of AF2 for protein structure prediction, together with its limitations and impacts on RNA virus protein characterization, and specifically address its low-confidence prediction scores, which largely capture IDRs. Key examples illustrate how biophysical-encoded preferences of generic sequence–ensemble relationships can help estimate the global RdRp structural diversity and RNA virus discovery. The quantitative perception we present also highlights the challenging magnitude of the emergent sequence-to-conformations relationships of proteins and illustrates more robust and accurate annotations of novel or divergent RdRps. Finally, the coarse-grained IDR-based structural depiction of RdRp conformations offers concrete perspectives on an integrative framework to directly generate innovative avenues to better understand viral replication in the early disease stages and the protein–protein affinities through the folding dynamics of these viral proteins. Overall, tapping into the current knowledge of RdRp conformational heterogeneity will serve further RNA virus discovery as similarities in the global RdRp landscape emerge with more clarity.
1 Introduction: structure and function of the hallmark RdRp module in viral replication
Based on recent abundant metagenomics and in-depth transcriptomics data, it has been unequivocally demonstrated that RNA viruses display striking genetic diversity. Hence, their sequence similarity is often too low to permit robust phylogenetic inferences, making it challenging to identify highly divergent viruses that embody the uncharted virosphere. Therefore, despite a fast expansion in the number of reported viruses following the advent of high-throughput sequencing (HTS), the identification and the annotation of novel viral phyla or highly divergent RNA viruses remain tedious (1–7).
RNA viruses (riboviruses) dominate the eukaryotic virome and are the most abundant biological entities on earth (2, 8–11). The accelerated emergence of new viral infections poses a significant global health concern, exerting tremendous burdens on economies and public health due to many cases arising with unknown etiology (12), thus emphasizing a pressing need to address faster advancements in RNA virus discovery. Most RNA viruses, however, encode a unique category of non-structural proteins named RNA-dependent RNA polymerases (RdRps). RdRps are classified as template-dependent nucleic acid polymerase proteins (13, 14). Upon host cell infection, transcription and replication are governed by the RdRp as a single-subunit module or are mediated by replication–transcription complexes (15). These complexes regulate viral RNA synthesis and are therefore, in the case of pathogenic viruses, proven potent druggable antiviral targets due to their higher evolutionary stability (16–19). To transcribe and replicate their genomes with optimized processivity (although with a relative fidelity) (20), RNA viruses use the RdRp together with a remarkably cooperative cohort of other replication enzymes and stimulating cofactors (21, 22). One can thus argue that RdRps are keystones to ensure translation by the host ribosomal machinery and to balance the spatiotemporal modulation of the viral cycle within the infected host cell (23, 24). This central enzyme therefore essentially coordinates with other viral modules and host factors for the production of both viral mRNAs and new genomes. Following its genomic RNA translation and proteolytic maturation, the RdRp initiates a relatively complex process of RNA synthesis and gene expression. This process is dependent on the interplay between the viral RNA and non-structural proteins on the one hand and the host cell proteins and membranes on the other hand. These major steps are assumed to be modulated by various accessory subunits required to orchestrate the template unwinding and switching, which are necessary for genomic RNA synthesis (25, 26). The multiple interactions between these subunits and their cognate regulatory viral RNA elements fine-tune the timely host- or niche-specific replication processivity of their genomic repertoires (27). Akin to other polymerases, RdRps use ribonucleoside triphosphates (NTPs) as precursor substrates to catalyze the incorporation of ribonucleoside monophosphates (NMPs) into the nascent RNA product with conserved integrity based on the genetic information provided by the template RNA (28–30). These replication-associated non-structural proteins are either directly implicated in nucleic acid metabolism or promote the efficient catalytic regulation of other non-structural proteins (31–33). Viral RNA synthesis relies on specific viral/host cofactor proteins. RdRps are essentially soluble in the cytoplasm, but they reside in the host cell as dynamic conformational ensembles that adapt to the functional requirements of the viral cycle. Thus, RdRps also participate in the formation of specific replication organelles, for example through their membranous anchoring as viral replication complexes (34, 35). Together with other cofactor proteins, RdRps are major players in driving virus–host adaptation (36, 37). Thus, providing better characterization of RdRps will enrich our knowledge of the RNA virome and evolution.
RdRps are indeed prime antiviral targets (38). However, designs in antiviral strategies are hindered by our incomplete understanding of viral RNA synthesis, particularly at the structural level. A few studies have focused on deciphering and contextualizing the distinct and manifold conformational features including their dynamic functioning at the interplay of RNA viral replication and transcription complexes (39, 40), while others are burgeoning (19, 41–44). Determining the conformational diversity of RdRps and their dynamic biophysical behavior can therefore provide essential knowledge for understanding the functional modulatory properties of this class of proteins. Computational and structural bioinformatics analyses are, thus far, based on the examination of 50 viral RdRps with experimentally determined well-folded structures reported in the Protein Data Bank (PDB) spanning 10 taxonomic viral families (45). They generally define the polymerase subunits into well-conserved sub-domains, including the terminal domain and an interface region with the conserved structured catalytic domain (core) of the RdRp (Figure 1). The core RdRp domain is, despite the large structural variability of the surrounding regions, delineated by a well-structured architecture analogous to the typical right-hand configuration, shared by all viral RdRps and composed of fingers, palm, and thumb. The residues responsible for nucleotide selection and RNA-specific catalysis are located on the inner interface of the core palm domain (45, 50). The active site is further subdivided into critical functional features with relative spacing and occasional permutations (51), known as catalytic motifs (A–G) (45, 52–57). Among them, motifs A–F exhibit strong conservation across all viral RdRps at the genus and family taxonomic ranks. Motif G is defined as a hallmark of primer-dependent RdRp in some positive-sense RNA viruses and interacts with the primer strand to initiate RNA synthesis (58). Most ABC motifs appear in a canonical suite within the primary sequence of most known RdRps. In certain evolutionarily divergent lineages, permutation in the active site sequence is observed into the CAB order (56, 59). Our discussion in this review will mainly cover a representative dataset of complete RdRps, including palmprint sequences and their defined canonical ABC motifs (with intervening variable segments V1 and V2; Figures 1A–C) (6, 48).
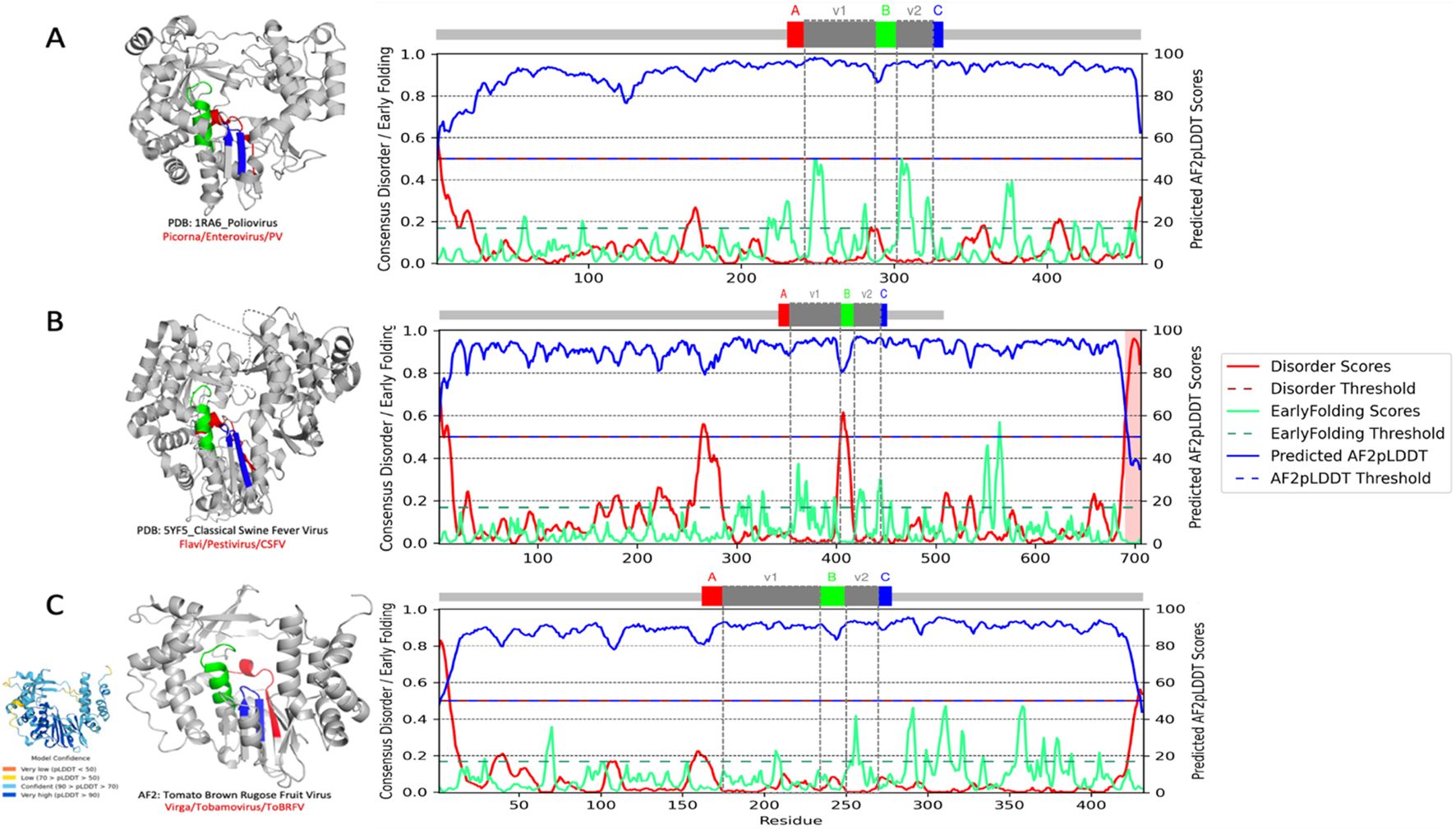
Figure 1. RNA-dependent RNA polymerase (RdRp) and conserved palmprint motifs from human, animal, and plant positive-strand RNA viruses. Full per-residue prediction scores for AF2--ppLDDT or metapredict-ppLDDT, blue) in relation to intrinsically disordered regions (IDRs, red) (46) and early folding (green) (47). The disorder and ppLDDT scores (metapredict-ppLDDT) are almost anti-correlated and correctly identify domain boundaries. The early folding prediction score indicates which residues in the sequences will form structures first through local interactions between amino acids. (A–C) The palmprint segment predicted by Serratus with the well-conserved ABC motifs in the active site of the polymerase domain and in their corresponding PDB (A, B) and AF2-predicted structure (C) (left). The intervening V1 and V2 segments (gray) are more variable regions. (A) Poliovirus (PDB:1RA6; Picornaviridae, Enterovirus, PV) cartoon representation with the conserved catalytic motifs colored from of the N to the C terminal: motif A (red), motif B (green), and motif C (blue). (B) Classical swine fever virus (PDB:5YF5; Flaviviridae, Pestivirus, CSFV). (C) Tomato brown rugose fruit virus (UniProt ID: A0A0S2T050; Virgaviridae, Tobamovirus, ToBRFV) (inlet: AF2 prediction with confidence scores). Cartoon representation with the ABC motifs and intervening V1/V2 regions color coded as indicated in the corresponding palmprint segment identified by Serratus (https://serratus.io/palmid) (48). Structure images were generated in PyMOL (49).
Current knowledge of the overall sequence–structure–function relationship of RdRps remains relatively scarce, which can impact our understanding of the biology and evolution of RNA viruses (60). Recent detailed studies using molecular dynamics have provided valuable insights into the stabilizing roles of these conserved structural motifs. They operate as sequence-specific conformational switches during the nucleotide incoming and positioning cycle (44, 45, 58, 61, 62). Motifs A and C contain two aspartic acid residues that contribute to coordinating interactions with two divalent metal ions essential for the phosphoryl transfer reaction. They are also required for other diversified classes of polymerases (63–65). In motif B, the backbone flexibility of a conserved glycine residue plays a critical role in recognizing the hydroxyl group of the NTP substrate, while the corresponding peptide bond flip accompanies an elusive conformational conversion of the NTP-induced RdRp active site closure. Motif C contains the critical catalytic residues, which reside in a turn loop connecting two adjacent strands. Motif F forms a fingertip that protrudes into the catalytic chamber and interacts with the finger extension loops and the thumb sub-domain to engage, via several of its highly conserved basic residues (lysine and arginine), with the triphosphate and base moieties of the NTP substrate.
Across the globally known virome, most known RdRp sequences are homologous throughout all phyletic lineages, thus enabling multiscale automated and multiple sequence alignment (MSA)-free comparative analysis. The features of RdRps, akin to all proteins, can also be investigated with biophysics-inspired tools to grasp a coarse-grained picture of their structural variability, which is particularly useful for novel or poorly characterized proteins (66–68). The overall variation of RdRps is indeed extreme among viruses, indicating potential heterogeneous and unannotated functional features. Their dynamic ensemble-driven conformational modulation to achieve their function is expected to be governed by a combination of sequence-encoded short- and long-range intramolecular interactions involving transient and topologically diverse motif-rich secondary structures (69–71). Defining the dynamic conformational behaviors and the biophysical landscape of their ensembles may therefore help in grasping and delineating the magnitude of this structural heterogeneity within the RdRp boundaries. Furthermore, identifying RdRp similarities between RNA viruses can address knowledge gaps on the functional roles of conformational ensembles populating the RdRp module. Linking the sequence conservation to the sequence-encoded conformational heterogeneity in their biophysical propensities can further provide vital understanding of the general molecular bases and functional modalities that govern this hallmark class of viral proteins, including non-canonical RdRps.
2 Coarse-grained sequence-based RdRp conformational landscape can catalyze data-driven discovery in the global RNA virosphere
In this review, we address the multiscale facets of the RdRp conformational properties as sequence–ensemble–function relationships and discuss their implications in RNA virus discovery. The main purpose of this review was therefore to place the current knowledge on RdRps in a broader conceptual context by considering both global and local protein structural flexibility and intrinsically disordered proteins (IDPs) and regions (IDRs) [collectively termed intrinsically disordered regions (IDRs) hereafter] (66, 71–75). These IDRs manifest as a repertoire of hypervariable, natively dynamic, interconverting heterogeneous conformations that can only be represented as conformational ensembles. IDRs lack a stable autonomous primary (3D) folding, but still mediate many functions (76), beyond the conserved catalytic core, through quantifiable ensemble features (77, 78). Exhaustive mapping of the IDRs and their features within RdRps can therefore help improve the overall understanding of the structure, function, and evolution of RdRps. By determining the biophysical landscape of RdRps in terms of global IDR dimensions, the distribution of the physicochemical properties and the inter-residue distances within the catalytic motifs, as well as deriving their local transient extended conformations, one can quantify the disorder-mediated conformational diversity of the RdRp catalytic module along its intramolecular interactions. These in turn serves as reference points to predict additional functional regions beyond the core catalytic module. The major strength of this conceptual approach (coined the “modulome”) (79) hinges on unifying sequence diversity and evolutionarily informed conformational states into a single data-driven machine learning-based quantitative sequence-to-function annotation. Inspired by seminal reviews (45, 80), we employed a computational framework that illustrates this pivotal concept via a comprehensive, representative RdRp-based metaproteomic dataset across taxonomical and evolutionary RNA viral lineages. Our framework integrates a suite of established bioinformatics toolkits to identify and annotate these sequences. By combining RdRp-specific biophysical propensities of structure and disorder-centric conformational heterogeneity, we used the modulome approach to survey the sequence-based conformational diversity of RdRps with respect to their taxonomical and evolutionary classification.
3 Methods
3.1 RdRp sequences and datasets
As a preliminary proof-of-concept, approximately 480,000 curated RdRp sequences from diverse published databases were included, covering all known or available evolutionary clades of the RNA viruses compiled in various recently published articles and publicly available databases (10, 48, 81), including representative RdRps assigned by the International Committee on Virus Taxonomy (ICTV). For sequence retrieval and storage, PALMdb (https://github.com/rcedgar/palmdb) served as the primary repository of RNA virus RdRp amino acid sequences, therefore providing the ground truth RdRp dataset (palmdb) (10). We also downloaded the protein sequence entries contained in UniRef90 (82) on October 2, 2022, from UniProt (83). To discover the diversity and evolutionary relationships of RNA viruses within individual virus clades, all previously documented RNA viruses from the palmID database were incorporated into the biophysical analyses, including RefSeq, UniProt, and PDB RdRp sequences (45, 84). The palmID web server (https://serratus.io/palmid) was used for searching, verifying, and classifying the amino acid sequences containing RdRps (Supplementary Material). Sequences that failed to cover these motifs were not considered. The palmprint segment, an ~100-amino acid (aa) region in the active site of the polymerase domain, was checked and collected. While motifs A, B, and C are well conserved, the intervening V1 and V2 regions are more variable. To verify the taxonomical assignation, the presence of the three key RdRp motifs (namely, the A, B, and C motifs) was verified through the built-in palmID alignment algorithm (https://serratus.io/palmid) to all RNA supergroups that possessed these motifs (i.e., motif A [DxxxxD], motif B [(S/T)Gxxx(T/G)xxxN], and motif C [(S/G/N)DD]) (see Supplementary Material).
3.2 Retrieval and processing of protein structures
The 3D structures of viral proteins were obtained from the PDB (https://www.rcsb.org). The selection criteria included high-resolution structures (≤3.0 Å) determined through X-ray crystallography or cryo-electron microscopy (cryo-EM), ensuring structural accuracy for comparative analyses. The protein structures were retrieved using their respective PDB identifiers and were selected based on biological relevance, structural integrity, and the presence of conserved catalytic motifs. The following representative structures were analyzed: poliovirus RNA-dependent RNA polymerase (PDB:1RA6; Picornaviridae, Enterovirus, PV) and the classical swine fever virus (PDB:5YF5; Flaviviridae, Pestivirus, CSFV). Each structure was downloaded in the PDB format (see Supplementary Material). To facilitate structural analysis, non-essential water molecules, ions, and ligands were removed. All other functions were used under default parameters.
3.3 AlphaFold structure predictions
All AlphaFold2 (AF2) 3D structures were either downloaded from the AlphaFoldDB database at the European Bioinformatics Institute (EBI) or generated using v1.5.5 and its MMseqs2 implementation (https://alphafold.com/) (85–87) on the Vlaams Supercomputer Centrum (VSC) infrastructure, with a cutoff date of February 15, 2024, for the templates used. The AlphaFold 3 (AF3) 3D structures were generated using the web server (https://alphafoldserver.com) under default settings. For each representative, the highest-ranking structural model, determined by the predicted local distance difference test (pLDDT) score, was retained.
3.3.1 AF3 with respect to stereochemistry, hallucinations, dynamics, and accuracy in RdRp predictions
AF3 (v3.0.1) (88) represents a significant advancement over AF2 in both the scope of biomolecular structure prediction and modeling accuracy. While AF2 is primarily designed for the prediction of the 3D structures of individual proteins and, to a more limited extent, protein–protein complexes, AF3 extends its capabilities to a broader range of biomolecular assemblies. Specifically, while both models address IDRs by typically producing unstructured, ribbon-like predictions where atomic coordinates are uncertain, AF3, however, occasionally introduces low-confidence secondary structural elements, such as spurious alpha helices, within these disordered regions. These predicted structures often have very low pLDDT scores and show poor reproducibility across prediction runs, suggesting that they are artifacts rather than biologically meaningful features. This behavior reflects the increased expressive power of AF3, which, while enabling broader modeling capabilities and parameters (https://github.com/google-deepmind/alphafold3), may also generate occasional noise in regions of structural ambiguity where unstructured regions are typically represented by long extended loops instead of compact structures. Indeed, while AF3 cross-distillation greatly reduced its hallucination behavior, the switch from the non-generative AF2 model to the diffusion-based AF3 model introduced the challenge of spurious structural order (hallucinations) in disordered regions (88). Default parameter settings were applied for both AF2 and AF3 implementations.
Moreover, structures were predicted using ESMFold through its online implementation (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/ESMFold.ipynb) (87). In addition, the pLDDT score was collected for each structure prediction as its per-residue estimate of the prediction confidence on a scale from 0 to 100. The structure of each representative sequence was then predicted using ColabFold based on its corresponding MSA-based sequence coverage and the AF2 model. Figures of the molecular structures were generated using PyMOL software v2.5.5 (The PyMOL Molecular Graphics System, Schrodinger, LLC; http://www.pymol.org/pymol) (89) and UCSF ChimeraX (90) (see Supplementary Material).
To evaluate the structural similarity between viral RdRp protein models, we employed the template modeling score (TM-score), a widely used metric for evaluating the quality of protein structure alignments by assessing their topological similarity (https://zhanggroup.org/TM-align/, https://zhanggroup.org/TM-score/) (91). The TM-score is particularly advantageous as it addresses two critical aspects of structural comparison.
i. Distance error sensitivity: Unlike root mean square deviation (RMSD), which treats all distance deviations equally, the TM-score assigns greater weight to smaller distance errors. This weighting enhances the sensitivity of the score to global structural similarity rather than local structural variations. As a result, the TM-score provides a more biologically relevant assessment of fold similarity, making it particularly useful for comparing protein structures with minor local variations but preserved overall topology.
ii. Length-dependent normalization: The TM-score incorporates a length-dependent scaling factor that normalizes distance errors based on protein size. This ensures that the score remains independent of sequence length when comparing random structure pairs.
Consequently, the TM-score allows for meaningful comparisons across proteins of different lengths without biasing the evaluation based on structural size. The TM-score ranges between 0 and 1, where 1 indicates a perfect structural match. This normalization and weighting strategy makes the TM-score a robust metric for determining the fold similarity between protein structures, making it particularly useful in structural bioinformatics and computational protein modeling.
3.4 Biophysical features and disorder predictions
On a subset of 250,081 RdRp target sequences, we primarily used the bio2Byte Tools (92, 93) with sequence-based predictors, namely, backbone dynamics (DynaMine; https://bio2byte.be/) (94), DisoMine (95), and EFoldMine (96). Related side-chain dynamics and conformational propensities (96) were predicted at the per-amino acid and per-sequence levels using default parameters. These methods are based on the per-residue characteristics (e.g., backbone dynamics) derived from nuclear magnetic resonance (NMR) chemical shift values and use a linear regression model for the prediction. Concretely, with regard to backbone dynamics, higher values denote that backbone movements are more likely to be limited. Values >1.0 indicate membrane spanning regions, >0.8 rigid conformations, and <0.69 flexible regions. Residues with 0.80–0.69 values are “context”-dependent and are capable of being either rigid or flexible. For rigidity of the side chain, higher values indicate that the side chain is more likely to be conformationally restricted. These values are highly dependent on the amino acid type (i.e., a Trp will be rigid and an Asp flexible). Early folding probabilities per residue were predicted using EFoldMine, which uses as input features the five previously mentioned DynaMine values for a five-residue fragment, resulting in a 25-dimensional feature vector that was trained using a support vector machine (SVM) on a set of high-resolution per-residue hydrogen–deuterium exchange (HDX) data from the NMR experiments. Early folding predictions indicate the likelihood that this residue will adopt a persistent conformation based on only local interactions with other amino acids. Values >0.169 indicate residues most likely to start the protein folding process. For the prediction of structure and disorder, the metapredict python package v2.4 and the online server (https://metapredict.netv3.0) (46) were used. Both the pLDDT predicted by metapredict (metapredict-ppLDDT, also referred to as the AF2-ppLDDT) and the disorder values from metapredict were integrated into a Python Pandas data frame. To predict disorder, metapredict was used as it is considered one of the most robust, accurate, and high-performance predictors of intrinsic disorder, which is also easy to install and quick to generate predictions. It uses a machine learning network to generate per-residue scores from amino acid sequences that reflect the likelihood of the residue being disordered. Metapredict v3 uses a new network to predict disorder that, in our benchmarks, is the most accurate version to date. Although metapredict v3 provides major improvements compared with metapredict v2, the default v2 network is, by all metrics, better for pLDDT prediction and is still recommended (https://metapredict.readthedocs.io/en/latest/getting_started.html). The original release of metapredict included the ability to predict the AF2 per-residue pLDDT scores. Metapredict v2 was trained by generating an initial hybrid score that combined the predicted AF2-pLDDT scores (AF2-ppLDDT, i.e., the metapredict-ppLDDT), used with consensus disorder along with some signal process algorithms to make a new structure/disorder consensus prediction. In the context of AF2, the pLDDT scores convey the confidence associated with a given structure prediction. Metapredict was trained on the original AF2 and the Critical Assessment of Protein Intrinsic Disorder (CAID) data to predict the pLDDT scores (AF2-ppLDDT) from the sequence alone. Although low pLDDT scores cannot strictly be interpreted directly as a given region having a high likelihood of being disordered, the AF2-pLDDT metric is nevertheless generally considered a reliable predictor of protein disorder, showing, to some extent, a correlation between the pLDDT scores and the low pLDDT scores’ tendency of amino acids to be generally disordered (97, 98). Moreover, the DisProt-PDB dataset was also used to train metapredict (v2) for accuracy analysis as it contains regions that have been experimentally determined to be either disordered or not disordered, allowing for the identification of true positive, true negative, false-positive, and false-negative predictions generated by metapredict v2 and v3 to integrate these predictions with either the predicted (v2.4, used here in this systematic review) or the actual (v3) (99) pLDDT obtained from AF2. The pLDDT scores represent confidence for the very local structural predictions obtained from a structure prediction model (in our case, AF2 or AF3).
When the predicted pLDDT scores for these proteins were examined (46), it was found that the metapredict-ppLDDT predictor did not give these regions predicted pLDDT scores as high as the actual AF2-generated pLDDT scores (46, 99). This suggests that the metapredict-ppLDDT predictor does not always produce high pLDDT scores for some disordered regions, even though the actual AF2-pLDDT scores for the same regions are relatively high. However, it remains noteworthy that this is not always the case, as metapredict authors were able to identify some known disordered protein regions where the metapredict-ppLDDT predictor generated high ppLDDT scores (46). Therefore, the metapredict-ppLDDT scores were suggested to provide an orthogonal mean to examine whether a protein region is likely to be disordered. The scores used to make metapredict (v2) were the predicted pLDDT scores (metapredict-ppLDDT scores), which were generated using a bidirectional recurrent neural network (BRNN) trained on the AF2-pLDDT scores from 21 proteomes. Thus, if there are any consistent circumstances where AF2 generates a high pLDDT score for a given disordered region or type of disordered region, metapredict (v2) will be unlikely to predict the region to be disordered. Indeed, a recent report highlighted a case where known disordered proteins or protein regions had high AF2-pLDDT scores, which was found to be at least in part due to the disordered regions undergoing conditional folding (100). Metapredict exploits ways of combining the predicted consensus disorder scores with the ppLDDT scores such that one could improve the accuracy of the predicted disordered regions. When evaluating the accuracy of metapredict v2 in comparison to 33 other disorder predictors (including legacy metapredict), metapredict v2 was found performing as the second most accurate disorder predictor, with the difference between metapredict v2 and the most accurate currently available predictors (46). In addition, metapredict is orders of magnitude faster in execution time compared with the other high-performing predictors (46). Nonetheless, metapredict (v2) still offers a substantial improvement in accuracy over legacy metapredict. Therefore, metapredict v2.4 was used to predict intrinsic protein disorder as metapredict-ppLDDT provides an ideal balance of dual metric speed, accuracy, and availability. To accurately benchmark and evaluate metapredict v2 sequence-based prediction of IDRs against the top state-of-the-art protein intrinsic disorder predictors, 44 metapredictors were utilized using an ensemble approach that averages the predictions from the top-performing neural networks, sorted by their optimized (on DISORDER-NOX) F1 binary scores threshold. These are freely available through the CAID prediction server (https://caid.idpcentral.org/portal, accessed April 18, 2025) (101–103).
In addition to the mapping of IDRs, we considered AF2 (85) and other recent structure-based prediction metadata to further contextualize the biophysical signals that underlie the possible modulating roles of RdRp-associated IDRs in functional viral replication. By focusing on hallmark residues of the RdRp module and referencing the available structural and functional data in the recent literature, we attempt to estimate the diversity of conformations adopted by this unique module across the evolutionary landscape. Since we cover a broad range of the RNA virome, this review may, in turn, serve as a reference basis for the investigation of novel unexplored functional RdRp features that go beyond their static structure representations and that may orchestrate host-specific functional replication in viruses. Although other functional non-structural proteins are also involved in replication, they are outside the scope of this review. We hope that the discussions presented hereafter may cast impetus for addressing future fundamental and applied innovative research lines in RNA virus discovery.
4 Results and discussion
4.1 RdRp intrinsically disordered regions as functional modulators of local conformational ensemble dynamics and their implications for computational structural inference
4.1.1 The biophysical foundations for RdRp structural malleability and function
AF2 has propelled RNA virus discovery into a new era where virologists can visualize hitherto unresolved protein folds at atomic-level accuracy, unlocking the RdRp structures, functions, and inter-residue flexibilities solely from their amino acid sequences (104). The sequence-based wealth of new RNA viruses often relies on RdRp sequences generated from HTS studies and metagenomics (105–108). However, fast and accurate standardized methods to support this approach are currently lacking. Moreover, RdRp functions emerge from a diverse set of function-determining structural motifs and their cognate conformational space. Nonetheless, not all RdRp proteins, or regions thereof, have a well-defined 3D structure. The functional dynamic behavior of flexible regions is indeed difficult to assess from experimentally determined 3D structures and molecular dynamic simulations using these folds (109).
Importantly, many highly divergent viral RdRps or remote homologs remain poorly detectable in a metagenomics context even with the use of more advanced sequence comparison or traditional computational methods such as BLAST (110–112) or custom-built hidden Markov models (113–115). The increased content in IDRs, which are linked to extensive conformational heterogeneity and sequence diversity, can contribute significantly to this major barrier (116). This can be mediated by taking into account the conservation of compositional biases that corroborate conserved composition–function relationships (117–119). AF2-generated models underscore the importance of mapping the properties of these IDRs with atomic-level accuracy to better interpolate the key sequence–function relationships and their function-bearing motifs (99, 120). In the context of the protein structure prediction of AlphaFold, the term “atomic-level accuracy” refers here to the high precision from the PDB from which AF2 has learned and is more likely to dominate in the overall structure prediction. While AF2 is thus capable of detecting a hard order/disorder boundary (97), it does not capture the dynamic propensities of a residue. This limitation is expected given its training data, which predominantly consisted of folded and crystallized proteins and analyzed under cryogenic conditions, thus blind to the conformational flexibility observed in solution (121). AF2 tends to predict the bound states of proteins if present in the training data, and while disordered regions are typically missing X-ray diffraction PDB structures, they are visible when adopting a single defined conformation upon interaction with another protein or ligand. With this distinction in mind, IDRs adopt structurally ubiquitous, interconvertible heterogeneous states of conformation (71, 122–125), reflecting extensive sequence variation and low hydrophobicity. Despite the lack of a well-defined stable and persistent 3D structure, IDRs are essential for viral processes ranging from transcriptional control to replication and viral assembly (126). Through their wide-ranging conformational tunability and biophysical adaptiveness, IDRs extend the repertoire of functional interactions by being readily malleable by their long-lived plasticity and hypervariable topology, making their underlying scattered charge decoration, chemical moieties, and hydropathic cores ultimate evolutionary drivers of molecular signaling and recognition (78, 127–133).
Perhaps more importantly, recent progress in protein science has deepened our understanding of the relationship between the linear protein sequence and the multiple dynamic conformational behaviors of IDRs (96, 118, 134, 135). However, the link between sequence and IDR function is less tangible and more difficult to define (136), with the magnitude of their potential roles in viral replication and pathogenesis demanding more systematic structural and conformational investigations. This is undeniably evident for RdRps, where the low-confidence regions predicted by AF2 often overlap with structurally transient regions that are predicted to be IDRs.
4.1.2 Conserved RdRp motifs have hypervariable IDR-mediated folding dynamics
Deep learning-driven metapredictors that enable accurate identification of IDRs (46, 76, 137–139) and their IDR-specific physicochemical propensities are now available and can, with reasonable accuracy, identify transitions such as disorder-to-order, order-to-disorder, and disorder-to-disorder upon chemical- or context-dependent coupled interactions (140, 141), including regions predicted to be enriched in intrinsic disorder and prone to liquid–liquid phase separation (142, 143). While the few experimentally determined RdRp structures are remarkably conserved and their globular 3D structures are well defined, the multifaceted functional ensemble nature and biophysical properties of IDRs in RdRps, including the extent of their context-sensitive modulation, remain poorly characterized. The RdRp folding dynamics is a complex process that can lead to the dysfunction of RdRp when it fails. Particularly ill characterized are the very early stages of RdRp folding within the core catalytic domain, which are likely defined, on one hand, by intrinsic local interactions between amino acids close to each other in the protein structure and, on the other hand, by the local interaction with already folded template regions. These local amino acid interactions are often governed by the initial conformational states of the backbone and side-chain dynamics and the secondary structure propensities as features. Accurate prediction of these statistically highly dynamic ensembles at atomic-level accuracy provides valuable insights into the overall folding process, complementing qualitative comparisons with independent experimental observations. In addition, locally involved structural elements are inclined to become the folding chain residues that are the most probable to interact within the folded structure or IDRs exhibit evolutionary preservation at the sequence level. From a mechanistic vantage, this context-sensitive process appears to be directed by an equilibrium between kinetically determined local residue interactions, which remain important in folded proteins (47, 96, 144), and its topological complexity.
To concretely showcase with clear examples, Figure 1 shows the predicted relationship between AF2-fueled structure metrics, the pLDDT. Throughout this review, two similar but distinct per-residue confidence scores of structural predictions are used and must be clearly distinguished: the pLDDT from AF2 (i.e., AF2-pLDDT) and the predicted pLDDT (AF2-ppLDDT, i.e., the metapredict-ppLDDT; used as in 46). The main difference between AF2-pLDDT and ppLDDT is that the latter is not a direct assessment of AF2 prediction and can be different from AF2-pLDDT. ppLDDT is a per-residue confidence score that is scaled between 0 and 100 and estimates how well the predicted structure would agree with the experimental structure. Together with the AF2-pLDDT, the disorder propensities of three different RdRps from human, animal, and plant single-stranded RNA viruses are shown, along with their predicted early folding fluctuations. In the RdRp sequence of poliovirus (PDB:1RA6; Picornaviridae, Enterovirus, PV) (Figure 1A), the VI and V2 variable regions within the RdRp palm domain exhibit two of the highest early folding predicted values in the vicinity of a low peak of predicted disorder at the 5′ start of motif B.
Comparatively, in the RdRp of the classical swine fever virus (PDB:5YF5; Flaviviridae, Pestivirus, CSFV) (Figure 1B), significantly more early folding propensities populate the corresponding V1 variable region, whereas the V2 region similarly exhibits two conserved peaks.
In this RdRp palm domain, a considerably higher AF2 anti-correlated IDR signal is predicted directly adjacent to motif B. In a last example, the AF2-predicted RdRp of the plant-infecting tomato brown rugose fruit virus (Virgaviridae, Tobamovirus, ToBRFV) (Figure 1C), the V1 and V2 variable regions within the RdRp palm domain similarly display two well-marked early folding peaks in the absence, as evidenced by the low score, of any detectable disorder propensity. More precisely, the local and global conformational heterogeneities that RdRp proteins might adopt through the intricate modulation of folding pathways during viral replication entangle the interpretation of the structural diversity across the RdRp evolutionary and taxonomical landscape (145). Furthermore, RdRps show comparatively salient disparities between the underlying fine-tuned dynamic features of early folding propensity and their secondary structure elements, which relates well to experimental observations (22, 62). Thus, it is expected that comparative analysis of the parameters describing the modulation of various RdRp folding pathways will bring to light additional elements for better clarification of the evolutionary and taxonomic landscape of these enzymes.
The evolution of the biophysical features governing protein folding may provide such insights (146), particularly given the evolutionary hallmark nature of the RdRp protein family. From that evolutionary vantage, it may be more informative to examine where related proteins adopt specific conformations based on local interactions rather than focusing on the conservation of individual amino acids. While early folding residues are more conserved than the non-early ones, this is largely due to their amino acid composition—residues such as Cys, Phe, and Trp, which are inherently more conserved, are enriched in early folding positions. Protein dynamics is another potential key feature. Although the relationship between both conservation and adaptation in protein dynamics and folding has been established (146), no information on RdRp early folding has been reported yet. Protein folds (tertiary structure) are often conserved across broad evolutionary distances, allowing folding pathways to become reliable hallmarks at higher taxonomic ranks, where structural constraints drive the folding routes. Therefore, predictions of the RdRp folding pathways are especially informative at the class or the phylum level, where structural conservation persists despite sequence divergence (primary structure). Even at shallower taxonomic levels, the evolutionary patterns in RdRp folding initiation can be captured, supporting functional biophysical annotation where sequence similarity alone may be insufficient. Predictions of early folding residues that are based on per-residue backbone dynamics estimated from experimental data on proteins in solution, capturing the full range of conformational behavior—from disordered to fully folded—including local events such as flexible loops or helix fraying, can help quantify these trends (96, 146). To summarize these aspects, the results (Supplementary Figure S1A) showed the distribution of the early folding mean values across various RNA virus phyla/classes, indicating variability in the early folding propensities among RdRp sequences. RdRp sequences with low early folding predicted scores may adopt diverse and often extended conformations, while those with high early folding scores are structurally more compact and constrained. Overall, taxonomic groups differ in their early folding profiles, suggesting evolutionary shaping of the folding pathways and structural compactness across viral lineages. Taxa such as Picornaviruses and Hypoviruses show lower early folding means, suggesting higher structural dynamic variability, whereas groups such as Tombus–Nodavirus exhibit higher values, suggesting a more restricted protein expansion. Such cautious interpretation is essential to improve our understanding of how context-dependent RdRp functional folding behavior is conserved across Riboviria lineages.
Our large-scale exploration of the folding initiation predictions for the RdRp palm domain revealed that early folding-prone amino acids i) tend to involve residues that make the most backbone interactions in the variable regions of the native fold, and ii) that because of their relative conservation, these are likely to be the results of co-evolution. Moreover, given the vicinity of context-sensitive IDRs, this also suggests that iii) the local intramolecular interactions in the RdRp palm domain are likewise highly context-specific, i.e., atomic-level interactions between residues can only be shaped in the presence of template- or taxon-dependent local structural elements. These unique RdRp features should greatly contribute to refined structural analyses of the diversity of RdRps. They should assist in unveiling emerging relationships between the dynamic folded and flexibility states, where allosteric characteristics might appear to be important additional RdRp features for function in viral replication (147–151). Finally, addressing the RdRp local conformational preferences of early folding sites is further expected to unravel uncharted relevant overall folding behaviors in the vicinity of the RdRp palm domain and their potential modulation of natively conserved folding/binding pathways. Taken together, we hypothesize that these functional modulators of RdRp will expand our structure/disorder-based knowledge of virus discovery and assist innovative strategies toward disruptive antiviral protein drug design.
4.1.3 Delineating biophysical flavors of RdRp protein intrinsic disorder: on the importance and implications of benchmarking ensemble-approach metapredictors
Predicting IDPs/IDRs is a challenging task, primarily because protein dynamics cannot be described by a limited set of fixed conformations (103). Several IDR prediction methods have been published, each with its own limitations and biases (152), and both the predicted and experimentally derived properties of IDRs, as well as the annotations related to their function, are stored in dedicated databases (76). Rather, proteins with multiple conformations, as observed for IDRs and IDPs, require ensemble-based representations to better characterize their structural heterogeneity (98). Despite this complexity, many accurate and robust disorder predictors have emerged over the years (101, 152). Early efforts in identifying IDRs have been driven by bioinformatics, with initial tools enabling the distinction between disordered and folded domains.
Experimental methods in structural biology—such as X-ray crystallography, circular dichroism, and NMR spectroscopy—have been used to derive information about intrinsic disorder (153). However, direct measurement of the highly dynamic behavior of IDRs remains very difficult (24, 101, 103, 138, 152, 154). IDRs, which do not adopt a fixed 3D fold in isolation under physiological conditions, are now a well-established concept in structural biology. They are characterized by their ability to sample a vast array of conformations, forming a continuum between fully disordered states and folded states with long dynamic regions (150). This complexity makes it difficult to establish a universal “ground truth” for IDRs. Moreover, intrinsic disorder is often context-dependent, with proteins undergoing order-to-disorder transitions in response to specific conditions, such as binding a partner molecule. This dynamic behavior distinguishes them from protein switches, which alternate between a limited number of conformations in response to defined stimuli. Adding to the complexity, IDRs also exhibit diverse functional mechanisms—often referred to as disorder “flavors”—including flexibility, folding-upon-binding, and conformational heterogeneity. These characteristics further complicate the development of accurate prediction models, particularly in the absence of a universally accepted definition or dataset encompassing all IDR variants. To tackle this, numerous computational methods have been developed to predict IDRs from protein sequences. Their performance is evaluated through initiatives such as the CAID (101). Over the past 25 years, disorder predictors have become increasingly accurate. In 2021, the first CAID competition was held, comparing various tools based on prediction accuracy and performance. CAID addresses the relatively simpler problem of identifying residues within protein sequences that are likely to be intrinsically disordered. Predictors are required to assign a probability score to each residue, which can be converted into binary predictions using a defined cutoff. The resulting predictions, along with experimentally validated IDRs and functional annotations, are stored in dedicated databases (97). While CAID evaluates both the predictive accuracy and software implementation, assessment of the true predictive power of tools remains a challenge. Many predictors are not publicly available, exist only as stand-alone executables, or are limited to web-based interfaces. Moreover, even when available, these methods lack standardization and often require expert knowledge for proper interpretation, which usually involves careful reading of the associated literature and nuanced understanding of the output formats. The CAID2 challenge highlighted varying performances across different predictors and benchmarks, reinforcing the need for more versatile and efficient prediction tools. Depending on the research context, users must balance accuracy with computational efficiency. For instance, methods based on AF2 have shown promise in predicting intrinsic disorder, but they tend to detect the absence of order rather than capture IDRs as defined in the DisProt database (155, 156). To facilitate access, CAID2 metapredictors are freely available through the CAID Prediction Portal (https://caid.idpcentral.org/portal), and CAID will serve as the official platform for future challenges. Nonetheless, the sheer variety of available predictors can overwhelm users, making it difficult for virologists to compare performance and make informed decisions. Moreover, measuring both local (e.g., helicity and NMR chemical shifts) and global (e.g., radius of gyration and end-to-end distance) IDR ensemble properties for the same IDR can be time-consuming and challenging. Instead, integrative biophysical studies—in which several methods measure distinct properties of a single IDR—have played key roles in enhancing our current understanding of sequence–ensemble relationships (71). Notwithstanding these substantial computational challenges, directly interpretable accurate and robust IDR predictors are essential to identifying and understanding the role of IDRs. These tools must be jointly considered to grasp the underlying functional and biophysical principles of divergent or newly sequenced RdRps. Finally, while substantial challenges remain in achieving highly accurate predictions across the conformational diversity of IDRs, it is crucial to emphasize the importance of using multiple or ensemble-based approaches of robust and accurate IDP metapredictors (Supplementary Figure S1B). These tools must provide strong coverage and generalization for all IDRs, both in well-characterized and newly discovered viral RdRps, and help virologists make informed choices and build new hypothesis.
4.2 AlphaFold3 et al.: predictors of static protein structures—potential advances and flurry of intractable limitations in capturing RdRp conformational heterogeneity
4.2.1 Potential misleading inferences from RdRp protein folding pathways
In the future, structural features of 3D protein folding captured by deep learning algorithms will likely generate new knowledge that enables the identification of key conformations from a single RdRp sequence. In the meantime, predicting and annotating RdRp structures with high accuracy remain critical challenges for the RdRp community, as for proteins in general for the broad life sciences. Overall, the AlphaFold deep learning models have provided a transformative advance to the field of structural biology and its community (88, 157–160). These neural networks use attention-based components to inject long-range contact information from the PDB and MSAs into vector representations (161), successfully constructing a complete single-state structure reaching near-perfect experimental coverage and providing virologists with a highly informative influx of biological insights into protein function that are extendable to large-scale structural evolutionary knowledge. The striking high-level prediction performance of AF2 persistently outperforms that of existing state-of-the-art computational tools (162), also demonstrating high accuracy in determining protein evolutionary covariations (163–169). Nonetheless, despite the outstanding performance of AF2, there are still some targets for which the prediction results are rather mixed, particularly in cases when the quality and depth of MSAs are insufficient for the proper modeling of inherent or induced protein IDRs (170).
As computational structure prediction is significantly impacted by the quality of MSAs, the superiority of template information does not guarantee the improvement of the final structure. Hence, template information refers to known structures of proteins (PDB or MSAs) that can serve as templates for the prediction of the structure of a related sequence (171). While template information is valuable, it is not always sufficient to guarantee accurate predictions, particularly if the MSAs used to generate these templates are of low quality or shallow in coverage. In other words, even if the template information is superior, if the underlying MSAs are flawed, the predicted 3D structure may still be of low resolution. Therefore, large margins still exist for further progress in deep neural network models, both in terms of the accuracy and quality of the predicted structures (172). Figure 2 shows a sampling of RdRp structures predicted by AF2. The regions with very low confidence, as indicated by the pLDDT scores, are particularly relevant. For example, in Figures 2A, B, F, the model coloring (by AF2-pLDDT) shows the majority of residues with >90, whereas the ppLDDT in the plots for the AF2-pLDDT confident regions indicates lower scores, such as with <80, and some even low–very low range. On the other hand, in Figure 2C, the high–very high confident regions indicated by the ppLDDT in the plot for aa 1–400 are inconsistent with the low AF2-pLDDT confidence coloring of the regions outside motifs A–C in the model. The BRNN (46) used to generate the ppLDDT scores was trained on the AF2-pLDDT scores from the proteomes of 21 organisms; however, viral proteomes were not mentioned (46). These differences and inconsistencies between AF2-pLDDT and ppLDDT highlight potential important limitations in the context of IDRs in viral proteins and, in particular here for the RdRps. i) Structural interpretation of IDRs: the AF2-pLDDT scores for IDRs are generally low due to their inherent flexibility and lack of fixed 3D structures. However, these low scores could reflect true intrinsic disorder or inaccurate modeling.
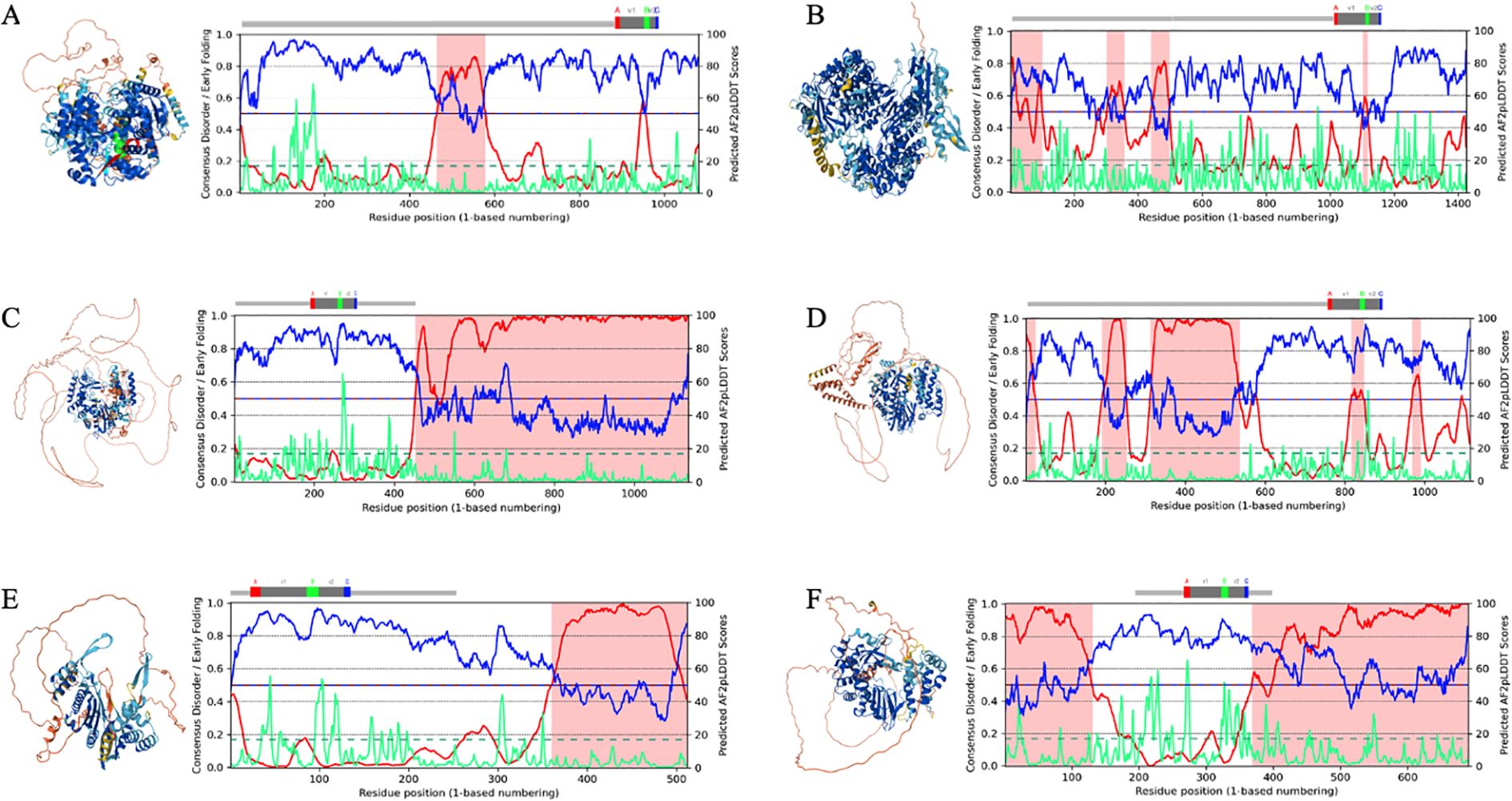
Figure 2. Illustrative examples of AlphaFold3 (AF3)-predicted RNA-dependent RNA polymerase (RdRp) structures with disorder-rich palmprint domains and long intrinsically disordered regions (IDRs) in newly discovered RNA viruses. Structures: AF3-derived structure prediction colored by the predicted local distance difference test (pLDDT) scores (see Methods). Plot: Full per-residue color coding based on the AlphaFold 2 (AF2) model confidence score (AF2-ppLDDT) along the sequence. Comparison of disorder (red) vs. AF2-ppLDDT predicted pLDDT (blue) (also termed Metapredict-ppLDDT; divided by 100 to place it on the same scale as in 46) and early folding (green). Regions highlighted in pink represent IDRs. Predictions provide a linear assessment of whether a residue falls within a disordered or a structured region (as described in Figure 1). RdRp sequences from publicly available repositories (see Supplementary Material for all RdRp sequences and palmprint analysis). (A) Beet chlorosis virus (BChV) (GenBank: AAK49956.1/RefSeq: NP_114361.2, Pisuviricota, Sobelivirales, Solemoviridae, Polerovirus). (B) Megalopteran phenui-related virus OKIAV286 (GenBank: QPL15334.1 Negarnaviricota, Bunyavirales, Phenuiviridae). (C) Picornavirales RdRp (SRR12149956, Nepovirus, Secoviridae, Picornavirales, AAL36026.1). (D) Weivirus RdRp (SRR7109325; closest palmID hit: Beihai weivirus-like virus 4, unclassified Riboviria, YP_009337162.1). (E) Yanvirus RdRp (SRR11679702; closest palmID hit: Tombusviridae sp., unclassified Kitrinoviricota). (F) Lenarviricota RdRp (SRR5215309; closest palmID hit: Apple ourmia-like virus 2, Lenarviricota, QIC52828.1). Structures were analyzed and figures were generated using PyMOL (49) and UCSF ChimeraX (173).
This ambiguity is compounded when the ppLDDT scores contradict the AF2-pLDDT coloring, as seen in Figure 2C. Then, the misclassification of IDRs as poorly modeled structured regions (or vice versa) can obscure functional insights into viral RdRps, where IDRs often play critical roles, such as RNA binding or replication complex assembly. ii) Training bias toward viral proteins: AF2 and AF2-ppLDDT predictors are primarily trained on structured protein datasets and non-viral proteomes. Viral proteins, including RdRps, often exhibit unique sequence features and functional intrinsic disorder, which are underrepresented or absent in these datasets. For example, the BRNN used for the ppLDDT score training (v2) does not mention the inclusion of viral proteomes (in contrast to its current version, v3), limiting its reliability when predicting residue confidence in viral proteins. In contrast, AF2 was trained on the PDB, which, while limited in viral protein representation, is not devoid of them. The discrepancies between the AF2-pLDDT and ppLDDT scores highlight critical limitations in confidence score interpretation for viral RdRps, particularly for ambiguous IDRs or low disorder regions, which imposes, as mentioned above, use of multiple or ensemble-based approaches of robust and accurate IDP metapredictors (see Supplementary Material for CAID benchmarking multiple disorder prediction methods on experimentally validated PDB structures; Supplementary Figures S3A–C). This is particularly significant for IDRs, where the inherent flexibility and lack of training on viral proteins introduce additional challenges.
Given these potential caveats, virologists should exercise greater caution when interpreting the AF2-pLDDT or ppLDDT scores in viral RdRps, complementing these predictions with disorder-aware predictors and experimental validation to mitigate these limitations and avoid misleading or inconsistent predictions.
In particular, enhancing the accuracy of side-chain modeling appears to be a cornerstone for more accurate modeling of the physiologically preferred low-energy state (174–176), especially in prevalent long IDRs or natively unfolded regions (Figures 2A–F). During the writing of this review, major efforts in structural biology have been made to improve the average prediction performance of AF2 in diverse manners since its publication by addressing diverse bottlenecks in its new implementation (see AF3) (88). One of the major advantages of AF3 is its capability to predict multiple types of biological molecules as a complex. In addition, AF3 improves the structure prediction in IDRs by addressing hallucinations (spurious structural order) through distillation training from AF2, which encourages ribbon-like predictions. This can significantly benefit the prediction of RdRps as it would assist in the enhanced identification of amino acid residues involved in substrate recognition, catalysis, and template binding.
4.2.2 Advancing protein structure prediction to account for the conformational space
One major path is to tackle the efficiency of training and inference speed. RoseTTAFold (177), ColabFold (86), and OpenFold (178) are the three initial works in this category. While ColabFold is a cloud-based, slightly re-implemented AF2 model with its own training system, OpenFold is a retrained version of AF2 with accelerated training/inference time. A number of works have attempted to build models that alleviate certain restrictions of AF2. ESMFold (87), a large language-based model (LLM), presents end-to-end MSA-free models that can predict complete structures without templates, which is impossible via AF2. Figure 3 illustrates the structural similarity prediction scores for RdRp from TuMV (Pisuviricota), a plant virus for which no experimentally determined RdRp structure is currently available, like for many plant viruses underrepresented in structural databases (such as PDB) compared with their animal or human counterparts, comparing the top-performing structure prediction methods from the aforementioned predictors. Palmprint domains, motifs, and the overall structure similarity predictions of the RdRp-TuMV predicted using AF2 (Figure 3A) were compared: AF2 vs. DeepFolding, AF2 vs. ESMFold, AF2 vs. PEZYFolding (DF), DeepFolding vs. ESMFold, PEZYFolding (DF) vs. DeepFolding, and PEZYFolding (DF) vs. ESMFold (Figures 3B–G). Metrics such as the global distance test for tertiary structure (GDT-TS), which is a standard measure of modeling accuracy, are used to evaluate the accuracy of the global backbone trace of the structure. In addition, the TM-score measures the similarity of the predicted structure to the native structure on a scale from 0 to 1, where higher scores indicate better structural alignment. The root mean square deviation for Cα atoms (RMSD-Cα) evaluates the average deviation of backbone Cα atomic positions between the predicted and the reference structures, with lower values indicating higher accuracy. ESMFold also exhibits potentially improved average performance (181). For example, when benchmarked against AF2 (Figures 3B, C), these LLM methods revealed promising performance in terms of accuracy and effectiveness (TM-score = 0.9386; GTS-TS-score = 0.9086), but still show relative limitations in contrast with AF2-based models (182, 183). More generally, another major hurdle of the top-performing machine learning-based predictors lies in their inability to account for folding kinetic pathways, specifically in some RdRps, where dynamic functions can be governed by stringent RNA template-dependent heterogeneous conformational states (43, 184–186).
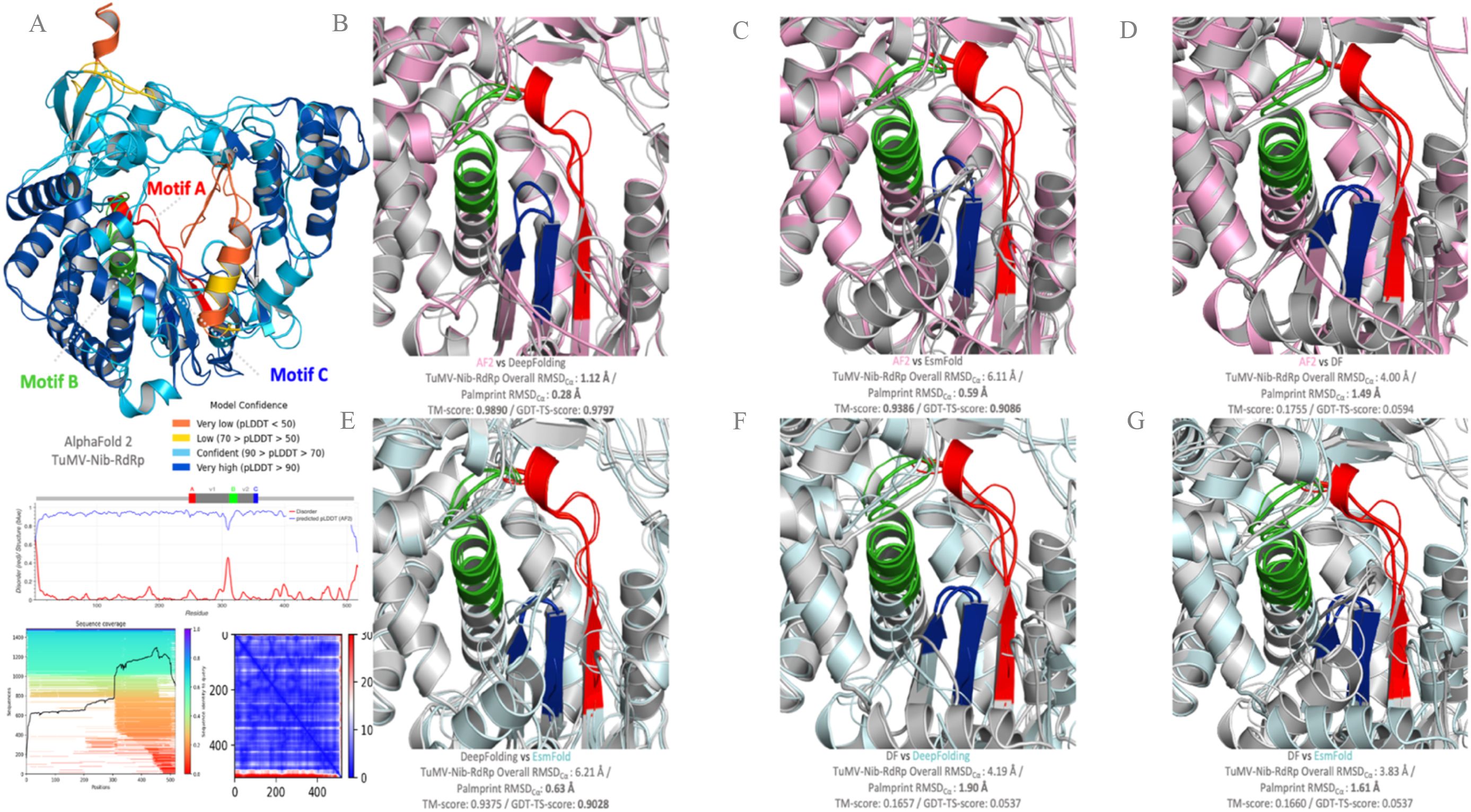
Figure 3. RNA-dependent RNA polymerase (RdRp) conformational heterogeneity within the conserved motifs predicted with AlphaFold2 (AF2) and benchmarked with other structural predictors and their 3D representations. AF2 prediction of the RdRp from Turnip mosaic virus (Pisuviricota, Patatavirales, Potyviridae, Potyvirus; TuMV-Nib, GenBank: BAA11836.1) with the per-residue local distance difference test (LDDT)-Cα and the predicted LDDT (pLDDT). The pLDDT score is colored by model confidence. (A) Palmprint domains, motifs, and overall structure similarity predictions of the RdRp-TuMV using AF2 vs. DeepFolding (B); AF2 vs. ESMFold (C); AF2 vs. PEZYFolding (DF) (D); DeepFolding vs. ESMFold (E); PEZYFolding (DF) vs. DeepFolding (F); and PEZYFolding (DF) vs. ESMFold (G). The globular domain is well predicted, but the extended interface exhibits low pLDDT and is incorrect apart from some of the secondary structure amino acids (orange and yellow). The global distance test for tertiary structure (GDT-TS) and the template modeling score (TM-score) were calculated using the TM-Align program. The AF2 predicted protein is shown in magenta color, with the motifs highlighted in colors. Predicted structures are superimposed over models with the greatest TM-scores. The references for each of the predictors used are as follows: Oda et al. (179) [PEZYFolding (DF)]; Zheng et al. (180) [Yang-Server (DF)]; Mirdita et al. (86) (ColabFold); Lin et al. (87) (ESMFold), Ahdritz et al. (178) (OpenFold); and Lee et al. (181) (DeepFold). The top scores in each metric are shown in bold. Protein structures were visualized in PyMOL (49).
More prominently, one of the overwhelming innovations for current computational structural biology is to combine natural language processing (NLP) algorithms to implement global (re-)optimization tools that capture the biophysical dynamics of the backbone foldability and their more accurate side-chain torsions including for IDRs at both local and global conformations.
Their biologically relevant states may foster learning how the linear amino acid sequence folds into space, which still lies out of reach (187). These pitfalls, which are likely to be resolved in the future, can currently lead to naive or erroneous mechanistic RdRp models with untestable hypotheses. Notwithstanding the tremendous advance recently witnessed in sequence homology and sequence-based protein structure prediction (188–190), the modeling accuracy of the ever-increasing number of viral proteins lacking fold homologs still requires improvements to better capture the IDR-mediated functional behaviors of dynamically heterogeneous multiple states. While these methods are currently unable to predict the effects of missense mutations on structural pathways (191, 192), it is conceivable that the incorporation of such physics-based experimental data from the known RdRp global mutational landscape will enable circumventing major unsolved optimization problems in future versions of protein structure prediction programs (193).
4.3 Emergent disorder-based sequence-to-ensemble relationships bring novel insights into the discernible conformational foldability dynamics of RdRps and their topology preferences
The RdRp functions are borne of the intimate set of dynamics and conformational fluctuations. Upon analysis of the palmprint database, our study revealed that the prediction of disorder propensities can exhibit substantial fluctuations across most RdRp functional domains (Figure 4) and is implicated in a variety of essential roles in viral molecular functions and diseases (78, 195). However, the observation of this variability depends on the specific criteria and algorithms used for disorder prediction (see Methods and Supplementary Material), which may yield variable outcomes. Therefore, the prediction and functional relevance of IDRs in viral RdRp proteins should be interpreted with caution, benchmarked accurately (101, 103, 152) and ideally validated through experimental approaches. The computational dissection of RdRp folds and IDR determinants—and, hence, their ensemble properties—can be deployed for the quantitative assessment of conformational magnitude and comparative sequence feature-based protein analysis to understand how functional evolution operates (119, 124, 196, 197). Arguably, one strategy for understanding hypervariability conservation in RdRps is by capturing the generic and emerging ensemble properties that can contribute to function, enabling the decoding of sequence–ensemble–function relationships (78, 99, 198).
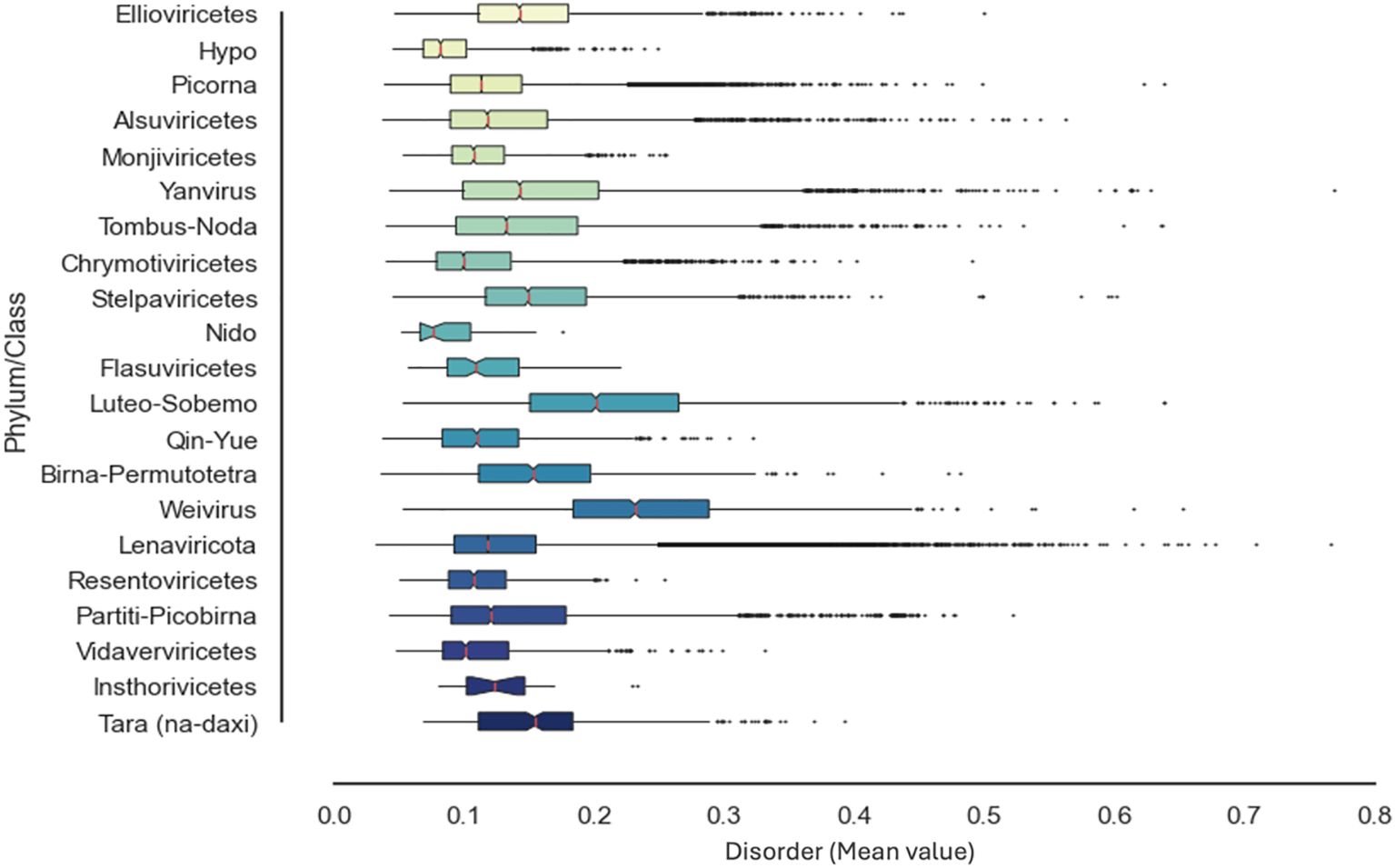
Figure 4. Distribution of the intrinsic disorder propensities in the functional RNA-dependent RNA polymerase (RdRp) domain across the RNA virus landscape. Per-sequence intrinsic disorder levels in the RdRp proteins from the Serratus RdRp palmprint database (48). Bars represent the mean protein-average disorder scores [the predicted local distance difference test predicted by AlphaFold 2 (AF2-ppLDDT, or the metapredict-ppLDDT)] in the corresponding RdRp lineages (n = 250,081sequences), whereas error dots reflect the corresponding standard deviations (see Supplementary Material for all RdRp sequences). All statistical analyses were performed using the Numpy and Scipy package in python (194).
4.3.1 Distinct patterns of sequence-to-conformational ensemble determinants in the RdRp module
Traditionally, our appreciation of how evolution has functionally diversified RdRps has relied on two main data sources: the primary protein amino acid sequence and the overall protein fold (199, 200). In contrast, the lack of knowledge about the conformational properties of RdRps is due to both the hypervariable fraction of disordered regions within proteins (201, 202) and the scarce record of RdRps that have been experimentally characterized (203, 204). Moreover, like most proteins in their cellular context (109, 205–208), part of the RdRp module is expected to bear highly dynamic structurally ambiguous ensembles of conformers embedded in intricate topologies. Although experimental structure models derived from NMR or electron paramagnetic resonance (EPR) data are currently the best means of providing a reasonably accurate experimental estimation of conformational ensembles (209, 210), RdRps still suffer from incomplete representation of their traditional rigidly fixed set of static coordinates and/or from the prediction of the most dominant single-folded AF2-assumed conformer. Similarly, residues that exist or are absent for the same protein in various X-ray structures exhibit a fractional or restricted disorder in distinctive experimental setups while being rarely statically disordered (109, 211). The overarching premise of our assumption to infer RdRp conformational diversity based on sequence–ensemble relationships is that, as amino acids co-evolve in the context of 3D dynamic (mis)folding in patterns reflective of their underlying structural functionality (212–214), then, sequence–ensemble relationships must also evolve in the context of the multiple conformational substates (conformers) that they adopt. While AF2 reached experimental accuracy at single-structure prediction, more dedicated efforts need to be garnered to improve its limited ability to predict and annotate multiple conformations of proteins (147, 215, 216). This is mainly because, for the reasons explained above, AF2 predictions find their root in complex experimental PDB data of proteins at cryogenic temperatures packed in crystals, from which it recognizes folding patterns, and do not incorporate the biophysical properties of prevalent multiple conformers (217, 218). Despite these hurdles, recent research has revealed that AF2 provided a single low-energy state for a given protein sequence can provide foldability information about the local regions of clustered proteins that may fold conditionally based on the CAID prediction dataset (97, 219, 220). This supports the relevance and impact of capturing alternative dynamic conformers through dedicated probabilistic biophysical analysis (78, 100, 221), as we are tackling here in our exploratory effort across the evolutionarily distributed RdRp landscape.
Notably, AF2-assigned low-to-very low per-residue confidence scores (pLDDT) are of particular biophysical interest as they may indicate IDRs that populate diverse sets of transiently formed structures; however, any low-confidence scores resulting from poor MSA should not be misinterpreted as IDRs (93, 121). One of the key innovations of AlphaFold is its ability to leverage evolutionary covariations derived from large-scale MSAs (222). However, the conformational “clouds” observed in AlphaFold predictions can be attributed to certain aspects of its underlying methodology (167). This is particularly important to acknowledge as IDRs pose a unique challenge due to their hypervariability across orthologs, making it difficult to uncover evolutionary constraints from alignments alone. When valid, MSAs play a critical role in the prediction of residue–residue distances. However, IDRs evolve much faster than structured regions, making their MSAs less reliable for structure prediction. This rapid evolution often results in alignments with large gaps or extended gaps due to the high variability and wide sequence lengths of orthologous IDRs.
Consequently, distance restraints within IDRs, as well as between IDRs and folded domains, are often poorly defined. The lower quality of information from MSAs for IDRs compared with intrinsically foldable domains likely contributes to the formation of diffuse “clouds” of predicted conformations around ordered domains in proteins with long IDRs. Such IDRs challenge the conventional sequence–structure–function relationships and limit our functional understanding as it is challenging to situate their molecular interactions across their evolutionary histories (78, 223). This lack of knowledge can be alleviated by exploiting biophysically informed computational tools used to model the emergent properties of marked or latent ambiguous protein behavior, such as secondary structure propensity, therefore adding relevant annotations to the AF2 predictions (224).
Delineating the diversity of RdRp conformers is a challenging task and necessarily entails an oversimplistic interpretation of complex and often elusive conformational states, including probabilistic descriptions of different interactions and (locally) diverse conformations at any given time point, prior or after various posttranslational modifications (225, 226). While accounting for these hurdles and the increasing amount of metagenomic data, we wanted to demarcate, in the most accurate and robust way possible, the spectrum of RdRp conformational behavior. Through an ensemble of machine learning-based methods that generate a wide set of biophysical features from protein sequences, combined with AF2 predictions, we attempted to identify conditional intrinsic disorder-mediated folding in order to survey the RdRp common properties and conformational specificities within a representative dataset (Figure 5A). Indeed, given the ensemble nature of proteins, RdRp conformers within their native ensemble could display similar backbone dynamics, yet diverge in the conformations of some IDR-promoting residues (196, 205, 227–229).
In terms of the overall properties and modes of intramolecular interactions of RdRps, we were interested in whether the biophysical dissection of RdRps via sequence–ensemble relationships could help us discern whether, beyond the per-residue local arrangements, certain sequence determinants are more dominant than others. For example, the relative positioning of hydrophobic and charged residues—referred to as patterning—is a key determinant of the ensemble properties in IDRs as it provides repulsive and attractive electrostatic interactions coupled with favorable free energies of solvation (230, 231; 232), involving the stabilization and possible regulation of RNA binding in RdRps (233). Order-promoting residues mostly include strong hydrophobic amino acids, which mainly enrich regular secondary structures and motifs that participate in the densely packed cores of globular domains and cellular membrane rearrangements (234, 235). More disordered segments, in contrast, are more often enforced by hydropathic residues, polar and/or charged residues. Examining the (non)foldability of pervasive peptides and the intramolecular hydrophobic topological patterning associated with their aliphatic residues therefore offers an efficient way to determine the impact of these emerging ensemble properties on disorder-based conformational signatures, even in remotely related sequences (236–238).
In line with this, capturing the regular distribution of hydrophobic motifs through systematic analysis can broaden our understanding of the driving roles of these transitory intramolecular interactions within regular transient secondary structures and reveal novel stabilization factors without prior knowledge of homogeneous sequences or consideration of pre-calculated properties (239). Hence, this offers useful proven elements to distinguish cases where AF2 low-confidence scores identify foldable IDRs from cryptic or ill-predicted structured regions (100, 240) and ultimately may help decipher the evolutionary constraints of RdRp conformational plasticity more effectively. We take advantage of this knowledge and our sequence-based RdRp ensemble approach to explore the correspondence between the clustering and patterning of these different residues and their impact on the structural diversity of RdRps (Figure 5B). This is consistent with previous studies on polymer scaling behavior (132, 231, 241–243), which combines hydropathy and charge patterning. These sequence features can enable the unique comparative characterization of the RdRp conformational distribution properties embedded in both conditionally foldable and transient linear IDR residues, therefore enabling in-depth analysis of the structural landscape of RdRps, beyond classical methods. In another example, shown in Figure 5C, interconnecting the RdRp backbone and side-chain dynamics with the propensity for polyproline II (PPII helices), as previously done for other proteins (244), can aid in understanding the molecular mechanisms of the structural plasticity of RdRps and their tethered IDR-based conformations. While side-chain dynamics provides a continuous and subtle picture of residue behavior with regard to their backbone rigidity and, by extension, to residue order and disorder (94), PPII helices have been frequently observed in disordered regions of proteins.
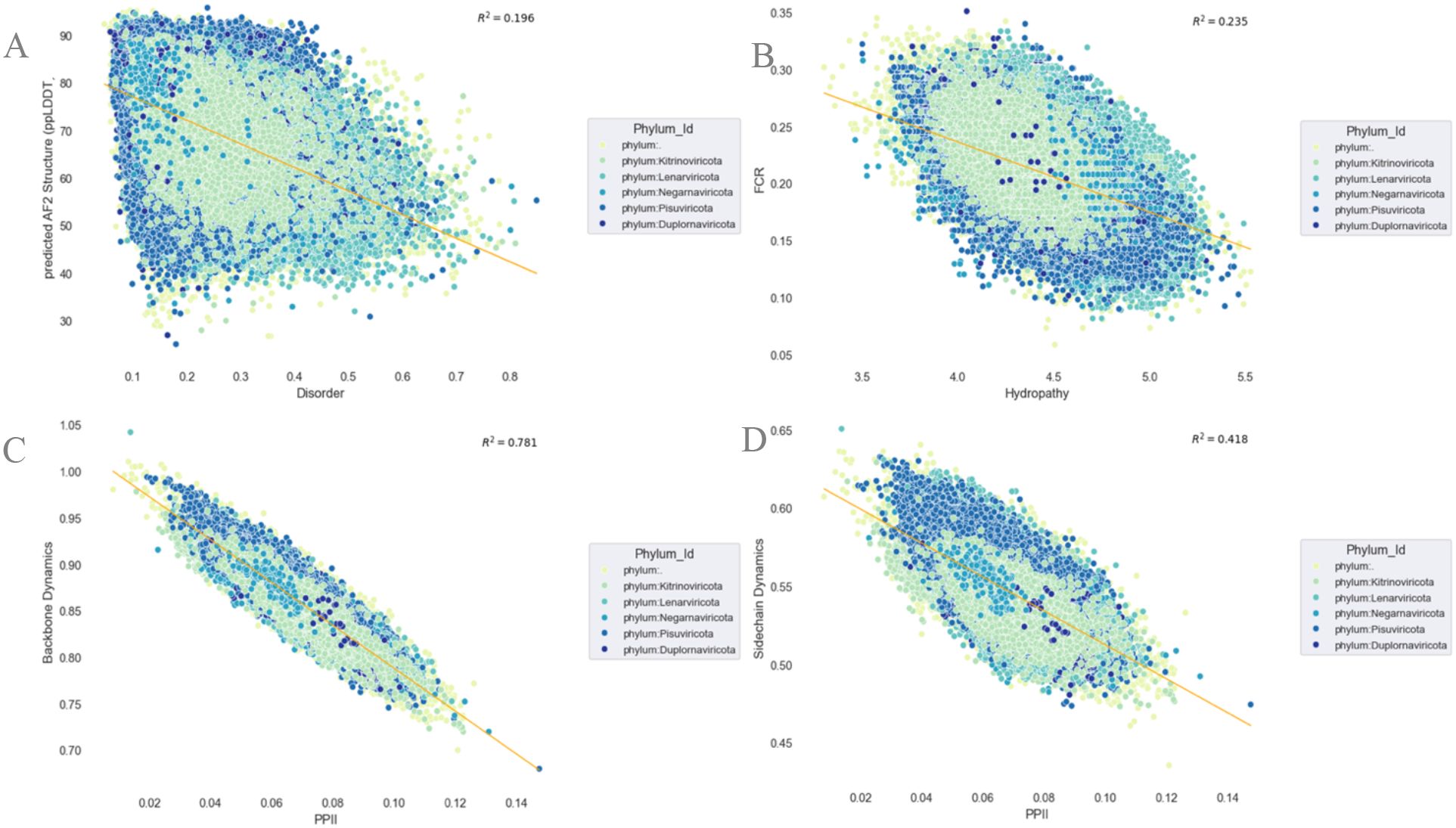
Figure 5. Emergent RNA-dependent RNA polymerase (RdRp) sequence-to-conformational ensemble relationships and their structural determinants across the RNA virus landscape. Per-sequence relations and propensity predictions between the predicted scores of the protein structure [the predicted local distance difference test predicted by AlphaFold 2 (AF2-ppLDDT, or metapredict-ppLDDT] (structure; see details in Figure 2) vs. protein intrinsic disorder (IDP) (A); fraction of charged residues (FCR) vs. hydropathy (B); backbone dynamics vs. polyproline II (PPII) (C); and side-chain dynamics vs. PPII (D) (see Supplementary Material for all RdRp sequences). All statistical analysis and R2 calculation were performed using the Numpy and Scipy package in python (194).
Unlike common helical structures, PPII helices have little to no hydrogen bonding capacity and have been observed to play a role in interactions between the protein domains. As natively unfolded peptides possess some degree of local order in their backbones, we examined the relationship between RdRp sequence-encoded PPII propensities and conformational dynamics. Proline possesses unique and distinct structural properties, such as the ability to disrupt the propagation of regular secondary structural elements, promoting α-helix nucleation and coil formation and locally contributing to backbone stiffening (245, 246). PPII is therefore an interesting multifaceted candidate for the detection of specific local conformational preferences across the RdRp sequence landscape of RNA viruses. The downstream responses of protein dynamics and related functions are also often dictated by signaling cascade cues that are triggered by posttranslational modifications, particularly within the proline-rich short linear motifs (184).
Thereby, as previously evidenced, the conformational properties of IDRs prior to and following multisite phosphorylation (Ser/Thr/Tyr) are directly relevant to disentangling the functions of RdRps. In particular, divergent RdRps folds that are globally well predicted by AF2, therefore capturing the main folded state, but that yet harbor enhanced conformational plasticity, remain difficult to examine using standard methods. We further speculate that the accessibility and downstream binding of RdRps to specific substrates is dictated by the sequence-encoded interplay of local and global conformational properties, including IDRs prior to and following phosphorylation (247–249).
Although the effects of (non-)local expansion and compaction involving synergistic long-range conformational relationships can leave global dimensions rather unperturbed, the manner in which various RdRp regions react to multisite phosphorylation is sequence context-dependent and strongly relies on the pattern of specific amino acid residues therein. As previously reported (231), no correlation between global PPII occupancy and the measurable global dimensions of protein conformations (e.g., Rg or polymer radius of gyration) can be found, advocating compensatory effects where sequences with similar PPII propensities can have very different effects on the ensemble average structure of the polymer (176). Remarkably, our exploratory survey revealed a strong anti-correlation between the RdRp side-chain dynamics and PPII (Figure 5D), which recapitulates the sequence-local effects of the interplay between backbone and side-chain dynamics motions that is often critical for optimizing protein–ligand interactions in the substrate-specific binding affinity of function-bearing motifs (250).
Notably, intrachain segregation patterns driven by repulsions are encoded by the RdRp amino acid sequence, which directs the overall conformational properties prior to and upon multisite phosphorylation. Interestingly, the proline-patterning and charged residue decorations with respect to all other residues change only slightly upon multisite phosphorylation (118). Such effects can be very important in the context of, for example, human health, where single mutations may lead to protein instabilities and malfunctions that cause human pathologies (251, 252).
In summary, our initial exploration highlighted the need for further investigation to fully characterize the effect of PPII-rich structures through experimental observables and to unravel the biological significance of these trends in viral replication. Through selected examples and a large sequence–ensemble dataset, this section only touched the surface of how the coexistence of charge decoration and hydrophobic cores, folding dynamics, and ensemble dimensions of well-folded regions and disordered contexts can constitute major factors in the cross-interaction-driven interplay of emerging synergistic or competing modulators of RdRp conformation. Furthermore, we anticipate that, with all recent major model updates of AF2/ColabFold to better capture cross-configuration (253, 254), the challenge in systematically relating the various computational tools and diverse data sources will be in this respect invaluable for the direct interpretation of RdRps spanning a diverse range of secondary structures affected by diverse conformational ensembles, as previously underlined. Consequently, understanding the hierarchical impacts of sequence determinants and structural contexts on the conformational properties of RdRps remains an open challenge. More crucially, accurate identification of fold-switching regions would aid in identifying new or cryptic (un)folded RdRp structures and promiscuous interactions that are targetable by antiviral therapeutics. Large-scale automated annotations and predictions will illuminate the foundational principles of context-sensitive RdRp folding dynamics, host-dependent functionalities, and trace molecular innovations over co-evolutionary histories that underlie the known and unknown RNA virosphere yet to be characterized.
5 Conclusion: outlooks on perspectives for RNA virus discovery and RdRp conformational diversity
The hallmark RdRps are among the best-studied proteins that modulate viral replication and many disease processes. RdRps, similarly to most native proteins, are often treated as rigid polypeptides, as observed in their crystal-packed conformation. However, this single homogeneous conformation does not capture the functional ensemble exploited by viruses and is not necessarily the most populated one in solution, with crystal structures likely altered by crystallization effects. The main thesis of this review is that RNA virus discovery can benefit from considering the evolution of the RdRp protein as structural ensembles—a continuum between multiple folded and disordered states under functional constraints (120)—which is a conformational sampling mechanism commonly exploited by viruses (77). Tapping into the broadly distributed ensembles of this conformational flexibility during replication provides viruses with selected adaptive solutions as a competent biological response to modulate cognate binding partners or disfavor unwanted interfaces, and more generally to adapt to their changing environment.
In our systematic review, we have underlined that structural and conformational variability analyses of the RdRp core domain and hypervariable IDRs are central to gaining a more nuanced probabilistic understanding of the mechanisms of RdRp-based RNA synthesis adopted by viruses of different taxonomical categories and evolutionary lineages. This also helps in capturing commonalities and generic themes defined by biophysical feature fluctuations of diverse magnitude.
In addition, these various prominent RdRp biophysical properties suggest other modulatory clues for the adaptive co-evolution of viruses in relation to hosts. Within this framework, we also succinctly reviewed the recent state-of-the-art computational progress in these domains, which allowed addressing the challenging new biological insights through large-scale structural analyses of RdRp sequences, elaborating on prior experimentally and computationally driven investigations in this landscape (255). Aware of its infancy, we expect rapid progress on multiple fronts, including virus discovery (84). On a first front, the prediction speed and memory efficiency remain bottlenecks, particularly when predicting multiple conformations, which must be applied at scale to assemble a comprehensive picture of the global RdRp modulome.
On another front, in many novel RdRps, little is known with regard to the repertoire of tolerated amino acids in proteins when only a few or no sequence homologs are available or that are MSA-recalcitrant. Although the surge of novel RdRp-based RNA virus discovery has expanded our understanding of the molecular mechanisms of viral replication and evolutionary diversity, the foundations of our current knowledge of these processes still remain primarily built on previous sequence-based research on RNA viruses and phylogenetic inference. The ability of AF2, and of artificial intelligence-based methods in general, to accurately provide an accounting of the conformational landscape of proteins remains limited. Recent appealing advances in a range of more proficient deep learning-based algorithms might emerge. More accurate, memory-cheap, and stable AF3-aware re-implementations could make it possible to tackle richer multipurpose representations of the RdRp sequence-to-conformation space. We stress that corroborating our understanding with more structural and conformation-driven information will likely reveal novel properties and processes shared between viral species, which are better informed and of potential value not only for novel AF3-modeled structural diversity (88) and evolutionary analysis but also for the design of efficient antiviral strategies.
Next, and of paramount importance, is the relevance of highlighting poorly understood biophysical properties that can have intricate and fluctuating effects on the spatiotemporal propensities that tune the function of RdRps in their evolving cellular context. For example, the drivers of dramatic conformational and structural switches are still largely understudied, including the repertoire of sequence-to-conformation determinants directly involved in operating the viral replication cycle. How these factors may direct, via spatial seclusion, dedicated virus-induced subcellular compartments and membranous vesicles to finally serve as replication factories is not clear, despite this feature being shared among many emerging or pandemic positive-strand RNA plant, animal, and human viruses during the earliest infection stages prior to viral propagation. Our review has some limitations. Among these, RdRp-based RNA virus discovery at diverse evolutionary scales is challenging due to the high sequence divergence, which makes RdRp sequence-to-conformation biophysics insufficient to reflect the true evolutionary histories of Riboviria. This highlights the importance of integrating complementary approaches to gain mechanistic residue-level insights into the RdRp sequence determinants.
Together, this systematic review emphasized, through multiple examples, emerging concepts on the complementary power of recent advances in sequence-based resources that are directly anchored in experimental data and closely connected fundamental principles of protein side-chain dynamics, early folding, and disorder-based conformational dimensions. To conclude, we hope to have provided better-informed and well-suited directly applicable investigational routes to infer the diversity features of RdRps across the Orthornavirae classification in light of their biological significance. This strengthens the foundations of bridging these concepts with the One Health outstanding challenges and paves ways for convenient generalizable routes to mitigate future viral outbreaks of uncharacterized or newly discovered viruses while anticipating the forthcoming vast expansion of the global RNA virosphere.
Data availability statement
Recent and publicly available datasets were utilized in this review. Data analysis used Python v.3.6 (https://www.python.org/), NumPy v.1.16.4 (https://github.com/numpy/numpy), SciPy v.1.2.1 (https://www.scipy.org/), seaborn v.0.11.1 (https://github.com/mwaskom/seaborn), scikit-learn v.0.24.0 (https://github.com/scikit-learn), Matplotlib v.3.3.4 (https://github.com/matplotlib/matplotlib), pandas v.1.1.5 (https://github.com/pandas-dev/pandas). All input data are freely available from public sources. The publicly available datasets analyzed in this study can be found here: 10 (Serratus); 4, 5, 81, 84. Raw sequence reads newly generated in this study are available at the NCBI Sequence Read Archive (SRA) database. Source code for the AF2 model, trained weights, and an inference script are available under an open-source license at https://github.com/deepmind/alphafold. Neural network analyses were carried out with TensorFlow v.1 (https://github.com/tensorflow/tensorflow). TM-align v.20190822 (https://zhanglab.dcmb.med.umich.edu/TM-align) was used for computing TM-scores. Structure analysis used Pymol v.2.3.0 (https://github.com/schrodinger/pymol-open-source). Due to size/format constraints [or specify reason], we are committed to sharing all per-residue data directly with interested researchers and keep it available upon request.
Author contributions
RT: Conceptualization, Writing – original draft, Writing – review & editing, Methodology, Data curation, Formal analysis, Visualization, Investigation, Supervision, Project administration. JC: Conceptualization, Writing – review & editing, Supervision. AD: Software, Methodology, Resources, Writing – original draft. KD: Writing – review & editing, Funding acquisition, Project administration. SM: Writing – review & editing, Supervision, Project administration. TM: Writing – review & editing. WV: Conceptualization, Resources, Writing – review & editing, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. RT was funded by the Belgian Federal Public Service Health, Food Chain Safety and Environment through the contract GenoPREDICT (RF 22/635). JC and TM were supported by the ID4VISA grant from the French National Research Agency (ANR-21-CE35-0009). WV acknowledges the Research Foundation Flanders (FWO) G.0328.16N project grant and the International Research Infrastructure grant I000323N.
Acknowledgments
RT wishes to thank Prof. A. Babaian, Dr. U. Neri, Prof. S. Nakagawa, Dr. S. Roux, and Prof. O. Peersen for insightful discussions during the RdRp Summit and before publication. RT also wishes to acknowledge access to computational resources from the Flemish Supercomputer Center (VSC–Vlaams Supercomputer Centrum), University of Brussels.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2025.1501616/full#supplementary-material
References
1. Wolf YI, Kazlauskas D, Iranzo J, Lucía-Sanz A, Kuhn JH, Krupovic M, et al. Origins and evolution of the global RNA virome. MBio. (2018) 9:10–1128. doi: 10.1128/mBio.02329-18
2. Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, et al. Redefining the invertebrate RNA virosphere. Nature. (2016) 540:539–43. doi: 10.1038/nature20167
3. Koonin EV, Dolja VV, Krupovic M, Varsani A, Wolf YI, Yutin N, et al. Global organization and proposed megataxonomy of the virus world. Microbiol Mol Biol Rev. (2020) 84:10–1128. doi: 10.1128/MMBR.00061-19
4. Neri U, Wolf YI, Roux S, Camargo AP, Lee B, Kazlauskas D, et al. Expansion of the global RNA virome reveals diverse clades of bacteriophages. Cell. (2022) 185:4023–4037.e18. doi: 10.1016/j.cell.2022.08.023
5. Zayed AA, Wainaina JM, Dominguez-Huerta G, Pelletier E, Guo J, Mohssen M, et al. Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science. (2022) 376:156–62. doi: 10.1126/science.abm5847
6. Charon J, Buchmann JP, Sadiq S, and Holmes EC. RdRp-scan: A bioinformatic resource to identify and annotate divergent RNA viruses in metagenomic sequence data. Virus Evol. (2022) 8:veac082. doi: 10.1093/ve/veac082
7. Dominguez-Huerta G, Wainaina JM, Zayed AA, Culley AI, Kuhn JH, and Sullivan MB. The RNA virosphere: How big and diverse is it? Environ Microbiol. (2023) 25:209–15. doi: 10.1111/1462-2920.16312
8. Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, et al. The evolutionary history of vertebrate RNA viruses. Nature. (2018) 556:197–202. doi: 10.1038/s41586-018-0012-7
9. Chen YM, Sadiq S, Tian JH, Chen X, Lin XD, Shen JJ, et al. RNA viromes from terrestrial sites across China expand environmental viral diversity. Nat Microbiol. (2022) 7:1312–23. doi: 10.1038/s41564-022-01180-2
10. Edgar RC, Taylor B, Lin V, Altman T, Barbera P, Meleshko D, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature. (2022) 602:142–7. doi: 10.1038/s41586-021-04332-2
11. Rivarez MPS, Pecman A, Bačnik K, Maksimović O, Vučurović A, Seljak G, et al. In-depth study of tomato and weed viromes reveals undiscovered plant virus diversity in an agroecosystem. Microbiome. (2023) 11:60. doi: 10.1186/s40168-023-01500-6
12. Yi B, Deng Q, Guo C, Li X, Wu Q, Zha R, et al. Evaluating the zoonotic potential of RNA viromes of rodents provides new insight into rodent-borne zoonotic pathogens in Guangdong, China. One Health (Amsterdam Netherlands). (2023) 17:100631. doi: 10.1016/j.onehlt.2023.100631
13. Koonin EV. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J Gen Virol. (1991) 72:2197–206. doi: 10.1099/0022-1317-72-9-2197
14. Mönenen HA, Ravantti JJ, Stuart DI, and Poranen MM. Automated structural comparisons clarify the phylogeny of the right-hand-shaped polymerases. Mol Biol Evol. (2014) 31:2741–52. doi: 10.1093/molbev/msu219
15. Iyer LM, Koonin EV, and Aravind L. Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct Biol. (2003) 3:1. doi: 10.1186/1472-6807-3-1
16. Appel N, Schaller T, Penin F, and Bartenschlager R. From structure to function: new insights into hepatitis C virus RNA replication. J Biol Chem. (2006) 281:9833–6. doi: 10.1074/jbc.r500026200
17. Mercorelli B, Palù G, and Loregian A. Drug Repurposing for Viral Infectious Diseases: How Far Are We? Trends Microbiol. (2018) 26:865–76. doi: 10.1016/j.tim.2018.04.004
18. Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. (2020) 368:779–82. doi: 10.1126/science.abb7498
19. Peng R, Xu X, Jing J, Wang M, Peng Q, Liu S, et al. Structural insight into arenavirus replication machinery. Nature. (2020) 579:615–9. doi: 10.1038/s41586-020-2114-2
20. Watkins CL, Kempf BJ, Beaucourt S, Barton DJ, and Peersen OB. Picornaviral polymerase domain exchanges reveal a modular basis for distinct biochemical activities of viral RNA-dependent RNA polymerases. J Biol Chem. (2020) 295:10624–37. doi: 10.1074/jbc.RA120.013906
21. Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, and Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. (2020) 584:154–6. doi: 10.1038/s41586-020-2368-8
22. Campagnola G, Govindarajan V, Pelletier A, Canard B, and Peersen OB. The SARS-coV nsp12 polymerase active site is tuned for large-genome replication. J Virol. (2022) 96:e0067122. doi: 10.1128/jvi.00671-22
23. Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, García-Sastre A, et al. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci. (2010) 107:11525–30. doi: 10.1073/pnas.1001984107
24. Liu W, Shi X, and Gong P. A unique intra-molecular fidelity-modulating mechanism identified in a viral RNA-dependent RNA polymerase. Nucleic Acids Res. (2018) 46:10840–54. doi: 10.1093/nar/gky848
25. Beerens N, Selisko B, Ricagno S, Imbert I, van der Zanden L, Snijder EJ, et al. De novo initiation of RNA synthesis by the arterivirus RNA-dependent RNA polymerase. J Virol. (2007) 81:8384–95. doi: 10.1128/jvi.00564-07
26. Appleby TC, Perry JK, Murakami E, Barauskas O, Feng J, Cho A, et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science. (2015) 347:771–5. doi: 10.1126/science.1259210
27. Wu R, Davison MR, Gao Y, Nicora CD, Mcdermott JE, Burnum-Johnson KE, et al. Moisture modulates soil reservoirs of active DNA and RNA viruses. Commun Biol. (2021) 4:.992. doi: 10.1038/s42003-021-02514-2
28. Choi KH, Groarke JM, Young DC, Kuhn RJ, Smith JL, Pevear DC, et al. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc Natl Acad Sci. (2004) 101:4425–30. doi: 10.1073/pnas.0400660101
29. Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, and Verdaguer N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J Biol Chem. (2004) 279:47212–21. doi: 10.1074/jbc.M405465200
30. Hengrung N, El Omari K, Serna Martin I, Vreede FT, Cusack S, Rambo RP, et al. Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nature. (2015) 527:114–7. doi: 10.1038/nature15525
31. Gong P and Peersen OB. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci. (2010) 107:22505–10. doi: 10.1073/pnas.1007626107
32. Gerlach P, Malet H, Cusack S, and Reguera J. Structural insights into bunyavirus replication and its regulation by the vRNA promoter. Cell. (2015) 161:1267–79. doi: 10.1016/j.cell.2015.05.006
33. Fan H, Walker AP, Carrique L, Keown JR, Serna Martin I, Karia D, et al. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature. (2019) 573:287–90. doi: 10.1038/s41586-019-1530-7
34. Puustinen P and Mäkinen K. Uridylylation of the potyvirus VPg by viral replicase NIb correlates with the nucleotide binding capacity of VPg. J Biol Chem. (2004) 279:38103–10. doi: 10.1074/jbc.M402910200
35. Lauber C, Zhang X, Vaas J, Klingler F, Mutz P, Dubin A, et al. Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates. PloS Pathog. (2024) 20:e1012163. doi: 10.1371/journal.ppat.1012163
36. Soh YS, Moncla LH, Eguia R, Bedford T, and Bloom JD. Comprehensive mapping of adaptation of the avian influenza polymerase protein PB2 to humans. eLife. (2019) 8:e45079. doi: 10.7554/eLife.45079
37. Tan CCS, van Dorp L, and Balloux F. The evolutionary drivers and correlates of viral host jumps. Nat Ecol Evol. (2024) 8(5):960–71. doi: 10.1038/s41559-024-02353-4
38. Yin W, Mao C, Luan X, Shen DD, Shen Q, Su H, et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. (2020) 368:1499–504. doi: 10.1126/science.abc1560
39. Yang X, Smidansky ED, Maksimchuk KR, Lum D, Welch JL, Arnold JJ, et al. Motif D of viral RNA-dependent RNA polymerases determines efficiency and fidelity of nucleotide addition. Structure (London England: 1993). (2012) 20:1519–27. doi: 10.1016/j.str.2012.06.012
40. Gunawardene CD, Donaldson LW, and White KA. Tombusvirus polymerase: Structure and function. Virus Res. (2017) 234:74–86. doi: 10.1016/j.virusres.2017.01.012
41. Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure. (1999) 7:1417–26. doi: 10.1016/s0969-2126(00)80031-3
42. Peng Q, Liu Y, Peng R, Wang M, Yang W, Song H, et al. Structural insight into RNA synthesis by influenza D polymerase. Nat Microbiol. (2019) 4:1750–9. doi: 10.1038/s41564-019-0487-5
43. Wang M, Li R, Shu B, Jing X, Ye HQ, and Gong P. Stringent control of the RNA-dependent RNA polymerase translocation revealed by multiple intermediate structures. Nat Commun. (2020) 11:2605. doi: 10.1038/s41467-020-16234-4
44. Malone B, Urakova N, Snijder EJ, and Campbell EA. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat Rev Mol Cell Biol. (2022) 23:21–39. doi: 10.1038/s41580-021-00432-z
45. Jia H and Gong P. A structure-function diversity survey of the RNA-dependent RNA polymerases from the positive-strand RNA viruses. Front Microbiol. (2019) 10:1945. doi: 10.3389/fmicb.2019.01945
46. Emenecker RJ, Griffith D, and Holehouse AS. Metapredict: a fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biophys J. (2021) 120:4312–9. doi: 10.1016/j.bpj.2021.08.039
47. Grau I, Nowé A, and Vranken W. Interpreting a black box predictor to gain insights into early folding mechanisms. Comput Struct Biotechnol J. (2021) 19:4919–30. doi: 10.1016/j.csbj.2021.08.041
48. Babaian A and Edgar R. Ribovirus classification by a polymerase barcode sequence. PeerJ. (2022) 10:e14055. doi: 10.7717/peerj.14055
50. Bruenn JA. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. (2003) 31:1821–9. doi: 10.1093/nar/gkg277
51. Ferrero DS, Buxaderas M, Rodríguez JF, and Verdaguer N. The structure of the RNA-dependent RNA polymerase of a permutotetravirus suggests a link between primer-dependent and primer-independent polymerases. PloS Pathog. (2015) 11:e1005265. doi: 10.1371/journal.ppat.1005265
52. Te Velthuis AJ, Arnold JJ, Cameron CE, Van Den Worm SH, and Snijder EJ. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. (2010) 38:203–14. doi: 10.1093/nar/gkp904
53. Te Velthuis AJ. Common and unique features of viral RNA-dependent polymerases. Cell Mol Life Sci. (2014) 71:4403–20. doi: 10.1007/s00018-014-1695-z
54. Te Velthuis AJ and Oymans J. Initiation, elongation, and realignment during influenza virus mRNA synthesis. J Virol. (2018) 92:10–1128. doi: 10.1128/JVI.01775-17
55. Gorbalenya AE, Koonin EV, Donchenko AP, and Blinov VM. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. (1989) 17:4847–61. doi: 10.1093/nar/17.12.4847
56. Gorbalenya AE, Pringle FM, Zeddam JL, Luke BT, Cameron CE, Kalmakoff J, et al. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J Mol Biol. (2002) 324:47–62. doi: 10.1016/s0022-2836(02)01033-1
57. Pan J, Vakharia VN, and Tao YJ. The structure of a birnavirus polymerase reveals a distinct active site topology. PNAS. (2007) 104:7385–90. doi: 10.1073/pnas.0611599104
58. Shu B and Gong P. Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. PNAS. (2016) 113:E4005–14. doi: 10.1073/pnas.1602591113
59. Sabanadzovic S, Ghanem-Sabanadzovic NA, and Gorbalenya AE. Permutation of the active site of putative RNA-dependent RNA polymerase in a newly identified species of plant alpha-like virus. Virology. (2009) 394:1–7. doi: 10.1016/j.virol.2009.08.006
60. Tian Z, Hu T, Holmes EC, Ji J, and Shi W. Analysis of the genetic diversity in RNA-directed RNA polymerase sequences: implications for an automated RNA virus classification system. Virus Evol. (2024) 10:veae059. doi: 10.1093/ve/veae059
61. Gibbs EB, Lu F, Portz B, Fisher MJ, Medellin BP, Laremore TN, et al. Phosphorylation induces sequence-specific conformational switches in the RNA polymerase II C-terminal domain. Nat Commun. (2017) 8:15233. doi: 10.1038/ncomms15233
62. Peersen OB. A comprehensive superposition of viral polymerase structures. Viruses. (2019) 11:745. doi: 10.3390/v11080745
63. Imbert I, Guillemot JC, Bourhis JM, Bussetta C, Coutard B, Egloff MP, et al. A second, non-canonical RNA-dependent RNA polymerase in SARS Coronavirus. EMBO J. (2006) 25:4933–42. doi: 10.1038/sj.emboj.7601368
64. Morin B, Kranzusch PJ, Rahmeh AA, and Whelan SP. The polymerase of negative-stranded RNA viruses. Curr Opin Virol. (2013) 3:103–10. doi: 10.1016/j.coviro.2013.03.008
65. Bi P, Shu B, and Gong P. Crystal structure of the coxsackievirus A16 RNA-dependent RNA polymerase elongation complex reveals novel features in motif A dynamics. Virologica Sin. (2017) 32:548–52. doi: 10.1007/s12250-017-4066-8
66. Wright PE and Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. (1999) 293:321–31. doi: 10.1006/jmbi.1999.3110
67. Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, and Tawfik DS. Do viral proteins possess unique biophysical features? Trends Biochem Sci. (2009) 34:53–9. doi: 10.1016/j.tibs.2008.10.009
68. Jiang Y, Yin W, and Xu HE. RNA-dependent RNA polymerase: Structure, mechanism, and drug discovery for COVID-19. Biochem Biophys Res Commun. (2021) 538:47–53. doi: 10.1016/j.bbrc.2020.08.116
69. Salmon L, Nodet G, Ozenne V, Yin G, Jensen MR, Zweckstetter M, et al. NMR characterization of long-range order in intrinsically disordered proteins. J Am Chem Soc. (2010) 132:8407–18. doi: 10.1021/ja101645g
70. Parigi G, Rezaei-Ghaleh N, Giachetti A, Becker S, Fernandez C, Blackledge M, et al. Long-range correlated dynamics in intrinsically disordered proteins. J Am Chem Soc. (2014) 136:16201–9. doi: 10.1021/ja506820r
71. Holehouse AS and Kragelund BB. The molecular basis for cellular function of intrinsically disordered protein regions. Nat Rev Mol Cell Biol. (2024) 25:187–211. doi: 10.1038/s41580-023-00673-0
72. Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, et al. Intrinsically disordered protein. J Mol Graphics Model. (2001) 19:26–59. doi: 10.1016/s1093-3263(00)00138-8
73. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, and Obradović Z. Intrinsic disorder and protein function. Biochemistry. (2002) 41:6573–82. doi: 10.1021/bi012159+
74. Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. (2002) 27:527–33. doi: 10.1016/s0968-0004(02)02169-2
75. Lemke EA, Babu MM, Kriwacki RW, Mittag T, Pappu RV, Wright PE, et al. Intrinsic disorder: A term to define the specific physicochemical characteristic of protein conformational heterogeneity. Mol Cell. (2024) 84:1188–90. doi: 10.1016/j.molcel.2024.02.024
76. Necci M, Piovesan D, and Tosatto SC. Critical assessment of protein intrinsic disorder prediction. Nat Methods. (2021) 18:472–81. doi: 10.1038/s41592-021-01117-3
77. González-Foutel NS, Glavina J, Borcherds WM, Safranchik M, Barrera-Vilarmau S, Sagar A, et al. Conformational buffering underlies functional selection in intrinsically disordered protein regions. Nat Struct Mol Biol. (2022) 29:781–90. doi: 10.1038/s41594-022-00811-w
78. Tesei G, Trolle AI, Jonsson N, Betz J, Knudsen FE, Pesce F, et al. Conformational ensembles of the human intrinsically disordered proteome. Nature. (2024) 626:897–904. doi: 10.1038/s41586-023-07004-5
79. Tahzima R, Haegeman A, Massart S, and Hébrard E. Flexible spandrels of the global plant virome – IPD at the virus host vector interplay. In: Dancing protein clouds, Chapter 11. The Netherlands. Amsterdam: Academic Press (2021). p. 166. Prog. Mol. Biol. Transl. Sci. doi: 10.1016/bs.pmbts.2021.06.007
80. De Farias ST, Dos Santos Junior AP, Rêgo TG, and José MV. Origin and evolution of RNA-dependent RNA polymerase. Front Genet. (2017) 8:125. doi: 10.3389/fgene.2017.00125
81. Olendraite I, Brown K, and Firth AE. Identification of RNA virus-derived rdRp sequences in publicly available transcriptomic data sets. Mol Biol Evol. (2023) 40:msad060. doi: 10.1093/molbev/msad060
82. Suzek BE, Huang H, McGarvey P, Mazumder R, and Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. (2007) 23:1282–8. doi: 10.1093/bioinformatics/btm098
83. UniProt Consortium. The UniProt: the universal protein knowledgebase in 2021. . Nucleic Acids Res. (2021) 49 :D480–9. doi: 10.1093/nar/gkaa1100
84. Hou X, He Y, Fang P, Mei SQ, Xu Z, Wu WC, et al. Using artificial intelligence to document the hidden RNA virosphere. Cell. (2024) 187:6929–6942.e16. doi: 10.1016/j.cell.2024.09.027
85. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
86. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. (2022) 19:679–82. doi: 10.1038/s41592-022-01488-1
87. Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science. (2023) 379:1123–30. doi: 10.1126/science.ade2574
88. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with AlphaFold-3. . Nat. (2024) 630(8016):493–500. doi: 10.1038/s41586-024-07487-w
89. DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific (2002). Available online at: http://www.pymol.org/ (Accessed September 2024).
90. Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. (2018) 27:14–25. doi: 10.1002/pro.3235
91. Zhang Y and Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. (2005) 33:2302–9. doi: 10.1093/nar/gki524
92. Kagami LP, Orlando G, Raimondi D, Ancien F, Dixit B, Gavaldá-García J, et al. b2bTools: online predictions for protein biophysical features and their conservation. Nucleic Acids Res. (2021) 49:W52–9. doi: 10.1093/nar/gkab425
93. Gavalda-Garcia J, Dixit B, Díaz A, Ghysels A, and Vranken W. Gradations in protein dynamics captured by experimental NMR are not well represented by AlphaFold2 models and other computational metrics. J Mol Biol. (2024) 437(2):168900. doi: 10.1016/j.jmb.2024.168900
94. Cilia E, Pancsa R, Tompa P, Lenaerts T, and Vranken WF. From protein sequence to dynamics and disorder with DynaMine. . Nat Commun. (2013) 4:2741. doi: 10.1038/ncomms3741
95. Orlando G, Raimondi D, Kagami LP, and Vranken WF. ShiCrypt: a web server to understand and biophysically align proteins through their NMR chemical shift values. Nucleic Acids Res. (2020) 48:W36–40. doi: 10.1093/nar/gkaa391
96. Raimondi D, Orlando G, Pancsa R, Khan T, and Vranken WF. Exploring the sequence-based prediction of folding initiation sites in proteins. Sci Rep. (2017) 7:8826. doi: 10.1038/s41598-017-08366-3
97. Piovesan D, Monzon AM, and Tosatto SC. Intrinsic protein disorder and conditional folding in AlphaFoldDB. Protein Sci. (2022) 31:e4466. doi: 10.1002/pro.4466
98. Gavalda-Garcia J, Dixit B, Díaz A, Ghysels A, and Vranken W. Gradations in protein dynamics captured by experimental NMR are not well represented by AlphaFold2 models and other computational metrics. J Mol Biol. (2025) 437:168900. doi: 10.1016/j.jmb.2024.168900
99. Lotthammer JM, Ginell GM, Griffith D, Emenecker R, and Holehouse AS. Direct prediction of intrinsically disordered protein conformational properties from sequence. Biophys J. (2024) 123:43a. doi: 10.1038/s41592-023-02159-5
100. Alderson TR, Pritisanac I, Kolaric D, Moses AM, and Forman-Kay JD. Systematic identification of conditionally folded intrinsically disordered regions by AlphaFold2 Proceedings. Natl Acad Sci. (2023) 120:e2304302120. doi: 10.1073/pnas.2304302120
101. Del Conte A, Bouhraoua A, Mehdiabadi M, Clementel D, Monzon AM, CAID predictors, et al. CAID prediction portal: a comprehensive service for predicting intrinsic disorder and binding regions in proteins. Nucleic Acids Res. (2023) 51:W62–9. doi: 10.1093/nar/gkad430
102. Wang K, Hu G, Basu S, and Kurgan L. flDPnn2: accurate and fast predictor of intrinsic disorder in proteins. J Mol Biol. (2024) 436:168605. doi: 10.1016/j.jmb.2024.168605
103. Han KS, Song SR, Pak MH, Kim CS, Ri CP, Del Conte A, et al. PredIDR: Accurate prediction of protein intrinsic disorder regions using deep convolutional neural network. Int J Biol macromolecules. (2025) 284:137665. doi: 10.1016/j.ijbiomac.2024.137665
104. Guo HB, Perminov A, Bekele S, Kedziora G, Farajollahi S, Varaljay V, et al. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci Rep. (2022) 12:10696. doi: 10.1038/s41598-022-14382-9
105. Sakaguchi S, Urayama SI, Takaki Y, Hirosuna K, Wu H, Suzuki Y, et al. NeoRdRp: A comprehensive dataset for identifying RNA-dependent RNA polymerases of various RNA viruses from metatranscriptomic data. . Microbes environments. (2022) , 37:ME22001. doi: 10.1264/jsme2.ME22001
106. Fontdevila Pareta N, Khalili M, Maachi A, Rivarez MPS, Rollin J, Salavert F, et al. Managing the deluge of newly discovered plant viruses and viroids: an optimized scientific and regulatory framework for their characterization and risk analysis. Front Microbiol. (2023) 14:1181562. doi: 10.3389/fmicb.2023.1181562
107. Nakagawa S, Sakaguchi S, Ogura A, Mineta K, Endo T, Suzuki Y, et al. Current trends in RNA virus detection through metatranscriptome sequencing data. FEBS Open Bio. (2023) 13:992–1000. doi: 10.1002/2211-5463.13626
108. Petrone ME, Parry R, Mifsud JCO, Van Brussel K, Vorhees IEH, Richards ZT, et al. Evidence for an ancient aquatic origin of the RNA viral order Articulavirales. Proc Natl Acad Sci United States America. (2023) 120:e2310529120. doi: 10.1073/pnas.2310529120
109. Roca-Martinez J, Lazar T, Gavalda-Garcia J, Bickel D, Pancsa R, Dixit B, et al. Challenges in describing the conformation and dynamics of proteins with ambiguous behavior. . Front Mol Biosci. (2022) 9:959956. doi: 10.3389/fmolb.2022.959956
110. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. (1997) 25:3389–402. doi: 10.1093/nar/25.17.3389
111. Altschul SF, Bundschuh R, Olsen R, and Hwa T. The estimation of statistical parameters for local alignment score distributions. Nucleic Acids Res. (2001) 29:351–61. doi: 10.1093/nar/29.2.351
112. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. (2010) 26:2460–1. doi: 10.1093/bioinformatics/btq461
113. Krogh A, Brown M, Mian IS, Sjölander K, and Haussler D. Hidden Markov models in computational biology: Applications to protein modeling. J Mol Biol. (1994) 235:1501–31. doi: 10.1006/jmbi.1994.1104
114. Skewes-Cox P, Sharpton TJ, Pollard KS, and DeRisi JL. Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PloS One. (2014) 9:e105067. doi: 10.1371/journal.pone.0105067
115. Oliveira LS and Gruber A. Rational design of profile hidden markov models for viral classification and discovery. In: Helder N, editor. Bioinformatics. Brisbane (AU): Exon Publications (2021). doi: 10.36255/exonpublications.bioinformatics.2021.ch9
116. Tan YB, Lello LS, Liu X, Law YS, Kang C, Lescar J, et al. Crystal structures of alphavirus nonstructural protein 4 (nsP4) reveal an intrinsically dynamic RNA-dependent RNA polymerase fold. Nucleic Acids Res. (2022) 50:1000–16. doi: 10.1093/nar/gkab1302
117. Mao AH, Lyle N, and Pappu RV. Describing sequence–ensemble relationships for intrinsically disordered proteins. Biochem J. (2013) 449:307–18. doi: 10.1042/BJ20121346
118. Das RK, Ruff KM, and Pappu RV. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr Opin Struct Biol. (2015) 32:102–12. doi: 10.1016/j.sbi.2015.03.008
119. Zarin T, Strome B, Nguyen Ba AN, Alberti S, Forman-Kay JD, and Moses AM. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. Elife. (2019) 8:e46883. doi: 10.7554/eLife.46883
120. Davey NE. The functional importance of structure in unstructured protein regions. Curr Opin Struct Biol. (2019) 56:155–63. doi: 10.1016/j.sbi.2019.03.009
121. Gavalda-Garcia J, Bickel D, Roca-Martinez J, Raimondi D, Orlando G, and Vranken W. Data-driven probabilistic definition of the low energy conformational states of protein residues. NAR Genomics Bioinf. (2024) 6:lqae082. doi: 10.1093/nargab/lqae082
122. Mao AH, Crick SL, Vitalis A, Chicoine CL, and Pappu RV. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc Natl Acad Sci. (2010) 107:8183–8. doi: 10.1073/pnas.0911107107
123. Lyle N, Das RK, and Pappu RV. A quantitative measure for protein conformational heterogeneity. J Chem Phys. (2013) 139(12):121907. doi: 10.1063/1.4812791
124. Cohan MC, Shinn MK, Lalmansingh JM, and Pappu RV. Uncovering Non-random Binary Patterns Within Sequences of Intrinsically Disordered Proteins. J Mol Biol. (2022) 434:167373. doi: 10.1016/j.jmb.2021.167373
125. Song J, Li J, and Chan HS. Small-angle X-ray scattering signatures of conformational heterogeneity and homogeneity of disordered protein ensembles. J Phys Chem B. (2021) 125:6451–78. doi: 10.1021/acs.jpcb.1c02453
126. Xue B, Blocquel D, Habchi J, Uversky AV, Kurgan L, Uversky VN, et al. Structural disorder in viral proteins. Chem Rev. (2014) 114:6880–911. doi: 10.1021/cr4005692
127. Fuxreiter M, Simon I, Friedrich P, and Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol. (2004) 338:1015–26. doi: 10.1016/j.jmb.2004.03.017
128. Gunasekaran K and Nussinov R. How different are structurally flexible and rigid binding sites? Sequence and structural features discriminating proteins that do and do not undergo conformational change upon ligand binding. . J Mol Biol. (2007) 365:257–73. doi: 10.1016/j.jmb.2006.09.062
129. Lindorff-Larsen K and Kragelund BB. On the potential of machine learning to examine the relationship between sequence, structure, dynamics and function of intrinsically disordered proteins. J Mol Biol. (2021) 433:167196. doi: 10.1016/j.jmb.2021.167196
130. Marsh JA and Forman-Kay JD. Sequence determinants of compaction in intrinsically disordered proteins. Biophys J. (2010) 98:2383–90. doi: 10.1016/j.bpj.2010.02.006
131. Bowman MA, Riback JA, Rodriguez A, Guo H, Li J, Sosnick TR, et al. Properties of protein unfolded states suggest broad selection for expanded conformational ensembles. Proc Natl Acad Sci. (2020) 117:23356–64. doi: 10.1073/pnas.2003773117
132. Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science. (2020) 367:694–9. doi: 10.1126/science.aaw8653
133. Hadarovich A, Chakravarty D, Tuzikov AV, Ben-Tal N, Kundrotas PJ, and Vakser IA. Structural motifs in protein cores and at protein–protein interfaces are different. Protein Sci. (2021) 30:381–90. doi: 10.1002/pro.3996
134. Mittag T and Forman-Kay JD. Atomic-level characterization of disordered protein ensembles. Curr Opin Struct Biol. (2007) 17:3–14. doi: 10.1016/j.sbi.2007.01.009
135. Tzavella K, Diaz A, Olsen C, and Vranken WF. Combining evolution and protein language models for an interpretab le cancer driver mutation prediction with D2Deep. bioRxiv. (2023) 26(1):bbae664. doi: 10.1093/bib/bbae664
136. Peng Z, Oldfield CJ, Xue B, Mizianty MJ, Dunker AK, Kurgan L, et al. A creature with a hundred waggly tails: intrinsically disordered proteins in the ribosome. Cell Mol Life sciences: CMLS. (2014) 71:1477–504. doi: 10.1007/s00018-013-1446-6
137. Katuwawala A, Oldfield CJ, and Kurgan L. Accuracy of protein-level disorder predictions. Briefings Bioinf. (2020) 21:1509–22. doi: 10.1093/bib/bbz100
138. Hatos A, Hajdu-Soltész B, Monzon AM, Palopoli N, Álvarez L, Aykac-Fas B, et al. DisProt: intrinsic protein disorder annotation in 2020. Nucleic Acids. (2020) 48:D269–76. doi: 10.1093/nar/gkz975
139. Dayhoff GW 2nd and Uversky VN. Rapid prediction and analysis of protein intrinsic disorder. Protein science: Publ Protein Soc. (2022) 31:e4496. doi: 10.1002/pro.4496
140. Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature. (2018) 555:61–6. doi: 10.1038/nature25762
141. Moses D, Yu F, Ginell GM, Shamoon NM, Koenig PS, Holehouse AS, et al. Revealing the hidden sensitivity of intrinsically disordered proteins to their chemical environment. J Phys Chem Lett. (2020) 11:10131–6. doi: 10.1021/acs.jpclett.0c02822
142. Chu X, Sun T, Li Q, Xu Y, Zhang Z, Lai L, et al. Prediction of liquid-liquid phase separating proteins using machine learning. BMC Bioinf. (2022) 23:72. doi: 10.1186/s12859-022-04599-w
143. Hardenberg M, Horvath A, Ambrus V, Fuxreiter M, and Vendruscolo M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc Natl Acad Sci United States America. (2020) 117:33254–62. doi: 10.1073/pnas.2007670117
144. Hu W, Kan ZY, Mayne L, and Englander SW. Cytochrome c folds through foldon-dependent native-like intermediates in an ordered pathway. Proc Natl Acad Sci. (2016) 113:3809–14. doi: 10.1073/pnas.1522674113
145. Černý J, Černá Bolfíková B, Valdés JJ, Grubhoffer L, and Růžek D. Evolution of tertiary structure of viral RNA dependent polymerases. PloS One. (2014) 9:e96070. doi: 10.1371/journal.pone.0096070
146. Pancsa R, Raimondi D, Cilia E, and Vranken WF. Early folding events, local interactions, and conservation of protein backbone rigidity. Biophys J. (2016) 110:572–83. doi: 10.1016/j.bpj.2015.12.028
147. Stevens AO and He Y. Benchmarking the accuracy of AlphaFold 2 in loop structure prediction. Biomolecules. (2022) 12:985. doi: 10.3390/biom12070985
148. Tompa P. Multisteric regulation by structural disorder in modular signaling proteins: an extension of the concept of allostery. Chem Rev. (2014) 114:6715–32. doi: 10.1021/cr4005082
149. Tompa P. The principle of conformational signaling. Chem Soc Rev. (2016) 45:4252–84. doi: 10.1039/c6cs00011h
150. Uversky VN. Protein intrinsic disorder and structure-function continuum. Prog Mol Biol Trans Sci. (2019) 166:1–17. doi: 10.1016/bs.pmbts.2019.05.003
151. Nussinov R. Introduction to protein ensembles and allostery. Chem Rev. (2016) 116:6263–6. doi: 10.1021/acs.chemrev.6b00283
152. Del Conte A, Mehdiabadi M, Bouhraoua A, Miguel Monzon A, Tosatto SCE, and Piovesan D. Critical assessment of protein intrinsic disorder prediction (CAID) - Results of round 2. Proteins. (2023) 91:1925–34. doi: 10.1002/prot.26582
153. Britt HM, Cragnolini T, and Thalassinos K. Integration of mass spectrometry data for structural biology. Chem Rev. (2021) 122:7952–86. doi: 10.1021/acs.chemrev.1c00356
154. Cozzeno D and Jones DT. The contribution of intrinsic disorder prediction to the elucidation of protein function. Curr Opin Struct Biol. (2013) 23:467–72. doi: 10.1016/j.sbi.2013.02.001
155. Aspromonte MC, Nugnes MV, Quaglia F, Bouharoua A, Consortium D, Tosatto SCE, et al. DisProt in 2024: improving function annotation of intrinsically disordered proteins. Nucleic Acids Res. (2024) 52:D434–41. doi: 10.1093/nar/gkad928
156. Quaglia F, Chasapi A, Nugnes MV, Aspromonte MC, Leonardi E, Piovesan D, et al. Best practices for the manual curation of intrinsically disordered proteins in DisProt. Database: J Biol Database Curation. (2024), 2024:baae009. doi: 10.1093/database/baae009
157. Service RF. ‘The game has changed.’ AI triumphs at protein folding. Science (USA, New York, N.Y.) (2020). doi: 10.1126/science.370.6521.1144.
158. Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, et al. Improved protein structure prediction using potentials from deep learning. Nature. (2020) 577:706–10. doi: 10.1038/s41586-019-1923-7
159. Pearce R and Zhang Y. Deep learning techniques have significantly impacted protein structure prediction and protein design. Curr Opin Struct Biol. (2021) 68:194–207. doi: 10.1016/j.sbi.2021.01.007
160. Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, et al. Highly accurate protein structure prediction for the human proteome. Nature. (2021) 596:590–6. doi: 10.1038/s41586-021-03828-1
161. Vaswani A, Shazeer N, Parmar N, Uszkoreit J, Jones L, Gomez AN, et al. Attention is all you need. In: Advances in neural information processing systems Cornell University,. (2017). p. 30. Available online at: https://papers.nips.cc/paper_files/paper/2017/hash/3f5ee243547dee91fbd053c1c4a845aa-Abstract.html.
162. Fersht AR. AlphaFold - A Personal Perspective on the Impact of Machine Learning. J Mol Biol. (2021) 433:167088. doi: 10.1016/j.jmb.2021.167088
163. AlQuraishi M. Protein-structure prediction revolutionized. Nature. (2021) 596 :487–8. doi: 10.1038/d41586-021-02265-4
164. AlQuraishi M. Machine learning in protein structure prediction. Curr Opin Chem Biol. (2021) 65:1–8. doi: 10.1016/j.cbpa.2021.04.005
165. Alley EC, Khimulya G, Biswas S, AlQuraishi M, and Church GM. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods. (2019) 16:1315–22. doi: 10.1038/s41592-019-0598-1
166. Chowdhury R, Bouatta N, Biswas S, Floristean C, Kharkar A, Roy K, et al. Single-sequence protein structure prediction using a language model and deep learning. Nat Biotechnol. (2022) 40:1617–23. doi: 10.1038/s41587-022-01432-w
167. Ruff KM and Pappu RV. AlphaFold and implications for intrinsically disordered proteins. J Mol Biol. (2021) 433:167208. doi: 10.1016/j.jmb.2021.167208
168. Buel GR and Walters KJ. Can AlphaFold2 predict the impact of missense mutations on structure? Nat Struct Mol Biol. (2022) 29:1–2. doi: 10.1038/s41594-021-00714-2
169. Bouatta N and AlQuraishi M. Structural biology at the scale of proteomes. Nat Struct Mol Biol. (2023) 30:129–30. doi: 10.1038/s41594-023-00924-w
170. Clark JJ, Benson ML, Smith RD, and Carlson HA. Inherent versus induced protein flexibility: comparisons within and between apo and holo structures. PloS Comput Biol. (2019) 15:e1006705. doi: 10.1371/journal.pcbi.1006705
171. Varadi M, Bertoni D, Magana P, Paramval U, Pidruchna I, Radhakrishnan M, et al. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. (2024) 52:D368–75. doi: 10.1093/nar/gkad1011
172. Perrakis A and Sixma TK. AI revolutions in biology: The joys and perils of AlphaFold. . EMBO Rep. (2021) 22:e54046. doi: 10.15252/embr.202154046
173. Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. (2023) 32:e4792. doi: 10.1002/pro.4792
174. Norn C, Wicky BI, Juergens D, Liu S, Kim D, Tischer D, et al. Protein sequence design by conformational landscape optimization. Proc Natl Acad Sci. (2021) 118:e2017228118. doi: 10.1073/pnas.2017228118
175. Strodel B. Energy landscapes of protein aggregation and conformation switching in intrinsically disordered proteins. J Mol Biol. (2021) 433:167182. doi: 10.1016/j.jmb.2021.167182
176. Koren G, Meir S, Holschuh L, Mertens HD, Ehm T, Yahalom N, et al. Intramolecular structural heterogeneity altered by long-range contacts in an intrinsically disordered protein. Proc Natl Acad Sci. (2023) 120:e2220180120. doi: 10.1073/pnas.2220180120
177. Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. (2021) 373:871–6. doi: 10.1126/science.abj8754
178. Ahdritz G, Bouatta N, Floristean C, Kadyan S, Xia Q, Gerecke W, et al. OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nat Methods. (2024) 21(8):1514–24. doi: 10.1038/s41592-024-02272-z
179. Oda T. Improving protein structure prediction with extended sequence similarity searches and deep-learning based refinement in CASP15. Proteins. (2023) 91:1712–23. doi: 10.1002/prot.26551
180. Zheng W, Wuyun Q, Freddolino L, and Zhang Y. Integrating deep learning, threading alignments, and a multi-MSA strategy for high-quality protein monomer and complex structure prediction in CASP15. Proteins. (2023) 91:1684–703. doi: 10.1002/prot.26585
181. Lee JW, Won JH, Jeon S, Choo Y, Yeon Y, Oh JS, et al. DeepFold: enhancing protein structure prediction through optimized loss functions, improved template features, and re-optimized energy function. Bioinformatics. (2023) 39:btad712. doi: 10.1093/bioinformatics/btad712
182. Michaud JM, Madani A, and Fraser JS. A language model beats alphafold2 on orphans. Nat Biotechnol. (2022) 40:1576–7. doi: 10.1038/s41587-022-01466-0
183. Bertoline LMF, Lima AN, Krieger JE, and Teixeira SK. Before and after AlphaFold2: An overview of protein structure prediction. Front Bioinf. (2023) 3:1120370. doi: 10.3389/fbinf.2023.1120370
184. Van Der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. (2014) 114:6589–631. doi: 10.1021/cr400525m
185. Milles S, Jensen MR, Lazert C, Guseva S, Ivashchenko S, Communie G, et al. An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci Adv. (2018) 4:eaat7778. doi: 10.1126/sciadv.aat7778
186. Wu J, Wang X, Liu Q, Lu G, and Gong P. Structural basis of transition from initiation to elongation in de novo viral RNA-dependent RNA polymerases. Proc Natl Acad Sci United States America. (2023) 120:e2211425120. doi: 10.1073/pnas.2211425120
187. Ben-Tal N and Kolodny R. Homologues not needed: Structure prediction from a protein language model. Structure. (2022) 30:1047–9. doi: 10.1016/j.str.2022.07.002
188. Edgar RC. Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat Commun. (2022) 13:6968. doi: 10.1038/s41467-022-34630-w
189. Bordin N, Sillitoe I, Nallapareddy V, Rauer C, Lam SD, Waman VP, et al. AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. Commun Biol. (2023) 6:160. doi: 10.1038/s42003-023-04488-9
190. van Kempen M, Kim SS, Tumescheit C, Mirdita M, Lee J, Gilchrist CLM, et al. Fast and accurate protein structure search with Foldseek. Nat Biotechnol. (2024) 42:243–6. doi: 10.1038/s41587-023-01773-0
191. Ben-Tal N, Kihara D, and Pandurangan AP. Computational approaches to study the impact of mutations on disease and drug resistance. Front Mol Biosci. (2021) 8:813552. doi: 10.3389/fmolb.2021.813552
192. Fernandez-Leiro R, Bhairosing-Kok D, Kunetsky V, Laffeber C, Winterwerp HH, Groothuizen F, et al. The selection process of licensing a DNA mismatch for repair. Nat Struct Mol Biol. (2021) 28:373–81. doi: 10.1038/s41594-021-00577-7
193. Casadevall G, Duran C, and Osuna S. AlphaFold2 and deep learning for elucidating enzyme conformational flexibility and its application for design. JACS Au. (2023) 3:1554–62. doi: 10.1021/jacsau.3c00188
194. Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. (2020) 17:261–72. doi: 10.1038/s41592-019-0686-2
195. Deryusheva E, Nemashkalova E, Galloux M, Richard CA, Eléouët JF, Kovacs D, et al. Does intrinsic disorder in proteins favor their interaction with lipids? Proteomics. (2019) 19:1800098. doi: 10.1002/pmic.201800098
196. Zarin T, Strome B, Peng G, Pritišanac I, Forman-Kay JD, and Moses AM. Identifying molecular features that are associated with biological function of intrinsically disordered protein regions. Elife. (2021) 10:e60220. doi: 10.7554/eLife.60220
197. Huihui J and Ghosh K. Intrachain interaction topology can identify functionally similar intrinsically disordered proteins. Biophys J. (2021) 120:1860–8. doi: 10.1016/j.bpj.2020.11.2282
198. Mittal A, Holehouse AS, Cohan MC, and Pappu RV. Sequence-to-conformation relationships of disordered regions tethered to folded domains of proteins. J Mol Biol. (2018) 430:2403–21. doi: 10.1016/j.jmb.2018.05.012
199. Zea JD, Miguel Monzon A, Fornasari MS, Marino-Buslje C, and Parisi G. Protein conformational diversity correlates with evolutionary rate. Mol Biol Evol. (2013) 30:1500–3. doi: 10.1093/molbev/mst065
200. Zea DJ, Monzon AM, Parisi G, and Marino-Buslje C. How is structural divergence related to evolutionary information? Mol Phylogenet Evol. (2018) 127:859–66. doi: 10.1016/j.ympev.06.033
201. Ghafouri H, Lazar T, Del Conte A, Tenorio Ku LG, Tompa P, Tosatto SC, et al. PED in 2024: improving the community deposition of structural ensembles for intrinsically disordered proteins. Nucleic Acids Res. (2024) 52:D536–44. doi: 10.1093/nar/gkad947
202. Lazar T, Martínez-Pérez E, Quaglia F, Hatos A, Chemes LB, Iserte JA, et al. PED in 2021: a major update of the protein ensemble database for intrinsically disordered proteins. Nucleic Acids Res. (2021) 49:D404–11. doi: 10.1093/nar/gkaa1021
203. Smith EC, Blanc H, Surdel MC, Vignuzzi M, and Denison MR. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PloS Pathog. (2013) 9:e1003565. doi: 10.1371/journal.ppat.1003565
204. Gitlin L, Hagai T, LaBarbera A, Solovey M, and Andino R. Rapid evolution of virus sequences in intrinsically disordered protein regions. PloS Pathog. (2014) 10:e1004529. doi: 10.1371/journal.ppat.1004529
205. Wei G, Xi W, Nussinov R, and Ma B. Protein ensembles: how does nature harness thermodynamic fluctuations for life The diverse functional roles of conformational ensembles in the cell. Chem Rev. (2016) 116:6516–51. doi: 10.1021/acs.chemrev.5b00562
206. Gerez JA, Prymaczok NC, and Riek R. In-cell NMR of intrinsically disordered proteins in mammalian cells. Intrinsically Disordered Proteins: Methods Protoc. (2020) 2141:873–93. doi: 10.1007/978-1-0716-0524-0_45
207. Bonucci A, Palomino-Schätzlein M, Malo de Molina P, Arbe A, Pierattelli R, Rizzuti B, et al. Crowding effects on the structure and dynamics of the intrinsically disordered nuclear chromatin protein NUPR1. Front Mol Biosci. (2021) 8:684622. doi: 10.3389/fmolb.2021.684622
208. Kinch LN, Pei J, Kryshtafovych A, Schaeffer RD, and Grishin NV. Topology evaluation of models for difficult targets in the 14th round of the critical assessment of protein structure prediction (CASP14). Proteins: Structure Function Bioinf. (2021) 89:1673–86. doi: 10.1002/prot.26172
209. Marino-Buslje C, Monzon AM, Zea DJ, Fornasari MS, and Parisi G. On the dynamical incompleteness of the Protein Data Bank. Briefings Bioinf. (2019) 20:356–9. doi: 10.1093/bib/bbx084
210. Akdel M, Pires DE, Pardo EP, Jänes J, Zalevsky AO, Mészáros B, et al. A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol. (2022) 29:1056–67. doi: 10.1038/s41594-022-00849-w
211. DeForte S and Uversky VN. Resolving the ambiguity: Making sense of intrinsic disorder when PDB structures disagree. Protein Sci. (2016) 25:676–88. doi: 10.1002/pro.2864
212. Daggett V and Fersht AR. Is there a unifying mechanism for protein folding? Trends Biochem Sci. (2003) 28:18–25. doi: 10.1016/s0968-0004(02)00012-9
213. Karplus M and Kuriyan J. Molecular dynamics and protein function. Proc Natl Acad Sci. (2005) 102:6679–85. doi: 10.1073/pnas.0408930102
214. Glazer DS, Radmer RJ, and Altman RB. Improving structure-based function prediction using molecular dynamics. Structure. (2009) 17:919–29. doi: 10.1016/j.str.2009.05.010
215. Huang YJ, Zhang N, Bersch B, Fidelis K, Inouye M, Ishida Y, et al. Assessment of prediction methods for protein structures determined by NMR in CASP14: Impact of AlphaFold2. Proteins: Structure Function Bioinf. (2021) 89:1959–76. doi: 10.1002/prot.26246
216. Saldaño T, Escobedo N, Marchetti J, Zea DJ, Mac Donagh J, Velez Rueda AJ, et al. Impact of protein conformational diversity on AlphaFold predictions. Bioinformatics. (2022) 38:2742–8. doi: 10.1093/bioinformatics/btac202
217. Chakravarty D and Porter LL. AlphaFold2 fails to predict protein fold switching. Protein Sci. (2022) 1:e4353. doi: 10.1002/pro.4353
218. Outeiral C, Nissley DA, and Deane CM. Current structure predictors are not learning the physics of protein folding. Bioinformatics. (2022) 38:1881–7. doi: 10.1093/bioinformatics/btab881
219. Liu S, Wu K, and Chen C. Obtaining protein foldability information from computational models of AlphaFold2 and RoseTTAFold. Comput Struct Biotechnol J. (2022) 20:4481–9. doi: 10.1016/j.csbj.2022.08.034
220. Wayment-Steele HK, Ojoawo A, Otten R, Apitz JM, Pitsawong W, Hömberger M, et al. Predicting multiple conformations via sequence clustering and AlphaFold2. Nature. (2024) 625:832–9. doi: 10.1038/s41586-023-06832-9
221. Alderson TR, Pritišanac I, Kolarić Đ., Moses AM, and Forman-Kay JD. Systematic identification of conditionally folded intrinsically disordered regions by AlphaFold2. Proc Natl Acad Sci. (2023) 120:e2304302120. doi: 10.1101/2022.02.18.481080
222. Hopf TA, Ingraham JB, Poelwijk FJ, Schärfe CP, Springer M, Sander C, et al. Mutation effects predicted from sequence co-variation. Nat Biotechnol. (2017) 35:128–35. doi: 10.1038/nbt.3769
223. Bugge K, Brakti I, Fernandes CB, Dreier JE, Lundsgaard JE, Olsen JG, et al. Interactions by disorder–a matter of context. Front Mol Biosci. (2020) 7:110. doi: 10.3389/fmolb.2020.00110
224. Thomasen FE and Lindorff-Larsen K. Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins. Biochem Soc Trans. (2022) 50:541–54. doi: 10.1042/BST20210499
225. Vu VL, Gevaert K, and De Smet I. Protein language: post-translational modifications talking to each other. Trends Plant Sci. (2018) 23:1068–80. doi: 10.1016/j.tplants.2018.09.004
226. Dixit B, Vranken W, and Ghysels A. Conformational dynamics of α-1 acid glycoprotein (AGP) in cancer: A comparative study of glycosylated and unglycosylated AGP. Proteins: Structure Function Bioinf. (2024) 92:246–64. doi: 10.1002/prot.26607
227. Monzon AM, Zea DJ, Fornasari MS, Saldaño TE, Fernandez-Alberti S, Tosatto SC, et al. Conformational diversity analysis reveals three functional mechanisms in proteins. PloS Comput Biol. (2017) 13(11):e1005398. doi: 10.1371/journal.pcbi.1005398
228. Ruff KM. Predicting conformational properties of intrinsically disordered proteins from sequence. Intrinsically Disordered Proteins: Methods Protoc. (2020) 2141(2020):347–89. doi: 10.1007/978-1-0716-0524-0_18
229. Wilson CJ, Choy WY, and Karttunen M. AlphaFold2: a role for disordered protein/region prediction? Int J Mol Sci. (2022) 23:4591. doi: 10.3390/ijms23094591
230. Das RK and Pappu RV. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. PNAS. (2013) 110:13392–7. doi: 10.1073/pnas.1304749110
231. Martin EW, Holehouse AS, Grace CR, Hughes A, Pappu RV, and Mittag T. Sequence determinants of the conformational properties of an intrinsically disordered protein prior to and upon multisite phosphorylation. J Am Chem Soc. (2016) 138:15323–35. doi: 10.1021/jacs.6b10272
232. Sørensen CS and Kjaergaard M. Effective concentrations enforced by intrinsically disordered linkers are governed by polymer physics. Proc Natl Acad Sci. (2019) 116:23124–31. doi: 10.1073/pnas.1904813116
233. Zaharias S, Zhang Z, Davis K, Fargason T, Cashman D, Yu T, et al. Intrinsically disordered electronegative clusters improve stability and binding specificity of RNA-binding proteins. J Biol Chem. (2021) 297:100945. doi: 10.1016/j.jbc.2021.100945
234. Urakova N, Strive T, and Frese M. RNA-dependent RNA polymerases of both virulent and benign rabbit caliciviruses induce striking rearrangement of golgi membranes. PloS One. (2017) 12:e0169913. doi: 10.1371/journal.pone.0169913
235. Smertina E, Urakova N, Strive T, and Frese M. Calicivirus RNA-dependent RNA polymerases: evolution, structure, protein dynamics, and function. Front Microbiol. (2019) 10:1280. doi: 10.3389/fmicb.2019.01280
236. Bitard-Feildel T, Lamiable A, Mornon JP, and Callebaut I. Order in disorder as observed by the “hydrophobic cluster analysis” of protein sequences. Proteomics. (2018) 18:1800054. doi: 10.1002/pmic.201800054
237. Lamiable A, Bitard-Feildel T, Rebehmed J, Quintus F, Schoentgen F, Mornon JP, et al. A topology-based investigation of protein interaction sites using hydrophobic cluster analysis. . Biochimie. (2019) 167:68–80. doi: 10.1016/j.biochi.2019.09.009
238. Bruley A, Mornon JP, Duprat E, and Callebaut I. Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules. (2022) 12:1467. doi: 10.3390/biom12101467
239. Perdigão N, Heinrich J, Stolte C, Sabir KS, Buckley MJ, Tabor B, et al. Unexpected features of the dark proteome. Proc Natl Acad Sci. (2015) 112:15898–903. doi: 10.1073/pnas.1508380112
240. Porta-Pardo E, Ruiz-Serra V, Valentini S, and Valencia A. The structural coverage of the human proteome before and after AlphaFold. PloS Comput Biol. (2022) 18:e1009818. doi: 10.1371/journal.pcbi.1009818
241. Beveridge R, Migas LG, Das RK, Pappu RV, Kriwacki RW, and Barran PE. Ion mobility mass spectrometry uncovers the impact of the patterning of oppositely charged residues on the conformational distributions of intrinsically disordered proteins. J Am Chem Soc. (2019) 141:4908–18. doi: 10.1021/jacs.8b13483
242. Zheng W, Dignon G, Brown M, Kim YC, and Mittal J. Hydropathy patterning complements charge patterning to describe conformational preferences of disordered proteins. J Phys Chem Lett. (2020) 11:3408–15. doi: 10.1021/acs.jpclett.0c00288
243. Zeng X, Ruff KM, and Pappu RV. Competing interactions give rise to two-state behavior and switch-like transitions in charge-rich intrinsically disordered proteins. . Proc Natl Acad Sci. (2022) , 119:e2200559119. doi: 10.1073/pnas.2200559119
244. Holehouse AS, Garai K, Lyle N, Vitalis A, and Pappu RV. Quantitative assessments of the distinct contributions of polypeptide backbone amides versus side chain groups to chain expansion via chemical denaturation. J Am Chem Soc. (2015) 137:2984–95. doi: 10.1021/ja512062h
245. Jephthah S, Pesce F, Lindorff-Larsen K, and Skepo M. Force field effects in simulations of flexible peptides with varying polyproline II propensity. J Chem Theory Comput. (2021) 17:6634–46. doi: 10.1021/acs.jctc.1c00408
246. Bakker MJ, Sørensen HV, and Skepo M. Exploring the role of globular domain locations on an intrinsically disordered region of p53: A molecular dynamics investigation. J Chem Theory Comput. (2024) 20:1423–33. doi: 10.1021/acs.jctc.3c00971
247. Eden JS, Sharpe LJ, White PA, and Brown AJ. Norovirus RNA-dependent RNA polymerase is phosphorylated by an important survival kinase, Akt. J Virol. (2011) 85:10894–8. doi: 10.1128/JVI.05562-11
248. Hu J, Zhang L, and Liu X. Role of post-translational modifications in influenza A virus life cycle and host innate immune response. Front Microbiol. (2020) 11:517461. doi: 10.3389/fmicb.2020.517461
249. Liu L, Madhugiri R, Saul VV, Bacher S, Kracht M, Pleschka S, et al. Phosphorylation of the PA subunit of influenza polymerase at Y393 prevents binding of the 5’-termini of RNA and polymerase function. Sci Rep. (2023) 13:7042. doi: 10.1038/s41598-023-34285-7
250. Narunsky A, Kessel A, Solan R, Alva V, Kolodny R, and Ben-Tal N. On the evolution of protein–adenine binding. Proc Natl Acad Sci. (2020) 117:4701–9. doi: 10.1073/pnas.1911349117
251. Ramasamy P, Turan D, Tichshenko N, Hulstaert N, Vandermarliere E, Vranken W, et al. Scop3P: a comprehensive resource of human phosphosites within their full context. J Proteome Res. (2020) 19:3478–86. doi: 10.1021/acs.jproteome.0c00306
252. Bludau I, Willems S, Zeng WF, Strauss MT, Hansen FM, Tanzer MC, et al. The structural context of posttranslational modifications at a proteome-wide scale. PloS Biol. (2022) 20:e3001636. doi: 10.1371/journal.pbio.3001636
253. Nagar N, Tubiana J, Loewenthal G, Wolfson HJ, Tal NB, and Pupko T. EvoRator2: predicting site-specific amino acid substitutions based on protein structural information using deep learning. J Mol Biol. (2023) 435:168155. doi: 10.1016/j.jmb.2023.168155
254. Ragonis-Bachar P, Axel G, Blau S, Ben-Tal N, Kolodny R, and Landau M. What can AlphaFold do for antimicrobial amyloids? Proteins: Structure Function Bioinf. (2024) 92:265–81. doi: 10.1002/prot.26618
Keywords: RNA-dependent RNA polymerase, RNA virus discovery, conformational ensembles, intrinsic disorder (IDP/IDR), structural biology, AlphaFold2 (AF2), folding dynamics, modulome and artificial intelligence
Citation: Tahzima R, Charon J, Diaz A, De Jonghe K, Massart S, Michon T and Vranken W (2025) Viral replication modulated by hallmark conformational ensembles: how AlphaFold-predicted features of RdRp folding dynamics combined with intrinsic disorder-mediated function enable RNA virus discovery. Front. Virol. 5:1501616. doi: 10.3389/fviro.2025.1501616
Received: 25 September 2024; Accepted: 01 July 2025;
Published: 22 August 2025.
Edited by:
So Nakagawa, Tokai University, JapanReviewed by:
Marco V. José, National Autonomous University of Mexico, MexicoPascal Mutz, National Institutes of Health (NIH), United States
Copyright © 2025 Tahzima, Charon, Diaz, De Jonghe, Massart, Michon and Vranken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachid Tahzima, cmFjaGlkLnRhaHppbWFAdWxpZWdlLmJl; cmFjaGlkLnRhaHppbWFAdnViLmJl